
Abstract
Pulmonary fibrosis is a progressive lung disorder with high mortality rate and limited successful treatment. This study was designed to assess the potential anti-oxidant and anti-fibrotic effects of aliskiren (Alsk) during bleomycin (BLM)-induced pulmonary fibrosis. Male Wistar rats were used as control untreated or treated with the following: a single dose of 2.5 mg/kg of BLM endotracheally and BLM and Alsk (either low dose 30 mg/kg/day or high dose 60 mg/kg/day), and another group was given Alsk 60 mg/kg/day alone. Alsk was given by gavage. Alsk anti-oxidant and anti-fibrotic effects were assessed. BLM significantly increased relative lung weight and the levels of lactate dehydrogenase and total and differential leucocytic count in bronchoalveolar lavage that was significantly ameliorated by high-dose Alsk treatment. As markers of oxidative stress, BLM caused a significant increase in the levels of lipid peroxides and nitric oxide accompanied with a significant decrease of superoxide dismutase and glutathione transferase enzymes. High-dose Alsk treatment restored these markers toward normal values. Alsk counteracted the overexpression of advanced glycation end products, matrix metalloproteinase-9 (MMP-9), and tissue inhibitor of metalloproteinases-1 in lung tissue induced by BLM. Fibrosis assessed by measuring hydroxyproline content, which markedly increased in the BLM group, was also significantly reduced by Alsk. These were confirmed by histopathological and immunohistochemical examination which revealed that Alsk attenuates signs of pulmonary fibrosis and decreased the overexpressed MMP-9 and transforming growth factor β1. Collectively, these findings indicate that Alsk has a potential anti-fibrotic effect beside its anti-oxidant activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pulmonary fibrosis is a chronic progressive lung disease. It is distinguished pathologically by deposition of extracellular matrix (ECM) with lung architectural remodeling (Todd et al. 2012). It is a deleterious sequel of discrete pulmonary syndromes as acute respiratory distress syndrome, idiopathic pulmonary fibrosis (IPF), occupational and environmental exposures, and drug reactions (Raghu et al. 2011).
Bleomycin (BLM) administration is a widely used model for understanding fibrotic lung remodeling (Latta et al. 2015). BLM, beside its anti-neoplastic properties, induces pulmonary fibrosis in experimental animals that resemble chronic fibrotic lung disease in human (Snider et al. 1978).
The oxidant/anti-oxidant imbalance (Daniil et al. 2008) and the dysregulated fibrogenesis (Selman et al. 2001) have emerged as critical components of the BLM-induced pulmonary fibrosis. Ample evidence pointed that reactive oxygen species (ROS) generated internally or exogenously by environmental exposure promoted the development of pulmonary fibrotic diseases (Kliment and Oury 2010; Faner et al. 2012; Liu et al. 2012; Liu et al. 2013). These oxidants can stimulate genes regulating cell growth, cell death, and fibroblast proliferation (Kinnula and Crapo 2003; Halliwell 2011) and thereby promote pulmonary fibrosis.
The advanced glycation end products (AGEs), the irreversible products of non-enzymatic glycation of proteins, nucleic acids, and lipids, are elevated during oxidative stress (Bohlender et al. 2005). Thus, AGEs are considered as a biomarker for local endogenous oxidative stress (Hudson et al. 2003). Literature demonstrated that AGEs contribute to the pathogenesis of different disorders (Ramasamy et al. 2005) including pulmonary fibrosis (Englert et al. 2008). Matsuse and his colleagues were the first to document the accumulation of enhanced AGE-modified proteins in alveolar macrophages in patients with IPF (Matsuse et al. 1998). Others have reported that AGEs can induce excessive ECM accumulation and pro-fibrotic cytokine expression, such as transforming growth factor β1 (TGF-β1) (Ohashi et al. 2004).
TGF-β1 is thought to be involved in the initiation and progression of fibrosis (Gauldie 2002; Keane and Strieter 2002). TGF-β1 is elevated in tissue samples of IPF seen in animal models (Coker et al. 1997) and patients (Coker et al. 2001). The three isoforms of TGF-β are inactive and attached to a latency association protein (LAP). ROS promotes TGF-β-induced fibrosis either by direct activating of latent TGF-β via oxidation of LAP with subsequent release of TGF-β or indirectly by ROS up-regulation of matrix metalloproteinases (MMPs) that eventually cleave LAP liberating active TGF-β (Liu and Gaston Pravia 2010; Cheresh et al. 2013).
MMPs, the mediators of matrix degradation, are a family of zinc endopeptidase. Different MMPs are activated in the IPF lungs. They contribute to remodeling of ECM and disruption of basement membrane seen in the disease pathogenesis (Pardo and Selman 2012). MMP-9 protein and gene expressions are up-regulated in both animal models and patients with lung fibrosis (Lemjabbar et al. 1999; Perez-Ramos et al. 1999). High levels of MMP-9 were observed in fibroblasts and alveolar macrophages isolated from lungs of IPF patient (Montano et al. 1989; Lemjabbar et al. 1999). Interestingly, MMP-9 was able to augment TGF-β promoting the increase of active TGF-β (Yu and Stamenkovic 2000).
Tissue inhibitors of metalloproteinases (TIMPs) inhibit MMP activities in vivo. However, disruption of the balance between MMPs and TIMP may evoke pathological processes, including pulmonary fibrosis. This was evidenced in fibroblasts derived from human fibrotic lung (Pardo et al. 1992).
Renin is a protease that cleaves angiotensinogen to form angiotensin I, which successively converted to angiotensin II by angiotensin-converting enzyme (Rokhsara et al. 2013). The tissue renin–angiotensin system (RAS) modulates the structure and function of different organs and systems via its autocrine and paracrine activities (Cooper et al. 2007).
Different studies revealed that RAS exerts a crucial role in fibrogenesis primarily through the actions of angiotensin II in different organs, including the lung (Li et al. 2003; Marshall et al. 2004; Bataller et al. 2005; Hartner et al. 2006).
Later, renin has been pointed to be a pro-fibrotic mediator of lung fibrosis functioning independently from angiotensin II. Its effects in human IPF lungs and fibroblasts resulted in overexpression of TGF-β and collagen. Renin gene silencing resulted in reduced expression of collagen and TGF-β1 in vitro (Montes et al. 2012).
Aliskiren (Alsk), a direct inhibitor of renin, has been shown in different studies to be effective in lowering blood pressure and to hold considerable potential for organ protection beyond blood pressure reduction. Alsk exhibited beneficial anti-fibrotic effects in different fibrotic models including peritoneal, renal, and cardiac fibrosis (Ke et al. 2010; Gross et al. 2011; Zhi et al. 2013).
Therefore, the present study aims to explore the potential anti-fibrotic and anti-oxidant effects of Alsk and try to elucidate the underlying mechanisms for modulating lung fibrosis induced by BLM.
