
Abstract
The use of carbon nanoparticles is shown for the detection and identification of different Shiga toxin-producing Escherichia coli virulence factors (vt1, vt2, eae and ehxA) and a 16S control (specific for E. coli) based on the use of lateral flow strips (nucleic acid lateral flow immunoassay, NALFIA). Prior to the detection with NALFIA, a rapid amplification method with tagged primers was applied. In the evaluation of the optimised NALFIA strips, no cross-reactivity was found for any of the antibodies used. The limit of detection was higher than for quantitative PCR (q-PCR), in most cases between 104 and 105 colony forming units/mL or 0.1–0.9 ng/μL DNA. NALFIA strips were applied to 48 isolates from cattle faeces, and results were compared to those achieved by q-PCR. E. coli virulence factors identified by NALFIA were in very good agreement with those observed in q-PCR, showing in most cases sensitivity and specificity values of 1.0 and an almost perfect agreement between both methods (kappa coefficient larger than 0.9). The results demonstrate that the screening method developed is reliable, cost-effective and user-friendly, and that the procedure is fast as the total time required is <1 h, which includes amplification.

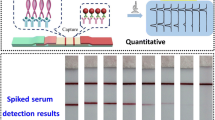
Results achieved with multi-analyte NALFIA for E. coli virulence factors. First strip: blank; second to sixth strip: each of the STEC factors; seventh strip: all factors
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Detection of pathogenic bacteria is key to the prevention and identification of problems related to health and safety. In spite of the real need for obtaining analytical results in the shortest time possible, traditional and standard bacterial detection methods, even if of sufficient sensitivity, are often expensive and too slow to be of any use; some may take up to 8 days to yield an answer [1, 2]. For this reason, over the recent years, a lot of effort has gone into the development of rapid biosensors of diverse nature. However, their performance is variable and still needs improvement [2]. Most of the reported methods for the detection of pathogenic bacteria are applied to Escherichia coli [1, 2].
There are many E. coli strains of which quite a number are part of the normal microbiota in the gut. Although most of them are harmless, some of them can cause severe illnesses; one of those is the Shiga toxin-producing E. coli (STEC, also called verotoxigenic E. coli or enterohemorrhagic E. coli) [3]. STEC has emerged as a food poisoning pathogen that can cause severe and potentially fatal illness like diarrhoea, hemorrhagic colitis or haemolytic uraemic syndrome. The most important reservoirs of STEC, and, consequently, causes of human infections, are domestic ruminants, especially cattle and sheep. Since STEC strains are found as part of the normal intestinal flora of these animals, transmission occurs through, e.g., consumption of undercooked meat, unpasteurised dairy products and vegetables or water contaminated by faeces of carriers [4, 5]. Even though many STEC serotypes have been associated with human illness, the vast majority of reported outbreaks and sporadic cases of STEC infection in humans have been associated with serotype O157:H7 [5]. Some of the STEC virulence factors are: cytotoxins (verotoxins) encoded by the genes vt1 or vt2; the outer membrane protein intimin encoded by the gene eae and involved in the attachment to intestine epithelial cells; and the enterohaemolysin encoded by the gene ehxA.
Monitoring and evaluation of E. coli virulence factors can be done by various methods that include laboratory culture, typing methods, serological methods, detection of DNA sequences and immunochemical methods [2, 5, 6]. The need for timely results has increased the interest in rapid screening methods. This interest is also growing due to the shift of pathogen testing from laboratories (governmental, reference, etc.) to on-site settings in, e.g. food-processing plants [1]. Most E. coli-specific methods have a detection limit between 103 and 105 cells/mL and some rely on the amplification of specific genes of the E. coli genome for specific identification [7, 8]. There are biosensors in the literature that can detect a lower amount of cells [2, 6], but in general, these systems have a moderate sensor-to-sensor reproducibility and poor batch-to-batch reproducibility [9], whilst others do not comply with the basic requirements for application in this field, i.e. a low price and ease of use [7]. Moreover, many methods do not distinguish between E. coli virulence factors/strains.
