
Abstract
Hepatoprotective effects of natural compounds have been frequently attributed to their antioxidant properties and the ability to mobilize endogenous antioxidant defense system. Because of involvement of oxidative stress in virtually all mechanisms of liver injury, it is a reasonable presumption that antioxidant properties of these compounds may play a key role in the mechanism of their hepatoprotective activity. Nevertheless, growing evidence suggests that other pharmacological activities of natural compounds distinct from antioxidant are responsible for their therapeutic effects. In this review, we discussed currently known molecular mechanisms of the hepatoprotective activity of 27 most intensively studied phytochemicals. These compounds have been shown to possess anti-inflammatory, antisteatotic, antiapoptotic, cell survival and antiviral activity through interference with multiple molecular targets and signaling pathways. Additionally, antifibrotic properties of phytochemicals have been closely associated with apoptosis of hepatic stellate cells and stimulation of extracellular matrix degradation. However, although these compounds exhibit a pronounced hepatoprotective effects in animal and cell culture models, the lack of clinical studies remains a bottleneck for their official acceptance by medical experts and physicians. Therefore, controlled clinical trials have an imperative in confirmation of the therapeutic activity of potentially hepatoprotective compounds. Understanding the principles of the hepatoprotective activity of phytochemicals could guide future drug development and help prevention of clinical trial failure. Also, the use of new delivery systems that enhances bioavailability of poorly water soluble compounds may improve the results already obtained. Most importantly, available data suggest that phytochemicals possess a various degree of modulation of specific signaling pathways, pointing out a need for usage of combinations of several hepatoprotective compounds in both experimental studies and clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The liver has a crucial role in the regulation of multiple metabolic functions and physiological processes, such as the metabolism of nutrients, bile secretion and synthesis of proteins, lipids and carbohydrates as well as vitamin storage. Its ability to detoxify xenobiotics makes it particularly important in the maintenance of body health. Hepatic diseases are among leading causes of morbidity and mortality worldwide. Unhealthy lifestyles, related to obesity and the excessive consumption of alcohol, drugs and soft drinks are a common cause of hepatic injury. Hepatic diseases can also be induced by biological factors (bacteria, virus and parasites) and autoimmune disorders (immune hepatitis and primary biliary cirrhosis) (Nseir et al. 2010). However, this is just one arm of the balance, which is counterweighted by liver’s capacity to metabolize toxic compounds and prevent liver impairment. Moreover, the self-healing and regenerative potential of the liver could result in an excessive accumulation of extracellular matrix (ECM) proteins such as collagen, followed by progressive tissue scarring, development of cirrhosis and loss of liver function.
Despite advances in modern medicine, there is no successful therapeutical approach regarding stimulation of hepatic function, liver protection or enhancement of hepatic cell regeneration (Madrigal-Santillan et al. 2014). Current drugs, such as pegylated interferon-alpha (IFN-α) and ribavirin used in the treatment of hepatitis virus infection, are not effective in all patients and some of them will not tolerate this therapy. Similarly, silymarin, the most known hepatoprotective substance, has shown limitations regarding treatment of chronic liver impairment such as cirrhosis. Thus, it is imperative to identify highly effective pharmaceuticals for the treatment of hepatic disorders, with the accent on their low toxicity. The use of natural products in the treatment of hepatic diseases has a long history. These products emerged as a promising source of relatively nontoxic hepatoprotective compounds. However, despite the numerous evidence of hepatoprotective effects of these compounds in vitro and in vivo, it should be emphasized that studies using animal models of diseases, although generally accepted by the scientific community, cannot be easily translated to humans due to differences between species, such as genomic response to acute inflammatory stresses (Seok et al. 2013) or the activity of hepatic metabolizing enzymes (Cheung and Gonzalez 2008). Similarly, in vitro studies could not completely reflect in vivo metabolic conditions. Therefore, controlled clinical trials have an imperative in defining the therapeutic activity of potentially hepatoprotective compounds.
In this review, we gathered data based on studies conducted in animal models of hepatic disease as well as liver cell cultures, which explored hepatoprotective properties of naturally occurring phytochemicals. We focused on the possible molecular mechanisms of hepatoprotective activity of pure substances and standardized formulas, such as silymarin, rather than crude plant extracts or their fractions, which contain numerous constituents, making it difficult to attribute biological activity and its mechanism to a specific compound. Data were collected by using search engines PubMed and Scopus, with keywords “compound name”[Title/Abstract] AND (liver*[Title/Abstract] OR hepato*[Title/Abstract] OR hepati*[Title/Abstract]). Also, we excluded studies on tumor-bearing animals or tumor cell lines, since we have not reviewed antitumor properties of natural compounds, although chemopreventive activity was discussed. Additional reason to exclude such studies was ambivalent use of human hepatocellular carcinoma (HCC) cell lines for studying both cytotoxic and cytoprotective effects of tested compounds, resulting in controversial results. Some studies demonstrated a beneficial effect of natural compounds through the proapoptotic effect or sensibilization of HCCs to apoptosis, promoting these compounds as “promising chemotherapeutic agents”(Abou El Naga et al. 2013; Gao et al. 2014; Nishikawa et al. 2006; Yan et al. 2015a) while others showed antiapoptotic activity and were suggested as “protective against hepatocyte apoptosis” (Wu et al. 2008; Jung et al. 2014; Wang et al. 2014a). This imposes a following question: is it a goal to destroy or preserve HCCs in the culture? Moreover, some studies demonstrated opposed effects of tested compounds on apoptosis induced in primary hepatocytes and HCC cells, suggesting the potential use of the compound as both “hepatoprotective drug and adjuvant in anti-cancer therapy” (Ansorena et al. 2002; Karimian et al. 2012). Similarly, HCCs are frequently used as a research model for study of hepatoprotective effect of natural compounds against oxidative stress-induced hepatocellular damage (Al-Sheddi et al. 2015; Wang et al. 2015b). However, oxidative stress, including increased generation of lipid peroxidation products, is involved in both cell proliferation and growth arrest of cancer cells (Barrera 2012). Moreover, manipulation with intracellular reactive oxygen species (ROS) level is proposed as a way to selectively kill cancer cells without causing a significant toxicity to normal cells (Schumacker 2006).
We also commented on the effect of natural compounds discussed in this review on the cytochrome P450 (CYP) induction. The cytochromes P450 (CYPs) are a multigene family of microsomal hemoproteins. These enzymes are most prominent in the liver, where they play the vital role in the biotransformation of exogenous compounds such as drugs, pesticides and carcinogens (Watkins et al. 1987). However, these enzymes are involved in metabolical activation of biologically inert compounds to electrophilic derivatives that can cause toxicity and liver pathologies from alcoholic liver disease to nonalcoholic steatohepatitis, but also cellular transformation which could result in cancer (Gonzalez 2005).
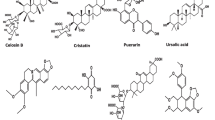
Virtually all classes of natural compounds have their representative compounds which exhibit a beneficial effect against liver diseases. We reviewed 27 most intensively studied natural compounds for the mechanisms of hepatoprotective activity, belonging to nine classes, including flavonoids, terpenoids, phenolic acids, stilbenes, alkaloids, antraquinones, curcuminoids, capsaicinoids and chromenes. The mechanisms of their hepatoprotective actions are summarized in Figs. 1, 2, 3 and 4.

Mechanisms of antioxidant and anti-inflammatory activities of phytochemicals in the liver. (1) luteolin, (2) baicalein, (3) baicalin, (4) genistein, (5) naringenin, (6) quercetin, (7) rutin, (8) troxerutin, (9) morin, (10) (–)-epigallocatechin-3-gallate, (11) silymarin, (12) thymoquinone, (13) andrographolide, (14) ginsenosides, (15) glycyrrhizin, (16) 18β-glycyrrhetinic acid, (17) betulinic acid, (18) ursolic acid, (19) chlorogenic acid, (20) salvianolic acid, (21) resveratrol, (22) berberine, (23) caffeine, (24) emodin, (25) curcumin, (26) capsaicin, (27) ellagic acid. Red circle denotes inhibitory effect; green circle denotes stimulatory effect. For abbreviations, see the abbreviation list

Mechanisms of antiapoptotic activity of phytochemicals in injured liver tissue. (1) luteolin, (2) baicalein, (3) baicalin, (4) genistein, (6) quercetin, (7) rutin, (8) troxerutin, (9) morin, (10) (–)-epigallocatechin-3-gallate, (11) silymarin, (13) andrographolide, (16) 18β-glycyrrhetinic acid, (17) betulinic acid, (18) ursolic acid, (19) chlorogenic acid, (20) salvianolic acid, (21) resveratrol, (22) berberine, (24) emodin, (25) curcumin, (26) capsaicin. Red circle denotes inhibitory effect; green circle denotes stimulatory effect. For abbreviations, see the abbreviation list

Mechanisms of antisteatotic activity of phytochemicals in fatty liver disease. (1) luteolin, (2) baicalein, (3) baicalin, (4) genistein, (5) naringenin, (6) quercetin, (7) rutin, (8) troxerutin, (9) morin, (10) (–)-epigallocatechin-3-gallate, (11) silymarin, (12) thymoquinone, (13) andrographolide, (14) ginsenosides, (16) 18β-glycyrrhetinic acid, (17) betulinic acid, (18) ursolic acid, (19) chlorogenic acid, (20) salvianolic acid, (21) resveratrol, (22) berberine, (23) caffeine, (24) emodin, (25) curcumin. Red circle denotes inhibitory effect, green circle denotes stimulatory effect. For abbreviations see the abbreviation list

Mechanisms of antifibrotic activity and HSCs apoptosis by phytochemicals in liver fibrosis. (1) luteolin, (2) baicalein, (3) baicalin, (4) genistein, (5) naringenin, (6) quercetin, (7) rutin, (9) morin, (10) (–)-epigallocatechin-3-gallate, (12) thymoquinone, (13) andrographolide, (14) ginsenosides, (16) 18β-glycyrrhetinic acid, (18) ursolic acid, (19) chlorogenic acid, (20) salvianolic acid, (21) resveratrol, (23) caffeine, (24) emodin, (25) curcumin, (26) capsaicin. Red circle denotes inhibitory effect; green circle denotes stimulatory effect. For abbreviations, see the abbreviation list
Flavonoids
Flavonoids are the largest group of naturally occurring phenolic compounds present in high concentrations in many foods, such as tea, apples, grapes and their processed beverages (Parvez et al. 2006). Flavonoids share a common 15-carbon skeleton consisting of two benzene rings linked via a heterocyclic pyrane ring. These compounds can be divided into several classes, including flavones, isoflavones, flavanones, flavonols (including flavan-3-ols and polymeric flavonolos, proanthocyanidins), flavanonols and anthocyanins. They are generally considered nontoxic for humans, exhibiting numerous pharmacological effects in vitro and in vivo. Still, their ability to modulate the activity of xenobiotic-metabolizing enzymes, particularly phase I enzymes, may have pharmacological and/or toxicological significance.
Flavones
Hepatoprotective activity of luteolin (3′,4′,5,7-tetrahydroxyflavone) (1), as well as virtually all hepatoprotective compounds reviewed in this article, has been associated with improvement in antioxidant defense, such as the increase in superoxid dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx) activity and glutathione (GSH) levels (Domitrovic et al. 2008, 2009a; Balamurugan and Karthikeyan 2012). It has been suggested that luteolin and other flavones, such chrysin and apigenin, inhibit hepatic oxidative stress through up-regulation of the extracellular signal-regulated kinase (ERK) 2/nuclear factor-erythroid-2-related factor 2 (Nrf2)/antioxidant response element (ARE) pathway (Huang et al. 2013). Luteolin has also been shown to modulate the expression of xenobiotic-induced phase I and phase II drug-metabolizing enzymes in several liver cell lines (Zhang et al. 2014f). Thus, luteolin inhibited the expression of NAD(P)H:quinone oxidoreductase-1 (NQO1) and aldo-keto reductases (AKRs) through the Nrf2 pathway and the expression of CYP1A1 and glutathione-S-transferase (GST) through the aryl hydrocarbon receptor (AhR) pathway. Moreover, luteolin inhibited CYP3A4 and CYP3A5 enzymes in human liver microsomes, suggesting a potential of pharmacokinetic interaction with co-administered drugs (Quintieri et al. 2008). Furthermore, this flavone attenuated chemically induced endoplasmic reticulum (ER) stress through suppression of activating transcription factor (ATF) 4 and CCAAT/enhancer-binding protein (C/EBP)-homologous protein (CHOP) signaling (Tai et al. 2015).
Hepatic fibrosis is a pathological condition that occurs as response to persistent liver injury, resulting in progression toward cirrhosis or resolution if an insult has been withdrawn. The reversion of hepatic fibrosis by luteolin has been accompanied by enhanced expression of matrix metalloproteinase (MMP)-9 and removal of collagen deposits, with attenuation of hepatic stellate cells (HSCs) activation (Domitrovic et al. 2009b). Li et al. (2015a) showed that luteolin may also induce apoptosis and G1 arrest in HSCs through inhibition of the platelet-derived growth factor (PDGF) and transforming growth factor-beta (TGF-β) signaling pathways, potent stimulators of fibrogenesis. Luteolin inhibited PDGF-BB-stimulated expression of phosphorylated Akt, its downstream target mammalian target of rapamycin (mTOR), as well as the mTOR substrate 70 kDa ribosomal S6 kinase (p70S6K). Additionally, luteolin attenuated TGF-β1-stimulated hepatic Smad2 phosphorylation, suggesting its potential to inhibit synthesis of fibrogenic mediators, such as connective tissue growth factor (CTGF). Moreover, these changes led to increased p53 expression and caspase-3 activity, with concomitant inhibition of the cell cycle through decrease in the expression of cyclin E and phosphorylated cyclin-dependent kinase (Cdk) 2, resulting in HSCs apoptosis and cell cycle arrest. On the other hand, luteolin protected against lipopolysaccharide (LPS)/d-galactosamine (GalN)-induced apoptotic liver damage in mice, which was accompanied by decrease in hepatic expression of tumor necrosis factor-alpha (TNF-α) receptor-associated death domain (TRADD) (Lee et al. 2011). Protection of hepatocytes was achieved by suppression of caspase-3 and caspase-8 activity and expression of proapoptotic B-cell lymphoma 2 (Bcl-2) family, including Bcl-2-associated X protein (Bax), Bcl-2 homology 3 (BH3) interacting-domain death agonist (Bid) and Bcl-2-interacting mediator of cell death (Bim).
The potential of luteolin to inhibit hepatic lipogenesis and improve hepatosteatosis was demonstrated in ethanol and high-fat diet fed (HFD) mice (Liu et al. 2014a; Kwon et al. 2015). Activation of peroxisome proliferator-activated receptor gamma (PPARγ) has emerged as a potential strategy for blocking HSCs activation and differentiation. In both models, luteolin increased PPARγ protein expression in adipose tissue and reduced hepatic expression of lipogenic genes, including sterol regulatory element-binding protein (SREBP)-1c, the master regulator of lipid synthesis, fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC) and stearoyl-CoA desaturase (SCD) 1. Luteolin abrogated ethanol-induced SREBP-1c phosphorylation via stimulation of 5′ AMP-activated protein kinase (AMPK) activity, the negative regulator of lipogenesis. In addition, luteolin improved hepatic insulin sensitivity in diet-induced obese mice by suppressing the expression of SREBP-1c and up-regulating insulin receptor (IR) substrate (IRS)-2 expression through its negative feedback (Kwon et al. 2015). Moreover, luteolin supplementation in diethylnitrosamine (DEN) and ethanol-intoxicated mice significantly reversed reduced silent mating type information regulation 2 homolog 1 (SIRT1) activity assessed by modulating the expression of forkhead box protein O (FOXO) 1 and SIRT1 target PPARγ coactivator 1 alpha (PGC1α), substantially preventing pre-neoplastic lesions in the liver (Rafacho et al. 2015). Usage of the phospholipid complex of luteolin has been proposed as an enhanced drug delivery system for oral administration of luteolin with increased bioavailability and hepatoprotective potential (Khan et al. 2015).
Flavones baicalein (5,6,7-trihydroxyflavone) (2) and baicalin (baicalein 7-O-glucuronide) (3) inhibited pro-oxidant-induced liver injury in vitro by scavenging ROS in hepatocytes (Zhao et al. 2006). Baicalein improved metabolic syndrome induced by HFD in mice through the inhibition of ERK1/2 and c-Jun N-terminal kinase (JNK)1/2/3 mitogen-activated protein kinases (MAPKs) phosphorylation and activation of the IRS-1/phosphoinositide 3-kinase (PI3K)/Akt pathway (Pu et al. 2012b). The lipid-lowering effect was attributed to the suppression of SREBP-1c, PPARγ and their target genes, including FAS, ACC, uncoupling protein (UCP) 2 and adipocyte fatty acid-binding protein (aP), with concomitant induction of lipolytic enzymes such as PPARα, cluster of differentiation (CD) 36 and carnitine palmitoyltransferase (CPT) 1. All these effects were dependent on AMPK activation. Interestingly, the same effect was observed by flavanone naringin (Pu et al. 2012a). Baicalein also prevented nonalcoholic steatohepatitis (NASH) through enhancement of Nrf2/heme oxygenase (HO)-1 pathway and suppression of NF-κB activation (Xin et al. 2014). In LPS/D-GalN-induced acute liver failure in mice, baicalein ameliorated gene expression of TNF-α, inducible nitric oxide synthase (iNOS), vascular endothelial growth factor (VEGF), VEGF receptor (VEGFR) and monocyte chemoattractant protein (MCP)-1, inhibiting hepatic inflammation and HSCs migration (Cheng et al. 2007; Wu et al. 2010; Chen et al. 2013a). Concomitantly, it inhibited hepatic apoptosis by suppressing phosphorylation of ERK and JNK (Wu et al. 2010). Moreover, baicalein activated cellular Fas-associated death domain (FADD)-like IL-1β-converting enzyme-inhibitory protein (c-FLIP), X-linked inhibitor of apoptosis protein (XIAP) and cellular inhibitor of apoptosis (cIAP) 2 proteins while reducing nuclear levels of RelA, suggesting attenuation of the NF-κB signaling. Similarly, the inhibition of MAPKs activation by this compound significantly improved the survival of mice with polymicrobial sepsis-induced liver injury (Liu et al. 2015a). Furthermore, baicalein prevented CCl4-induced liver fibrosis in rats and inhibited HSCs activation and proliferation by down-regulation of PDGF receptor beta (PDGFRβ) (Sun et al. 2010). In addition, baicalein increased gene expression of TGF-α, hepatocyte growth factor (HGF) and epidermal growth factor (EGF), well-known regulators of liver regeneration.
Similarly to baicalein, its glucoronide conjugate, baicalin, exhibited protection against oxidative liver injury (Kim et al. 2010c; Zhang et al. 2012). Baicalin also suppressed toll-like receptor (TLR) 4-mediated inflammatory response after ischemia/reperfusion-induced liver injury in rats (Kim and Lee 2012) which was associated with the inhibition of myeloid differentiation factor 88 (MyD88) protein expression and the nuclear translocation of NF-κB. Additionally, baicalin up-regulated cytoprotective HO-1 expression, which is suggesting the activation of the Nrf2 pathway (Kim et al. 2010c). Moreover, baicalin suppressed apoptosis in concanavalin A-treated mice livers by inhibiting TNF-α-induced JNK phosphorylation and suppressing caspase-3 and caspase-9 activation (Liu et al. 2007). Studying the antifibrotic potential of baicalin, several authors showed attenuation of CCl4-intoxication in rats through suppression of TGF-β1 and PPARγ activation (Peng et al. 2009; Qiao et al. 2011). Yang et al. (2012) showed that the mechanism of PPARγ-mediated suppression of fibrotic response by baicalin, as well as rosmarinic acid, could be mediated through the suppression of signaling by canonical Wnts. Furthermore, Guo et al. (2009) demonstrated that baicalin had beneficial effect on development of hepatic steatosis by reducing hepatic lipid accumulation through enhancement of AMPK phosphorylation and down-regulation of genes involved in lipogenesis, including FAS and its upstream regulator SREBP-1c. Additionally, baicalin could attenuate HFD-induced obesity and fatty liver disease through the inhibition of Ca2+-calmodulin dependent protein kinase kinase (CAMKK)/AMPK/ACC pathway (Xi et al. 2015).
Both compounds, baicalein and baicalin, were able to modulate the activity of hepatic CYP system. Baicalein inhibited CYP1A2 and CYP3A4 activity in human liver microsomes and CYP2E1 and CYP3A expression in mice liver (Ueng et al. 2000; Kim et al. 2002). Baicalin inhibited hepatic CYP3A and CYP2D activity in rats (Tian et al. 2013; Gao et al. 2014) and CYP1A2 in both human and rat livers (Cheng et al. 2014). These findings suggest that baicalein and baicalin may modulate the activity of drug-metabolizing enzymes.
Isoflavones
The inhibition of NF-κB and MAPK signaling was suggested as a mechanism of the anti-inflammatory activity of genistein (4′,5,7-trihydroxyisoflavone) (4) (Ji et al. 2011; Lin et al. 2014b; Saleh et al. 2014; Ganai et al. 2015). This soy estrogenic isoflavone showed the ability to improve liver fibrosis induced by chronic CCl4 administration through the increase in expression and proteolytic activity of urokinase-type plasminogen activator (uPA), a potent inductor of collagenases and ECM degradation (Salas et al. 2007). Antifibrotic activity of genistein has also been associated with reduced levels of PDGF-BB (Demiroren et al. 2014), which acts via its cell surface tyrosine kinase receptors, inducing proliferation of HSCs via PI3K/Akt and ERK pathways (Fang et al. 2013). In hepatic fibrosis induced by chronic administration of alcohol in rats, genistein promoted extracellular matrix (ECM) degradation by reducing tissue inhibitor of matrix metalloproteinase (TIMP)-1 and increasing MMP-2 mRNA levels, with concomitant inhibition of TGF-β1 expression. In addition, genistein exhibited potency to attenuate PDGF-induced c-Fos, c-Jun and cyclin D1 expression and inhibit HSCs proliferation (Liu et al. 2002d). Genistein also acted as a selective agonist for the beta isoform of the estrogen receptor (ESRβ), which is highly expressed by active HSCs (McCarty et al. 2009).
Genistein has also been shown to possess inhibitory effect on hepatic steatosis in HFD-induced nonalcoholic fatty liver disease (NAFLD) in mice through down-regulation of genes associated with lipogenesis, including SREBP-1c, liver X receptor alpha (LXRα), retinoid X receptor alpha (RXRα), PPARγ and ACC2, with concomitant up-regulation of genes involved in β-oxidation, including AMPK and PPARα (Kim et al. 2010b). Additionally, genistein augmented the expression of anti-steatohepatitic adiponectin, an adipocyte-derived hormone, and reduced levels of pro-inflammatory cytokine TNF-α. Moreover, this isoflavone inhibited expression of TLR4, a member of a class of proteins involved in regulation of IkappaB kinase (IKK) activity and downstream NF-κB activation (Ambade and Mandrekar 2012), leading to attenuation of production of proinflammatory cytokines in diet-induced NAFLD (Yoo et al. 2015). Concomitant reduction in CHOP, X-box binding protein 1 (XBP-1) and ER chaperone immunoglobulin binding protein (BiP) mRNA expression suggested attenuation of ER stress in hepatocytes. Genistein-treated younglings showed lower hepatic expression of FAS and SREBP-1, but higher expression of PPARα, indicating lower rates of lipid synthesis and higher rates of β-oxidation (Huang et al. 2011). Moreover, genistein was able to sensitize hepatic insulin signaling by improving insulin-stimulated tyrosine phosphorylation of IR-β and IRS-1 as well as downstream PI3K and Akt phosphorylation in the mouse model of metabolic syndrome (Arunkumar et al. 2013). Genistein treatment also resulted in the increase in AMPK and the decrease in p70S6K phosphorylation, suggesting its beneficial effect in amelioration of both hepatic steatosis and metabolic syndrome.
Nevertheless, caution with intake of higher doses of genistein has been suggested, since they can produce several undesirable effects by affecting multiple cellular pathways (Okazaki et al. 2002; Singh et al. 2014). Specific tyrosine kinase inhibitory properties of genistein resulted in dose-dependent inhibition of the recombinant human HGF-mediated stimulation of DNA synthesis by hepatocytes, which could have negative impact on liver repair and regeneration (Arakaki et al. 1992; Kaibori et al. 2006). The potential for diet–drug interactions of genistein and several other dietary flavonoids has also been demonstrated. Thus, these flavonoids acted as modulators of organic anion-transporting polypeptide (OATP) 1B1, a liver-specific uptake transporter important in hepatic drug disposition (Wang et al. 2005). In addition, genistein was shown to inhibit the activity of several CYPs, including CYP1A2 and CYP2E1, in a noncompetitive manner (Roberts-Kirchhoff et al. 1999).
Flavanones
The most intensively studied citrus flavonoid, naringenin (4′,5,7-trihydroxyflavanone) (5), exhibited protective activity against hepatic oxidative injury (Pari and Gnanasoundari 2006; Lv et al. 2013). Naringenin has been showing a potential to attenuate inflammation in several experimental models of liver injury. The suppression of CCl4- and cholesterol-induced hepatic inflammation in rats was mediated by down-regulation of proinflammatory genes, including NF-κB, TNF-α, IL-1 β, IL-6, iNOS and EGF-like module-containing mucin-like hormone receptor 1 (EMR1, the macrophage-specific gene) (Esmaeili and Alilou 2014; Chtourou et al. 2015). In addition, naringenin up-regulated the cytoprotective Nrf2/HO-1 pathway. Several studies showed that naringenin may be useful in preventing the ECM accumulation and development of hepatic fibrosis. This flavone inhibited gene expression of the antifibrotic targets MMP-2 and MMP-9 and prevented ECM remodeling (Liu et al. 2006). Mechanistically, the antifibrotic activity of naringenin was accompanied by inhibition of TGF-β1-induced production of Smad3 at both mRNA and protein levels. In HFD-fed mice, naringenin reduced hepatic triglyceride concentration and gene expression of hepatic lipogenic enzymes and up-regulated hepatic fatty acid oxidation genes, suggesting inhibition of SREBP-1c and induction of SIRT1/PGC-1α pathways (Assini et al. 2015). In docking study, naringenin and quercetin exhibited minimum binding energy with nonstructural hepatitis C virus (HCV) NS2 protease, suggesting inhibitory potential against the viral replication (Lulu et al. 2015). As seen in similar studies, usage of naringenin-loaded nanoparticles system could improve hepatoprotective effects of the flavonoid after oral administration (Yen et al. 2009). Nanoformulation improved the bioavailability of the compound, which resulted in more potent antioxidant and antiapoptotic activities.
The ability of naringenin to modulate the activity of CYPs has also been demonstrated. Naringenin was potent inhibitor of CYP1A1, CYP1A2, CYP2B1, CYP2D6, CYP2E1 and CYP3A1/2 and relatively weak inhibitor of CYP3A4 activity in vitro (Fuhr et al. 1993; Bear and Teel 2000; Ho et al. 2001; Motawi et al. 2014; Arinc et al. 2015). Thus, the possibility of modulation of drug and other xenobiotic metabolism exists, which should be further investigated, particularly in light of protection against xenobiotic-induced carcinogenesis.
Flavonols
Quercetin (3,3′,4′,5,7-pentahydroxyflavone) (6) is one of the most studied natural products, showing numerous beneficial effects, such as hepatoprotection. It has been found that quercetin could ameliorate oxidative livery injury by reducing lipid peroxidation and increasing GSH levels and the activity of antioxidant enzymes (Casella et al. 2014; Surapaneni and Jainu 2014; El-Shafey et al. 2015). Several studies demonstrated that protective activity of quercetin against oxidative hepatocyte injury was mediated through activation of the Nrf2/HO-1 pathway (Liu et al. 2012, 2015b; Li et al. 2013c). The mechanism of induction involved p38 and ERK activation and nuclear translocation of Nrf2. Antioxidant activity of quercetin was also associated with expression of other oxidative stress-related genes, including peroxiredoxin (Prx) 1, 2, 3, 5, 6, thioredoxin reductase (TrxR) 1 and 2 and thioredoxin (Trx) 1 and 2 (Zhang et al. 2014c). We showed that expression of Nrf2 and HO-1 was more potently suppressed by rutin, quercetin 3-O-rutinoside, than its aglycone quercetin (Domitrovic et al. 2012). In contrast, quercetin more potently attenuated the expression of TGF-β1, suggesting strong impact of structure–activity relationship between these two compounds on amelioration of liver injury. Several studies also demonstrated antiapoptotic activity of quercetin in chemically intoxicated rats (de David et al. 2011; Sekaran et al. 2012; Sarkar and Sil 2014). The mechanism involved down-regulation of p53-inducible apoptotic proteins (Bax, caspase-9, caspase-3) and up-regulation of the pro-survival ERK1/2 pathway. In contrast, quercetin initiated proapoptotic events in DMN-induced hepatocarcinogenesis in rat livers (Casella et al. 2014) and induced the cell cycle arrest by reducing expression of cyclins and Cdk1.
Quercetin suppressed nuclear import of NF-κB in diet-induced hepatic inflammation in mice (Das et al. 2013; Sikder et al. 2014). The inhibition of NF-κB was accompanied by suppression of TNF-α, iNOS, cyclooxygenase-2 (COX-2), interleukin (IL)-1β and C-reactive protein (CRP), as well as suppression of IKKα and prevention of IkappaBalpha (IκBα) degradation, both involved in NF-κB activation (Dias et al. 2005; Martínez-Flórez et al. 2005; García-Mediavilla et al. 2007; Ponmari et al. 2014). Moreover, quercetin has been shown to decrease protein expression of TLR4, an upstream inductor of NF-κB (Marcolin et al. 2013). Quercetin also suppressed arsenite-induced expression of COX-2 and PGE2 production by blocking the activation of the PI3K/Akt/p70S6K signaling pathway and phosphorylation of ERK1/2 (Lee et al. 2010b). The inhibition of heavy metal-induced hepatic injury could be, at least in part, attributed to inhibition of the p38/STAT1/NF-κB signaling pathway (Liu et al. 2015b). Furthermore, quercetin suppressed nonalcoholic steatohepatitis in mice induced by methionine/choline-deficient diet, which was associated with the reduction of the mRNA levels of inflammatory mediators and the decrease in protein expression of JNK and p65 NFκB subunit (Marcolin et al. 2012). Zhang et al. (2015b) demonstrated that quercetin could ameliorate high-fructose diet-induced caspase-1 expression in rat hepatocytes and activation of proinflammatory cytokines, such as IL-18 and IL-1β, through inhibition of thioredoxin-interacting protein (TXNIP)-mediated NACHT, LRR and PYD domains-containing protein 3 (NALP-3) inflammasome. In addition, quercetin inhibited activation of janus-activated kinase 2/signal transducers and activators of transcription 3 (JAK2/STAT3)-mediated inflammatory signaling and altered the expression of lipid metabolism-related enzymes, including PPARα, SREBP1 and SCD1, thus preventing lipid accumulation in the liver. An amelioration of metabolic syndrome in rats by this phenolic was mediated through the suppression of NF-κB and caspase-3 activation, with increased expression of Nrf2, HO-1 and CPT1 (Panchal et al. 2012).
