
Abstract
Fluoride is ubiquitously present throughout the world. It is released from minerals, magmatic gas, and industrial processing, and travels in the atmosphere and water. Exposure to low concentrations of fluoride increases overall oral health. Consequently, many countries add fluoride to their public water supply at 0.7–1.5 ppm. Exposure to high concentrations of fluoride, such as in a laboratory setting often exceeding 100 ppm, results in a wide array of toxicity phenotypes. This includes oxidative stress, organelle damage, and apoptosis in single cells, and skeletal and soft tissue damage in multicellular organisms. The mechanism of fluoride toxicity can be broadly attributed to four mechanisms: inhibition of proteins, organelle disruption, altered pH, and electrolyte imbalance. Recently, there has been renewed concern in the public sector as to whether fluoride is safe at the current exposure levels. In this review, we will focus on the impact of fluoride at the chemical, cellular, and multisystem level, as well as how organisms defend against fluoride. We also address public concerns about fluoride toxicity, including whether fluoride has a significant effect on neurodegeneration, diabetes, and the endocrine system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Chemical properties of fluoride
The element fluorine has the highest electronegativity and the second highest electron affinity, making it highly reactive. At room temperature, fluorine exists as the gas F2, which reacts explosively with many elements. Fluorine is so reactive that it can form complexes with noble gases, most notably xenon (Holloway 1966). Due to its low stability, isolated fluorine is never found in nature. Instead, fluorine is either found as a complex or in its ionized form, fluoride.
Fluoride interacts with many cations, including hydrogen and a wide variety of metals. It is the only halide with a positive pKa (3.2), and therefore exists in acidic environments as its protonated form (HF). HF, commonly released as industrial or volcanic fumes, turns gaseous above 20 ℃. Fluoride is most toxic in its protonated form, and vertebrates that reside in areas near HF production often show symptoms of lung damage and fluoride toxicity.
Fluoride readily associates with metals. This affinity is driven by three factors: a negative Gibbs free energy for formation, a high stability constant, and poor solubility of the metal–fluoride complex. The energy state and stability of metallo-fluorides are much greater than that of other metallo-halides, and as such fluoride can often displace metal interacting partners in nature. The most favorable metal reactions with fluoride are aluminum, calcium, and magnesium; the most stable are aluminum, iron, and beryllium. Overall, the most favorable metal to bind fluoride is aluminum (Skelton 1971; Martin 1996). Aluminum is also the most abundant metal on the earth’s crust, and only micromolar levels of aluminum are necessary to complex with fluoride (Mullenix 2014; Berger et al. 2015). In terms of biological relevancy, calcium and magnesium are at much higher abundance and form complexes with fluoride in vivo (Marier 1980; Spencer et al. 1980). Interestingly, both of these metals are highly insoluble when complexed with fluoride. As calcium- or magnesium-fluoride precipitates out of solution, these complexes do not readily associate with other elements.
The abundancy of fluoride in nature
Fluoride gradually accumulates in the environment from volcanic emissions, dissolution of minerals, and industrial byproducts. Fluoride is present at roughly 300–900 ppm throughout the earth’s crust, and is estimated to be 200-fold more abundant in the mantle (Weinstein and Davison 2004; Koga and Rose-Koga 2018). Therefore, magma and magmatic gas contain fluoride, both unbound and complexed with hydrogen, silicon, and ammonium (Das and Behera 2008). These molecules settle as ash into the nearby soil and groundwater. Monitoring of fluoride particles in ash from the 1991 eruption of Mt. Hudson in Chile found that fluoride was highest in ash deposited furthest from the volcano; fluoride completely dissolved out of the ash and seeped into the surrounding soil and water sources after the first rainfall (Rubin et al. 1994). Historically, organisms in areas with high volcanic activity have displayed signs of fluoride poisoning (Olsen and Fruchter 1986; Witham et al. 2005; Fluek and Smith-Fluek 2013). Today, volcanic regions with high fluoride include Sicily, New Zealand, Iceland, the East African Rift, China, and South India (Cronin et al. 2003; Bellomo 2006; D'Alessandro 2006).
Fluorine can also be found in 296 different species of minerals, the most abundant of which are fluorspar (CaF2), fluorapatite (Ca5(PO4)3F), topaz (Al2(SiO4)(F,OH)2), and cryolite (Na3AlF6) (Mineralogy Database; Garcia and Borgnino 2015). These occur as vein deposits associated with igneous rocks, especially metallic minerals. Some of the largest deposits of fluoro-minerals are found in China, Mexico, Mongolia, South Africa, and Russia; this occurrence correlates strongly with areas whose populations suffer from endemic fluoride toxicity (Fig. 1) (Kilgore and Pelham 1987; Gupta and Ayoob 2016). Endemic fluoride toxicity in humans occurs from the chronic consumption of over 1.5 ppm (75 µM) fluoride.

Global distribution of fluoride. Shown here are areas with a < 1.5 ppm fluoride in the groundwater (purple), b mining of fluoro-compounds (blue squares -fluorspar, brown circles—fluorapatite, green triangles—topaz, and black diamonds—cryolite), c percent of the population given government-regulated fluoridated water (light teal 0–33%, teal 34–67%, black 68–100% of population), and d endemic fluoride toxicity (red) (Qian et al. 1999; WHO 2004; Gupta and Ayoob 2016, British Geological Survey, and the USGS Database) (color figure online)
Demineralization leads to an increased concentration of fluoride in the soil and water. Over time, minerals naturally break down through weathering and erosion. Most fluoride–metal complexes, especially fluorspar and fluorapatite, are poorly soluble in water (Moreno et al. 1974; Pan and Darvell 2007). Without further dissolution, the fluoride–metal complexes distribute into the soil where they are absorbed by plants and microbes. However, alkaline groundwater (pH > 8) can solubilize fluoride from minerals. The East African Rift Valley has several alkaline lakes near volcanic sites; there fluoride concentrations in groundwater range from 2.1 to 9.0 ppm (100–500 µM) (Malago et al. 2017). Fluoride dissolution from minerals is also favored under conditions with geothermal water, low concentrations of calcium and other metals, and areas with strong evaporation (Jha et al. 2013; Bouzourra et al. 2015; Batabyal and Gupta 2017). Acid rain has similar effects. The acidity releases sodium bicarbonates into the water, which in turn react with and dissolve fluoride minerals (Nath and Dutta 2010; Salifu et al. 2012). Over time, fluoride eventually deposits into the ocean. Oceanic water naturally contains fluoride, which cycles in and out of the atmosphere through the water cycle (Carpenter 1969). This fluoride is thought to be from the breakdown of marine sediments, such as phosphate rock. Free fluoride is present in the ocean at 1.2–1.4 ppm (60–80 µM) (WHO 2004).
Fluoride is also released into the air, water, and soil during mining and industrial processes. Fluoride is either used in a reaction, such as in aluminum smelting or glass production, or released as a byproduct, such as in phosphate fertilizer production, ceramic production and coal burning (Monfort et al. 2008; Gouider et al. 2010; Seixas et al. 2010). From these processes, excess fluoride is released into the environment as fumes or in groundwater (Roy et al. 2017). Phosphate fertilizer is a particular problem for widespread fluoride groundwater contamination, as fluoride is both released during the breakdown of phosphate ore, and makes up an additional 1.5–3% of the final fertilizer, which then enters the environment (McLaughlin et al. 1996; Li et al. 2015). For fluoride released as fumes, coal burning poses a substantial health risk. Fluoride is emitted as fumes upon coal combustion. In areas with indoor coal usage, many individuals show signs of fluoride toxicity (Li and Cao 1994; Ando et al. 2001). In 1997, an estimated 31 Mio. people in China showed signs of fluoride toxicity from coal intake (Ando et al. 2001). Fluoride travels efficiently through the air before precipitating back into the soil and water, so fluoride emissions can lead to higher fluoride exposure in areas far from the original industrial site (Walna et al. 2013).
Fluoride exposure can also vary based on diet. All foods contain fluoride. Some of the highest concentrated food sources occur from fluoride-accumulating plants, such as tomato, spinach, grapes, tea, and elderberry. The Hitchcock lab found that tomatoes grown in 10 mM NaF (190 ppm) accumulated 900 ppm fluoride (dry weight) in their leaves (Jacobsen et al. 1966). However, the amount of fluoride in plants depends strongly on the fluoride concentration in the soil; U.S.-grown tomatoes typically contain around 0.02 ppm fluoride. Plants such as tea accumulate fluoride with age. In populations with heavy tea consumption, fluoridated tea is believed to be the primary mechanism of adult fluoride toxicity (Cao et al. 2003). In many developed countries, a large majority of fluoride exposure comes from government-instituted fluoridated water (0.7–1.5 ppm), toothpaste (typically 1000–2500 ppm), dental gel (12,300 ppm) and varnish (23,000 ppm) (Cappelli and Mobley 2008; Pretty 2016).
Benefits of water fluoridation
The practice of adding fluoride to the public water supply has, in general, increased overall dental health. The first documentation of fluoride’s effects began with three independent reports from Italy, America, and the U.K. in the early 1900s noting that individuals with mottled, brown-stained teeth had lower incidences of dental caries (Eager 1901; McKay 1917; Ainsworth 1933). H.V. Churchill, who originally attributed the effect to high aluminum exposure, later found fluoride to be the causative agent preventing caries and triggering the skeletal defect later known as “fluorosis” (Churchill 1931). Fluoride was first added to the public water supply in Grand Rapids, Michigan in 1945 as a method of caries prevention. As of 2019, 24 countries participate in water fluoridation. An estimated 30–60% of caries incidences have been reduced because of water fluoridation (Armfield 2010). Because of this advancement, the CDC named water fluoridation as one of the top ten greatest public health achievements of the twentieth century (CDC 1999).
Controlled exposure to fluoride increases overall teeth quality through enamel replacement and the killing of plaque-causing bacteria. Fluoride reacts with tooth enamel because of its high affinity to metals. Tooth enamel is comprised mostly of the mineral hydroxyapatite (Ca10(PO4)6)(OH)2). Fluoride readily displaces the hydroxide to form fluorapatite (Ca5(PO4)3)F) (Fig. 2). Under normal conditions, bacteria like Streptococcus mutans ferment along the enamel, producing acid that gradually dissociates the hydroxyapatite (critical pH 5.5) (Featherstone 2008). When fluoride is present, it scavenges excess phosphate and calcium in the saliva for partial tooth remineralization, as well as displaces the hydroxyl group in remaining enamel (Featherstone 1999; Amaechi and van Loveren 2013). The resulting fluorapatite is more resistant to acidity (critical pH 4.5) than normal enamel, and as such, individuals exposed to fluoride have less tooth decay and more enamel than individuals with no fluoride exposure (Slade et al. 2018). For this reason, fluoride is added to the water sources of many countries.

Fluoride interactions in vitro.a Fluoride is the most similar in terms of size and charge to hydroxide, but has a much higher affinity for metals. b Unit cells of crystallized hydroxyapatite (Ca5(PO4)3OH) and fluorapatite (Ca5(PO4)3F) (Minerology Database). c Crystal structures of fluoride bound to urease and d phosphoserine phosphatase. Structures were generated on PyMOL using RCSB PDB c 4GOA and d 1L7N
Toxicity from the interaction of fluoride with metals
While low doses of fluoride are beneficial for overall teeth integrity, high doses of fluoride lead to a myriad of toxicity phenotypes. Broadly, fluoride triggers oxidative stress, cell cycle arrest, and apoptosis. While the exact mechanism of fluoride toxicity is unknown, the stress phenotypes are generally attributed to the inhibition of proteins, organelle disruption, altered pH, and electrolyte imbalance (Adamek et al. 2005; Barbier et al. 2010; Agalakova and Gusev 2011). These four mechanisms occur to varying degrees depending on the concentration of fluoride, its route of administration in multicellular organisms, and each cell’s surrounding environment. There is not a complete consensus over which downstream stress phenotype is linked to each mechanism, as each stressor can independently trigger oxidative stress and apoptosis. However, most papers attribute the primary mechanism of fluoride toxicity with its ability to inhibit metalloproteins (Adamek et al. 2005; Agalakova and Gusev 2011). A common practice in the study of fluoride toxicity is to look for reduced activity in an organelle or pathway under fluoride exposure, and then find essential metalloproteins within that organelle/pathway. However, many pathways show altered rates at lower fluoride exposure than that needed to inhibit essential proteins in vitro. This suggests either a still unidentified primary target, or that fluoride initiates toxicity through a combination of many targets.
Inhibition of metalloproteins by ionized fluoride
Fluoride’s properties as a protein inhibitor were established prior to the discovery of its properties in preventing dental caries. Starting in the late 1800s, sodium fluoride was identified as a lipase inhibitor (Tappeiner 1889; Loevenhart and Peirce 1906). Later fluoride was found to inhibit a range of phosphatases, kinases, hydrolases, and other metalloproteins in vitro. As of 2018, the protein databank has collected over 700 proteins crystallized in complex with fluoride (RCSB). Over 100 of these have had separate, independent enzymatic studies for fluoride inhibition in vitro (Adamek et al. 2005). While fluoride has been reported in a few select cases to bind directly to amino acids or to displace essential hydroxides, the majority of noted protein interactions are through either (1) fluoride binding to an essential metal in a metalloprotein’s active site, or (2) the complexing of fluoride with metal and acting as a substrate mimic (Edwards et al. 1984; Adamek et al. 2005; Schenk et al. 2008).
