
Abstract
Key message
Glycine soja germplasm can be used to successfully introduce new alleles with the potential to add valuable new genetic diversity to the current elite soybean gene pool.
Abstract
Given the demonstrated narrow genetic base of the US soybean production, it is essential to identify beneficial alleles from exotic germplasm, such as wild soybean, to enhance genetic gain for favorable traits. Nested association mapping (NAM) is an approach to population development that permits the comparison of allelic effects of the same QTL in multiple parents. Seed yield, plant maturity, plant height and plant lodging were evaluated in a NAM panel consisting of 392 recombinant inbred lines derived from three biparental interspecific soybean populations in eight environments during 2016 and 2017. Nested association mapping, combined with linkage mapping, identified three major QTL for plant maturity in chromosomes 6, 11 and 12 associated with alleles from wild soybean resulting in significant increases in days to maturity. A significant QTL for plant height was identified on chromosome 13 with the allele increasing plant height derived from wild soybean. A significant grain yield QTL was detected on chromosome 17, and the allele from Glycine soja had a positive effect of 166 kg ha−1; RIL’s with the wild soybean allele yielded on average 6% more than the lines carrying the Glycine max allele. These findings demonstrate the usefulness and potential of alleles from wild soybean germplasm to enhance important agronomic traits in a soybean breeding program.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soybean [Glycine max (L.) Merr] is one of the most important oilseed crops produced worldwide (FAOSTAT 2018). Over the past three decades, the total global production of soybean has tripled to > 300 million metric tons in 2016/17 (FAOSTAT 2018). Genetic improvements in the yield potential of modern soybean varieties and the implementation of improved management practices have enabled enormous increases in productivity from 700 kg ha−1 in 1924 to more than 3000 kg ha−1 in 2017 (USDA 2018). Recent studies in the USA have estimated genetic gains in soybean yield range from 12 to 27 kg ha−1 year−1 over the last century, close to 1% year−1 (Rowntree et al. 2013; Rincker et al. 2014; Rogers et al. 2015), while average yield increases worldwide were 1.3% per year (Ray et al. 2013). A pedigree analysis indicated that most modern US public cultivars could be traced back to 35 ancestral lines (Gizlice et al. 1994). Therefore, the lack of genetic diversity in modern soybean breeding programs is of major concern and focus for ensuring continued genetic gains in soybean grain yield well into the future.
Soybean was domesticated from its wild progenitor (Glycine soja Sieb. and Zucc.) 3000–5000 years ago in China (Carter et al. 2004; Doebley et al. 2006). During the domestication process, more than half of the genetic diversity found in G. soja was lost, decreasing pairwise diversity (π) from 2.94 × 10−3 in Glycine soja to 1.05 × 10−3 in modern cultivars (Hyten et al. 2006; Zhou et al. 2015). Currently, it is estimated that ~ 8500 unique accessions of G. soja and 45,000 accessions of Asian landraces of G. max are maintained in germplasm banks around the world (Hyten et al. 2006; Wen et al. 2009). The USDA Germplasm Resources Information Network (GRIN) collection contains ~ 1100 G. soja accessions (Song et al. 2015). However, only a small fraction of the publicly available G. soja germplasm has been used in commercial breeding aimed at improving elite soybean variety development.
Wild soybean exhibits diverse undesirable agronomic characteristics such as excessive lodging, late flowering, small black seeds, hard seed coat reducing uniform germination and emergence, a lack of uniformity for plant maturity and excessive pod shattering (Liu et al. 2007). Despite the many unfavorable traits found in G. soja germplasm, useful genetic diversity have been reported within G. soja germplasm including those associated with resistance to soybean aphid (Aphis glycines) (Zhang et al. 2017c) and soybean cyst nematode (Heterodera glycines) (Wang et al. 2001; Winter et al. 2007; Zhang et al. 2017b) and with tolerance to salt and drought (Lee et al. 2009; Chen et al. 2013; Ning et al. 2017; Nisa et al. 2017), and with improved seed composition such as increased protein content and improved fatty acid and amino acid profiles (Ha et al. 2014; Yan et al. 2014; Leamy et al. 2017; La et al. 2019).
Although much work has been done recently to understand the implications of domestication as well as the genetic architecture of wild soybean (Liu et al. 2007; Zhou et al. 2015), little research has been done to investigate the potential of wild soybean to be used for improving grain yield. Ertl and Fehr (1985) evaluated two populations derived from G. soja × G. max backcrosses across two years and four environments and observed no significant grain yield increases when comparing recombinant inbred lines (RILs) to the G. max recurrent parent. Akpertey et al. (2014) reported G. soja-derived lines that were not significantly different in yield form the G. max parent. In another study using a similar approach, Concibido et al. (2003) mapped QTL associated with grain yield from G. soja on chromosome 14. The yield QTL significantly increased yield by 9.4% when averaged across all test environments. Li et al. (2008) utilized two backcrossed populations from G. max × G. soja and identified a QTL associated with grain yield on chromosome 5 (Satt511). Grain yield of the lines carrying the G. soja allele were averaged across all environments, and those lines demonstrated a 6.3% yield increase over lines carrying the G. max recurrent parent allele. Thus, a small amount of evidence exists that G. soja germplasm may harbor useful genes for improving the agronomic performance of modern day soybean cultivars, but more research is clearly needed.