The use of nucleic acid lateral flow immunoassays (NALFIA) as biosensors for the detection of specific amplified nucleic acid genes is not new [10]. In NALFIA, nucleic acids can be captured on the lateral flow test strips either in an antibody-independent or antibody-dependent manner [11, 12]. In the first one, a hybridization step is required upon running the sample through the membrane [11–15]. The second and simpler approach uses immobilised antibodies to directly recognise specific tag-labelled amplicons [11, 12, 16, 17]. Some of the advantages of using lateral flow strips are: one-step assay, simple, fast, low cost, versatile and a prolonged shelf life (with no need for refrigeration) [11, 12, 18–20]. Moreover, compared with traditional electrophoresis detection, NALFIA has other advantages like shorter response time and no need for hazardous reagents [16, 17]. NALFIA strips comply with most of the requirements for biosensors for bacterial detection: low detection limit, species selectivity, strain selectivity, assay time for a single test between 5 and 10 min, no reagent addition needed (except buffer), direct measurement, highly automated format, no operator skills needed to carry out the assay, and also compact size, portable, handheld, and designed for field application [7]. In addition, they comply with two other characteristics: low price and easy manufacture. Another interesting feature is that they are disposable; hence, regeneration time is eliminated. Finally, if NALFIA strips are used with coloured nanoparticles, a visual detection can be easily performed; in this sense, this combination overcomes one of the major shortcomings in the application of immunosensor techniques, which is the cost of the required equipment [8].
The aim of this work was to present the use of carbon nanoparticles in NALFIAs. Carbon nanoparticles were chosen for their high signal-to-noise ratio (black to a white background) [17, 21] and their excellent sensitivity, i.e. low picomolar by visual inspection [18]. NALFIA was applied to the rapid detection and identification of genes encoding various STEC virulence factors (vt1, vt2, eae and ehxA) and of the gene coding for E. coli 16S rRNA as a control. Genes were amplified with specific tag-labelled primers, and amplicons were sandwiched between neutravidin coated onto carbon nanoparticles and specific anti-tag antibodies immobilised on the NALFIA nitrocellulose membrane. The limit of detection of the NALFIA strips was determined, and the strips were tested with DNA from 48 field strains originating from cattle faeces. Performance of the NALFIA strips was evaluated by comparing the results with those achieved by quantitative PCR (q-PCR).
Material and methods
Reagents
Borate buffer (BB) 100 mM, pH 8.8, was prepared by mixing 100 mM solutions of H3BO3 (Merck, Darmstadt, Germany) and Na2B4O7⋅10 H2O (Sigma-Aldrich Chemie BV, Zwijndrecht, the Netherlands). BB (5 mM) was obtained diluting 100 mM BB appropriately. With these two buffers, bovine serum albumin (BSA, Sigma-Aldrich Chemie BV) and Tween 20 (Merck, Amsterdam, the Netherlands), several buffers were prepared: washing buffer (WB, 5 mM BB, 1% (w/v) BSA), storage buffer (SB, 100 mM BB, 1% (w/v) BSA) and running buffer (RB, 100 mM BB, 1% (w/v) BSA, 0.05% (v/v) Tween 20). NaN3 was added to all buffers to a final concentration of 0.02% (w/v).
Polyclonal antibodies against digoxigenin (DIG, Roche, Penzberg, Germany), 2,4-dinitrophenol (DNP, Invitrogen Corporation, Camarillo, CA, USA), fluorescein isothiocyanate (FITC, AbD-Serotec, Oxford, UK) and Texas Red (TxR, Invitrogen Corporation) were used. A monoclonal antibody against Cy5 (Sigma-Aldrich Chemie BV) was used for the 16S primer anti-tag antibody. As control line, biotinylated anti-rabbit antibody (Pierce Biotechnology, Rockford, IL, USA) was used.
E. coli strains and DNA isolation
E. coli strains used as controls are listed in Table 1. These strains were selected for having different sets of genes coding for virulence factors. The strains were grown overnight aerobically at 37 °C on heart infusion agar (Difco Laboratories, Detroit, MI, USA) supplemented with 5% sheep blood (HIS plates). DNA was isolated by harvesting bacteria from a plate with one streak of an inoculation loop and subsequent resuspension in 300 μL 6% Chelex 100 (Bio-Rad Laboratories, Hercules, CA, USA). The suspension was heated at 56 °C for 15 min, then at 100 °C for 8 min, and centrifuged at 14,000×g for 5 min. Supernatants containing genomic DNA were transferred to a new vessel and stored at −20 °C until use.