Materials and methods
Drugs and chemicals
Aliskiren (Alsk) was supplied as formulated drug product manufactured by Novartis Pharma AG, Basel, Switzerland. Bleomycin (BLM) was supplied as ampoules (Bleocin) (Nippon Kayaku Co. Ltd., Tokyo, Japan). Each ampoule contains 15 mg BLM-HCl as lyophilized powder. Both dissolved in normal saline. Other chemicals and reagents were of analytical grade.
Animals
Sixty male Wistar rats weighing 200–250 g were purchased from the Egyptian Organization for Biological Products and Vaccines (VACSERA, Helwan, Egypt). The rats were housed at room temperature 24 ± 1 °C with 12-h light–dark cycle and allowed free access to food and water. The rats were acclimated for 1 week before the start of experiment. All animal procedures complied with the criteria outlined in European Directive 2010/63/EU and guidelines of Ethical Committee, Faculty of Medicine, Ain Shams University.
Experimental design
Animals were classified into five groups (12 animals per group). To induce pulmonary fibrosis, animals received endotracheally, a single sublethal dose of 2.5 mg /kg of BLM dissolved in 0.25 ml saline (0.9 % NaCl) (Boyaci et al. 2006). Control animals received the same volume of endotracheal saline instead of BLM. Tracheal instillation was carried out under halothane (1–2 %) anesthesia. The third and fourth groups were given both of BLM and Alsk (either low dose 30 mg/kg/day or high dose 60 mg/kg/day, respectively). The fifth group was given Alsk 60 mg/kg/day alone (Alsk was given by gavage) (Zhang et al. 2014). Fourteen days after endotracheal instillation of BLM, the body weights of animals were recorded and then sacrificed by a lethal injection of sodium pentobarbital (100 mg/kg ip). The lung weights were weighed. Both lungs were rapidly dissected out and washed with ice-cold saline. For six animals, one of two lungs was used for biochemical assessment of oxidative stress markers including lipid peroxidation; nitric oxide (NO) and the anti-oxidant enzyme activity of superoxide dismutase (SOD) and glutathione transferase (GST); and analysis of collagen, advanced glycation end products (AGEs), matrix metalloproteinase-9 (MMP-9), and tissue inhibitor of metalloproteinase-1 (TIMP-1), while the other lung was used for histopathological examination and immunohistochemical detection of MMP-9 and transforming growth factor β1 (TGF-β1). For the other six animals, the lung was used for the assessment of lung injury markers in bronchoalveolar lavage fluid (BALF) including lactate dehydrogenase (LDH) and total and differential leucocytic count.
Assessment of lung injury markers
After dissection of the lung and trachea, three successive 5-ml 0.9 % saline solutions were infused and slowly withdrawn from the lungs through a cannula inserted into the trachea for three successive times. Recovered BALF volumes were collected. BALF was centrifuged at 500×g for 10 min at 4 °C. The supernatant was collected and stored at −80 °C for assay of LDH. The cell pellets were resuspended in saline, and total cell count was counted with a hemocytometer. Differential leukocyte cell counts were obtained by preparing smear slides and staining them with Giemsa solution. Differential leukocyte cell counts were obtained from a count of 300 cells per smear.
LDH was determined using a commercial kit (Stanbio Laboratory, Inc., San Antonio, TX, USA) in accordance with the manufacturer’s instructions. Relative lung weight was calculated: lung weight / body weight.
Assessment of lipid peroxides and NO levels as well as the anti-oxidant enzyme activities of SOD and GST in the lung
Lipid peroxidation, measured as malondialdehyde (MDA), was assayed using the thiobarbituric acid test as described by Cragg (1998). SOD activity was determined in lung homogenate in accordance to the method of El-Dakhakhny (1965). Total NO content was determined in lung homogenate in accordance to the method of Paino et al. (2011). GST activity was determined in lung homogenate in accordance to the method of Gilani et al. (2001).
Assessment of lung fibrosis
Assessment of lung hydroxyproline and AGEs
Hydroxyproline content, as an index of collagen, was assayed in the lung homogenate according to the method described by Ramadan et al. (2003). AGE level in the lung homogenate was determined using rat AGE ELISA kit (Cusabio Biotech Co., Ltd) in accordance with the manufacturer’s instructions.
Assessment of lung MMP-9 level
Lung levels of MMP-9, as well as that of TIMP-1, were measured in lung tissue homogenates using MMP-9 ELISA kit (Cusabio Biotech Co., Ltd) and TIMP-1 ELISA kit (Ray Biotech, Inc.), respectively, according to the manufacturer’s instructions.
Microscopic examination
For microscopic examination, the lungs from the rats were fixed in 10 % formalin for 8 h, processed as usual, and embedded in paraffin. Three-micrometer serial sections were stained with hematoxylin-eosin (H&E). The extent of the fibrotic pathology was assessed using the Ashcroft scoring system (Ashcroft et al. 1988). The fibrotic pathology, as determined by Masson’s trichrome stain for collagen, consists of highly cellular interstitial infiltrates with areas of diffuse fibrosis, adjacent to areas of unaffected tissue. For histological analysis of the inflammation using a modified semiquantitative method, a scale from 1 to 5 (0 = absent, 1 = minimal, 2 = less than 25 %, 3 = 25 to 49 %, 4 = 50 to 75 %, and 5 = greater than 75 %) was applied to measure the extent and intensity of cellularity in the (interstitial) alveolar wall (Cherniack et al. 1991).
Immunohistochemical examination
Four-micrometer sections of formalin-fixed and paraffin-embedded samples of all lung specimens were prepared. Immunohistochemical staining was performed using primary antibodies: MMP-9 (ready to use, CLG4B, 82 kDa MMP-9, collagenase type 4 beta, GELB, macrophage gelatinase, MANDP2, type V collagenase, antibody, biotin conjugate (167599); US Biological Life Sciences Inc., Fremont, CA) and mouse monoclonal antibody against TGF-β1 (ready to use, TGF β-1 polyclonal antibody; Catalog Number PA1-9574; Thermo Fisher Scientific Inc., Fremont, CA). Avidin-biotin immune-peroxidase complex technique was used by applying the super sensitive detection kit (Biogenex, CA, USA). The prepared tissue sections were fixed on poly-l-lysine-coated slides overnight at 37 °C. They were deparaffinized and rehydrated through graded alcohol series. Then, the sections were heated in a microwave oven in 10 mM citrate buffer (pH 6.0) for 10 min. After the blocking of endogenous peroxidase and incubation in Protein Block Serum-Free Solution (DakoCytomation) for 20 min, the sections were incubated with MMP-9 antibody and with TGF-β1 according to the manufacturer’s instructions. Biotinylated anti-mouse immunoglobulin and streptavidin conjugated to horseradish peroxidase were then added. Finally 3, 3′-diaminobenzidine as the substrate or chromogen was used to form an insoluble brown product. Finally, the sections were counterstained with hematoxylin and mounted. Sections of ulcerative colitis and breast carcinoma were used as positive control for MMP-9 and TGF-β1, respectively. Negative control sections were incubated with normal mouse or rabbit serum instead of the primary antibody.
