
Abstract
Catecholamines are endogenous neurosignalling mediators and hormones. They are integral in maintaining homeostasis by promptly responding to any stressor. Their synthetic equivalents are the current mainstay of treatment in shock states to counteract myocardial depression and/or vasoplegia. These phenomena are related in large part to decreased adrenoreceptor sensitivity and altered adrenergic signalling, with resultant vascular and cardiomyocyte hyporeactivity. Catecholamines are predominantly used in supraphysiological doses to overcome these pathological consequences. However, these adrenergic agents cause direct organ damage and have multiple ‘off-target’ biological effects on immune, metabolic and coagulation pathways, most of which are not monitored or recognised at the bedside. Such detrimental consequences may contribute negatively to patient outcomes. This review explores the schizophrenic ‘Jekyll-and-Hyde’ characteristics of catecholamines in critical illness, as they are both necessary for survival yet detrimental in excess. This article covers catecholamine physiology, the pleiotropic effects of catecholamines on various body systems and pathways, and potential alternatives for haemodynamic support and adrenergic modulation in the critically ill.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Impact of inflammatory shock on the cardiovascular system
Recognition of pathogen-associated molecular patterns (PAMPs) related to microorganisms and/or release of intracellular damage-associated molecular patterns (DAMPs) from injured cells, such as mitochondria, heat shock proteins and intracellular cytokines, triggers a systemic inflammatory host response [1]. Indeed, DAMPs act through similar receptors to those that recognise PAMPs [2, 3]. This inflammatory response modulates multiple downstream pathways ranging from immune to cardiovascular, hormonal to coagulation, metabolic to bioenergetic [4]. When inflammation is excessive and/or dysregulated, macro- and microcirculatory abnormalities ensue [5]. Myocardial depression, excessive vasodilation and increased capillary leak, resulting in hypovolaemia and tissue oedema, may all impede delivery of sufficient oxygen and substrate to meet cellular metabolic demands. This will be compounded by mitochondrial dysfunction that further compromises ATP production [6]. Cells may defend themselves by reducing metabolic activity to lessen the risk of activating death pathways, but at the cost of a decreased functionality [7]. Therefore, ‘inflammatory’ shock constitutes the hallmark of sepsis, but also a final common pathway of any form of severe, protracted tissue hypoperfusion or cellular poisoning.
Therapeutic interventions targeting microcirculatory and mitochondrial dysfunction are currently lacking, so management of inflammatory shock focuses on treating the macrocirculatory abnormalities while correcting/removing the underlying trigger event. Hypovolaemia is ubiquitous during the early stages of inflammatory shock, due to both external losses and capillary leak. However, even after volume expansion, patients often remain haemodynamically compromised due to myocardial depression and vasoplegia.
Myocardial dysfunction is commonplace during shock states. Systolic and diastolic dysfunction occurs in up to 50 and 25 % of patients with septic shock, respectively [8, 9]. Serum troponin and natriuretic peptides are elevated [10, 11] indicative of both myocardial injury and dysfunction, and both prognosticate for poor outcomes. Myocardial dysfunction is usually reversible in survivors of sepsis, with little or no obvious long-term consequences on cardiac function [12]. Several mechanisms contribute to myocardial depression [8], including reduced numbers and functionality of β1-adrenoreceptors, voltage-activated calcium (Ca2+) channels and ryanodine receptors, resulting in decreased intracellular Ca2+ and less actin–myosin cross-bridge formation. In addition, the sarcoplasmic reticulum has reduced Ca2+ reuptake affecting diastolic relaxation, while myofibrils show reduced Ca2+ sensitivity, and mitochondrial dysfunction makes less energy available for the contraction–relaxation process.
Vascular dysfunction is a hallmark of acute critical illness. Vascular tone and often blood pressure are compromised despite high levels of endogenous and exogenous vasopressors. Mechanisms contributing to vasoplegia include overproduction of vasodilatory mediators, such as nitric oxide and eicosanoids; alterations in the main hormonal axes, with catecholamine hyporesponsiveness, vasopressin deficiency, dysfunction of the hypothalamic–pituitary–adrenal axis and renin–angiotensin–aldosterone system; decreased Ca2+-sensitivity; and activation of vascular smooth muscle ATP-sensitive potassium channels [13–15].
Although the pathogenesis of inflammatory shock is multifactorial and not yet fully understood, it does not include catecholamine deficiency. Endogenous epinephrine and norepinephrine levels in serum are markedly elevated in septic patients [16, 17]. However, catecholamines exert a plethora of other non-haemodynamic effects. They are a key component of the stress response, a finely tuned cardiovascular, metabolic, immune and neurobehavioural process preserved through the course of evolution [18]. While integral to coping with acutely demanding situations, the stress response—and thus catecholamine overload—may be detrimental if its magnitude and/or duration is excessive.
Physiological effects of catecholamines
To better understand how persistently supraphysiological endogenous and/or exogenous catecholamine levels can produce maladaptation in stressful disease states, it is useful to first describe their pleiotropic actions in normal physiology.
Catecholamines function as both neurotransmitters when released into the synaptic space, and hormones when released into the bloodstream. They are produced from tyrosine hydroxylation to DOPA (l-3,4-dihydroxyphenylalanine), with subsequent cell-specific reactions producing dopamine, norepinephrine and epinephrine (Fig. 1). Catecholamines are stored in cytosolic granules and released via a Ca2+-dependent mechanism triggered by the action potential in adrenergic synapses and by sympathetic discharges in the adrenal medulla. Adrenergic receptors are G-protein coupled and comprise α, β and γ subunits. The α-subunit determines the signal transduction pathway, with receptors classified depending upon which α-subunit they contain. Gs and Gi receptors stimulate and inhibit, respectively, the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) pathway, ultimately leading to phosphorylation (Gs) or de-phosphorylation (Gi) of target proteins. Gq receptors stimulate the inositol 1,4,5-triphosphate/diacylglycerol (IP3/DAG) pathway, ultimately increasing intracellular Ca2+ (Fig. 2) [19].

