
Abstract
Fluoride is present everywhere in nature. The primary way that individuals are exposed to fluoride is by drinking water. It’s interesting to note that while low fluoride levels are good for bone and tooth growth, prolonged fluoride exposure is bad for human health. Additionally, preclinical studies link oxidative stress, inflammation, and programmed cell death to fluoride toxicity. Moreover, mitochondria play a crucial role in the production of reactive oxygen species (ROS). On the other hand, little is known about fluoride’s impact on mitophagy, biogenesis, and mitochondrial dynamics. These actions control the growth, composition, and organisation of mitochondria, and the purification of mitochondrial DNA helps to inhibit the production of reactive oxygen species and the release of cytochrome c, which enables cells to survive the effects of fluoride poisoning. In this review, we discuss the different pathways involved in mitochondrial toxicity and dysfunction induced by fluoride. For therapeutic approaches, we discussed different phytochemical and pharmacological agents which reduce the toxicity of fluoride via maintained by imbalanced cellular processes, mitochondrial dynamics, and scavenging the ROS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fluoride is an essential component for the development and growth that are typical for animals; however, an excessive amount of fluoride can cause the body of the animal to become damaged. Dental fluorosis [1], hypertension [2], skeletal fluorosis [3], reproductive disorders [4], and dementia [5] have been identified as the usual manifestations of fluorosis, and a huge number of studies indicate [1]. Fluoride exists in large quantities in our environment in a variety of various forms, and it’s having ability to damage the health and natural environment. Fluorosis has been spotted in a significant number of people all over the world. The fluorine pollution in soil is a major cause for concern all over the world, but especially in China because it is having a bigger detrimental effect on human health as well as the ecological environment [6]. Over 260 million people worldwide have fluorosis, according to studies on the condition’s prevalence in 20 different countries, which is brought on by exposure to a variety of fluorine sources [7]. Fluoride can be found in various food and beverages, such as fish, milk, and brick tea, as well as in water, toothpaste, and mouthwashes [8].
Fluoride is most commonly consumed by humans through drinking water, which can be contaminated by rocks and fluoride-containing minerals in the groundwater. For humans, this is only the major origin of fluoride element [3]. The levels of fluoride that are acceptable in consumable water are set by various organisations which have been shown in Table 1 [9].
Following consumption, around 75–90% of the soluble fluoride is rapidly absorbed, making its way into the bloodstream and making a contribution to the plasma fluoride levels [10]. Fluoride creates insoluble complexes with cations like calcium (Ca2+) and magnesium (Mg2+) which are then removed from the body through faeces, because of its instability and high binding affinities. The absorption distribution of fluoride in different body parts has been shown in Fig. 1.

Every ecosystem has the element fluoride (F), which is present in the soil, groundwater, vegetation, and animals. Diet and accidental ingestion of fluoride via dental products (toothpaste and mouth wash) are the two most important contributors to an individual’s fluoride intake. Eighty to ninety percent of the ingested F is absorbed by passive diffusion from the gastrointestinal system. Then get into the plasma accumulated in different part of body via excrete out the different route.
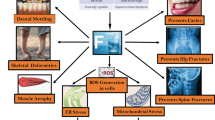
A significant amount of fluoride that is dispersed throughout the body bond with Ca2+ in the body’s hard tissues [11], where it takes the place of hydroxyl ions in the enamel that is naturally present (hydroxyapatite). When an individual consumes an excessive amount of fluoride, their digestive system is largely affected. This may cause nausea, nausea or vomiting, abdominal cramp, and diarrhoea [12]. Fluorosis, also known as fluoride poisoning, is caused by prolonged exposure to fluoride (more than 1.5 mg/L). This condition is frequently connected with the progressive degradation of bones and teeth. In addition, there is evidence that fluoride poisoning can have a negative impact on soft tissues, including the lungs [13, 14], liver [15, 16], heart [17, 18], kidney [19], spleen [20], and brain [21,22,23], which has been shown in Fig. 1. Both the placenta and the blood-brain barrier (BBB) can be penetrated by fluoride in the form of non-ionic hydrogen fluoride (HF) [24, 25]. It is believed that children’s BBB are undeveloped or underdeveloped, making them more susceptible to poisons like fluoride [26, 27]. Fluoride transport over the BBB has negative impact on neuron metabolism, enzyme, neurotransmitters, and oxidation homeostasis, which eventually results in impaired mental status [28, 29]. Fluoride has a high level of chemical and biological activity, which is related to its toxicity. Fluoride readily and quickly crosses biological membranes when the pH of nearby bodily fluid compartments changes, most frequently by passively non-ionic diffusion in the form of HF [30]. Depending on the kind of cell, the concentration, and the length of exposure, fluoride can have different effects on cellular metabolism and physiology (Fig. 2) [31]. For example, fluoride micromolar concentrations in tooth and bone tissues create potentially advantageous effects by stimulating cell division and development, but fluoride millimolar dosages decrease cell multiplication and cause programed cell death. Various literatures have been revealed that higher exposure of fluoride levels results in cell death by apoptosis in ameloblasts [32,33,34], odontoblasts [35], and osteoblasts [36,37,38]. Fluoride has a number of detrimental consequences, such as inflammatory responses, cell contractile responses, suppression of cell growth and cell cycle arrest, and DNA oxidative stress [31]. Apoptosis is a sophisticated and tightly controlled event that is essential for the elimination of unneeded or damaged cells as well as for a number of regular physiological events such as cell multiplication and proliferation, tissue homeostasis, and ageing [39,40,41]. Studies show that high fluoride ingestion causes cell dysplasia and reduces cell proliferation and differentiation. Fluoride alters the mitochondrial morphology of cells and prevents the mechanism of the respiratory chain complex, leading to drop in adenosine tri phosphate and an elevation in ROS (Fig. 2). In this review, we have explained how fluoride accumulation occurs in body and pathways involved in fluorosis via a different source, which cause mitochondrial dysfunction and responsible to raise the level of ROS. In this review, we discuss the different mechanisms involved in mitochondrial dysfunction via fluorosis. How fluoride acts on the different metabolic activities of mitochondria alters the energy production. In this review, we discuss therapeutic approaches for fluoride toxicity and different antioxidant, phytochemical, and pharmacological modulators which act on the different targets and neutralised the toxicity of fluoride.

Schematic representation of environmental fluoride from different source can easily crass the biological membrane and bound with intracellular calcium to form insoluble fluoride and accumulated in intracellular environment and damage the mitochondria, disturb the ETC which cause oxidant and antioxidant imbalance, and increase the level ROS.