The expression of the MMP-9 antibodies was semiquantitatively evaluated through the Allred score, modified as follows: Grade 0, 1, 2, and 3 were determined according to the numbers of the positive cells (0 = <50 positive cells; 1 = 50–100 positive cells; 2 = 101–200 positive cells; 3 = >200 positive cells), and the intensity was evaluated as 1, 2, and 3 (1 = weakly positive; 2 = moderately positive; 3 = strongly positive). Immunohistochemical score was obtained by multiplying these two indices. All these examinations were done on five different high-power fields (×400) (Allred et al. 1993). The intensity of immune-staining of TGF-β1 was graded from − (absent) to +++ (strongly positive) (Bergeron et al. 2003).
All the histopathological steps, including the microscopic examination, special stain results, and subsequent immunohistochemical scoring, were performed in a blinded manner; i.e., the pathologist was unaware of the different groups in the current experimental work; including the study group.
Data analysis
Biochemical data were presented as mean ± SD. Multiple comparisons were performed using one-way ANOVA followed by Bonferroni’s multiple comparison test as a post hoc test. A significance level of P < 0.05 was used in all tests. Statistical analyses were performed using GraphPad Prism version 5.00 for Windows 7 (GraphPad Software, San Diego, CA, USA). Variables of histopathological examinations were expressed as mean ± SD and median and interquartile range. Kruskal-Wallis and Mann-Whitney tests were used to compare non-parametric data. A significance level of P < 0.05 was used in all tests. Statistical procedures were carried out using SPSS version 15 for Windows (SPSS Inc., Chicago, IL, USA). The interpreted data of BLM and the Alsk-alone groups were always in comparison to the control group where the treated groups (Alsk-H and Alsk-L groups) were always in comparison to the BLM group. Sample size is determined by using GraphPad StatMate, software program, Version 1.01i Jan. 16, 1998. Sample size was kept at the minimum required to provide the power sufficient for statistical comparisons.
Results
Lung injury markers
As shown in Table 1, the relative lung weight was significantly (P < 0.05) increased and body weight was significantly (P < 0.05) decreased in BLM group compared to the control group. Concurrent treatment of animals with Alsk high dose (Alsk-H) and BLM significantly (P < 0.05) counteracted the effect of BLM on relative lung weight and final body weight near the normal level. BALF assessment revealed that BLM induced a significant (P < 0.05) increase in the total leucocytic count and differential cell counts compared to the control group. Furthermore, BLM induced a significant increase (P < 0.05) in LDH activity as compared to the control group. The BLM-induced changes on total and differential cells were significantly (P < 0.05) reversed by concurrent Alsk-H. Alsk-H effect was more obvious on some parameters as the total cell count, neutrophils, and eosinophils. Alsk low-dose (Alsk-L) concurrent treatment compared to the BLM group and Alsk-alone treatment compared to control group did not show any significant changes in all markers. Non-significant (P < 0.05) change was found between all compared groups in lung weight.
Oxidative stress markers
BLM-induced oxidative stress was evaluated by assessing lipid peroxides and NO levels as well as the anti-oxidant enzyme activity of SOD and GST. As shown in Table 2, BLM induced a significant (P < 0.05) increase in lung lipid peroxides and NO levels as compared to the control values. On the other hand, BLM significantly (P < 0.05) decreased the anti-oxidant enzyme activity of GST and SOD as compared to the control values.
Only treatment of animals with Alsk-H concomitantly with BLM significantly (P < 0.05) restored levels of anti-oxidant enzyme activities, NO and lipid peroxides close to levels of control group. Concurrent Alsk-L treatment showed non-significant changes compared to BLM group. Moreover, animals treated with Alsk alone did not show any significant changes in the oxidative stress markers as compared to the control group (Table 2).
Hydroxyproline content and AGE level of lung homogenates
Both lung hydroxyproline content and AGE level were significantly (P < 0.05) increased in BLM group compared to the control group. Treatment with concurrent Alsk-H but not Alsk-L significantly (P < 0.05) reduced the hydroxyproline content and AGE level compared to BLM group. Alsk-alone treatment did not significantly alter the lung hydroxyproline content or AGE level compared to the control group (Fig. 1).

Effects of Alsk on a hydroxyproline content expressed as milligram per gram of lung homogenates in rats induced by BLM and b AGE expressed as microgram per milligram protein of lung homogenates in rats induced by BLM. Alsk-H (60 mg kg−1 day−1); Alsk-L (30 mg kg−1 day−1). Data represent means ± SD of six rats. *P < 0.05 compared with control group; #P < 0.05 compared with BLM group using one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test as a post hoc test
MMP-9 and TIMP-1 levels of lung homogenates
ELISA analysis of the lung homogenates in BLM-treated rats revealed significantly (P < 0.05) higher MMP-9 and TIMP-1 levels than that of the control group. Both lung MMP-9 and TIMP-1 overexpressions were significantly (P < 0.05) decreased by concurrent Alsk-H treatment compared to the BLM group, whereas the effect of concurrent Alsk-L treatment on both of these was non-significant. Alsk-alone treatment showed no significant change in the lung MMP-9 and TIMP-1 levels compared to the control group (Fig. 2).

a Effects of Alsk on MMP-9 expressed as picogram per milligram protein of lung homogenates in rats induced by BLM. b Effects of Alsk on TIMP-1 expressed as nanogram per milligram protein of lung homogenates in rats induced by BLM. Alsk-H (60 mg kg−1 day−1); Alsk-L (30 mg kg−1 day−1). Data represent means ± SD of six rats. *P < 0.05 compared with control group; #P < 0.05 compared with BLM group using one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test as a post hoc test
Histopathological examination and lung fibrosis markers
Hematoxylin-eosin and Masson’s trichrome stains findings
In control and Alsk-alone-treated groups (Table 3 and Figs. 3a, b and 4a, b), only focal mild lymphocytic infiltration around the small vessels was detected with a mean inflammatory score of the control group 1.5 ± 0.5 and that of Alsk-alone-treated group was 1.3 ± 0.5 with non-significant difference among both groups. Fibrosis was scarce. The mean fibrosis score of the control group was 1.3 ± 0.8 and that of Alsk-alone-treated group was 1.3 ± 0.8 with non-significant difference among both groups.