The catecholamine (red) synthesis pathway, with involved enzymes (green) and coenzymes/group donors (blue). The last biosynthetic step is restricted to some adrenergic neurons and to chromaffin cells in the adrenal medulla, and requires the presence of glucocorticoids (adapted from Wurtman [109])

Catecholamines stimulate α1-, α2- and β-adrenoreceptors (red), which are coupled with Gq, Gi and Gs proteins (green), respectively. Signal transduction pathways are exemplified: effector enzymes are shown in orange, second messengers in purple, and green and red arrows indicate stimulation and inhibition, respectively. PLC-β phospholipase C-β, PIP 2 phosphatidylinositol 4,5-bisphosphate, IP 3 inositol 1,4,5-triphosphate, DAG diacylglycerol, PKC protein kinase C, AC adenylate cyclase, AMP adenosine monophosphate, cAMP cyclic adenosine monophosphate, PKA protein kinase A
Central nervous system
Neurons located in the locus coeruleus and the lateral tegmental field represent the core of the noradrenergic system. These receive inputs from, and send outputs to, virtually every region of the central nervous system. All adrenoreceptor subtypes are found within the central nervous system, but α1-receptors predominate. The noradrenergic system is crucial for many physiological (sensory perception and anti-nociception, muscle tone and contraction, modulation of the autonomic nervous system, regulation of body temperature and hormone secretion, sleep–wake cycle) and cognitive (arousal and attention, memory storage and recall, learning and behavioural adaptation) functions. Its alterations are implicated in psychiatric disorders including anxiety, depression and post-traumatic stress [20].
Autonomic nervous system and adrenal medulla
The sympathetic division of the autonomic nervous system originates from the intermediolateral column of the thoracolumbar spinal cord. Axons (preganglionic fibres) leave the spinal cord and enter paravertebral sympathetic ganglia. Here, they stimulate ganglionic neurons, whose axons (postganglionic fibres) form plexuses around the body’s main arteries, entering target organs alongside the vascular supply. At the organ level, they release norepinephrine that binds to α- and β-receptors of smooth muscle and glandular epithelial cells, the ultimate target of the autonomic nervous system. The adrenal medulla constitutes the inner portion of the adrenal gland and is an ectopic sympathetic ganglion; indeed, it is innervated by preganglionic fibres from the 7th–9th thoracic segments. In response to sympathetic stimulation, chromaffin cells release epinephrine and norepinephrine into the circulation at a ratio of 85:15 [21].
Cardiovascular system
Catecholamines increase cardiac output through increasing heart rate and stroke volume via cardiac β1-receptors, and increasing venous return via venous α1-receptors. Vascular tone alters through activation of arteriolar constricting α1-receptors or dilating β2-receptors. Blood pressure, the product of cardiac output and vascular resistance, changes accordingly.
Chronotropism
Catecholamines modulate heart rate through the sinoatrial and atrioventricular nodes. Stimulation of β1-receptors on nodal cells leads to phosphorylation of the sodium (Na+) and Ca2+ channels responsible for the inward “funny” current (I f), leading to an influx of Na+ and Ca2+ and an increased frequency of cell firing.
Inotropism
Activation of cardiomyocyte β1-receptors increases the amount of Ca2+ that enters the cardiomyocyte. Here, Ca2+ binds to troponin C, inducing a conformational change in the troponin complex, allowing actin and myosin to bind. A higher Ca2+ concentration increases the number of actin–myosin bonds, ultimately increasing the force of heart contraction.
Myocardial energetic requirements
Ca2+ entering the cardiomyocyte during each depolarisation must be pumped back outside the cell or into the sarcoplasmic reticulum. As this transport occurs against both electrical and chemical gradients, it requires energy. ATP is also consumed to “re-load” the myosin heads. ATP turnover in cardiomyocytes is extremely high; the heart renews 6 kg of ATP (20 times its own weight) daily. Indeed, cardiomyocytes contain more mitochondria (one-third of their volume) than any other cell type [22]. Catecholamines increase myocardial energy and therefore oxygen requirements as they increase both the amount of ATP required per beat (inotropism) and the number of beats per minute (chronotropism). Catecholamine overload induces cardiomyocyte death in human and animal models, both in vitro and in vivo [23, 24].
Peripheral circulation
As with cardiomyocytes, vascular smooth muscle cell contraction is driven by myosin “loading” and “springing back”. In smooth muscle cells myosin activity is regulated by phosphorylation, provided by myosin light-chain kinase (MLCK). Catecholamines induce either vasoconstriction or vasodilation depending on the receptor they bind to, and, ultimately, upon their effect on MLCK. The α1-adrenoreceptors increase intracellular Ca2+ which, in turn, activates MLCK, thereby inducing contraction. The β2-adrenoreceptors induce production of cAMP, activation of PKA and phosphorylation of MLCK, inducing relaxation. Some vascular beds are relatively insensitive to catecholamines, either because they have fewer adrenoreceptors, or different mechanisms and mediators prevail locally. These beds can self-regulate blood flow over a wide range of blood pressures (e.g. cerebral and renal circulations), or couple flow to cellular metabolic demands (e.g. cerebral and coronary circulations). However, the hepato-splanchnic, muscular and cutaneous circulations depend on mean arterial pressure and local vascular resistance for their perfusion. The effect of catecholamines on a regional circulation depends on the balance between increased cardiac output and systemic arterial pressure on the one hand and regional arteriolar tone on the other.