Antifibrotic potential of quercetin has been shown in rat common bile duct ligation (BDL) and CCl4 models of hepatic damage (Bona et al. 2012; Lin et al. 2014a). Quercetin attenuated BDL-induced NF-κB activation and the expression of MyD88 and TGF-β-activated kinase 1 (TAK1), critical for linking TLR and NF-κB. Quercetin also reduced Smad2/3 activity critical for the fibrogenic potential of TGF-β1 and HSCs activation (Lin et al. 2014a; Wan et al. 2014). In addition, this compound down-regulated the expression of inflammatory genes and proteins related to precancerous conditions, such as glioma-associated oncogenes (GLI)-1 and -2 (Cuevas et al. 2011). Suppression of amphiregulin (AR)/EGF receptor (EGFR) axis was associated with the inhibition of Akt and ERK1/2, major survival signals. In CCl4-induced cirrhosis model in rats, quercetin was shown to decrease MMP-2 levels (Bona et al. 2012) and ameliorate HSCs activation by reducing the levels of inflammatory cytokines. Moreover, quercetin decreased the hepatic gene expression of PDGF-BB, HSCs mitogen, CTGF and TGF-β1, potent stimulators of ECM production, and the EGFR ligand AR, suggesting inhibition of multiple profibrotic gene pathways (Marcolin et al. 2012). Further, quercetin induced G1 arrest and impaired the proliferation of activated rat HSCs by increasing levels of p53, p21 and p27 and decreasing expression of cyclins D1, D2, A and E (Wu et al. 2011). Additionally, quercetin prevented HSCs activation by reducing the levels of inflammatory cytokines, including chemokine (C-X-C motif) ligand (CXCL)-1 and midkine. Quercetin also stimulated HSCs apoptosis, which was accompanied by increased expression of Fas/Fas ligand (FasL) and caspase-3 activity. Previously, Kawada et al. (1998) showed that resveratrol, and more potently, quercetin, may suppress inositol phosphate (IP3) production, receptor-tyrosine kinase phosphorylation and ERK1/2 activation in PDGF/BB-stimulated HSCs, indicating a complex role of quercetin in hepatoprotection.
Further, quercetin showed inhibitory effect on HCV replication in vitro, which was attributed to inhibition of HCV nonstructural protein (NS) protease activity and reduction of HSP40 and HSP70 expression (Gonzalez et al. 2009; Bachmetov et al. 2012). The HCV genome is translated through an internal ribosome entry site (IRES) before its conversion into individual mature viral proteins and NS5A has been implicated in the regulation of viral genome replication. Quercetin has been shown to reduce IRES activity and its augmentation by NS5A (Gonzalez et al. 2009). Interestingly, inhibition of the PI3K/Akt/LXRα-dependent lipogenesis also contributed to suppression of the viral replication (Pisonero-Vaquero et al. 2014).
It should be mentioned that some studies indicated that quercetin may exhibit prooxidant activity and exacerbate toxic liver injury (Meng et al. 2013; Pietsch et al. 2014). Nevertheless, doses used in these studies were relatively high compared to average dietary intake. Quercetin has been mostly shown to inhibit the activity of various CYPs, including CYP1A1, CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19 and CYP3A4 (Walsky et al. 2005; He et al. 2006; Tiong et al. 2010; Rastogi and Jana 2014; Arinc et al. 2015). It also reduced CYP2E1 levels and activity and decreased CYP1B1 expression (Choi et al. 2012; Tang et al. 2013; Surapaneni et al. 2014). The inhibitory potential against gene expression of CYP2B9, CYP2B10, CYP2B13 and CYP3A59 in mice livers has also been demonstrated (Marcolin et al. 2012). In addition, among several flavonoids tested, including resveratrol and curcumin, only quercetin induced accumulation of CYP3A4 mRNA in primary human hepatocytes (Raucy 2003), indicating the potential for adverse reactions by this compound.
Rutin, a quercetin-rutinoside (7), exhibited the hepatoprotective effect through modulation of the antioxidant genes expression and improvement in antioxidant status in several models of liver injury (Khan et al. 2012; Al-Rejaie et al. 2013). In mice fed high-cholesterol diet, rutin, as well as quercetin, attenuated the increase in expression of redox sensitive transcription factor NF-κB and inflammatory markers such as CRP and TNF-α (Sikder et al. 2014). Similarly, inflammatory response in cyclophosphamide-intoxicated mice was inhibited through down-regulation of COX-2 and p38 MAPK expression (Nafees et al. 2015). Rutin administration also attenuated the increase in expression of hepatic iNOS in ischemia-/reperfusion-injured rat livers, while increasing endothelial nitric oxide synthase (eNOS) expression (Lanteri et al. 2007) and nitric oxide (NO) production (Yagnik et al. 2002). Moreover, rutin supplementation completely reversed CCl4-induced increase in the gene expression of EGF, FADD, Bcl-xL, IL-6, STAT3 and JAK and decrease in Bcl-2 expression, suggesting attenuation of hepatotoxicity through several signaling pathways (Hafez et al. 2015). The beneficial effects of rutin in cholestatic liver injury were associated with down-regulation of inflammatory NF-κB and profibrotic TGF-β1/Smad2/3 signaling pathways, most likely via suppression of ERK and AMPK activation as well as enhancement of Nrf2 and HO-1 expression (Pan et al. 2014). Additional suggestion for antifibrotic mechanism of rutin came from another study, which demonstrated that rutin, as well as curcumin, may stimulate microtubule-associated protein light chain 3 (LC3) protein expression and autophagy of activated HSCs via stimulation of the PI3K/Akt/mTOR-dependent pathway (Lee et al. 2014c). The hepatoprotective effects of rutin against fatty acid-induced inflammation and obesity were associated with the suppression of transcriptional activation of SREBP-1c and CD36 in the liver and amelioration of fatty liver disease (Gao et al. 2013a).
Troxerutin, a trihydroxyethylated derivative of rutin (8), has been shown to exert hepatoprotective activity through amelioration of hepatic oxidative stress and inflammation by restoring the activity of antioxidant enzymes and suppressing NF-κB, iNOS and COX-2 expression (Zhang et al. 2009, 2015e). Troxerutin could reduce hepatic oxidative stress-mediated NAD+-depletion in intoxicated mice and restored SIRT1 protein expression and activity (Zhang et al. 2015e), which was accompanied by suppression of NF-κB nuclear translocation and inflammatory genes induction. The same authors demonstrated that troxerutin could inhibit hepatocyte apoptosis by alleviating oxidative stress-mediated proteasome dysfunction and restoring ER stress-mediated apoptotic pathway (Zhang et al. 2015d). Mechanistically, troxerutin blocked TNF-α receptor (TNFR)-associated factor (TRAF) 2/apoptosis signal-regulating kinase (ASK) 1/JNK and CHOP-mediated signaling. Troxerutin could also prevent liver steatosis by blocking oxidative stress and restoring dysfunction of lipin 1 signaling in high-fat diet-treated mice, improving lipid homeostasis by enhancing fatty acid oxidation and triglyceride secretion while suppressing lipogenesis (Zhang et al. 2014g). The authors demonstrated that troxerutin suppressed oxidative stress-mediated NAD+-depletion by increasing nicotinamide phosphoribosyltransferase (NAMPT) protein expression and decreasing poly (ADP-ribose) polymerase (PARP)-1 protein expression and activity in mice livers. Consequently, troxerutin restored hepatic SIRT1 protein expression and activity, thus promoting AMPK activation and subsequent inhibition of mTOR complex 1 (mTORC1) signaling. This resulted in reduced cytoplasmic and increased nuclear localization of lipin 1, where it could serve as a component of a transcriptional complex with PPARα and PGC-1α to regulate fatty acid metabolism in liver (Finck et al. 2006).
Morin (3,5,7,2′,4′-pentahydroxyflavone) (9), a flavonoid isolated from herbs of the Moraceae family, showed hepatoprotective activity by reducing hepatic oxidative stress (Shankari et al. 2010; Merwid-Lad et al. 2014). Morin prevented acute liver damage induced by CC4 by inhibiting the expression of NF-κB and the production of inflammatory cytokines (Heeba and Mahmoud 2014). High-glucose-induced oxidative stress and apoptosis in primary rat hepatocytes, evidenced by increased Bax, caspase-3 and caspase-9 expression and cytochrome c cytoplasmic translocation, were attenuated by morin (Kapoor and Kakkar 2012). Morin could also be employed as a chemopreventive compound in attenuation of fibrogenesis. Morin demonstrated the ability to inhibit hepatic fibrosis in rats and induce apoptosis of HSCs by suppressing canonical NF-κB signaling (MadanKumar et al. 2015). Those authors demonstrated the inhibition of DEN-induced liver fibrosis in rats by this compound through the down-regulation of glycogen synthase kinase-3 beta (GSK-3β), β-catenin and cyclin D1 expressions, which was accompanied by the inhibition of Wnt signaling (MadanKumar et al. 2014). Using the same model of hepatic fibrogenesis, other authors also showed the suppression of TGF-β1 expression by this compound (Lee et al. 2009). Moreover, oral supplementation of morin down-regulated NF-κB nuclear translocation and MMP-2 and MMP-9 expression in DEN-intoxicated rats, indicating the potential of morin to prevent inflammation and carcinogenesis (Sivaramakrishnan and Niranjali Devaraj 2009). Morin administration also resulted in inhibition of the PI3K/Akt-mediated suppression of GSK-3β (Sivaramakrishnan and Devaraj 2010).
Further, morin attenuated hepatosteatosis in high-fructose fed rats by inhibiting NF-κB activation (Wang et al. 2013). Fructose feeding activated sphingosine kinase 1 (SphK1)/sphingosine-1-phosphate (S1P) signaling pathway, which in turn caused activation of the NF-κB pathway and inflammation in rat livers. Subsequently, hepatic leptin and insulin signaling impairment resulted in liver lipid accumulation. Morin restored high-fructose-induced changes in mice by increasing phosphorylation of IR, IRS-1(Tyr), ERK, JAK and reducing phosphorylation of the long form of leptin receptor (Ob-RL), STAT3, suppressor of cytokine signaling (SOCS) 3 and IRS-1(Ser), with the decrease in SphK1 activity and S1P production. Additionally, the level of lipolytic PPARα and CPT1 was increased by morin treatment. Taken together, these data showed the potential of morin to ameliorate hepatic inflammation and metabolic syndrome.
Flavan-3-ols
(–)-Epigallocatechin-3-gallate ((–)-cis-3,3′,4′,5,5′,7-hexahydroxy-flavane-3-gallate, EGCG) (10), the major catechin found in green tea, Camellia sinensis L., showed protective effects against oxidative damage in hepatic cells through the increase in SOD, CAT, GPx and GR activity and vitamin C and E levels (Kaviarasan et al. 2008; Ren et al. 2011; An et al. 2014), which was attributed to activation of the Nrf2 pathway (Wang et al. 2015a). Numerous studies indicated potentially therapeutic role of EGCG against various kinds of impairment of liver function. Thus, EGCG reduced caspase-3 expression and apoptosis in the ischemia/reperfused rat livers through down-regulation of NF-κB and c-Jun expression (Giakoustidis et al. 2010). Pretreatment with EGCG reduced LPS-induced production of inflammatory mediators, including TNF-α, Rantes, MCP-1, intracellular adhesion molecule (ICAM)-1, VEGF, NO and MMP-2 by inhibiting NF-κB, Akt and MAPK pathways (Liu et al. 2014c).
EGCG prevented NASH-related preneoplastic lesions in obese and hypertensive rats by decreasing the mRNA expression of angiotensin-converting enzyme (ACE) and angiotensin II receptor type 1 (AT1R), components of the renin–angiotensin system (RAS), which is closely associated with liver fibrosis and carcinogenesis (Kochi et al. 2014). Additionally, EGCG attenuated the mRNA expression of liver fibrosis-related MMP-2, MMP-9, TIMP-1, TIMP-2, alpha-smooth muscle actin (α-SMA), TGF-β1 and plasminogen activator inhibitor type 1 (PAI-1). In addition, this compound decreased levels of hepatic phospho-JNK and reduced mRNA expression of inflammatory mediators. EGCG also prevented DEN-induced liver steatosis and tumorigenesis in obese and diabetic mice by activating AMPK and inhibiting the insulin growth factor (IGF)/IGF 1 receptor (IGF-1R) axis (Shimizu et al. 2011). EGCG inhibited the phosphorylation of IGF-1R, ERK, Akt, GSK-3β, STAT3 and JNK in mice livers. Furthermore, EGCG ameliorated hepatic preneoplastic lesions induced by DEN and HFD by decreasing hepatic gene expression of proinflammatory mediators and cyclin D1 and suppressing hepatocyte proliferation (Sumi et al. 2013). In rat liver epithelial cells, the gap junctional intercellular communication (GJIC), which is strongly associated with carcinogenesis, particularly the tumor promotion process, was inhibited by EGCG (Kang et al. 2008). EGCG treatment increased phosphorylation of ERK1/2 and connexin 43 (Cx43), the major regulator of GJIC, whereas inhibition of ERK by a pharmacological inhibitor U0126 completely reverted inhibition of GJIC. Interestingly, the GJIC inhibitory properties of EGCG were attributed to its prooxidant activity. EGCG has also been reported as an in vitro inhibitor of several CYP isoforms, including CYP1A1, CYP1A2, CYP2B1/2, CYP2B6, CYP2C8 and CYP3A (Yun et al. 2007; Weng et al. 2012; Misaka et al. 2013), which could contribute to its cytoprotective potential.
Treatment with EGCG attenuated hepatic inflammation and fibrosis in the CCl4, BDL and NASH models in vivo and HSCs in vitro (Zhen et al. 2007; Tipoe et al. 2010; Xiao et al. 2014; Yu et al. 2015; Ding et al. 2015). EGCG ameliorated expression of inflammatory and fibrotic markers such as NOS, COX-2, TNF-α, collagen α1(I), MMP-2, MMP-9, TIMP-1 and α-SMA, which was associated with down-regulation of the TGF-β1/Smad2/3, PI3K/Akt/FOXO1 and NF-κB pathways. Further, EGCG ameliorated the mRNA expression of IGF-1R and the mRNA and protein levels of PDGFRβ in the liver of fibrotic rats (Yasuda et al. 2009). Moreover, EGCG interrupted TGF-β signaling by reducing the gene expression of TGF-β receptors (Tβ-R) I and II and Smad4, resulting in reduced the mRNA levels of CTGF, collagen and fibronectin, which was dependent on the induction of de novo synthesis of GSH (Fu et al. 2008b; Yumei et al. 2006). This compound also inhibited HSCs growth, which was attributed to the suppression of the tyrosine phosphorylation and the gene expression of PDGFRβ by blocking the activation of transcription factors AP-1 and NF-κB (Chen and Zhang 2003). In cultured human HSCs, EGCG inhibited the PDGF-BB-induced HSCs proliferation and collagen α1(I) and (IV) mRNA expression (Sakata et al. 2004). Furthermore, EGCG regulated HSCs growth through Rho-signaling pathway, which is implicated in activation and proliferation of HSCs (Higashi et al. 2005). Activated Rho (the GTP-bound state) was inhibited by ECGC, which was accompanied by suppression of phosphorylation of focal adhesion kinase (FAK), an regulator of Rho-signaling pathways. Moreover, EGCG was found to suppress HSCs invasiveness, through inhibition of MMP-2 activation (Zhen et al. 2006). EGCG also interrupted EGF signaling in activated HSCs by reducing the trans-activation of early growth response protein (EGR)-1 and suppressing gene expression of EGFR (Fu and Chen 2006; Hirsova et al. 2012), which plays an important role in differentiation and mitogenesis.
EGCG administration to hypercholesterolemic rats diminished induction of acyl-CoA:cholesterol acyltransferase ACAT and SREBP-1 mRNA and raised reduced levels of ATP-binding cassette transporter (ABC) G5 and G8 (Hirsova et al. 2012). Moreover, EGCG supplementation reduced the TNF-α-mediated Ca2+-dependent nuclear factor of activated T-cell (NFAT) expression and its downstream targets, including ICAM-1 and E-selectin, and NF-κB-mediated downstream targets such as VCAM-1 and P-selectin, thus inhibiting high-cholesterol-induced macrophage infiltration and hepatic steatosis (Krishnan et al. 2014). Similarly, the beneficial effects of EGCG on HFD-induced fatty liver in mice were associated with reduced levels of inflammatory cytokines (Chen et al. 2011) and down-regulation of lipogenesis-related genes, including ACC, FAS and SCD1 (Friedrich et al. 2012). The reduction of hepatosteatosis was accompanied by increased expression of LC3-I/II and phosphorylation of AMPK, one of major regulators of autophagy (Zhou et al. 2014). In addition, EGCG was shown to down-regulate uncoupling protein (UCP) 2 expression (Jamal et al. 2015), which is involved in development of steatohepatitis.
EGCG has been shown to suppress the expression of HBV surface antigen (HBsAg) and hepatitis B e antigen (HBeAg), the hepatitis B virus (HBV) antigens, and to reduce extracellular HBV DNA production and intracellular HBV DNA replication in vitro, although it had weaker efficacy compared to green tea extract (Xu et al. 2008). EGCG also demonstrated inhibitory activity on HCV viral protein, NS5B, which possesses the key function of replicating HCV viral RNA (Roh and Jo 2011). Moreover, EGCG opposed HBV-induced incomplete autophagy via enhancing lysosomal acidification, which seems to be unfavorable for HBV replication (Zhong et al. 2015). EGCG also inhibited HCV attachment to hepatocytes, disrupting the initial step of cell entry (Ciesek et al. 2011), which was attributed to impairment of HCV envelope glycoproteins E1 and E2 (Calland et al. 2012). Other catechins, such as (+)-epicatechin and (–)-epicatechin, also showed significant anti-HCV activity, protecting against HCV replication and attenuating virus-induced inflammation (Lin et al. 2013).
Recent study of Church et al. (2015) indicated that hepatotoxicity reports of green tea extract consumption in humans could be related to differences in sensitivity to EGCG, which emerge from the genetic background. Research conducted by James et al. (2015) may also partly explain the observed variation in hepatotoxic response to EGCG and green tea-containing dietary supplements. Thus, dietary pretreatment with EGCG may limit the bioavailability and hepatotoxicity of successive oral bolus doses of the catechin. Most importantly, randomized, double-blind, placebo-controlled study demonstrated that oral doses of EGCG of up to 800 mg per day for 10 days were safe and very well tolerated (Ullmann et al. 2004). Interestingly, the authors found the increase in accumulation factor for EGCG in the high-dosage group, suggesting dose-dependent saturation of capacity-limited excretion routes or an increase of hepato-duodenal recirculation, which should be taken into consideration.
Flavonolignans
Silymarin (11) is a natural substance isolated from Silybum marianum L., commonly known as Milk thistle. This substance contains several flavolignans including silybin, isosilybin, silychristin and silydianin, and flavonoids such as taxifolin and quercetin (Zhu et al. 2013). Silymarin has been extensively studied for its hepatoprotective effects in the last decade. However, surprisingly small number of studies investigated the underlying mechanisms of hepatoprotective activity of silymarin or its components. Generally, silymarin has been shown to modulate enzymatic and nonenzymatic markers of oxidative injury in the liver (Pradeep et al. 2007; Tzeng et al. 2013) and induce Nrf2 expression (Kim et al. 2012). Silymarin also exhibited anti-inflammatory activity in several models of liver damage. In the alcoholic fatty liver model in rats silymarin down-regulated expression of NF-κB, ICAM-1 and IL-6 (Zhang et al. 2013a). Silymarin also attenuated chemically induced hepatocellular damage by increasing the expression of antiapoptotic Bcl-xL protein and reducing p53 expression, caspase-3 activity, PARP activity and DNA fragmentation (Patel et al. 2010; Sherif and Al-Gayyar 2013). Inhibition of TGF-β1 and PDGF signaling has also been involved in the hepatoprotective effects of silymarin (Chen et al. 2012a; Clichici et al. 2015). In chronic liver fibrosis in mice silymarin down-regulated hepatic TGF-β1, MMP-2, MMP-13, TIMP-1, TIMP-2, AP-1, tumor suppressor Krueppel-like factor 6 (KLF6) and collagen α1 expression with significant reduction of hepatic hydroxyproline content (Chen et al. 2012a; El-Lakkany et al. 2012). Moreover, Polyak et al. (2010) demonstrated that silymarin could inhibit HCV cell infection through suppression of TNF-α-induced activation of NF-κB and its nuclear translocation.
Major constituent of silymarin, silybin, exhibited antioxidant effect in nonalcoholic steatohepatitis (Haddad et al. 2011). Silibinin, a mixture of silybin A and silybin B, and more bioavailable silibinin-phosphatidylcholine complex, significantly inhibited IL-1β-induced production of proinflammatory markers PGE2, IL-8 and MCP-1 in hepatocytes (Au et al. 2011). Mechanistically, both compounds attenuated NF-κB activation and its nuclear translocation. Yao et al. (2011) suggested that inhibition of oxidative stress, stabilization of mitochondrial membrane and improved insulin resistance may be the key mechanisms for the hepatoprotective effect of silibinin against NAFLD.
Silymarin has a good safety profile and is well tolerated by patients (National Toxicology 2011). In individuals occupationally exposed to hydrogen sulfide, treatment with 140 mg of silymarin, three times per day for 1 month, resulted in a significant decrease in serum aminotransferases and alkaline phosphatase levels (Mandegary et al. 2013). Nevertheless, the effectiveness of silymarin was modulated by the TNF-α polymorphisms. Schrieber et al. (2008) showed the correlation between the severance of hepatic disease (HCV noncirrhosis, NAFLD and HCV cirrhosis) and the level of total silymarin flavonolignans in the blood. Current research provides optimistic data regarding improvement of physicochemical property of silymarin. Silymarin-loaded solid lipid nanoparticles have been proposed as a useful system for the delivery of poorly water-soluble compounds such as silymarin, showing enhanced antioxidant and hepatoprotective activity compared to a crude silymarin (Hsu et al. 2012; Hwang et al. 2014; Cengiz et al. 2015).
Interaction between silymarin or its components and CYPs activity has not been intensively studied. A few data available suggested that silybin ameliorated CYP3A expression and activity in thioacetamide-injured rat liver (Xie et al. 2013). Studying the effect of the anti-Alzheimer drug tacrine and silibinin, Chen et al. (2012c) showed that the co-drug administration diminished tacrine-induced hepatotoxicity and induction of CYPs. These studies indicated that silymarin or its component may not be involved in CYP activation.
Terpenoids
Monoterpenoids
Thymoquinone (12), a monoterpenoid quinone, the major active compound derived from the Nigella sativa L. seeds, has been reported to protect experimental animals against oxidative hepatic injury by improving hepatic antioxidant status (Sayed-Ahmed et al. 2010; Prabhakar et al. 2014). In addition, thymoquinone treatment has been shown to significantly suppress CYP1A2, CYP3A4 but not CYP2E1 activity in rabbits (Elbarbry et al. 2012).
Chemically induced hepatic fibrosis and inflammation in mice were attenuated by thymoquinone through suppression of protein and mRNA expression of collagen I and TIMP-1 and reduction of ECM accumulation (Bai et al. 2014; Ghazwani et al. 2014). Thymoquinone down-regulated the expression of TLR4 and decreased proinflammatory cytokine levels (Bai et al. 2014). In addition, it also inhibited PI3K phosphorylation, enhanced the phosphorylation AMPK and liver kinase B (LKB)-1. In rats injected with cisplatin, thymoquinone reduced the expression of NF-κB and proinflammatory proteins TNF-α, iNOS and IL-1β (Al-Malki and Sayed 2014). Investigating the mechanism of antifibrotic activity in several HSC lines, Ghazwani et al. (2014) showed that the inhibition of LPS-induced mRNA expression of IL-6 and MCP-1 was associated with the inactivation of NF-κB pathway and down-regulation of mRNA expression of several fibrosis-related genes. This quinone also showed the inhibitory potential toward TLR4 and PI3K/Akt signaling pathways in activated HSCs and proapoptotic activity, as shown by decreased XIAP and c-FLIP expression (Bai et al. 2013). Moreover, thymoquinone administration in rats fed HFD diminished metabolic syndrome by preventing reduction in hepatic mRNA levels of PPAR-α and PPAR-γ (Prabhakar et al. 2014).
Diterpenoids
Andrographolide
Andrographolide (13) is a diterpenoid lactone isolated from Andrographis paniculata L., which possesses antioxidant effects against chemically induced liver injury (Trivedi et al. 2007; Chen et al. 2014a). It up-regulated protein and gene expression of oxidative stress response genes such as hypoxia-inducible factor-1 alpha (HIF-1α), SOD-1, HO-1 and GST, which was associated with increased nuclear Nrf2 content and its DNA-binding activity (Ye et al. 2011; Shi et al. 2012; Chen et al. 2014a). Similarly, the anti-HCV activity of andrographolide has been mediated by up-regulation of HO-1 via the p38 MAPK/Nrf2 pathway (Lee et al. 2014a).
Andrographolide suppressed thioacetamide-induced hepatic inflammation, angiogenesis and fibrosis in mice (Lee et al. 2014a, d). Andrographolide treatment decreased TNF-α and COX-2 expression and reduced liver hypoxia, as shown by the down-regulation of hypoxia-inducible genes, such as VEGF. In addition, andrographolide treatment resulted in the decrease in Tβ-RI expression and hepatic fibrogenesis. Similarly, andrographolide attenuated hepatic apoptosis and fibrosis after BDL in rats (Lee et al. 2010c). The compound decreased serum levels of TNF-α and IL-1β and hepatic expression of TGF-β, cannabinoid receptor type 1 (CBR1) and Bax. The effect was mediated by suppression of JNK and ERK phosphorylation. Andrographolide was also able to down-regulate cellular lipid accumulation in HFD-fed mice (Ding et al. 2014). The compound reduced the hepatic protein and mRNA levels of SREBP-1 and its downstream targets, such as FAS, ACC and SCD-1 as well as SREBP-2 and its target genes involved in cholesterol biosynthesis.
Nevertheless, andrographolide showed a potential to induce CYP1A1, CYP2A4, CYP2B9 and CYP2B10 expression in mice hepatocytes (Chatuphonprasert et al. 2009). In a similar way, andrographolide decreased CYP2C and CYP3A expression and activity in human hepatocytes, but without the effect on CYP2E1 (Pekthong et al. 2008, 2009), suggesting a potential to interact with drug metabolism.
Triterpenoids
Ginsenosides (14) are the major steroid compounds in ginseng root, which belong to the genus Panax of the family Araliaceae. These compounds reduced ROS generation in chemically injured hepatocytes by increasing SOD, GPx and CAT activity and restoring GSH levels (Park et al. 2012; Li et al. 2014b), while suppressing ERK and JNK MAPKs. Ginsenoside Rg3, but not other tested ginsenoides (Rb1, Rc and Rg1), increased acetaminophen-induced GSTA2 protein expression and the transcriptional activation by the multiple cellular signaling, including PI3K, JNK and protein kinase A (PKA) (Gum and Cho 2013b). Ginsenoside Rg3 ameliorated chemical toxicity in hepatic cells by inducing Nrf2-mediated gene expression of multidrug resistance protein (MRP) 1 and 3, involved in a detoxification process (Gum and Cho 2013a). Further, ginsenosides were effective in attenuation of hepatic fibrosis induced by alcohol and CCl4. Ginsenoside Rb1 attenuated liver inflammation and fibrosis by suppressing hepatic PGE2 and TIMP-1 expression (Hou et al. 2014). Similarly, ginsenoide Rg1 protected against hepatic fibrosis by increasing nuclear translocation of Nrf2 and the expression of antioxidant enzymes, including HO-1 and NQO1 (Li et al. 2014b). In addition, ginsenoside Rg1 down-regulated the expression of PDGFRβ in cultured HSCs by reducing the NF-κB activity, which resulted in the inhibition of PDGF-BB-stimulated cell proliferation and activation (Geng et al. 2010). In H2O2-activated HSCs, ginsenoside Rb1 suppressed the mRNA and protein expression of TGF-β1, MMP-2, TIMP-1 and collagen (Lo et al. 2011), suggesting that Rb1 may exert an antifibrotic effect by inhibiting HSCs activation and proliferation.
Ginsenoside Rb1 reduced fatty liver in obese rats by activating hepatic AMPK (Shen et al. 2013). In primary rat hepatic cells, activation of AMPK consistently stimulated the expression of genes encoding fatty acid oxidative enzymes, including PGC-1α, PPARα, and peroxisomal acyl-coenzyme A oxidase 1 (ACOX1) and increased the activity of CPT1, a key enzyme in fatty acid β-oxidation. In a similar fashion, ginsenoside Re lowered hepatic lipid levels via activation of the AMPK pathway and protected against hepatic steatosis in HFD-fed mice (Quan et al. 2012).
Furthermore, ginsenoides were shown to suppress hepatic inflammation through the inhibition of NF-κB signaling in several experimental models. Ginsenosides Rd1 and Rg1 protected mouse liver against ischemia–reperfusion injury through down-regulation of NF-κB expression and reduced production of proinflammatory cytokines, including TNF-α and ICAM-1 (Wang et al. 2008; Tao et al. 2014). Similarly, ginsenoside Rg1 attenuated concanavalin A-induced hepatitis in mice through the inhibition of IκBα and p65 phosphorylation as well as ICAM-1 and CXCL-10 mRNA expression and cytokine secretion (Cao et al. 2013). Ginsenoides Rd and Rg3 also attenuated hepatic NF-κB, COX-2 and iNOS protein expression in LPS-challenged murine (Kang et al. 2007; Kim et al. 2013). Moreover, ginsenoside Rd suppressed NO production and PGE2 synthesis (Kim et al. 2013).
Ginsenoside Rg1 and Rb1 showed the potential to decrease hepatitis A virus (HAV) titer in infected hepatocytes (Lee et al. 2013b). Moreover, ginsenoside Rg3 was shown to attenuate HBV replication (Kang et al. 2013). Rg3 inhibited the expression of TRAF6 and TAK1, adaptor molecules that signal through the TLR/MyD88-dependent pathway. Furthermore, Rg3 inhibited MAPK signaling in hepatic cells by inhibiting JNK phosphorylation. In addition, it reduced the expression of AP-1 transcription factors c-Jun and JunB and inhibited AP-1 promoter activity, resulting in reduced gene and protein expression of proinflammatory cytokines.
It has been shown that ginsenoside Rd, as well as quercetin, have a potential to inhibit CYP2C9 and CYP3A4 in human liver microsomes (He and Edeki 2004). However, available data suggest that ginsenosides Rb1, Rb2, Rc and Rd are not likely to interact with conventional medicines that are metabolized by CYP2C19 and CYP2D6 (He et al. 2006). Similarly, in another study, ginsenosides Rb1, Rb2, Rc, Rd, Re, Rf and Rg1 showed only weak inhibition of CYP1A1, CYP1A2 and CYP1B1 activity (Chang et al. 2002). These data suggest that ginsenosides have relatively low CYP-modulating effect.