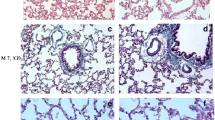
Photomicrographs of lung sections stained by H&E (×200). a Control group: showing normal architecture, b Alsk group, and c, d BLM group: showing variable inflammation and fibrosis scores. Tissue sections display signs of acute lung injury, edema fluid, and inflammation in the alveolar spaces. Marked alveolar epithelial injury and formation of cyst-like structures (arrows). The inset showed bronchiolization with nearby mature collagen (arrow and chevron inside inset, respectively). e BLM + Alsk-L group: showing decreased fibrosis and inflammation score. f BLM + Alsk-H group: showing highly significant decrease of inflammation and fibrosis score

Photomicrographs of lung sections stained by Masson’s trichrome stain. a Control group (×100), b Alsk group (×100), c, d BLM group: showing positive staining of the fibroblasts is clearly visible within areas of increased cellularity and focal diffuse collagen fiber deposition (×200) (inset ×400), e BLM + Alsk-L group, and f BLM + Alsk-H group
On the other hand, in the BLM group (Table 3 and Figs. 3c, d and 4c, d), prominent inflammation was detected in the lung parenchyma. Septal inflammatory infiltrate was specifically prominent with a characteristic feature so-called “bronchiolization,” the ingrowth of cuboidal cells from adjacent bronchioles to alveoli, forming a tubular structure. Inflammatory score in this group was 4.8 ± 0.4. In regard to fibrosis, immature-appearing collagen fibers were variably deposited around the arterioles, bronchioles, and alveolar septae where the inflammatory changes were present. Intra-alveolar fibrosis was apparent, with multiple fibrotic foci and mature collagen deposition. The mean fibrosis score was 7.2 ± 1.2. These were stained deep blue on Masson trichrome. A significant (P < 0.05) difference in inflammation and fibrosis between the control and BLM groups was detected.
The treatment effect was evident in concurrent Alsk-H-treated group (Table 3 and Figs. 3e, f and 4e, f) in the form of significant (P < 0.05) decrease in inflammation and fibrosis scores (median = 1.5 and 1, respectively) with near basal scores as seen in control group. Concurrent Alsk-L treatment led to mild decrease in scores of inflammation and fibrosis (median 4 and 5, respectively).
Immunohistochemical stain findings
In control and Alsk-alone-treated groups (Table 3 and Fig. 5a), MMP-9 was expressed in mild degrees in smooth muscle cells in the arteriolar wall and weakly expressed in a few alveolar macrophages and in some lymphocytes around the arterioles and bronchioles. The mean score of MMP-9 expression was 1.5 ± 0.5 each.

Immunostaining. a–d Representative sections of the MMP immunostaining in the following: a control group: negative immunostaining (×100); b BLM group: positive immunostaining in bronchial epithelium (inset), in the fibrous tissue, and in the inflammatory score (IHC, ×200); c BLM + Alsk-L group: decreased immunostaining score (×200); and d BLM + Alsk-H group: highly significant decrease of the MMP score (×200). e–h Representative section of the TGF-β1 immunostaining in the following: e control group: focal positivity around the bronchioles and blood vessel wall (×200); f BLM group: positive immunostaining in the alveolar pneumocytes, fibrous tissue, and occasional inflammatory cells (IHC, ×200); g BLM + Alsk-L group: decreased immunostaining score (×200); and h BLM + Alsk-H group: highly significant weak immunostaining (×200)
TGF-β1 labeling (Table 3 and Fig. 5e) was also seen in the layer of fibrous connective tissue surrounding bronchi and bronchioles and vascular smooth muscle with negative expression in bronchial/bronchiolar epithelial cells. The mean score of TGF-β1 in control and Alsk alone-treated group expression was 1.3 ± 0.5 and 1.2 ± 0.4, respectively. The mean combined expression of both markers was 2.8 ± 0.8 and 2.5 ± 0.8, respectively.
In the BLM group, the cells exhibiting features of cellular injury, activation, and/or repair particularly showed more prominent expression of both MMP-9 and TGF-β1. The inflammatory cells were also strongly positive. The MMP-9 was also expressed in the intra-alveolar edema, the pneumocytes, and the macrophages and at the fibrotic foci. The mean score of MMP-9 was 5.5 ± 0.8 which was significantly (P < 0.05) higher compared to the control group (mean 1.5 ± 0.5) (Table 3 and Fig. 5b).
TGF-β1 was expressed at higher levels in the fibroblastic foci. In addition, interstitial fibroblasts were present in the areas of parenchymal destruction. In those sections of lungs in BLM group in which there was a concurrent inflammatory response, there was a weak expression of TGF-β1 by macrophages or neutrophils. The mean score of TGF-β1 was 1.3 ± 0.8 significantly (P < 0.05) higher compared to the control groups (mean 1.3 ± 0.5 and 1.2 ± 0.4). The mean combined expression of both markers was 8.3 ± 0.8 (Table3 and Fig. 5f).
By estimating the changes of the expression of MMP-9 and TGF-β1 at different doses in concurrent Alsk-treated groups, a gradual decrease of both marker expression scores compared to the BLM group with a significant (P < 0.05) change in Alsk-H treatment was found (median = 1.5, IQR = 1–2 and median = 1, IQR = 1–2). The mean combined expression of both markers in Alsk-H group was 3.5 ± 1.2. While in the Alsk-L group, the mean combined expression of both markers was 6 ± 1.1 (Table 3 and Fig. 5c, d, g, h, respectively).
Discussion
In the present study, an experimental model of BLM-induced lung fibrosis was used to assess two aims: first, the potential anti-fibrotic effects of the direct renin inhibitor Alsk in this model and, second, trying to elucidate the underlying mechanisms by studying the effect of Alsk on oxidative stress and lung fibrosis markers as well as the expression of AGEs, MMP- 9, TIMP-1, and TGF-β1.
BLM-induced pulmonary fibrosis has been documented in studies (Wang et al. 1991; Tzurel et al. 2002). BLM causes lung injury and lead to oxidant-induced inflammatory and fibrotic lesions (Hay et al. 1991). BLM selectively affects the lung, as it lacks an enzyme that hydrolyzes BLM (Filderman et al. 1988). This animal model of pulmonary fibrosis resembles that seen in humans (Hyde and Giri 1990) and so has been widely used for studying the mechanisms involved in the progression of pulmonary fibrosis (Yara et al. 2001; El-Khatib 2002). It was suggested that the most suitable time point for assessing lung fibrosis in this model is 14 days after intra-tracheal instillation of BLM, as animals developed extensive fibrosis, with less mortality rates (Izbicki et al. 2002).
In order to detect the anti-fibrotic effect of Alsk, its effects on different biochemical and histological changes triggered in lung tissue in cases of pulmonary fibrosis induced by BLM were assessed. As expected, BLM caused a significant elevation of lung injury markers measured in BALF including LDH and total and differential leucocytic count in addition to a significant elevation in the relative lung weight compared to the control group; all these effects confirm the induction of lung injury. Concurrent treatment of animals with high-dose Alsk significantly counteracted the deleterious effects of BLM.