Quantitative trait loci (QTL) mapping and genome-wide association studies (GWAS) have been used extensively to dissect the genetic architecture of agronomic traits (Holland 2007). One drawback of QTL mapping is that the estimated QTL interval is usually relatively large, due to the relatively large and extensive linkage disequilibrium (LD) present in these soybean populations, and the traditionally low marker density throughout the genome (Xu et al. 2017). QTLs are often population- and environment-specific, which can generate non-reproducible results in non-related environments, populations and individuals (Kang et al. 2009; Bernardo 2010). As an alternative, GWAS relies on historical LD to increase the analysis power (Zhu et al. 2008) and to detect marker associations. GWAS studies have identified nucleotide variants associated with complex traits in soybeans including seed oil and protein content (Hwang et al. 2014; Cao et al. 2017), flowering time and maturity (Zhang et al. 2015; Mao et al. 2017) and soybean cyst nematode resistance (Heterodera glycines) (Zhang et al. 2016). However, this method has a relatively low power to detect associations with rare alleles and can be confounded by population structure (Price et al. 2010; Auer and Lettre 2015). Accordingly, maize geneticists developed an approach to overcome some of the pitfalls of both conventional GWAS and linkage mapping techniques (Yu et al. 2008). The design consisted of multi-cross recombinant inbred lines (RILs) where each family has a unique parent crossed with a common “hub” parent, (or “founder”). The NAM design combines aspects of two genetic approaches: linkage mapping through genetic recombination and association mapping through historical linkage disequilibrium presented in the populations, improving the resolution and the power for detection of rare alleles in the analysis (Yu et al. 2008).
The SoyNAM project (http://www.soybase.org/SoyNAM) has the objective of understanding the genetic architecture of seed yield in soybean using NAM RILs derived from crosses between IA3023 (hub parent) and 40 different G. max adapted cultivars specifically selected for improved grain yield. A significant QTL for canopy coverage identified on chromosome 19 was associated with an increase in grain yield of ~ 50 kg ha−1 (Xavier et al. 2017). In another study using the same NAM panel, Xavier et al. (2018) identified a genomic region positively associated with grain yield stability across a range of environments located on chromosome 18, and insight was gained about the QTL × environment interaction prevalent in most reported QTL for grain yield. Again using the same soybean NAM panel, Diers et al. (2018) identified 23 significant regions associated with seed yield and 29 associated with seed weight, demonstrating the complexity of grain yield-related traits. NAM populations have also been used to dissect complex traits in other important crop species such as rice (Oryza sativa) (Fragoso et al. 2017), barley (Hordeum vulgare) (Sharma et al. 2018) and wheat (Triticum aestivum) (Bajgain et al. 2016). Although the large G. max NAM panel contained exotic germplasm in the population structure, there continues to be a need also to investigate the potential of alleles from wild soybean utilizing modern population structures. The objectives of this study were to identify genomic regions associated with complex agronomic traits in wild soybean germplasm and to identify alleles from G. soja accessions that may be used to improve agronomic traits in soybean using a nested association mapping panel.
Materials and methods
Germplasm development and phenotyping
Our NAM panel contains 392 F4-derived RILs, developed from three biparental cross-combinations between the G. max hub parent Williams 82 (Bernard and Cremeens 1988) and three G. soja plant introductions PI458536, PI464890B and PI522226. The G. soja accessions were among eight diverse accessions selected for crossing that year based on origin and extreme values for plant and seed composition traits. PI458536 (MG 0) was from Heilongjiang, China, and had a protein concentration greater than 53 g kg−1 and moderate resistance to Soybean Mosaic Virus. PI464890B (MG I) originated from Jilin, China, and had moderate resistance to Soybean Mosiac Virus. PI522226 (MG 000) came from Primorye, Russia, and had extremely narrow leaflets. The four parental lines were obtained from the USDA Soybean Germplasm Collection. The crosses were made in Urbana, IL, during the summer of 2010, and the true F1 hybrid plants were grown at the same environment in 2011 and bulk harvested. Approximately, 10,000 F2 plants were grown at the Bradford Research Center in Columbia, MO, during the summer of 2012 for each of the three populations, and F2 plants were bulk harvested. In 2013, approximately, 100,000 F3 plants were grown at the Bradford Research Center in Columbia, MO, for each of the three populations, and ~ 500 single plants were harvested separately from each F3 population. The 500 F3 single plants were selected during the fall of 2013 within each population based on a visual selection of appropriate maturity date, upright plant growth and resistance to pod shattering. The F3:4 plant rows were grown at the Bay Farm Research Facility in Columbia, MO, during 2014, and a single plant was harvested from each row. Single plants within a row were selected based on critical agronomic traits needed for yield evaluation including maturity date, erect plant growth, pod shattering resistance and resistance to soybean Bacterial Pustule (Xanthomonas axonopodis pv. glycines). The F4:5 plant rows were grown at the Bay Farm Research Facility in Columbia, MO, during the summer of 2015, and entire plant rows were again selected based on agronomic characteristics that would enable high-quality yield evaluations. Each plant row was bulk harvested for development of the F4-derived RILs. Only lines with sufficient seed for multi-environment testing were carried forward, and RILs were randomly chosen to represent each of the three populations to reduce the NAM panel size to a manageable field experiment.