Different Apoptotic Pathways Involved In Mitochondrial Disturbance via Fluorosis And There Therapeutic Intervention
In Energy Metabolic Pathway
Different energy metabolic pathways are used by immune system cells to create intermediates that enable cellular growth and proliferation as well as provide ATP to support cellular functions and survival [42, 43]. These bioenergetic requirements are principally satisfied by three metabolic processes that are interconnected: glycolysis, the tricarboxylic acid cycle, and OXPHOS. Glycolysis takes place in the cytoplasm, which is in contrast to the mitochondria, which are the exclusive locations for the OXPHOS and TCA cycles. Glucose is the first taken in from the outside environment by glucose transporters, where it is then phosphorylated by hexokinases to G6P. G6P is converted into pyruvate during the glycolytic pathway, converting NAD+ to NADH and producing two molecules of ATP in the process. G6P can also take part in the PPP, which is a byproduct of glycolysis that produces the reducing equivalents of NADPH that are necessary for the NOX-controlled microbicidal pathways and the riboses that are necessary for the synthesis of nucleotides. G6P can participate in both of these processes. During conditions of normoxia, the pyruvate that is created as a byproduct of glycolysis is converted into acetyl-CoA. This acetyl-CoA is then oxidised as part of the TCA cycle events, which leads in the formation of NADH and FADH2. These molecules are responsible for the transmission of electrons to the ETC so that OXPHOS can be fuelled and for the more efficient production of 30–36 molecules of ATP for each molecule of glucose. Fluoride prevents the transfer of nutrients, cell respiration, and glycolysis by inhibiting metalloproteins [44,45,46]. Long-term exposure to fluoride causes metabolic disturbance, production of stress signals, inhibition of transmembrane proteins, aberrant mitochondria, and electrolyte imbalance at the molecular level [47,48,49]. Chronic exposure of fluoride causes damage in mitochondria permanently and leads in an aberrant respiratory chain. This is mostly because anaerobic glycolysis produces less ATP (ATP ADP+AMP+H+), which releases protons and causes intracellular acidification and oxidative stress [44, 46, 50].
Role of fluoride in SIRT1
Sirtuins are NAD+-dependent class III deacetylases, human homologues of the yeast gene sir2, which stands for silent information regulator-2 [51,52,53]. The acetyl groups removing from cellular protein, such as histones and transcription regulators, by sirtuins, which controls the post-translational biological functions of the protein substrates [54], is how sirtuins exert their influence. There are seven different proteins that make up the sirtuin family in mammals (SIRT1 to SIRT7) [55]. SIRT1 is related to the Sir2 gene that is found in Saccharomyces cerevisiae [34, 56], and SIRT1 itself is regulated posttranscriptionally by the process of phosphorylation [57,58,59]. cyclinB/Cdk1 is responsible for phosphorylating residues Thr530 and Ser540 [58], whereas c-Jun N-terminal kinase 1 (JNK1) is responsible for phosphorylating residues Ser27, Ser47, and Thr530 [59]. In comparison to its version that has not been phosphorylated, phosphorylated SIRT1 (p-SIRT) functions as an active deacetylase [58]. SIRT1 helps the cell resist the stress induced by oxidative stress, caloric restriction (CR), and endoplasmic reticulum (ER) stress [60,61,62,63] by deacetylating target substrates such as FOXOs, PGC-1, and p53. SIRT1 is in responsible for controlling cellular activities involved in maintaining homeostasis and responding to stress; it is clear that SIRT1 is favourable to the continued existence of cells. During times of cell stress, SIRT1 is responsible for regulating autophagy [64, 65]. Macroautophagy, or autophagy as it is more widely known, is an intracellular catabolic process that is phylogenetically conserved and allows the breakdown of cytoplasmic content [66,67,68], such as defective proteins and organelles. Autophagy is often referred to as the term “autophagy.” It has been discovered that sirtuins mostly have an anti-apoptotic property. In the traumatic brain damage mouse model, SIRT1 activation downregulates the pro-apoptotic protein Bax and upregulates the anti-apoptotic protein Bcl-2, which is consistent with decreased neuronal death [69]. By increasing autophagy during oocyte ageing, SIRT2 inhibition exacerbated cell apoptosis and supported the anti-apoptotic function of SIRT2 [70]. SIRT3 was discovered to inhibit COX-1 deacetylation-induced cell death in response to oxidative stress, which could be beneficial in preventing cerebral ischemia/reperfusion injury [71]. Additionally, pancreatic cell apoptosis and dysfunction brought on by the injection of too much palmitate and glucose were reduced by overexpression of SIRT5 [72]. In vitro exposure of podocytes to high glucose levels resulted in SIRT6 suppressing Notch signalling in a way dependent upon deacetylation activity, as demonstrated by [73]. Additionally, SIRT7 increased cardiomyocytes’ resistance to apoptosis, possibly through deacetylating p53 [74]. Sirtuins may play pro-apoptotic roles in some situations, despite the fact that numerous research have focused on their anti-apoptotic functions. For instance, SIRT5 can increase tumour cell death by acting as a tumour suppressor through its desuccinylase activity [75]. IDH1 mutation-induced R-2-hydroxyglutarate (R-2HG) accumulation results in mitochondrial hypersuccinylation, which promotes carcinogenesis and apoptosis resistance. However, desuccinylase SIRT5 overexpression can reverse these effects. The varied enzymatic activities of sirtuins and their numerous downstream targets may help to explain these seemingly contradicting actions of sirtuins in controlling cellular death. Therefore, more research is required to demonstrate the sirtuins’ complex functions in apoptosis.
Fluoride was found to activate SIRT1 and autophagy, which are adaptive reaction that prevent cells from cell stress [76]. Many studies confirmed that fluoride has this effect. SIRT1 is controlled by a number of different variables in response to cellular stress [77]. SIRT1 expression can be boosted, for instance, by transcription factors such as peroxisome proliferator-activated receptors (PPARs) [78, 79] and cAMP response element binding (CREB) [80].
For therapeutic approach, since SIRT1 is known to regulate crucial metabolic pathways and has been linked to both oncogenic and chemotherapeutic processes, its activation may be advantageous in the fight against metabolic disorders. In recent years, scientists who were interested in finding SIRT1 modulators were able to find new small molecules that target SIRT1 activity. Many studies have shown that different natural phytochemicals, like resveratrol, fisetin, curcumin, quercetin, and berberine, are natural non-polyphenolic substances which are good for your health [81]. Natural polyphenols are possible ways to prevent and treat oxidative stress diseases. They are present in many plants and fruits. Long-term consumption is linked to health benefits and reduced the toxicity of fluoride (Fig. 3).