Hydroxyproline is the collagen precursor; it is considered a good biochemical index of collagen content (Ali and Mann 2004). Compared to the control group, the BLM group exhibited a significant increase in lung hydroxyproline content. This result is in accordance with previous studies, which also reported remarkable increase in lung hydroxyproline content in BLM-induced pulmonary fibrosis models (El-Medany et al. 2005; Gazdhar et al. 2007; Zhao et al. 2010). Excess or abnormal collagen deposition is a characteristic of lung fibrosis as reported by many while trying to elucidate the mechanisms behind BLM-induced lung fibrosis (Daba et al. 2002; Serrano-Mollar et al. 2003; Pardo et al. 2003). Similarly, the present biochemical indication of BLM-induced lung fibrosis was further confirmed histopathologically. Sections from BLM-induced lung fibrosis revealed multifocal areas of accentuated peribronchiolar fibrosis with bronchiolization and occasional honeycombing. Inflammatory changes are in the form of moderate interstitial mixed inflammatory infiltration. The microscopic observation of the Masson’s trichrome-stained histological sections confirmed collagen accumulation and deposition in peribronchial and perialveolar tissues that obliterated alveolar spaces as tiny fibrils. However, the lung tissues from high-dose Alsk-concurrent-treated rats showed significantly ameliorated histopathological characteristics for lung fibrosis induced by BLM observed by H&E and Masson’s trichrome stain with near normal collagen deposition, indicating that Alsk’s ameliorative role against BLM induced pulmonary fibrosis.
These results confirming the anti-fibrotic effect of Alsk are supported by the results of previous studies in which Alsk was able to ameliorate signs of fibrosis in different animal models including peritoneal, renal, and cardiac fibrosis (Ke et al. 2010; Gross et al. 2011; Zhi et al. 2013).
Although low-dose Alsk was able to exhibit some beneficial modulation regarding BLM-induced pulmonary fibrosis, these effects were not statistically enough to lead to a significant change compared to the BLM-induced pulmonary fibrosis group. Results of different studies showed that lower doses of Alsk were able to ameliorate many diseases in different animal models, however, the needed doses of Alsk to do so may change from one disease to another (Ke et al. 2010; Asker et al. 2015). The variation in the results of our study compared to those results may be due to the difference in duration of administration (acute or chronic) and the change in the efficacy of Alsk in different diseases or reflect the differences in the inhibitory activity of human renin inhibitors between species (Jensen et al. 2008).
The second aim of the study was to try to elucidate the underlying mechanisms by which Alsk was able to exhibit some anti-fibrotic potential by studying the effect of Alsk on oxidative stress and lung fibrosis markers.
It is known that ROS play an important role in the development of fibrotic responses in the lung upon BLM challenge. BLM catalyzes the formation of ROS (Sriram et al. 2009) which target different biomacromolecules leading to damage of the lung (Liang et al. 2011). In the results of our study, signs of oxidative stress were observed as exemplified by a significant increase of lipid peroxides and NO associated with a significant decrease in the activities of anti-oxidant enzymes GST and SOD compared to the control group. High-dose Alsk concurrent treatment significantly ameliorates these changes. These results supported the previous studies that described the crucial role of oxidative stress in BLM-induced pulmonary fibrosis and the beneficial effects of anti-oxidant drugs in treating pulmonary fibrosis (Oury et al. 2001; El-Demerdash 2011). Additionally, it supported the anti-oxidant effect of Alsk (Virdis et al. 2012; Santuzzi et al. 2015) which so can guard against the effects of oxidative stress that result in initiation of fibrosis.
The pathologic effects of AGEs are correlated to their ability to promote inflammation and oxidative stress by binding to its receptor (Schmidt et al. 1999). Results of many experimental studies indicate AGEs as an effective stimulator in fibrogenesis (Zhou et al. 2004; Lohwasser et al. 2006). Blockade of AGE formation attenuates BLM-induced pulmonary fibrosis in rats, and this was found to be implicated in inhibition of TGF-β expression (Chen et al. 2009). The current results found that BLM caused a significant elevation of AGEs compared to the control group. Concurrent treatment of animals with high-dose Alsk significantly reduced AGE level.
Another target for ROS generated by BLM are the MMPs. Different studies revealed that enhanced oxidative stress activates MMPs and increases their transcription (Nelson and Melendez 2004; Kinnula et al. 2005). MMPs are matrix-degrading proteinases involved in the pathophysiological process of pulmonary fibrosis and are implicated in the abnormal remodeling of the ECM (Kunugi et al. 2001; Pardo and Selman 2012). A growing body of evidence supports the role of MMP-9, in particular, in pulmonary fibrosis. McKeown and his colleagues (2009) report an increase in MMP-9 in BALF from IPF patients. Moreover, animal studies show similar results with increases in MMP-9 in the fibrotic phase of BLM-induced fibrosis (Tan et al. 2006). The catalytic activity of MMPs is accomplished primarily by a specific family of inhibitors named TIMPs. An imbalance of MMPs and TIMPs plays a pivotal role in fibrogenesis (Woessner 1994). In the present study, it was found that BLM caused a significant increase in lung MMP-9 and TIMP-1 compared to the control group. Concurrent treatment of animals with Alsk significantly reduced the overexpressed MMP-9 and TIMP-1. Similar observations made by others give credence to the present notion (Zhang et al. 2014).
Recent studies have shown that the release and activation of several key growth factors involved in pulmonary fibrosis occur through the action of MMPs; these include TGF-β (Bhattacharyya et al. 2007). Yu and Stamenkovic (2000) demonstrated that MMP-9 is involved in the activation of TGF-β complexes.
In the current study, immunohistochemical detection of MMP-9 was detected in the parenchymal cells, such as bronchiolar epithelial cells and type II pneumocytes mainly the ones surrounding the fibrotic foci, in addition to the inflammatory cells. Additionally, it is detected in the alveolar epithelial cells, alveolar fibroblasts, and alveolar macrophages. These findings are in concordance with previous studies (Sasaki et al. 2000; John et al. 2002; Chung 2005; Kim et al. 2009).
On the other hand, the examined lung specimens demonstrated that TGF-β1 was expressed in the alveolar epithelial cells, myofibroblastic foci, alveolar macrophages, and epithelial cells. TGF-β1, the most potent fibrogenic cytokine, has been reported to increase in pulmonary fibrosis and to be expressed in higher levels even in later phase of pulmonary fibrosis (Broekelmann et al. 1991; Bonniaud et al. 2005).
The overexpression of both MMP-9 and TGF-β1 proved in previous studies was supported in our study by the concomitant increase of both markers’ scores and the positive correlation of both with the experimental fibrosis and inflammatory scores.