Gastrointestinal tract
Catecholamines can affect virtually every cell within the gastrointestinal tract. Neurally released norepinephrine influences the enteric nervous system located within the submucosa and muscularis of the splanchnic organs. This can act independently of autonomic control to finely modulate epithelial, smooth muscular and immune cells [25].
The gut also produces catecholamines. Being in part gut-derived, norepinephrine is highly concentrated within the portal circulation [26]. Kupffer cells and hepatocytes are thus exposed to high catecholamine levels, norepinephrine induces cytokine production by Kupffer cells [27] and hepatocellular dysfunction via α2-receptors [28]. Catecholamines also modulate blood flow to the gut and are important mediators in diverting blood flow away from the splanchnic district towards other more needy organs such as the brain, heart and skeletal muscle during, for example, exercise.
Metabolism
Catecholamines induce a catabolic state that is integral to the fight-or-flight response. They promote breakdown of glycogen and triglyceride stores to generate glucose, fatty acids and ketone bodies as ready fuel for heart, brain and skeletal muscle. Catecholamines stimulate lactate release from muscle to provide fuel source for varied organs including brain, liver, heart and kidney [29].
Haemostasis
Sympathetic activation affects haemostasis through inducing release of von Willebrand factor and factor VIII (mediated by β-receptors), and by promoting platelet activation, aggregation and secretion (mediated by both α- and β-receptors). This translates into significantly accelerated blood clotting. Catecholamines stimulate the amplification phase of clot formation and stabilisation so, strictly speaking, they are not prothrombotic but rather induce faster thrombus generation. Thrombus generation has been implicated in the pathogenesis of cardiovascular disease and is likely to occur during critical illness; however, the extent of the phenomenon and its clinical relevance have yet to be determined [30].
Immune system
Adrenergic agents influence virtually every aspect of the innate and adaptive immune response. Immune cells are targeted by the nervous system via exposure to circulating catecholamines, but also via sympathetic innervation of lymphoid organs: bone marrow, lymph nodes, thymus and spleen [31]. Almost all immune cells express (mainly β2-) adrenergic receptors; moreover, they produce considerable amounts of catecholamines, especially when exposed to pathogens [32]. Activation of the sympathetic and parasympathetic nervous systems are, in general, inhibitory on innate immune responses at both systemic and regional levels. On the other hand, peripheral nervous system activation will often amplify local innate immune responses [33]. Catecholamines also modulate proliferation, differentiation and apoptosis of the adaptive immune system cells, as well as cytokine production (see below).
Pathological effects of catecholamines and impact on outcomes
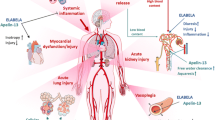
The previous section highlights the crucial role that catecholamines play in health. This can however spil over into harm affecting multiple organ systems. However, among all the pleiotropic actions of catecholamines mentioned above and summarised in Fig. 3, only their cardiovascular effects are routinely monitored and targeted in critically ill patients.

Pleiotropic effects of neurally released (via the sympathetic nervous system) and circulating (produced by the adrenal medulla) catecholamines
The effects of neural activation on the immune system illustrate the potential negativity of excess catecholamines in critical illness. Severe infection represents an obvious stressful state and the innate immune response relies mainly upon non-specific inflammation and phagocyte recruitment to eliminate pathogens. However, catecholamines inhibit the phagocytic capacity of both neutrophils and macrophages in vitro, and impair the ability of neutrophils to generate a respiratory burst [34]. Overall, the in vitro effect of catecholamines can be summarised as an inhibition of adaptive immunity, characterised by generalised lymphopenia—due to inhibition of proliferation of T helper, T cytotoxic and B cells—and a shift in Th1/Th2 balance towards Th2 polarisation, as demonstrated by low Th1/Th2 cell, TNF-α/IL-4 and IFN-γ/IL-4 ratios [35, 36]. If these effects are translated to the in vivo situation, these would appear to be counter-intuitive in combatting infection. On similar lines, catecholamines can promote growth of virtually every bacterial species [37–39], perhaps through increasing iron availability [40]. In addition, they augment bacterial virulence by promoting biofilm formation and virulence-related gene transcription [41], and bacterial recovery following an antibiotic challenge [42]. Catecholamines can mimic bacterial signalling molecules termed “autoinducers” [43]; these operate within the context of bacterial collective decision-making (quorum sensing). Depending upon environmental conditions, bacterial behaviour can change from beneficial or neutral (commensal/saprophytic) to organised host attack (pathogenic) [44]. The interplay between the adrenergic and immune systems and bacteria is indeed highly complex. Indeed, a picture of lymphopenia, a low Th1/Th2 ratio and bacterial overproliferation identical to that induced by catecholamines in vitro are found in vivo in both animal models and patients with stroke-associated infections [45, 46]. High catecholamine levels are associated with more severe lymphopenia, and a greater risk of infection and death [46, 47]. In murine models, β-adrenergic blockade could reverse these immunological and microbiological alterations and improve survival [45]. In critically ill patients, lymphopenia and a low Th1/Th2 ratio are poor prognostic biomarkers [48].