An estimated 30–50% of proteins require metal; consequently, there are thousands of potential targets for fluoride inhibition (Ascone et al. 2003). Fluoride is negatively charged, and associates with positive sites on proteins. In the case of metalloproteins, fluoride interacts with the essential metals, forming a highly stable, often insoluble complex. Fluoride can also form ternary complex with metal and phosphate, which has even greater stability than metallo-fluoride (Qin et al. 2006). Many pathways, particularly glycolysis, nutrient transport, and cellular respiration are inhibited during fluoride exposure, presumably through metalloprotein inhibition (Feig et al. 1971; Fina et al. 2014; Rogalska et al. 2017).
One of the most cited protein targets for fluoride inhibition is enolase. This enzyme catalyzes the penultimate step of glycolysis, converting 3-phosphoglycerate to phosphoenolpyruvate. The ability of fluoride to bind enolase was discovered accidentally by Warburg and Christian (Warburg and Christian 1941). Their laboratory in Germany happened to use water high in fluoride while working to crystallize enolase. During structural analysis, they found fluoride bound to phosphate and magnesium at enolase’s active site. Later, enzymatic analysis found that enolase has one of the lowest KDs for fluoride, at 1–10 mM (20–200 ppm) depending on species (Cimasoni 1972; Shahed et al. 1980; Maurer and Nowak 1981; Qin et al. 2006). Although enolase inhibition is often referenced as the primary target of fluoride, several papers have offered data against this hypothesis. Fluoride-resistant species of bacteria had no significant change in enolase activity or sequence compared to wild type (Van Loveren et al. 2008; Mitsuhata et al. 2014; Liao et al. 2015). RNA-Seq analyses of mammalian and plant cells exposed to fluoride reported no changes in enolase expression (Li et al. 2017; Pereira et al. 2018). Overexpression of enolase in fluoride-sensitized yeast produced no change in fluoride resistance (Johnston and Strobel 2019). Clinical researchers, using fluoride to inhibit glucose consumption in stored blood, found that metabolic activity decreased only after the induction of stress signaling from high fluoride (Montagnana and Lippi 2017). One alternative explanation is that metabolism is inhibited during fluoride exposure as a side effect of stress induction, such as oxidative stress or intracellular acidification. In agreement with this hypothesis, several studies found that acidification alone reduced glycolysis to a greater degree than fluoride treatment (Boink et al. 1994; Belli et al. 1995; Gambino et al. 2009). Regardless of the mechanism, glycolysis is consistently inhibited across organism models during fluoride toxicity.
Inhibition of proteins by metallo-fluoride substrate mimics
Fluoride toxicity is greatly enhanced when complexed with metal. Among the most toxic (and most studied) complexes are aluminum- (AlF3,4) and beryllium fluoride (BeF3,4). These compounds are isomorphous to phosphate, and consequently able to inhibit phosphoryl-transfer enzymes (Chabre 1990). Nonetheless, the affinity for each metallo-fluoride to various enzymes depends on the pH and interaction with essential positively charged amino acids (Schlichting and Reinstein 1999; Strunecka et al. 2002). Over 100 enzymes have been crystallized with aluminum- or beryllium fluoride, the majority of which are classified as either ATPases, GTPases, or kinases (Berman et al. 2000).
Aluminum- and beryllium fluoride can also alter the phosphorylation state of various proteins, particularly through GTP mimicry. Many proteins, including G-proteins, actin, and microtubules, are regulated by GTP binding; GDP-bound proteins are in an “off” state, while GTP-bound proteins are “on”. In the case of aluminum- or beryllium-fluoride exposure, both metallo-fluorides bind GDP to mimic the bound γ-phosphate of GTP (Bigay et al. 1987; Antonny and Chabre 1991). AlFx and BeFx stabilize the transition state for the “on” conformation. Consequently, proteins activated by metallo-fluoride are more stable than those activated naturally, and stay in the “on” conformation (Li 2003). Because of the far-reaching roles of GTPase regulation, their non-selective activation by metallo-fluorides leads to wide dysregulation of functions including cell signaling, transport, and cytoskeleton integrity (Muhlrad et al. 1994; Loweth et al. 1996; Li 2003; Agalakova and Gusev 2011).
Free fluoride can also activate protein and pathway activity by altering phosphorylation states. Rho GTPase-binding proteins have been shown to bind to GTPases and stabilize the transition state in a fluoride-, but not aluminum-dependent manner (Antonny et al. 1993; Vincent et al. 1998). Instead, in both cases, magnesium was found to be essential, and could theoretically form a magnesium-fluoride phosphate mimic.
Fluoride-induced pH and electrolyte imbalance
At both the single- and multi-cellular levels, fluoride exposure causes acidification and electrolyte imbalance. The exact mechanism is unknown. Prolonged exposure of vertebrates to high fluoride results in the loss of calcium and magnesium from the plasma, and an excess of potassium (Dalamaga et al. 2008). Complementary to this finding, fluoride exposure in single cells results in an influx of calcium and magnesium, and a loss of potassium (Johnston and Strobel 2019). This effect has been proposed to be due to either downstream stress signaling, or the binding of fluoride to metals (Boink et al. 1994; Giachini and Pierleoni 2004). Regardless of mechanism, the imbalance of electrolytes in organisms from fluoride exposure has far reaching implication in cell homeostasis and signaling disruption.
Fluoride exposure is also associated with a drop in intracellular pH. Fluoride is a weak acid that enters cells as HF and dissociates, thus releasing one proton per fluoride. Consequently, the more fluoride that enters a cell, the more acidic the cytoplasm becomes. However, fluoride causes intracellular acidification to a larger degree than can be explained by proton shuttling (Kawase and Suzuki 1989; Belli et al. 1995; Guha-Chowdhury et al. 1996; Marquis et al. 2003; Gassowska et al. 2013). Many hypotheses have been put forward to explain this observation. Among these, the most common are metabolic disruption, perturbation of the mitochondria, transmembrane protein inhibition, and induction of stress signaling (Belli et al. 1995; Marquis et al. 2003; Gassowska et al. 2013).
Fluoride inhibits metabolism through an unclear mechanism, the downstream effects include reduction in intracellular ATP and damage to the mitochondria. ATP is reduced both in cells containing mitochondria—which display signs of permanent damage and reduced respiration after fluoride exposure—and in erythrocytes (red blood cells), which lack mitochondria and produce ATP solely through anaerobic glycolysis (Feig et al. 1971; Agalakova and Gusev 2011; Fina et al. 2014). ATP depletion leads to the hydrolysis of ATP into ADP and AMP along with the release of protons, consequently leading to intracellular acidification. Damage to the mitochondria releases free radicals, resulting in oxidative stress. This in turn causes DNA damage, metabolic disruption, ATP hydrolysis, protein inhibition, and intracellular acidification (Boonstra and Post 2004). However, just as free radicals are known to disrupt metabolism, acidify the cell, and activate stress signaling, each of these phenotypes also activate the release of free radicals from the mitochondria (Chen et al. 2003; Liu et al. 2003; Berezhnov et al. 2016). As such, the order in which each known phenomenon occurs is difficult to determine.
Another potential explanation for the drop in pH is the inhibition of transmembrane proton transporters by fluoride. Key among them are the Na+/H+ symporters, ATPases, and G-coupled proteins. Fluoride generally inhibits transmembrane proteins by changing the protein’s confirmation to its transitional state, such as metallo-fluoride forcing Na+/K+-ATPase into its E2P state (Montes et al. 2015; Faraj et al. 2019). Na+/H+ symporters, ATPases, and G-coupled proteins regulate a wide range of cellular processes, including pH homeostasis (Loweth et al. 1996; Palmgren et al. 2010; Syrovatkina et al. 2016). In the case of the Na+/H+ symporter, inhibition by fluoride would theoretically accumulate extracellular protons. However, the opposite occurs (Kawase and Suzuki 1989). Kawase and Suzuki proposed that this extracellular proton buildup would result in increased fluoride protonation, which would, therefore, increase overall fluoride toxicity and stress signaling. It could also be that while this particular protein inhibition raises extracellular pH, the overall inhibition of transmembrane transporters leads to a net accumulation of protons in the cytoplasm.
Organelle disruption by fluoride
Prolonged exposure to high levels of fluoride leads to widespread organelle damage. This damage is both time- and concentration-dependent. While the sensitivity of each organelle to fluoride varies slightly by organism, in general, fluoride disrupts the cell surface, mitochondria, endoplasmic reticulum, Golgi, and nucleus (Fig. 3).

Intracellular fluoride toxicity. General scheme of downstream organelle damage after prolonged exposure to high fluoride, conserved across eukaryotes
Fluoride can irreversibly damage the cell surface. Organisms that have calcium-pectate in their cell walls, such as plants, are prone to calcium depletion upon fluoride exposure (Tsunoda and Yu 1985). The plasma membrane during fluoride exposure is prone to lipid peroxidation and cytoskeleton rearrangement (Wang et al. 2004; Liang et al. 2015; Chen et al. 2017). This effect is not specific to fluoride; many stressors are known to induce membrane remodeling (Farah et al. 2011; Brandao et al. 2014; Westman et al. 2019). While most membrane changes are attributed to either oxidative stress, apoptotic signaling or lipid peroxidation, free or metallo-fluoride can also directly bind actin and change its polymerization (Combeau and Carlier 1989; Allen et al. 1996). Regardless of membrane damage, cell cycle arrest during fluoride exposure is enhanced with an increased uptake in fluoride. This suggests that fluoride’s principal mechanism of toxicity at high doses is intracellular (Ji et al. 2014).
The organelle most widely reported to be inhibited by fluoride is the mitochondria. Many mitochondrial proteins are metalloproteins and have been linked to fluoride inhibition, including respiratory complexes I–IV and F-ATPase (Batenburg and van den Bergh 1972; Sutton et al. 1987; Fina et al. 2014; Zhao et al. 2019). Either directly or indirectly, fluoride exposure damages mitochondrial membrane integrity (Yan et al. 2015). This reduces the overall activity of the mitochondria, inhibits cellular respiration, and triggers leakage of free radicals and cytochrome c, ultimately inducing oxidative stress (Miller and Miller 1974; Anuradha et al. 2001; Jothiramajayam et al. 2014). Not surprisingly, the addition of antioxidants partially rescues from fluoride toxicity (Basha and Sujitha 2011). The last few years have seen renewed interest in mitochondrial inactivation under fluoride exposure as the primary mechanism of fluoride toxicity, as much of the known adverse effects can be attributed to free radicals (Farrugia and Balzan 2012; Yan et al. 2015; Lu et al. 2017).
Fluoride-exposed cells undergo intensive DNA damage, presumably through free radical oxidation. Fluoride triggers both single- and double-stranded DNA damage following oxidative stress (Podder et al. 2015). Due to DNA damage, high fluoride eventually leads to S-phase cell cycle arrest (Wang et al. 2004). Typically, studies that expose mice or rats to high concentrations of fluoride for weeks to months report DNA damage. The necessity of a long incubation time suggests that DNA damage is a downstream stress effect rather than a direct target of fluoride.
The endoplasmic reticulum (ER) activates stress signaling under fluoride exposure. The mechanism by which fluoride causes ER stress is unclear. Prolonged fluoride exposure damages the ER membrane, as well as induces the unfolded protein response pathway (Matsuo et al. 2000; Kubota et al. 2005). Fluoride also disrupts Ca2+ homeostasis in the ER (Borke and Whitford 1999; Zhang et al. 2016). These phenotypes could either be through the direct scavenging of Ca2+ by fluoride, the interaction of fluoride with ER proteins such as Ca2+-ATPase, or the oxidative stress signaling pathway, which involves the release of Ca2+ into the cytoplasm (Murphy and Coll 1992; Borke and Whitford 1999; Ermak and Davies 2002). In support of the hypothesis that ER stress is linked with oxidative stress, the addition of ER stress inhibitors reduced DNA damage upon fluoride exposure (Kubota et al. 2005).
Fluoride inhibits Golgi stacking, although this effect is reversible. Aluminum fluoride (AlF4−) has been shown to activate G-proteins by mimicking the gamma-phosphate of GTP (Bigay et al. 1987). As G-proteins function in signal transduction, their activation has far-reaching implications, especially in vesicle-mediated exocytosis and Golgi function. Activation of plasma membrane G-proteins by aluminum fluoride leads to a calcium-dependent activation of exocytosis (Elferink et al. 1980). G-proteins are also found on the Golgi, and are essential for Golgi stacking (van Hook 2015). The Rothman lab demonstrated that addition of aluminum fluoride to a cell-free system inhibited protein transport between Golgi stacks (Melancon et al. 1987). Aluminum-fluoride also alters the protein coating assembly along the Golgi (Finazzi et al. 1994; Tomas et al. 2010). Later studies in neuroendocrine cells found that free fluoride also inhibited Golgi stacking by interfering with matrix and cisternae protein assembly, and that this effect was reversed within 2 h of recovery (Back et al. 2004). However, the fluoride may still have been in a metal complex; a study by the Lowe lab demonstrated that in the absence of aluminum, fluoride will combine with magnesium to bind G-proteins (Graham et al. 1999). As a whole, fluoride reversibly inhibits Golgi function, although this might only happen when fluoride is complexed with metal.