The different expression of both MMPs and TGF-β1 in the studied experimental groups was observed. In contrast to the BLM group, in Alsk high-dose-treated group, the typical patchy areas of fibrosis and structural remodeling were less frequent, where MMP and TGF-β1 immunoreactivities were near to that of normal lung parenchyma of control tissue samples. Our data indicate that Alsk is capable of abrogating the effects of both MMP-9 and, consequently, TGF-β1, key mediators in lung fibrogenesis. This agrees with the results of study done by Asker et al. (2015) which reported that renin inhibition by Alsk attenuated pulmonary fibrosis through decreasing TGF-β1.
These results support the beneficial anti-fibrotic effect of Alsk in ameliorating pulmonary fibrosis. Interestingly, comparing these effects with the effects of both pirfenidone and nintedanib, recently approved to be used in current practice for treatment of pulmonary fibrosis, may highlight and support the potential beneficial effects of Alsk in pulmonary fibrosis. These two drugs were recommended to be used in pulmonary fibrosis depending on the results of studies which pointed to their anti-inflammatory activity, demonstrated by reduced lymphocyte and neutrophil counts in the BALF, in addition to their anti-fibrotic activity, demonstrated by reduced fibrosis in the histologic analysis and by diminished lung collagen, reflecting reduced ECM production and deposition, and also their ability to reduce TIMP-1, a key factor in the fibrogenic response to BLM in addition to their ability improve in forced vital capacity (FVC) compared with placebo (Wollin et al. 2014; Inomata et al. 2015).
Using BLM model led to a pulmonary injury characterized by an inflammatory response followed by fibrosis resembling what is happening in humans. It has been suggested that if the test compounds work by merely inhibiting the initial inflammation, they should prove active when given over the whole period, whereas drugs with anti-fibrotic activity should exhibit effectiveness irrespective of the treatment mode (Chaudhary et al. 2006). Although the results obtained in this study confirm the potential anti-fibrotic beneficial effect of Alsk in the reduction of the BLM-induced pulmonary fibrosis and its reflection on the markers of oxidative stress, inflammation, and fibrosis, this needs further investigation especially regarding timing of administration.
In conclusion, it seems that Alsk, by attenuating oxidative stress, may consequently decrease the associated inflammation and also decrease the activation of TGF-β1 either directly or indirectly by decreasing AGEs and MMP-9 in the BLM-induced pulmonary fibrosis model. Additional investigations are necessary to elucidate Alsk full anti-fibrotic potentials, especially with regard to timing of Alsk administration in addition to the potential for Alsk administration as a concomitant therapy for patients with lung fibrosis, including that produced during BLM treatment.
References
Ali S, Mann DA (2004) Signal transduction via the NF-kappa B pathway: a targeted treatment modality for infection, inflammation and repair. Cell Biochem Funct 22:67–79
Allred DC, Clark GM, Elledge R, et al. (1993) Association of p53 protein expression with tumor cell proliferation rate and clinical outcome in node-negative breast cancer. J Natl Cancer Inst 85:200–206
Ashcroft T, Simpson JM, Timbrell V (1988) Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol 41(4):467–470
Asker SA, Mazroa SA, Boshra V, et al. (2015) Biochemical and histological impact of direct renin inhibition byaliskiren on myofibroblasts activation and differentiation inbleomycin induced pulmonary fibrosis in adult mice. Tissue Cell. doi:10.1016/j.tice.2015.05.001
Bataller R, Sancho-Bru P, Gine’s P, et al. (2005) Liver fibrogenesis: a new role for the renin-angiotensin system. Antioxid Redox Signal 7:1346–1355
Bergeron A, Soler P, Kambouchner M, et al. (2003) Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-b and IL-10. Eur Respir J 22:69–76
Bhattacharyya P, Acharya D, Roychowdhury S (2007) Role of matrix metalloproteinases in the pathophysiology of idiopathic pulmonary fibrosis. Lung India 24:61–65
Bohlender JM, Franke S, Stein G, et al. (2005) Advanced glycation end products and the kidney. Am J Physiol Renal Physiol 289:F645–F659
Bonniaud P, Margetts PJ, Ask K, et al. (2005) TGF-beta and Smad3 signaling link inflammation to chronic fibro genesis. J Immunol 175:5390–5395
Boyaci H, Maral H, Turan G, et al. (2006) Effects of erdosteine on bleomycin-induced lung fibrosis in rats. Mol Cell Biochem 281(1–2):129–137
Broekelmann TJ, Limper AH, Colby TV, et al. (1991) Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc Natl Acad Sci U S A 88:6642–6646
Chaudhary NI, Schnapp A, Park JE (2006) Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am J Respir Crit Care Med 173:769–776
Chen L, Wang T, Wang X, et al. (2009) Blockade of advanced glycation end product formation attenuates bleomycin-induced pulmonary fibrosis in rats. Respir Res 10:55. doi:10.1186/1465-9921-10-55
Cheresh P, Kim S, Tulasiram S, et al. (2013) Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta 1832:1028–1040
Cherniack RM, Colby TV, Flint A, et al. (1991) Quantitative assessment of lung pathology in idiopathic fibrosis. Am J Respir Dis 144:892–900
Chung KF (2005) Inflammatory mediators in chronic obstructive pulmonary disease. Curr Drug Targets Inflamm Allergy 4:619–625
Coker RK, Laurent GJ, Jeffery PK, et al. (2001) Localization of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax 56:549–556
Coker RK, Laurent GJ, Shahzeidi S, et al. (1997) Transforming growth factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am J Pathol 150:981–991
Cooper SA, Whaley-Connell A, Habibi J, et al. (2007) Renin–angiotensin–aldosterone system and oxidative stress in cardiovascular insulin resistance. Am J Physiol Heart Circ Physiol 293:H2009–H2023
Cragg GM (1998) Paclitaxel (Taxol): a success story with valuable lessons for natural product drug discovery and development. Med Res Rev 18:315–331
Daba MA, Abdel-Aziz AH, Moustafa AM, et al. (2002) Effects of L carnitine and Ginkgo biloba extract (EGb 761) in experimental bleomycin-induced lung fibrosis. Pharmacol Res 45(6):461–467