During the growing seasons of 2016 and 2017, RILs were planted in four-row plots with a row spacing of 76 cm, a row length of 3.6 m and a 1.2 m fallow alley. The environments included the Hundley-Whaley Research Center in Albany, MO (40° 14′ 53″ N 94° 19′ 51″ W), the Bay Farm Research Facility in Columbia, MO (38° 57′ 50″ N 92° 19′ 42″ W), the Greenley Research Center in Novelty, MO (39° 57′ 28.23″ N 92° 10′ 40.45″ W) and the Graves-Chapple Research Center in Rock Port, MO (40° 16′ 18.09″ N 95° 28′ 26.38″ W) (Supplementary Fig. 1). The plots were arranged in an augmented incomplete block experimental design with one replication in 2016 and two replications in 2017, with two check cultivars per block in both years, IA3023 for MG III and IA4005 for MG IV. Genotypes were randomly assigned to a block and replication, and blocks were randomly ordered in the field across all environments. The experiment was divided into two field blocks at each environment for MG III and MG IV RILs to accommodate combine harvesting.
Grain yield was measured by harvesting the two center rows of each four-row plot with an Almaco R1 or an Almaco SPC-40 plot combine (Nevada, Iowa). The total seed weight and seed moisture were measured in real time on the combine, and the final grain yield was calculated as kg ha−1 on a 13% moisture basis. Plant maturity was recorded as the day when 95% of the pods in the center two rows of each plot reached final color (R8 stage) (Fehr et al. 1971) and considering September 1 as day one. Lodging was rated for the center two rows of each plot at R8 stage according to the following score: (1) all plants erect; (2) 5–25% of the plants prostrate; (3) 25–50% of the plants prostrate; (4) 50–80% of the plants prostrate; (5) all plants prostrate. Plant height was recorded as the average distance (cm) between the top node and the soil surface of plants in the center two rows of each plot.
Genotyping and SNP analysis
Genomic DNA was extracted from young trifoliate leaf tissue of ten random plants for each RIL and the respective parents during the V4/V5 growth stage. A Qiagen Dneasy Plant 96 kit (QIAGEN, Valencia, CA) was used for high-quality DNA extraction, and the protocol given for the kit was followed. DNA samples were submitted to the Soybean Genomics and Improvement Laboratory, USDA-ARS, for genotyping using the Illumina Infinium BARCSoySNP6K BeadChip (Song et al. 2013). Genotypes were called using the software Genome Studio (Illumina, San Diego, California USA). SNPs with less than 90% call rate were excluded, and SNPs were imputed using the LD-KNNi method implemented in version 5 of Tassel software (Bradbury et al. 2007). A total of 5786 SNPs remained for subsequent quality control, association mapping and linkage analysis. The R package ‘ParentOffspring’ (Abdel-Haleem et al. 2013), and a principal component analysis using version 5 of the TASSEL software (Bradbury et al. 2007) were used to eliminate RILs that were found to exhibit genotypes inconsistent with expected genetic similarity (≥ 80%) to the respective parents.
Only the filtered SNP marker data were used to measure the linkage disequilibrium (LD) in the NAM panel using the sliding window method implemented in version 5 of TASSEL software. The LD SNPs were estimated using the pairwise squared correlation (r2), and then they were plotted against SNP pair distances to obtain the rate of the LD decay in the heterochromatin and euchromatin region (Song et al. 2013). We calculated the fixation index (FST) among the three populations in the NAM panel using the unbiased estimator (Weir and Cockerham 1984) implemented in the NAM package (Xavier et al. 2015).
Nested association mapping
Best linear unbiased estimators (BLUE) were calculated to account for non-genetic sources of variance associated with environmental variation. BLUE values were then used in the nested association mapping according to the model adopted by Jarquín et al. (2014): \(y_{ijkl} = \mu + g_{i} + cb_{l} + l_{j} + gl_{ij} + r_{k(j)} + b_{l(k)} + e_{ijk}\), where \(\mu\) is the trait mean, \(g_{i}\) is the genetic effect of the genotypes, \(cb_{k}\) is the interaction effect between the checks and incomplete blocks, \(l_{j}\) is the effect of environments, \(gl_{ij}\) is the interaction effect between the genotypes and environments, \(r_{k(j)}\) is the effect of the replications nested in environment, \(b_{l(k)}\) is the incomplete block effect nested within replicate, and \(e_{ijkl}\) is the residual. Broad-sense heritability (H2) on an entry-mean basis was calculated as follows: \(H^{2} = \sigma_{\text{g}}^{2} /(\sigma_{\text{g}}^{2} + \sigma_{\text{ge}}^{2} /l + \sigma_{\text{e}}^{2} /rl)\), where \(\sigma_{\text{g}}^{2}\), \(\sigma_{\text{ge}}^{2}\) and \(\sigma_{\text{e}}^{2}\) are the genetic variance, genotype × environment variance and error variance; \(l\) is the number of environments and \(r\) is the number of replications (Fehr 1991). Narrow-sense heritability was calculated using the genomic relationship matrix from GBLUP model (de los Campos et al. 2015) as follows: \(h^{2} = \sigma_{\text{a}}^{2} /\sigma_{\text{y}}^{2}\), where \(\sigma_{\text{a}}^{2}\) is the additive variance estimated from GBLUP model, and \(\sigma_{\text{y}}^{2}\) is the phenotypic variance (Tan et al. 2017).
Nested association mapping was conducted using the R package NAM (Xavier et al. 2015) developed from the SoyNAM project. The mixed linear model designed for multiple parent intercross populations was used for the SNP and haplotype-based association (Wei and Xu 2016): \(y = \mu + X\alpha + g + \text{cov} + e\), where \(\mu\) is the intercept, \(X\) is the allele matrix from SNP/haplotype data and family information, \(\alpha\) is the SNP/haplotype effects, \(g\) is the population structure effect, cov is the covariate, and \(e\) is the residual effect. The R8 date was used as a covariate in the NAM analysis to account for maturity effects on grain yield, plant height and lodging. A false discovery rate (FDR) threshold at \(\alpha\) ≤ 0.05 level was used to declare SNP significant in the association mapping.