With respect to metabolism, excess catecholamines induce insulin resistance, increase hepatic glycogenolysis and gluconeogenesis, and inhibit glycogen synthesis in skeletal muscle, all of which induce hyperglycaemia [49]. This provides a ready source of glucose substrate in acute stress, but is detrimental if prolonged. The β3-receptors on adipose cells mediate the lipolytic effects of catecholamines by stimulating hormone-sensitive lipase, which breaks down triglycerides to glycerol and fatty acids that are subsequently released into the circulation. Free fatty acids represent an important energy source for the heart; however, their accumulation has both pro-inflammatory [50] and cardiotoxic [51] effects.
The splanchnic circulation is an important vascular bed jeopardised during shock states [52]. Catecholamines, most notably epinephrine, are potent mesenteric vasoconstrictors. While helping to preserve ‘vital’ organ perfusion, they can induce or aggravate gut ischaemia [53] and perhaps contribute to decreased barrier function, with translocation of bacteria and/or toxins [54]. Circulating catecholamines promote leukocyte influx to the intestinal mucosa [55], bacterial–epithelium adhesion [56], bacterial internalisation [57] and virulence (see above).
A hyperadrenergic state is responsible for the reversible myocardial depression that characterises both phaeochromocytoma crisis [58] and the stress-related (Takotsubo) cardiomyopathy [59]. This latter “broken heart” syndrome can be triggered by a physical or emotional upset and is characterised by very high plasma levels of catecholamines and cardiac injury/dysfunction biomarkers such as troponin and B-type natriuretic peptide, echocardiographic abnormalities such as apical ballooning, and variable electrocardiographic changes yet normal coronary arteries. Stress cardiomyopathy can mimic acute coronary syndromes and may lead to heart failure; it is also recognised after isolated brain injury, perhaps representing the ultimate effort of the damaged brain to ensure its own perfusion at any cost [60].
In many other clinical conditions not primarily caused by an adrenergic surge, a persistent stress response can be identified. In fact, numerous examples can be found where adrenergic excess, both endogenous and exogenous, is associated with poor outcome. Catecholaminergic overload is associated with a poor prognosis in acute coronary syndromes, heart failure, liver cirrhosis and acute cerebrovascular disease [61–64]. High catecholamine levels prognosticate worse outcomes in patients with trauma and infection [65, 66] regardless of disease severity, and even in otherwise healthy, high-functioning elderly subjects [67].
Notwithstanding this association with adverse outcomes, adrenergic agonists remain the mainstay of cardiovascular support. Norepinephrine is the current recommended first-line agent for low vascular resistance states, while dobutamine is recommended for myocardial dysfunction [68]. Epinephrine has both inotropic and pressor properties that can be used as an alternative to either [69]. It is likely that these exogenous catecholamines will add further to the endogenous stress response, therefore increasing total adrenergic stress. After adjustments for propensity scoring, dobutamine administration was independently associated with increased mortality in acute heart failure and after cardiac surgery [70, 71]. High levels of endogenous [72] and exogenous [73] catecholamines as well as a persistently high heart rate [74] predict poor patient outcomes in sepsis. While high catecholamine levels could simply be a marker of disease severity, they may also be a perpetrator of further organ dysfunction. Indeed, increasing catecholamine doses were associated with increasing mortality, independent of effects on blood pressure [75]. Even in the setting of cardiac arrest, epinephrine use and dose are independent predictors of poor recovery [76, 77].
Alternatives to catecholamines
The potential iatrogenic contribution of catecholamine administration to poor outcomes demands further study. While useful and even life-saving for short-term restoration of tissue perfusion or correction of life-threatening hypotension, catecholamines—like any drug—can be poisonous when given in excess. Attempting to minimise catecholamine dosing by selecting an appropriate blood pressure target for the individual patient, optimising sedation and other hypotensive/myocardial depressant agents, optimising fluid loading and using alternative approaches should all be given due consideration.
The first step towards reducing adrenergic (over)load is to not necessarily target “normal” or “supranormal” haemodynamic values. While too low a blood pressure or cardiac output may compromise tissue perfusion and oxygenation, neither increasing blood pressure >65 mmHg [78] nor targeting “supranormal” values of cardiac output [79] translated into an overall survival benefit. Indeed, previously normotensive patients trended to worse outcomes when a higher blood pressure was targeted [75]. Similarly, many patients with critical illness have often unrecognized diastolic dysfunction and this may be compromised further by the use of catecholamines [9]. In spite of this evidence, catecholamine overuse is still commonplace, even when the mean arterial pressure is well above the declared targets. In a recent randomised controlled trial, most patients had mean arterial pressure values well above the target range, yet were still receiving high dose of catecholamines despite the study protocol prompting their rapid de-escalation [78].
A variety of non-adrenergic inotropes and vasopressors, and adjunct therapies have been investigated for myocardial depression and vasoplegia in both preclinical and clinical studies (Table 1). These agents also have their own side-effect profiles. Thus, none have yet conclusively demonstrated a clear benefit over adrenergic equivalents, and some studies were stopped prematurely because of harm [80, 81]. However, post hoc analyses do suggest benefit in certain subsets of patients. Options for vasoplegia include vasopressin and its analogues, nitric oxide and eicosanoid modulation [82, 83], angiotensin II [84], inhibition of vascular smooth muscle potassium channels [85], and fever control by external cooling [86]. Despite no overall outcome benefit compared to norepinephrine, low dose AVP reduced catecholamine requirements and offered improved survival rates in patients receiving lower doses of norepinephrine at baseline [87]. Myocardial depression has also been treated with levosimendan or glucose–insulin–potassium therapy; preclinical or small patient studies demonstrate short-term benefits [88, 89]. A randomised controlled trial of 516 patients assessing levosimendan in septic shock is shortly to complete enrolment [90]. In terms of adjunct therapy, corticosteroid therapy has been extensively studied in septic shock; corticosteroids increase adrenergic receptor transcription and thus cardiac [91] and vascular [92] responsiveness to catecholamines, and many critically ill patients have adrenal dysfunction which is prognostically relevant [93]. Clinical trials demonstrated that stress-dose glucocorticoids led to a quicker resolution of shock [94]. While there was no overall survival effect, a benefit was seen in patients with vasopressor-resistant shock, for which corticosteroids are currently recommended [68].