Glycyrrhizin (15), a triterpenoid glycoside isolated from the roots of licorice plant (Glycyrrhiza glabra L.), has been shown to increase antioxidant defense in the liver (Rahman and Sultana 2006; Orazizadeh et al. 2014). Glycyrrhizin may also protect against liver injury by reducing the expression of high-mobility group protein box 1 (HMGB1), an early mediator of inflammation (Mabuchi et al. 2009; Ogiku et al. 2011) and interrupting HMGB1 binding to GSTO1 promoter region (Kuroda et al. 2014). Glycyrrhizin and its metabolite, glycyrrhetinic acid, inhibited collagen αI(I) gene expression and progression of liver fibrosis induced by CCl4 (Moro et al. 2008). The compounds significantly decreased mRNA expression of TGF-β1, Smad2/3 and specificity protein-1 (SP-1) in the liver (Qu et al. 2015). Further, glycyrrhizin down-regulated proinflammatory mediators and induced expression of HO-1 (Lee et al. 2007a). The potential of this compound to accelerate recovery from hepatic injury has been demonstrated in vitro. Glycyrrhizin suppressed activation of HSCs and induced their apoptosis by blocking nuclear translocation of NF-κB (Qu et al. 2012). Importantly, glycyrrhizin and its metabolites may induce growth of hepatocytes by binding to EGFR and stimulating ERK2-mediated hepatocyte DNA synthesis and proliferation (Kimura et al. 2001), which could contribute to acceleration of liver regeneration.
Antiviral activity of glycyrrhizin has been demonstrated both in vitro and in vivo. Glycyrrhizin treatment of HCV-infected hepatic cells resulted in reduced release of infectious HCV particles through inhibitory effect on phospholipase A2 (PLA2), whereas a co-treatment with glycyrrhizin augmented antiviral effect of IFN-α (Matsumoto et al. 2013). A similar effect on HAV antigen expression and infectivity has also been reported (Crance et al. 1990). Moreover, glycyrrhizin modified the intracellular transport and suppressed sialylation of HBsAg in vitro (Takahara et al. 1994), which was also observed in patients with chronic HBV infection (Sato et al. 1996).
Although mechanistically modestly investigated, glycyrrhizin has been intensively studied in clinical trials. In patients who failed previous IFN-α-based therapy, intravenous administration of glycyrrhizin significantly reduced serum alanine transaminase (ALT) level after 12 weeks of therapy and improved necro-inflammation and fibrosis after 52-week treatment (Manns et al. 2012). In another study, a 6-month co-treatment with IFN-α2b and glycyrrhizin was less effective in reducing ALT levels compared to IFN-α2b and ribavirin co-administration; however, the treatment was associated with significantly lower frequencies of leukopenia and anemia (Acharya et al. 2012). Interestingly, the decrease in ALT levels after 26 months treatment in a smaller group of patients treated with glycyrrhizin did not translate in a significant histological improvement (Orlent et al. 2006). Nevertheless, glycyrrhizin injection therapy showed effectiveness in prevention of progression from HCV-related cirrhosis to HCC, particularly in a group of older patients (Ikeda 2007; Ikeda et al. 2014). Similar effect of this compound was observed in another study on 1093 patients nonresponding to IFN (Veldt et al. 2006). Importantly, usage of the suppositories of glycyrrhizin improved quality of life for chronic hepatitis C patients similarly to intravenously treated patients, with greater benefit in those who did not respond to IFN therapy (Fujioka et al. 2003).
18β-Glycyrrhetinic acid (16), a hydrolytic product of glycyrrhizin, has also been shown to possess a wide range of pharmacological and biological activities, including hepatoprotection (Hasan et al. 2015). 18β-Glycyrrhetinic acid decreased oxidative stress and expression of inflammatory markers both in vivo and in vitro models of hepatocyte injury, which coincided with down-regulation of NF-κB and up-regulation of Nrf2 target genes (Chen et al. 2013b; Hasan et al. 2015). Importantly, this compound inhibited hepatic expression and activity of CYP2E1 in CCl4-intoxicated mice (Jeong et al. 2002) and the activity of CYP2C9, CYP2C19 and CYP3A4 in rat and human liver microsomes (Zhao et al. 2012). 18β-Glycyrrhetinic acid but not glycyrrhizin inhibited PARP, caspase-3, caspase-9 and caspase-10 activation and suppressed phosphorylation of JNK in a model of cholestatic liver injury, protecting hepatocytes against necrosis and apoptosis (Gumpricht et al. 2005). In human and rat HSCs, the compound inhibited mRNA and protein expression of collagen type I and III, which was attributed to down-regulation of Smad3, up-regulation of Smad7 and inhibition of DNA-binding activities of NF-κB as well as SP-1 and AP-1, both involved in the synthesis of ECM (Moro et al. 2008; Zong et al. 2012). Additionally, proliferation of activated HSCs was inhibited and apoptosis was induced, as evidenced by decreased expression of cyclin D1 and the pro-survival Bcl-2 homolog A1/Bfl-1, with simultaneous increase in Bax and PPARγ-mediated expression of p27 (Zong et al. 2013).
Betulinic acid (17), a natural pentacyclic lupane-type triterpenoid found in various plants, especially bark of the birch tree (genus Betula, family Betulaceae), showed hepatoprotection against ethanol-induced oxidative stress and steatohepatitis in mice (Wan et al. 2013b; Yi et al. 2014). The amelioration of steatohepatitis involved decrease in TLR4 expression and increased phosphorylation of STAT3, signaling molecule involved in regulation of cell survival (Wan et al. 2013b). Furthermore, the potential role of betulinic acid in prevention of hepatic inflammation and fibrosis has also been demonstrated in thioacetamide-intoxicated rats (Wan et al. 2012). Mechanistically, betulinic acid induced suppression of the TLR4/MyD88/NF-κB signaling pathway.
Other studies showed that betulinic acid and betulin could inhibit ethanol-induced activation of HSCs at different levels (Szuster-Ciesielska et al. 2011; Wan et al. 2013b). Both compounds inhibited the production of ROS by HSCs, inhibited their migration and down-regulated ethanol-induced TIMP-1, TIMP-2 and MMP-2 activity (Szuster-Ciesielska et al. 2011). Additionally, betulin inhibited the activation of the p38 MAPK and the JNK transduction pathways, while betulinic acid inhibited the JNK transduction pathway only. Nevertheless, both compounds inhibited phosphorylation of IκB and Smad3 and attenuated the activation of TGF-β1 and NF-κB transduction signaling. Betulinic acid also showed several other hepatoprotective activities. This compound was also able to prevent hepatic apoptosis induced by LPS/D-GalN through suppression of apoptosis-related JNK1/2 and ERK1/2 signaling (Zheng et al. 2011). Further, the inhibitory effect of betulinic acid on lipid accumulation and amelioration of NAFLD in mice involved suppression of SREBP-1 activity via down-regulation of the CAMKK/AMPK/mTOR/p70S6K signaling pathway (Quan et al. 2013b). Moreover, betulinic acid exhibited antiviral properties. In hepatocytes from HBV-transgenic mice, betulinic acid mediated inhibitory effect on HBV replication through down-regulation of manganese SOD expression via attenuation of cAMP-response element-binding protein (CREB) phosphorylation and inhibition of its transcriptional activity (Yao et al. 2009). Moreover, betulinic acid inhibited HCV replication, acting synergistically with IFN-α and NS5B polymerase inhibitor. The compound down-regulated HCV-induced COX-2 expression through reducing the phosphorylation of NF-κB and ERK1/2 (Lin et al. 2015a), suggesting betulinic acid as a promising compound for treatment of hepatitis virus-infected patients.
Ursolic acid (18), a pentacyclic triterpenoid acid found in various plants, suppressed oxidative stress in various models of liver injury (Kazmi et al. 2013; Ma et al. 2014). Ursolic acid modestly inhibited CYP2C19 in vitro, while other CYPs, including CYP2C8, CYP2C9, CYP3A4, CYP2E1, CYP1A2 and CYP2D6, showed weak inhibition by this triterpene or no inhibition at all (Kim et al. 2004), suggesting low potential of interaction with drugs and activation of pro-carcinogens.
Ursolic acid was shown to activate autophagy in mice model of hepatic steatosis in the NAFLD model in rodents, by inducing the expression of LC3-II and beclin 1 (Jia et al. 2015). It also activated PPARα and expression of genes involved in fatty acid uptake and β-oxidation, such as fatty acid transport protein 4 (FATP4), acetyl-CoA synthetase 1 (ACS1), CPT1 and peroxisomal acyl-coenzyme A oxidase 1 (ACOX1), while down-regulating genes involved in lipogenesis, such as SREBP-1c, FAS and ACC1 (Li et al. 2014e; Sundaresan et al. 2014; Jia et al. 2015). Moreover, ursolic acid treatment significantly decreased hepatic steatosis in db/db mice by modulating β-oxidation and ER stress in the liver (Li et al. 2015b). Mechanistically, it reduced expression of the unfolded protein response sensor inositol-requiring enzyme-1alpha (IRE-1α) expression and activation of ERK, JNK and CHOP, while increasing PPARα levels. In addition, ursolic acid decreased palmitic acid-induced intracellular lipid accumulation in L02 cells, with concomitant inhibition of ATF6, IRE-1α and CHOP gene expression.
Wang et al. (2011) and Yang et al. (2015) demonstrated the beneficial effect of ursolic acid against hepatic fibrosis. In culture-activated HSCs, ursolic acid activated caspase-3 and caspase-9, decreased phosphorylation of Akt and diminished nuclear localization of NF-κB (Wang et al. 2011), suggesting their apoptosis and suppression of survival mechanisms. Treatment of hepatocytes with ursolic acid in the presence of LPS dose-dependently inhibited ROS production and NF-κB activation. The improvement of liver functions in mice was associated with activation of the LKB1/AMPK pathway, involved in cell growth and control of metabolism. Ursolic acid also prevented CCl4-induced hepatotoxicity and fibrosis in mice, at least in part, through modulation of the Nrf2/ARE signaling pathway (Ma et al. 2015a). In addition, this compound suppressed hepatic production of proinflammatory and proapoptotic markers, including TNF-α, IL-1β, COX-2 and caspase-3, while increasing antiapoptotic Bcl-2 expression (Ma et al. 2014). The underlying mechanisms of ursolic acid action involved suppression of MAPKs activation and the suppression of immunoregulatory transcription factor NF-κB. Ursolic acid also enhanced hepatic cell proliferation in partially hepatectomized mice, which was associated with increased cyclin D1, cyclin E and C/EBPβ protein expression (Jin et al. 2012), suggesting its potential to facilitate liver regeneration.
Phenolic acids
Chlorogenic acid (5-O-caffeoylquinic acid) (19) ameliorated liver injury in several models of experimentally induced oxidative stress (Shi et al. 2013a; Koriem and Soliman 2014). It should be also mentioned that chlorogenic acid may exhibit in vitro inhibitory effect on the activity of hepatic metabolizing enzymes, including CYP1A1, CYP1A2 and CYP2B (Baer-Dubowska et al. 1998).
Treatment of animals with this compound inhibited acetaminophen-induced activation of caspase-3 and caspase-7, ERK1/2, JNK and p38 MAPKs upstream molecular signals, including ASK1, c-Raf and mitogen-activated protein kinase kinases MEK1/2, MKK4 and MKK3/6 (Ji et al. 2013), suggesting inhibition of apoptosis. Concomitantly, chlorogenic acid restored glutamate-cysteine ligase, catalytic subunit (GCLC), Trx1/2 and TrxR1 expression (Ji et al. 2013). Several studies demonstrated anti-inflammatory action of chlorogenic acid in the liver (Xu et al. 2010; Yun et al. 2012; Park et al. 2015). Chlorogenic acid inhibited various TLR agonist-, IL-1α- and HMGB1-stimulated activation of IL-1R-associated kinase 4 (IRAK4) in mice peritoneal macrophages of LPS-intoxicated mice via directly affecting the kinase activity of IRAK4, a signal transducer in the TLR/MyD88-mediated signaling cascade (Park et al. 2015). In addition, it significantly suppressed the expression of phospho-IκBα, phospho-TAK1, phospho-JNK1/2 and phospho-p38. This resulted in reduction of protein and mRNA levels of NF-κB/AP-1 target genes encoding TNF-α, IL-1α, IL-6 and HMGB-1, resulting in attenuation of acute hepatic inflammation. Chlorogenic acid also suppressed hepatic expression of TLR3 and TLR4-dependent nuclear translocation of NF-κB in inflammatory liver injury (Yun et al. 2012; Zheng et al. 2015). Concomitantly, the compound restored mRNA level of transcriptional coactivator PGC-1α and inhibited activity of IFN regulatory factor-1 (IRF-1), with enhanced Nrf2 nuclear translocation and HO-1 expression. Chlorogenic acid ameliorated the development of HFD-induced hepatic steatosis and insulin resistance in mice by suppressing the expression of genes for fatty acid-binding protein (FABP), PPARγ and CD36, a scavenger receptor that mediates internalization of low-density lipoprotein (LDL) particles (Ma et al. 2015b). In addition, treatment by chlorogenic acid attenuated hepatic inflammation by decreasing the mRNA levels of macrophage inflammatory genes. The gene expression of PPARα in diet-induced hypercholesterolemia in rats was up-regulated by this compound, suggesting decreased risk for development of complications such as NAFLD (Wan et al. 2013a). Moreover, the expression of adiponectin receptors (AdipoR) 1/2, the phosphorylation of AMPK and the mRNA and protein levels of PPARα in the liver were significantly higher in chlorogenic acid-treated diabetic db/db mice, suggesting a potential to alleviate obesity-related metabolic syndrome (Jin et al. 2015).
Chlorogenic acid has also been shown to attenuate CCl4-induced liver inflammation and fibrosis in rats via inhibition of the TLR4/MyD88/NF-κB pathway, which coincided with the inhibition of collagen I, α-SMA, iNOS and COX-2 expression and concomitant increase in bone morphogenetic protein and activin membrane-bound inhibitor (Bambi) expression, which is expressed in quiescent HSCs, blocking Tβ-RI activity (Shi et al. 2013b). In addition, chlorogenic acid suppressed mRNA levels of profibrotic inductors such as VEGF and TGF-β1 (Shi et al. 2009). In cultured HSCs, this compound inhibited LPS-induced IκBα phosphorylation, nuclear translocation of NF-κB and the gene expression of inflammatory mediators (Shi et al. 2013a). Chlorogenic acid, as well as quinic acid and caffeic acid, also showed inhibitory potential against HBV DNA replication and HBsAg production (Wang et al. 2009a), suggesting a potential against viral hepatitis infection.
A combined nutraceutical containing chlorogenic acid, berberine and tocotrienols improved a large number of metabolic and liver parameters in a double-blind cross-over trial in 40 overweight subjects with mixed hyperlipidaemia (Cicero et al. 2015).
The antioxidant activity of salvianolic acid (20) (Salvia miltiorrhiza L. active component), a rosmarinic acid dimmer, has been associated with amelioration of drug-induced hepatotoxicity (Gao et al. 2012; Lin et al. 2015b). Salvianolic acid B attenuated hepatocyte apoptosis by regulating ER stress, death receptor-mediators and mitochondrial pathways (Yan et al. 2015b). The compound markedly decreased TNF-α/D-GalN-induced levels of phospho-eukaryotic initiation factor (eIF) 2α, ATF4, 78 kDa glucose-regulated protein (GRP78), CHOP, cleaved-caspase-3 and caspase-9, apoptosis-inducing factor (AIF) and apoptotic protease-activating factor 1 (Apaf-1), with concomitant increase in cytochrome c release. In addition, salvianolic acid B reduced TNFR1 and restored Bcl-2 expression in apoptotic cells (Yan et al. 2010). In acetaminophen-intoxicated mice, salvianolic acid B pretreatment induced the expression of Nrf2 and phase II enzymes via activation of the PI3K and protein kinase C (PKC) pathways (Lin et al. 2015b). Further, pretreatment with salvianolic acid A and B ameliorated ethanol and concanavalin A-induced liver injury in murine by SIRT1-dependent mechanism (Xu et al. 2013; Li et al. 2014d). Mechanistically, salvianolic acid B increased the expression of SIRT1, a NAD+-dependent deacetylase which plays an important role in protection against acute hypoxia damage and metabolic liver diseases through modulation of NF-κB and p53 expression. The increase in SIRT1 by salvianolic acid B was accompanied by the decrease in acetyl-p53 expression (Li et al. 2014d) and down-regulation of the p66 isoform of the growth factor adapter Shc (Xu et al. 2013). Additionally, both salvianolic acid A and B decreased cytotoxic cytokine levels and abrogated the increase in NF-κB and cleaved caspase-3 as well as decrease in Bcl-xL expression. Interestingly, in both type 1 and type 2 diabetic animals, salvianolic acid A activated AMPK phosphorylation through CaMKKβ/AMPK signaling pathway, independent of LKB1/AMPK pathway (Qiang et al. 2015).
Both salvianolic acid A and B showed the inhibitory effect on the PDGF-BB-induced signaling in HSCs through different signaling pathways (Tsai et al. 2011). Thus, salvianolic acid A and B diminished PDGF-induced activation of protein kinase D (PKD), Nrf2, Trx and HO-1. In addition, salvianolic acid A inhibited an expression of Akt, p70S6K and related proteins such as eIF-4E and translation repressor eIF-4E-binding protein 1 (4E-BP1). In contrast, salvianolic acid B attenuated PDGF-induced JNK, p38 and PKC-δ phosphorylation. Further, salvianolic acid A attenuated PDGF-BB-stimulated proliferation of HSCs by inducing the cell cycle inhibitory proteins p21 and p27, down-regulating cyclins D1 and E and suppressing Akt and PDGFRβ phosphorylation (Lin et al. 2006). The inhibition of TGF-β1 signaling by the compound in isolated rat HSCs was associated with down-regulation of Smad2/3 protein expression and suppression of collagen production (Liu et al. 2002a; Zhao et al. 2004). The down-regulation of Tβ-RI activity in vitro and TGF-β1 autosecretion were contributing mechanism of salvianolic B antifibrotic activity (Liu et al. 2002c; Tao et al. 2013). In primary rat HSCs, this compound inhibited ERK1/2 and p38 MAPK pathways via inhibition of MEK and MKK3/6 phosphorylation (Lv et al. 2010). Interestingly, the effect was independent on TGF-β1 stimulation. The antifibrotic effect of salvianolic acid B was mediated by direct inhibition of p38 signaling and inhibition the cross-talk between the Smad and ERK signaling (Lv and Xu 2012). Furthermore, salvianolic acid B abrogated DMN-induced hepatic fibrosis in rats, which coincided with down-regulation of angiotensin (Ang) II-stimulated HSCs activation and proliferation (Li et al. 2012). The mechanism involved down-regulation of TGF-β gene and AT1R protein expression, as well as suppression of ERK1/2 and c-Jun phosphorylation. Moreover, salvianolic acid B reduced portal hypertension in rats with DMN-induced cirrhosis (Xu et al. 2012). In vitro study showed that this compound decreased endothelin (ET) 1-induced contraction of activated HSCs by reducing intracellular Ca2+ increase, the actin cytoskeleton regulator RhoA expression and activation of Rho-associated coiled coil-forming protein kinase (ROCK) II, a major downstream effector of the RhoA. This resulted in reduced phosphorylation of the myosin phosphatase target subunit 1 (MYPT1), which is involved in regulation of the contraction and relaxation of vascular smooth musculature. In addition, salvianolic acid B reduced CCl4-induced hepatic fibrosis in rats by inhibiting nuclear translocation of NF-κB (Wang et al. 2012b).
In the double-blind randomized study, salvianolic acid B reversed serum markers of liver fibrosis in patients with chronic hepatitis B, more effectively than IFN-γ after 6 months of treatment, with no obvious side effects (Liu et al. 2002b). The drug delivery system based on salvianolic acid B loaded mesoporous silica nanoparticles was significantly more effective than free salvianolic acid B in suppressing the ROS level and proliferative activity of LX-2 cells (He et al. 2010), suggesting a promising novel application route of this compound in patients. Salvianoic acid B was found as a relatively weak inhibitor of CYP1A2 (Qiu et al. 2008), suggesting low impact on hepatic drug-metabolizing enzymes.
Stilbenes
Resveratrol (3,5,4′-trihydroxy-trans-stilbene) (21), well known as grape polyphenol, has been shown to possess hepatoprotective activity through attenuation of oxidative stress in the liver (Das 2011; Dalaklioglu et al. 2013; Ahmad and Ahmad 2014). Resveratrol protected hepatocytes against oxidative injury by modulating the expression of nuclear transcription factors Nrf2 and NF-κB and down-regulating HO-1 and iNOS gene expression (Sahin et al. 2012), which led to increased expression of antioxidant and phase II enzymes (Rubiolo et al. 2008; Cerny et al. 2009). Interestingly, resveratrol behaved as an antioxidant during the dark span and as a pro-oxidant during the light span, suggesting a day/night rhythm-dependent antioxidant activity of the compound (Gadacha et al. 2009). Wong et al. (2009) demonstrated that resveratrol treatment attenuated oxidative stress in mice tissues with prominent age-related oxidative damage accumulation, but prolonged treatment resulted in nephrotoxicity.
Resveratrol possesses the ability to modulate a number of signaling pathways involved in liver diseases, emerging as a promising therapeutic agent for prevention or treatment of hepatic disorders in humans.
Pretreatment with resveratrol was able to ameliorate the pathologic effects of concanavalin A-induced autoimmune hepatitis, significantly inhibiting proinflammatory cytokines such as IL-2, IL-6 and TNF-α (Zhou et al. 2015). In addition sonic hedgehog (Shh), patched (Ptch) and GLI-1 expression was decreased, suggesting suppression of the Shh-Ptch-GLI pathway that is involved in regulation of the cell cycle, apoptosis and angiogenesis. Administration of resveratrol resulted in decreased accumulation of bile acids through up-regulation of hepatic transporters gene expression, including farnesoid X receptor (FXR) and MRP2, which play a central role in the bile acid metabolism (Wang et al. 2014c). The protective effect of resveratrol in cholestatic liver injury was mediated by up-regulation of autophagy and suppression of proapoptotic proteins Bax and caspase-3 and caspase-8 (Chan et al. 2011; Lin et al. 2012b; Chan et al. 2014). Further, resveratrol, a well-known SIRT1 agonist, ameliorated hepatic inflammation in vivo and in vitro by reducing NF-κB activation (Andrade et al. 2014). Resveratrol also enhanced SIRT1-mediated suppression of HMGB1 translocation, which is an essential step in response to sepsis-induced liver injury (Xu et al. 2014b). Further, hepatotoxicity induced by isoniazid and rifampicin was reduced with resveratrol by modulating SIRT1 mRNA expression in mice liver, which was accompanied by decreased hepatic oxidative stress, cytokine production and PPARγ gene expression (Nicoletti et al. 2014). Resveratrol also prevented acetaminophen-induced hepatotoxicity through induction of SIRT1expression and negative regulation of p53 signaling, inducing cyclin D1, Cdk4 and proliferating cell nuclear antigen (PCNA) expression and promoting hepatocyte proliferation and liver regeneration (Wang et al. 2015d). Moreover, resveratrol modulated HFD-induced alterations in SIRT pathway and activation of NALP-3 inflammasome, involved in maturation of proinflammatory cytokines (Yang and Lim 2014). Interestingly, resveratrol reduced COX-2 expression and mRNA levels of NALP-3 inflammasome components, including NALP-1, NALP-3, apoptosis-associated speck-like protein containing a carboxy-terminal CARD (ASC) and its downstream target caspase-1 in old but not young mice (Tung et al. 2015), suggesting its beneficial effect in elderly population. Other researchers showed that prevention of NAFLD development by resveratrol in rats fed a HFD was associated with up-regulated expression of hepatic UCP2, the inhibitor of mitochondrial ROS production and oxidative stress regulator (Poulsen et al. 2012). In human hepatocytes, resveratrol ameliorated ethanol-induced activation of signaling pathways associated with ER stress, including GRP78, IRE-1α, eIF2α, translational regulator RNA-dependent protein kinase R (PKR)-like ER kinase (PERK) and ATF4 (Liu et al. 2014b). In addition, resveratrol inhibited apoptosis by decreasing cleaved caspase-3 as well as CHOP and Bax expression, which was associated with restoration of SIRT1 levels and suppression of phosphodiesterase (PDE) activity. Similarly, resveratrol prevented hepatic steatosis and dyslipidemia by attenuating ER stress in rat livers (Pan et al. 2015). Mechanistically, the compound down-regulated HFD-induced increase in ATF4, CHOP and BiP levels.
Resveratrol also showed a potential to inhibit cell growth of activated HSCs by inducing cell cycle arrest in sub-G1 phase and to modulate the SIRT1/PPARγ ratio (de Souza et al. 2015). This compound reduced collagen I, TGF-β and NF-κB mRNA expression in cirrhotic rat livers (Di Pascoli et al. 2013) and decreased the secretion of MMP-2 in human liver myofibroblasts (Godichaud et al. 2000), suggesting the ability to deactivate HSCs. Nevertheless, the up-regulation of profibrotic genes in human HSCs by resveratrol in the presence of free fatty acids, suggests the possibility of fibrogenic activity in obese patients (Bechmann et al. 2009).
SIRT-1 has also been identified as a regulator of hepatocyte lipid metabolism via activation of AMPK. Activation of the SIRT1/LKB1/AMPK pathway has been suggested as a key mechanism by which resveratrol counteracts hepatic lipid accumulation, hyperlipidemia and atherosclerosis (Hou et al. 2008). Resveratrol ameliorated HFD-induced hepatic steatosis and decrease in gene expression of SIRT1 in mice liver, with a concomitant suppression of liver lipogenic genes, including ACC, PPARγ and SREBP-1 (Andrade et al. 2014). The SREBP-1 inhibitory effect of resveratrol was accompanied by activation of PGC-1α, increased circulating levels of adiponectin and enhanced mRNA expression of hepatic AdipoR1/2 (Ajmo et al. 2008). Jin et al. (2013) demonstrated that resveratrol may inhibit SREBP-1c-inducing ability of LXRα, a regulator of de novo fatty acid synthesis, consequently impairing the expression of target genes and hepatic steatosis. This effect was not dependent on AMPK and SIRT1 but involved up-regulation of sestrin 2 (SESN2) expression. Interestingly, in normally fed mice, resveratrol treatment did not change the expression of LXR target genes but activated AMPK and increased phosphorylation of ACC (Gao and Liu 2013). In contrast to steatosis, resveratrol treatment had no consistent therapeutic effect on amelioration of experimental steatohepatitis (Heeboll et al. 2015). Furthermore, resveratrol showed the ability to inhibit the expression of SREBP1 via SIRT1-FOXO1 signaling pathway in vitro (Wang et al. 2009b). Members of the FOXO family of transcription factors have been shown to regulate the expression of numerous genes involved in the cell cycle, apoptosis, differentiation and cellular response to oxidative stress. Resveratrol treatment could attenuate hepatic steatosis and lipid metabolic disorder in mice by up-regulating the levels of SIRT1, phospho-AMPK and phospho-FOXO1 expression (Zhu et al. 2014). It should be mentioned that treatment with this compound enhanced ethanol-induced expression of autophagy-related genes in mouse hepatocytes via increased deacetylation of FOXO3a, which seems to play a critical role in ethanol-induced autophagy (Ni et al. 2013). Experimental NAFLD in a murine was also improved through regulation of autophagic mediator UNC-51-like kinase 1 (ULK1) and NF-κB pathways, which coincided with amelioration of inflammation and insulin resistance (Li et al. 2014c). Moreover, most recent study demonstrated resveratrol-mediated induction of autophagy in the steatotic liver via the cAMP-PKA-AMPK-SIRT1 signaling pathway (Zhang et al. 2015c).
In HBV X protein (HBX) transgenic mice, resveratrol exhibited chemopreventive activity by reducing the incidence of HBV-associated HCC (Lin et al. 2012b). It inhibited LXRα expression and down-regulated lipogenic genes, with concomitant stimulation of AMPK and SIRT1 activity. In human hepatic cells HBX up-regulated β-catenin, which plays important role in cell–cell adhesion and maintenance of epithelial cell layers, by sequestering SIRT1 deacetylase (Srisuttee et al. 2012). Interestingly, resveratrol significantly enhanced HCV RNA replication and attenuated antiviral effects of IFN-α2b and ribavirin (Nakamura et al. 2010), suggesting a need for further investigation of HCV therapeutic potential in patients.
Finally, resveratrol was found to exert a chemopreventive activity against DEN-induced rat liver tumorigenesis. It reduced gene expression and production of hepatic inflammatory cytokines (Mbimba et al. 2012), while up-regulating protein and mRNA expression of Nrf2 (Bishayee et al. 2010). The stilbenoid also attenuated HSCs activation and reduced hydroxyproline content and TGF-β1 gene expression in DEN-treated rats (Lee et al. 2010a). Administration of resveratrol after warm ischemia and reperfusion in rat liver attenuated gene expression of HIF-1α and VEGF, associated with development of tumors and their metastases (Zhang et al. 2014d). Moreover, resveratrol blocked phosphorylation of ERK1/2 and ERK1/2-induced connexin 43 (Cx43), a critical regulator of GJIC, which has been involved in carcinogenesis (Kim et al. 2009). Interestingly, resveratrol has been shown to prevent deregulation of GJIC by toxicants that act only through MEK1/2- or phosphatidylcholine-specific phospholipase C (PC-PLC)-dependent pathways (Sovadinova et al. 2015). The inhibitory effect of resveratrol on chemically induced hepatocarcinogenesis may be also attributed to down-regulation of CYPs, such as CYP2E1 (Wu et al. 2013). Interestingly, Kim et al. (2009) showed that protective effect of resveratrol against H2O2-induced inhibition of GJIC in rat epithelial cells is not mediated through its free radical-scavenging activity.
The inhibition of CYP activities as a common feature of resveratrol has been demonstrated in a number of studies. Among several flavonoids, including quercetin, naringenin, hesperidin and rutin, resveratrol was the most potent inhibitor of CYP1A1 in vivo (Arinc et al. 2015). Resveratrol suppressed pregnane X receptor (PXR)-mediated CYP3A4 and CYP3A11 mRNA and protein expression in primary mouse hepatocytes (Deng et al. 2014) and CYP2D2 activity in isolated rat livers (Zendulka et al. 2009). It also inhibited CYP1A1 and CYP1A2 mRNAs in primary human hepatocytes (Dvorak et al. 2008). The inhibition of expression of CYP1A1 and CYP1B1 by resveratrol was mediated by inhibition of the recruitment of AhR and aryl hydrocarbon nuclear translocator (ARNT) (Beedanagari et al. 2009). Resveratrol inhibited chemically induced human CYP1 enzymes in vitro by two distinct mechanisms: direct inhibition of CYP1B1 and CYP1A1 and irreversible NADPH-dependent inactivation in case of CYP1A2 (Chang et al. 2001). Interestingly, resveratrol showed over 50-fold selectivity for CYP1A1 over CYP1A2 (Chun et al. 1999).