Fluoride exposure inhibits protein synthesis, an effect linked to both stress-signaling and ribosome inhibition. Stress pathways, particularly oxidative stress, are known to inhibit protein synthesis (Sheikh and Fornace 1999; Liu and Qian 2014). This effect also occurs during ATP depletion, a well-established fluoride effect (Freudenberg and Mager 1971). Unsurprisingly then, fluoride exposure has been shown to reduce the turnover of new proteins (Hongslo and Holland 1979; Sharma et al. 2008). Additionally, fluoride exposure in vitro causes the reversible inhibition of polyribosome formation, and “NaF ribosomes”: ribosomes thought to either contain an extra 40S subunit, or a deacetylated tRNAMet bound to the 40S complex (making a 43S subunit) (Ravel et al. 1966; Bishop 1968; Culp et al. 1970; Sameshima et al. 1972; Godchaux III and Atwood IV 1976; Holland 1979; Hoerz and McCarty 1969). Overall, the exposure to fluoride results in inhibition of polypeptide chain initiation (Ravel et al. 1966; Mosteller et al. 1967; O’Rourke and Godchaux III 1975). The research on fluoride’s interaction with ribosomes effect was mostly conducted in the 1960–1970s; consequently, the direct action of fluoride on ribosomes has been largely forgotten. Fluoride has been widely recorded to halt protein translation, but this effect is generally linked to apoptotic signaling. Recent RNA-Seq data have reported strongly repressed ribosomal subunit expression during fluoride exposure, indicating that this underappreciated phenotype may play a role in fluoride toxicity (Melo et al. 2017; Li et al. 2017; Johnston and Strobel 2019).
Fluoride exposure and toxicity in vertebrates
Most vertebrates are exposed to fluoride through their diet. Fluoride is present in food and water, where after ingestion fluoride passes along the gastrointestinal tract and into the plasma. Over time, fluoride accumulates in soft tissue such as the spleen, kidney, and especially the bone, leading to potential chronic toxicity.
Acute fluoride toxicity has only been reported in individuals exposed to very high concentrations of fluoride. Typically, symptoms include nausea, diarrhea, headaches, and gastric pain after ingestion of 5–8 mg/kg bodyweight (Ullah et al. 2017). Chronic exposure to high fluoride has been correlated with inflammatory bowel disease, disruption of tissue lining, and nerve damage along the gastrointestinal tract (Das et al. 1994; Follin-Arbelet and Moum 2016; de Oliveira et al. 2017; Melo et al. 2017).
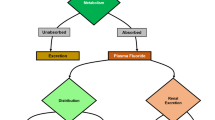
Exposure of tissues to fluoride depends on the surrounding pH. Protonated fluoride (HF) readily passes through biological membranes, and can spread to tissues far from the gastrointestinal tract. Consequently, organisms in contact with HF gas can experience cellular toxicity in tissues far from the original area of exposure (Sheridan et al. 1995). The mouth, esophagus, and upper stomach are at neutral pH (6.5–7.5, 5.0–7.0 and 4.0–6.5, respectively) (Fallingborg 1999; Tutuian and Castell 2006; Baliga et al. 2013). However, the lower stomach is highly acidic (pH 1.5–3.5). Given that fluoride has a pKa of 3.2, 67–96% of fluoride forms HF and can dissociate in equilibrium across the stomach lining. Nonetheless, only 25% of ingested fluoride is thought to be absorbed by the stomach and pass on to other cells; the rest of the fluoride is moved to the intestines (Kanduti et al. 2016). The majority of ingested fluoride is absorbed by the small intestine. Just ~ 10% of consumed fluoride is never absorbed by the body and excreted through feces (Whitford 1994; Buzalaf et al. 2015). Fluoride has been hypothesized to pass through the small intestines as its anion form F− (Nopakun and Messer 1990). Because of fluoride’s reactivity, the nutrients ingested along with fluoride, especially those rich in calcium, influence the amount of fluoride absorbed by the intestines (Whitford 1994). Once absorbed, fluoride travels throughout the body via the blood stream, before being filtered by the kidney and excreted in urine. Approximately 45–60% of ingested fluoride is excreted in urine, with the rest re-circulated into the plasma or deposited into the bone (Buzalaf et al. 2015).
Dental fluorosis
The risk for developing adverse symptoms from fluoride exposure is dose dependent. Typically, the concentration of fluoride in water is regulated to be between 0.7 and 1.2 ppm (40–60 µM) (USDHHS 2015). Chronic toxicity to low doses of fluoride occurs after prolonged exposure to > 1.5 ppm (75 µM) fluoride. This toxicity, known as dental fluorosis, is characterized by mottled and discolored teeth. Fluorosis generally correlates with a high degree of fluorapatite formation. Fluorapatite is mechanically weaker than hydroxyapatite, and the replacement of natural enamel with fluorapatite increases the brittleness of teeth (DenBesten and Li 2011). Fluoride most readily forms fluorapatite as fresh enamel is developing; as such, vertebrates are most sensitive to dental fluorosis during tooth development. For humans, juveniles up to 8 years of age (with particular sensitivity during the first 2 years) are susceptible to fluorosis (Hong et al. 2006; Bhagavatula et al. 2016).
Ameloblast toxicity
Although it is well established that fluoride triggers fluorapatite formation in teeth and that excess fluoride leads to fluorosis, the full mechanism of dental fluorosis is unknown. Several mechanisms besides remineralization have been put forward, most notably that fluorosis is caused by ameloblast damage (Fejerskov et al. 1977). Ameloblasts, or cells that function in depositing enamel, are present in the gums of organisms developing new teeth. Fluoride exposure to ameloblasts in cell culture causes endoplasmic reticulum stress, DNA fragmentation, cytoskeleton defects, protein synthesis inhibition, reduced secretion of enamel matrix proteins, and eventual apoptosis (Kubota et al. 2005; Li et al. 2005; Hassanuma et al. 2007; Bronckers et al. 2009). However, it is important to note that these studies are typically conducted at much higher fluoride (100–150 ppm) than levels causing dental fluorosis in humans (> 1.5 ppm).
Skeletal fluorosis
Skeletal fluorosis is the most severe form of chronic fluoride toxicity. The underlying mechanics of skeletal fluorosis are similar to dental fluorosis: high fluoride exposure leads to a change in mineral formation, as well as stress to surrounding cells. The uptake of fluoride into bone results in the conversion of the bone mineral hydroxyapatite into fluorapatite, which alters the general bone lattice and reduces its overall strength (Grynpas 1990). As such, vertebrates exposed to high fluoride have mechanically weaker bones (Evans and Wood 1976; Sogaard et al. 1995). This increased brittleness is associated with skeletal dysmorphia and higher risk of fracture.
The onset of skeletal fluorosis in humans varies by individual. The reported dosage required for developing skeletal fluorosis varies widely, and is affected by age, metabolic rate, genetic disposition, and overall health of the individual (Marier et al. 1963; Krishnamachari 1986; USDHHS 1991; Pramanik and Saha 2017). It is estimated that for the average person, 6–10 mg per day of fluoride ingestion for at least 10 years leads to skeletal fluorosis (Whitford 1996). Children are the most susceptible, and accumulate fluoride at a greatly accelerated rate compared with adults (Teotia et al. 1998). Patients with kidney disorders are also at high risk for skeletal fluorosis, given that kidneys are essential for filtering fluoride (Gerster et al. 1983; Krishnamachari 1986; Bansal and Tiwari 2006).
Skeletal fluorosis affects surrounding osteoblasts. Low fluoride concentrations (5–1000 µM, or 0.1–20 ppm in vitro), stimulates osteoblasts (Hall 1987; Lau and Baylink 1998; Qu et al. 2008). The exact mechanism by which fluoride causes osteoblast proliferation is unclear, although it has been shown that fluoride causes an uptake in phosphate, increased alkaline phosphatase activity, decreased acid phosphatase activity, and an activation of the MAPK pathway (Farley et al. 1983; Lau et al. 1989, 2002; Selz et al. 1991). The stimulation of osteoblasts triggers new bone formation and an increase in overall bone density in patients with over 0.2% fluoride content in bones (Aaron et al. 1991). However, the bone matrix is disrupted by fluoride, and the improved bone mass is of lower quality and more likely to fracture. Fluoride above 1 mM (20 ppm) results in osteoblast cytotoxicity, particularly to the nucleus and endoplasmic reticulum (Qu et al 2008; Zhou et al. 2013; Liu et al. 2015). As such, there is an overall bimodal trend of low fluoride stimulating bone mass, while high fluoride decreases bone mass.
Neurotoxicity
There are many claims as to fluoride’s long-term toxicity in the human body that lack thorough scientific studies, but hold a sympathetic belief in the public. Probably, the most well known is the concern that fluoride exposure leads to neuronal damage, including Parkinson’s disease, Alzheimer’s disease, and a reduced IQ. While there have been several studies to explore this connection, the evidence is insufficient or even anti-correlative.
For a molecule to cause neurotoxicity, it must pass through the blood–brain barrier. The blood–brain barrier only allows passive diffusion of small, uncharged, lipid soluble compounds, or molecules that can pass through selective channels, pumps, or vesicles (Wong et al. 2013). In the case of fluoride, the ion is highly electronegative and would only pass through the blood–brain barrier as HF. As blood has a pH between 7.3 and 7.45, the majority of fluoride would remain in its unprotonated form (Kellum 2000). Consequently, the levels of fluoride detected in brain tissue are typically much lower than the concentration of fluoride in serum, and generally lower than all other tissues in the body (Whitford 1996).
Many studies have found correlations between low IQ and high fluoride exposure. Over 60 studies have been conducted in areas with high fluoridated groundwater, and most of these studies have reported a lower average IQ in the children of those regions compared with children in areas with normal fluoride exposure (Tang et al. 2008; Aravind et al. 2016; Green et al. 2019). Several other studies have been conducted and found either no correlation between fluoride and IQ, or that high fluoride correlated with higher IQs (Spittle et al. 1998; He and Zhang 2010; Li et al. 2010; Soto-Barreras et al. 2019). Each of these studies do not fully take into consideration socioeconomic factors, unconscious biases, as well as other possible toxicants in groundwater that could be affecting IQ. The areas of interest—primarily China, India, and Mexico—frequently have groundwater high in other neurotoxicants such as arsenic and mercury (Wang et al. 2007; UNEP 2013). In a systematic review of fluoride-IQ studies, the Grandjean lab concluded that if fluoride is a neurotoxicant, it would be over 1000 times less potent than other known neurotoxicants (Choi et al. 2012). Because of each of these factors, the correlation of high fluoride exposure and lower IQ does not necessarily imply causation.
In a laboratory setting, high doses of fluoride are toxic to neuronal cells. Neuronal cell lines exposed to ≥ 60 ppm (3 mM) NaF undergo DNA damage, oxidative stress, mitochondrial agglutination, and cytoskeleton damage (Zhang et al. 2008; Chen et al. 2017; Tu et al. 2018). Because the primary function of neurons is synaptic signaling, membrane defects from fluoride exposure reduce the overall activity of neurons. Rats and mice exposed to high fluoride (≥ 50 ppm, or 2.5 mM) showed decreased nicotinic acetylcholine receptor expression, lowered acetyl cholinesterase activity, and damaged myelin and microtubules (Long et al. 2002; Basha and Sujitha 2012; Niu et al. 2015, 2018). Nonetheless, tests done with ≤ 50 ppm (2.5 mM) fluoride showed no significant brain damage (Varner et al. 1998; Shivarajashankara et al. 2002).
Several studies have argued that fluoride damages neuronal cells through an indirect mechanism. The Rigalli lab suggested that the alteration of glucose and insulin homeostasis in the blood during fluoride exposure could lead to nutrient depletion and downstream stress in the nervous system (Lombarte et al. 2016). However, the Nowak lab reported an enhancement in glucose uptake in brain tissue upon treatment of rats with 50 ppm (2.5 mM) fluoride (Rogalska et al. 2017). Others have argued that the release of free radicals by either the intake of ≥ 60 ppm (3 mM) NaF, or the intragastric injection of ≥ 20 mg/kg body weight NaF—about four times more concentrated than that needed to cause acute gastrointestinal toxicity in humans—could have negative effects on neuronal tissue, particularly the hippocampus (Bhatnagar et al. 2002; Pan et al. 2015; Shanmugam et al. 2018). Again, these studies rely on exposure to high doses of fluoride, well above that typically found in fluoridated water.