Finally, significant attention has been stimulated by a recent single-centre study from Rome [95] assessing the role of beta-adrenergic blockade in a poor prognosis subset of patients with septic shock, i.e. requiring high doses of catecholamines after 24 h and with a concurrent tachycardia. Those patients randomised to esmolol demonstrated significant reductions in mortality, time on vasopressors, and renal and myocardial injury compared to the control group.
The stress response is highly preserved in different species. From an evolutionary point of view, the organism must be able to cope with physically or psychologically demanding situations. However, as critical illness and management in a critical care unit are characterised by a severe and abnormally prolonged stressor response, this response may become maladaptive. Given this premise, attenuation of an excessive adrenergic component of the stress reaction is a tempting therapeutic option during sepsis and other critically ill states. Pretreatment with β-blockers reduced mortality in animal models [96], while β-blocker use before hospital admission was associated with increased survival rates [97, 98]. During established sepsis in animal models, beta blockade controlled heart rate without reducing stroke volume or blood pressure [99]; furthermore, improved cardiac function, decreased inflammation, preserved intestinal barrier function and improved survival have all been demonstrated [96, 100–103]. In patient studies, titration of β-blocker dosing to a target heart rate appears feasible without compromising haemodynamics in most patients; stroke volume usually increases while catecholamine requirements decrease [95, 104]. Possible mechanisms include improved ventricular filling and ventricular-arterial coupling; restoration of adrenergic receptor density, which may have been reduced by excessive catecholamine stimulation [101, 105]; and a decrease in the systemic inflammatory response [106, 107]. More investigation is required to confirm benefit from beta blockade in sepsis and other critical illness states. Patient selection and close monitoring are likely to be crucial in this setting because of the risk of worsening myocardial dysfunction. Fixed-dose (i.e. not titrated to individual needs) beta blockade can be detrimental [108].
Conclusions
Although some degree of sympathetic activation is required for survival of a patient or animal under the stressful conditions of sepsis, adrenergic overload has several underappreciated side effects that may impact negatively on final outcome. Several strategies exist to avoid catecholamine overstimulation during critical illness, including acceptance of abnormal haemodynamic values that remain compatible with adequate organ perfusion, use of non-catecholamine vasopressors and inotropes, and β-adrenergic blockade. The last of these is a promising therapeutic tool that requires further investigation in order to identify those subset(s) of patients who may either benefit or be harmed from such an intervention.
References
Beutler B, Hoebe K, Du X et al (2003) How we detect microbes and respond to them: the Toll-like receptors and their transducers. J Leukoc Biol 74:479–485
Tang D, Kang R, Coyne CB et al (2012) PAMPs and DAMPs: signals that spur autophagy and immunity. Immunol Rev 249:158–175
Shi Y, Evans JE, Rock KL (2003) Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425:516–521
Abraham E, Singer M (2007) Mechanisms of sepsis-induced organ dysfunction. Crit Care Med 35:2408–2416
Spronk PE, Zandstra DF, Ince C (2004) Bench-to-bedside review: sepsis is a disease of the microcirculation. Crit Care 8:462–468
Brealey D, Brand M, Hargreaves I et al (2002) Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360:219–223
Hochachka PW, Buck LT, Doll CJ et al (1996) Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA 93:9493–9498
Rudiger A, Singer M (2007) Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med 35:1599–1608
Landesberg G, Gilon D, Meroz Y et al (2012) Diastolic dysfunction and mortality in severe sepsis and septic shock. Eur Heart J 33:895–903
Spies C, Haude V, Fitzner R et al (1998) Serum cardiac troponin T as a prognostic marker in early sepsis. Chest 113:1055–1063
Maeder M, Fehr T, Rickli H, Ammann P (2006) Sepsis-associated myocardial dysfunction: diagnostic and prognostic impact of cardiac troponins and natriuretic peptides. Chest 129:1349–1366
Parker MM, Shelhamer JH, Bacharach SL et al (1984) Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med 100:483–490
Kimmoun A, Ducroq N, Levy B (2013) Mechanisms of vascular hyporesponsiveness in septic shock. Curr Vasc Pharmacol 11:139–149
Landry DW, Levin HR, Gallant EM et al (1997) Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation 95:1122–1125
Bucher M, Ittner KP, Hobbhahn J et al (2001) Downregulation of angiotensin II type 1 receptors during sepsis. Hypertension 38:177–182
Woolf PD, Hamill RW, Lee LA et al (1988) Free and total catecholamines in critical illness. Am J Physiol 254:E287–E291
Lin IY, Ma HP, Lin AC et al (2005) Low plasma vasopressin/norepinephrine ratio predicts septic shock. Am J Emerg Med 23:718–724