Double-blind, randomized, placebo-controlled trial in relatively small group of patients indicated that resveratrol supplementation decreased serum alanine aminotransferase, HOMA-IR, TNF-α, cytokeratin 18 fragment and FGF21 levels, while increasing adiponectin level, suggesting a beneficial effect in amelioration of NALFD in humans (Chen et al. 2015).
Alkaloids
Berberine (22), a quaternary ammonium salt from the protoberberine group of isoquinoline alkaloids, possesses antioxidant properties which could suppress oxidative stress in the liver (Li et al. 2014a; Othman et al. 2014). DEN- and CCl4-induced HCC development was prevented by berberine and even more potently with a combination of berberine and S-allyl-cysteine (Sengupta et al. 2014). Carcinogen-stimulated induction of Akt-dependent mouse double minute 2 homolog (Mdm2)/histone deacetylase 1 (HDAC1) interaction led to p53 deacetylation and its subsequent degradation. This resulted in increased expression of antiapoptotic Bcl-xL and decreased levels of proapoptotic p53-up-regulated modulator of apoptosis (PUMA), Bax and Bak proteins. Berberine alone or in combination with S-allyl-cysteine diminished these effects and induced apoptosis by stimulating protein phosphatase 2A (PP2A) and inhibiting JNK activation. Furthermore, amelioration of the early phase of DEN and phenobarbital-induced hepatocarcinogenesis by berberine was accompanied by suppression of iNOS expression and inhibition of CYP2E1 and CYP1A2 activities (Zhao et al. 2008). Berberine also ameliorated apoptosis in ischemia-/reperfusion-injured rat livers by increasing the Bcl-2/Bax ratio and inhibiting caspase-3 cleavage. The mechanism of its action involved up-regulation of Akt, with concomitant inhibition of mTOR expression (Sheng et al. 2015). Moreover, berberine protected against ethanol-induced steatosis in mice by restoring PPARα/PGC-1α and hepatocyte nuclear factor 4 alpha (HNF4α)/microsomal triglyceride transfer protein (MTTP) pathways, involved in secretion of lipoproteins (Zhang et al. 2014e).
Berberine pretreatment in LPS-induced inflammation in mice reduced the expression of hepatic proprotein convertase subtilisin/kexin type 9 (PCSK9), a cholesterol homeostasis regulator, and decreased IFN-γ, TNF-α, IL-1α and 8-isoprostane levels (Xiao et al. 2012). CCl4-induced acute liver injury was ameliorated by berberine through suppression of TNF-α, COX-2 and iNOS expression, with concomitant attenuation of oxidative/nitrosative stress (Domitrovic et al. 2011). In experimental liver fibrosis, berberine decreased TGF-β1 expression, increased MMP-2 levels and stimulated elimination of fibrous deposits (Domitrovic et al. 2013). Moreover, berberine treatment attenuated liver fibrosis via activation of AMPK and decreased expression of NOX4 and phosphorylated Akt (Li et al. 2014a).
In hyperlipidemic patients with HBV, HCV and liver cirrhosis, treatment with 500 mg of berberine hydrochloride orally twice a day for 3 months has been shown to markedly improve serum indicators of liver injury and lipid parameters (Zhao et al. 2008), suggesting its beneficial potential in hepatic viral infections.
Caffeine (1,3,7-trimethylxanthine) (23), is a well-known ingredient of coffee, a brewed drink prepared from roasted coffee (genus Coffea) beans. Caffeine has been recognized as a protector against hepatic oxidative damage since 1990s (Devasagayam et al. 1996). It has been shown that conventional coffee and caffeine intake provide better beneficial effects than decaffeinated coffee against liver injury in male animal model (Furtado et al. 2012). However, the hepatoprotective effects of coffee cannot be directly attributed to caffeine but also to other compounds present in the coffee, such as diterpenes kahweol and cafestol (Lee et al. 2007b). Thus, inflammatory cytokine IL-1β gene expression in the liver of mice fed HFD was reduced by simultaneous intake of either regular of decaffeinated coffee (Fukushima et al. 2009). Most of the studies investigating hepatoprotection by this compound were focused on its antifibrotic potential and prevention of steatohepatitis. Second line of studies was oriented toward CYP-modulating effects of caffeine and a possible risk of indicating activation of carcinogens. Caffeine has been frequently reported as CYP1A2 inductor in mice and rats (Goasduff et al. 1996; Kuribayashi et al. 2006); however, this induction was not observed in human hepatocytes (Vaynshteyn and Jeong 2012). Interestingly, a recent study in mice demonstrated that caffeine acted as an inhibitor of CYP1A2 activity and an enhancer of CYP3A activity, decreasing APAP hepatotoxicity (Wolf et al. 2005).
Caffeine protected against alcoholic liver injury by attenuating production of ROS and TNF-α in Kupffer cells (Lv et al. 2010). It also inhibited TNF-α-induced apoptosis of hepatocytes but had no significant effect on anti-Fas antibody-induced hepatitis and apoptosis, which suggest that caffeine differentially affects TNFR- and Fas-mediated liver injury in mice (Sugiyama et al. 2001). Further, caffeine reduced hepatosteatosis in mice fed a HFD and concomitantly stimulated the autophagy-lysosomal pathway. Caffeine inhibited mTOR and its downstream targets p70S6K and 4E-BP1 (Sinha et al. 2014). 4E-BP1 acts as a repressor of mRNA translation, suggesting restoration of transcriptional activity in hepatic cells. Inhibition of mTOR was closely associated with increased LC3-II levels and enhanced lipid accumulation in autolysosomes. Caffeine also attenuated lipid accumulation in hepatic cells by inhibiting lipogenesis and stimulating lipolysis through modulation of the AMPK signaling pathway. Mechanistically, caffeine increased phosphorylation of AMPK and decreased the mRNA level of lipogenesis-associated genes, including SREBP-1c, SREBP-2, FAS, SCD and HMG-CoA reductase (HMGCR) (Quan et al. 2013a). Caffeine could also improve high-energy diet-induced hepatic steatosis by promoting lipid metabolism via the cAMP/CREB/SIRT3/AMPK/ACC pathway (Zhang et al. 2015a).
Caffeine was found to increase HSCs apoptosis and cyclic adenosine monophosphate (cAMP) expression in human HSCs (Shim et al. 2013). However, although pharmacological activity of caffeine was usually attributed to inhibition of phosphodiesterase enzymes and elevation of cAMP levels, some studies have shown that caffeine may act via inhibition of receptor-stimulated inositol 3-phosphate (IP3) formation and direct inhibition of the IP3-sensitive Ca2+-release channel in a cAMP-independent way (Combettes et al. 1994). Other mechanisms of hepatoprotective actions of caffeine were also investigated. Thus, caffeine protected against alcohol-induced liver fibrosis, which was associated with inhibition of the cAMP/PKA-dependent activation of CREB, one of the major stimulators of liver fibrosis (Wang et al. 2015c). CREB acts as a regulator of PPARγ expression, which in turn down-regulates CTGF expression. Caffeine down-regulates TGF-β-induced expression of CTGF in hepatocytes by stimulating degradation of TGF-β effector Smad2, inhibition of Smad1/3 phosphorylation and up-regulation of PPARγ-receptor (Gressner et al. 2008). Study in rats demonstrated that caffeine could also diminish thioacetamide-induced liver fibrosis by impairing the expression of profibrogenic and proinflammatory genes such as TNF-α, PDGF and TGF-β1 (Gordillo-Bastidas et al. 2013; Arauz et al. 2014). Concomitantly, caffeine reduced expression of transcriptional factor Snail1, which is involved in fibrotic processes. Further, caffeine inhibited activation of HSCs and reduced levels of collagen mRNA, with concomitant inhibition of MMP-2 and MMP-9, main tissue remodelators (Arauz et al. 2014). Moreover, this compound inhibited HSCs activation via adenosine A2A receptor (A2AR) mediated by the cAMP/PKA/Src/ERK1/2 and p38 MAPK signaling pathways (Wang et al. 2014b), which play an important role in the pathogenesis of hepatic cirrhosis (Chan et al. 2006).
Investigating dietary behavior in NAFLD patients, an association between caffeine intake and lower risk for NAFLD (Birerdinc et al. 2012), reduced liver fibrosis among patients with NASH or chronic hepatitis C virus infection (Modi et al. 2010; Molloy et al. 2012; Khalaf et al. 2015), as well as reduced risk of HCC (Johnson et al. 2011), the severity of chronic hepatitis C (Costentin et al. 2011) or chronic liver disease (Ruhl and Everhart 2005) was established. Hepatoprotective effect of caffeine was clearly dependent on daily intake, showing beneficial effects in patients consuming more than two cups of coffee per day.
In HBV-infected HCC cells, ROS accumulation induced DNA damage that activated the ATM-Chk2 pathway, resulting in the inhibitory phosphorylation of Cdc25C phosphatase and Cdk1 (Kim et al. 2015), suggesting the inhibitory potential of caffeine against the viral replication.
Antraquinones
Emodin (1,3,8-trihydroxy-6-methyl-anthraquinone) (24), is an anthraquinone derivative isolated from Rheum species. Emodin protected against acetaminophen and CCl4-induced oxidative stress and acute liver injury in rats (Dang et al. 2008; Bhadauria 2010). Attenuation of fulminant hepatitis and proinflammatory response by this compound was associated with inhibition of the p38 MAPK/NF-κB pathway and blockade of TLR4/myeloid differentiation factor (MD) 2 complex expression (Yin et al. 2014; Xue et al. 2015). Emodin partially protected against cholestatic injury in rats (Zhao et al. 2009) by exerting anti-inflammatory activity through decreased nuclear translocation of NF-κB (Ding et al. 2008). Emodin also alleviated alcohol-mediated oxidative stress and liver steatosis in mice by down-regulating hepatic CYP2E1 expression (Liu et al. 2014d).
Emodin has been shown to ameliorate dyslipidemia in HFD-fed rats by activating AMPK and its downstream targeting enzyme, ACC (Tzeng et al. 2012). Concomitant up-regulation of gene expression of CPT1 and down-regulation of SREBP-1 and FAS protein levels in hepatocytes suggested attenuation of lipid accumulation by decreasing lipogenesis and increasing fatty acid β-oxidation. Emodin also ameliorated NAFLD in rats induced with a high caloric diet by suppressing GRP78-mediated SREBP-1c pathway in the liver and restoring reduced expression of PPARγ gene expression (Dong et al. 2005; Li et al. 2015c). In steatotic hepatic cells, emodin down-regulated HMGCR and diacylgycerol acyltransferase 1 (DGAT1), key enzymes in the synthesis of cholesterol and triglycerides, while up-regulating expression of CYP7A, involved in hepatic bile acid biosynthesis (Wang et al. 2014d).
Protection against CCl4-induced fibrogenesis by emodin was mediated by the reduction of the mRNA levels of TGF-β1 and Smad4 and inhibition of myofibroblastic differentiation (Dong et al. 2009). In immortalized rat HSCs, emodin reduced AP-1 DNA-binding activities and attenuated JunD mRNA expression (Gui et al. 2007). Emodin also markedly inhibited TGF-β1-induced ERK1/2 phosphorylation as well as the expression of TIMP-1.
However, some concerns arise from available data on chronic usage of this compound. A comprehensive study by National Toxicology (2001) showed that chronic administration of emodine resulted in sex and species-dependent low but significant carcinogenic activity of emodin and increased incidences of nephropathy in female mice. CYP-modulating activity of emodin was not intensively studied. However, one available study demonstrated activation of the metabolism of midazolam, a CYP3A4/5 substrate, in human liver microsomes by emodin (Li et al. 2014f). Biotransformation of emodin by the microsomal enzymes, which resulted in several metabolites, demonstrated that at least one of them, 2-hydroxyemodin (1,2,3,8-tetrahydroxy-6-methyl-anthraquinone), possessed a direct mutagenic activity to the test strain, Salmonella typhimurium (Masuda and Ueno 1984). These findings suggest potential adverse effects of emodin by long-term exposure in patients. In addition, emodin showed severe cytotoxicity against human liver cell line L-02 (Yu et al. 2011).
Curcuminoids
Curcumin, diferuloylmethane (25), is a major polyphenolic compound of the spice turmeric obtained from rhizomes of Curcuma species. The mechanism of hepatoprotection seems to be related at least in part to antioxidant activity and activation of the Nrf2/Kelch-like ECH-associated protein 1 (Keap1)/ARE pathway, with concomitant induction of phase II detoxifying/antioxidant enzymes such as HO-1 and NQO1 (Farombi et al. 2008; Gao et al. 2013b; Garcia-Nino and Pedraza-Chaverri 2014; Xu et al. 2014a). Moreover, curcumin administration in diet-induced oxidative stress reduced CYP2E1 as well as Prx1 expression, while up-regulating Prx6 expression (Lee et al. 2015). Oxidative stress induced with hepatotoxins is closely related to hepatic inflammatory response through activation of several signaling pathways, including MAPKs, NFκB and STAT3 (Ambade and Mandrekar 2012). Nevertheless, numerous studies demonstrated decreased hepatic expression of NF-κB and its downstream targets by curcumin (Bisht et al. 2011; Tu et al. 2012b; Xu et al. 2014a). It has also been shown that curcumin could decrease the expression of TLR2 and TLR4 and their ligand molecule HMGB1 in the rat model of fibrogenesis (Tu et al. 2012b) and T-cell-mediated hepatitis in concanavalin A-challenged mice (Tu et al. 2012a, 2013), suggesting a potential to attenuate inflammatory processes in the liver. Moreover, curcumin could ameliorate LPS/D-GalN-induced liver injury through reduction of hepatic mRNA levels of SIRT1 (Zhang et al. 2014b).
Furthermore, curcumin treatment resulted in alleviation of hepatic inflammation in steatohepatitis. Fructose is a dietary compound known to decrease tyrosine phosphorylation of insulin-induced IRS1 and inhibit activation of Akt and ERK1/2 in peripheral tissues. Administration of curcumin in rats increased phosphorylation of hepatic JAK2 and stimulated Akt and ERK1/2 activation in the model of fructose diet-induced steatohepatitis (Li et al. 2010). Overexpression and hyperactivity of hepatic protein tyrosine phosphatase 1B (PTP1B) was reduced by curcumin, with subsequent improvement of insulin and leptin signaling. Further, this compound suppressed HFD-mediated increase in SREBP-1, ACC1, FAS and CD36 expression, thus attenuating hepatic steatosis (Um et al. 2013). Curcumin also inhibited gene expression of receptor for advanced glycation end-products (RAGE) in HSCs in vitro by elevating the PPARγ activity and attenuating oxidative stress, leading to elimination of the AGE effects on HSCs activation (Lin et al. 2012a). Moreover, it decreased the hepatic gene expression of inflammatory cytokines, procollagen I and TIMP-1 in experimental steatohepatitis in mice (Vizzutti et al. 2010).
Curcumin has been shown to target multiple pathways to inhibit hepatic fibrosis. Hedgehog (Hh) signaling becomes activated in chronic liver injury and plays a role in the pathogenesis of hepatic fibrosis. Therefore, targeting Hh signaling may represent a novel therapeutic strategy for treatment of liver fibrosis. HSCs are Hh-responsive cells and activation of the Hh pathway may promote transdifferentiation of HSCs into myofibroblasts. Curcumin down-regulated Ptch and smoothened (Smo), two key elements in Hh signaling, simultaneously restoring Hhip expression in fibrotic rat livers and cultured HSCs (Lian et al. 2015). Curcumin also prevented the nuclear translocation, DNA binding, and transcription activity of GLI-1. Additionally, this compound arrested the cell cycle, induced mitochondrial apoptosis, restored lipid accumulation, reduced fibrotic gene expression and inhibited invasion and migration in HSCs. Further, curcumin inhibited HSCs activation in CCl4-intoxicated rat livers by activating PPARγ and reducing expression of mRNA and proteins involved in cell proliferation, including PDGF, PDGFRβ and EGFR, as well as genes related to fibrogenesis, including TGF-β, Tβ-RI/II and fibronectin (Fu et al. 2008a). As the result, curcumin inhibited proliferation of HSCs in the G2/M phase of the cell cycle (Shu et al. 2009). Furthermore, this compound activated AMPK and increased expression of PGC-1α, a coactivator for PPARγ, resulting in increased PPARγ activity and reduced collagen α1(I) gene expression (Zhai et al. 2015). In addition, curcumin has been shown to down-regulate Wnt signaling pathway, including the expression of Axin2 and Fra1 (Shin et al. 2009), both involved in HSCs activation (Jiang et al. 2006). During hepatic fibrogenesis, suppression of PPARγ is negatively associated with PDGF and EGF mitogenic signaling in HSCs. Curcumin could interrupt PDGF and EGF signaling by inhibiting gene expression and phosphorylation of PDGFRβ and EGFR and suppression PI3K, ERK and JNK signaling (Zhou et al. 2007; Fu et al. 2008a; Lin and Chen 2008). Curcumin also impaired production of ECM proteins in alcohol-stimulated HSCs and CCl4-induced liver by suppressing the TGF-β/Smad2/3 signaling and inducing Smad7 (Chen et al. 2014b). Activation of PPARγ by curcumin was required for inhibition of both VEGF expression and angiogenic properties of HSCs (Zhang et al. 2014a). VEGF plays a crucial role in the initial stages of new vessel formation and endothelial cell proliferation (Carmeliet 2005). Mechanistically, curcumin inhibited PDGFRβ/ERK and PI3K/Akt/mTOR pathways in activated HSCs via PPARγ signaling, thus inhibiting VEGF mRNA and protein expression (Zhang et al. 2014a). Inhibition of PDGFRβ also blocked FAK/RhoA cascade, resulting in reduced HSCs motility. Moreover, curcumin ameliorated hepatic angiogenesis and sinusoidal capillarization in fibrotic livers through suppression of proangiogenic factors, including HIF-1α, VEGFR-1, placental growth factor (PGF) and COX-2 (Yao et al. 2013). The inhibition of HIF-1α expression was at least in part mediated through inhibition of the ERK pathway (Zhao et al. 2014). Induction of CTGF in activated HSCs is another mechanism for increased production of ECM during fibrogenesis. Activation of PPARγ by curcumin resulted in interruption of TGF-β signaling by suppressing gene expression of Tβ-Rs, ultimately leading to inhibition of CTGF gene expression and decreased production of ECM proteins (Zheng and Chen 2006). Moreover, this compound inhibited CTGF expression in activated HSCs via inhibition of TLR-mediated NF-κB activation (Chen and Zheng 2008). Curcumin not only could impair HSCs activation and proliferation, but also blocked antiapoptotic protein c-FLIP and increased Bax expression, resulting in caspase-3 activation and induction of HSCs apoptosis (Priya and Sudhakaran 2008; Shin et al. 2009; Bisht et al. 2011). Curcumin also protected HSCs against leptin-induced activation in vitro and increased AMPK activity, leading to increased expression of genes relevant to lipid accumulation, including PPARγ, SREBP-1 and C/EBPα (Tang and Chen 2010). Finally, the inhibition of CBR1 can be added to a broad spectrum of antifibrotic activities of curcumin (Zhang et al. 2013b). Curcumin reduced the mRNA and protein abundance of CBR1 in cultured HSCs and inhibited ECM production. This compound also alleviated hepatic injury in parasitic infestation. In hamsters infected with a trematode parasite Opisthorchis viverrini, curcumin decreased the mRNA expression of TIMP-1, TIMP-2 and TNF-α, while increasing MMP-13 and MMP-7 levels (Pinlaor et al. 2010), suggesting degradation of ECM and improvement of liver fibrosis.
DEN-induced hepatocarcinogenesis was another pathological process which was prevented by curcumin administration. Beneficial effect of curcumin was associated with decreased levels of oncogenic p21 and p53 as well as cell cycle-related proteins, including PCNA, cyclin E and Cdc2 but not Cdk2 or cyclin D1 (Chuang et al. 2000a, b). In DNE-induced HCC, mice treatment with doxorubicin and curcumin co-delivery lipid nanoparticles (doxorubicin/curcumin-NPs) induced increase in caspase-3 activation and Bax/Bcl-2 ratio, and PCNA and VEGF (Zhao et al. 2015). On the other hand, doxorubicin/curcumin-NPs exhibited the synergistic effect on the apoptosis, proliferation and angiogenesis of HCC by decreasing the levels of multidrug resistance protein 1 (MDR1), P-gp, Bcl-2 and HIF-1α, indicating that curcumin might reverse multidrug resistance through these pathways. These findings suggested that doxorubicin/curcumin-NPs may be a promising treatment for HCC. Furthermore, curcumin treatment protected against acetaminophen-induced hepatocyte apoptosis via decreasing expression of pro-apoptotic genes Bax and caspase-3, while inducing antiapoptotic genes such as Bcl-xL and increasing Bcl-2/Bax ratio (Bulku et al. 2012; Li et al. 2013a). Interestingly, curcumin was able to up-regulate p53 protein expression and down-regulate Bcl-2 mRNA expression in thioacetamide-induced cytotoxicity, forcing the damaged cells to undergo apoptosis, thus suppressing hepatic inflammatory response and fibrogenesis (Wang et al. 2012a).
Additionally, curcumin protected mice against human cytomegalovirus (HCMV) infection through its anti-inflammatory and antioxidant effects (Lv et al. 2014). Curcumin also inhibited Rift Valley fever virus replication in infected human cells by inhibiting phosphorylation of NFκB p65 subunit via inhibition of kinase activity of the IKKβ2 complex (Narayanan et al. 2012). Curcumin could also inhibit HBV and HCV replication via down-regulation of metabolic coactivator PGC-1α and the Akt/SREBP-1 pathway, respectively (Kim et al. 2010a; Rechtman et al. 2010). Additionally, it inhibited HCV entry independently of the genotype in primary human hepatocytes, without effect on HCV RNA replication or viral assembly/release (Anggakusuma et al. 2014). Nevertheless, another study demonstrated inhibition of HCV replication through suppression of PI3K/Akt and induction of HO-1 (Chen et al. 2012b).
Administration of curcumin has been shown to decrease activity of CYP2B1/2 and CYP1A1 in mice liver (Sehgal et al. 2013) and inhibit activation of CYP2E1 in chronic alcohol and high-fat diet-induced liver injury in mice (Lee et al. 2013a). Similarly, microsomal CYP2C and CYP3A activities in bovine hepatocytes were inhibited by treatment with curcumin (Lemley and Wilson 2010). It has also been reported that curcumin inhibited activation of carcinogens metabolized by rat CYP isozymes, namely, CYP1A1, 1A2 and 2B1 (Thapliyal and Maru 2001). Interestingly, minimal to no induction of CYP1A2, CYP2B6, CYP3A4, CYP2C8/2C9 or CYP2D6 in human hepatocytes studies was observed (Price et al. 2008; Mach et al. 2010), with no effect on CYP2B1 and CYP2E1-mediated activation of carcinogenic N-nitrosamines in mice liver, kidney or intestine (Mori et al. 2006). Taken together, available data suggest low potential for CYP-mediated drug interactions at physiological serum concentrations of curcumin.
Capsaicinoids
Capsaicin (8-methyl-N-vanillyl-6-nonenamide) (26) protected against liver injury by reducing hepatic oxidative stress (Manjunatha and Srinivasan 2007; Hassan et al. 2012). However, capsaicin was able to induce CYP3A4 expression via PXR and C/EBPβ activation in rat livers and in human liver microsomes (Takanohashi et al. 2010; Han et al. 2012), suggesting a potential of causing drug–drug interactions.
Several studies revealed that capsaicin efficiently reduced liver fibrosis, inhibited HSCs proliferation and promoted cell apoptosis. Thus, capsaicin reduced gene expression of TGF-β1 and TIMP-1 in HSCs (Yu et al. 2014). Moreover, this compound induced apoptosis of HSCs, which was associated with increased expression of cytochrome c, Bax and caspase-3 and reduced levels of Bcl-2. Capsaicin also inhibited culture-induced activation of mouse HSCs by preventing the up-regulation of several activation markers such MMP-2, MMP-9 and TIMP-1 (Bitencourt et al. 2012). In addition, capsaicin inhibited PDGF-induced chemotaxis and proliferation of HSCs. Moreover, mice receiving capsaicin after BDL showed a significant improvement of liver fibrosis accompanied by a decrease in collagen deposition (Bitencourt et al. 2015). In CCl4-intoxicated mice, capsaicin prophylactically inhibited up-regulation of profibrogenic markers, but it could not attenuate already established fibrosis. Additionally, capsaicin inhibited autophagic process during HSCs activation. The same authors have shown induction of quiescent phenotype in HSCs via PPARγ activation, with the decrease in COX-2 and type I collagen mRNA expression (Bitencourt et al. 2012). These events preceded the suppression of TGF-β1 and collagen secretion.
Further, capsaicin stimulated hepatic lipolysis by increasing levels of phosphorylated hormone-sensitive lipase (HSL), CPT1 and PPARδ in mice liver (Li et al. 2013b). Activation of transient receptor potential vanilloid subfamily, member 1 (TRPV1), a capsaicin-specific receptor, and a concomitant PPARδ activation, prevented NAFLD through induction of autophagy-related proteins, such as LC3-II, Beclin1, Atg5 and Atg7. In addition, capsaicin suppressed inflammatory responses in obese mice fed a HFD by decreasing mRNA and proteins levels of TNF-α, MCP-1 and IL-6 in adipose and liver tissue (Kang et al. 2010). Concomitantly, capsaicin increased hepatic PGC-1α and TRPV1 expression in adipose tissue.
Chromenes
Ellagic acid (2,3,7,8-tetrahydroxy-chromeno[5,4,3-cde]chromene-5,10-dione) (27), a natural phenol antioxidant, also exhibited protection against hepatic oxidative injury (Pari and Sivasankari 2008; Girish et al. 2009). Administration of ellagic acid to rats increased NQO1, CAT, GPX and GST activity in the liver, while reducing CYP1A, 2B, 2C and 2E activity, suggesting impact on the metabolism of chemical carcinogens and drugs by affecting the activity of enzymes involved in xenobiotic activation and detoxification (Celik et al. 2013). Further, administration of ellagic acid prevented experimental liver cancer induced by DEN through activation of Bax and caspase-9 (Srigopalram et al. 2014). Concomitant down-regulation of NF-κB, cyclin D1, cyclin E1, MMP-2, MMP-9 and PCNA expression suggested inhibition of the cell cycle and activation of tissue remodeling process. Ellagic acid also decreased the expression of MMPs and TIMP-2 induced by alcohol consumption, resulting in amelioration of hepatic fibrosis (Devipriya et al. 2007).
Further, ellagic acid decreased concanavalin A-induced expression of TLR2 and TLR4 at both mRNA and protein level. This led to decrease in phosphorylation of JNK, ERK1/2 and p38 and suppression of NF-κB activation (Lee et al. 2014b). In addition, ellagic acid treatment decreased the expression of proinflammatory cytokines, including TNF-α, IL-6 and IL-1β, suggesting protection against T-cell-mediated hepatitis through the inhibition of TLR/MAPK/NF-κB signaling pathway. The suppression of metabolic syndrome by ellagic acid was also mediated through inhibition of NF-κB activation and induction of hepatoprotective Nrf2, HO-1 and CPT1 enzymes (Panchal et al. 2013). In addition, ellagic acid reduced serum resistin level and up-regulated mRNA expression of lipolytic genes, leading to improvement in hepatic steatosis (Yoshimura et al. 2013).
Treatment with ellagic acid has also been shown to overcome host immune tolerance induced by HBeAg during HBV infection (Kang et al. 2006) and block HBeAg secretion in infected hepatic cells, suggesting its beneficial effect on immune tolerance in HBV-infected individuals (Shin et al. 2005). Moreover, ellagic acid blocked the HCV NS3/4A protease activity in vitro. Structural analysis showed that ellagic acid interacted with the catalytic and substrate binding residues of NS3/4A protease, leading to its inhibition (Reddy et al. 2014).
Conclusion and future perspectives
Although all reviewed compounds showed a clear potential to alleviate different liver pathologies through multiple signaling pathways that reach beyond their antioxidant activity, the lack of clinical studies could not promote them as the hepatoprotective drugs. Interestingly, although silymarin gained a reputation as the hepatoprotective gold standard, glycyrrhizin has been a clinically most investigated natural compound, with a considerable amount of mechanistical evidence which support its hepatoprotective activity.
It is known that natural compound may directly bind to cellular molecules such as proteins or DNA (Walle et al. 2003), which could mediate their cellular actions. It has been found that quercetin binds directly to the BH3 domain of Bcl-2 and Bcl-xL proteins, inhibiting their activity (Primikyri et al. 2014). The molecular docking studies with quercetin showed bonded interaction within iNOS active site region, although quercetin analogues exhibited more favorable interaction than quercetin (Singh and Konwar 2012). Some natural compounds, such as genistein, have been shown to function as inhibitors of tyrosine protein kinases, which play an important role in the activation and proliferation of HSCs (Liu et al. 2002d). Resveratrol has been shown to directly interact with numerous protein molecules involved in signal transduction, such as PI3K, IKK and COX-2 (Pirola and Frojdo 2008). Dietary flavonoids luteolin, naringenin, eriodictyol and daidzein may stimulate the DNA-binding activity of NodD1 transcriptional regulator (Peck et al. 2006). Similarly, in silico docking studies indicated that morin was a better PI3K inhibitor than the classical inhibitor LY294002 (Sivaramakrishnan and Devaraj 2010). Therefore, the use of computational docking studies could lead to development of more potent hepatoprotective compounds.
The optimistic data based on the current knowledge of the mechanisms of hepatoprotective activity of natural compounds may result in therapeutic approaches with enhanced bioavailability and increased effectiveness of these compounds. Nevertheless, these compounds have to be evaluated in pre-clinical and clinical assays to determine their safety for humans. Generally, natural compounds have a low potential of interaction with drugs and activation of pro-carcinogens. However, since interaction between these compounds and pharmaceutical drugs has not been thoroughly examined, particularly at the level of CYPs, co-administration of these compounds by health-care practitioners requires caution.