Daniil ZD, Papaqeorqiou E, Koutsokera K, et al. (2008) Serum levels of oxidative stress as a marker of disease severity in idiopathic pulmonary fibrosis. Pulm Pharmacol Ther 21(1):26–31
El-Dakhakhny M (1965) Studies on the Egyptian Nigella sativa L IV Some pharmacological properties of the seeds’ active principle in comparison to its dihydro compound and its polymer. Arzneimittelforschung 15:1227–1229
El-Demerdash E (2011) Anti-inflammatory and anti-fibrotic effects of methyl palmitate. Toxicol Appl Pharmacol 254:238–244
El-Khatib AS (2002) Possible modulatory role of nitric oxide in lung toxicity induced in rats by chronic administration of bleomycin. Chemotherapy 48(4–5):244–251
El-Medany A, Hagar HH, Moursi M, et al. (2005) Attenuation of bleomycin induced lung fibrosis in rats by mesna. Eur J Pharmacol 509(1):61–70
Englert JM, Hanford LE, Kaminski N, et al. (2008) A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol 172:583–591. doi:10.2353/ajpath.2008.070569
Faner R, Rojas M, Macnee W, et al. (2012) Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 186:306–313
Filderman AE, Genovese LA, Lazo JS (1988) Alterations in pulmonary protective enzymes following systemic bleomycin treatment in mice. Biochem Pharmacol 37(6):1111–1116
Gauldie J (2002) Inflammatory mechanisms are a minor component of the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 165:1205–1206
Gazdhar A, Fachinger P, Van Leer C, et al. (2007) Gene transfer of hepatocyte growth factor by electroporation reduces bleomycin-induced lung fibrosis. Am J Physiol: Lung Cell Mol Physiol 292:L529–L536
Gilani AH, Aziz N, Khurram IM, et al. (2001) Bronchodilator, spasmolytic and calcium antagonist activities of Nigella sativa seeds (Kalonji): a traditional herbal product with multiple medicinal uses. J Pak Med Assoc 51:115–120
Gross O, Girgert R, Rubel D, et al. (2011) Renal protective effects of aliskiren beyond its antihypertensive property in a mouse model of progressive fibrosis. Am J Hypertens 24:355–361
Halliwell B (2011) Free radicals and antioxidants—quo vadis? Trends Pharmacol Sci 32:125–130
Hartner A, Porst M, Klanke B, et al. (2006) Angiotensin II formation in the kidney and nephrosclerosis in Ren-2 hypertensive rats. Nephrol Dial Transplant 21:1778–1785
Hay J, Shahzeidi S, Laurent G (1991) Mechanisms of bleomycin-induced lung damage. Arch Toxicol 65:81–94
Hudson BI, Bucciarelli LG, Wendt T, et al. (2003) Blockade of receptor for advanced glycation endproducts: a new target for therapeutic intervention in diabetic complications and inflammatory disorders. Arch Biochem Biophys 419(1):80–88
Hyde DM, Giri SN (1990) Polyinosinic–polycytidylic acid, an interferon inducer, ameliorates bleomycin-induced lung fibrosis in mice. Exp Lung Res 16:533–546
Inomata M, Nishioka Y, Azuma A (2015) Nintedanib: evidence for its therapeutic potential in idiopathic pulmonary fibrosis. Core Evid 10:89–98
Izbicki G, Segel MJ, Christensen TG, et al. (2002) Time course of bleomycin-induced lung fibrosis. Int J Exp Pathol 83:111–119
Jensen C, Herold P, Brunner HR (2008) Aliskiren: the first renin inhibitor for clinical treatment. Nat Rev Drug Discov 7:399–410
John M, Oltmanns U, Fietze I, et al. (2002) Increased production of matrix metalloproteinase-2 in alveolar macrophages and regulation by interleukin-10 in patients with acute pulmonary sarcoidosis. Exp Lung Res 28:55–68
Ke CY, Lee CC, Lee CJ, et al. (2010) Aliskiren ameliorates chlorhexidine digluconate induced peritoneal fibrosis in rats. Eur J Clin Investig 40(4):301–309
Keane MP, Strieter RM (2002) The importance of balanced pro-inflammatory and anti-inflammatory mechanisms in diffuse lung disease. Respir Res 3:5
Kim JY, Choeng HC, Ahn C, et al. (2009) Early and late changes of MMP-2 and MMP-9 in bleomycin-induced pulmonary fibrosis. Yonsei Med J 50:68–77
Kinnula VL, Crapo JD (2003) Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med 167:1600–1619
Kinnula VL, Fattman CL, Tan RJ, et al. (2005) Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med 172(4):417–422
Kliment CR, Oury TD (2010) Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Rad Biol Med 49:707–717
Kunugi S, Fukuda Y, Ishizaki M, et al. (2001) Role of MMP-2 in alveolar epithelial cell repair after bleomycin administration in rabbits. Lab Investig 81:1309–1318
Latta VD, Cecchettini A, Del Ry S, et al. (2015) Bleomycin in the setting of lung fibrosis induction: from biological mechanisms to counteractions. Pharmacol Res. doi:10.1016/j. phrs.2015.04.012
Lemjabbar H, Gosset P, Lechapt-Zalcman E, et al. (1999) Overexpression of alveolar macrophage gelatinase B (MMP-9) in patients with idiopathic pulmonary fibrosis: effects of steroid and immunosuppressive treatment. Am J Respir Cell Mol Biol 20:903–913
Li X, Rayford H, Uhal BD (2003) Essential roles for angiotensin receptor AT1a in bleomycin-induced apoptosis and lung fibrosis in mice. Am J Pathol 163:2523–2530
Liang X, Tian Q, Wei Z, et al. (2011) Effect of feining on bleomycin induced pulmonary injuries in rats. J Ethno Pharmacol 134(3):971–976
Liu RM, Gaston Pravia KA (2010) Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic Biol Med 48:1–15
Liu G, Cheresh P, Kamp DW (2013) Molecular basis of asbestos-induced lung disease. Annu Rev Pathol Mech Dis 8:161–187
Liu RM, Vayalil PK, Ballinger C, et al. (2012) Transforming growth factor beta suppresses glutamate-cysteine ligase gene expression and induces oxidative stress in a lung fibrosis model. Free Rad Biol Med 53:554–563
Lohwasser C, Neureiter D, Weigle B, et al. (2006) The receptor for advanced glycation end products is highly expressed in the skin and upregulated by advanced glycation end products and tumor necrosis factor-alpha. J Invest Dermatol 126(2):291–299
Marshall RP, Gohlke P, Chambers RC, et al. (2004) Angiotensin II and the fibro proliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol 286:L156–L164
Matsuse T, Ohga E, Teramoto S, et al. (1998) Immunohistochemical localization of advanced glycation end products in pulmonary fibrosis. J Clin Pathol 51(7):515–519
McKeown S, Richter AG, O’Kane C, et al. (2009) MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur Respir J 33:77–84
Montano M, Ramos C, Gonzalez G, et al. (1989) Lung collagenase inhibitors and spontaneous and latent collagenase activity in idiopathic pulmonary fibrosis and hypersensitivity pneumonitis. Chest 96:1115–1119