While free fluoride is unlikely to pass through the blood–brain barrier, metallo-fluoride, specifically aluminum fluoride, can cause neurotoxicity. Aluminum fluoride (AlF3 or AlF4) is a phosphate mimic, and could theoretically cross the blood–brain barrier through phosphate transporters (Strunecka et al. 2002). Rats exposed to 10 ppm NaF and 100 ppm AlCl3 in combination for 30 days showed neuronal shrinkage and inhibition of acetylcholinesterase activity (Akinrinade et al. 2015). Furthermore, aluminum fluoride was found to cause more histopathological changes to brain tissue than sodium fluoride alone, particularly in the neocortex and hippocampus (Varner et al. 1998; NRC 2006). However, it is also possible that the neurotoxicity is due to free aluminum. Several studies of brain defects found the highest association with aluminum exposure, not fluoride levels (Forbes et al. 1991; Kraus and Forbes 1992; Jacqmin et al. 1994). Free aluminum acts as a neurotoxicant, disrupting the cell membrane integrity of the blood–brain barrier, activating the innate immune response, and potentially increasing dementia (Banks and Kastin 1989; Armstrong et al. 1996). As such, aluminum could be at least partially responsible for fluoride neurotoxicity.
Diabetes
Given that fluoride alters cellular metabolism, there have been concerns about the effect of fluoride on sugar homeostasis and diabetes. Prolonged exposure to high fluoride inhibits glycolysis and ATP production. Cells respond to fluoride stress, as well as to many acids, by increasing glucose uptake (Hay and Paul 1967; Rogalska et al. 2017). The direct intraperitoneal injection of mammals with high fluoride, resulting in at least 0.1 mg/L total fluoride in the blood, can lead to higher blood glucose (McGown and Suttie 1977; Suketa et al. 1985; NRC 2006). However, studies with rats fed 15–50 ppm (0.8–3 mM) fluoride showed either no change, or a decrease in serum glucose levels (Lupo et al. 2011; Lobo et al. 2015; Malvezzi et al. 2019).
Fluoride exposure has also been demonstrated to alter insulin concentrations in the blood. Fluoride reversibly inhibits insulin secretion, leading to an overall reduced insulin concentration in the serum (Rigalli et al. 1990; Menovo et al. 2005). However, exposure to low (10 ppm, or 0.5 mM) doses of fluoride enhances insulin sensitivity (Lobo et al. 2015). In all, chronic exposure to high fluoride may partially contribute to diabetes, while low fluoride exposure may be protective against diabetes.
Endocrine disruption
Fluoride was officially classified by the National Research Council in 2006 as an endocrine disruptor for its ability to inhibit the thyroid at high concentrations. However, its mechanism of action remains unknown (NRC 2006). Exposure to fluoride can lead to a decrease in the thyroid hormones triiodothyronine (T3) and thyroxine (T4). Studies that have reported a significant decrease of T3 and T4 by fluoride typically involve either patients with dental fluorosis, or mammals exposed to 30–80 ppm (2–4 mM) fluoride for ≥ 2 months (Bobek et al. 1976; Jiang et al. 2015). On a cellular level, the thyroid undergoes DNA damage, membrane disruption, mitochondrial and endoplasmic reticulum stress, and oxidative stress signaling during fluoride exposure (Sundstrom 1971; Liu et al. 2016; Abdelaleem et al. 2018). Cells exposed to both excessive fluoride and iodide showed synergistic triggering of endoplasmic stress, IRE1 signaling, and DNA damage (Liu et al. 2014, 2016; Jiang et al. 2015). The resulting hypothyroidism alters the body’s ability to regulate temperature, metabolism, and heart rate, which has far-reaching implications for patients with severe fluorosis. Given that fluoride does not accumulate in the thyroid, this toxicity is attributed to high fluoride in the blood (Galletti and Joyet 1958; Singh et al. 2014).
In 2015, the public nominated fluoride for investigation by the United States National Toxicology Program (NTP) for its potential role in non-thyroidal endocrine disruption, cancer, and neurological disorders. After conducting a systematic review, the NTP cited insufficient research on low doses of fluoride to investigate the real risk of these effects (NTP 2017). Research conducted with high dosages of fluoride reports direct and indirect inhibitory effects on the endocrine system, independent of the thyroid. The most notable targets are the pineal gland, adrenal glands, and the parathyroid. The pineal gland lies outside of the blood–brain barrier and has the highest calcification rate of any organ in the body (Tan et al. 2018). Fluoride has been shown to gradually accumulate in the pineal gland along with calcium (Luke 2001; Kalisinska et al. 2014). Few studies have been conducted on whether fluoride accumulation significantly disrupts pineal gland activity. The key study, conducted in 1997, reported that prepubescent gerbils fed 40 ppm (2 mM) NaF daily had lower melatonin production by the pineal gland than the controls, but that melatonin was restored to normal concentrations when the gerbils reached adulthood (Luke 1997). Patients with skeletal fluorosis (the most severe form of fluoride toxicity), are sometimes found to have secondary hyperparathyroidism (Teotia and Teotia 1973). This has been suggested as the body’s attempt to restore calcium and phosphate homeostasis, rather than the direct disruption of the parathyroid by fluoride (Faccini 1969; Krishnamachari 1986).
Resistance to fluoride
Many organisms have evolved defense mechanisms against fluoride. The first discovery of a fluoride-specific defense pathway occurred in 2012, when the Breaker lab identified a region of RNA, known as a riboswitch, conserved in many bacteria and archaea (Baker et al. 2012). Riboswitches are located on some mRNA and control downstream gene expression upon binding to ligands. In the case of the fluoride riboswitch, the RNA coordinates with three magnesium ions to bind fluoride at a KD of 60 uM (1.1 ppm) (Ren et al. 2012). Over 2000 examples of the fluoride riboswitch were identified, and found to control expression of many genes linked to fluoride resistance (Baker et al. 2012). Included in this list were genes functioning in oxidative stress, DNA repair, and intracellular acidification. There were also genes for proteins known to be inhibited by fluoride, including enolase, Na+/H+ antiporters, and pyrophosphatase. Third, there were two newly discovered fluoride channels: EriCF and Fluc.
The transporters EriCF and Fluc confer significant fluoride resistance. EriCF is a member of the Clc family of membrane proteins, and acts as a F−/H+ antiporter (Lim et al. 2013). Fluc is believed to be a channel whose driving force for fluoride efflux is the electrochemical gradient of the bacterial plasma membrane (Stockbridge et al. 2013; Ji et al. 2014). Fluoride exporters are an essential part in mediating fluoride’s toxic effects, conferring a 200-fold increase in resistance to bacterial growth arrest. In yeast, this resistance is even more pronounced, with over 1000-fold increased resistance to fluoride (Li et al. 2013). While the fluoride transporter is conserved across many species of eukaryotes and prokaryotes, no homolog has been identified in vertebrates. Nonetheless, fluoride sensitivity varies across species and tissue type, suggesting there is an as yet undiscovered mechanism of defense.
Microbes have evolved multiple mechanisms of fluoride resistance. These resistance factors are generally found by either isolating organisms from areas with high fluoride, or exposing cells to fluoride in a laboratory. Several studies identified fluoride-resistant bacteria that express higher copies of fluoride transporters, as well as higher copies of known fluoride targets (Liao et al. 2015, 2016; Liu et al. 2017). In one of the few reports that did not find an altered fluoride channel, fluoride-resistant S. mutans adjusted their composition of fatty acids and had enhanced general acid resistance (Zhu et al. 2012). This correlation of fluoride resistance with acid resistance has been widely observed, although the mechanism has never been found (Sheng and Liu 2000; Marquis et al. 2003). A DNA microarray of a resistant strain of A. ferrooxidans offered one of the most complete pictures of fluoride tolerance, showing a change in expression of genes related to metabolism, protein synthesis, and cell membrane maintenance (Ma et al. 2016). As a whole, microbes appear to gain fluoride resistance by increasing expression of fluoride transporters and protein targets of fluoride inhibition, most notably ATPases and glycolytic enzymes (Fig. 4).

Global network of cellular processes involved in the resistance of bacteria to fluoride. a Conserved molecular functions and cellular components of (A) genes regulated by the fluoride riboswitch, as reported by Weinberg et al. (2010), and b altered genes in fluoride resistant bacteria (Zhu et al. 2012; Liao et al. 2015, 2016; Ma et al. 2016; Liu et al. 2017). Genes were converted to their E. coli homologs, and duplicates were discarded. Data was analyzed on Cytoscape using ClueGO. Node size corresponds with the number of genes per category, and similar colored nodes denote a similar cluster in function
Mammalian cells are also capable of gaining fluoride resistance. A study comparing mice that were either resistant or sensitive to developing fluorosis showed differences on chromosomes 2 and 11, although these differences were comprised of nearly 2000 genes (Everett et al. 2009). An RNA-Seq study investigating gene expression differences in a fluoride-resistant mouse adipose cell line showed increased expression in genes related to general stress response, protein synthesis, and cell membrane maintenance (Ran et al. 2017). Studies on mammalian resistance to fluoride have not yet found a link with overexpressing glycolytic enzymes, although RNA-Seq of rats after 20–60 day exposure to 50 ppm (3 mM) NaF showed an increased expression of genes related to glucose uptake (Pereira et al. 2018). Continued investigation into fluoride toxicity and the concurrent mechanism of resistance across species is a much-needed avenue for fully understanding the biological effects of fluoride exposure.
Concluding remarks
The issue of whether fluoride is safe depends on the sensitivity of the organism, the concentration of fluoride, and the conditions by which fluoride is administered. Fluoride has both positive and negative effects. Low fluoride levels decrease cavities and partially restore the minerals in teeth. High levels of fluoride lead to protein inhibition, a release of free radicals, disruption of metal homeostasis, and tissue damage. The question then becomes how much fluoride an organism will encounter during their lifetime.
Most organisms are in regions with low- to mid-range fluoride, and are at low risk to experience fluoride toxicity. Consequently, the majority of the world’s population have no visible signs of fluorosis. However, the inhabitants of certain regions around the world, including in India, China, and Africa, have to be particularly aware of the fluoride levels to which they are exposed. In these areas, there is emerging interest on the safe removal of fluoride from the groundwater and air.
References
Aaron JE, de Vernejoul MC, Kanis JA (1991) The effect of sodium fluoride on trabecular architecture. Bone 12:307–310. https://doi.org/10.1016/8756-3282(91)90015-B
Abdelaleem MM, El-Tahawy NFG, Abozaid SMM, Abdel-Hakim SA (2018) Possible protective effect of curcumin on the thyroid gland changes induced by sodium fluoride in albino rats: light and electron microscopic study. Endocr Regul 52:59–68. https://doi.org/10.2478/enr-2018-0007
Adamek E, Palowska-Goral K, Bober K (2005) In vitro and in vivo effects of fluoride ions on enzyme activity. Ann Acad Med Stetin 51:69–85
Agalakova NI, Gusev GP (2011) Molecular mechanisms of cytotoxicity and apoptosis induced by inorganic fluoride. Int Sch Res Not 2012:16. https://doi.org/10.5402/2012/403835
Ainsworth NJ (1933) Mottled teeth. Brit. Dent J 55:233–250
Akinrinade ID, Memudu AE, Ogundele OM (2015) Fluoride and aluminum disturb neuronal morphology, transport functions, cholinesterase, lysosomal and cell cycle activities. Pathophysiol 22:105–115. https://doi.org/10.1016/j.pathophys.2015.03.001
Allen PG, Laham LE, Way M, Janmey PA (1996) Binding of phosphate, aluminum fluoride, or beryllium fluoride to f-actin inhibits severing by gelsolin. JBC 271:4665–4670. https://doi.org/10.1074/jbc.271.9.4665
Amaechi BT, van Loveren C (2013) Fluorides and non-fluoride remineralization systems. Monogr Oral Sci 23:15–26. https://doi.org/10.1159/000350458
Ando M, Tadano M, Yamamoto S et al (2001) Health effects of fluoride pollution caused by coal burning. Sci Total Environ 271:107–116. https://doi.org/10.1016/S0048-9697(00)00836-6
Antonny B, Chabre M (1991) Characterization of the aluminum and beryllium fluoride species which activate transducin. JBC 267:6710–6718
Antonny B, Sukumar M, Bigay J, Chabre M, Higashijima T (1993) The mechanism of aluminum-independent G-protein activation by fluoride and magnesium. JBC 268:2393–2402
Anuradha CD, Kanno S, Hirano S (2001) Oxidative damage to mitochondria is a preliminary step to caspase-3 activation in fluoride-induced apoptosis in HL-60 cells. Free Radic Bio Med 31:367–373. https://doi.org/10.1016/S0891-5849(01)00591-3
Aravind A, Dhanya RS, Narayan A et al (2016) Effect of fluoridated water on intelligence in 10–12-year-old school children. Int Soc Prev Community Dent 6:237–242. https://doi.org/10.4103/2231-0762.197204
Armstrong RA, Winsper SJ, Blair JA (1996) Aluminium and Alzheimer's disease:review of possible pathogenic mechanisms. Dement Geriatr Cogn Dis 7:1–9. https://doi.org/10.1159/000106845
Ascone I, Fourme R, Hasnain SS (2003) Introductory overview: X-ray absorption spectroscopy and structural genomics. J Synchotron Radiat 10:1–3. https://doi.org/10.1107/s0909049502022434
Armfield JM (2010) Community effectiveness of public water fluoridation in reducing children's dental disease. Publ Health Rep 125:655–664. https://doi.org/10.1177/003335491012500507
Back N, Litonius E, Mains RE, Eipper BA (2004) Fluoride causes reversible dispersal of Golgi cisternae and matrix in neuroendocrine cells. Eur J Cell Biol 83:389–402. https://doi.org/10.1078/0171-9335-00405
Baker JL, Sudarsan N, Weinberg Z et al (2012) Widespread genetic switches and toxicity resistance proteins for fluoride. Science 335:233–235. https://doi.org/10.1126/science.1215063
Baliga S, Muglikar S, Kale R (2013) Salivary pH:a diagnostic biomarker. J Ind Soc Periodontol 17:461–465. https://doi.org/10.4103/0972-124X.118317
Banks WA, Kastin AJ (1989) Aluminum-induced neurotoxicity:alterations in membrane function at the blood-brain barrier. Neurosci Behav Rev 13:47–53. https://doi.org/10.1016/S0149-7634(89)80051-X
Bansal R, Tiwari SC (2006) Back pain in chronic renal failure. Nephrol Dial Transplant 21:2331–2332
Barbier O, Arreola-Mendoza L, Del Razo LM (2010) Molecular mechanisms of fluoride toxicity. Chem Bio Interact 188:319–333. https://doi.org/10.1016/j.cbi.2010.07.011
Basha PM, Sujitha NS (2011) Chronic fluoride toxicity and myocardial damage: antioxidant offered protection in second generation rats. Toxiol Int 18:99–104. https://doi.org/10.4103/0971-6580.84260
Basha PM, Sujitha NS (2012) Combined impact of exercise and temperature in learning and memory performance of fluoride toxicated rats. Biol Trace Elem Res 150:306–313. https://doi.org/10.1007/s12011-012-9489-3
Batabyal AK, Gupta S (2017) Fluoride-contaminated groundwater of Birbhum district, West Bengal, India:interpretation of drinking and irrigation suitability and major geochemical processes using principal component analysis. Environ Monit Assess 189:369. https://doi.org/10.1007/s10661-017-6041-0
Batenburg JJ, van den Bergh SG (1972) The mechanism of inhibition by fluoride of mitochondrial fatty acid oxidation. BBA 280:495–505. https://doi.org/10.1016/0005-2760(72)90129-4
Belli E, Buckley DH, Marquis RE (1995) Weak acid effects and fluoride inhibition of glycolysis by Streptococcus mutans GS-5. Can J Microbiol 41:785–791. https://doi.org/10.1139/m95-108
Bellomo S (2006) Environmental impact of magmatic fluorine emission in the Mt. Etna Area. Dissertation, University of Cambridge.