To isolate DNA for quantitative purposes, EDL 933 was grown aerobically at 37 °C in 10 mL Bacto Heart Infusion Broth (Becton Dickinson, Sparks, MD, USA). A quantity of 1 mL containing 8.4 × 107 colony forming units (cfu) was taken from a culture in the exponential growth phase for DNA isolation using Chelex, resulting in a DNA preparation with 775 ng/μL.
Polymerase chain reaction
DNA was amplified prior to detection with NALFIA strips. Primers used in the amplification of the genes coding for four STEC virulence factors (vt1, vt2, eae and ehxA) and a 16S control specific for E. coli are indicated in Table 2. Forward primers were labelled with specific tags TxR (vt1), FITC (vt2), DIG (eae), DNP (ehxA) or Cy5 (16S), whereas all reverse primers were labelled with biotin (Eurogentec, Seraing, Belgium). To achieve an amplification reaction in the shortest time possible with economical equipment, PCR reactions were carried out with Phire Hot Start DNA Polymerase (Finnzymes) and a PIKO Thermal cycler (Finnzymes, Espoo, Finland) using extra thin vessels that allow an increase in heat transfer speed. Since annealing temperature may be affected by the use of tags coupled to primers [22], amplification reactions were optimised with the final aim of using identical conditions for all genes.
The protocol applied had a total length of 30 min and consisted of an initial denaturation at 98 °C (30 s), followed by 30 cycles with a denaturation step at 98 °C (5 s), an annealing step at 61 °C (5 s) and an extension step at 72 °C (5 s), with a final extension step at 72 °C (1 min) followed by a lowering of temperature to 4 °C. The reaction tube contained 2 μL template and 0.4 μL polymerase (as provided in the kit) in a total volume of 20 μL, with final concentrations of 1.5 mM MgCl2, 0.25 μM dNTPs and 0.9 μM of each primer (forward and reverse). Positive and negative controls were included in all runs. Amplified DNA was directly analysed with a NALFIA strip or stored at −20 °C until use.
Carbon conjugate
Carbon nanoparticles bind proteins non-covalently and, if optimal conditions have been applied, without changing their bioactivity [17, 21, 25, 26]. Therefore, carbon nanoparticles were labelled with neutravidin biotin-binding protein (Pierce Biotechnology) following O’Keefe et al. [27]. Briefly, the procedure to obtain the carbon conjugate was: 10 mg of carbon (Spezial Schwartz 4, Degussa AG, Frankfurt, Germany) was suspended in 1 mL of MilliQ water and sonicated for 5 min (Branson model 250 Sonifier, Danbury, CT, USA). The resulting 1% (w/v) carbon suspension was fivefold diluted in 5 mM BB and sonicated for 5 min. Next, 350 μg neutravidin was added to 1 mL of the diluted carbon suspension and stirred for 3 h at room temperature. This suspension was first centrifuged at 13,636×g for 15 min; the supernatant was removed and the pellet washed with WB to remove unbound protein. To achieve this, 1 mL of WB was added to the pellet and the mixture centrifuged at 13,636×g for 15 min. Subsequently, the supernatant was removed and the pellet was resuspended in 1 mL WB. This process was repeated twice. After the final wash, the pellet was resuspended in 1 mL SB and stored at 4 °C until use. The resulting homogeneous suspension contained 0.2% (w/v) carbon conjugate.
Nucleic acid lateral flow immunoassay
NALFIA strips were manufactured using nitrocellulose (NC) membranes cut to a width of 2.5 cm (HiFlow Plus HFB135, Millipore, Billerica, MA, USA). A Linomat IV TLC dispenser (Camag, Berlin, Germany) was used to dispense the specific antibody (test line) and the biotinylated antibody (control line) on the NC membrane at a dose of 1 μL/5 mm (diluted in 5 mM BB). Distance between lines was 5 mm. The NC membranes were dried overnight at 37 °C. Then, the NC membranes were fixed on a plastic backing along with a cellulose absorbent pad (Schleicher and Schuell, ’s-Hertogenbosch, the Netherlands). Finally, strips were cut at a width of 5 mm using a Bio-Dot Cutter CM4000 (Irvine, CA, USA), used immediately, or packaged in aluminium pouches (with a silica desiccation pad), sealed and stored at room temperature until use.