A representation in schematic form of the apoptosis caused by fluoride and mediated by the SIRT1-p53 pathway. Fluoride is responsible for the formation of intracellular ROS, which in turn activates the cytosolic SIRT1. It is able to attach to deacetylated p53 and prevent the protein from moving into the nucleus. The deacetylation of p53 causes it to go into the OMM, where it then releases the pro-apoptotic proteins BCL2 and BAX. When BAX is activated, this process causes cytochrome c (Cyt C) to be released from the mitochondria into the cytoplasm. Fluoride is transported into the mitochondria from the cytoplasm, where it then induces an increase in the ROS concentration of the mitochondria. The increases in ROS both cause acetylation of SOD2 and decrease the expression of the sirt-1 protein in mitochondria. The decrease in mitochondrial sirt-1 leads to an increase in acetylation of PGC1, which in turn causes mitochondria to become dysfunctional. Apoptosis is commonly caused by mitochondria that are not functioning properly. The structural integrity of mitochondria is preserved, its translocation is facilitated, and its transcriptional activity is carried out as a result of resveratrol’s activation of the AMPK pathway. In addition, active AMPK is responsible for the phosphorylation of PGC-1, which allows the protein to enter the nucleus and be deacetylated by SIRT1. After that, PGC-1 will support Nrf2, which will result in a rise in the expression of antioxidative genes, which will eventually lead to a reduction in oxidative stress. Resveratrol triggers the activation of AMPK, which in turn promotes SIRT1, which in turn inhibits MAPK signalling pathways, which ultimately leads to autophagy.
Reactive oxygen species pathway
Reactive oxygen species are formed when O2-derived free radicals such as hydroxyl radicals (HO•), peroxyl (RO2•), superoxide anions (O2•), and alkoxyl (RO•) as well as O2-derived non-radical species like hydrogen peroxide (H2O2) combine (ROS)[82]. A significant intracellular generator of ROS is the mitochondrion. According to research, 1–2% of the mitochondrial O2 used is diverted to production of ROS, particularly at complex I and complex III of the mitochondrial membrane [83, 84]. This is thought to vary on the tissue and species [83, 84]. In the presence of metallic ions, manganese antioxidant enzyme transforms mitochondrial O2• to H2O2 and produces highly reactive HO•, damaging cellular components, phospholipid, and DNA. Approximately 10 putative ROS-generating mechanisms in mitochondria have been found thus far [85]. Krebs cycle enzyme complexes, including pyruvate dehydrogenase and -ketoglutarate dehydrogenase (-KGDH), have been mentioned as important mitochondrial superoxide anion radical and H2O2 sources among these [86]. It is important to note that higher levels of nicotinamide adenine dinucleotide (NADH) are connected with higher levels of H2O2 synthesis by mitochondria -KGDH. This increased oxidant strain results in more ROS generation from mitochondrial complex I, which accelerates the death of cells [87]. Other mitochondrial ROS generators include p66Shc [88], the outer membrane enzyme monoamine oxidase [85], the modified membrane potential of the mitochondria [89], and the pH of the matrix [90]. Due to their role as significant ROS producers, mitochondria are frequently exposed to high levels of ROS, which can have harmful effects such oxidative DNA damage [91, 92]. The mechanism by which mitochondrial DNA damage drives apoptotic signals is not fully understood and should be a promising path for further research. Elevated O2• and HO• linked to mtDNA damage cause apoptosis (Fig. 4) [93]. Peroxisomes are known to be sources of cytosolic H2O2 under physiological conditions; however, the removal of peroxisomal H2O2 is compartmentalised due to the structural arrangement of peroxisomes. During this time, H2O2 that has been catalysed by urate oxidase is being transferred into the cytoplasm by means of crystalloid core tubules [94], and the H2O2 that is produced in the matrix is removed by catalase. NADPH oxidase-derived ROS at the cellular membranes signal cells. Endoplasmic reticular (ER) some protein, such as cytochrome P450, are causes the large levels generation of cellular hydroxyl radicals and oxygen radicals, all of which contribute to the promotion of peroxidation, calcium metabolism, mitochondrial dysfunction, and cell death [95, 96]. ROS, which are connected to the development and spread of cancer, are the cause of nearly all types of nuclear damage [97] as well as base alterations, strand breakage, genomic cross-linking, and peptides; ROS damages proteins. However, the potential of ROS to trigger apoptotic, which are a major technique in the treatment of cancer [98,99,100], presents a conundrum in the context of biological systems, ROS interrupt the electron transport chain, open the permeability transition pore (PTP), damage the mitochondrial membrane, and release Cyt c. Together with Apaf-1 and procaspase-9, cyt-c generates “apoptosomes” in the cytoplasm, which stimulate caspase-9, which stimulates caspase-3, leading to protein breakage and apoptotic cell death [101,102,103,104]. In point of fact, one of them is hydrogen peroxide (H2O2), which is both a direct inducer of apoptosis and a major ROS that ranks among the most significant ROS (Fig. 4) [105]. The ability of antioxidant phytochemicals to reduce the formation of reactive oxygen species (ROS) by scavenging free radicals [51,52,53,54, 106,107,108] and inhibiting pro-apoptotic signals [55,56,57,58,59, 109,110,111,112] is what is thought to be responsible for the protective effect that antioxidant phytochemicals have against diseases that are related to oxidative stress.

The electron transport chain is responsible for the release of ROS. Complexes I and III of the mitochondrial matrix are responsible for the release of reactive oxygen species (ROS). Complex III is responsible for the release of reactive oxygen species into the intermembrane gap. UQ, ubiquinone; Cyt C, cytochrome c
Fluoride-induced oxidative damage and apoptosis through use of natural and synthesised phytochemicals that neutralise ROS-mediated harm in Body. Different phytochemical likes gallic acid, Panax ginseng, blackberry, Tamarindus indica, curcumin, silymarin, lycopene, quercetin, Terminalia arjuna, thymoquinone, epigallocatechin gallate, and proanthocyanidin. Other ingredients include epigallocatechin gallate, thymoquinone, and proanthocyanidin. Others phytochemicals such as aloe vera [26, 61, 62, 113, 114], Ocimum sanctum [62], fisetin [60, 113], genistein, and pomegranate [63, 115] are showing antioxidant property and neutralise the toxicity of fluoride.