Linkage mapping analysis
We used the consensus genetic linkage map constructed by Song et al. (2016) based on 21,478 SNPs mapped in the Williams 82 × PI479752 (G. soja) population to define SNP positions. QTL mapping was performed using the composite interval mapping (CIM) functionality qtl package in R (Broman and Sen 2009) individually in each population of the NAM panel. To fit multiple QTL models, we used the function fitqtl which drops one QTL at a time. The logarithm of the odds (LOD) for defining QTL significance was determined by a 1000 permutation test analysis at \(\alpha\) ≤ 0.05 significant level and estimated confidence intervals of QTL via used 1.5-LOD support intervals. Additive allelic effects for NAM and CIM analysis were estimated by family relative to the common parent (Williams 82).
We searched for candidate genes, published genes, and published QTLs using the soybean reference genome available at SoyBase (www.soybase.org) in the Wm82.a2.v1 assembly. The SNPs and QTL intervals that were highly associated with traits were considered as a potential region for candidate genes. SoyBase was also used to search for QTLs previously found to affect the traits.
Results
Phenotypic variation, linkage disequilibrium and population structure
The analysis of variance (ANOVA) results for all traits evaluated in this study are shown in Supplementary Table 1. Significant genetic differences were found for all measured traits, and both environmental effects and RIL × environment interaction were significant (p < 0.01) for all the traits. The entry-mean-based estimates of heritability (h2) were 0.51–0.64 for grain yield (GY), 0.87–0.93 for plant maturity (PM), 0.82–0.93 for plant height (PH) and 0.85–0.91 for lodging score (LDG).
There was a broad, continuous and normal distribution for all phenotypic traits (Supplementary Table 1 and Supplementary Fig. 2) measured in this study, and transgressive segregation was observed in both directions when comparing the phenotypic values of the hub parent (Williams 82) and RILs, suggesting the effect of multiple genes controlling the traits and appropriate distribution of alleles among hub parent and G. soja parents to move forward with subsequent mapping efforts to identify marker associations for significant QTL.
Linkage disequilibrium (LD) decay varied between euchromatin and heterochromatin regions (Fig. 1a). The LD decayed to half its maximum r2 value (0.44) at an average 619.5 kb in euchromatin and 892.6 kb in heterochromatin. Since we estimated LD in experimental biparental populations, a greater extent of LD is expected when comparing expected LD to natural soybean population (Zhou et al. 2015) due to the limited amount of recombination events and selection in experimental population development (Hyten et al. 2007). Principal component analysis (PCA) was used to evaluate the variation in genotypic data of the NAM population (Fig. 2b). The PCA could not distinguish clearly the subpopulation structures likely due to the half-sib family relationship among the three subpopulations, yet PC1 explained 10.1% of the variation, while PC2 explained 6.3% of the genotypic variation.

Linkage disequilibrium (LD) decay of the NAM panel (a), distribution of the NAM RILs under PC1 and PC2 (b), and fixation index of the NAM panel over the entire genome (c), where the red line represents the 99th percentile (0.29)

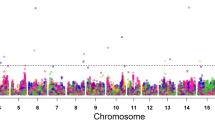
Manhattan plots of nested association mapping (NAM) analysis for grain yield, plant maturity, plant height and lodging plotted against positions on each of the 20 chromosomes, across four environments within 2016 (red dots) and 2017 (blue dots). The significant SNPs were distinguished by the FDR 0.05 (3.8 × 10−5) threshold line (color figure online)
More than 70% of the SNPs had an estimated FST value smaller than 0.1 (Fig. 1c). Just 0.02% of the SNPs had an FST value larger than the 99th percentile (red line). To distinguish the SNPs under selection and potential genetic drift in the NAM population, we utilized the smoothing kernel (blue lines) procedure implemented in the NAM package (Flori et al. 2009; Xavier et al. 2015). The results revealed significant FST values on chromosomes 3, 5, 6, 9, 11 and 20. The SNPs ss715594391, ss715593858 and ss715594468 on chromosome 6 had the highest FST values 0.67, 0.66 and 0.63, respectively. ss715593858 is ~ 400 kb from the region of E1 (Glyma06g23026) (Xia et al. 2012), a well-known photoperiod response/plant maturity gene. This is likely a result of fixing the maturity date during the inbreeding process.
Nested association mapping
A QTL region was determined by the marker with the greatest LOD score presented in the region associated with a trait. Additive allelic effects were estimated by family relative to the hub parent (Williams 82) where a positive effect represents an increase in the value of the trait as a result of substituting the hub parent (G. max) allele with the respective G. soja allele, and a negative effect decreases the value of the trait when the hub parent allele is substituted with the G. soja allele. Four SNPs on Chr 16 were significantly associated with grain yield (GY) in 2017 across the average of four environments (Fig. 2a), with allelic effects ranging from − 116.2 to 59.4 kg ha−1. This marker and the associated locus has been identified to harbor (~ 200 kb) the gene responsible for regulating pod shattering, Pdh1 (Funatsuki et al. 2014). Hence, the relatively large negative allelic effects on grain yield.