Following PCR, amplicons were detected by means of NALFIA: The double-tagged amplicons were sandwiched between neutravidin (bound to carbon nanoparticles) and an antibody against the discriminating tag (immobilised on the NC membrane, capture ligand) (see Electronic supplementary material (ESM) Fig. S1). To achieve this, NALFIA assays were performed in a low-binding 96-well microplate. Once reagents were introduced in a well and mixed thoroughly, a strip was dipped vertically and reagents moved upwards through the strip by capillary force. Strips used for qualitative results (presence/absence of virulence factors) were visually examined after 10 min; a positive result (+) was recorded when a line was distinguishable from the background, and a negative result (−) when no line was seen. For quantitative results, strips were dried at 37 °C, and the pixel grey intensity of the test lines was evaluated following conventional flatbed scanning [28].
Immunoassays need to be optimised for each antibody with respect to performance and for the set of antibodies used. To accomplish this, several experiments were performed to find the optimum amount of antibody sprayed on the NC membranes and the amount of PCR product to be used. For this, strips were manufactured with serial dilutions of each of the antibodies and tested with different amounts of PCR product. For one of the antibodies (specific to Cy5), the addition of 2% sucrose (Merck, Darmstadt, Germany) remarkably improved the line quality [29].
Cross-reactivities were studied by testing all amplified genes (using EDL 933 diluted 1/100 as template) on each of the specific NALFIA strips. With the aim of identifying several STEC genes simultaneously, a series of strips was prepared onto which all antibodies were printed. In this case, the width of the NC membrane used was 5 cm and the distance between lines 3 mm.
To evaluate the analytical performance of the NALFIA strips developed, DNA of E. coli strain EDL 933 (775 ng/μL, corresponding with 8.4 × 107 cfu/mL) was subjected to tenfold serial dilutions and divided into six aliquots. Three aliquots were used in the optimised PCR amplification procedure, and for each aliquot, two NALFIA strips were used. For comparison, the remaining three aliquots were analysed using the reference method, q-PCR. For each factor, dried strips were scanned, data (in pixel grey volumes) were transformed to percentage of the maximum, and the resulting percentages were adjusted to a four-parameter logistic curve (Sigmaplot 11, Systat Software, Inc., San Jose, CA, USA). The limit of detection (LOD) was calculated as the signal of the blank plus three times the standard deviation of the blank (SD). For simplicity of interpretation, all quantitative results will be given in colony forming units per millilitre.
Quantitative PCR
To identify whether an isolate is an STEC, q-PCR can be used to detect the various virulence factors and was used as the reference method in this work. Reaction mixtures for q-PCR were assembled in 20-μL quantities using the TaqMan Fast Universal PCR Master mix according to the manufacturer’s instructions (Applied Biosystems, Nieuwerkerk a⁄d IJssel, the Netherlands) with 2-μL template DNA. PCRs were performed using the same primers as used for PCR preceding NALFIA (see above; Table 2) and TaqMan probes labelled with the fluorescent dyes 6-FAM or VIC at the 5′ end and a quencher at the 3′ end, i.e. TAMRA for the vt1, vt2, eae and ehxA probes (Applied Biosystems) and BHQ for the 16S probe (Eurogentec, Table 2). The PCRs were performed on an Applied Biosystems 7500 Fast Real-Time PCR System in Fast 7500 run mode using the standard thermal cycler protocol. Positive and negative controls were included in all runs; the cutoff was set at a Ct of 32 based on results with negative controls. All PCRs were performed in duplex reactions (vt1/vt2, eae/ehxA) or in a single reaction (16S).