Effected the autophagy and apoptosis mechanism
Elevated ROS levels are a result of the cellular process known as autophagy, which involves the destruction of proteins and organelles. Autophagy regulation by ROS has been researched extensively [116,117,118]. Autophagy has a variety of outcomes, including as pathogen removal, infection prevention, and the death of defective cellular organelles in individual cells. These effects suggest that ROS may have the ability to function as a signalling target in autophagy-related survival [119,120,121]. Recently, the focus of study has shifted to determining whether ROS-derived autophagy may be used to treat cancer [122,123,124]. The modulation of the induction of autophagy in malignant tumours has also been demonstrated to directly correlate with the intracellular ROS levels [125, 126]. The process of autophagy is triggered when H2O2 causes the enzyme known as 4A cysteine peptidase (ATG4) to get oxidised. This step is required for the de-lipidation of the ATG8 protein, which is the end result of autophagy being triggered. The process of autophagy is triggered when H2O2 causes the enzyme known as 4A cysteine peptidase (ATG4) to get oxidised. This step is required for the de-lipidation of the ATG8 protein, which is the end result of autophagy being triggered. This H2O2-induced oxidation then renders ATG4 inactive, which causes a rise in the formation of LC3-associated autophagosomes [125]. However, the AMP-activated pathway also contributes significantly to the control of ROS-related autophagy. AMPK activation suppresses mTORC1, causing autophagy. Oxidative stress stimulates AMPKK to control the AMPK pathway (AMPK kinase). This increases the production of H2O2 and leads to the induction of death in the cells. This AMPK activation suppresses mTORC1, which in turn causes autophagy to occur. Additionally, a variety of transcription factors, such as NF-B, have the ability to change the regulation of genes associated by autophagy such as ATG6/Beclin1, which affects ROS-induced autophagy in cancer cells. [127, 128]. In preliminary studies, SOD overproduction suppressed selenite-induced cytotoxic effects (autophagy) in malignant glioma cells [128]. ATG6/7 (autophagy related genes 6 and 7) were also reported to be knocked down by tiny miRNA, which decreased the cytotoxicity caused by selenite [121, 129, 130]. These results have led to the hypothesis that increased ROS levels, and their regulation causes malignant cells to activate autophagy (Fig. 5). Now for therapeutic approaches, many research targeting inhibitions of mTOR signalling attenuate NaF-induced apoptosis and promote viability of neuronal cells by activation of autophagy. Rapamycin probably alleviates this impairment by decreasing the expression of p62, thereby preventing autophagy defects. The protein kinase known as mammalian target of rapamycin (mTOR) is responsible for regulating cell survival, proliferation, and growth. There is a significant amount of interest in the research and development of medications that target the enzyme mTOR, which is commonly elevated in cancer.

Autophagy and apoptosis is induced by NaF through the inhibition of PI3K and mTOR activity. This increased the regulation of AKT, which in turn upregulates the LC3 and Beclin1, while decreasing the level of expression for p62.
Fluoride disturbs mitochondrial dynamic cycle: fission and fusion
Mitochondrial dynamics, or building a network through fusion and fission, is related to autophagy, cell signal transcription factors, and iron metabolism. Mitochondrial dynamics is the mechanism by which mitochondria made a network together. Fluorosis can cause mitochondrial dysfunction by disrupting the balance between fission and fusion, leading to abnormal mitochondrial morphology [131]. Fluoride’s neurotoxicity is linked to mitochondrial damage [132]. To keep mitochondria functioning properly, it is necessary to have a grasp of the dynamics of mitochondrial fusion and fission [133]. Mitochondrial division requires multiple protein like Drp1, MID49, MID51, Fis-1, Mff, and Dyn2. Mitochondrial members have dynamic protein group known as Mfn1 and Mfn2 which is responsible for mediating the process of mitochondrial outer membrane fusion. One of the proteins of the dynamic’s family proteins is responsible for mediating the process by which the inner membranes of mitochondria fuse together. That protein is known as fusion protein OPA1. Under certain physiological settings or when mitochondrial are exposed to detrimental external forces, Drp1 accumulates on the mitochondrial outer membrane, forming a ring that squeezes mitochondria [134]. After then, Dyn2 and Drp1 work together to regulate the last stage of the mitochondrial division process [135]. The fact that a Drp1 deletion can block mitochondrial division and boost mitochondrial networking to generate big mitochondria lends credence to the idea that Drp1 plays a critical role in the process of mitochondrial fission [136]. Overexpression of Drp1 can speed up the process of mitochondrial fission and result in substantial damage to mitochondria. It is abundantly obvious that cells devoid of Dyn2 do not divide because of mitochondrial membranes which are unable to contract in the appropriate manner (Fig. 6) [135]. In rats with persistent fluorosis, the mitochondrion of cortical neurons split and migrated. According to research, the quantities of mRNA and protein produced by Fis1 were decreased by NaF, whereas the levels of Mfn1 and Mfn2 were increased. Increasing drp1 mRNA lowered drp1 protein. An excess of fluoride can cause aberrant mitochondrial division, resulting in a rise in total mitochondria and severe damage to this cytosol. When mitochondria are injured, mPTP opens, which causes the release of cytochrome C from the mitochondria inner membrane into the cytosol [137]. Following this, the intermediate recruiting motif is responsible for activating the Pro-Caspase-9 enzyme. Caspase-9 needs to first cleave and activate the executioner enzyme, Caspase-3, before it can start the apoptotic destruction process [138]. In a dose-dependent way, Cyt C, Caspase-9, and Caspase-3 mRNA expression levels increased as a response to fluoride stimulation. This effect was most pronounced at 100 mg/L. In addition to this, NaF caused a significant shift in the proportion of fusion proteins to fission proteins in the hippocampal of newborn rats, which suggests that fluoride may contribute to an imbalance in the proportion of fission proteins to fusion proteins [139, 140]. When division and fusion equilibrium is disturbed, mitochondria experience some morphological as well as functional alterations. Several studies have been shown that the mitochondrial dynamics fission mechanism inhibition that is generated by NaF in human neuroblastoma cells is mostly responsible for the mitochondrial irregularities, autophagy deficiencies, increased apoptosis, and neuronal damage that are caused by NaF [129]. Contempt the partial retrieval of autophagy, the mechanistically generated mitochondrial abnormalities and cell death caused by NaF are made worse by the pharmacological suppression of mitochondrial fission. This results in an increase in the rate of apoptosis [134]. It has been hypothesised that being exposed to fluoride levels that are typical of the environment can impair memory and learning in addition to causing morphological changes in the mitochondria of the hippocampus. These changes include fission inhibition and accelerated fusion, in addition to deficiencies in autophagy, excessive programmed cell death, and neuronal loss [132]. Children who are subjected to fluoride in drinking water for an extended period of time are at a greater risk of experiencing mental decline. This risk is linked to mitochondrial fission/fusion molecular cycling. In most cases, inhibition of mitochondrial fission leads to abnormalities in the mitochondria membrane, which is lead to improper apoptosis and autophagy which ultimately result in the death of neuron [133]. As a result, targeting the mitochondrial dynamics regulator may provide a possible therapeutic strategy for the treatment of fluoride toxicity. In autoimmune disorders like rheumatoid arthritis, an anti-inflammatory medication called leflunomide can modulate the responses of T cells quite effectively. Its ability to produce pyrimidine pool exhaustion is traditionally credited as being the function that fulfils this role. Leflunomide causes a rise in the quantities of MFN2 and MFN1 transcripts as well as proteins in HeLa and C2C12 muscle cells. This leads to mitochondrial elongation but has no effect on the rate of respiration. Another pharmacological agent, such as mitofusion peptide, SAM-A, epigallocatechin gallate, and liquiritigenin, a flavonoid with cytoprotective effects, was found to be an inducer of mitochondrial fusion in a neuroblastoma cell line. This agent was successful in rescuing mitochondrial fragmentation that was observed in cells lacking Mfn1, Mfn2, and OPA1 (Fig. 6). Importantly, it was demonstrated that liquiritigenin may inhibit mitochondrial fragmentation and cytotoxicity generated by amyloid, so giving a viable therapeutic method for Alzheimer’s disease.