Abbreviations
- 4E-BP:
-
eIF4E-binding protein
- A2AR:
-
Adenosine A2A receptor
- AA:
-
Arachidonic acid
- ABC:
-
ATP-binding cassette transporter
- ACAT:
-
Acyl-CoA:cholesterol acyltransferase
- ACC:
-
Acetyl-CoA carboxylase
- ACE:
-
Angiotensin-converting enzyme
- ACOX:
-
Acyl-coenzyme A oxidase
- ACS:
-
Acetyl-CoA synthetase
- AdipoR:
-
Adiponectin receptor
- AhR:
-
Aryl hydrocarbon receptor
- AIF:
-
Apoptosis-inducing factor
- AKR:
-
Aldo–keto reductase
- ALT:
-
Alanine transaminase
- AMPK:
-
5′ AMP-activated protein kinase
- Ang:
-
Angiotensin
- AP:
-
Activator protein
- aP:
-
Adipocyte fatty acid-binding protein
- Apaf:
-
Apoptotic protease-activating factor
- AR:
-
Amphiregulin
- ARE:
-
Antioxidant response element
- ARNT:
-
Aryl hydrocarbon nuclear translocator
- ASC:
-
Apoptosis-associated speck-like protein containing a carboxy-terminal CARD
- ASK:
-
Apoptosis signal-regulating kinase
- AT1R:
-
Angiotensin II type 1 receptor
- ATF:
-
Activating transcription factor
- BA:
-
Bile acid
- Bambi:
-
Bone morphogenetic protein and activin membrane-bound inhibitor
- Bax:
-
Bcl-2-associated X protein
- Bcl-2:
-
B-cell lymphoma 2
- BDL:
-
Common bile duct ligation
- Bid:
-
Bcl-2 homology 3 (BH3) interacting-domain death agonist
- Bim:
-
Bcl-2-interacting mediator of cell death
- BiP:
-
Immunoglobulin binding protein
- C/EBP:
-
CCAAT/enhancer-binding protein
- CAMKK:
-
Ca2+-calmodulin dependent protein kinase kinase
- cAMP:
-
Cyclic adenosine monophosphate
- casp:
-
Caspase
- CAT:
-
Catalase
- CBR:
-
Cannabinoid receptor type
- CD:
-
Cluster of differentiation
- Cdk:
-
Cyclin-dependent kinase
- c-FLIP:
-
(FADD-like IL-1β-converting enzyme)-inhibitory protein
- CHOP:
-
C/EBP homologous protein
- cIAP:
-
Cellular inhibitor of apoptosis
- CPT:
-
Carnitine palmitoyltransferase
- CREB:
-
CAMP-response element-binding protein
- CRP:
-
C-reactive protein
- CTGF:
-
Connective tissue growth factor
- Cx43:
-
Connexin 43
- CXCR:
-
Chemokine (CXC motif) receptor
- CXCL:
-
Including chemokine (C-X-C motif) ligand
- CYP:
-
Cytochrome P450
- DAG:
-
Diacylglycerol
- DEN:
-
Diethyl nitrosamine
- DGAT:
-
Diacylgycerol acyltransferase
- ECM:
-
Extracellular matrix
- EGF:
-
Epidermal growth factor
- EGFR:
-
EGF receptor
- EGR:
-
Early growth response protein
- eIF:
-
Eukaryotic initiation factor
- EMR:
-
EGF-like module-containing mucin-like hormone receptor
- eNOS:
-
Endothelial nitric oxide synthase
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular regulated kinase
- ESR:
-
Estrogen receptor
- ET:
-
Endothelin
- FA:
-
Fatty acid
- FABP:
-
Fatty acid-binding protein
- FADD:
-
Cellular Fas-associated death domain
- FAK:
-
Focal adhesion kinase
- FAS:
-
Fatty acid synthase
- FasL:
-
Fas ligand
- FATP:
-
Fatty acid transport protein
- FOXO:
-
Forkhead box protein O
- FXR:
-
Farnesoid X receptor
- GalN:
-
d-Galactosamine
- GCLC:
-
Glutamate-cysteine ligase, catalytic subunit
- GJIC:
-
The gap junctional intercellular communication
- GLI:
-
Glioma-associated oncogenes
- GPx:
-
Glutathione peroxidase
- GR:
-
Glutathione reductase
- GRP78:
-
78 kDa glucose-regulated protein
- GSH:
-
Glutathione
- GSK:
-
Glycogen synthase kinase
- GST:
-
Glutathione-S-transferase
- H2O2 :
-
Hydrogen peroxide
- HAV:
-
Hepatitis A virus
- HBeAg:
-
Hepatitis B e antigen
- HBsAg:
-
HBV surface antigen
- HBV:
-
Hepatitis B virus
- HCC:
-
Hepatocellular carcinoma
- HCMV:
-
Human cytomegalovirus
- HCV:
-
Hepatitis C virus
- HDAC:
-
Histone deacetylase
- HFD:
-
High-fat diet fed
- HGF:
-
Hepatocyte growth factor
- HIF:
-
Hypoxia-inducible factor
- HMGB:
-
High-mobility group protein box
- HMGCR:
-
HMG-CoA reductase
- HNF:
-
Hepatocyte nuclear factor
- HO:
-
Heme oxygenase
- HSCs:
-
Hepatic stellate cells
- HSL:
-
Hormone-sensitive lipase
- ICAM:
-
Intracellular adhesion molecule
- IFN:
-
Interferon
- IGF:
-
The insulin growth factor
- IGF-1R:
-
IGF 1 receptor
- IKK:
-
IkappaB kinase
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- IP3 :
-
Inositol 3-phosphate
- IR:
-
Insulin receptor
- IRAK:
-
IL-1R-associated kinase
- IRE:
-
Inositol-requiring enzyme
- IRES:
-
Internal ribosome entry site
- IRF:
-
IFN regulatory factor
- IRS:
-
IR substrate
- IκBα:
-
IkappaBalpha
- JAK:
-
Janus-activated kinase
- JNK:
-
c-Jun N-terminal kinase
- Keap:
-
Kelch-like ECH-associated protein
- KLF:
-
Krueppel-like factor
- LC3:
-
Microtubule-associated protein light chain 3
- LDL:
-
Low-density lipoprotein
- LDLR:
-
LDL receptor
- LKB:
-
Liver kinase B
- LPS:
-
Lipopolysaccharide
- LXR:
-
Liver X receptor
- MAPK:
-
Mitogen-activated protein kinase
- MCP:
-
Monocyte chemoattractant protein
- MD:
-
Myeloid differentiation factor
- Mdm2:
-
Mouse double minute 2 homolog
- MDR:
-
Multidrug resistance protein
- MEK:
-
Mitogen-activated protein/extracellular signal-regulated kinase kinase
- MKK:
-
Mitogen-activated protein kinase kinase
- MMP:
-
Matrix metalloproteinase
- MRP:
-
Multidrug resistance protein
- mTOR:
-
Mammalian target of rapamycin
- MT:
-
Metallothionein
- MTTP:
-
Microsomal triglyceride transfer protein
- MyD:
-
Myeloid differentiation factor
- MYPT:
-
Myosin phosphatase target subunit
- NAFLD:
-
Nonalcoholic fatty liver disease
- NALP:
-
NACHT, LRR and PYD domains-containing protein
- NAMPT:
-
Nicotinamide phosphoribosyltransferase
- NASH:
-
Nonalcoholic steatohepatitis
- NFAT:
-
Nuclear factor of activated T cells
- NO:
-
Nitric oxide
- NQO:
-
NAD(P)H:quinone oxidoreductase
- Nrf:
-
Nuclear factor-erythroid-2-related factor
- NS:
-
Nonstructural protein
- OATP:
-
Organic anion-transporting polypeptide
- p70S6K :
-
70 kDa ribosomal S6 kinase
- PAI:
-
Plasminogen activator inhibitor
- PARP:
-
Poly (ADP-ribose) polymerase
- PCNA:
-
Proliferating cell nuclear antigen
- PC-PLC:
-
Phosphatidylcholine-specific phospholipase C
- PCSK:
-
Proprotein convertase subtilisin/kexin
- PDE:
-
Phosphodiesterase
- PDGF:
-
Platelet-derived growth factor
- PDGFRβ:
-
PDGF receptor beta
- PERK:
-
PKR-like ER kinase
- PG:
-
Prostaglandin
- PGC:
-
PPARγ coactivator
- PGF:
-
Placental growth factor
- PI3K:
-
Phosphoinositide 3-kinase
- PKA:
-
Protein kinase A
- PKC:
-
Protein kinase C
- PKD:
-
Protein kinase D
- PKR:
-
Protein kinase R
- PLA:
-
Phospholipase A
- PP2A:
-
Protein phosphatase 2A
- PPAR:
-
Peroxisome proliferator-activated receptor
- Prx:
-
Peroxiredoxin
- PTP:
-
Protein tyrosine phosphatase
- PUMA:
-
p53-up-regulated modulator of apoptosis
- PXR:
-
Pregnane X receptor
- RAGE:
-
Receptor for advanced glycation end-products
- RAS:
-
Renin–angiotensin system
- ROCK:
-
Rho-associated coiled coil-forming protein kinase
- ROS:
-
Reactive oxygen species
- RSK:
-
Ribosomal S6 kinase
- RXR:
-
Retinoid X receptor
- S1P:
-
Sphingosine-1-phosphate
- SCD:
-
Stearoyl-CoA desaturase
- SESN:
-
Sestrin
- SIRT:
-
Silent mating type information regulation 2 homolog
- Smo:
-
Smoothened
- SOCS:
-
Suppressor of cytokine signaling
- SOD:
-
Superoxid dismutase
- SP:
-
Specificity protein
- SphK:
-
Sphingosine kinase
- SREBP:
-
Sterol regulatory element-binding protein
- STAT:
-
Signal transducers and activators of transcription
- TAB:
-
TGF-β-activated kinase
- TAG:
-
Triacylglycerol
- TAK:
-
TGF-β-activated kinase
- TGF:
-
Transforming growth factor
- TIMP:
-
Tissue inhibitor of matrix metalloproteinase
- TLR:
-
Toll-like receptor
- TNFR:
-
TNF-α receptor
- TNF:
-
Tumor necrosis factor
- TRADD:
-
Receptor-associated death domain
- TRAF:
-
TNFR-associated factor
- TRPV:
-
Transient receptor potential vanilloid subfamily
- Trx:
-
Thioredoxin
- TrxR:
-
Thioredoxin reductase
- TXNIP:
-
Thioredoxin-interacting protein
- Tβ-R:
-
TGF-β receptor
- UCP:
-
Uncoupling protein
- ULK:
-
UNC-51-like kinase
- uPA:
-
Urokinase-type plasminogen activator
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
VEGF receptor
- XBP:
-
X-box binding protein
- XIAP:
-
X-linked inhibitor of apoptosis protein
- SMA:
-
Smooth muscle actin
References
Abou El Naga RN, Azab SS, El-Demerdash E et al (2013) Sensitization of TRAIL-induced apoptosis in human hepatocellular carcinoma HepG2 cells by phytochemicals. Life Sci 92:555–561
Acharya SK, Sreenivas V, Gupta SD et al (2012) Treatment of chronic hepatitis due to hepatitis C virus (CH-C) in India: a randomized controlled trial comparing daily interferon-alfa-2b and ribavirin with daily interferon-alfa-2b and glycyrrhizin—a multicenter study. J Clin Exp Hepatol 2:10–18
Ahmad A, Ahmad R (2014) Resveratrol mitigate structural changes and hepatic stellate cell activation in N′-nitrosodimethylamine-induced liver fibrosis via restraining oxidative damage. Chem Biol Interact 221:1–12
Ajmo JM, Liang X, Rogers CQ, Pennock B, You M (2008) Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol 295:G833–G842
Al-Malki AL, Sayed AA (2014) Thymoquinone attenuates cisplatin-induced hepatotoxicity via nuclear factor kappa-beta. BMC Complement Altern Med 14:282
Al-Rejaie SS, Aleisa AM, Sayed-Ahmed MM et al (2013) Protective effect of rutin on the antioxidant genes expression in hypercholestrolemic male Westar rat. BMC Complement Altern Med 13:136
Al-Sheddi ES, Farshori NN, Al-Oqail MM et al (2015) Protective effect of Lepidium sativum seed extract against hydrogen peroxide-induced cytotoxicity and oxidative stress in human liver cells (HepG2). Pharm Biol. doi:10.3109/13880209.2015.1035795
Ambade A, Mandrekar P (2012) Oxidative stress and inflammation: essential partners in alcoholic liver disease. Int J Hepatol 2012:853175
An Z, Qi YM, Huang DJ et al (2014) EGCG inhibits Cd2+-induced apoptosis through scavenging ROS rather than chelating Cd2+ in HL-7702 cells. Toxicol Mech Method 24:259–267
Andrade JMO, Paraiso AF, de Oliveira MVM et al (2014) Resveratrol attenuates hepatic steatosis in high-fat fed mice by decreasing lipogenesis and inflammation. Nutrition 30:915–919
Anggakusuma ColpittsCC, Schang LM et al (2014) Turmeric curcumin inhibits entry of all hepatitis C virus genotypes into human liver cells. Gut 63:1137–1149
Ansorena E, Garcia-Trevijano ER et al (2002) S-adenosylmethionine and methylthioadenosine are antiapoptotic in cultured rat hepatocytes but proapoptotic in human hepatoma cells. Hepatology 35:274–280
Arakaki N, Hirono S, Kawakami S et al (1992) Effects of protein-kinase inhibitors on the mitogenic activity of human hepatocyte growth-factor on rat hepatocytes in primary culture. Biochem Biophys Res Commun 185:22–28
Arauz J, Zarco N, Segovia J et al (2014) Caffeine prevents experimental liver fibrosis by blocking the expression of TGF-β. Eur J Gastroenterol Hepatol 26:164–173
Arinc E, Yilmaz D, Bozcaarmutlu A (2015) Mechanism of inhibition of CYP1A1 and glutathione S-transferase activities in fish liver by quercetin, resveratrol, naringenin, hesperidin, and rutin. Nutr Cancer 67:137–144
Arunkumar E, Karthik D, Anuradha CV (2013) Genistein sensitizes hepatic insulin signaling and modulates lipid regulatory genes through p70 ribosomal S6 kinase-1 inhibition in high-fat-high-fructose diet-fed mice. Pharm Biol 51:815–824
Assini JM, Mulvihill EE, Burke AC et al (2015) Naringenin prevents obesity, hepatic steatosis, and glucose intolerance in male mice independent of fibroblast growth factor 21. Endocrinology 156:2087–2102
Au AY, Hasenwinkel JM, Frondoza CG (2011) Silybin inhibits interleukin-1β-induced production of pro-inflammatory mediators in canine hepatocyte cultures. J Vet Pharmacol Ther 34:120–129
Bachmetov L, Gal-Tanamy M, Shapira A et al (2012) Suppression of hepatitis C virus by the flavonoid quercetin is mediated by inhibition of NS3 protease activity. J Viral Hepat 19:E81–E88
Baer-Dubowska W, Szaefer H, Krajka-Kuzniak V (1998) Inhibition of murine hepatic cytochrome P450 activities by natural and synthetic phenolic compounds. Xenobiotica 28:735–743
Bai T, Lian LH, Wu YL, Wan Y, Nan JX (2013) Thymoquinone attenuates liver fibrosis via PI3K and TLR4 signaling pathways in activated hepatic stellate cells. Int Immunopharmacol 15:275–281
Bai T, Yang Y, Wu YL et al (2014) Thymoquinone alleviates thioacetamide-induced hepatic fibrosis and inflammation by activating LKB1-AMPK signaling pathway in mice. Int Immunopharmacol 19:351–357
Balamurugan K, Karthikeyan J (2012) Evaluation of luteolin in the prevention of N-nitrosodiethylamine-induced hepatocellular carcinoma using animal model system. Indian J Clin Biochem 27:157–163
Barrera G (2012) Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol 2012:137289
Bear WL, Teel RW (2000) Effects of citrus phytochemicals on liver and lung cytochrome P450 activity and on the in vitro metabolism of the tobacco-specific nitrosamine NNK. Anticancer Res 20:3323–3329
Bechmann LP, Zahn D, Gieseler RK et al (2009) Resveratrol amplifies profibrogenic effects of free fatty acids on human hepatic stellate cells. Hepatol Res 39:601–608
Beedanagari SR, Bebenek I, Bui P, Hankinson O (2009) Resveratrol inhibits dioxin-induced expression of human CYP1A1 and CYP1B1 by inhibiting recruitment of the aryl hydrocarbon receptor complex and RNA polymerase II to the regulatory regions of the corresponding genes. Toxicol Sci 110:61–67
Bhadauria M (2010) Dose-dependent hepatoprotective effect of emodin against acetaminophen-induced acute damage in rats. Exp Toxicol Pathol 62:627–635
Birerdinc A, Stepanova M, Pawloski L, Younossi ZM (2012) Caffeine is protective in patients with non-alcoholic fatty liver disease. Aliment Pharm Ther 35:76–82
Bishayee A, Barnes KF, Bhatia D, Darvesh AS, Carroll RT (2010) Resveratrol suppresses oxidative stress and inflammatory response in diethylnitrosamine-initiated rat hepatocarcinogenesis. Cancer Prev Res 3:753–763
Bisht S, Khan MA, Bekhit M et al (2011) A polymeric nanoparticle formulation of curcumin (NanoCurc) ameliorates CCl4-induced hepatic injury and fibrosis through reduction of pro-inflammatory cytokines and stellate cell activation. Lab Invest 91:1383–1395
Bitencourt S, de Mesquita FC, Caberlon E et al (2012) Capsaicin induces de-differentiation of activated hepatic stellate cell. Biochem Cell Biol 90:683–690
Bitencourt S, Stradiot L, Verhulst S et al (2015) Inhibitory effect of dietary capsaicin on liver fibrosis in mice. Mol Nutr Food Res 59:1107–1116
Bona S, Filippin LI, Di Naso FC et al (2012) Effect of antioxidant treatment on fibrogenesis in rats with carbon tetrachloride-induced cirrhosis. ISRN Gastroenterol 2012:762920
Bulku E, Stohs SJ, Cicero L et al (2012) Curcumin exposure modulates multiple pro-apoptotic and anti-apoptotic signaling pathways to antagonize acetaminophen-induced toxicity. Curr Neurovasc Res 9:58–71
Calland N, Albecka A, Belouzard S et al (2012) (–)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology 55:720–729
Cao LJ, Zou Y, Zhu JL, Fan XH, Li JB (2013) Ginsenoside Rg1 attenuates concanavalin A-induced hepatitis in mice through inhibition of cytokine secretion and lymphocyte infiltration. Mol Cell Biochem 380:203–210
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438:932–936
Casella ML, Parody JP, Ceballos MP et al (2014) Quercetin prevents liver carcinogenesis by inducing cell cycle arrest, decreasing cell proliferation and enhancing apoptosis. Mol Nutr Food Res 58:289–300
Celik G, Semiz A, Karakurt S et al (2013) A comparative study for the evaluation of two doses of ellagic acid on hepatic drug metabolizing and antioxidant enzymes in the rat. Biomed Res Int 2013:358945
Cengiz M, Kutlu HM, Burukoglu DD, Ayhanci A (2015) A comparative study on the therapeutic effects of silymarin and silymarin-loaded solid lipid nanoparticles on D-GaIN/TNF-alpha-induced liver damage in Balb/c mice. Food Chem Toxicol 77:93–100
Cerny D, Canova NK, Martinek J et al (2009) Effects of resveratrol pretreatment on tert-butylhydroperoxide induced hepatocyte toxicity in immobilized perifused hepatocytes: involvement of inducible nitric oxide synthase and hemoxygenase-1. Nitric Oxide 20:1–8
Chan ES, Montesinos MC, Fernandez P et al (2006) Adenosine A(2A) receptors play a role in the pathogenesis of hepatic cirrhosis. Brit J Pharmacol 148:1144–1155
Chan CC, Cheng LY, Lin CL et al (2011) The protective role of natural phytoalexin resveratrol on inflammation, fibrosis and regeneration in cholestatic liver injury. MMol Nutr Food Res 55:1841–1849
Chan CC, Lee KC, Huang YH et al (2014) Regulation by resveratrol of the cellular factors mediating liver damage and regeneration after acute toxic liver injury. J Gastroenterol Hepatol 29:603–613
Chang TK, Chen J, Lee WB (2001) Differential inhibition and inactivation of human CYP1 enzymes by trans-resveratrol: evidence for mechanism-based inactivation of CYP1A2. J Pharmacol Exp Ther 299:874–882
Chang TK, Chen J, Benetton SA (2002) In vitro effect of standardized ginseng extracts and individual ginsenosides on the catalytic activity of human CYP1A1, CYP1A2, and CYP1B1. Drug Metab Dispos 30:378–384
Chatuphonprasert W, Jarukamjorn K, Kondo S, Nemoto N (2009) Synergistic increases of metabolism and oxidation-reduction genes on their expression after combined treatment with a CYP1A inducer and andrographolide. Chem Biol Interact 182:233–238
Chen AP, Zhang L (2003) The antioxidant (–)-epigallocatechin-3-gallate inhibits rat hepatic stellate cell proliferation in vitro by blocking the tyrosine phosphorylation and reducing the gene expression of platelet-derived growth factor-beta receptor. J Biol Chem 278:23381–23389
Chen A, Zheng S (2008) Curcumin inhibits connective tissue growth factor gene expression in activated hepatic stellate cells in vitro by blocking NF-κB and ERK signalling. Brit J Pharmacol 153:557–567
Chen YK, Cheung C, Reuhl KR et al (2011) Effects of green tea polyphenol (–)-epigallocatechin-3-gallate on newly developed high-fat/Western-style diet-induced obesity and metabolic syndrome in mice. J Agric Food Chem 59:11862–11871
Chen IS, Chen YC, Chou CH et al (2012a) Hepatoprotection of silymarin against thioacetamide-induced chronic liver fibrosis. J Sci Food Agric 92:1441–1447
Chen MH, Lee MY, Chuang JJ et al (2012b) Curcumin inhibits HCV replication by induction of heme oxygenase-1 and suppression of AKT. Int J Mol Med 30:1021–1028
Chen XY, Zenger K, Lupp A et al (2012c) Tacrine-silibinin codrug shows neuro- and hepato protective effects in vitro and pro-cognitive and hepatoprotective effects in vivo. J Med Chem 55:5231–5242
Chen HJ, Liang TM, Lee IJ, Huang YT, Lin YL (2013a) Scutellariae radix suppresses LPS-induced liver endothelial cell activation and inhibits hepatic stellate cell migration. J Ethnopharmacol 150:835–842
Chen S, Zou L, Li L, Wu T (2013b) The protective effect of glycyrrhetinic acid on carbon tetrachloride-induced chronic liver fibrosis in mice via upregulation of Nrf2. PLoS One 8:e53662
Chen HW, Huang CS, Li CC et al (2014a) Bioavailability of andrographolide and protection against carbon tetrachloride-induced oxidative damage in rats. Toxicol Appl Pharmacol 280:1–9
Chen N, Geng Q, Zheng J et al (2014b) Suppression of the TGF-beta/Smad signaling pathway and inhibition of hepatic stellate cell proliferation play a role in the hepatoprotective effects of curcumin against alcohol-induced hepatic fibrosis. Int J Mol Med 34:1110–1116
Chen S, Zhao X, Ran L et al (2015) Resveratrol improves insulin resistance, glucose and lipid metabolism in patients with non-alcoholic fatty liver disease: a randomized controlled trial. Dig Liver Dis 47:226–232
Cheng PY, Lee YM, Wu YS et al (2007) Protective effect of baicalein against endotoxic shock in rats in vivo and in vitro. Biochem Pharmacol 73:793–804
Cheng ZY, Tian X, Gao J et al (2014) Contribution of baicalin on the plasma protein binding displacement and CYP3A activity inhibition to the pharmacokinetic changes of nifedipine in rats in vivo and in vitro. PLoS One 9:e87234
Cheung C, Gonzalez FJ (2008) Humanized mouse lines and their application for prediction of human drug metabolism and toxicological risk assessment. J Pharmacol Exp Ther 327:288–299
Choi EJ, Kim T, Kim GH (2012) Quercetin acts as an antioxidant and downregulates CYP1A1 and CYP1B1 against DMBA-induced oxidative stress in mice. Oncol Rep 28:291–296
Chtourou Y, Fetoui H, Jemai R et al (2015) Naringenin reduces cholesterol-induced hepatic inflammation in rats by modulating matrix metalloproteinases-2, 9 via inhibition of nuclear factor kappaB pathway. Eur J Pharmacol 746:96–105
Chuang SE, Cheng AL, Lin JK, Kuo ML (2000a) Inhibition by curcumin of diethylnitrosamine-induced hepatic hyperplasia, inflammation, cellular gene products and cell-cycle-related proteins in rats. Food Chem Toxicol 38:991–995
Chuang SE, Kuo ML, Hsu CH et al (2000b) Curcumin-containing diet inhibits diethylnitrosamine-induced murine hepatocarcinogenesis. Carcinogenesis 21:331–335
Chun YJ, Kim MY, Guengerich FP (1999) Resveratrol is a selective human cytochrome P450 1A1 inhibitor. Biochem Bioph Res Commun 262:20–24
Church RJ, Gatti DM, Urban TJ et al (2015) Sensitivity to hepatotoxicity due to epigallocatechin gallate is affected by genetic background in diversity outbred mice. Food Chem Toxicol 76:19–26
Cicero AF, Rosticci M, Parini A et al (2015) Short-term effects of a combined nutraceutical of insulin-sensitivity, lipid level and indexes of liver steatosis: a double-blind, randomized, cross-over clinical trial. Nutr J 14:30
Ciesek S, von Hahn T, Colpitts CC et al (2011) The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology 54:1947–1955
Clichici S, Olteanu D, Nagy AL, Oros A, Filip A et al (2015) Silymarin inhibits the progression of fibrosis in the early stages of liver injury in CCl4-treated rats. J Med Food 18:290–298
Combettes L, Berthon B, Claret M (1994) Caffeine inhibits cytosolic calcium oscillations induced by noradrenaline and vasopressin in rat hepatocytes. Biochem J 301:737–744
Costentin CE, Roudot-Thoraval F, Zafrani ES et al (2011) Association of caffeine intake and histological features of chronic hepatitis C. J Hepatol 54:1123–1129
Crance JM, Biziagos E, Passagot J, van Cuyck-Gandre H, Deloince R (1990) Inhibition of hepatitis A virus replication in vitro by antiviral compounds. J Med Virol 31:155–160
Cuevas MJ, Tieppo J, Marroni NP, Tunon MJ, Gonzalez-Gallego J (2011) Suppression of amphiregulin/epidermal growth factor receptor signals contributes to the protective effects of quercetin in cirrhotic rats. J Nutr 141:1299–1305
Dalaklioglu S, Genc GE, Aksoy NH, Akcit F, Gumuslu S (2013) Resveratrol ameliorates methotrexate-induced hepatotoxicity in rats via inhibition of lipid peroxidation. Hum Exp Toxicol 32:662–671
Dang SS, Zhang X, Jia XL et al (2008) Protective effects of emodin and Astragalus polysaccharides on chronic hepatic injury in rats. Chin Med J 121:1010–1014
Das A (2011) Heat stress-induced hepatotoxicity and its prevention by resveratrol in rats. Toxicol Mech Method 21:393–399
Das N, Sikder K, Bhattacharjee S et al (2013) Quercetin alleviates inflammation after short-term treatment in high-fat-fed mice. Food Funct 4:889–898
de David C, Rodrigues G, Bona S et al (2011) Role of quercetin in preventing thioacetamide-induced liver injury in rats. Toxicol Pathol 39:949–957
de Souza IC, Meira Martins LA, de Vasconcelos M et al (2015) Resveratrol regulates the quiescence-like induction of activated stellate cells by modulating the PPARγ/SIRT1 ratio. J Cell Biochem. doi:10.1002/jcb.25181
Demiroren K, Dogan Y, Kocamaz H et al (2014) Protective effects of L-carnitine, N-acetylcysteine and genistein in an experimental model of liver fibrosis. Clin Res Hepatol Gastroenterol 38:63–72
Deng RR, Xu CS, Chen X et al (2014) Resveratrol suppresses the inducible expression of CYP3A4 through the pregnane X receptor. J Pharmacol Sci 126:146–154
Devasagayam TPA, Kamat JP, Mohan H, Kesavan PC (1996) Caffeine as an antioxidant: inhibition of lipid peroxidation induced by reactive oxygen species. Biochim Biophys Acta 1282:63–70
Devipriya N, Sudheer AR, Srinivasan M, Menon VP (2007) Effect of ellagic acid, a plant polyphenol, on fibrotic markers (MMPs and TIMPs) during alcohol-induced hepatotoxicity. Toxicol Mech Method 17:349–356
Di Pascoli M, Divi M, Rodriguez-Vilarrupla A et al (2013) Resveratrol improves intrahepatic endothelial dysfunction and reduces hepatic fibrosis and portal pressure in cirrhotic rats. J Hepatol 58:904–910
Dias AS, Porawski M, Alonso M et al (2005) Quercetin decreases oxidative stress, NF-κB activation, and iNOS overexpression in liver of streptozotocin-induced diabetic rats. J Nutr 135:2299–2304
Ding Y, Zhao L, Mei H et al (2008) Exploration of emodin to treat alpha-naphthylisothiocyanate-induced cholestatic hepatitis via anti-inflammatory pathway. Eur J Pharmacol 590:377–386
Ding L, Li J, Song B et al (2014) Andrographolide prevents high-fat diet-induced obesity in C57BL/6 mice by suppressing the sterol regulatory element-binding protein pathway. J Pharmacol Exp Ther 351:474–483
Ding Y, Sun X, Chen Y, Deng Y, Qian K (2015) Epigallocatechin gallate attenuated non-alcoholic steatohepatitis induced by methionine- and choline-deficient diet. Eur J Pharmacol. doi:10.1016/j.ejphar.2015.05.005
Domitrovic R, Jakovac H, Grebic D, Milin C, Radosevic-Stasic B (2008) Dose- and time-dependent effects of luteolin on liver metallothioneins and metals in carbon tetrachloride-induced hepatotoxicity in mice. Biol Trace Elem Res 126:176–185
Domitrovic R, Jakovac H, Milin C, Radosevic-Stasic B (2009a) Dose- and time-dependent effects of luteolin on carbon tetrachloride-induced hepatotoxicity in mice. Exp Toxicol Pathol 61:581–589
Domitrovic R, Jakovac H, Tomac J, Sain I (2009b) Liver fibrosis in mice induced by carbon tetrachloride and its reversion by luteolin. Toxicol Appl Pharmacol 241:311–321
Domitrovic R, Jakovac H, Blagojevic G (2011) Hepatoprotective activity of berberine is mediated by inhibition of TNF-alpha, COX-2, and iNOS expression in CCl4-intoxicated mice. Toxicology 280:33–43
Domitrovic R, Jakovac H, Marchesi VV et al (2012) Differential hepatoprotective mechanisms of rutin and quercetin in CCl4-intoxicated BALB/cN mice. Acta Pharmacol Sin 33:1260–1270
Domitrovic R, Jakovac H, Marchesi VV, Blazekovic B (2013) Resolution of liver fibrosis by isoquinoline alkaloid berberine in CCl4-intoxicated mice is mediated by suppression of oxidative stress and upregulation of MMP-2 expression. J Med Food 16:518–528
Dong H, Lu FE, Gao ZQ et al (2005) Effects of emodin on treating murine nonalcoholic fatty liver induced by high caloric laboratory chaw. World J Gastroenterol 11:1339–1344
Dong MX, Jia Y, Zhang YB et al (2009) Emodin protects rat liver from CCl4-induced fibrogenesis via inhibition of hepatic stellate cells activation. World J Gastroenterol 15:4753–4762
Dvorak Z, Vrzal R, Henklova P et al (2008) JNK inhibitor SP600125 is a partial agonist of human aryl hydrocarbon receptor and induces CYP1A1 and CYP1A2 genes in primary human hepatocytes. Biochem Pharmacol 75:580–588