Forty-nine SNPs significantly associated with plant maturity (PM) were identified on chromosomes 6, 11 and 12 in 2016 and 2017 (Fig. 2b). These regions were consistently associated with the trait across environments, and SNPs associated with PM on Chr 6 and 12 were observed in all the eight environments, and in Albany 2016 (ALB2016), Columbia 2016 (CLM2016) and Novelty (NOV2016) for the region on Chr 11 (Supplementary Fig. 3b). The allelic effect estimates for PM ranged from − 2.0 to 5.7 days, depending on the SNP associated and population (Table 1 and Supplementary Table 2). A higher frequency of estimated positive PM alleles was evident from the G. soja founder parents. The three most significant SNPs associated with PM on Chr 6 were ss715594029, ss715593853 and ss715594119. These SNPs were co-located in the same region (~ 350 kb apart) as the known E1 photoperiod response/plant maturity gene (Xia et al. 2012). Also, the genomic region associated with PM on Chr 11 and 12 were reported to be correlated with flowering/maturity time (Zhang et al. 2015; Fang et al. 2017; Mao et al. 2017), and two homologs GmPRR3A (~ 30 kb) and GmPRR3B (~ 100 kb) have recently been cloned which underlie these regions (Li et al. 2019).
The three regions associated with PM (ss715593853 on Chr 6, ss715608829 on Chr 11 and ss715613171 on Chr 12) explained 36%, 16% and 18% of the genetic variation, respectively (Fig. 3a–c). The RIL carrying the genotype CC (G. soja) at ss715593853 exhibited a positive increment of 4.3 days in PM compared to the TT (G. max) genotype (Fig. 3a). A similar trend was observed for ss715608829, and ss715613171, where the lines carrying the G. soja allele were 3.6 and 3.9 days higher PM than the lines carrying G. max allele, respectively (Fig. 3b, 3c). The allelic effect for the SNPs on Chr 6, 11 and 12 varies among G. soja parents.

Difference in genotypic values (a–c) between lines segregating at SNP ss715593853 (a), ss715608829 (b), and ss715613171 (c) associated with plant maturity in the NAM panel. The difference in genotypic values of plant height associated with SNP ss715616047 (d), and lodging associated with SNP ss715602804 (e), between lines in the NAM panel across eight environments. The difference in genotypic values between lines segregating at SNP ss715625973 for grain yield at Pop3-Williams 82 × PI522226 across eight environments (f). The least square difference (LSD) and differences in mean (∆m), and the probability of a greater F statistic (Pr > F) and R2 are in the figures
Seven SNPs were found to be associated with plant height (PH) across four environments in 2016 and 2017 (Fig. 2c), one SNP on Chr 10 and six on Chr 13. The region on Chr 13 was identified in five of the eight environments across years (Supplementary Fig. 3c), with an allelic effect ranging from − 2.86 to 13.07 cm (Supplementary Table 2). The most significant SNP was ss715616047, which explained 12% of the genetic variation and exhibited an increase in PH of 19.6 cm when comparing lines carrying the AA (G. soja) genotype to the CC (G. max) genotype. The region on Chr 13 was located ~ 200 Kb of qPH13.1; a confirmed QTL associated with increased plant height in G. soja (Zhang et al. 2018).
A novel region on Chr 8 (ss715602804 and ss715602662) was significantly associated with lodging (LDG) in 2016. In 2017, four SNPs were associated with LDG, ss715579524 on Chr 1, ss715602804 on Chr 8, ss715630642 on Chr 18 and ss715633186 on Chr 19 (Table 1). The QTL region associated with ss715602804 was more stable across environments and was also identified at Novelty 2016 and Columbia 2017. The region on Chr 8 was responsible for explaining 17% of the phenotypic variation, and lines carrying the TT allele (G. soja) exhibited a significant increase in 0.46 in LDG score when compared to the lines with the CC allele (G. max) (Fig. 3e).
Linkage mapping
Twelve QTLs across eleven chromosomes in three biparental populations were identified using the composite interval mapping (CIM) in 2016 and 2017 (Fig. 4). Three novel QTL were associated with GY on Chr 17, 18 and 20 (Fig. 4a); four QTL with PM on Chr 6, 11, 12 and 20 (Fig. 4b); three QTL with PH on Chr 1, 2 and 13 (Fig. 4c); and one QTL associated with LDG on Chr 9 (Fig. 4d).

Graphical display of significant QTL detected in three populations from composite interval mapping (CIM), for a grain yield, b plant maturity, c plant height and (d) lodging across eight environments. Pop1-Williams 82 × PI464890B; Pop2-Williams 82 × PI458536; Pop3-Williams 82 × PI522226. The dashed dark-red horizontal line indicates the threshold logarithm of the odds (LOD) based on a 1000 permutation test
Four QTL were detected for PM in multiple environments in 2016 and 2017 (Supplementary Fig. 4c-d). A significant QTL on Chr 6 was detected in all three populations, and the phenotypic variance ranged from 28.3 to 52.6% dependent on the specific G. soja parental line. The additive effect of qPM-6-1, qPM-6-2 and qPM-6-3 ranged from 2.34 to 5.5 days (Table 2). Three minor QTLs were detected in specific populations; qPM-11 on Chr 11 in Williams 82 × PI464890B (Pop1), qPM-12 on Chr 12 in Williams 82 × PI522226 (Pop3) and qPM-20 on Chr 20 in Pop1. The confidence interval for the QTL qPM-6-1, qPM-6-2, qPM-6-3, qPM-11 and qPM-12 overlapped with the SNPs associated with PM on chromosomes 6, 11 and 12 in the NAM analysis (Table 1). Additive effect represents the impact of substituting the G. max allele with the G. soja allele for each respective SNP. The QTL qPM-20 was responsible for 12.5% of the phenotypic variation with a negative effect of − 2.87 days on the trait. qPM-20 was found close to the reported position of the E4 gene (Liu et al. 2008). Also, an epistatic interaction was significant between qPM-6-1 × qPM-20 in 2017, accounting for more than 5.6% of the phenotypic variation for PM.