Field samples
To test field samples, DNA was obtained from 48 E. coli isolates originating from cattle faeces [30]. The isolated DNA was subjected (in duplicate) to the 30-min PCR amplification and subsequently to a NALFIA test strip. The result achieved for each single sample (i.e. the presence or absence of a virulence factor) was compared with the result achieved by q-PCR. When results were identical, it was considered as a true positive (or negative), and when they differed it was considered a false positive (or negative). With these values, several reliability parameters were calculated to evaluate the performance of the NALFIA strips [31–33]: sensitivity, specificity, positive (and negative) predictive values, and efficiency. At present, there is no agreement which of these previous parameters is the most useful one, and there are no guidelines to evaluate results achieved. For this reason, the kappa coefficient (k) was also calculated. This statistic coefficient takes into account the possibility of chance agreement, and Landis and Koch [34] provided a starting point to evaluate the strength of the agreement. The use of k does not exclude the use of other parameters since predictive values are interpretable as indices of test performance (how well a sample is classified), whereas k is interpretable as an index of test quality (how well a test makes use of evidence in classifying samples) [33].
The mathematical equations to calculate all these reliability parameters are given in ESM Table S1, and an Excel spreadsheet specifically developed for evaluating diagnostic tests was used [35].
Results and discussion
Nucleic acid lateral flow immunoassay
NALFIA strips were manufactured with serial dilutions of each of the antibodies and were used with different amounts of PCR product. Line intensity increased with increasing amounts of antibody and PCR product (until a maximum plateau value was reached). The selected final assay conditions were those that achieved good line intensity using the lowest amount of antibody (to make the strips as cost-effective as possible) and the lowest volume of PCR product. For convenience, the final PCR volume used was the same for all factors tested. Examples regarding the selection of optimal conditions are given in the ESM attached.
The selected optimal amount of antibody sprayed on the NC membrane was: 100 μg/mL for TxR (vt1), DIG (eae) and biotinylated IgG (control); 125 μg/mL for DNP (ehxA); 250 μg/mL for FITC (vt2); and 300 μg/mL (in 2% (w/v) sucrose) for Cy5 (16S). Strips were finally used in microplate wells that contained a mixture of 1 μL of PCR product (sample), 1 μL carbon–neutravidin conjugate and 98 μL RB.
To verify that there was no cross-reactivity, optimised strips for the identification of each of the genes were used with all other amplicons, and no cross-reactivity was found. NALFIA strips were also tested with different E. coli strains (Table 1) and a blank (water). Once samples had been randomly identified by a number, they were submitted to PCR and their products applied to the optimised NALFIA strips. All genes coding for the virulence factors were correctly identified, allowing the identification of the six E. coli strains used (see ESM Table S2). These results were very promising with respect to testing real field samples.
When NALFIA strips were used with different E. coli dilutions (submitted to PCR amplification), maximum intensity values was around 105 pixel grey volumes. When data were transformed to percentage of the maximum signal, differences in the performance of NALFIA strips having different antibodies were noticeable (Fig. 1). For all systems, the regression values for the adjustment of these data to a four-parameter logistic curve were larger than 0.97, and the LOD increased in the following order: vt2<ehxA<16S<eae<vt1 (Table 3), being around 105 cfu/mL (0.9 ng/μL), which would correspond to 200 cfu in the PCR tube. The lowest LOD was achieved for vt2 with a value of 6.7 104 cfu/mL (0.6 ng/μL). LODs of the NALFIA strips developed are similar to those achieved by other systems for the detection of E. coli [8], but in our study, virulence factors were specifically identified.

Sensitivity of the various NALFIAs for each of the E. coli virulence factors tested. Average ± SD (n = 6)
When comparing the LODs of NALFIA strips with those of q-PCR, the LOD achieved by q-PCR in most cases was (at least) two orders of magnitude lower than for NALFIA (Table 3). The sole exception is vt2; the LOD achieved in q-PCR (5.9 105 cfu/mL or 5.4 ng/μL) was higher than the one for NALFIA (6.7 104 cfu/mL or 0.6 ng/μL). As can be noted, the order of sensitivity achieved by q-PCR is almost contrary to the one achieved by NALFIA strips. These differences in LOD will explain the performance of the NALFIA strips with real samples (presence of false results; see “Field samples”).