Chrousos GP (2009) Stress and disorders of the stress system. Nat Rev Endocrinol 5:374–381
Krasel C, Vilardaga JP, Bünemann M et al (2004) Kinetics of G-protein-coupled receptor signalling and desensitization. Biochem Soc Trans 32:1029–1031
Berridge CW, Waterhouse BD (2003) The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev 42:33–84
Perlman RL, Chalfie M (1977) Catecholamine release from the adrenal medulla. Clin Endocrinol Metab 6:551–576
Schaper J, Meiser E, Stämmler G (1985) Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ Res 56:377–391
Ellison GM, Torella D, Karakikes I et al (2007) Acute beta-adrenergic overload produces myocyte damage through calcium leakage from the ryanodine receptor 2 but spares cardiac stem cells. J Biol Chem 282:11397–11409
Karch SB (1987) Resuscitation-induced myocardial necrosis. Catecholamines and defibrillation. Am J Forensic Med Pathol 8:3–8
De Jonge WJ (2013) The gut’s little brain in control of intestinal immunity. ISRN Gastroenterol 2013:630159
Yang S, Koo DJ, Zhou M, Chaudry IH et al (2000) Gut-derived norepinephrine plays a critical role in producing hepatocellular dysfunction during early sepsis. Am J Physiol Gastrointest Liver Physiol 279:G1274–G1281
Zhou M, Das P, Simms HH, Wang P (2005) Gut-derived norepinephrine plays an important role in up-regulating IL-1beta and IL-10. Biochim Biophys Acta 1740:446–452
Yang S, Zhou M, Chaudry IH, Wang P (2001) Norepinephrine-induced hepatocellular dysfunction in early sepsis is mediated by activation of alpha2-adrenoceptors. Am J Physiol Gastrointest Liver Physiol 281:G1014–G1021
Adeva-Andany M, López-Ojén M, Funcasta-Calderón R et al (2014) Comprehensive review on lactate metabolism in human health. Mitochondrion 17:76–100
Von Känel R, Dimsdale JE (2000) Effects of sympathetic activation by adrenergic infusions on hemostasis in vivo. Eur J Haematol 65:357–369
Mignini F, Streccioni V, Amenta F (2003) Autonomic innervation of immune organs and neuroimmune modulation. Auton Autacoid Pharmacol 23:1–25
Flierl MA, Rittirsch D, Nadeau BA, Chen AJ et al (2007) Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature 449:721–725
Sternberg EM (2006) Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol 6:318–328
Wenisch C, Parschalk B, Weiss A et al (1996) High-dose catecholamine treatment decreases polymorphonuclear leukocyte phagocytic capacity and reactive oxygen production. Clin Diagn Lab Immunol 3:423–428
Kohm AP, Sanders VM (2001) Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev 53:487–525
Huang HW, Tang JL, Han XH et al (2013) Lymphocyte-derived catecholamines induce a shift of Th1/Th2 balance toward Th2 polarization. Neuroimmunomodulation 20:1–8
Lyte M, Freestone PP, Neal CP et al (2003) Stimulation of Staphylococcus epidermidis growth and biofilm formation by catecholamine inotropes. Lancet 361:130–135
Freestone PP, Haigh RD, Lyte M (2007) Specificity of catecholamine-induced growth in Escherichia coli O157:H7, Salmonella enterica and Yersinia enterocolitica. FEMS Microbiol Lett 269:221–228
Freestone PP, Hirst RA, Sandrini SM et al (2012) Pseudomonas aeruginosa–catecholamine inotrope interactions: a contributory factor in the development of ventilator-associated pneumonia? Chest 142:1200–1210
Messenger AJ, Barclay R (1983) Bacteria, iron and pathogenicity. Biochem Educ 11:54–62
Sandrini S, Alghofaili F, Freestone PP et al (2014) Host stress hormone norepinephrine stimulates pneumococcal growth, biofilm formation and virulence gene expression. BMC Microbiol 14:180
Freestone PP, Haigh RD, Lyte M (2008) Catecholamine inotrope resuscitation of antibiotic-damaged staphylococci and its blockade by specific receptor antagonists. J Infect Dis 197:1044–1052
Karavolos MH, Winzer K, Williams P et al (2013) Pathogen espionage: multiple bacterial adrenergic sensors eavesdrop on host communication systems. Mol Microbiol 87:455–465
Cogan TA, Thomas AO, Rees LE et al (2007) Norepinephrine increases the pathogenic potential of Campylobacter jejuni. Gut 56:1060–1065
Prass K, Meisel C, Hoflich C et al (2003) Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med 198:725–736
Chamorro A, Urra X, Planas AM (2007) Infection after acute ischemic stroke: a manifestation of brain-induced immunodepression. Stroke 38:1097–1103
Chamorro A, Amaro S, Vargas M et al (2007) Catecholamines, infection, and death in acute ischemic stroke. J Neurol Sci 252:29–35
Wu HP, Chung K, Lin CY, Jiang BY et al (2013) Associations of T helper 1, 2, 17 and regulatory T lymphocytes with mortality in severe sepsis. Inflamm Res 62:751–763
Rizza RA, Cryer PE, Haymond MW et al (1980) Adrenergic mechanisms of catecholamine action on glucose homeostasis in man. Metabolism 29:1155–1163
Savary S, Trompier D, Andréoletti P, Le Borgne F et al (2012) Fatty acids - induced lipotoxicity and inflammation. Curr Drug Metab 13:1358–1370
Kjekshus JK, Mjos OD (1972) Effect of free fatty acids on myocardial function and metabolism in the ischemic dog heart. J Clin Invest 51:1767–1776
Treggiari MM, Romand JA, Burgener D et al (2002) Effect of increasing norepinephrine dosage on regional blood flow in a porcine model of endotoxin shock. Crit Care Med 30:1334–1339