A representation in schematic developing neuronal damage caused by fluoride exposure. Inhibition of mitochondrial fission contributes to embryonic fluoride neurotoxicity by causing aberrant autophagy and apoptosis associated with mitochondrial abnormalities. These mitochondrial defects are a cause of developmental fluoride neurotoxicity. (1) On the surface of the mitochondria, the expression of Mff, Fis1, and MiD49 are upregulated, and Drp1 protein was recruited from cytoplasm as well as endoplasmic reticulum. The mitochondria are compressed as a result of a significant number of Drp1 molecules forming a circular configuration in the centre of the mitochondria (2). Upregulation of Dyn2 and Drp1 occurs concurrently in order to complete the mitochondrial division process. (3) Excessive mitosis leads to mitochondrial structural damage, which ultimately results in an increased expression of cytochrome c from IMM (inner mitochondrial membrane). Different pharmacological agents like leflunomide stimulate the fusion by increasing the expression of Mfn1, Mfn2, and OPA-1, while some pharmacological agents like SAMBA and liquiritigenin inhibited the fission by suppressing the expression of fis1 and Drp1.
Fluoride binds with calcium and form insoluble fluoride
Fluorine affects bones. Fluorine circulates fast through the bloodstream and accumulates in calcium-rich organs like teeth and bones. Fluoride causes collagen fibres in the tibia to be become loose, heterogeneous, and curved, enlarging the space of bone fossa, decreasing bone flexibility, diminishing tolerance of bone, and increasing fracture risk. Recently, it was shown that the proper calcium intake can treat fluorosis [141]. Numerous academic studies have demonstrated the high affinity between F and Ca. By interacting with F in the colon, supplemental Ca creates the new complex known as insoluble CaF2, which lessens the toxicity and absorption of fluoride [142]. High serum calcium in bone tissue causes hyperosteogeny, hypocalcemia, and increased bone mineral density. In fluorosis, the rate of osteoblast proliferation decreased and apoptosis rose. The mitochondrial route is one of the most crucial apoptosis pathways. The primary energy centres and the primary generator of ATP and ROS are mitochondria. Ca2+ has strong control over how they behave. Mitochondrial Ca2+ uptake is essential to meet energy needs during fluoride toxicity while maintaining antioxidant capacity to avoid excessive ROS production (Fig. 2) [143]. According to several studies, cells exposed to fluoride had higher fluoride levels and Ca2+ concentrations. This could be the result of fluoride just diffusing into the cell [144]. Fluoride accumulation may promote intracellular Ca2+ release, harming cells. TSCE lowered Ca2+ and fluoride levels in fluoride-exposed cells. Through the mitochondrial Ca2+ unidirectional receptor, mitochondria absorb Ca2+. Unipolar mitochondrial calcium which are the principal mediators of calcium flows into mitochondria, which controls the metabolic process of energy production in the cell. ROS synthesis and programmed cell death are all necessary for fluorosis [143]. According to studies, fluoride may increase endoparasitic reticulum stress, which in turn activates the endoparasitic reticulum pathway generated by calcium, leading to osteoblast apoptosis and mitochondrial malfunction [14]. Ca2+ is primary signalling messenger of the endoplasmic reticulum and is involved in practically all physiological processes. The primary pool of calcium in the osteoblasts is the (ER) endoplasmic reticulum, and endoplasmic reticulum stress can cause an increase in intracellular Ca2+ level. Calpain and Caspase-12 are divided and activated by Ca2+, which also activates Ca2+-dependent enzymes. When Caspase-12 is activated, it interacts with Caspase-9’s promoter and Caspase-3’s and Caspase-7’s effectors to cause apoptosis [145]. Recent research has specifically shown the connection between the endoplasmic reticulum, mitochondria autophagy, and calcium-apoptotic process. During mitochondrial-associated Caspase activation, pro-apoptotic BCL-2 family members (Bax and Bak) successfully convey the increase in intracellular Ca2+ level to mitochondria, functioning unwittingly as a significant upregulating signal of apoptosis mechanism. The production of pro-apoptotic molecules like CtyC was increased by the rise in mitochondrial calcium levels [143]. In addition, multiple Caspase-3 and Caspase-7 were separated in the cytoplasmic and reticulum lumen as a consequence of CtyC and APAF-1 treating pro-Caspase-9 and producing apoptotic bodies. This occurred as a result of the treatment of pro-Caspase-9 by CtyC and APAF-1 [145].
Discussion
The potential molecular mechanisms of steady-state caused fluorosis, such as ROS production, energy metabolism, cell damage, and apoptosis, were the main topics of this review. These pathways are related to. It was found that fluorosis can cause significant damage to the configuration of a cell’s mitochondria; disrupt the dynamic equilibrium between fusion and fission; block the transmission of the mitochondrial electron transport chain; let a few free electrons escape from the respiratory chain; alter the potential of a mitochondrial inner membrane; enhance the levels of ATP, ROS, and Ca2+; and initiate a cascade reaction and ultimately result in cell apoptosis. Utilising molecular mechanisms such as energy metabolism pathway, mitochondrial dynamics, generation of reactive oxygen species, cell death, and other pathways, it is feasible to explore the likely molecular mechanism by which fluorosis causes damage to cells. This can be done in a number of ways. It has been discovered that the specific mechanism of the common regulation route of mitophagy following fluorosis cannot be substantiated when considering the cytotoxicity action of fluoride from the standpoint of mitochondrial malfunction. This was discovered after it was found that fluorosis prevented validation of this mechanism. At this moment, it is also unknown what molecular targets will be used in fluorosis diagnosis, therapy, and interventional procedures. The interplay between various trade-off mechanisms in mitochondrial cycle, oxidative stress, mitochondrial biogenesis, death of cells, and mitochondrial homeostasis—all of which are controlled by calcium ion homeostasis—is obscure, and the precise mechanisms require further study. Leflunomide is promote the fusion mechanism while liquiritigenin, BGP-15 are suppressing the fission that result showing protective effect on the mitochondria its dynamic and biogenesis. Rapamycin is a drug which is used as anticancer and suppresses the immune system; they are helpful in treatment of fluoride toxicity by inhibiting the mTOR activity. Some other antioxidant and phytochemical like festin, curcumin, and quercetin helped in neutralising the fluoride toxicity if includes in diet.
Data Availability
We collected all the data from indexed journal and available online.