Field samples
DNA of each of the 48 cattle faeces isolates was amplified by PCR (30 min) and the resulting amplicons applied to NALFIA tests. The qualitative result achieved for each sample (presence/absence) was compared to the result achieved by q-PCR (considered in this work as the reference method). The results achieved for each virulence factor tested are summarised in Table 4 (see ESM Table S1 for clarification). The presence of FP and FN results is justified by the different performance (LOD) of NALFIA and q-PCR for each of the amplified virulence factors. The three FN found for vt1 and the one found for ehxA are due to the higher LOD of the NALFIA strips (compared to q-PCR); E. coli was present, but in a concentration that was not detected [36, 37]. On the other hand, the two FP achieved for vt2 can be explained if the lower LOD for NALFIA is taken into account (one order of magnitude lower than for q-PCR); that is, in this particular sample, this factor was not detected by q-PCR.
Reliability parameters of the NALFIA strips developed are indicated in Table 5 (standard error values are given in ESM Table S3). For all factors tested, sensitivity and specificity values were very high (larger than 0.85, and in most cases equal to 1.00). In all cases, positive and negative predictive values (i.e. how well a sample is classified) were larger than 0.90. Moreover, efficiency values (i.e. correct answers) were 0.92 or greater, meaning that more than 90% of the answers were the same as the reference procedure. The kappa coefficient was also calculated, giving values larger than 0.80 and in some cases equal to 1.00. According to Landis and Koch [34], this indicates that there is an almost perfect agreement between NALFIA and the standard used (q-PCR) for vt1, vt2 and ehxA, and a perfect agreement for eae and 16S. This excellent agreement between the NALFIA strips developed and the reference system used demonstrates that the proposed analytical procedure has been proven to be fit for purpose, i.e. it is valuable for fast screening of E. coli virulence factors.
A preliminary multi-analyte NALFIA was prepared with five test lines (one for each of the specific antibodies: TxR, FITC, DIG, DNP and Cy5) and one control line (biotinylated anti-rabbit IgG) with the conditions indicated previously. These NALFIA strips were tested for cross-reactivities (Fig. 2) by running a blank sample (first strip), each of the STEC factors (second to sixth strip), and a sample with all factors (seventh strip). As previously noted, no cross-reactivity was found and all specific amplicons were correctly identified, showing the potential of NALFIA strips for the simultaneous detection of multiple virulence factors.

Results achieved with multi-analyte NALFIA for E. coli virulence factors. Samples: blank (first strip), each of the STEC factors (second to sixth strip), and all factors (seventh strip)
Conclusions
The performance of the NALFIA strips developed in this work (with a preceding 30-min PCR reaction step) has shown that this system is feasible and reliable and that it allows the specific identification of the four E. coli virulence genes (vt1, vt2, eae and ehxA) and a marker specific for E. coli in approximately 40 min (once DNA is isolated). Results achieved for the detection of E. coli virulence factors in real samples by NALFIA (following DNA isolation and PCR amplification) were in very good agreement with those achieved by q-PCR. Most discrepancies can be explained by the different LODs for the various genes in both methods. Nevertheless, NALFIA strips developed have very good sensitivity and specificity, and results achieved by the strips are in very good agreement with the reference method used (q-PCR). When comparing both systems, q-PCR could have the advantage that (in most cases) the LOD is lower than NALFIA and that the identification is done in one step. However, it has several drawbacks: The first one is the price of the equipment, the second one is that specific knowledge is needed to understand and interpret the results, and the third one is the need of a laboratory setting. When comparing the number of factors that can be analysed, for q-PCR, only a limited number of fluorescent dyes are available, and multiplexing is complicated by the spectral overlap of the various dyes, allowing only a limited combination of dyes in one run (generally up to six). However, as verified in this work, NALFIA strips can also be manufactured with six lines (five test lines and one control line), having several advantages over more sophisticated instrumentation as listed above. Moreover, the same carbon–neutravidin nanoparticle conjugate is presently used to sandwich tag-labelled amplicons in antibody microarrays (results from Wageningen UR Food & Biobased Research, manuscript in preparation). In this format, the number of discrete targets can be easily increased by introducing other tag–antibody combinations. The results of this approach, a 30-min amplification followed by a one-step incubation for 1 h on the microarray, can be obtained by automated data processing following the digitisation of the spots by flatbed scanning or digital photography (10-min total processing time). Finally, if the price per test would be compared, NALFIA is less costly than q-PCR. The price for a duplex q-PCR test (e.g. virulence gene and 16S control) was calculated to be €1.16 (provided multiple samples are tested in the 96-well microtiter plate format). On the other hand, cost calculation for a two-line NALFIA (duplex PCR) resulted in €0.41. In conclusion, the NALFIA technology enables a larger multiplexing than q-PCR, is substantially less demanding with respect to laboratory facilities and has a lower price per test.