Montes E, Ruiz V, Checa M, et al. (2012) Renin is an angiotensin-independent profibrotic mediator: role in pulmonary fibrosis. Eur Respir J 39:141–148
Nelson KK, Melendez JA (2004) Mitochondrial redox control of matrix metalloproteinases. Free Radic Biol Med 37(6):768–784
Ohashi S, Abe H, Takahashi T, et al. (2004) End products increase collagen-specific chaperone protein in mouse diabetic nephropathy. J Biol Chem 279(19):19816–19823
Oury TD, Thakker K, Menache M, et al. (2001) Attenuation of bleomycin-induced pulmonary fibrosis by a catalytic antioxidant metalloporphyrin. Am J Respir Cell Mol Biol 25:164–169
Paino IM, Miranda JC, Marzocchi-Machado CM, et al. (2011) Phagocytosis and nitric oxide levels in rheumatic inflammatory states in elderly women. J Clin Lab Anal 25:47–51
Pardo A, Selman M (2012) Role of matrix metaloproteases in idiopathic pulmonary fibrosis. Fibrogenesis Tissue Repair 5(1):S9
Pardo A, Ruiz V, Arreola JL, et al. (2003) Bleomycin-induced pulmonary fibrosis is attenuated in 훾-glutamyl transpeptidase deficient mice. Am J Respir Crit Care Med 167(6):925–932
Pardo A, Selman M, Ramirez R, et al. (1992) Production of collagenase and tissue inhibitor of metalloproteinases by fibroblasts derived from normal and fibrotic human lung. Chest 102:1085–1089
Perez-Ramos J, Segura-Valdez M, Vanda B, et al. (1999) Matrix metalloproteinases 2, 9, and 13, and tissue inhibitors of metalloproteinases 1 and 2 in experimental lung silicosis. Am J Respir Crit Care Med 160:1274–1282
Raghu G, Collard HR, Egan JJ, et al. (2011) ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183:788–824
Ramadan MF, Kroh LW, Morsel JT (2003) Radical scavenging activity of black cumin (Nigella sativa L.), coriander (Coriandrum sativum L.), and niger (Guizotia abyssinica Cass.) crude seed oils and oil fractions. J Agric Food Chem 51:6961–6969
Ramasamy R, Vannucci SJ, Yan SS, et al. (2005) Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15(7):16R–28R
Rokhsara R, Maya M, Timothy E, et al. (2013) A review of current and novel therapies for idiopathic pulmonary fibrosis. J Thorac Dis 5(1)
Santuzzi CH, Tiradentes RV, Mengal V, et al. (2015) Combined aliskiren and L-arginine treatment has antihypertensive effects and prevents vascular endothelial dysfunction in a model of renovascular hypertension. Braz J Med Biol Res 48(1):65–76
Sasaki M, Kashima M, Ito T, et al. (2000) Differential regulation of metalloproteinase production, proliferation and chemotaxis of human lung fibroblasts by PDGF, interleukin-1beta and TNF-alpha. Mediat Inflamm 9:155–160
Schmidt AM, Yan SD, Wautier JL, et al. (1999) Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ Res 84(5):489–497
Selman M, King TE, Pardo A (2001) Idiopathic pulmonary fibrosis: prevailing evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 134:136–151
Serrano-Mollar A, Closa D, Prats N, et al. (2003) In vivo antioxidant treatment protects against bleomycin-induced lung damage in rats. Br J Pharmacol 138(6):1037–1048
Snider GL, Celli BR, Goldstein RH, et al. (1978) Chronic interstitial pulmonary fibrosis produced in hamsters by endotracheal bleomycin. Lung volumes volume–pressure relations, carbon monoxide uptake, and arterial blood gas studied. Am Rev Respir Dis 117:289–297
Sriram N, Kalayarasan S, Sudhandiran G (2009) Epigallocatechin-3-gallate augments antioxidant activities and inhibits inflammation during bleomycin-induced experimental pulmonary fibrosis through Nrf2-Keap1 signaling. Pulm Pharmacol Ther 22(3):221–236
Tan RJ, Fattman CL, Niehouse LM, et al. (2006) Matrix metalloproteinases promote inflammation and fibrosis in asbestos-induced lung injury in mice. Am J Respir Cell Mol Biol 35(3):289–297
Todd NW, Luzina IG, Atamas SP (2012) Molecular and cellular mechanisms of pulmonary Fibrosis. Fibrogenesis & Tissue Repair 5:11. treatment modality for infection, inflammation and repair. Cell Biochem Funct 22:67–79
Tzurel A, Segel J, Or R, et al. (2002) Halofuginone does not reduce fibrosis in bleomycin-induced lung injury. Life Sci 7(14):1599–1606
Ulrich P, Cerami A (2001) Protein glycation, diabetes, and aging. Recent Prog Horm Res 56:1–21
Virdis A, Ghiadoni L, Qasem A, et al. (2012) Effect of aliskiren treatment on endothelium dependent vasodilation and aortic stiffness in essential hypertensive patients. Eur Heart J 33:1530–1538
Wang Q, Giri SN, Hyde DM, et al. (1991) Amelioration of bleomycin-induced pulmonary fibrosis in hamsters by combined treatment with taurine and niacin. Biochem Pharmacol 42(5):1115–1122
Woessner JF (1994) The family of matrix metalloproteinase family. Ann N Y Acad Sci 732:11–21
Wollin L, Maillet I, Quesniaux V, et al. (2014) Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther 349:209–220
Yara S, Kawakami K, Kudeken N, et al. (2001) FTS reduces bleomycin-induced cytokine and chemokine production and inhibits pulmonary fibrosis in mice. Clin Exp Immunol 124:77–85
Yu Q, Stamenkovic I (2000) Cell surface localized matrixmetalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev 14:163–176
Zhang W, Han Y, Meng G, et al. (2014) Direct renin inhibition with aliskiren protects against myocardial ischemia/reperfusion injury by activating nitric oxide synthase signaling in spontaneously hypertensive rats. J Am Heart Assoc 3:e000606. doi:10.1161/JAHA.113.000606
Zhao L, Wang X, Chang Q, et al. (2010) Neferine, a bisbenzylisoquinline alkaloid attenuates bleomycin-induced pulmonary fibrosis. Eur J Pharmacol 627(1–3):304–312
Zhi H, Luptak I, Alreja G, et al. (2013) Effects of direct renin inhibition on myocardial fibrosis and cardiac fibroblast function. PLoS One 8(12):e81612. doi:10.1371/journal.pone.0081612
Zhou GH, Li C, Cai L (2004) Advanced glycation end products induce connective tissue growth factor-mediated renal fibrosis predominantly through transforming growth factor beta independent pathway. Am J Pathol 165(6):2033–2043
Acknowledgments
This research was supported by the Medical Research Service of Ain Shams University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Abuelezz, S.A., Hendawy, N. & Osman, W.M. Aliskiren attenuates bleomycin-induced pulmonary fibrosis in rats: focus on oxidative stress, advanced glycation end products, and matrix metalloproteinase-9. Naunyn-Schmiedeberg's Arch Pharmacol 389, 897–909 (2016). https://doi.org/10.1007/s00210-016-1253-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-016-1253-3