Three QTL were identified for PH in multiple environments on Chr 1, 2 and 13 (Fig. 4c). qPH-1 on Chr 1 was observed in the population Williams 82 × PI458536 (Pop2), qPH-2 on chromosome 2 was identified in the population Williams 82 × PI464890B (Pop2), qPH-1 accounted for 18.7% of the variation, while qPH-2 accounted for 15.2% to 20.1% of the phenotypic variation (Table 2). The G. soja alleles for both qPH-1 and qPH-2 showed a negative effect on PH, from − 8.9 to − 14.2 cm. qPH-13 was identified in the population Williams 82 × PI522226 (Pop3), and the phenotypic variance explained was 23.3% with the G. soja alleles having a positive effect of 11.87 cm. The qPH-13 interval overlapped with the SNPs previously associated with PH on chromosome 13 in the NAM analysis (Table 1) that region had G. soja alleles associated with greater plant height (Fig. 3d).
One QTL on chromosome 9 was associated with LDG in the population Williams 82 × PI458536 (Pop2) (Fig. 4d and Table 2). The phenotypic variation explained for qLDG-9 was 14.3%, and the additive effect for the G. soja alleles was 0.24 on the 1–5 scale. The qLDG-9 interval region overlaps a QTL (Lodging 5–9) reported by Lee et al. (1996).
Three QTL associated with GY were identified on chromosomes 17, 18 and 20 explained 24.4%, 21.2% and 17.5% of the total phenotypic variance, with additive effects for the G. soja alleles ranging from − 297.2 to 166.1 kg ha−1 (Table 2). Only for qGY-17 did G. soja alleles have a positive impact on GY, and this was found across two environments in 2016: Columbia and Albany. Lines carrying the G. soja allele (BB) presented an increment of 197.1 kg ha−1 or 6% in grain yield when comparing to the lines carrying G. max allele (AA) across all environments (Fig. 3f).
Discussion
In this study, we evaluated a nested association mapping panel by using both linkage mapping in three biparental populations, as well as association mapping with the entire panel of RILs. The NAM analysis identified more markers associated with traits than the CIM approach. The NAM design has been successfully used for gaining an understanding of the genetic architecture for several traits in soybean, as well as in other crops (Yu et al. 2008; Fragoso et al. 2017; Li et al. 2017; Diers et al. 2018; Xavier et al. 2018). One advantage of the NAM design is that it allows for comparing allelic effects for the same marker from multiple parents, in relation to a common parent, enabling the distinction of allelic effects in our study for each G. soja parent relative to the Williams 82 (hub parent) (Xavier et al. 2015). The allelic effect varies between G. soja parents for markers associated with traits in the NAM panel, similar to the results observed in other NAM studies (Diers et al. 2018; Chen et al. 2019). In general, the parent-specific additive effects are smaller for the NAM analysis than the effects observed using the CIM method. It is important to mention that the CIM method comprises a relatively small number of RILs compared to the full NAM panel, which likely increases the risk of overestimating the individual QTL effect (Maurer et al. 2017; Vatter et al. 2017). From that perspective, it is important to evaluate the wild soybean allelic effects across the NAM panel and within each family for a better understanding of the genetic background and environment effect, and a truer estimate of allelic effects relative to the hub parent.
QTL associated with plant maturity
Four QTL associated with PM in the NAM panel were identified. The region associated with PM on Chr 6 (E1 gene-ss715593853/qPM-6-1, 6-2, 6-3) (Xia et al. 2012) was identified in all individual populations and in the NAM panel across all environments, and showed the largest allelic effects (Fig. 3b, Supplementary Fig. 4c-d, and Table 1). When comparing the allelic effects of ss715593853, the differences between G. soja allele (E1) and G. max (e1-as) were 4.3 days. The minor allele frequency (MAF) for the significant SNPs was relatively high, ranging from 0.15 to 0.50. The NAM design was robust enough to detect associations and dissect the genetic architecture of complex traits as compared to standard GWAS (Buckler et al. 2009; Diers et al. 2018). Also, the E1 gene was found to be under strong selection in our NAM panel, revealed by the highest FST values by SNPs surrounding the gene locus (Fig. 2c). E1 is part of the phytochrome A signaling pathway and down-regulated GmFT2a and GmFT5a, orthologs of Arabidopsis FLOWERING LOCUS T responsible for early flowering (Kong et al. 2010). Xia et al. (2012), through positional cloning located the position of E1 (Glyma.06g207800), and characterize E1 as functional, and two non-functional alleles e1-fs and e1-nl, and one not fully functional e1-as. These results impact breeding strategies for maturity and soybean adaptation in different regions in the US. Langewisch et al. (2017) evaluated a series of combinations of E1 and e1-as from maturity group (MG) 000 to X and showed a higher frequency of e1-as allele in MG IV and below and a higher allele frequency of E1 in MG V and above. E1 promotes late flowering and may be used in the southern US and e1-as promotes earlier flowering and may be used in the northern US in MG 0 to IV (Wolfgang and An 2017; Langewisch et al. 2017).