Martikainen TJ, Tenhunen JJ, Giovannini I et al (2005) Epinephrine induces tissue perfusion deficit in porcine endotoxin shock: evaluation by regional CO2 content gradients and lactate-to-pyruvate ratios. Am J Physiol Gastrointest Liver Physiol 288:G586–G592
Coopersmith CM, Stromberg PE, Davis CG et al (2003) Sepsis from Pseudomonas aeruginosa pneumonia decreases intestinal proliferation and induces gut epithelial cell cycle arrest. Crit Care Med 31:1630–1637
Vlisidou I, Lyte M, van Diemen PM et al (2004) The neuroendocrine stress hormone norepinephrine augments Escherichia coli O157:H7-induced enteritis and adherence in a bovine ligated ileal loop model of infection. Infect Immun 72:5446–5451
Chen C, Lyte M, Stevens MP, Vulchanova L et al (2006) Mucosally-directed adrenergic nerves and sympathomimetic drugs enhance non-intimate adherence of Escherichia coli O157:H7 to porcine cecum and colon. Eur J Pharmacol 539:116–124
Green BT, Lyte M, Kulkarni-Narla A et al (2003) Neuromodulation of enteropathogen internalization in Peyer’s patches from porcine jejunum. J Neuroimmunol 141:74–82
Whitelaw BC, Prague JK, Mustafa OG et al (2014) Phaeochromocytoma crisis. Clin Endocrinol 80:13–22
Wittstein IS, Thiemann DR, Lima JA et al (2005) Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 352:539–548
Guglin M, Novotorova I (2011) Neurogenic stunned myocardium and takotsubo cardiomyopathy are the same syndrome: a pooled analysis. Congest Heart Fail 17:127–132
Ostrowski SR, Pedersen SH, Jensen JS et al (2013) Acute myocardial infarction is associated with endothelial glycocalyx and cell damage and a parallel increase in circulating catecholamines. Crit Care 17:R32
Venugopalan P, Argawal AK (2003) Plasma catecholamine levels parallel severity of heart failure and have prognostic value in children with dilated cardiomyopathy. Eur J Heart Fail 5:655–658
Tage-Jensen U, Henriksen JH, Christensen E et al (1988) Plasma catecholamine level and portal venous pressure as guides to prognosis in patients with cirrhosis. J Hepatol 6:350–358
Feibel JH, Hardy PM, Campbell RG et al (1977) Prognostic value of the stress response following stroke. JAMA 238:1374–1376
Johansson PI, Stensballe J, Rasmussen LS et al (2012) High circulating adrenaline levels at admission predict increased mortality after trauma. J Trauma Acute Care Surg 72:428–436
Ostrowski SR, Gaïni S, Pedersen C et al (2015) Sympathoadrenal activation and endothelial damage in patients with varying degrees of acute infectious disease: an observational study. J Crit Care 2015(30):90–96
Reuben DB, Talvi SLA, Rowe JW et al (2000) High urinary catecholamine excretion predicts mortality and functional decline in high-functioning, community-dwelling older persons: MacArthus Studies of Successful Aging. J Gerontol A Biol Sci Med Sci 55:M618–M624
Dellinger RP, Levy MM, Rhodes A et al (2013) Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med 39:165–228
Myburgh JA, Higgins A, Jovanovska A et al (2008) A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med 34:2226–2234
Abraham WT, Adams KF, Fonarow GC et al (2005) In-hospital mortality in patients with acute decompensated heart failure requiring intravenous vasoactive medications: an analysis from the Acute Decompensated Heart Failure National Registry (ADHERE). J Am Coll Cardiol 46:57–64
Shahin J, DeVarennes B, Tse CW et al (2011) The relationship between inotrope exposure, six-hour postoperative physiological variables, hospital mortality and renal dysfunction in patients undergoing cardiac surgery. Crit Care 15:R162
Boldt J, Menges T, Kuhn D et al (1995) Alterations in circulating vasoactive substances in the critically ill: a comparison between survivors and non-survivors. Intensive Care Med 21:218–225
Brown SM, Lanspa MJ, Jones JP et al (2013) Survival after shock requiring high-dose vasopressor therapy. Chest 143:664–671
Leibovici L, Gafter-Gvili A, Paul M et al (2007) Relative tachycardia in patients with sepsis: an independent risk factor for mortality. QJM 100:629–634
Dünser MW, Ruokonen E, Pettilä V et al (2009) Association of arterial blood pressure and vasopressor load with septic shock mortality: a post hoc analysis of a multicenter trial. Crit Care 13:R181
Hagihara A, Hasegawa M, Abe T et al (2012) Prehospital epinephrine use and survival among patients with out-of-hospital cardiac arrest. JAMA 307:1161–1168
Dumas F, Bougouin W, Geri G et al (2014) Is epinephrine during cardiac arrest associated with worse outcome in resuscitated patients? J Am Coll Cardiol 64:2360–2367
Asfar P, Meziani F, Hamel JF et al (2014) High versus low blood-pressure target in patients with septic shock. N Engl J Med 370:1583–1593
Gattinoni L, Brazzi L, Pelosi P et al (1995) A trial of goal-oriented hemodynamic therapy in critically ill patients. SvO2 Collaborative Group. N Engl J Med 333:1025–1032
López A, Lorente JA, Steingrub J et al (2004) Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med 32:21–30
Vincent JL, Privalle CT, Singer M et al (2015) Multicenter, randomized, placebo-controlled phase III study of pyridoxalated hemoglobin polyoxyethylene in distributive shock (PHOENIX). Crit Care Med 43:57–64