References
Stangvaltaite-Mouhat L et al (2021) Fluoride in the drinking water and dental caries experience by tooth surface susceptibility among adults. BMC Oral Health 21(1):234
Davoudi M et al (2021) Relationship of fluoride in drinking water with blood pressure and essential hypertension prevalence: a systematic review and meta-analysis. Int Arch Occup Environ Health 94(6):1137–1146
Srivastava S, Flora SJS (2020) Fluoride in drinking water and skeletal fluorosis: a review of the global impact. Curr Environ Health Rep 7(2):140–146
Liang C et al (2020) Fluoride induced mitochondrial impairment and PINK1-mediated mitophagy in Leydig cells of mice: In vivo and in vitro studies. Environ Pollut 256:113438
Ge QD et al (2019) Differential expression of miRNAs in the hippocampi of offspring rats exposed to fluorine combined with aluminum during the embryonic stage and into adulthood. Biol Trace Elem Res 189(2):463–477
Tang Z et al (2021) Mangiferin prevents the impairment of mitochondrial dynamics and an increase in oxidative stress caused by excessive fluoride in SH-SY5Y cells. J Biochem Mol Toxicol 35(4):e22705
Herath H, Kawakami T, Tafu M (2018) Repeated heat regeneration of bone char for sustainable use in fluoride removal from drinking water. Healthcare (Basel) 6(4)
Urbansky ET (2002) Fate of fluorosilicate drinking water additives. Chem Rev 102(8):2837–2854
Khichar M, Kumbhat S (2015) Defluoridation-a review of water from aluminium and alumina based compound. International Journal of chemical studies 2:04–11
Dhar V, Bhatnagar M (2009) Physiology and toxicity of fluoride. Indian J Dent Res 20(3):350–355
O'Mullane DM et al (2016) Fluoride and oral health. Community Dent Health 33(2):69–99
Kanduti D, Sterbenk P, Artnik B (2016) Fluoride: a review of use and effects on health. Mater Soc 28(2):133–137
Ameeramja J, Perumal E (2018) Possible modulatory effect of tamarind seed coat extract on fluoride-induced pulmonary inflammation and fibrosis in rats. Inflammation 41(3):886–895
Ameeramja J, Perumal E (2017) Pulmonary fluorosis: a review. Environ Sci Pollut Res Int 24(28):22119–22132
Kanagaraj VV et al (2015) Caffeic acid, a phyto polyphenol mitigates fluoride induced hepatotoxicity in rats: a possible mechanism. Biofactors 41(2):90–100
Ekambaram P et al (2010) Therapeutic efficacy of Tamarindus indica (L) to protect against fluoride-induced oxidative stress in the liver of female rats. Fluoride 43:134–140
Panneerselvam L, Raghunath A, Perumal E (2017) Acute fluoride poisoning alters myocardial cytoskeletal and AMPK signaling proteins in rats. Int J Cardiol 229:96–101
Panneerselvam L et al (2019) Acute fluoride exposure alters myocardial redox and inflammatory markers in rats. Mol Biol Rep 46(6):6155–6164
Dharmaratne RW (2019) Exploring the role of excess fluoride in chronic kidney disease: a review. Hum Exp Toxicol 38(3):269–279
Podder S et al (2010) Histopathology and cell cycle alteration in the spleen of mice from low and high doses of sodium fluoride. Fluoride 43:237–245
Paul V, Ekambaram P, Jayakumar AR (1998) Effects of sodium fluoride on locomotor behavior and a few biochemical parameters in rats. Environ Toxicol Pharmacol 6(3):187–191
Ekambaram P, Paul V (2002) Modulation of fluoride toxicity in rats by calcium carbonate and by withdrawal of fluoride exposure. Pharmacol Toxicol 90(2):53–58
Bhatnagar M et al (2011) Effects of fluoride in drinking water on NADPH-diaphorase neurons in the forebrain of mice: a possible mechanism of fluoride neurotoxicity. Fluoride 44:195–209
Niu R et al (2015) Effects of fluoride on microtubule ultrastructure and expression of Tubα1a and Tubβ2a in mouse hippocampus. Chemosphere 139:422–427
Shalini B, Sharma JD (2015) Beneficial effects of Emblica officinalis on fluoride-induced toxicity on brain biochemical indexes and learning-memory in rats. Toxicol Int 22(1):35–39
Madhusudhan N et al (2010) Effect of maternal fluoride exposure on developing CNS of rats: protective role of Aloe vera, Curcuma longa and Ocimum sanctum. Indian J Exp Biol 48(8):830–836
Needham LL et al (2011) Partition of environmental chemicals between maternal and fetal blood and tissues. Environ Sci Technol 45(3):1121–1126
Vani M, Reddy K (2000) Effects of fluoride accumulation on some enzymes of brain and gastrocnemius muscle of mice. Fluoride 33
Shivarajashankara YM et al (2002) Histological changes in the brain of young fluoride-intoxicated rats. Fluoride 35:12–21
Whitford GM (1994) Intake and metabolism of fluoride. Adv Dent Res 8(1):5–14
Barbier O, Arreola-Mendoza L, Del Razo LM (2010) Molecular mechanisms of fluoride toxicity. Chem Biol Interact 188(2):319–333
Kubota K et al (2005) Fluoride induces endoplasmic reticulum stress in ameloblasts responsible for dental enamel formation. J Biol Chem 280(24):23194–23202
Yan Q et al (2007) Micromolar fluoride alters ameloblast lineage cells in vitro. J Dent Res 86(4):336–340
Jacinto-Alemán LF et al (2010) In vitro effect of sodium fluoride on antioxidative enzymes and apoptosis during murine odontogenesis. J Oral Pathol Med 39(9):709–714
Karube H et al (2009) NaF activates MAPKs and induces apoptosis in odontoblast-like cells. J Dent Res 88(5):461–465
Qu WJ et al (2008) Sodium fluoride modulates caprine osteoblast proliferation and differentiation. J Bone Miner Metab 26(4):328–334
Yan X et al (2009) Effects of sodium fluoride treatment in vitro on cell proliferation, apoptosis and caspase-3 and caspase-9 mRNA expression by neonatal rat osteoblasts. Arch Toxicol 83(5):451–458
Yang S et al (2011) Sodium fluoride induces apoptosis and alters bcl-2 family protein expression in MC3T3-E1 osteoblastic cells. Biochem Biophys Res Commun 410(4):910–915
Wyllie AH (2010) “Where, O death, is thy sting?” A brief review of apoptosis biology. Mol Neurobiol 42(1):4–9
Ola MS, Nawaz M, Ahsan H (2011) Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem 351(1-2):41–58
Mason EF, Rathmell JC (2011) Cell metabolism: an essential link between cell growth and apoptosis. Biochim Biophys Acta 1813(4):645–654
Ganeshan K, Chawla A (2014) Metabolic regulation of immune responses. Annu Rev Immunol 32:609–634
O'Neill LA, Kishton RJ, Rathmell J (2016) A guide to immunometabolism for immunologists. Nat Rev Immunol 16(9):553–565
Fina BL et al (2014) Fluoride increases superoxide production and impairs the respiratory chain in ROS 17/2.8 osteoblastic cells. PLoS One 9(6):e100768
Li QS et al (2017) Effect of fluoride treatment on gene expression in tea plant (Camellia sinensis). Sci Rep 7(1):9847
Kim JW et al (2021) Blood hemoglobin, in-vivo Alzheimer pathologies, and cognitive impairment: a cross-sectional study. Front Aging Neurosci 13:625511
Gassowska M et al (2013) Effect of fluoride on sodium-proton exchanger activity, intracellular pH and calcium concentration in human non-stimulated platelets. Ann Acad Med Stetin 59(2):54–61