Berezhnov AV, Soutar MPM, Fedotova EI et al (2016) Intracellular pH modulates autophagy and mitophagy. JBC 291:8701–8708. https://doi.org/10.1074/jbc.M115.691744
Berger T, Mathurin F, Gustagsson JP, Peltola P, Astrom ME (2015) The impact of fluoride on Al abundance and speciation in boreal streams. Chem Geol 419:118–124. https://doi.org/10.1016/j.chemgeo.2015.05.013
Berman HM, Westbrook J, Feng Z et al (2000) The Protein Data Bank. Nucl Acids Res 28:235–242. https://doi.org/10.1093/nar/28.1.235
Bhagavatula P, Levy SM, Broffitt B, Weber-Gasparoni K, Warren JJ (2016) Timing of fluoride intake and dental fluorosis on late-erupting permanent teeth. Community Dent Oral Epidemiol 44:32–45. https://doi.org/10.1111/cdoe.12187
Bhatnagar M, Rhao P, Jain S (2002) Neurotoxicity of fluoride: neurodegeneration in hippocampus of female mice. Ind J Exp Biol 40:546–554
Bigay J, Deterre P, Pfister C, Chabre M (1987) Fluoride complexes of aluminum or beryllium act on G-proteins as reversibly bound analogues of the gamma phosphate of GTP. EMBO J 6:2907–2913
Bishop JO (1968) Effect of puromycin and sodium fluoride on reticulocyte ribosomal monomers and subribosomal particles. Arch Biochem Biophys 125:449–451. https://doi.org/10.1016/0003-9861(68)90601-2
Bobek S, Kahl S, Ewy Z (1976) Effect of long-term fluoride administration on thyroid hormones level blood in rats. Endocrinol Exp 10:289–295
Boink AB, Wemer J, Meulenbelt J, Vaessen HA, de Wildt DJ (1994) The mechanism of fluoride-induced hypocalcaemia. Hum Exp Toxicol 13:149–155. https://doi.org/10.1177/096032719401300302
Boonstra J, Post JA (2004) Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 337:1–13. https://doi.org/10.1016/j.gene.2004.04.032
Borke JL, Whitford GM (1999) Chronic fluoride ingestion decreases 45Ca uptake by rat kidney membranes. J Nutr 129:1209–1213. https://doi.org/10.1093/jn/129.6.1209
Bouzourra H, Bouhlila R, Elango L, Slama F, Ouslati N (2015) Characterization of mechanisms and processes of groundwater salinization in irrigated coastal area using statistics, GIS, and hydrogeochemical investigations. Environ Sci Pollut Res 22:2643–2660. https://doi.org/10.1007/s11356-014-3428-0
Brandao RL, Rosa JCC, Nicoli JR et al (2014) Investigating acid stress response in different Saccharomyces strains. J Mycol 2014:9. https://doi.org/10.1155/2014/178274
Bronckers ALJJ, Lyaruu DM, DenBesten PK (2009) The impact of fluoride on ameloblasts and the mechanisms of enamel fluorosis. J Dent Res 88:877–893. https://doi.org/10.1177/0022034509343280
Buzalaf CP, Leite ADL, Buzalaf MA (2015) Fluoride metabolism. In: Preedy VR (ed) Fluorine: chemistry, analysis, function and effects. Royal Society of Chemistry, Cambridge, pp 54–72
Cao J, Zhao Y, Liu J, Xirao R, Danzeng S, Daji D, Yan Y (2003) Brick tea fluoride as a main source of adult fluorosis. Food Chem Toxicol 41:535–542. https://doi.org/10.1016/s0278-6915(02)00285-5
Cappelli DP, Mobley CC (2008) Prevention in clinical oral health care. Elsevier, St. Louis, Missouri
Carpenter R (1969) Factors controlling the marine geochemistry of fluorine. Geochem Cosmochim Acta 33:1153–1167. https://doi.org/10.1016/0016-7037(69)90038-6
CDC (1999) Ten great public health achievements. MMWR 48:241–243
Chabre M (1990) Aluminofluoride and beryllofluoride complexes: new phosphate analogs in enzymology. Trends Biochem Sci 15:6–10. https://doi.org/10.1016/0968-0004(90)90117-T
Chen L, Ning H, Yin Z et al (2017) The effects of fluoride on neuronal function occur via cytoskeleton damage and decreased signal transmission. Chemosphere 185:589–594. https://doi.org/10.1016/j.chemosphere.2017.06.128
Chen Q, Chai YC, Mazumder S et al (2003) The late increase in intracellular free radical oxygen species during apoptosis is associated with cytochrome c release, caspase activation, and mitochondrial dysfunction. Cell Death Diff 10:323–334. https://doi.org/10.1038/sj.cdd.4401148
Choi AL, Sun G, Zhang Y, Grandjean P (2012) Developmental fluoride neurotoxicity: a systematic review and meta-analysis. Environ Health Perspect 120:1362–1368. https://doi.org/10.1289/ehp.1104912
Churchill HV (1931) Occurrence of fluorides in some waters of the United States. Indus Eng Chem 23:996–998. https://doi.org/10.1021/ie50261a007
Cimasoni G (1972) The inhibition of enolase by fluoride in vitro. Caries Res 6:93–102. https://doi.org/10.1159/000259782
Combeau C, Carlier MF (1989) Characterization of the aluminum and beryllium fluoride species bound to F-actin and microtubules at the site of the y-phosphate of the nucleotide. JBC 264:19017–19021
Cronin S, Neall VE, Lecointre JA, Hedley MJ, Loganathan P (2003) Environmental hazards of fluoride in volcanic ash: a case study from Ruapehu volcano, New Zealand. J Volcan Geotherm Res 121:271–291. https://doi.org/10.1016/S0377-0273(02)00465-1
Culp W, Morrisey J, Hardesty B (1970) Initiator tRNA for the synthesis of globin peptides. Biochem Biophys Res Commun 40:777–785. https://doi.org/10.1016/0006-291X(70)90970-8
D'Alessandro W (2006) Human fluorosis related to volcanic activity: a review. Nat Inst Geophys Volcan 10:21–30. https://doi.org/10.2495/ETOX06003
Dalamaga M, Karmaniolas K, Nikolaidou A, Papadavid E (2008) Hypocalcemia, hypomagnesemia, and hypokalemia following hydrofluoric acid chemical injury. J Burn Bare Res 29:541–543. https://doi.org/10.1097/BCR.0b013e3181711152
Das TK, Susheela AK, Gupta IP, Dasarathy S, Tandon RK (1994) Toxic effects of chronic fluoride ingestion on the upper gastrointestinal tract. J Clin Gastroenterol 18:194–199
Das RC, Behera DK (2008) Environmental science: principles and practice. Prentice Hall, New Delhi, India
de Oliveira FA, MacVinish LJ, Amin S et al (2017) The effect of fluoride on the structure, function, and proteome of intestinal epithelia. Environ Toxicol 33:63–71. https://doi.org/10.1002/tox.22495
DenBesten P, Li W (2011) Chronic fluoride toxicity: dental fluorosis. Monogr Oral Sci 22:81–96. https://doi.org/10.1159/000327028
Eager JM (1901) Denti di chiaie (Chiaie teeth). Publ Health Rep 16:284–285
Edwards SL, Poulos TL, Kraut J (1984) The crystal structure of fluoride-inhibited cytochrome c peroxidase. JBC 259:12984–12988
Elferink JGR, Alsbach EJJ, Riemersma JC (1980) The interaction of fluoride with rabbit polymorphonuclear leukocytes: induction of exocytosis and cytolysis. Biochem Pharm 29:3051–3057. https://doi.org/10.1016/0006-2952(80)90445-1
Ermak G, Davies KJ (2002) Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol 38:713–772
Evans FG, Wood JL (1976) Mechanical properties and density of bone in a case of severe endemic fluorosis. Acta Orthop Scand 47:489–495. https://doi.org/10.3109/17453677608988726
Everett ET, Yan D, Weaver M et al (2009) Detection of dental fluorosis-associated quantitative trait loci on mouse chromosomes 2 and 11. Cells Tissues Org 189:212–218. https://doi.org/10.1159/000151383
Faccini J (1969) Fluoride and bone Calc Tiss Res 3:1–16
Fallingborg J (1999) Intraluminal pH of the human gastrointestinal tract. Dan Med Bull 46:183–196
Farah ME, Sirotkin V, Haarer B, Kakhniashvili D, Amberg DC (2011) Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton 68:340–354. https://doi.org/10.1002/cm.20516
Faraj S, Centeno M, Rossi RC, Montes MR (2019) A kinetic comparison between E2P and the E2P-like state induced by a beryllium fluoride complex in the Na, K-ATPase. Biochim Biophys Acta 1861:355–365. https://doi.org/10.1016/j.bbamem.2018.10.020
Farley JR, Wergedal JE, Baylink DJ (1983) Fluoride directly stimulates proliferation and alkaline phosphatase activity of bone-forming cells. Science 222:330–332. https://doi.org/10.1126/science.6623079
Farrugia G, Balzan R (2012) Oxidative stress and programmed cell death in yeast. Front Oncol 2:64. https://doi.org/10.3389/fonc.2012.00064
Featherstone JDB (1999) Prevention and reversal of dental caries: role of low-level fluoride. Community Dent Oral Epidem 27:31–40
Featherstone JDB (2008) Dental caries: a dynamic disease process. Aust Dent J 53:286–291. https://doi.org/10.1111/j.1834-7819.2008.00064.x
Feig SA, Shohet SB, Nathan DG (1971) Energy metabolism in human erythrocytes: effects of sodium fluoride. J Clin Invest 50:1731–1737. https://doi.org/10.1172/JCI106662
Fejerskov O, Thylstrup A, Larsen MJ (1977) Clinical and structural features and possible pathogenic mechanisms of dental fluorosis. Scand J Dent Res 85:510–534. https://doi.org/10.1111/j.1600-0722.1977.tb02110.x
Fina BL, Lombarte M, Rigalli JP, Rigalli A (2014) Fluoride increases superoxide production and impairs the respiratory chain in ROS 17/2.8 osteoblastic cells. PLoS ONE 9:e100768. https://doi.org/10.1371/journal.pone.0100768
Finazzi D, Cassel D, Donaldson JG, Klausner RD (1994) Aluminum fluoride acts on the reversibility of ARF1-dependent coat protein binding to Golgi membranes. JBC 269:13325–13330
Fluek W, Smith-Fluek JA (2013) Severe dental fluorosis in juvenile deer linked to recent volcanic eruption in Patagonia. J Wildl Dis 49:355–366. https://doi.org/10.7589/2012-11-272
Follin-Arbelet B, Moum B (2016) Fluoride: a risk factor for inflammatory bowel disease? Scand J Gastroenter 51:1019–1024. https://doi.org/10.1080/00365521.2016.1177855
Forbes WF, Hayward LM, Agwani N (1991) Dementia, aluminum, and fluoride. Lancet 338:1592–1593
Freudenberg H, Mager J (1971) Studies on the mechanism of the inhibition of protein synthesis induced by intracellular ATP depletion. Biochim Biophys Acta 232:537–555. https://doi.org/10.1016/0005-2787(71)90608-3
Galletti PM, Joyet G (1958) Effect of fluorine on thyroidal iodine metabolism in hyperthyroidism. J Clin Endocrinol Metab 18:1102–1110
Gambino R, Piscitelli J, Ackattupathil TA et al (2009) Acidification of blood is superior to sodium fluoride alone as an inhibitor of glycolysis. Clin Chem 55:1019–1021. https://doi.org/10.1373/clinchem.2008.121707
Garcia MG, Borgnino L (2015) Fluorine: chemistry, analysis, function, and effects. RSC Publishing, Cambridge
Gassowska M, Gutowska I, Baranowska-Bosiacka I, Chlubek D (2013) Effect of fluoride on sodium-proton exchanger activity, intracellular pH and calcium concentration in human non-stimulated platelets. Ann Acad Med Stetin 59:54–61
Gerster JC, Charhon SA, Jaeger P et al (1983) Bilateral fractures of femoral neck in patients with moderate renal failure receiving fluoride for spinal osteoporosis. Br Med J 287:723–725. https://doi.org/10.1136/bmj.287.6394.723
Giachini M, Pierleoni F (2004) Fluoride toxicity. Minerva Stomatol 53:171–177
Godchaux W III, Atwood KC IV (1976) Structure and function of initiation complexes which accumulate during inhibition of protein synthesis by fluoride ion. JBC 251:292–301
Gouider M, Feki M, Sayadi S (2010) Bioassay and use in irrigation of untreated and treated wastewaters from phosphate fertilizer industry. Ecotoxicol Environ Saf 73:932–938. https://doi.org/10.1016/j.ecoenv.2009.12.021
Graham DL, Eccleston JF, Chung CW, Lowe PN (1999) Magnesium fluoride-dependent binding of small G proteins to their GTPase-activating proteins. Biochem J 38:14981–14987
Green R, Lanphear B, Hornung R et al (2019) Association between maternal fluoride exposure during pregnancy and IQ scores in offspring in Canada. JAMA Ped. https://doi.org/10.1001/jamapediatrics.2019.1729