In this work, it has been demonstrated that the use of NALFIA strips in combination with a quick PCR amplification step comprises most of the requirements for alternative rapid methods [6]: accurate, with very good sensitivity and specificity, reliable, and with a detection limit that enables the correct discrimination of the different E. coli virulence factors. The test has also been validated against a standard test (q-PCR), its speed is high (<40 min, once the enrichment step is performed), and the cost of the whole system (strips, reagents) is low, requiring only one instrument (the thermocycler). Other characteristics that this system meets are that NALFIA strips are easily manufactured, have a long shelf life, can be stored under ambient conditions, and are very easy to use. In addition, whilst for q-PCR often small fragments are chosen (100–150 bp), the size of amplicon targets is not critical in NALFIA. This would allow using larger fragments if the variation in a particular region would require so.
Potentially, the developed NALFIA system can be used in the field, near the animal that has to be tested on (zoonotic) pathogens. In that case, transportation of infected samples to testing laboratories would not be necessary, thereby reducing the risk of contamination outside the place of identification. The user friendliness of the procedure can be further improved by applying ready-to-use amplification reagents combined with isothermal amplification protocols and on-tissue amplification protocols that would make DNA isolation superfluous. No expensive apparatus is needed to perform the test procedure, and the number of specific amplicons that can be tested simultaneously is only dependent on the availability of tag–antibody combinations and on the possibility to develop a multiplex PCR protocol. Moreover, the detection system is broadly applicable since any molecular biological diagnostic question can be translated in primer sets having biotin on one of the primers and a discriminating tag on the other primer. A carbon conjugate with neutravidin (or streptavidin) will always recognise double-labelled amplicons having a biotin on one end of the double-strand chain, and specific anti-tag antibodies on the membrane will bind to their respective tags, thereby revealing the identity of the specific amplicons. Integration of the NALFIA technology in a field-applicable device with which amplification can be done, would further decrease hands-on time and simplify the overall detection procedure.
In conclusion, we have demonstrated that the use of carbon nanoparticles to detect amplified double-strand genetic material by NALFIA results in a screening method that is reliable, simple to use, cost-effective and fast; once DNA is isolated, results are achieved within 40 min, including the amplification step.
References
Alocilja EC, Radke SM (2003) Biosens Bioelectron 18:841–846
Lazcka O, Campo FJD, Muñoz FX (2007) Biosens Bioelectron 22:1205–1217
Nataro JP, Kaper JB (1998) Clin Microbiol Rev 11:142–201
Meng J, Doyle MP (1998) Bull Inst Pasteur 96:151–163
Karmali MA, Gannon V, Sargeant JM (2010) Vet Microbiol 140:360–370
de Boer E, Beumer RR (1999) Int J Food Microbiol 50:119–130
Ivnitski D, Abdel-Hamid I, Atanasov P, Wilkins E (1999) Biosens Bioelectron 14:599–624
Tokarskyy O, Marshall DL (2008) Food Microbiol 25:1–12
Chemburu S, Wilkins E, Abdel-Hamid I (2005) Biosens Bioelectron 21:491–499
Rule G, Montagna R, Durst R (1996) Clin Chem 42:1206–1209
Ngom B, Guo Y, Wang X, Bi D (2010) Anal Bioanal Chem 397:1113–1135
Posthuma-Trumpie GA, Korf J, van Amerongen A (2009) Anal Bioanal Chem 393:569–582