Cai JN et al (2018) Sucrose challenges to Streptococcus mutans biofilms and the curve fitting for the biofilm changes. FEMS Microbiol Ecol 94(7)
Han Y (2021) Effects of brief sodium fluoride treatments on the growth of early and mature cariogenic biofilms. Sci Rep 11(1):18290
Wei M et al (2022) Effect of fluoride on cytotoxicity involved in mitochondrial dysfunction: a review of mechanism. Front Vet Sci 9:850771
Landry J et al (2000) The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci U S A 97(11):5807–5811
Imai S et al (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403(6771):795–800
Smith JS et al (2000) A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci U S A 97(12):6658–6663
Blander G, Guarente L (2004) The Sir2 family of protein deacetylases. Annu Rev Biochem 73:417–435
Morris BJ (2013) Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med 56:133–171
Frye RA (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273(2):793–798
Beausoleil SA et al (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci U S A 101(33):12130–12135
Sasaki T et al (2008) Phosphorylation regulates SIRT1 function. PLoS One 3(12):e4020
Nasrin N et al (2009) JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One 4(12):e8414
Kume S et al (2010) Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest 120(4):1043–1055
Hasegawa K et al (2008) Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun 372(1):51–56
Ghosh HS, Reizis B, Robbins PD (2011) SIRT1 associates with eIF2-alpha and regulates the cellular stress response. Sci Rep 1:150
Han MK et al (2008) SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell 2(3):241–251
Hariharan N et al (2010) Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res 107(12):1470–1482
Lee IH et al (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 105(9):3374–3379
Ohsumi Y (2001) Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2(3):211–216
Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306(5698):990–995
Lum JJ, DeBerardinis RJ, Thompson CB (2005) Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 6(6):439–448
Wei G et al (2021) Sirtuin 1 alleviates neuroinflammation-induced apoptosis after traumatic brain injury. J Cell Mol Med 25(9):4478–4486
Xu D et al (2019) SIRT2 functions in aging, autophagy, and apoptosis in post-maturation bovine oocytes. Life Sci 232:116639
Tu LF et al (2019) Sirt3-dependent deacetylation of COX-1 counteracts oxidative stress-induced cell apoptosis. FASEB J 33(12):14118–14128
Wang Y et al (2018) Sirtuin 5 overexpression attenuates glucolipotoxicity-induced pancreatic β cells apoptosis and dysfunction. Exp Cell Res 371(1):205–213
Liu M et al (2017) Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nat Commun 8(1):413
Vakhrusheva O et al (2008) Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res 102(6):703–710
Li F et al (2015) NADP(+)-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol Cell 60(4):661–675
Suzuki M, Bartlett JD (2014) Sirtuin1 and autophagy protect cells from fluoride-induced cell stress. Biochim Biophys Acta 1842(2):245–255
Nogueiras R et al (2012) Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev 92(3):1479–1514
Hayashida S et al (2010) Fasting promotes the expression of SIRT1, an NAD+ -dependent protein deacetylase, via activation of PPARalpha in mice. Mol Cell Biochem 339(1-2):285–292
Han L et al (2010) SIRT1 is regulated by a PPAR {γ}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res 38(21):7458–7471
Noriega LG et al (2011) CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO Rep 12(10):1069–1076
McCubrey JA et al (2017) Effects of resveratrol, curcumin, berberine and other nutraceuticals on aging, cancer development, cancer stem cells and microRNAs. Aging (Albany NY) 9(6):1477–1536
Halliwell B, Cross CE (1994) Oxygen-derived species: their relation to human disease and environmental stress. Environ Health Perspect 102(Suppl 10):5–12
Ames BN, Shigenaga MK, Hagen TM (1993) Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci U S A 90(17):7915–7922
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552(Pt 2):335–344
Andreyev AY, Kushnareva YE, Starkov AA (2005) Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 70(2):200–214
Starkov AA et al (2004) Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci 24(36):7779–7788
Tretter L, Adam-Vizi V (2004) Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci 24(36):7771–7778
Migliaccio E, Giorgio M, Pelicci PG (2006) Apoptosis and aging: role of p66Shc redox protein. Antioxid Redox Signal 8(3-4):600–608
Korshunov SS, Skulachev VP, Starkov AA (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 416(1):15–18
Lambert AJ, Brand MD (2004) Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J 382(Pt 2):511–517
Circu ML et al (2009) Contribution of glutathione status to oxidant-induced mitochondrial DNA damage in colonic epithelial cells. Free Radic Biol Med 47(8):1190–1198
Rachek LI et al (2009) Troglitazone, but not rosiglitazone, damages mitochondrial DNA and induces mitochondrial dysfunction and cell death in human hepatocytes. Toxicol Appl Pharmacol 240(3):348–354
Ricci C et al (2008) Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am J Phys Cell Phys 294(2):C413–C422
Fritz R et al (2007) Compartment-dependent management of H (2)O(2) by peroxisomes. Free Radic Biol Med 42(7):1119–1129
Zangar RC, Davydov DR, Verma S (2004) Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol Appl Pharmacol 199(3):316–331
Caro AA, Cederbaum AI (2006) Role of cytochrome P450 in phospholipase A2- and arachidonic acid-mediated cytotoxicity. Free Radic Biol Med 40(3):364–375
Dumitru CA et al (2007) Ceramide: a novel player in reactive oxygen species-induced signaling? Antioxid Redox Signal 9(9):1535–1540
Woo CC et al (2013) Thymoquinone inhibits tumor growth and induces apoptosis in a breast cancer xenograft mouse model: the role of p38 MAPK and ROS. PLoS One 8(10):e75356
Dai X et al (2017) A novel benzimidazole derivative, MBIC inhibits tumor growth and promotes apoptosis via activation of ROS-dependent JNK signaling pathway in hepatocellular carcinoma. Oncotarget 8(8):12831–12842
Kim C et al (2018) Formononetin-induced oxidative stress abrogates the activation of STAT3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer Lett 431:123–141
Giorgio M et al (2005) Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122(2):221–233
Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116(2):205–219
Simon HU, Haj-Yehia A, Levi-Schaffer F (2000) Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis : an international journal on programmed cell death 5:415–418
Redza-Dutordoir M, Averill-Bates DA (2016) Activation of apoptosis signalling pathways by reactive oxygen species. Biochimica et Biophysica Acta (BBA) - Molecular. Cell Res 1863(12):2977–2992
Giorgio M et al (2007) Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 8(9):722–728