Grynpas MD (1990) Fluoride effects on bone crystals. J Bone Miner Res 5:169–175. https://doi.org/10.1002/jbmr.5650051362
Guha-Chowdhury N, Iwami Y, Yamada T (1996) Effect of low levels of fluoride on proton excretion and intracellular pH in glycolysing streptococcal cells under strictly anaerobic conditions. Caries Res 31:373–378
Gupta AK, Ayoob S (2016) Fluoride in drinking water: status, issues, and solutions. Florida, Boca Raton
Hall BK (1987) Sodium fluoride as an initiator of osteogenesis from embryonic mesenchyme in vitro. Bone 8:111–116
Hassanuma RM, Filho EVZ, Ceolin DS et al (2007) Ultrastructural and immunohistochemical study of the influence of fluoride excess on the development of rat incisor tooth buds. J Appl Oral Sci 15:292–298. https://doi.org/10.1590/S1678-77572007000400010
Hay RJ, Paul J (1967) Factors influencing glucose flux and the effect of insulin in cultured human cells. J Gen Physiol 50:1663–1680. https://doi.org/10.1085/jgp.50.6.1663
He M, Zhang C (2010) Investigation of children’s intelligence quotient and dental fluorosis in drinking water-type of endemic fluorosis area in Pucheng county Shaanxi province before and after drinking water change. Chin J Epidem 29:547–548
Hoerz W, McCarty KS (1969) Evidence for a proposed initiation complex for protein synthesis in reticulocyte polyribosome profiles. PNAS 63:1206–1213. https://doi.org/10.1073/pnas.63.4.1206
Holland RI (1979) Fluoride inhibition of protein synthesis. Cell Biol Int Rep 3:701–705. https://doi.org/10.1016/0309-1651(79)90074-2
Holloway J (1966) The photochemical reaction of xenon with fluorine at room temperature: a demonstration of the reactivity of xenon. J Chem Educ 43:202. https://doi.org/10.1021/ed043p202
Hong L, Levy SM, Broffitt B et al (2006) Timing of fluoride intake in relation to development of fluorosis on maxillary central incisors. Community Dent Oral Epidemiol 34:299–309
Hongslo JK, Holland RI (1979) Effect of sodium fluoride on protein and DNA synthesis, ornithine decarboxylase activity, and polyamine content in LS cells. Acta Pharmacol Toxicol 44:350–353
Jacobsen JS, Weinstein LJ, McCune DC, Hitchcock AE (1966) The accumulation of fluorine by plants. J Air Pollut Control Assoc 16:412–417. https://doi.org/10.1080/00022470.1966.10468494
Jacqmin H, Commenges D, Letenneur L, Barberger-Gateau P, Dartigues JF (1994) Components of drinking water and risk of cognitive impairment in the elderly. Am J Epidemiol 139:48–57. https://doi.org/10.1093/oxfordjournals.aje.a116934
Jha SK, Singh RK, Damodaran T et al (2013) Fluoride in groundwater:toxicological exposure and remedies. J Toxicol Environ Health B Crit Rev 16:52–66. https://doi.org/10.1080/10937404.2013.769420
Ji C, Stockbridge RB, Miller C (2014) Bacterial fluoride resistance, Fluc channels, and the weak acid accumulation effect. J Gen Physiol 144:257–261. https://doi.org/10.1085/jgp.201411243
Jiang Y, Guo X, Sun Q, Shan Z, Teng W (2015) Effects of excess fluoride and iodide on thyroid function and morphology. Biol Trace Elem Res 170:382–389. https://doi.org/10.1007/s12011-015-0479-0
Johnston NR, Strobel SA (2019) Nitrate and phosphate transporters rescue fluoride toxicity in yeast. Chem Res Toxicol 32:2305–2319. https://doi.org/10.1021/acs.chemrestox.9b00315
Jothiramajayam M, Sinha S, Ghosh M et al (2014) Sodium fluoride promotes apoptosis by generation of reactive oxygen species in human lymphocytes. J Toxicol Environ Health 77:1269–1280. https://doi.org/10.1080/15287394.2014.928658
Kalisinska E, Bosiacka-Baranowska I, Lanocha N et al (2014) Fluoride concentrations in the pineal gland, brain and bone of goosander (Mergus merganser) and its prey in Odra River estuary in Poland. Environ Geochem Health 36:1063–1077. https://doi.org/10.1007/s10653-014-9615-6
Kanduti D, Sterbenk P, Artnik B (2016) Fluoride: a review of use and effects on health. Mater Sociomed 28:133–137. https://doi.org/10.5455/msm.2016.28.133-137
Kawase T, Al S (1989) Fluoride-induced cytoplasmic acidification: possible role of protein kinase c in BCECF-loaded L929 cells. Pharm Toxicol 64:426–428
Kellum JA (2000) Determinants of blood pH in health and disease. Crit Care 4:6–14. https://doi.org/10.1186/cc644
Kilgore CC, Pelham L (1987) The worldwide availability of fluorspar. Nat Resour Forum 11:127–140
Koga K, Rose-Koga E (2018) Fluorine in the earth and the solar system, where does it come from and can it be found? C R Chim 21:749–756. https://doi.org/10.1016/j.crci.2018.02.002
Kraus AS, Forbes WF (1992) Aluminum, fluoride and the prevention of Alzheimer's disease. Eur PMC 83:97–100
Krishnamachari KA (1986) Skeletal fluorosis in humans: a review of recent progress in the understanding of the disease. Prog Food Nutr Sci 10:279–314
Kubota K, Lee DH, Tsuchiya M et al (2005) Fluoride induces endoplasmic reticulum stress in ameloblasts responsible for dental enamel formation. JBC 280:23194–23202. https://doi.org/10.1074/jbc.M503288200
Lau KH, Farley JR, Freeman TK, Baylink DJ (1989) A proposed mechanism of the mitogenic action of fluoride on bone cells: Inhibition of the activity of an osteoblastic acid phosphatase. Metabolism 38:858–868. https://doi.org/10.1016/0026-0495(89)90232-1
Lau KH, Baylink DJ (1998) Molecular mechanism of action of fluoride on bone cells. JBMR 13:1660–1667. https://doi.org/10.1359/jbmr.1998.13.11.1660
Lau KH, Goodwin C, Arias M, Mohan S, Baylink DJ (2002) Bone cell mitogenic action of fluoroaluminate and aluminum fluoride but not that of sodium fluoride involves upregulation of the insulin-like growth factor system. Bone 30:705–711. https://doi.org/10.1016/S8756-3282(02)00671-3
Li JX, Cao SR (1994) Recent studies on endemic fluorosis in China. Fluoride 27:125–128
Li L (2003) The biochemistry and physiology of metallic fluoride: action, mechanism, and implications. Crit Rev Oral Biol Med 14:100–114
Li QS, Lin XM, Qiao RY et al (2017) Effect of fluoride treatment on gene expression in tea plant (Camellia sinensis). Nat Sci Rep 7:9847. https://doi.org/10.1038/s41598-017-08587-6
Li S, Smith KD, Davis JH et al (2013) Eukaryotic resistance to fluoride toxicity mediated by a widespread family of fluoride export proteins. PNAS 110:19018–19023. https://doi.org/10.1073/pnas.1310439110
Li X, Hou G, Yu B et al (2010) Investigation and analysis of children's IQ and dental fluorosis in a high fluoride area. Chin J Pest Control 26:230–231
Li Y, Decker S, Yuan Z et al (2005) Effects of sodium fluoride on the actin cytoskeleton of murine ameloblasts. Arch Oral Biol 50:681–688. https://doi.org/10.1016/j.archoralbio.2004.11.021
Li Y, Zhang H, Zhang Z, Shao L, He P (2015) Treatment and resource recovery from inorganic fluoride-containing waste produced by the pesticide industry. J Environ Sci 1:21–29. https://doi.org/10.1016/j.jes.2014.10.016
Liang S, Zhao MH, Ock SA, Kim NH, Cui XS (2015) Fluoride impairs oocyte maturation and subsequent embryonic development in mice. Environ Toxicol 31:1486–1495. https://doi.org/10.1002/tox.22153
Liao Y, Chen J, Brandt BW et al (2015) Identification and functional analysis of genome mutations in a fluoride-resistant Streptococcus mutans strain. PLoS ONE 10:e0122630. https://doi.org/10.1371/journal.pone.0122630
Liao Y, Brandt BW, Zhang M et al (2016) A single nucleotide change in the promoter Mutp enhances fluoride resistance of Streptococcus mutans. Antimicrob Agents Chemother 60:7509–7512. https://doi.org/10.1128/AAC.01366-16
Lim H, Stockbridge RB, Miller C (2013) Fluoride-dependent interruption of the transport cycle of a CLC Cl−/H+ antiporter. Nat Chem Biol 9:721–725. https://doi.org/10.1038/nchembio.1336
Liu B, Qian SB (2014) Translational reprogramming in stress response. Wiley Interdiscip Rev RNA 5:301–305. https://doi.org/10.1002/wrna.1212
Liu H, Zeng Q, Cui Y et al (2014) The effects and underlying mechanism of excessive iodide on excessive fluoride-induced thyroid cytotoxicity. Environ Toxicol Pharm 38:332–340. https://doi.org/10.1016/j.etap.2014.06.008
Liu H, Hou C, Zeng Q et al (2016) Role of endoplasmic reticulum stress-induced apoptosis in rat thyroid toxicity caused by excess fluoride and/or iodide. Environ Toxicol Pharmacol 46:277–285. https://doi.org/10.1016/j.etap.2016.08.007
Liu L, Zhang Y, Gu H, Zhang K, Ma L (2015) Fluorosis induces endoplasmic reticulum stress and apoptosis in osteoblasts in vivo. Biol Trace Elem Res 164:64–71. https://doi.org/10.1007/s12011-014-0192-4
Liu X, Tian J, Liu L et al (2017) Identification of an operon involved in fluoride resistance in Enterobacter cloacae FRM. Nat Sci Rep 7:6786. https://doi.org/10.1038/s41598-017-06988-1
Liu Y, Song XD, Liu W, Zhang TY, Zuo J (2003) Glucose deprivation induces mitochondrial dysfunction and oxidative stress in PC12 cell line. J Cell Mol Med 7:49–56. https://doi.org/10.1111/j.1582-4934.2003.tb00202.x
Lobo JG, Leite AL, Pereira HA, Fernandes MS, Peres-Buzalaf C, Sumida DH, Rigalli A, Buzalaf MA (2015) Low-level fluoride exposure increases insulin sensitivity in experimental diabetes. J Dent Res 94:990–997. https://doi.org/10.1177/0022034515581186
Loevenhart AS, Peirce G (1906) The inhibiting effect of sodium fluoride on the action of lipase. JBC 2:397–413
Lombarte M, Fina BL, Lupion PM, Lupo M, Rigalli A (2016) In vivo measurement of fluoride effects on glucose homeostasis: an explanation for the decrease in intelligence quotient and insulin resistance induced by fluoride. Fluoride 49:204–210
Long YG, Wang YN, Chen J et al (2002) Chronic fluoride toxicity decreases the number of nicotinic acetylcholine receptors in rat brain. Neurotoxicol Teratol 24:751–757. https://doi.org/10.1016/s0892-0362(02)00273-8
Loweth AC, Williams GT, Scarpello JH, Morgan NG (1996) Heterotrimeric G-proteins are implicated in the regulation of apoptosis in pancreatic beta-cells. Exp Cell Res 229:69–76. https://doi.org/10.1006/excr.1996.0344
Lu Y, Luo Q, Cui H et al (2017) Sodium fluoride causes oxidative stress and apoptosis in the mouse liver. Aging 9:1623–1639. https://doi.org/10.18632/aging.101257
Luke J (1997) The effect of fluoride on the physiology of the pineal gland. Thesis, University of Surrey, Guildford
Luke J (2001) Fluoride deposition in the aged human pineal gland. Caries Res 35:125–128. https://doi.org/10.1159/000047443
Lupo M, Buzalaf MA, Rigalli A (2011) Effect of fluoridated water on plasma insulin levels and glucose homeostasis in rats with renal deficiency. Biol Trace Elem Res 140:198–207. https://doi.org/10.1007/s12011-010-8690-5
Ma L, Li Q, Shen L et al (2016) Insights into the fluoride-resistant regulation mechanism of Acidithiobacillus ferrooxidans ATCC 23270 based on whole genome microarrays. J Ind Microbiol Biotechnol 43:1441–1453. https://doi.org/10.1007/s10295-016-1827-6
Malago J, Makoba E, Muzuka ANN (2017) Fluoride levels in surface and groundwater in Africa: a review. Am J Water Sci Eng 3:1–17. https://doi.org/10.11648/j.ajwse.20170301.11
Malvezzi MAPN, Pereira HABS, Dionizio A, Araujo TT, Buzalaf NR, Sabino-Arias IT, Fernandes MS, Grizzo LT, Magalhaes AC, Buzalaf MAR (2019) Low-level fluoride exposure reduces glycemia in NOD mice. Ecotoxicol Environ Saf 168:198–204. https://doi.org/10.1016/j.ecoenv.2018.10.055
Marier JR (1980) Observations and implications of the Mg-vs-F in interrelations in biosystems: a review. Proc Finn Dent Soc 76:93–102