The PM QTL on Chr 12 (ss715613171) was identified in seven of eight environments, and in both mapping methods. The allelic effects for the association analysis were higher in the Pop3, ranging from 0.62 to 1.30 days. In the CIM analysis, qPM-12 was specific to Pop3 and explained ~ 22% of the phenotypic variation presented in the population for the trait. SNPs associated with the first flower were reported in the same region by Mao et al. (2017) and Fang et al. (2017). The PM QTL on Chr 11 (ss715608829/qPM-11) was detected in the association analysis and for Pop1 in the CIM analysis. The largest allelic effects were observed on Pop1 in 2016 (1.14–1.35 days). Li et al. (2017) reported a QTL for days to flowering (qDTF-11-4) using a NAM panel in the same region with an additive effect of 1.87 days. Two homologs GmPRR3A (Glyma11g15580) and GmPRR3B (Glyma12g07861) controlling growth period of soybean have recently been cloned which underlie regions associated with PM on chromosomes 11 and 12 (Li et al. 2019).
A QTL on chromosome 20 (qPM-20) was significantly associated with PM in the CIM analysis, explained 12.5% of the PM variation, and the allele form the G. soja parent had a negative effect of − 2.9 days. QTLs are many times population specific which makes the results less reproducible in diverse genetic background (Bernardo 2010). The qPM-20 is ~ 4 Mb apart from the maturity gene E4 that encodes the phytochrome protein GmphyA2 (Glyma.20g090000) (Liu et al. 2008), and recessive alleles of E4 provide earlier flowering (Xu et al. 2013), which is the phenotype observed in RILs which inherited the G. soja alleles of this QTL.
QTL associated with plant height and plant lodging
Wild soybeans are well-known for expressing excessive branching, extreme lodging and a prostrate growth habit. Until now, more than 239 and 87 QTLs have been reported to be associated with plant height (PH) and lodging (LDG) in soybean, respectively (http://www.soybase.org, “SoyBase browser,” verified 01/20/2019). We identified a significant QTL for PH on Chr 13 in both mapping methods (ss715616047/qPH-13). The lines carrying the G. soja allele showed a significant increase in PH compared to the lines carrying the G. max allele (Fig. 3d). Our findings were supported by Zhang et al. (2018), as they fine mapped a QTL from G. soja in the same region of ss715616047/qPH-13, which exhibited a greater PH for the lines that carried the wild allele. RT-PCR showed that Glyma.13g249400 demonstrated higher expression in genotypes that expressed higher PH. Also, sequence data identified a difference in six amino acids from Glyma.13g249400 between parents (Zhang et al. 2018).
Two SNPs were associated with LDG on Chr 8. The SNP ss715602804 showed a significant increase of 0.46 units in the LDG score in the lines that carried the G. soja allele (Fig. 2e). Another QTL was identified in our linkage analysis on Chr 9 (Fig. 4d and Table 2), qLDG-9 expressed a positive allelic effect of 0.24 units in the LDG score. Introgression of wild soybean alleles in currently elite germplasm may result in linkage drag of unfavorable traits, such as excessive plant growth and lodging (Zhang et al. 2017a). However, this issue could be fixed with backcrossing with more adaptable germplasm and more cycles of selection (Ertl and Fehr 1985; Concibido et al. 2003).
QTL associated with grain yield
The QTL associated with GY on Chr 16 harbors the shattering gene Pdh1 (Funatsuki et al. 2014), with the G. soja allele associated with adverse effects on the trait. Pdh1 increases pod dehiscence by promoting the torsion of the pod decreasing yield (Funatsuki et al. 2014). Breeding for shattering resistance (pdh1) allowed soybean production to expand to more arid conditions (Bandillo et al. 2017), and the selection for pdh1 soybean varieties was crucial for the expansion and the success of the industrialized soybean system (Funatsuki et al. 2014).
A novel QTL associated with grain yield (qGY-17) showed a significant 6% increase in GY for the G. soja allele as compared to the G. max allele when averaged across all RIL’s and environments, although the QTL was only significant in Pop3 (Williams 82 × PI522226) and across two environments. Similar results were found by Concibido et al. (2003) using backcross populations from G. soja × G. max, which identified positive effect of G. soja allele in two of six populations evaluated. Also, a QTL (qYIE-D2-1) in the same region was detected by Wang et al. (2014), and SNP markers were identified for pod number and seed weight (Hu et al. 2014; Yan et al. 2017). Further evaluation using different genetic background and environments will be necessary to understand better the impact of the qGY-17 on grain yield. Assessment of large yield testing in G. soja × G. max populations is challenging due to the presence of numerous deleterious agronomic traits (shattering, lodging, vine-grown) which result in difficulties in mechanic harvesting of plots.
Moreover, an impressive number of G. soja × G. max RILs yielded significantly more than the G. max parent (Williams 82) (Fig. 5 and Supplementary Table 3). Among those SA15-109, SA15-66476, SA15-63422, SA15-66599, SA15-63423, SA15-66563 and SA15-63650 yielded between 4 and 17% more than the Williams 82 (depending on the environment). The high yielding line SA15-109, from Williams 82 × PI458536, yielded 400 kg ha−1 more and matured one day later than Williams 82 across four environments in 2017. In Rock Port 2017, this line yielded 725 kg ha−1 more and matured on the same day as Williams 82. Another high yielding line SA15-66476, from the population Williams 82 × PI522226, yielded 420 kg ha−1 more and matured two days earlier than Williams 82 in 2016 and 2017 in the combined analysis (Fig. 5). Ertl and Fehr (1985) and Wang et al. (2004) were unsuccessful in increasing the yield potential in interspecific crosses using G. soja. On the other hand, Concibido et al. (2003) and Li et al. (2008) identified positive grain yield QTLs from G. soja crosses. A major outcome of this study was the identified positive transgressive segregates for grain yield using G. soja in the pedigree, and the best line significantly outperformed the Glycine max parent (Williams 82) by 400 kg ha−1 across all environments. Similar results were expected to be achieved using G. max × G. max crosses; however, using single crosses of G. max × G. soja is quite a surprise.