De Cruz SJ, Kenyon NJ, Sandrock CE (2009) Bench-to-bedside review: the role of nitric oxide in sepsis. Expert Rev Respir Med 3:511–521
Aronoff DM (2012) Cyclooxygenase inhibition in sepsis: is there life after death? Mediat Inflamm 2012:696897
Chawla LS, Busse L, Brasha-Mitchell E et al (2014) Intravenous angiotensin II for the treatment of high-output shock (ATHOS trial): a pilot study. Crit Care 18:534–539
Lange M, Morelli A, Westphal M (2008) Inhibition of potassium channels in critical illness. Curr Opin Anaesthesiol 21:105–110
Schortgen F, Clabault K, Katsahian S et al (2012) Fever control using external cooling in septic shock: a randomized controlled trial. Am J Respir Crit Care Med 185:1088–1095
Russell JA, Walley KR, Singer J et al (2008) Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 358:877–887
Morelli A, De Castro S, Teboul JL et al (2005) Effects of levosimendan on systemic and regional hemodynamics in septic myocardial depression. Intensive Care Med 31:638–644
Hamdulay SS, Al-Khafaji A, Montgomery H (2006) Glucose–insulin and potassium infusions in septic shock. Chest 129:800–804
Orme RM, Perkins GD, McAuley DF et al (2014) An efficacy and mechanism evaluation study of Levosimendan for the Prevention of Acute oRgan Dysfunction in Sepsis (LeoPARDS): protocol for a randomized controlled trial. Trials 15:199
Saito T, Takanashi M, Gallagher E et al (1995) Corticosteroid effect on early beta-adrenergic down-regulation during circulatory shock: hemodynamic study and beta-adrenergic receptor assay. Intensive Care Med 21:204–210
Sakaue M, Hoffman BB (1991) Glucocorticoids induce transcription and expression of the alpha 1B adrenergic receptor gene in DTT1 MF-2 smooth muscle cells. J Clin Invest 88:385–389
Annane D, Bellissant E (2000) Prognostic value of cortisol response in septic shock. JAMA 284:308–309
Sprung CL, Annane D, Keh D (2008) Hydrocortisone therapy for patients with septic shock. N Engl J Med 358:111–124
Morelli A, Ertmer C, Westphal M et al (2013) Effect of heart rate control with esmolol on hemodynamic and clinical outcomes in patients with septic shock: a randomized clinical trial. JAMA 310:1683–1691
Ackland GL, Yao ST, Rudiger A et al (2010) Cardioprotection, attenuated systemic inflammation, and survival benefit of beta1-adrenoceptor blockade in severe sepsis in rats. Crit Care Med 38:388–394
Christensen S, Johansen MB, Tønnesen E et al (2011) Preadmission beta-blocker use and 30-day mortality among patients in intensive care: a cohort study. Crit Care 15:R87
Macchia A, Romero M, Comignani PD et al (2012) Previous prescription of β-blockers is associated with reduced mortality among patients hospitalized in intensive care units for sepsis. Crit Care Med 40:2768–2772
Aboab J, Sebille V, Jourdain M et al (2011) Effects of esmolol on systemic and pulmonary hemodynamics and on oxygenation in pigs with hypodynamic endotoxin shock. Intensive Care Med 37:1344–1351
Hagiwara S, Iwasaka H, Maeda H et al (2009) Landiolol, an ultrashort-acting beta1-adrenoceptor antagonist, has protective effects in an LPS-induced systemic inflammation model. Shock 31:515–520
Suzuki T, Morisaki H, Serita R et al (2005) Infusion of the beta-adrenergic blocker esmolol attenuates myocardial dysfunction in septic rats. Crit Care Med 33:2294–2301
Mori K, Morisaki H, Yajima S et al (2011) Beta-1 blocker improves survival of septic rats through preservation of gut barrier function. Intensive Care Med 37:1849–1856
Wilson J, Higgins D, Hutting H, Serkova N et al (2013) Early propranolol treatment induces lung heme-oxygenase-1, attenuates metabolic dysfunction, and improves survival following experimental sepsis. Crit Care 17:R195
Morelli A, Donati A, Ertmer C et al (2013) Microvascular effects of heart rate control with esmolol in patients with septic shock: a pilot study. Crit Care Med 41:2162–2168
Heilbrunn SM, Shah P, Bristow MR et al (1989) Increased beta-receptor density and improved hemodynamic response to catecholamine stimulation during long-term metoprolol therapy in heart failure from dilated cardiomyopathy. Circulation 79:483–490
Berk JL, Hagen JF, Dunn JM (1970) The role of beta adrenergic blockade in the treatment of septic shock. Surg Gynecol Obstet 130:1025–1034
Gore DC, Wolfe RR (2006) Hemodynamic and metabolic effects of selective beta1 adrenergic blockade during sepsis. Surgery 139:686–694
Schmitz D, Wilsenack K, Lendemanns S et al (2007) Beta-adrenergic blockade during systemic inflammation: impact on cellular immune functions and survival in a murine model of sepsis. Resuscitation 72:286–294
Wurtman RJ (1966) Control of epinephrine synthesis by the pituitary and adrenal cortex: possible role in the pathophysiology of chronic stress. Recent Adv Biol Psychiatry 9:359–368
Acknowledgments
The authors are thankful to Fabio Zugni, MD, for invaluable technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Andreis, D.T., Singer, M. Catecholamines for inflammatory shock: a Jekyll-and-Hyde conundrum. Intensive Care Med 42, 1387–1397 (2016). https://doi.org/10.1007/s00134-016-4249-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-016-4249-z