Marquis RE, Clock SA, Mota-Meira M (2003) Fluoride and organic weak acids as modulators of microbial physiology. FEMS 26:493–510. https://doi.org/10.1111/j.1574-6976.2003.tb00627.x
Marier JR, Rose D, Boulet M (1963) Accumulation of skeletal fluoride and its implications. Arch Environ Health 6:664–671. https://doi.org/10.1080/00039896.1963.10663458
Martin RB (1996) Ternary complexes of Al3+ and F− with a third ligand. Coord Chem Rev 149:23–32. https://doi.org/10.1016/S0010-8545(96)90008-9
Matsuo S, Nakagawa H, Kiyomiya K, Kurebe M (2000) Fluoride-induced ultrastructural changes in exocrine pancreas cells of rats: fluoride disrupts the export of zymogens from the rough endoplasmic reticulum (rER). Arch Toxicol 73:611–617
Maurer PJ, Nowak T (1981) Fluoride inhibition of yeast enolase. Biochem 20:6894–6900
McGown EL, Suttie JW (1977) Mechanism of fluoride-induced hyperglycemia in the rat. Toxicol Appl Pharm 40:83–90. https://doi.org/10.1016/0041-008X(77)90119-3
McKay FS (1917) Investigation of mottled enamel and brown stain. JADA 4:273–278. https://doi.org/10.14219/jada.archive.1917.0054
McLaughlin MJ, Tiller KG, Naidu R, Stevens DP (1996) Review: the behavior and environmental impact of contaminants in fertilizers. Aust J Soil Res 34:1–54. https://doi.org/10.1071/SR9960001
Melancon P, Glick BS, Malhotra V et al (1987) Involvement of GTP-binding "G" proteins in transport through the Golgi stack. Cell 51:1053–1062
Melo CGS, Perles JVCM, Zanoni JN et al (2017) Enteric innervation combined with proteomics for the evaluation of the effects of chronic fluoride exposure on the duodenum of rats. Nat Sci Rep 7:1070. https://doi.org/10.1038/s41598-017-01090-y
Menovo I, Rigalli A, Puche RC (2005) Effect of fluoride on the secretion of insulin in the rat. Arzneimittelforschung 55:455–460
Miller JE, Miller GW (1974) Effects of fluoride on mitochondrial activity in higher plants. Physiol Plant 32:115–121. https://doi.org/10.1111/j.1399-3054.1974.tb03737.x
Minerology Database. http://www.webmineral.com/. Accessed 3 September 2019
Mitsuhata C, Puteri MM, Ohara Y, Tatsukawa N, Kozai K (2014) Possible involvement of enolase in fluoride resistance in Streptococcus mutans. Pediatr Dent J 24:12–16. https://doi.org/10.1016/j.pdj.2013.10.002
Monfort E, Garcia-Ten J, Celades I, Gazulla MF, Gomar S (2008) Evolution of fluorine emissions during the fast firing of ceramic tile. Appl Clay Sci 38:250–258. https://doi.org/10.1016/j.clay.2007.03.001
Montagnana M, Lippi G (2017) Overcoming preanalytical issues for diagnosing diabetes with fasting plasma glucose. Ann Transl Med 5:257. https://doi.org/10.21037/atm.2017.05.19
Montes MR, Ferreira-Gomes MS, Centeno M, Rossi RC (2015) The E2P-like state induced by magnesium fluoride complexes in the Na, K-ATPase. Biochim Biophys Acta 1848:1514–1523. https://doi.org/10.1016/j.bbamem.2015.03.023
Moreno EC, Kresak M, Zahradnik RT (1974) Fluoridated hydroxyapatite solubility and caries formation. Nature 247:64–65. https://doi.org/10.1038/247064a0
Mosteller RD, Culp WJ, Boyd H (1967) A reaction associated with nonenzymatic binding in the reticulocyte transfer system. PNAS 57:1817–1824. https://doi.org/10.1073/pnas.57.6.1817
Muhlrad A, Cheung P, Phan BC, Miller C, Reisler E (1994) Dynamic properties of actin: structural changes induced by beryllium fluoride. JBC 269:11852–11858
Mullenix PJ (2014) A new perspective on metals and other contaminants in fluoridation chemicals. Int J Occup Environ Health 20:157–166. https://doi.org/10.1179/2049396714Y.0000000062
Murphy AJ, Coll RJ (1992) Fluoride is a slow, tight-binding inhibitor of the calcium ATPase of sarcoplasmic reticulum. JBC 267:5229–5235
National Toxicology Program (2017) Protocol for systematic review of effects of fluoride exposure on neurodevelopment. US DHHS.
Nath SK, Dutta R (2010) Fluoride removal from water using crushed limestone. Indian J Chem Technol 17:120–125
National Research Council (2006) Fluoride in drinking water: a scientific review of EPA's standards. The National Academies Press, Washington DC
Niu R, Xue X, Zhao Y et al (2015) Effects of fluoride on microtubule ultrastructure and expression of Tubα1a and Tubβ2a in mouse hippocampus. Chemosphere 139:422–427. https://doi.org/10.1016/j.chemosphere.2015.07.011
Niu R, Chen H, Manthari RK et al (2018) Effects of fluoride on synapse morphology and myelin damage in mouse hippocampus. Chemosphere 194:628–633. https://doi.org/10.1016/j.chemosphere.2017.12.027
Nopakun J, Messer HH (1990) Mechanism of fluoride absorption from the rat small intestine. Nutr Res 10:771–779. https://doi.org/10.1016/S0271-5317(05)80826-7
Olsen K, Fruchter J (1986) Identification of the physical and chemical characteristics of volcanic hazards. AJPH 76:45–52
O'Rourke JC, Godchaux W III (1975) Fluoride inhibition of the initiation of protein synthesis in the reticulocyte lysate cell-free system. JBC 250:3443–3450
Palmgren MG, Baekgaard L, Lopez-Marques RL, Fuglsang AT (2010) Plasma membrane ATPases. Plant Plasma Membr Berlin. https://doi.org/10.1007/978-3-642-13431-9_7
Pan HB, Darvell BW (2007) Solubility of calcium fluoride and fluorapatite by solid titration. Arch Oral Biol 52:861–868. https://doi.org/10.1016/j.archoralbio.2007.03.002
Pan Y, Lu P, Yin L, Chen K, He Y (2015) Effect of fluoride on the proteomic profile of the hippocampus in rats. Z Naturforsch C J Biosci 70:151–157. https://doi.org/10.1515/znc-2014-4158
Pereira H, Dionizio AS, Araujo TT et al (2018) Proposed mechanism for understanding the dose- and time-dependency of the effects of fluoride in the liver. Toxicol Appl Pharmacol 358:68–75. https://doi.org/10.1016/j.taap.2018.09.010
Podder S, Ghoshal N, Banerjee A et al (2015) Interaction of DNA-lesions induced by sodium fluoride and radiation and its influence in apoptotic induction in cancer cell lines. Toxicol Rep 2:461–471. https://doi.org/10.1016/j.toxrep.2015.02.001
Pramanik S, Saha D (2017) The genetic influence in fluorosis. Environ Toxicol Pharmacol 56:157–162. https://doi.org/10.1016/j.etap.2017.09.008
Pretty IA (2016) High fluoride concentration toothpastes for children and adolescents. Caries Res 50:9–14. https://doi.org/10.1159/000442797
Qian J, Susheela AK, Mudgal A, Keast G (1999) Fluoride in water: an overview. Unicef: Waterfront 13:11–13
Qin J, Chai G, Brewer JM, Lovelace LL, Lebioda L (2006) Fluoride inhibition of enolase: crystal structure and thermodynamics. Biochem 45:793–800. https://doi.org/10.1021/bi051558s
Qu WJ, Zhong DB, Wu PF, Wang JF, Han B (2008) Sodium fluoride modulates caprine osteoblast proliferation and differentiation. J Bone Miner Metab 26:328–334. https://doi.org/10.1007/s00774-007-0832-2
Ran S, Sun N, Liu Y et al (2017) Fluoride resistance capacity in mammalian cells involves complex global gene expression changes. FEBS 7:968–980. https://doi.org/10.1002/2211-5463.12236
Ravel JM, Mosteller RD, Hardesty B (1966) NaF inhibition of the initial binding of aminoacyl-sRNA to reticulocyte ribosomes. PNAS 56:701–708. https://doi.org/10.1073/pnas.56.2.701
Ren A, Rajashankar KR, Patel DJ (2012) Fluoride ion encapsulation by Mg2+ and phosphates in a fluoride riboswitch. Nature 486:85–89. https://doi.org/10.1038/nature11152
Rigalli A, Ballina JC, Roveri E, Puche RC (1990) Inhibitory effect of fluoride on the secretion of insulin. Calcif Tissue Int 46:333–338
Rogalska A, Kutler K, Zalazko A et al (2017) Fluoride alteration of [3H] glucose uptake in Wistar rat brain and peripheral tissues. Neurotox Res 31:436–443. https://doi.org/10.1007/s12640-017-9709-x
Roy A, Sengupta S, Das P (2017) Integral approach of adsorption and chemical treatment of fluoride containing wastewater: batch and optimization using RSM. J Environ Chem Eng 5:274–282. https://doi.org/10.1016/j.jece.2016.12.003
Rubin CH, Noji EK, Seligman PJ et al (1994) Evaluating a fluorosis hazard after a volcanic eruption. Arch Environ Health 49:395–401. https://doi.org/10.1080/00039896.1994.9954992
Salifu A, Petrusevski B, Ghebremichael K, Buamah R, Amy G (2012) Multivariate statistical analysis for fluoride occurrence in groundwater in the Northern region of Ghana. J Contam Hydrol 140–141:34–44. https://doi.org/10.1016/j.jconhyd.2012.08.002
Sameshima M, Ito K, Iwabuchi M (1972) Effect of sodium fluoride on the amount of polyribosomes, single ribosomes and ribosomal subunits in a cellular slime mold, Dictyostelium discoideum. Biochim Biophys Acta 281:79–85. https://doi.org/10.1016/0005-2787(72)90189-X
Seixas NS, Cohen M, Zevenbergen B, Cotey M, Carter S, Kaufman J (2010) Urinary fluoride as an exposure index in aluminum smelting. Am Ind Hyg Assoc 61:89–94. https://doi.org/10.1080/15298660008984520
Schenk G, Elliott TW, Leung E et al (2008) Crystal structures of a purple acid phosphatase, representing different steps of this enzyme's catalytic cycle. BMC Struct Biol. https://doi.org/10.1186/1472-6807-8-6
Schlichting I, Reinstein J (1999) pH influences fluoride coordination number of the AlFx phosphoryl transfer transition state analog. Nat Struct Biol 6:721–723. https://doi.org/10.1038/11485
Selz T, Caverzasio J, Bonjour JP (1991) Fluoride selectively stimulates Na-dependent phosphate transport in osteoblast-like cells. Am J Physiol 260:E833–838. https://doi.org/10.1152/ajpendo.1991.260.6.E833
Shahed AR, Miller A, Allmann DW (1980) Effect of fluorine containing compounds on the activity of glycolytic enzymes in rat hepatocytes. Biochem Biophys Res Comm 94:901–908. https://doi.org/10.1016/0006-291X(80)91320-0
Shanmugam T, Abdulla S, Yakulasamy V, Selvaraj M, Mathan R (2018) A mechanism underlying the neurotoxicity induced by sodium fluoride and its reversal by epigallocatechin gallate in the rat hippocampus: involvement of NrF2/Keap-1 signaling pathway. J Basic Appl Zool 79:17. https://doi.org/10.1186/s41936-018-0020-z
Sharma R, Tsuchiya M, Bartlett JD (2008) Fluoride induces endoplasmic reticulum stress and inhibits protein synthesis and secretion. Environ Health Perspect 116:1142–1146. https://doi.org/10.1289/ehp.11375
Sheikh MS, Fornace AJ (1999) Regulation of translation initiation following stress. Oncogene 18:6121–6128
Sheng JY, Liu Z (2000) Acidogenicity and acidurance of fluoride-resistant Streptococcus sobrinus. Chin J Dent Res 3:7–14
Sheridan RL, Ryan CM, Quinby WC Jr et al (1995) Emergency management of major hydrofluoric acid exposures. Burns 21:62–64. https://doi.org/10.1016/0305-4179(95)90785-X
Shivarajashankara YM, Shivashankara AR, Bhat PG, Rao SM, Rao SH (2002) Histological changes in the brain of young fluoride-intoxicated rats. Fluoride 35:12–21
Singh N, Verma KG, Verma P, Sidhu GK, Sachdeva S (2014) A comparative study of fluoride ingestion levels, serum thyroid hormone & TSH level derangements, dental fluorosis status among school children from endemic and non-endemic fluorosis areas. Springerplus 3:7. https://doi.org/10.1186/2193-1801-3-7
Slade GD, Grider WB, Maas WR, Sanders AE (2018) Water fluoridation and dental caries in U.S. children and adolescents. J Dent Res 97:1122–1128. https://doi.org/10.1177/0022034518774331
Skelton WHJ (1971) Free energy changes for metal, metal fluoride displacement reactions in the solid state. Dissertation, Iowa State University.