Carter DJ, Cary RB (2007) Nucleic Acids Res 35:e74
Kalogianni DP, Goura S, Aletras AJ, Christopoulos TK, Chanos MG, Christofidou M, Skoutelis A, Ioannou PC, Panagiotopoulos E (2007) Anal Biochem 361:169–175
Litos IK, Ioannou PC, Christopoulos TK, Traeger-Synodinos J, Kanavakis E (2009) Biosens Bioelectron 24:3135–3139
Blažková M, Koets M, Rauch P, van Amerongen A (2009) Eur Food Res Technol 229:867–874
Mens PF, van Amerongen A, Sawa P, Kager PA, Schallig HD (2008) Diagn Microbiol Infect Dis 61:421–427
Gordon J, Michel G (2008) Clin Chem 54:1250–1251
Aldus CF, van Amerongen A, Ariens RM, Peck MW, Wichers JH, Wyatt GM (2003) J Appl Microbiol 95:380–389
Capps KL, McLaughlin EM, Murray AW, Aldus CF, Wyatt GM, Peck MW, van Amerongen A, Ariens RM, Wichers JH, Baylis CL, Wareing DR, Bolton FJ (2004) J AOAC Int 87:68–77
van Amerongen A, Koets M (2005) In: van Amerongen A, Barug D, Lauwaars M (eds) Rapid methods for biological and chemical contaminants in food and feed. Wageningen Academic Publishers, Wageningen, pp 105–216
Moreira BG, You Y, Behlke MA, Owczarzy R (2005) Biochem Biophys Res Commun 327:473–484
Nielsen EM, Andersen MT (2003) J Clin Microbiol 41:2884–2893
Huijsdens XW, Linskens RK, Mak M, Meuwissen SGM, Vandenbroucke-Grauls CMJE, Savelkoul PHM (2002) J Clin Microbiol 40:4423–4427
Koets M, Sander I, Bogdanovic J, Doekes G, van Amerongen A (2006) J Environ Monit 8:942–946
van Amerongen A, Wichers JH, Berendsen LBJM, Timmermans AJM, Keizer GD, van Doorn AWJ, Bantjes A, van Gelder WMJ (1993) J Biotechnol 30:185–195
O’Keeffe M, Crabbe P, Salden M, Wichers J, van Peteghem C, Kohen F, Pieraccini G, Moneti G (2003) J Immunol Methods 278:117–126
Posthuma-Trumpie GA, Korf J, van Amerongen A (2008) Anal Bioanal Chem 392:1215–1223
Chang LL, Shepherd D, Sun J, Ouellette D, Grant KL, Tang XC, Pikal MJ (2005) J Pharm Sci 94:1427–1444
Geue L, Segura-Alvarez M, Conraths FJ, Kuczius T, Bockemuhl J, Karch H, Gallien P (2002) Epidemiol Infect 129:173–185
Eurachem/CITAC (2003) EURACHEM/CITAC Guide: The expression of uncertainty in qualitative testing. LGC, Teddington, Middlesex, UK, p 22
Ellison SLR, Fearn T (2005) Trac-Trends Anal Chem 24:468–476
Gilchrist JM (2009) J Clin Microbiol 62:1045–1053
Landis JR, Koch GG (1977) Biometrics 33:159–174
Mackinnon A (2000) Comput Biol Med 30:127–134
Mil’man BL, Konopel’ko LA (2004) J Anal Chem 59:1128–1141
AOAC (2006) Final report and executive summaries from the AOAC International Presidential Task Force on best practices in microbiological methodology. AOAC International, Gaithersburg, Maryland, USA, p 201
Acknowledgments
This work was partially supported by the Generalitat Valenciana (BEST/2009/026), the Universidad Politécnica de Valencia (PAID-00-09-2837), and by the Dutch Ministry of Agriculture, Nature and Food Quality (KennisBasis 6 programme). The authors would like to thank Dr. Eva Møller Nielsen at the Danish Veterinary Institute (Copenhagen, Denmark) for providing E. coli control strains and Dr. Lutz Geue (Friedrich-Loeffler-Institut, Wusterhausen, Germany) and Dr. Dörte Döpfer (School of Veterinary Medicine, University of Wisconsin, Madison, WI, USA) for field isolates.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM 1
Analytical and bioanalytical chemistry (PDF 229 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Noguera, P., Posthuma-Trumpie, G.A., van Tuil, M. et al. Carbon nanoparticles in lateral flow methods to detect genes encoding virulence factors of Shiga toxin-producing Escherichia coli . Anal Bioanal Chem 399, 831–838 (2011). https://doi.org/10.1007/s00216-010-4334-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-4334-z