Elbarbry F, Ragheb A, Marfleet T, Shoker A (2012) Modulation of hepatic drug metabolizing enzymes by dietary doses of thymoquinone in female New Zealand White rabbits. Phytother Res 26:1726–1730
El-Lakkany NM, Hammam OA, El-Maadawy WH et al (2012) Anti-inflammatory/anti-fibrotic effects of the hepatoprotective silymarin and the schistosomicide praziquantel against Schistosoma mansoni-induced liver fibrosis. Parasit Vectors 5:9
El-Shafey MM, Abd-Allah GM, Mohamadin AM, Harisa GI, Mariee AD (2015) Quercetin protects against acetaminophen-induced hepatorenal toxicity by reducing reactive oxygen and nitrogen species. Pathophysiology 22:49–55
Esmaeili MA, Alilou M (2014) Naringenin attenuates CCl4-induced hepatic inflammation by the activation of an Nrf2-mediated pathway in rats. Clin Exp Pharmacol Physiol 41:416–422
Fang L, Zhan SX, Huang C et al (2013) TRPM7 channel regulates PDGF-BB-induced proliferation of hepatic stellate cells via PI3K and ERK pathways. Toxicol Appl Pharmacol 272:713–725
Farombi EO, Shrotriya S, Na HK, Kim SH, Surh YJ (2008) Curcumin attenuates dimethylnitrosamine-induced liver injury in rats through Nrf2-mediated induction of heme oxygenase-1. Food Chem Toxicol 46:1279–1287
Finck BN, Gropler MC, Chen Z et al (2006) Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARα regulatory pathway. Cell Metab 4:199–210
Friedrich M, Petzke KJ, Raederstorff D, Wolfram S, Klaus S (2012) Acute effects of epigallocatechin gallate from green tea on oxidation and tissue incorporation of dietary lipids in mice fed a high-fat diet. Int J Obes 36:735–743
Fu Y, Chen A (2006) The phyto-chemical (–)-epigallocatechin gallate suppresses gene expression of epidermal growth factor receptor in rat hepatic stellate cells in vitro by reducing the activity of Egr-1. Biochem Pharmacol 72:227–238
Fu Y, Zheng S, Lin J, Ryerse J, Chen A (2008a) Curcumin protects the rat liver from CCl4-caused injury and fibrogenesis by attenuating oxidative stress and suppressing inflammation. Mol Pharmacol 73:399–409
Fu Y, Zheng S, Lu SC, Chen A (2008b) Epigallocatechin-3-gallate inhibits growth of activated hepatic stellate cells by enhancing the capacity of glutathione synthesis. Mol Pharmacol 73:1465–1473
Fuhr U, Klittich K, Staib AH (1993) Inhibitory effect of grapefruit juice and its bitter principal, naringenin, on CYP1A2 dependent metabolism of caffeine in man. Brit J Clin Pharmacol 35:431–436
Fujioka T, Kondou T, Fukuhara A et al (2003) Efficacy of a glycyrrhizin suppository for the treatment of chronic hepatitis C: a pilot study. Hepatol Res 26:10–14
Fukushima Y, Kasuga M, Nakao K, Shimomura I, Matsuzawa Y (2009) Effects of coffee on inflammatory cytokine gene expression in mice fed high-fat diets. J Agric Food Chem 57:11100–11105
Furtado KS, Prado MG, Silva MAAE et al (2012) Coffee and caffeine protect against liver injury induced by thioacetamide in male Wistar rats. Basic Clin Pharmacol Toxicol 111:339–347
Gadacha W, Ben-Attia M, Bonnefont-Rousselot D et al (2009) Resveratrol opposite effects on rat tissue lipoperoxidation: pro-oxidant during day-time and antioxidant at night. Redox Rep 14:154–158
Ganai AA, Khan AA, Malik ZA, Farooqi H (2015) Genistein modulates the expression of NF-kappa B and MAPK (p-38 and ERK1/2), thereby attenuating d-galactosamine induced fulminant hepatic failure in Wistar rats. Toxicol Appl Pharmacol 283:139–146
Gao M, Liu D (2013) Resveratrol suppresses T0901317-induced hepatic fat accumulation in mice. AAPS J 15:744–752
Gao HY, Li GY, Lou MM et al (2012) Hepatoprotective effect of Matrine salvianolic acid B salt on carbon tetrachloride-induced hepatic fibrosis. J Inflamm 9:16
Gao M, Ma Y, Liu D (2013a) Rutin suppresses palmitic acids-triggered inflammation in macrophages and blocks high fat diet-induced obesity and fatty liver in mice. Pharm Res 30:2940–2950
Gao S, Duan XX, Wang X et al (2013b) Curcumin attenuates arsenic-induced hepatic injuries and oxidative stress in experimental mice through activation of Nrf2 pathway, promotion of arsenic methylation and urinary excretion. Food Chem Toxicol 59:739–747
Gao N, Qi B, Liu FJ et al (2014) Inhibition of baicalin on metabolism of phenacetin, a probe of CYP1A2, in human liver microsomes and in rats. PLoS One 9:e89752
García-Mediavilla V, Crespo I, Collado PS et al (2007) The anti-inflammatory flavones quercetin and kaempferol cause inhibition of inducible nitric oxide synthase, cyclooxygenase-2 and reactive C-protein, and down-regulation of the nuclear factor kappaB pathway in Chang Liver cells. Eur J Pharmacol 557:221–229
Garcia-Nino WR, Pedraza-Chaverri J (2014) Protective effect of curcumin against heavy metals-induced liver damage. Food Chem Toxicol 69:182–201
Geng JW, Peng W, Huang YG, Fan H, Li SD (2010) Ginsenoside-Rg1 from Panax notoginseng prevents hepatic fibrosis induced by thioacetamide in rats. Eur J Pharmacol 634:162–169
Ghazwani M, Zhang Y, Gao X et al (2014) Anti-fibrotic effect of thymoquinone on hepatic stellate cells. Phytomedicine 21:254–260
Giakoustidis DE, Giakoustidis AE, Iliadis S et al (2010) Attenuation of liver ischemia/reperfusion induced apoptosis by epigallocatechin-3-gallate via down-regulation of NF-κB and c-Jun expression. J Surg Res 159:720–728
Girish C, Koner BC, Jayanthi S et al (2009) Hepatoprotective activity of picroliv, curcumin and ellagic acid compared to silymarin on paracetamol induced liver toxicity in mice. Fund Clin Pharmacol 23:735–745
Goasduff T, Dreano Y, Guillois B, Menez JF, Berthou F (1996) Induction of liver and kidney CYP1A1/1A2 by caffeine in rat. Biochem Pharmacol 52:1915–1919
Godichaud S, Krisa S, Couronne B et al (2000) Deactivation of cultured human liver myofibroblasts by trans-resveratrol, a grapevine-derived polyphenol. Hepatology 31:922–931
Gonzalez FJ (2005) Role of cytochromes P450 in chemical toxicity and oxidative stress: studies with CYP2E1. Mutat Res 569:101–110
Gonzalez O, Fontanes V, Raychaudhuri S et al (2009) The heat shock protein inhibitor quercetin attenuates hepatitis C virus production. Hepatology 50:1756–1764
Gordillo-Bastidas D, Oceguera-Contreras E, Salazar-Montes A et al (2013) Nrf2 and Snail-1 in the prevention of experimental liver fibrosis by caffeine. World J Gastroenterol 19:9020–9033
Gressner OA, Lahme B, Rehbein K et al (2008) Pharmacological application of caffeine inhibits TGF-beta-stimulated connective tissue growth factor expression in hepatocytes via PPARγ and SMAD2/3-dependent pathways. J Hepatol 49:758–767
Gui M, Zhang YF, Xiao ZY et al (2007) Inhibitory effect of emodin on tissue inhibitor of metalloproteinases-1 (TIMP-1) expression in rat hepatic stellate cells. Dig Dis Sci 52:200–207
Gum SI, Cho MK (2013a) The amelioration of N-acetyl-p-benzoquinone imine toxicity by ginsenoside Rg3: the role of Nrf2-mediated detoxification and Mrp1/Mrp3 transports. Oxid Med Cell Longev 2013:957947
Gum SI, Cho MK (2013b) Korean red ginseng extract prevents APAP-induced hepatotoxicity through metabolic enzyme regulation: the role of ginsenoside Rg3, a protopanaxadiol. Liver Int 33:1071–1084
Gumpricht E, Dahl R, Devereaux MW, Sokol RJ (2005) Licorice compounds glycyrrhizin and 18beta-glycyrrhetinic acid are potent modulators of bile acid-induced cytotoxicity in rat hepatocytes. J Biol Chem 280:10556–10563
Guo HX, Liu DH, Ma Y et al (2009) Long-term baicalin administration ameliorates metabolic disorders and hepatic steatosis in rats given a high-fat diet. Acta Pharmacol Sin 30:1505–1512
Haddad Y, Vallerand D, Brault A, Haddad PS (2011) Antioxidant and hepatoprotective effects of silibinin in a rat model of nonalcoholic steatohepatitis. Evid Based Complement Altern Med 2011:nep164
Hafez MM, Al-Harbi NO, Al-Hoshani AR et al (2015) Hepato-protective effect of rutin via IL-6/STAT3 pathway in CCl4-induced hepatotoxicity in rats. Biol Res 48:30
Han EH, Kim HG, Choi JH et al (2012) Capsaicin induces CYP3A4 expression via pregnane X receptor and CCAAT/enhancer-binding protein beta activation. Mol Nutr Food Res 56:797–809
Hasan SK, Khan R, Ali N et al (2015) 18-beta Glycyrrhetinic acid alleviates 2-acetylaminofluorene-induced hepatotoxicity in Wistar rats: Role in hyperproliferation, inflammation and oxidative stress. Hum Exp Toxicol 34:628–641
Hassan MH, Edfawy M, Mansour A, Hamed AA (2012) Antioxidant and antiapoptotic effects of capsaicin against carbon tetrachloride-induced hepatotoxicity in rats. Toxicol Ind Health 28:428–438
He N, Edeki T (2004) The inhibitory effects of herbal components on CYP2C9 and CYP3A4 catalytic activities in human liver microsomes. Am J Ther 11:206–212
He N, Xie HG, Collins X, Edeki T, Yan Z (2006) Effects of individual ginsenosides, ginkgolides and flavonoids on CYP2C19 and CYP2D6 activity in human liver microsomes. Clin Exp Pharmacol Physiol 33:813–815
He Q, Zhang J, Chen F et al (2010) An anti-ROS/hepatic fibrosis drug delivery system based on salvianolic acid B loaded mesoporous silica nanoparticles. Biomaterials 31:7785–7796
Heeba GH, Mahmoud ME (2014) Therapeutic potential of morin against liver fibrosis in rats: modulation of oxidative stress, cytokine production and nuclear factor kappa B. Environ Toxicol Pharmacol 37:662–671
Heeboll S, Thomsen KL, Clouston A et al (2015) Effect of resveratrol on experimental non-alcoholic steatohepatitis. Pharmacol Res 95–96:34–41
Higashi N, Kohjima M, Fukushima M et al (2005) Epigallocatechin-3-gallate, a green-tea polyphenol, suppresses Rho signaling in TWNT-4 human hepatic stellate cells. J Lab Clin Med 145:316–322
Hirsova P, Kolouchova G, Dolezelova E et al (2012) Epigallocatechin gallate enhances biliary cholesterol secretion in healthy rats and lowers plasma and liver cholesterol in ethinylestradiol-treated rats. Eur J Pharmacol 691:38–45
Ho PC, Saville DJ, Wanwimolruk S (2001) Inhibition of human CYP3A4 activity by grapefruit flavonoids, furanocoumarins and related compounds. J Pharm Pharm Sci 4:217–227
Hou XY, Xu SQ, Maitland-Toolan KA et al (2008) SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem 283:20015–20026
Hou YL, Tsai YH, Lin YH, Chao JCJ (2014) Ginseng extract and ginsenoside Rb1 attenuate carbon tetrachloride-induced liver fibrosis in rats. BMC Complement Altern Med 14:415
Hsu WC, Ng LT, Wu TH et al (2012) Characteristics and antioxidant activities of silymarin nanoparticles. J Nanosci Nanotechno 12:2022–2027
Huang CF, Qiao XB, Dong B (2011) Neonatal exposure to genistein ameliorates high-fat diet-induced non-alcoholic steatohepatitis in rats. Brit J Nutr 106:105–113
Huang CS, Lii CK, Lin AH et al (2013) Protection by chrysin, apigenin, and luteolin against oxidative stress is mediated by the Nrf2-dependent up-regulation of heme oxygenase 1 and glutamate cysteine ligase in rat primary hepatocytes. Arch Toxicol 87:167–178
Hwang DH, Kim YI, Cho KH et al (2014) A novel solid dispersion system for natural product-loaded medicine: silymarin-loaded solid dispersion with enhanced oral bioavailability and hepatoprotective activity. J Microencapsul 31:619–626
Ikeda K (2007) Glycyrrhizin injection therapy prevents hepatocellular carcinogenesis in patients with interferon-resistant active chronic hepatitis C. Hepatol Res 37:S287–S293
Ikeda K, Kawamura Y, Kobayashi M et al (2014) Prevention of disease progression with anti-inflammatory therapy in patients with HCV-related cirrhosis: a Markov model. Oncology 86:295–302
Jamal MH, Ali H, Dashti A et al (2015) Effect of epigallocatechin gallate on uncoupling protein 2 in acute liver injury. Int J Clin Exp Pathol 8:649–654
James KD, Forester SC, Lambert JD (2015) Dietary pretreatment with green tea polyphenol, (–)-epigallocatechin-3-gallate reduces the bioavailability and hepatotoxicity of subsequent oral bolus doses of (–)-epigallocatechin-3-gallate. Food Chem Toxicol 76:103–108
Jeong HG, You HJ, Park SJ et al (2002) Hepatoprotective effects of 18beta-glycyrrhetinic acid on carbon tetrachloride-induced liver injury: inhibition of cytochrome P450 2E1 expression. Pharmacol Res 46:221–227
Ji GY, Yang QH, Hao J et al (2011) Anti-inflammatory effect of genistein on non-alcoholic steatohepatitis rats induced by high fat diet and its potential mechanisms. Int Immunopharmacol 11:762–768
Ji L, Jiang P, Lu B, Sheng Y, Wang X, Wang Z (2013) Chlorogenic acid, a dietary polyphenol, protects acetaminophen-induced liver injury and its mechanism. J Nutr Biochem 24:1911–1919
Jia Y, Kim S, Kim J et al (2015) Ursolic acid improves lipid and glucose metabolism in high-fat-fed C57BL/6J mice by activating peroxisome proliferator-activated receptor alpha and hepatic autophagy. Mol Nutr Food Res 59:344–354
Jiang F, Parsons CJ, Stefanovic B (2006) Gene expression profile of quiescent and activated rat hepatic stellate-cells implicates Wnt signaling pathway in activation. J Hepatol 45:401–409
Jin YR, Jin JL, Li CH, Piao XX, Jin NG (2012) Ursolic acid enhances mouse liver regeneration after partial hepatectomy. Pharm Biol 50:523–528
Jin SH, Yang JH, Shin BY et al (2013) Resveratrol inhibits LXR alpha-dependent hepatic lipogenesis through novel antioxidant Sestrin2 gene induction. Toxicol Appl Pharmacol 271:95–105
Jin S, Chang C, Zhang L et al (2015) Chlorogenic acid improves late diabetes through adiponectin receptor signaling pathways in db/db mice. PLoS One 10:e0120842
Johnson S, Koh WP, Wang RW et al (2011) Coffee consumption and reduced risk of hepatocellular carcinoma: findings from the Singapore Chinese Health Study. Cancer Cause Control 22:503–510
Jung TW, Hwang HJ, Hong HC et al (2014) Resolvin D1 reduces ER stress-induced apoptosis and triglyceride accumulation through JNK pathway in HepG2 cells. Mol Cellr Endocrinol 391:30–40
Kaibori M, Yanagida H, Nakanishi H et al (2006) Hepatocyte growth factor stimulates the induction of cytokine-induced neutrophil chemoattractant through the activation of NF-κB in rat hepatocytes. J Surg Res 130:88–93
Kang EH, Kown TY, Oh GT et al (2006) The flavonoid ellagic acid from a medicinal herb inhibits host immune tolerance induced by the hepatitis B virus-e antigen. Antivir Res 72:100–106
Kang KS, Kim HY, Yamabe N, Park JH, Yokozawa T (2007) Preventive effect of 20(S)-ginsenoside Rg3 against lipopolysaccharide-induced hepatic and renal injury in rats. Free Radic Res 41:1181–1188
Kang NJ, Lee KM, Kim JH et al (2008) Inhibition of gap junctional intercellular communication by the green tea polyphenol (–)-epigallocatechin gallate in normal rat liver epithelial cells. J Agric Food Chem 56:10422–10427
Kang JH, Goto T, Han IS et al (2010) Dietary capsaicin reduces obesity-induced insulin resistance and hepatic steatosis in obese mice fed a high-fat diet. Obesity 18:780–787
Kang LJ, Choi YJ, Lee SG (2013) Stimulation of TRAF6/TAK1 degradation and inhibition of JNK/AP-1 signalling by ginsenoside Rg3 attenuates hepatitis B virus replication. Int J Biochem Cell B 45:2612–2621
Kapoor R, Kakkar P (2012) Protective role of morin, a flavonoid, against high glucose induced oxidative stress mediated apoptosis in primary rat hepatocytes. PLoS One 7:e41663
Karimian G, Buist-Homan M, Faber KN, Moshage H (2012) Pertussis toxin, an inhibitor of G(αi) PCR, inhibits bile acid- and cytokine-induced apoptosis in primary rat hepatocytes. PLoS One 7:e43156
Kaviarasan S, Sundarapandiyan R, Anuradha CV (2008) Epigallocatechin gallate, a green tea phytochemical, attenuates alcohol-induced hepatic protein and lipid damage. Toxicol Mech Method 18:645–652
Kawada N, Seki S, Inoue M, Kuroki T (1998) Effect of antioxidants, resveratrol, quercetin, and N-acetylcysteine, on the functions of cultured rat hepatic stellate cells and Kupffer cells. Hepatology 27:1265–1274
Kazmi I, Narooka AR, Afzal M et al (2013) Anticancer effect of ursolic acid stearoyl glucoside in chemically induced hepatocellular carcinoma. J Physiol Biochem 69:687–695
Khalaf N, White D, Kanwal F et al (2015) Coffee and caffeine are associated with decreased risk of advanced hepatic fibrosis among patients with hepatitis C. Clin Gastroenterol Hepatol. doi:10.1016/j.cgh.2015.01.030
Khan RA, Khan MR, Sahreen S (2012) CCl4-induced hepatotoxicity: protective effect of rutin on p53, CYP2E1 and the antioxidative status in rat. BMC Complement Altern Med 12:178
Khan J, Saraf S, Saraf S (2015) Preparation and evaluation of luteolin-phospholipid complex as an effective drug delivery tool against GalN/LPS induced liver damage. Pharm Dev Technol. doi:10.3109/10837450.2015.1022786
Kim SJ, Lee SM (2012) Effect of baicalin on toll-like receptor 4-mediated ischemia/reperfusion inflammatory responses in alcoholic fatty liver condition. Toxicol Appl Pharmacol 258:43–50
Kim JY, Lee SY, Kim DH et al (2002) Effects of flavonoids isolated from Scutellariae radix on cytochrome P-450 activities in human liver microsomes. J Toxicol Env Heal A 65:373–381
Kim KA, Lee JS, Park HJ et al (2004) Inhibition of cytochrome P450 activities by oleanolic acid and ursolic acid in human liver microsomes. Life Sci 74:2769–2779
Kim JH, Choi SH, Kim J et al (2009) Differential regulation of the hydrogen-peroxide-induced inhibition of gap-junction intercellular communication by resveratrol and butylated hydroxyanisole. Mutat Res 671:40–44
Kim K, Kim KH, Kim HY et al (2010a) Curcumin inhibits hepatitis C virus replication via suppressing the Akt-SREBP-1 pathway. FEBS Lett 584:707–712
Kim MH, Kang KS, Lee YS (2010b) The inhibitory effect of genistein on hepatic steatosis is linked to visceral adipocyte metabolism in mice with diet-induced non-alcoholic fatty liver disease. Brit J Nutr 104:1333–1342
Kim SJ, Moon YJ, Lee SM (2010c) Protective effects of baicalin against ischemia/reperfusion injury in rat liver. J Nat Prod 73:2003–2008
Kim M, Yang SG, Kim JM et al (2012) Silymarin suppresses hepatic stellate cell activation in a dietary rat model of non-alcoholic steatohepatitis: analysis of isolated hepatic stellate cells. Int J Mol Med 30:473–479
Kim DH, Chung JH, Yoon JS et al (2013) Ginsenoside Rd inhibits the expressions of iNOS and COX-2 by suppressing NF-kappaB in LPS-stimulated RAW264.7 cells and mouse liver. J Ginseng Res 37:54–63
Kim S, Lee HS, Ji JH et al (2015) Hepatitis B virus X protein activates the ATM-Chk2 pathway and delays the cell cycle progression. J Gen Virol. doi:10.1099/vir.0.000150
Kimura M, Inoue H, Hirabayashi K, Natsume H, Ogihara M (2001) Glycyrrhizin and some analogues induce growth of primary cultured adult rat hepatocytes via epidermal growth factor receptors. Eur J Pharmacol 431:151–161
Kochi T, Shimizu M, Terakura D et al (2014) Non-alcoholic steatohepatitis and preneoplastic lesions develop in the liver of obese and hypertensive rats: suppressing effects of EGCG on the development of liver lesions. Cancer Lett 342:60–69
Koriem KM, Soliman RE (2014) Chlorogenic and caftaric acids in liver toxicity and oxidative stress induced by methamphetamine. J Toxicol 2014:583494
Krishnan TR, Velusamy P, Srinivasan A et al (2014) EGCG mediated downregulation of NF-AT and macrophage infiltration in experimental hepatic steatosis. Exp Gerontol 57:96–103
Kuribayashi M, Asamoto M, Suzuki S et al (2006) Lack of modification of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx) rat hepatocarcinogenesis by caffeine, a CYP1A2 inducer, points to complex counteracting influences. Cancer Lett 232:289–299
Kuroda N, Inoue K, Ikeda T et al (2014) Apoptotic response through a high mobility box 1 protein-dependent mechanism in LPS/GalN-induced mouseliver failure and glycyrrhizin-mediated inhibition. PLoS One 9:e92884
Kwon EY, Jung UJ, Park T, Yun JW, Choi MS (2015) Luteolin attenuates hepatic steatosis and insulin resistance through the interplay between the liver and adipose tissue in mice with diet-induced obesity. Diabetes 64:1658–1669
Lanteri R, Acquaviva R, Di Giacomo C et al (2007) Rutin in rat liver ischemia/reperfusion injury: effect on DDAH/NOS pathway. Microsurgery 27:245–251
Lee CH, Park SW, Kim YS et al (2007a) Protective mechanism of glycyrrhizin on acute liver injury induced by carbon tetrachloride in mice. Biol Pharm Bull 30:1898–1904
Lee KJ, Choi JH, Jeong HG (2007b) Hepatoprotective and antioxidant effects of the coffee diterpenes kahweol and cafestol on carbon tetrachloride-induced liver damage in mice. Food Chem Toxicol 45:2118–2125
Lee HS, Jung KH, Park IS et al (2009) Protective effect of morin on dimethylnitrosamine-induced hepatic fibrosis in rats. Dig Dis Sci 54:782–788
Lee ES, Shin MO, Yoon S, Moon JO (2010a) Resveratrol inhibits dimethylnitrosamine-induced hepatic fibrosis in rats. Arch Pharm Res 33:925–932
Lee KM, Hwang MK, Lee DE, Lee KW, Lee HJ (2010b) Protective effect of quercetin against arsenite-induced COX-2 expression by targeting PI3K in rat liver epithelial cells. J Agric Food Chem 58:5815–5820
Lee TY, Lee KC, Chang HH (2010c) Modulation of the cannabinoid receptors by andrographolide attenuates hepatic apoptosis following bile duct ligation in rats with fibrosis. Apoptosis 15:904–914
Lee WC, Jung HA, Choi JS, Kim YS, Lee SM (2011) Protective effects of luteolin against apoptotic liver damage induced by d-galactosamine/lipopolysaccharide in mice. J Nat Prod 74:1916–1921
Lee HI, McGregor RA, Choi MS et al (2013a) Low doses of curcumin protect alcohol-induced liver damage by modulation of the alcohol metabolic pathway, CYP2E1 and AMPK. Life Sci 93:693–699
Lee MH, Lee BH, Lee S, Choi C (2013b) Reduction of hepatitis A virus on FRhK-4 cells treated with Korean red ginseng extract and ginsenosides. J Food Sci 78:M1412–M1415
Lee JC, Tseng CK, Young KC et al (2014a) Andrographolide exerts anti-hepatitis C virus activity by up-regulating haeme oxygenase-1 via the p38 MAPK/Nrf2 pathway in human hepatoma cells. Brit J Pharmacol 171:237–252
Lee JH, Won JH, Choi JM et al (2014b) Protective effect of ellagic acid on concanavalin A-induced hepatitis via toll-like receptor and mitogen-activated protein kinase/nuclear factor kappaB signaling pathways. J Agric Food Chem 62:10110–10117
Lee KW, Thiyagarajan V, Sie HW et al (2014c) Synergistic effect of natural compounds on the fatty acid-induced autophagy of activated hepatic stellate cells. J Nutr Biochem 25:903–913
Lee TY, Chang HH, Wen CK, Huang TH, Chang YS (2014d) Modulation of thioacetamide-induced hepatic inflammations, angiogenesis and fibrosis by andrographolide in mice. J Ethnopharmacol 158(Pt A):423–430
Lee SJ, Kang JH, Iqbal W, Kwon OS (2015) Proteomic analysis of mice fed methionine and choline deficient diet reveals marker proteins associated with steatohepatitis. PLoS One. doi:10.1371/journal.pone.0120577
Lemley CO, Wilson ME (2010) Effect of cytochrome P450 and aldo-keto reductase inhibitors on progesterone inactivation in primary bovine hepatic cell cultures. J Dairy Sci 93:4613–4624
Li JM, Li YC, Kong LD, Hu QH (2010) Curcumin inhibits hepatic protein-tyrosine phosphatase 1B and prevents hypertriglyceridemia and hepatic steatosis in fructose-fed rats. Hepatology 51:1555–1566
Li S, Wang LN, Yan XC et al (2012) Salvianolic acid B attenuates rat hepatic fibrosis via downregulating angiotensin II signaling. Evid Based Complement Altern Med. doi:10.1155/2012/160726
Li G, Chen JB, Wang C et al (2013a) Curcumin protects against acetaminophen-induced apoptosis in hepatic injury. World J Gastroenterol 19:7440–7446
Li Q, Li L, Wang F et al (2013b) Dietary capsaicin prevents nonalcoholic fatty liver disease through transient receptor potential vanilloid 1-mediated peroxisome proliferator-activated receptor delta activation. Pflug Arch Eur J Phys 465:1303–1316
Li Y, Gao C, Shi Y et al (2013c) Carbon monoxide alleviates ethanol-induced oxidative damage and inflammatory stress through activating p38 MAPK pathway. Toxicol Appl Pharmacol 273:53–58
Li J, Pan Y, Kan M et al (2014a) Hepatoprotective effects of berberine on liver fibrosis via activation of AMP-activated protein kinase. Life Sci 98:24–30
Li JP, Gao Y, Chu SF et al (2014b) Nrf2 pathway activation contributes to anti-fibrosis effects of ginsenoside Rg1 in a rat model of alcohol-and CCl4-induced hepatic fibrosis. Acta Pharmacol Sin 35:1031–1044
Li L, Hai J, Li Z et al (2014c) Resveratrol modulates autophagy and NF-κB activity in a murine model for treating non-alcoholic fatty liver disease. Food Chem Toxicol 63:166–173
Li MZ, Lu Y, Hu Y et al (2014d) Salvianolic acid B protects against acute ethanol-induced liver injury through SIRT1-mediated deacetylation of p53 in rats. Toxicol Lett 228:67–74
Li S, Liao X, Meng F et al (2014e) Therapeutic role of ursolic acid on ameliorating hepatic steatosis and improving metabolic disorders in high-fat diet-induced non-alcoholic fatty liver disease rats. PLoS One 9:e86724
Li X, Hu J, Wang B et al (2014f) Inhibitory effects of herbal constituents on P-glycoprotein in vitro and in vivo: herb-drug interactions mediated via P-gp. Toxicol Appl Pharmacol 275:163–175
Li J, Li XX, Xu WH et al (2015a) Antifibrotic effects of luteolin on hepatic stellate cells and liver fibrosis by targeting AKT/mTOR/p70S6K and TGF beta/Smad signalling pathways. Liver Int 35:1222–1233
Li JS, Wang WJ, Sun Y, Zhang YH, Zheng L (2015b) Ursolic acid inhibits the development of nonalcoholic fatty liver disease by attenuating endoplasmic reticulum stress. Food Funct 6:1643–1651
Li X, Xu Z, Wang S et al (2015c) Emodin ameliorates hepatic steatosis through endoplasmic reticulum stress-sterol regulatory element binding protein 1 c pathway in liquid-fructose feeding rats. Hepatol Res. doi:10.1111/hepr.12538
Lian N, Jiang Y, Zhang F et al (2015) Curcumin regulates cell fate and metabolism by inhibiting hedgehog signaling in hepatic stellate cells. Lab Invest 95:790–803
Lin J, Chen A (2008) Activation of peroxisome proliferator-activated receptor-gamma by curcumin blocks the signaling pathways for PDGF and EGF in hepatic stellate cells. Lab Invest 88:529–540
Lin YL, Lee TF, Huang YJ, Huang YT (2006) Antiproliferative effect of salvianolic acid A on rat hepatic stellate cells. J Pharm Pharmacol 58:933–939
Lin CM, Lee JF, Chiang LL et al (2012a) The Protective effect of curcumin on ischemia-reperfusion-induced liver injury. Transpl Proc 44:974–977
Lin TK, Huang LT, Huang YH et al (2012b) The effect of the red wine polyphenol resveratrol on a rat model of biliary obstructed cholestasis: involvement of anti-apoptotic signalling, mitochondrial biogenesis and the induction of autophagy. Apoptosis 17:871–879
Lin YT, Wu YH, Tseng CK et al (2013) Green Tea phenolic epicatechins inhibit hepatitis C virus replication via cycloxygenase-2 and attenuate virus-induced inflammation. PLoS One. doi:10.1371/journal.pone.0054466
Lin SY, Wang YY, Chen WY et al (2014a) Beneficial effect of quercetin on cholestatic liver injury. J Nutr Bioch 25:1183–1195
Lin X, Zhang SJ, Huang RB et al (2014b) Protective effect of genistein on lipopolysaccharide/d-galactosamine-induced hepatic failure in mice. Biol Pharm Bull 37:625–632