Spencer H, Kramer L, Osis D, Wiatrowski E, Norris C, Lender M (1980) Effect of calcium, phosphorus, magnesium, and aluminum on fluoride metabolism in man. Ann NY Acad Sci 355:1810194. https://doi.org/10.1111/j.1749-6632
Spittle B, Ferguson D, Bouwer C (1998) Intelligence and fluoride exposure in New Zealand children. Fluoride 31:S13
Sogaard CH, Mosekild L, Schwartz W et al (1995) Effects of fluoride on rat vertebral body biomechanical competence and bone mass. Bone 16:163–169. https://doi.org/10.1016/8756-3282(95)80028-O
Soto-Barreras U, Escalante-Villalobos KY, Holguin-Loya B et al (2019) Effect of fluoride in drinking water on dental caries and IQ in children. Fluoride 3:474–482
Stockbridge RB, Robertson JL, Kolmakova-Partensky L, Miller C (2013) A family of fluoride-specific ion channels with dual-topology architecture. eLife 2:e01084. https://doi.org/10.7554/eLife.01084.001
Strunecka A, Strunecka O, Patocka J (2002) Fluoride plus aluminum: useful tools in laboratory investigations, but messengers of false information. Physiol Res 51:557–564
Suketa Y, Asao Y, Kanamoto Y, Sakashita T, Okada S (1985) Changes in adrenal function as a possible mechanism for elevation of serum glucose by a single large dose of fluoride. Toxicol Appl Pharm 80:199–205
Sundstrom B (1971) Thyroidal c-cells and short term, experimental fluorosis in the rat. Acta Pathol Microbiol 79A:407–408. https://doi.org/10.1111/j.1699-0463.1971.tb01838.x
Sutton SV, Bender GR, Marquis RE (1987) Fluoride inhibition of proton-translocating ATPases of oral bacteria. Infect Immun 55:2597–2603
Syrovatkina V, Alegre KO, Dey R, Huang XY (2016) Regulation, signaling and physiological functions of g-proteins. J Mol Biol 428:3850–3868. https://doi.org/10.1016/j.jmb.2016.08.002
Tang QQ, Du J, Ma HH, Jiang SJ, Zhou XJ (2008) Fluoride and children's intelligence: a meta-analysis. Biol Trace Elem Res 126:115–120. https://doi.org/10.1007/s12011-008-8204-x
Tan DX, Xu B, Zhou X, Reiter RJ (2018) Pineal calcification, melatonin production, aging, associated health consequences and rejuvenation of the pineal gland. Molecule 23:E301. https://doi.org/10.3390/molecules23020301
Tappeiner H (1889) Contribution to the knowledge of the action of sodium fluoride. Arch Exp Path Parmakol 25:203–204
Teotia M, Teotia SPS, Sing KP (1998) Endemic chronic fluoride toxicity and dietary calcium deficiency interaction syndromes of metabolic bone disease and deformities in India. Indian J Pediatr 65:371–381
Teotia SPS, Teotia M (1973) Secondary hyperparathyroidism in patients with endemic skeletal fluorosis. Br Med J 1:637–640. https://doi.org/10.1136/bmj.1.5854.637
Tomas M, Martinez-Alonso E, Ballesta J, Martinez-Menarguez JA (2010) Regulation of ER–Golgi intermediate compartment tubulation and mobility by COPI coats, motor proteins and microtubules. Traffic 11:616–625. https://doi.org/10.1111/j.1600-0854.2010.01047.x
Tsunoda H, Yu MH (1985) Fluoride research. International Society for Fluoride Research, Elsevier, Morioka, Japan
Tu W, Zhang Q, Liu Y et al (2018) Fluoride induces apoptosis via inhibiting SIRT1 activity to activate mitochondrial p53 pathway in human neuroblastoma SH-SY5Y cells. Toxicol Appl Pharmacol 15:60–69. https://doi.org/10.1016/j.taap.2018.03.030
Tutuian R, Castell DO (2006) Gastroesophageal reflux monitoring: pH and impedance. GI Motil Online. https://doi.org/10.1038/gimo31
Ullah R, Zafar MS, Shahani N (2017) Potential fluoride toxicity from oral medicaments: a review. Iran J Basic Med Sci 20:841–848. https://doi.org/10.22038/IJBMS.2017.9104
United Nations Environment Program (2013) Global mercury assessment: sources, emissions, releases, and environmental transport. https://wedocs.unep.org/handle/20.500.11822/7984. https://wedocs.unep.org/bitstream/handle/20.500.11822/7984/-Global%20Mercury%20Assessment-201367.pdf?sequence=3&isAllowed=y
US Department of Health and Human Services (1991) Review of Fluoride: Benefits and Risks. https://www.dentalwatch.org/fl/phs_1991.pdf
US Department of Health and Human Services (2015) U.S. public health service recommendation for fluoride concentration in drinking water for the prevention of dental caries. Publ Health Rep 130:318–333. https://doi.org/10.1177/003335491513000408
van Hook AM (2015) G protein activation at the Golgi. Sci Signal 8:109. https://doi.org/10.1126/scisignal.aab4170
van Loveren C, Hoogenkamp MA, Deng DM, ten Cate JM (2008) Effects of different kinds of fluoride on enolase and ATPase activity of a fluoride-sensitive and fluoride-resistant Streptococcus mutans strain. Caries Res 42:429–434. https://doi.org/10.1159/000159606
Varner JA, Jensen KF, Horvath W, Isaacson RL (1998) Chronic administration of aluminum–fluoride or sodium–fluoride to rats in drinking water: alterations in neuronal and cerebrovascular integrity. Brain Res 784:284–298. https://doi.org/10.1016/S0006-8993(97)01336-X
Vincent S, Brouns M, Hart MJ, Settleman J (1998) Evidence for distinct mechanisms of transition state stabilization of GTPases by fluoride. PNAS 95:2210–2215. https://doi.org/10.1073/pnas.95.5.2210
Walna B, Kurzyc I, Bednorz E, Kolendowicz L (2013) Fluoride pollution of atmospheric precipitation and its relationship with air circulation and weather patterns. Environ Monit Assess 185:5497–5514. https://doi.org/10.1007/s10661-012-2962-9
Wang AG, Xia T, Chu QL et al (2004) Effects of fluoride on lipid peroxidation, DNA damage and apoptosis in human embryo hepatocytes. Biomed Environ Sci 17:217–222
Wang SX, Wang ZH, Cheng XT et al (2007) Arsenic and fluoride exposure in drinking water: children's IQ and growth in Shanyin county, Shanxi province, China. Environ Health Perspect 115:643–647
Warburg O, Christian W (1941) Isolation and crystallization of enolase. Biochem Zeitschrift 310:384–421
Weinberg Z, Wang JX, Bogue J, Yang J, Corbino K, Moy RH, Breaker RR (2010) Comparative genomics reveals 104 candidate structured RNAs from bacteria, archaea, and their metagenomes. Genome Biol 11:R31. https://doi.org/10.1186/gb-2010-11-3-r31
Weinstein LH, Davison A (2004) Fluorides in the environment: effects on plants and animals. CABI, Wallingford, CT
Westman J, Berhard H, Fairn GD (2019) Integrity under stress: host membrane remodeling and damage by fungal pathogens. Cell Microbiol 21:e13016. https://doi.org/10.1111/cmi.13016
Whitford GM (1994) Effects of plasma fluoride and dietary calcium concentrations on GI absorption and secretion of fluoride in the rat. Calcif Tissue Int 54:421–425
Whitford GM (1996) The metabolism and toxicity of fluoride. Monogr Oral Sci 16:1–153
WHO (2004) Fluoride. Rolling revision of the WHO guidelines for drinking water quality. https://www.who.int/water_sanitation_health/dwq/nutfluoride.pdf
Witham CS, Oppenheimer C, Horwell CJ (2005) Volcanic ash-leachates: a review and recommendations for sampling methods. J Volcan Geotherm Res 141:299–326. https://doi.org/10.1016/j.jvolgeores.2004.11.010
Wong AD, Ye M, Placone A et al (2013) The blood-brain barrier: an engineering perspective. Neuroengineering. https://doi.org/10.3389/fneng.2013.00007
Yan X, Yang X, Hao X et al (2015) Sodium fluoride induces apoptosis in H9c2 cardiomyocytes by altering mitochondrial membrane potential and intracellular ROS level. Biol Trace Elem Res 166:210–215. https://doi.org/10.1007/s12011-015-0273-z
Zhang M, Wang A, Xia T, He P (2008) Effects of fluoride on DNA damage, S-phase cell-cycle arrest and the expression of NF-kappaB in primary cultured rat hippocampal neurons. Toxicol Lett 179:1–5. https://doi.org/10.1016/j.toxlet.2008.03.002
Zhang Y, Zhang KQ, Ma L et al (2016) Fluoride induced endoplasmic reticulum stress and calcium overload in ameloblasts. Arch Oral Biol 69:95–101. https://doi.org/10.1016/j.archoralbio.2016.05.015
Zhao WP, Wang HW, Liu J et al (2019) Mitochondrial respiratory chain complex abnormal expressions and fusion disorder are involved in fluoride-induced mitochondrial dysfunction in ovarian granulosa cells. Chemosphere 215:619–625. https://doi.org/10.1016/j.chemosphere.2018.10.043
Zhou Y, Shi H, Li X et al (2013) Role of endoplasmic reticulum stress in aberrant activation of fluoride-treated osteoblasts. Biol Trace Elem Res 154:448–456. https://doi.org/10.1007/s12011-013-9752-2
Zhu L, Zhang Z, Liang J (2012) Fatty-acid profiles and expression of the fabM gene in a fluoride-resistant strain of Streptococcus mutans. Arch Oral Biol 57:10–14. https://doi.org/10.1016/j.archoralbio.2011.06.011
Acknowledgements
We thank Dave Hiller and other members of the Strobel lab for their valuable discussions and insight. This work was supported by funding provided by the N.I.H. Chemical Biology Interface training grant (to N.R.J.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Johnston, N.R., Strobel, S.A. Principles of fluoride toxicity and the cellular response: a review. Arch Toxicol 94, 1051–1069 (2020). https://doi.org/10.1007/s00204-020-02687-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-020-02687-5