Salar RK, Seasotiya L (2011) Free radical scavenging activity, phenolic contents and phytochemical evaluation of different extracts of stem bark of Butea monosperma (Lam.) Kuntze. Frontiers in Life Science 5(3-4):107–116
Benmehdi H et al (2017) Free radical scavenging activity, kinetic behaviour and phytochemical constituents of Aristolochia clematitis L. roots. Arab J Chem 10:S1402–S1408
Thatoi HN, Patra JK, Das SK (2014) Free radical scavenging and antioxidant potential of mangrove plants: a review. Acta Physiol Plant 36(3):561–579
Gad FA, Farouk SM, Emam MA (2021) Antiapoptotic and antioxidant capacity of phytochemicals from Roselle (Hibiscus sabdariffa) and their potential effects on monosodium glutamate-induced testicular damage in rat. Environ Sci Pollut Res Int 28(2):2379–2390
Dutta K, Ghosh D, Basu A (2009) Curcumin protects neuronal cells from Japanese encephalitis virus-mediated cell death and also inhibits infective viral particle formation by dysregulation of ubiquitin-proteasome system. J NeuroImmune Pharmacol 4(3):328–337
Sobeh M et al (2017) Senna singueana: antioxidant, hepatoprotective, antiapoptotic properties and phytochemical profiling of a methanol bark extract. Molecules 22(9)
Choi YJ et al (2003) Polyphenolic flavonoids differ in their antiapoptotic efficacy in hydrogen peroxide-treated human vascular endothelial cells. J Nutr 133(4):985–991
Inkielewicz-Stepniak I, Radomski MW, Wozniak M (2012) Fisetin prevents fluoride- and dexamethasone-induced oxidative damage in osteoblast and hippocampal cells. Food Chem Toxicol 50(3-4):583–589
Sujatha K (2016) Hematobiochemical and antioxidant evaluation of Aloe vera whole leaf extract on fluoride induced toxicity in Wistar albino rats. SOJ Veterinary Sciences 2(1):1–5
Bouasla A et al (2016) Prophylactic effects of pomegranate (Punica granatum) juice on sodium fluoride induced oxidative damage in liver and erythrocytes of rats. Can J Physiol Pharmacol 94(7):709–718
Scherz-Shouval R, Shvets E, Elazar Z (2007) Oxidation as a post-translational modification that regulates autophagy. Autophagy 3(4):371–373
Scherz-Shouval R, Elazar Z (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36:30–38
Ferro F et al (2020) Autophagy and mitophagy in cancer metabolic remodelling. Semin Cell Dev Biol 98:129–138
Scherz-Shouval R, Elazar Z (2007) ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 17(9):422–427
Ciccarone F, Castelli S, Ciriolo MR (2019) Oxidative stress-driven autophagy across onset and therapeutic outcome in hepatocellular carcinoma. Oxidative Med Cell Longev 2019:6050123
Azad MB, Chen Y, Gibson SB (2009) Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal 11(4):777–790
Lim SD et al (2005) Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 62(2):200–207
Ghavami S et al (2008) Brevinin-2R(1) semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J Cell Mol Med 12(3):1005–1022
Cai J et al (2008) Emodin-induced generation of reactive oxygen species inhibits RhoA activation to sensitize gastric carcinoma cells to anoikis. Neoplasia 10(1):41–51
Poillet-Perez L et al (2015) Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol 4:184–192
Rodríguez-Vargas JM, Oliver-Pozo FJ, Dantzer F (2019) PARP1 and poly (ADP-ribosyl) ation signaling during autophagy in response to nutrient deprivation. Oxidative Med Cell Longev 2019:2641712
Boyer-Guittaut M et al (2014) The role of GABARAPL1/GEC1 in autophagic flux and mitochondrial quality control in MDA-MB-436 breast cancer cells. Autophagy 10(6):986–1003
Lin Y et al (2019) Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother 118:109249
Kim YS et al (2007) TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell 26(5):675–687
Li L et al (2015) ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol 35(5):615–621
Adebayo M et al (2021) Mitochondrial fusion and fission: the fine-tune balance for cellular homeostasis. FASEB J 35(6):e21620
Puty B et al (2021) Human cultured IMR-32 neuronal-like and U87 glial-like cells have different patterns of toxicity under fluoride exposure. PLoS One 16(6):e0251200
Di Pietro V et al (2017) Fusion or fission: the destiny of mitochondria in traumatic brain injury of different severities. Sci Rep 7(1):9189
Oshima Y et al (2021) Parkin-independent mitophagy via Drp1-mediated outer membrane severing and inner membrane ubiquitination. J Cell Biol 220(6)
Lee JE et al (2016) Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540(7631):139–143
Valera-Alberni M et al (2021) Crosstalk between Drp1 phosphorylation sites during mitochondrial remodeling and their impact on metabolic adaptation. Cell Rep 36(8):109565
Feng Z et al (2019) Effects of Fluoride on Autophagy in Mouse Sertoli Cells. Biol Trace Elem Res 187(2):499–505
Ommati MM et al (2018) Is immunosuppression, induced by neonatal thymectomy, compatible with poor reproductive performance in adult male rats? Andrology 6(1):199–213
Chen Y et al (2020) Mitochondrial fusion and fission in neuronal death induced by cerebral ischemia-reperfusion and its clinical application: a mini-review. Med Sci Monit 26:e928651
Zhou BH et al (2020) Drp1/Mff signaling pathway is involved in fluoride-induced abnormal fission of hepatocyte mitochondria in mice. Sci Total Environ 725:138192
Song B et al (2021) Methionine deficiency affects liver and kidney health, oxidative stress, and ileum mucosal immunity in broilers. Front Vet Sci 8:722567
AlRefeai MH et al (2021) Assessment of bond integrity, durability, and degree of conversion of a calcium fluoride reinforced dentin adhesive. Polymers (Basel) 13(15)
Aulestia FJ et al (2020) Fluoride exposure alters Ca(2+) signaling and mitochondrial function in enamel cells. Sci Signal 13(619)
Mohamed NE (2016) The role of calcium in ameliorating the oxidative stress of fluoride in rats. Biol Trace Elem Res 170(1):128–144
Lezmy J et al (2021) Astrocyte Ca(2+)-evoked ATP release regulates myelinated axon excitability and conduction speed. Science 374(6565):eabh2858
Data availability
All the data used from publicly available search engines namely PubMed, Scopus, Web of Science. These articles are available online.
Funding
We thank Indian Council of Medical Research for providing Extra mural research funding including fellowship support to Mr. Sachindra Kumar (Grant Number: - 5/8-4/6/Env/2020-NCD-II)
Author information
Authors and Affiliations
Contributions
Conceptualisation: Sachindra Kumar, Smita Shenoy, Ravindra Shantakumar Swamy, V. Ravichandiran, Nitesh Kumar. Manuscript writing: Sachindra Kumar, Nitesh Kumar. Figures: Sachindra Kumar, Nitesh Kumar. Review: Sachindra Kumar, Smita Shenoy, Ravindra Shantakumar Swamy, V. Ravichandiran, Nitesh Kumar
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kumar, S., Shenoy, S., Swamy, R.S. et al. Fluoride-Induced Mitochondrial Dysfunction and Approaches for Its Intervention. Biol Trace Elem Res 202, 835–849 (2024). https://doi.org/10.1007/s12011-023-03720-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-023-03720-1