Lin CK, Tseng CK, Chen KH et al (2015a) Betulinic acid exerts anti-hepatitis C virus activity via the suppression of NF-κB and MAPK-ERK1/2-mediated cyclooxygenase-2 expression. Brit J Pharmacol. doi:10.1111/bph.13233
Lin M, Zhai X, Wang G et al (2015b) Salvianolic acid B protects against acetaminophen hepatotoxicity by inducing Nrf2 and phase II detoxification gene expression via activation of the PI3K and PKC signaling pathways. J Pharm Sci 127:203–210
Liu C, Liu P, Hu Y, Zhu D (2002a) Effects of salvianolic acid-B on TGF-β1 stimulated hepatic stellate cell activation and its intracellular signaling. Zhonghua Yi Xue Za Zhi 82:1267–1272
Liu P, Hu YY, Liu C et al (2002b) Clinical observation of salvianolic acid B in treatment of liver fibrosis in chronic hepatitis B. World J Gastroenterol 8:679–685
Liu P, Liu CH, Wang HN, Hu YY, Liu C (2002c) Effect of salvianolic acid B on collagen production and mitogen-activated protein kinase activity in rat hepatic stellate cells. Acta Pharmacol Sin 23:733–738
Liu XJ, Yang L, Mao YQ et al (2002d) Effects of the tyrosine protein kinase inhibitor genistein on the proliferation, activation of cultured rat hepatic stellate cells. World J Gastroenterol 8:739–745
Liu X, Wang W, Hu H et al (2006) Smad3 specific inhibitor, naringenin, decreases the expression of extracellular matrix induced by TGF-β1 in cultured rat hepatic stellate cells. Pharm Res 23:82–89. doi:10.1007/s11095-005-9043-5
Liu LL, Gong LK, Wang H et al (2007) Baicalin protects mouse from concanavalin A-induced liver injury through inhibition of cytokine production and hepatocyte apoptosis. Liver Int 27:582–591
Liu S, Hou W, Yao P et al (2012) Heme oxygenase-1 mediates the protective role of quercetin against ethanol-induced rat hepatocytes oxidative damage. Toxicol In Vitro 26:74–80
Liu G, Zhang Y, Liu C et al (2014a) Luteolin alleviates alcoholic liver disease induced by chronic and binge ethanol feeding in mice. J Nutr 144:1009–1015
Liu LQ, Fan ZQ, Tang YF, Ke ZJ (2014b) The resveratrol attenuates ethanol-induced hepatocyte apoptosis via inhibiting ER-related caspase-12 activation and PDE activity in vitro. Alcohol Clin Exp Res 38:683–693
Liu Q, Qian Y, Chen F et al (2014c) EGCG attenuates pro-inflammatory cytokines and chemokines production in LPS-stimulated L02 hepatocyte. Acta Biochim Biophys Sin 46:31–39
Liu Y, Chen XL, Qiu MC et al (2014d) Emodin ameliorates ethanol-induced fatty liver injury in mice. Pharmacology 94:71–77
Liu AD, Wang WJ, Fang HS et al (2015a) Baicalein protects against polymicrobial sepsis-induced liver injury via inhibition of inflammation and apoptosis in mice. Eur J Pharmacol 748:45–53
Liu CM, Ma JQ, Xie WR et al (2015b) Quercetin protects mouse liver against nickel-induced DNA methylation and inflammation associated with the Nrf2/HO-1 and p38/STAT1/NF-κB pathway. Food Chem Toxicol 82:19–26
Lo YT, Tsai YH, Wu SJ, Chen JR, Chao JCJ (2011) Ginsenoside Rb1 inhibits cell activation and liver fibrosis in rat hepatic stellate cells. J Med Food 14:1135–1143
Lulu SS, Thabitha A, Vino S, Priya AM, Rout M (2015) Naringenin and quercetin—potential anti-HCV agents for NS2 protease targets. Nat Prod Res. doi:10.1080/14786419.2015.1020490
Lv Z, Xu L (2012) Salvianolic acid B inhibits ERK and p38 MAPK signaling in TGF-β1-stimulated human hepatic stellate cell line (LX-2) via distinct pathways. Evid Based Complement Altern Med 2012:960128
Lv X, Chen Z, Li J et al (2010) Caffeine protects against alcoholic liver injury by attenuating inflammatory response and oxidative stress. Inflamm Res 59:635–645
Lv Y, Zhang B, Xing G, Wang F, Hu Z (2013) Protective effect of naringenin against acetaminophen-induced acute liver injury in metallothionein (MT)-null mice. Food Funct 4:297–302
Lv Y, Lei N, Wang D et al (2014) Protective effect of curcumin against cytomegalovirus infection in Balb/c mice. Environ Toxicol Pharmacol 37:1140–1147
Ma JQ, Ding J, Zhang L, Liu CM (2014) Ursolic acid protects mouse liver against CCl4-induced oxidative stress and inflammation by the MAPK/NF-κB pathway. Environ Toxicol Pharmacol 37:975–983
Ma JQ, Ding J, Zhang L, Liu CM (2015a) Protective effects of ursolic acid in an experimental model of liver fibrosis through Nrf2/ARE pathway. Clin Res Hepatol Gastroenterol 39:188–197
Ma Y, Gao M, Liu D (2015b) Chlorogenic acid improves high fat diet-induced hepatic steatosis and insulin resistance in mice. Pharm Res 32:1200–1209
Mabuchi A, Wake K, Marlini M, Watanabe H, Wheatley AM (2009) Protection by glycyrrhizin against warm ischemia-reperfusion-induced cellular injury and derangement of the microcirculatory blood flow in the rat liver. Microcirculation 16:364–376
Mach CM, Chen JH, Mosley SA, Kurzrock R, Smith JA (2010) Evaluation of liposomal curcumin cytochrome p450 metabolism. Anticancer Res 30:811–814
MadanKumar P, NaveenKumar P, Manikandan S, Devaraj H, NiranjaliDevaraj S (2014) Morin ameliorates chemically induced liver fibrosis in vivo and inhibits stellate cell proliferation in vitro by suppressing Wnt/beta-catenin signaling. Toxicol Appl Pharmacol 277:210–220
MadanKumar P, NaveenKumar P, Devaraj H, NiranjaliDevaraj S (2015) Morin, a dietary flavonoid, exhibits anti-fibrotic effect and induces apoptosis of activated hepatic stellate cells by suppressing canonical NF-κB signaling. Biochimie 110:107–118
Madrigal-Santillan E, Madrigal-Bujaidar E, Alvarez-Gonzalez I et al (2014) Review of natural products with hepatoprotective effects. World J Gastroenterol 20:14787–14804
Mandegary A, Saeedi A, Eftekhari A, Montazeri V, Sharif E (2013) Hepatoprotective effect of silyamarin in individuals chronically exposed to hydrogen sulfide; modulating influence of TNF-α cytokine genetic polymorphism. Daru 21:28
Manjunatha H, Srinivasan K (2007) Hypolipidemic and antioxidant effects of curcumin and capsaicin in high-fat-fed rats. Can J Physiol Pharmacol 85:588–596
Manns MP, Wedemeyer H, Singer A et al (2012) Glycyrrhizin in patients who failed previous interferon alpha-based therapies: biochemical and histological effects after 52 weeks. J Viral Hepat 19:537–546
Marcolin E, San-Miguel B, Vallejo D et al (2012) Quercetin treatment ameliorates inflammation and fibrosis in mice with nonalcoholic steatohepatitis. J Nutr 142:1821–1828
Marcolin E, Forgiarini LF, Rodrigues G et al (2013) Quercetin decreases liver damage in mice with non-alcoholic steatohepatitis. Basic Clin Pharmacol Toxicol 112:385–391
Martínez-Flórez S, Gutiérrez-Fernández B, Sánchez-Campos S, González-Gallego J, Tuñón MJ (2005) Quercetin attenuates nuclear factor-kappaB activation and nitric oxide production in interleukin-1beta-activated rat hepatocytes. J Nutr 135:1359–1365
Masuda T, Ueno Y (1984) Microsomal transformation of emodin into a direct mutagen. Mutat Res 125:135–144
Matsumoto Y, Matsuura T, Aoyagi H et al (2013) Antiviral activity of glycyrrhizin against hepatitis C virus in vitro. PLoS One 8:e68992
Mbimba T, Awale P, Bhatia D et al (2012) Alteration of hepatic proinflammatory cytokines is involved in the resveratrol-mediated chemoprevention of chemically-induced hepatocarcinogenesis. Curr Pharm Biotechnol 13:229–234
McCarty MF, Barroso-Aranda J, Contreras F (2009) Genistein and phycocyanobilin may prevent hepatic fibrosis by suppressing proliferation and activation of hepatic stellate cells. Med Hypotheses 72:330–332
Meng B, Gao W, Wei J et al (2013) Quercetin reduces serum homocysteine level in rats fed a methionine-enriched diet. Nutrition 29:661–666
Merwid-Lad A, Trocha M, Chlebda-Sieragowska E et al (2014) The impact of morin, a natural flavonoid, on cyclophosphamide-induced changes in the oxidative stress parameters in rat livers. Adv Clin Exp Med 23:505–509
Misaka S, Kawabe K, Onoue S et al (2013) Effects of green tea catechins on cytochrome P450 2B6, 2C8, 2C19, 2D6 and 3A activities in human liver and intestinal microsomes. Drug Metab Pharmacokinet 28:244–249
Modi AA, Feld JJ, Park Y et al (2010) Increased caffeine consumption is associated with reduced hepatic fibrosis. Hepatology 51:201–209
Molloy JW, Calcagno CJ, Williams CD et al (2012) Association of coffee and caffeine consumption with fatty liver disease, nonalcoholic steatohepatitis, and degree of hepatic fibrosis. Hepatology 55:429–436
Mori Y, Tatematsu K, Koide A et al (2006) Modification by curcumin of mutagenic activation of carcinogenic N-nitrosamines by extrahepatic cytochromes P-4502B1 and 2E1 in rats. Cancer Sci 97:896–904
Moro T, Shimoyama Y, Kushida M et al (2008) Glycyrrhizin and its metabolite inhibit Smad3-mediated type I collagen gene transcription and suppress experimental murine liver fibrosis. Life Sci 83:531–539
Motawi TK, Teleb ZA, El-Boghdady NA, Ibrahim SA (2014) Effect of simvastatin and naringenin coadministration on rat liver DNA fragmentation and cytochrome P450 activity: an in vivo and in vitro study. J Physiol Biochem 70:225–237
Nafees S, Rashid S, Ali N, Hasan SK, Sultana S (2015) Rutin ameliorates cyclophosphamide induced oxidative stress and inflammation in Wistar rats: role of NFkappaB/MAPK pathway. Chem Biol Interact 231:98–107
Nakamura M, Saito H, Ikeda M et al (2010) An antioxidant resveratrol significantly enhanced replication of hepatitis C virus. World J Gastroenterol 16:184–192
Narayanan A, Kehn-Hall K, Senina S et al (2012) Curcumin inhibits Rift Valley fever virus replication in human cells. J Biol Chem 287:33198–33214
National Toxicology P (2001) NTP toxicology and carcinogenesis studies of EMODIN (CAS NO. 518-82-1) feed studies in F344/N rats and B6C3F1 mice. Natl Toxicol Program Tech Rep Ser 493:1–278
National Toxicology P (2011) Toxicology and carcinogenesis studies of milk thistle extract (CAS No. 84604-20-6) in F344/N rats and B6C3F1 mice (feed studies). Natl Toxicol Program Tech Rep Ser 565:1–177
Ni HM, Du K, You M, Ding WX (2013) Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am J Pathol 183:1815–1825
Nicoletti NF, Rodrigues-Junior V, Santos AA Jr et al (2014) Protective effects of resveratrol on hepatotoxicity induced by isoniazid and rifampicin via SIRT1 modulation. J Nat Prod 77:2190–2195
Nishikawa T, Nakajima T, Moriguchi M et al (2006) A green tea polyphenol, epigalocatechin-3-gallate, induces apoptosis of human hepatocellular carcinoma, possibly through inhibition of Bcl-2 family proteins. J Hepatol 44:1074–1082
Nseir W, Nassar F, Assy N (2010) Soft drinks consumption and nonalcoholic fatty liver disease. World J Gastroenterol 16:2579–2588
Ogiku M, Kono H, Hara M, Tsuchiya M, Fujii H (2011) Glycyrrhizin prevents liver injury by inhibition of high-mobility group box 1 production by Kupffer cells after ischemia-reperfusion in rats. J Pharmacol Exp Ther 339:93–98
Okazaki K, Okazaki S, Nakamura H et al (2002) A repeated 28-day oral dose toxicity study of genistein in rats, based on the ‘Enhanced OECD Test Guideline 407’ for screening endocrine-disrupting chemicals. Arch Toxicol 76:553–559
Orazizadeh M, Fakhredini F, Mansouri E, Khorsandi L (2014) Effect of glycyrrhizic acid on titanium dioxide nanoparticles-induced hepatotoxicity in rats. Chem Biol Interact 220:214–221
Orlent H, Hansen BE, Willems M et al (2006) Biochemical and histological effects of 26 weeks of glycyrrhizin treatment in chronic hepatitis C: a randomized phase II trial. J Hepatol 45:539–546
Othman MS, Safwat G, Aboulkhair M, Abdel Moneim AE (2014) The potential effect of berberine in mercury-induced hepatorenal toxicity in albino rats. Food Chem Toxicol 69:175–181
Pan PH, Lin SY, Wang YY et al (2014) Protective effects of rutin on liver injury induced by biliary obstruction in rats. Free Radic Biol Med 73:106–116
Pan QR, Ren YL, Liu WX et al (2015) Resveratrol prevents hepatic steatosis and endoplasmic reticulum stress and regulates the expression of genes involved in lipid metabolism, insulin resistance, and inflammation in rats. Nutr Res. doi:10.1016/j.nutres.2015.05.006
Panchal SK, Poudyal H, Brown L (2012) Quercetin ameliorates cardiovascular, hepatic, and metabolic changes in diet-induced metabolic syndrome in rats. J Nutr 142:1026–1032
Panchal SK, Ward L, Brown L (2013) Ellagic acid attenuates high-carbohydrate, high-fat diet-induced metabolic syndrome in rats. Eur J Nutr 52:559–568
Pari L, Gnanasoundari M (2006) Influence of naringenin on oxytetracycline mediated oxidative damage in rat liver. Basic Clin Pharmacol Toxicol 98:456–461
Pari L, Sivasankari R (2008) Effect of ellagic acid on cyclosporine A-induced oxidative damage in the liver of rats. Fundam Clin Pharmacol 22:395–401
Park HM, Kim SJ, Mun AR et al (2012) Korean red ginseng and its primary ginsenosides inhibit ethanol-induced oxidative injury by suppression of the MAPK pathway in TIB-73 cells. J Ethnopharmacol 141:1071–1076
Park SH, Baek SI, Yun J et al (2015) IRAK4 as a molecular target in the amelioration of innate immunity-related endotoxic shock and acute liver injury by chlorogenic acid. J Immunol 194:1122–1130
Parvez S, Tabassum H, Rehman H, Banerjee BD, Athar M, Raisuddin S (2006) Catechin prevents tamoxifen-induced oxidative stress and biochemical perturbations in mice. Toxicology 225:109–118
Patel N, Joseph C, Corcoran GB, Ray SD (2010) Silymarin modulates doxorubicin-induced oxidative stress, Bcl-xL and p53 expression while preventing apoptotic and necrotic cell death in the liver. Toxicol Appl Pharmacol 245:143–152
Peck MC, Fisher RF, Long SR (2006) Diverse flavonoids stimulate NodD1 binding to nod gene promoters in Sinorhizobium meliloti. J Bacteriol 188:5417–5427
Pekthong D, Martin H, Abadie C et al (2008) Differential inhibition of rat and human hepatic cytochrome P450 by Andrographis paniculata extract and andrographolide. J Ethnopharmacol 115:432–440
Pekthong D, Blanchard N, Abadie C et al (2009) Effects of Andrographis paniculata extract and andrographolide on hepatic cytochrome P450 mRNA expression and monooxygenase activities after in vivo administration to rats and in vitro in rat and human hepatocyte cultures. Chem Biol Interact 179:247–255
Peng XD, Dai LL, Huang CQ, He CM, Chen LJ (2009) Correlation between anti-fibrotic effect of baicalin and serum cytokines in rat hepatic fibrosis. World J Gastroenterol 15:4720–4725
Pietsch C, Hollender J, Dorusch F, Burkhardt-Holm P (2014) Cytotoxic effects of pentachlorophenol (PCP) and its metabolite tetrachlorohydroquinone (TCHQ) on liver cells are modulated by antioxidants. Cell Biol Toxicol 30:233–252
Pinlaor S, Prakobwong S, Hiraku Y et al (2010) Reduction of periductal fibrosis in liver fluke-infected hamsters after long-term curcumin treatment. Eur J Pharmacol 638:134–141
Pirola L, Frojdo S (2008) Resveratrol: one molecule, many targets. IUBMB Life 60:323–332
Pisonero-Vaquero S, Garcia-Mediavilla MV, Jorquera F et al (2014) Modulation of PI3K-LXRα-dependent lipogenesis mediated by oxidative/nitrosative stress contributes to inhibition of HCV replication by quercetin. Lab Invest 94:262–274
Polyak SJ, Morishima C, Lohmann V et al (2010) Identification of hepatoprotective flavonolignans from silymarin. Proc Natl Acad Sci USA 107:5995–5999
Ponmari G, Annamalai A, Gopalakrishnan VK, Lakshmi PT, Guruvayoorappan C (2014) NF-κB activation and proinflammatory cytokines mediated protective effect of Indigofera caerulea Roxb. on CCl4 induced liver damage in rats. Int Immunopharmacol 23:672–680
Poulsen MM, Larsen JO, Hamilton-Dutoit S et al (2012) Resveratrol up-regulates hepatic uncoupling protein 2 and prevents development of nonalcoholic fatty liver disease in rats fed a high-fat diet. Nutr Res 32:701–708
Prabhakar P, Reeta KH, Maulik SK, Dinda AK, Gupta YK (2014) Protective effect of thymoquinone against high-fructose diet-induced metabolic syndrome in rats. Eur J Nutr. doi:10.1007/s00394-014-0788-7
Pradeep K, Mohan CV, Gobianand K, Karthikeyan S (2007) Silymarin modulates the oxidant-antioxidant imbalance during diethylnitrosamine induced oxidative stress in rats. Eur J Pharmacol 560:110–116
Price RJ, Scott MP, Giddings AM et al (2008) Effect of butylated hydroxytoluene, curcumin, propyl gallate and thiabendazole on cytochrome P450 forms in cultured human hepatocytes. Xenobiotica 38:574–586
Primikyri A, Chatziathanasiadou MV, Karali E et al (2014) Direct binding of Bcl-2 family proteins by quercetin triggers its pro-apoptotic activity. ACS Chem Biol 9:2737–2741
Priya S, Sudhakaran PR (2008) Curcumin-induced recovery from hepatic injury involves induction of apoptosis of activated hepatic stellate cells. Indian J Biochem Biophys 45:317–325
Pu P, Gao DM, Mohamed S et al (2012a) Naringin ameliorates metabolic syndrome by activating AMP-activated protein kinase in mice fed a high-fat diet. Arch Biochem Biophys 518:61–70
Pu P, Wang XA, Salim M et al (2012b) Baicalein, a natural product, selectively activating AMPKα(2) and ameliorates metabolic disorder in diet-induced mice. Mol Cell Endocrinol 362:128–138
Qiang G, Yang X, Shi L et al (2015) Antidiabetic effect of salvianolic acid a on diabetic animal models via AMPK Activation and mitochondrial regulation. Cell Physiol Biochem 36:395–408
Qiao HX, Han HC, Hong DS et al (2011) Protective effects of baicalin on carbon tetrachloride induced liver injury by activating PPARγ and inhibiting TGFβ1. Pharm Biol 49:38–45
Qiu FR, Zhang R, Sun JG et al (2008) Inhibitory effects of seven components of danshen extract on catalytic activity of cytochrome P450 enzyme in human liver microsomes. Drug Metab Dispos 36:1308–1314
Qu Y, Chen WH, Zong L, Xu MY, Lu LG (2012) 18 alpha-Glycyrrhizin induces apoptosis and suppresses activation of rat hepatic stellate cells. Med Sci Monit 18:Br24–Br32
Qu Y, Zong L, Xu M, Dong Y, Lu L (2015) Effects of 18alpha-glycyrrhizin on TGF-β1/Smad signaling pathway in rats with carbon tetrachloride-induced liver fibrosis. Int J Clin Exp Pathol 8:1292–1301
Quan HY, Yuan HD, Jung MS et al (2012) Ginsenoside Re lowers blood glucose and lipid levels via activation of AMP-activated protein kinase in HepG2 cells and high-fat diet fed mice. Int J Mol Med 29:73–80
Quan HY, Kim do Y, Chung SH (2013a) Caffeine attenuates lipid accumulation via activation of AMP-activated protein kinase signaling pathway in HepG2 cells. BMB Rep 46:207–212
Quan HY, Kim do Y, Kim SJ et al (2013b) Betulinic acid alleviates non-alcoholic fatty liver by inhibiting SREBP1 activity via the AMPK-mTOR-SREBP signaling pathway. Biochem Pharmacol 85:1330–1340
Quintieri L, Palatini P, Nassi A, Ruzza P, Floreani M (2008) Flavonoids diosmetin and luteolin inhibit midazolam metabolism by human liver microsomes and recombinant CYP 3A4 and CYP3A5 enzymes. Biochem Pharmacol 75:1426–1437
Rafacho BP, Stice C, Liu C et al (2015) Inhibition of diethylnitrosamine-initiated alcohol-promoted hepatic inflammation and precancerous lesions by flavonoid luteolin is associated with increased sirtuin 1 activity in mice. Hepatobiliary Surg Nutr 4:124–134
Rahman S, Sultana S (2006) Chemopreventive activity of glycyrrhizin on lead acetate mediated hepatic oxidative stress and its hyperproliferative activity in Wistar rats. Chem Biol Interact 160:61–69
Rastogi H, Jana S (2014) Evaluation of inhibitory effects of caffeic acid and quercetin on human liver cytochrome p450 activities. Phytother Res 28:1873–1878
Raucy JL (2003) Regulation of CYP3A4 expression in human hepatocytes by pharmaceuticals and natural products. Drug Metab Dispos 31:533–539
Rechtman MM, Har-Noy O, Bar-Yishay I et al (2010) Curcumin inhibits hepatitis B virus via down-regulation of the metabolic coactivator PGC-1α. FEBS Lett 584:2485–2490
Reddy BU, Mullick R, Kumar A et al (2014) Small molecule inhibitors of HCV replication from pomegranate. Sci Rep 4:5411
Ren Y, Deng F, Zhu H et al (2011) Effect of epigallocatechin-3-gallate on iron overload in mice with alcoholic liver disease. Mol Biol Rep 38:879–886
Roberts-Kirchhoff ES, Crowley JR, Hollenberg PF, Kim H (1999) Metabolism of genistein by rat and human cytochrome P450s. Chem Res Toxicol 12:610–616
Roh C, Jo SK (2011) (–)-Epigallocatechin gallate inhibits hepatitis C virus (HCV) viral protein NS5B. Talanta 85:2639–2642
Rubiolo JA, Mithieux G, Vega FV (2008) Resveratrol protects primary rat hepatocytes against oxidative stress damage: activation of the Nrf2 transcription factor and augmented activities of antioxidant enzymes. Eur J Pharmacol 591:66–72
Ruhl CE, Everhart JE (2005) Coffee and tea consumption are associated with a lower incidence of chronic liver disease in the United States. Gastroenterol 129:1928–1936
Sahin K, Orhan C, Akdemir F et al (2012) Resveratrol protects quail hepatocytes against heat stress: modulation of the Nrf2 transcription factor and heat shock proteins. J Anim Physiol Anim Nutr (Berl) 96:66–74
Sakata R, Ueno T, Nakamura T et al (2004) Green tea polyphenol epigallocatechin-3-gallate inhibits platelet-derived growth factor-induced proliferation of human hepatic stellate cell line LI90. J Hepatol 40:52–59
Salas AL, Ocampo G, Farina GG, Reyes-Esparza J, Rodriguez-Fragoso L (2007) Genistein decreases liver fibrosis and cholestasis induced by prolonged biliary obstruction in the rat. Ann Hepatol 6:41–47
Saleh DO, Jaleel GARA, El-Awdan SA, Oraby F, Badawi M (2014) Thioacetamide-induced liver injury: protective role of genistein. Can J Physiol Pharm 92:965–973
Sarkar A, Sil PC (2014) Iron oxide nanoparticles mediated cytotoxicity via PI3K/AKT pathway: role of quercetin. Food Chem Toxicol 71:106–115
Sato H, Goto W, Yamamura J et al (1996) Therapeutic basis of glycyrrhizin on chronic hepatitis B. Antivir Res 30:171–177
Sayed-Ahmed MM, Aleisa AM, Al-Rejaie SS et al (2010) Thymoquinone attenuates diethylnitrosamine induction of hepatic carcinogenesis through antioxidant signaling. Oxid Med Cell Longev 3:254–261
Schrieber SJ, Wen ZM, Vourvahis M et al (2008) Pharmacokinetics of silymarin is altered in patients with hepatitis C virus and nonalcoholic fatty liver disease and correlates with plasma caspase-3/7 activity. Drug Metab Dispos 36:1909–1916
Schumacker PT (2006) Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell 10:175–176
Sehgal A, Kumar M, Jain M, Dhawan DK (2013) Modulatory effects of curcumin in conjunction with piperine on benzo(a)pyrene-mediated DNA adducts and biotransformation enzymes. Nutr Cancer 65:885–890
Sekaran S, Kandaswamy S, Gunasekaran K et al (2012) Protective role of quercetin on polychlorinated biphenyls (Aroclor-1254) induced oxidative stress and apoptosis in liver of adult male rats. J Biochem Mol Toxicol 26:522–532
Sengupta D, Chowdhury KD, Sarkar A, Paul S, Sadhukhan GC (2014) Berberine and S allyl cysteine mediated amelioration of DEN + CCl4 induced hepatocarcinoma. Biochim Biophys Acta 1840:219–244
Seok J, Warren HS, Cuenca AG et al (2013) Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA 110:3507–3512
Shankari SG, Karthikesan K, Jalaludeen AM, Ashokkumar N (2010) Hepatoprotective effect of morin on ethanol-induced hepatotoxicity in rats. J Basic Clin Physiol Pharmacol 21:277–294
Shen L, Xiong Y, Wang DQH et al (2013) Ginsenoside Rb1 reduces fatty liver by activating AMP-activated protein kinase in obese rats. J Lipid Res 54:1430–1438
Sheng M, Zhou Y, Yu W et al (2015) Protective effect of Berberine pretreatment in hepatic ischemia/reperfusion injury of rat. Transpl Proc 47:275–282
Sherif IO, Al-Gayyar MM (2013) Antioxidant, anti-inflammatory and hepatoprotective effects of silymarin on hepatic dysfunction induced by sodium nitrite. Eur Cytokine Netw 24:114–121
Shi H, Dong L, Bai Y et al (2009) Chlorogenic acid against carbon tetrachloride-induced liver fibrosis in rats. Eur J Pharmacol 623:119–124
Shi G, Zhang Z, Zhang R et al (2012) Protective effect of andrographolide against concanavalin A-induced liver injury. Naunyn Schmiedebergs Arch Pharmacol 385:69–79
Shi H, Dong L, Dang X et al (2013a) Effect of chlorogenic acid on LPS-induced proinflammatory signaling in hepatic stellate cells. Inflamm Res 62:581–587
Shi H, Dong L, Jiang J et al (2013b) Chlorogenic acid reduces liver inflammation and fibrosis through inhibition of toll-like receptor 4 signaling pathway. Toxicology 303:107–114
Shim SG, Jun DW, Kim EK et al (2013) Caffeine attenuates liver fibrosis via defective adhesion of hepatic stellate cells in cirrhotic model. J Gastroenterol Hepatol 28:1877–1884
Shimizu M, Sakai H, Shirakami Y et al (2011) Preventive effects of (–)-epigallocatechin gallate on diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BL/KsJ-db/db mice. Cancer Prev Res 4:396–403
Shin MS, Kang EH, Lee YI (2005) A flavonoid from medicinal plants blocks hepatitis B virus-e antigen secretion in HBV-infected hepatocytes. Antivir Res 67:163–168
Shin HW, Park SY, Lee KB, Jang JJ (2009) Down-regulation of Wnt signaling during apoptosis of human hepatic stellate cells. Hepatol Gastroenterol 56:208–212
Shu JC, He YJ, Lv X et al (2009) Effect of curcumin on the proliferation and apoptosis of hepatic stellate cells. Braz J Med Biol Res 42:1173–1178
Sikder K, Kesh SB, Das N, Manna K, Dey S (2014) The high antioxidative power of quercetin (aglycone flavonoid) and its glycone (rutin) avert high cholesterol diet induced hepatotoxicity and inflammation in Swiss albino mice. Food Funct 5:1294–1303
Singh SP, Konwar BK (2012) Molecular docking studies of quercetin and its analogues against human inducible nitric oxide synthase. SpringerPlus 1:69
Singh P, Sharma S, Rath SK (2014) Genistein Induces deleterious effects during its acute exposure in Swiss mice. Biomed Res Int. doi:10.1155/2014/619617
Sinha RA, Farah BL, Singh BK et al (2014) Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology 59:1366–1380
Sivaramakrishnan V, Devaraj SN (2010) Morin fosters apoptosis in experimental hepatocellular carcinogenesis model. Chem Biol Interact 183:284–292
Sivaramakrishnan V, Niranjali Devaraj S (2009) Morin regulates the expression of NF-κB-p65, COX-2 and matrix metalloproteinases in diethylnitrosamine induced rat hepatocellular carcinoma. Chem Biol Interact 180:353–359
Sovadinova I, Babica P, Boke H et al (2015) Phosphatidylcholine specific PLC-induced dysregulation of gap junctions, a robust cellular response to environmental toxicants, and prevention by resveratrol in a rat liver cell model. PLoS One 10:e0124454
Srigopalram S, Jayraaj IA, Kaleeswaran B et al (2014) Ellagic acid normalizes mitochondrial outer membrane permeabilization and attenuates inflammation-mediated cell proliferation in experimental liver cancer. Appl Biochem Biotechnol 173:2254–2266
Srisuttee R, Koh SS, Kim SJ et al (2012) Hepatitis B virus X (HBX) protein upregulates beta-catenin in a human hepatic cell line by sequestering SIRT1 deacetylase. Oncol Rep 28:276–282
Sugiyama K, Noda Y, He P (2001) Suppressive effect of caffeine on hepatitis and apoptosis induced by tumor necrosis factor-alpha, but not by the anti-Fas antibody, in mice. Biosci Biotechnol Biochem 65:674–677
Sumi T, Shirakami Y, Shimizu M et al (2013) (–)-Epigallocatechin-3-gallate suppresses hepatic preneoplastic lesions developed in a novel rat model of non-alcoholic steatohepatitis. SpringerPlus 2:690
Sun H, Che QM, Zhao X, Pu XP (2010) Antifibrotic effects of chronic baicalein administration in a CCl4 liver fibrosis model in rats. Eur J Pharmacol 631:53–60
Sundaresan A, Radhiga T, Pugalendi KV (2014) Effect of ursolic acid and rosiglitazone combination on hepatic lipid accumulation in high fat diet-fed C57BL/6J mice. Eur J Pharmacol 741:297–303
Surapaneni KM, Jainu M (2014) Comparative effect of pioglitazone, quercetin and hydroxy citric acid on the status of lipid peroxidation and antioxidants in experimental non-alcoholic steatohepatitis. J Physiol Pharmacol 65:67–74
Surapaneni KM, Priya VV, Mallika J (2014) Pioglitazone, quercetin and hydroxy citric acid effect on cytochrome P450 2E1 (CYP2E1) enzyme levels in experimentally induced non alcoholic steatohepatitis (NASH). Eur Rev Med Pharmacol Sci 18:2736–2741
Szuster-Ciesielska A, Plewka K, Kandefer-Szerszen M (2011) Betulin, betulinic acid and butein are inhibitors of acetaldehyde-induced activation of liver stellate cells. Pharmacol Rep 63:1109–1123
Tai M, Zhang J, Song S et al (2015) Protective effects of luteolin against acetaminophen-induced acute liver failure in mouse. Int Immunopharmacol 271:164–170
Takahara T, Watanabe A, Shiraki K (1994) Effects of glycyrrhizin on hepatitis B surface antigen: a biochemical and morphological study. J Hepatol 21:601–609
Takanohashi T, Isaka M, Ubukata K, Mihara R, Bernard BK (2010) Studies of the toxicological potential of capsinoids, XIII: inhibitory effects of capsaicin and capsinoids on cytochrome P450 3A4 in human liver microsomes. Int J Toxicol 29:22S–26S
Tang Y, Chen A (2010) Curcumin protects hepatic stellate cells against leptin-induced activation in vitro by accumulating intracellular lipids. Endocrinology 151:4168–4177
Tang Y, Tian H, Shi Y et al (2013) Quercetin suppressed CYP2E1-dependent ethanol hepatotoxicity via depleting heme pool and releasing CO. Phytomedicine 20:699–704
Tao YY, Wang QL, Shen L, Fu WW, Liu CH (2013) Salvianolic acid B inhibits hepatic stellate cell activation through transforming growth factor beta-1 signal transduction pathway in vivo and in vitro. Exp Biol Med 238:1284–1296
Tao TZ, Chen F, Bo LL et al (2014) Ginsenoside Rg1 protects mouse liver against ischemia-reperfusion injury through anti-inflammatory and anti-apoptosis properties. J Surg Res 191:231–238
Thapliyal R, Maru GB (2001) Inhibition of cytochrome P450 isozymes by curcumins in vitro and in vivo. Food Chem Toxicol 39:541–547
Tian X, Cheng ZY, He J, Jia LJ, Qiao HL (2013) Concentration-dependent inhibitory effects of baicalin on the metabolism of dextromethorphan, a dual probe of CYP2D and CYP3A, in rats. Chem Biol Interact 203:522–529
Tiong KH, Yiap BC, Tan EL, Ismail R, Ong CE (2010) In vitro modulation of naturally occurring flavonoids on cytochrome P450 2A6 (CYP2A6) activity. Xenobiotica 40:458–466
Tipoe GL, Leung TM, Liong EC et al (2010) Epigallocatechin-3-gallate (EGCG) reduces liver inflammation, oxidative stress and fibrosis in carbon tetrachloride (CCl4)-induced liver injury in mice. Toxicology 273:45–52
Trivedi NP, Rawal UM, Patel BP (2007) Hepatoprotective effect of andrographolide against hexachlorocyclohexane-induced oxidative injury. Integr Cancer Ther 6:271–280
Tsai MK, Lin YL, Huang YT (2011) Differential inhibitory effects of salvianolic acids on activation of rat hepatic stellate cells by platelet-derived growth factor. Planta Med 77:1495–1503
Tu CT, Han B, Yao QY, Zhang YA, Liu HC, Zhang SC (2012a) Curcumin attenuates concanavalin a-induced liver injury in mice by inhibition of toll-like receptor (TLR) 2, TLR4 and TLR9 expression. Int Immunopharmacol 12:151–157
Tu CT, Yao QY, Xu BL et al (2012b) Protective effects of curcumin against hepatic fibrosis induced by carbon tetrachloride: modulation of high-mobility group box 1, toll-like receptor 4 and 2 expression. Food Chem Toxicol 50:3343–3351
Tu CT, Yao QY, Xu BL, Zhang SC (2013) Curcumin protects against concanavalin A-induced hepatitis in mice through inhibiting the cytoplasmic translocation and expression of high mobility group box 1. Inflammation 36:206–215
Tung BT, Rodriguez-Bies E, Talero E et al (2015) Anti-inflammatory effect of resveratrol in old mice liver. Exp Gerontol 64:1–7
Tzeng TF, Lu HJ, Liou SS, Chang CJ, Liu IM (2012) Emodin, a naturally occurring anthraquinone derivative, ameliorates dyslipidemia by activating AMP-activated protein kinase in high-fat-diet-fed rats. Evid Based Complement Altern Med 2012:781812
Tzeng JI, Chen MF, Chung HH, Cheng JT (2013) Silymarin decreases connective tissue growth factor to improve liver fibrosis in rats treated with carbon tetrachloride. Phytother Res 27:1023–1028
Ueng YF, Shyu CC, Lin YL et al (2000) Effects of baicalein and wogonin on drug-metabolizing enzymes in C57BL/6J mice. Life Sci 67:2189–2200
Ullmann U, Haller J, Decourt JD et al (2004) Plasma-kinetic characteristics of purified and isolated green tea catechin epigallocatechin gallate (EGCG) after 10 days repeated dosing in healthy volunteers. Int J Vitam Nutr Res 74:269–278
Um MY, Hwang KH, Ahn J, Ha TY (2013) Curcumin attenuates diet-induced hepatic steatosis by activating AMP-activated protein kinase. Basic Clin Pharmacol Toxicol 113:152–157
Vaynshteyn D, Jeong H (2012) Caffeine induces CYP1A2 expression in rat hepatocytes but not in human hepatocytes. Drug Metab Lett 6:116–119
Veldt BJ, Hansen BE, Ikeda K et al (2006) Long-term clinical outcome and effect of glycyrrhizin in 1093 chronic hepatitis C patients with non-response or relapse to interferon. Scand J Gastroenterol 41:1087–1094
Vizzutti F, Provenzano A, Galastri S et al (2010) Curcumin limits the fibrogenic evolution of experimental steatohepatitis. Lab Invest 90:104–115
Walle T, Vincent TS, Walle UK (2003) Evidence of covalent binding of the dietary flavonoid quercetin to DNA and protein in human intestinal and hepatic cells. Biochem Pharmacol 65:1603–1610
Walsky RL, Gaman EA, Obach RS (2005) Examination of 209 drugs for inhibition of cytochrome P450 2C8. J Clin Pharmacol 45:68–78
Wan Y, Wu YL, Lian LH et al (2012) The anti-fibrotic effect of betulinic acid is mediated through the inhibition of NF-κB nuclear protein translocation. Chem Biol Interact 195:215–223
Wan CW, Wong CN, Pin WK et al (2013a) Chlorogenic acid exhibits cholesterol lowering and fatty liver attenuating properties by up-regulating the gene expression of PPAR-alpha in hypercholesterolemic rats induced with a high-cholesterol diet. Phytother Res 27:545–551
Wan Y, Jiang S, Lian LH et al (2013b) Betulinic acid and betulin ameliorate acute ethanol-induced fatty liver via TLR4 and STAT3 in vivo and in vitro. Int Immunopharmacol 17:184–190
Wan Y, Tang MH, Chen XC et al (2014) Inhibitory effect of liposomal quercetin on acute hepatitis and hepatic fibrosis induced by concanavalin A. Braz J Med Biol Res 47:655–661
Wang XD, Wolkoff AW, Morris ME (2005) Flavonoids as a novel class of human organic anion-transporting polypeptide OATP1B1 (OATP-C) modulators. Drug Metab Dispos 33:1666–1672
Wang J, Qiao L, Li Y, Yang G (2008) Ginsenoside Rb1 attenuates intestinal ischemia-reperfusion- induced liver injury by inhibiting NF-κB activation. Exp Mol Med 40:686–698
Wang GF, Shi LP, Ren YD et al (2009a) Anti-hepatitis B virus activity of chlorogenic acid, quinic acid and caffeic acid in vivo and in vitro. Antivir Res 83:186–190
Wang GL, Fu YC, Xu WC et al (2009b) Resveratrol inhibits the expression of SREBP1 in cell model of steatosis via Sirt1-FOXO1 signaling pathway. Biochem Biophys Res Commun 380:644–649
Wang X, Ikejima K, Kon K et al (2011) Ursolic acid ameliorates hepatic fibrosis in the rat by specific induction of apoptosis in hepatic stellate cells. J Hepatol 55:379–387
Wang ME, Chen YC, Chen IS et al (2012a) Curcumin protects against thioacetamide-induced hepatic fibrosis by attenuating the inflammatory response and inducing apoptosis of damaged hepatocytes. J Nutr Biochem 23:1352–1366
Wang R, Yu XY, Guo ZY, Wang YJ, Wu Y, Yuan YF (2012b) Inhibitory effects of salvianolic acid B on CCl4-induced hepatic fibrosis through regulating NF-κB/IκBα signaling. J Ethnopharmacol 144:592–598
Wang X, Zhang DM, Gu TT et al (2013) Morin reduces hepatic inflammation-associated lipid accumulation in high fructose-fed rats via inhibiting sphingosine kinase 1/sphingosine 1-phosphate signaling pathway. Biochem Pharmacol 86:1791–1804
Wang F, Liu S, Shen Y et al (2014a) Protective effects of -acetylcysteine on cisplatin-induced oxidative stress and DNA damage in HepG2 cells. Exp Ther Med 8:1939–1945
Wang H, Guan WJ, Yang WZ et al (2014b) Caffeine inhibits the activation of hepatic stellate cells induced by acetaldehyde via adenosine A2A receptor mediated by the cAMP/PKA/SRC/ERK1/2/P38 MAPK signal pathway. PLoS One 9:e92482
Wang T, Zhou ZX, Sun LX et al (2014c) Resveratrol effectively attenuates alpha-naphthyl-isothiocyanate-induced acute cholestasis and liver injury through choleretic and anti-inflammatory mechanisms. Acta Pharmacol Sin 35:1527–1536
Wang W, He Y, Lin P et al (2014d) In vitro effects of active components of Polygonum multiflorum radix on enzymes involved in the lipid metabolism. J Ethnopharmacol 153:763–770
Wang D, Wang Y, Wan X, Yang CS, Zhang J (2015a) Green tea polyphenol (–)-epigallocatechin-3-gallate triggered hepatotoxicity in mice: responses of major antioxidant enzymes and the Nrf2 rescue pathway. Toxicol Appl Pharmacol 283:65–74
Wang P, Peng X, Wei ZF et al (2015b) Geraniin exerts cytoprotective effect against cellular oxidative stress by upregulation of Nrf2-mediated antioxidant enzyme expression via PI3K/AKT and ERK1/2 pathway. Biochim Biophys Acta. doi:10.1016/j.bbagen.2015.04.010
Wang Q, Dai XF, Yang WZ et al (2015c) Caffeine protects against alcohol-induced liver fibrosis by dampening the cAMP/PKA/CREB pathway in rat hepatic stellate cells. Int Immunopharmacol 25:340–352
Wang Y, Jiang Y, Fan X et al (2015d) Hepato-protective effect of resveratrol against acetaminophen-induced liver injury is associated with inhibition of CYP-mediated bioactivation and regulation of SIRT1-p53 signaling pathways. Toxicol Lett 236:82–89
Watkins PB, Wrighton SA, Schuetz EG, Molowa DT, Guzelian PS (1987) Identification of glucocorticoid-inducible cytochromes P-450 in the intestinal mucosa of rats and man. J Clin Invest 80:1029–1036
Weng Z, Greenhaw J, Salminen WF, Shi Q (2012) Mechanisms for epigallocatechin gallate induced inhibition of drug metabolizing enzymes in rat liver microsomes. Toxicol Lett 214:328–338
Wolf KK, Wood SG, Hunt JA et al (2005) Role of the nuclear receptor pregnane X receptor in acetaminophen hepatotoxicity. Drug Metab Dispos 33:1827–1836
Wong YT, Gruber J, Jenner AM et al (2009) Elevation of oxidative-damage biomarkers during aging in F2 hybrid mice: protection by chronic oral intake of resveratrol. Free Radic Biol Med 46:799–809
Wu X, Zhang L, Gurley E et al (2008) Prevention of free fatty acid-induced hepatic lipotoxicity by 18beta-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology 47:1905–1915
Wu YL, Lian LH, Wan Y, Nan JX (2010) Baicalein inhibits nuclear factor-kappaB and apoptosis via c-FLIP and MAPK in D-GalN/LPS induced acute liver failure in murine models. Chem Biol Interact 188:526–534
Wu LC, Lu IW, Chung CF, Wu HY, Liu YT (2011) Antiproliferative mechanisms of quercetin in rat activated hepatic stellate cells. Food Funct 2:204–212
Wu X, Li C, Xing G, Qi X, Ren J (2013) Resveratrol downregulates CYP2E1 and attenuates chemically induced hepatocarcinogenesis in SD rats. J Toxicol Pathol 26:385–392
Xi Y, Wu M, Li H et al (2015) Baicalin attenuates high fat diet-induced obesity and liver dysfunction: dose-response and potential role of CaMKKbeta/AMPK/ACC pathway. Cell Physiol Biochem 35:2349–2359
Xiao HB, Sun ZL, Zhang HB, Zhang DS (2012) Berberine inhibits dyslipidemia in C57BL/6 mice with lipopolysaccharide induced inflammation. Pharmacol Rep 64:889–895
Xiao J, Ho CT, Liong EC et al (2014) Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3K/Akt/FoxO1, and NF-κB pathways. Eur J Nutr 53:187–199
Xie Y, Hao HP, Wang H, Wang ZX, Wang GJ (2013) Reversing effects of silybin on TAA-induced hepatic CYP3A dysfunction through PXR regulation. Chin J Nat Med 11:645–652
Xin HG, Zhang BB, Wu ZQ et al (2014) Treatment with baicalein attenuates methionine-choline deficient diet-induced non-alcoholic steatohepatitis in rats. Eur J Pharmacol 738:310–318
Xu J, Wang J, Deng F, Hu ZH, Wang HL (2008) Green tea extract and its major component epigallocatechin gallate inhibits hepatitis B virus in vitro. Antivir Res 78:242–249
Xu Y, Chen J, Yu X et al (2010) Protective effects of chlorogenic acid on acute hepatotoxicity induced by lipopolysaccharide in mice. Inflamm Res 59:871–877
Xu H, Zhou Y, Lu C, Ping J, Xu LM (2012) Salvianolic acid B lowers portal pressure in cirrhotic rats and attenuates contraction of rat hepatic stellate cells by inhibiting RhoA signaling pathway. Lab Invest 92:1738–1748
Xu XM, Hu Y, Zhai XH et al (2013) Salvianolic acid A preconditioning confers protection against concanavalin A-induced liver injury through SIRT1-mediated repression of p66shc in mice. Toxicol Appl Pharmacol 273:68–76
Xu D, Hu L, Su C et al (2014a) Tetrachloro-p-benzoquinone induces hepatic oxidative damage and inflammatory response, but not apoptosis in mouse: the prevention of curcumin. Toxicol Appl Pharmacol 280:305–313
Xu W, Lu Y, Yao J et al (2014b) Novel role of resveratrol: suppression of high-mobility group protein box 1 nucleocytoplasmic translocation by the upregulation of sirtuin 1 in sepsis-induced liver injury. Shock 42:440–447
Xue JH, Chen F, Wang J et al (2015) Emodin protects against concanavalin a-induced hepatitis in mice through inhibiting activation of the p38 MAPK-NF-kappa B signaling pathway. Cell Physiol Biochem 35:1557–1570
Yagnik GP, Takahashi Y, Tsoulfas G et al (2002) Blockade of the l-arginine/NO synthase pathway worsens hepatic apoptosis and liver transplant preservation injury. Hepatology 36:573–581
Yan X, Zhou T, Tao Y et al (2010) Salvianolic acid B attenuates hepatocyte apoptosis by regulating mediators in death receptor and mitochondrial pathways. Exp Biol Med (Maywood) 235:623–632
Yan CM, Chai EQ, Cai HY, Miao GY, Ma W (2015a) Oleuropein induces apoptosis via activation of caspases and suppression of phosphatidylinositol 3-kinase/protein kinase B pathway in HepG2 human hepatoma cell line. Mol Med Rep 11:4617–4624
Yan X, Jiang Z, Bi L, Yang Y, Chen W (2015b) Salvianolic acid A attenuates TNF-α- and D-GalN-induced ER stress-mediated and mitochondrial-dependent apoptosis by modulating Bax/Bcl-2 ratio and calcium release in hepatocyte LO2 cells. Naunyn Schmiedebergs Arch Pharmacol 388:817–830
Yang SJ, Lim Y (2014) Resveratrol ameliorates hepatic metaflammation and inhibits NLRP3 inflammasome activation. Metabolism 63:693–701
Yang MD, Chiang YM, Higashiyama R et al (2012) Rosmarinic acid and baicalin epigenetically derepress peroxisomal proliferator-activated receptor gamma in hepatic stellate cells for their antifibrotic effect. Hepatology 55:1271–1281
Yang Y, Zhao Z, Liu Y et al (2015) Suppression of oxidative stress and improvement of liver functions in mice by ursolic acid via LKB1-AMP-activated protein kinase signaling. J Gastroenterol Hepatol 30:609–618
Yao DC, Li HW, Gou YL et al (2009) Betulinic acid-mediated inhibitory effect on hepatitis B virus by suppression of manganese superoxide dismutase expression. FEBS J 276:2599–2614
Yao JY, Zhi M, Chen MH (2011) Effect of silybin on high-fat-induced fatty liver in rats. Braz J Med Biol Res 44:652–659
Yao Q, Lin Y, Li X, Shen X, Wang J, Tu C (2013) Curcumin ameliorates intrahepatic angiogenesis and capillarization of the sinusoids in carbon tetrachloride-induced rat liver fibrosis. Toxicol Lett 222:72–82
Yasuda Y, Shimizu M, Sakai H et al (2009) (–)-Epigallocatechin gallate prevents carbon tetrachloride-induced rat hepatic fibrosis by inhibiting the expression of the PDGFRbeta and IGF-1R. Chem Biol Interact 182:159–164
Ye JF, Zhu H, Zhou ZF et al (2011) Protective mechanism of andrographolide against carbon tetrachloride-induced acute liver injury in mice. Biol Pharm Bull 34:1666–1670
Yen FL, Wu TH, Lin LT et al (2009) Naringenin-loaded nanoparticles improve the physicochemical properties and the hepatoprotective effects of naringenin in orally-administered rats with CCl4-induced acute liver failure. Pharm Res 26:893–902
Yi J, Xia W, Wu J et al (2014) Betulinic acid prevents alcohol-induced liver damage by improving the antioxidant system in mice. J Vet Sci 15:141–148
Yin X, Gong X, Jiang R et al (2014) Emodin ameliorated lipopolysaccharide-induced fulminant hepatic failure by blockade of TLR4/MD2 complex expression in d-galactosamine-sensitized mice. Int Immunopharmacol 23:66–72
Yoo NY, Jeon S, Nam Y et al (2015) Dietary supplementation of genistein alleviates liver inflammation and fibrosis mediated by a methionine–choline–deficient diet in db/db mice. J Agric Food Chem 63:4305–4311
Yoshimura Y, Nishii S, Zaima N, Moriyarha T, Kawamura Y (2013) Ellagic acid improves hepatic steatosis and serum lipid composition through reduction of serum resistin levels and transcriptional activation of hepatic ppara in obese, diabetic KK-A(y) mice. Biochem Biophys Res Commun 434:486–491
Yu J, Xie J, Mao XJ et al (2011) Hepatoxicity of major constituents and extractions of Radix Polygoni Multiflori and Radix Polygoni Multiflori Praeparata. J Ethnopharmacol 137:1291–1299
Yu FX, Teng YY, Zhu QD, Zhang QY, Tang YH (2014) Inhibitory effects of capsaicin on hepatic stellate cells and liver fibrosis. Biochem Cell Biol 92:406–412
Yu DK, Zhang CX, Zhao SS et al (2015) The anti-fibrotic effects of epigallocatechin-3-gallate in bile duct-ligated cholestatic rats and human hepatic stellate LX-2 cells are mediated by the PI3K/Akt/Smad pathway. Acta Pharmacol Sin 36(4):473–482
Yumei F, Zhou YJ, Zheng SZ, Chen AP (2006) The antifibrogenic effect of (–)-epigallocatechin gallate results from the induction of de novo synthesis of glutathione in passaged rat hepatic stellate cells. Lab Invest 86:697–709
Yun JW, Kim YK, Lee BS et al (2007) Effect of dietary epigallocatechin-3-gallate on cytochrome P450 2E1-dependent alcoholic liver damage: enhancement of fatty acid oxidation. Biosci Biotechnol Biochem 71:2999–3006
Yun N, Kang JW, Lee SM (2012) Protective effects of chlorogenic acid against ischemia/reperfusion injury in rat liver: molecular evidence of its antioxidant and anti-inflammatory properties. J Nutr Biochem 23:1249–1255
Zendulka O, Totusek J, Sulcova A (2009) Intersexual differences in inhibitory influence of trans-resveratrol on activity of cytochrome P450 2D2 in rats. Neuroendocrinol Lett 30:88–91
Zhai X, Qiao H, Guan W et al (2015) Curcumin regulates peroxisome proliferator-activated receptor-gamma coactivator-1alpha expression by AMPK pathway in hepatic stellate cells in vitro. Eur J Pharmacol 746:56–62
Zhang ZF, Fan SH, Zheng YL et al (2009) Troxerutin protects the mouse liver against oxidative stress-mediated injury induced by d-galactose. J Agric Food Chem 57:7731–7736
Zhang Y, Huang Y, Deng XR et al (2012) Iron overload-induced rat liver injury: Involvement of protein tyrosine nitration and the effect of baicalin. Eur J Pharmacol 680:95–101
Zhang W, Hong RT, Tian TL (2013a) Silymarin’s protective effects and possible mechanisms on alcoholic fatty liver for rats. Biomol Ther 21:264–269
Zhang ZL, Guo Y, Zhang S et al (2013b) Curcumin modulates cannabinoid receptors in liver fibrosis in vivo and inhibits extracellular matrix expression in hepatic stellate cells by suppressing cannabinoid receptor type-1 in vitro. Eur J Pharmacol 721:133–140
Zhang F, Zhang ZL, Chen L et al (2014a) Curcumin attenuates angiogenesis in liver fibrosis and inhibits angiogenic properties of hepatic stellate cells. J Cell Mol Med 18:1392–1406
Zhang J, Xu L, Zhang L, Ying Z, Su W, Wang T (2014b) Curcumin attenuates d-galactosamine/lipopolysaccharide-induced liver injury and mitochondrial dysfunction in mice. J Nutr 144:1211–1218
Zhang JQ, Shi L, Xu XN et al (2014c) Therapeutic detoxification of quercetin against carbon tetrachloride-induced acute liver injury in mice and its mechanism. J Zhejiang Univ Sci B 15:1039–1047
Zhang M, Li W, Yu L, Wu S (2014d) The suppressive effect of resveratrol on HIF-1alpha and VEGF expression after warm ischemia and reperfusion in rat liver. PLoS One 9:e109589
Zhang P, Ma D, Wang Y et al (2014e) Berberine protects liver from ethanol-induced oxidative stress and steatosis in mice. Food Chem Toxicol 74:225–232
Zhang TS, Kimura Y, Jiang SY et al (2014f) Luteolin modulates expression of drug-metabolizing enzymes through the AhR and Nrf2 pathways in hepatic cells. Arch Biochem Biophys 557:36–46
Zhang ZF, Fan SH, Zheng YL et al (2014g) Troxerutin improves hepatic lipid homeostasis by restoring NAD+-depletion-mediated dysfunction of lipin 1 signaling in high-fat diet-treated mice. Biochem Pharmacol 91:74–86
Zhang SJ, Li YF, Wang GE et al (2015a) Caffeine ameliorates high energy diet-induced hepatic steatosis: sirtuin 3 acts as a bridge in the lipid metabolism pathway. Food Funct. doi:10.1039/c5fo00247h
Zhang X, Zhang JH, Chen XY et al (2015b) Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid Redox Signal 22:848–870
Zhang Y, Chen ML, Zhou Y et al (2015c) Resveratrol improves hepatic steatosis by inducing autophagy through the cAMP signaling pathway. Mol Nutr Food Res. doi:10.1002/mnfr.201500016
Zhang ZF, Shan Q, Zhuang J et al (2015d) Troxerutin inhibits 2,2′,4,4′-tetrabromodiphenyl ether (BDE-47)-induced hepatocyte apoptosis by restoring proteasome function. Toxicol Lett 233:246–257
Zhang ZF, Zhang YQ, Fan SH et al (2015e) Troxerutin protects against 2,2′,4,4′-tetrabromodiphenyl ether (BDE-47)-induced liver inflammation by attenuating oxidative stress-mediated NAD+-depletion. J Hazard Mater 283:98–109
Zhao JF, Liu CH, Hu YY et al (2004) Effect of salvianolic acid B on Smad3 expression in hepatic stellate cells. Hepatobiliary Pancreat Dis Int 3:102–105
Zhao Y, Li H, Gao Z, Gong Y, Xu H (2006) Effects of flavonoids extracted from Scutellaria baicalensis Georgi on hemin–nitrite–H2O2 induced liver injury. Eur J Pharmacol 536:192–199
Zhao X, Zhang JJ, Wang X et al (2008) Effect of berberine on hepatocyte proliferation, inducible nitric oxide synthase expression, cytochrome P450 2E1 and 1A2 activities in diethylnitrosamine- and phenobarbital-treated rats. Biomed Pharmacother 62:567–572
Zhao YL, Wang JB, Zhou GD, Shan LM, Xiao XH (2009) Investigations of free anthraquinones from rhubarb against alpha-naphthylisothiocyanate-induced cholestatic liver injury in rats. Basic Clin Pharmacol Toxicol 104:463–469
Zhao K, Ding M, Cao H, Cao ZX (2012) In vitro metabolism of glycyrrhetinic acid by human and rat liver microsomes and its interactions with six CYP substrates. J Pharm Pharmacol 64:1445–1451
Zhao YL, Ma X, Wang JB et al (2014) Curcumin protects against CCl4-induced liver fibrosis in rats by inhibiting HIF-1α through an ERK-dependent pathway. Molecules 19:18767–18780
Zhao X, Chen Q, Li Y et al (2015) Doxorubicin and curcumin co-delivery by lipid nanoparticles for enhanced treatment of diethylnitrosamine-induced hepatocellular carcinoma in mice. Eur J Pharm Biopharm 93:27–36
Zhen MC, Huang XH, Wang Q et al (2006) Green tea polyphenol epigallocatechin-3-gallate suppresses rat hepatic stellate cell invasion by inhibition of MMP-2 expression and its activation. Acta Pharmacol Sin 27:1600–1607
Zhen MC, Wang Q, Huang XH et al (2007) Green tea polyphenol epigallocatechin-3-gallate inhibits oxidative damage and preventive effects on carbon tetrachloride-induced hepatic fibrosis. J Nutr Bioch 18:795–805
Zheng SZ, Chen AP (2006) Curcumin suppresses the expression of extracellular matrix genes in activated hepatic stellate cells by inhibiting gene expression of connective tissue growth factor. Am J Physiol Gastrointest Liver Physiol 290:G883–G893
Zheng ZW, Song SZ, Wu YL et al (2011) Betulinic acid prevention of d-galactosamine/lipopolysaccharide liver toxicity is triggered by activation of Bcl-2 and antioxidant mechanisms. J Pharm Pharmacol 63:572–578
Zheng Z, Sheng Y, Lu B, Ji L (2015) The therapeutic detoxification of chlorogenic acid against acetaminophen-induced liver injury by ameliorating hepatic inflammation. Chem Biol Interact 238:93–101
Zhong L, Hu J, Shu W, Gao B, Xiong S (2015) Epigallocatechin-3-gallate opposes HBV-induced incomplete autophagy by enhancing lysosomal acidification, which is unfavorable for HBV replication. Cell Death Dis 6:e1770
Zhou Y, Zheng S, Lin J, Zhang QJ, Chen A (2007) The interruption of the PDGF and EGF signaling pathways by curcumin stimulates gene expression of PPARγ in rat activated hepatic stellate cell in vitro. Lab Invest 87:488–498
Zhou J, Farah BL, Sinha RA, Wu Y, Singh BK (2014) Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, stimulates hepatic autophagy and lipid clearance. PLoS One 9:e96884
Zhou Y, Chen K, He L et al (2015) The protective effect of resveratrol on concanavalin-A-induced acute hepatic injury in mice. Gastroenterol Res Pract 2015:506390
Zhu HJ, Brinda BJ, Chavin KD et al (2013) An assessment of pharmacokinetics and antioxidant activity of free silymarin flavonolignans in healthy volunteers: a dose escalation study. Drug Metab Dispos 41:1679–1685
Zhu W, Chen S, Li Z et al (2014) Effects and mechanisms of resveratrol on the amelioration of oxidative stress and hepatic steatosis in KKAy mice. Nutr Metab 11:35
Zong L, Qu Y, Xu MY, Dong YW, Lu LG (2012) 18 alpha-glycyrrhetinic acid down-regulates expression of type I and III collagen via TGF-β1/Smad signaling pathway in human and rat hepatic stellate cells. Int J Med Sci 9:370–379
Zong L, Qu Y, Xu MY, Dong YW, Lu LG (2013) 18alpha-glycyrrhetinic acid extracted from Glycyrrhiza radix inhibits proliferation and promotes apoptosis of the hepatic stellate cell line. J Dig Dis 14:328–336
Acknowledgments
This research was supported by grants from the University of Rijeka (Project 13.06.1.2.24). The authors thank Nika Kosanovic for manuscript proofreading.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Domitrović, R., Potočnjak, I. A comprehensive overview of hepatoprotective natural compounds: mechanism of action and clinical perspectives. Arch Toxicol 90, 39–79 (2016). https://doi.org/10.1007/s00204-015-1580-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-015-1580-z