Selected G. soja-derived RILs exhibiting significantly higher grain yield than the NAM hub parent Williams 82 for test MG III and MG IV combined over four environments in 2016 (a), 2017 (b) and both years (2016 + 2017) (c)
Summary
A total of 17 QTLs with alleles from G. soja that are associated with increased and decreased grain yield, extended plant maturity, increased plant height, and increased lodging were identified by the nested association mapping and composite interval mapping. A novel QTL for grain yield on chromosome 17 from G. soja was detected in the CIM analysis, and lines carrying the wild soybean allele presented an increase of 6% in grain yield. There have been very few reports in the long history of soybean genetics of QTL associated with increased grain yield derived from interspecific crosses with G. soja, and our results suggest further investigation is warranted to understand the potential utility of this QTL on breeding. Regions associated with plant maturity were identified on Chr 11, and 12 and the photoperiod response/plant maturity gene E1 was confirmed on Chr 6. A significant QTL associated with PH was identified on Chr 13, exhibiting increased plant height. The use of wild soybean germplasm for commercial breeding purposes is still quite rare for breeders, mainly due to difficulties in working with undomesticated plant germplasm. Our results indicate breeding with G. soja germplasm can successfully introduce new alleles and add valuable new beneficial alleles with the potential to increase valuable new genetic diversity to the current elite soybean gene pool.
References
Abdel-Haleem H, Ji P, Boerma HR, Li Z (2013) An R package for SNP marker-based parent-offspring tests. Plant Methods 9:44. https://doi.org/10.1186/1746-4811-9-44
Akpertey A, Belaffif M, Graef GL et al (2014) Effects of selective genetic introgression from wild soybean to soybean. Crop Sci 54:2683–2695. https://doi.org/10.2135/cropsci2014.03.0189
Auer PL, Lettre G (2015) Rare variant association studies: considerations, challenges and opportunities. Genome Med 7:16. https://doi.org/10.1186/s13073-015-0138-2
Bajgain P, Rouse MN, Tsilo TJ et al (2016) Nested association mapping of stem rust resistance in wheat using genotyping by sequencing. PLoS ONE 11:e0155760. https://doi.org/10.1371/journal.pone.0155760
Bandillo NB, Anderson JE, Kantar MB et al (2017) Dissecting the genetic basis of local adaptation in soybean. Sci Rep 7:17195. https://doi.org/10.1038/s41598-017-17342-w
Bernard RL, Cremeens CR (1988) Registration of ‘Williams 82’ soybean. Crop Sci 28:1027–1028. https://doi.org/10.2135/cropsci1988.0011183X002800060049x
Bernardo R (2010) Breeding for quantitative traits in plants, 2nd edn. Stemma Press, Woodbuty
Bradbury PJ, Zhang Z, Kroon DE et al (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Broman KW, Sen S (2009) A guide to QTL mapping with R/qtl. Springer, New York
Buckler ES, Holland JB, Bradbury PJ et al (2009) The genetic architecture of maize flowering time. Science 325(80):714–718. https://doi.org/10.1126/science.1174276
Campos G, Sorensen D, Gianola D (2015) Genomic heritability: what is it? PLoS Genet 11:1–21. https://doi.org/10.1371/journal.pgen.1005048
Cao Y, Li S, Wang Z et al (2017) Identification of major quantitative trait loci for seed oil content in soybeans by combining linkage and genome-wide association mapping. Front Plant Sci 8:1–10. https://doi.org/10.3389/fpls.2017.01222
Carter TE, Hymowitz T, Nelson RL (2004) Biogeography, local adaptation, Vavilov, and genetic diversity in soybean. In: Werner D (ed) Biological resources and migration. Springer, Berlin, pp 47–59
Chen P, Yan K, Shao H, Zhao S (2013) Physiological mechanisms for high salt tolerance in wild soybean (Glycine soja) from Yellow River Delta, China: photosynthesis, osmotic regulation, ion flux and antioxidant capacity. PLoS ONE 8:e83227. https://doi.org/10.1371/journal.pone.0083227
Chen Q, Yang CJ, York AM et al (2019) TeoNAM: a nested association mapping population for domestication and agronomic trait analysis in Maize. bioRxiv 647461. https://doi.org/10.1534/genetics.119.302594
Concibido V, La Vallee B, Mclaird P et al (2003) Introgression of a quantitative trait locus for yield from Glycine soja into commercial soybean cultivars. Theor Appl Genet 106:575–582. https://doi.org/10.1007/s00122-002-1071-5
Diers BW, Specht J, Rainey KM et al (2018) Genetic architecture of soybean yield and agronomic traits. G3 Genes Genomes Genet. https://doi.org/10.1534/g3.118.200332
Doebley JF, Gaut BS, Smith BD (2006) The molecular genetics of crop domestication. Cell 127:1309–1321. https://doi.org/10.1016/j.cell.2006.12.006
Ertl DS, Fehr WR (1985) Agronomic performance of soybean genotypes from Glycine max × Glycine soja Crosses1. Crop Sci 25:589–592. https://doi.org/10.2135/cropsci1985.0011183X002500040003x
Fang C, Ma Y, Wu S et al (2017) Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol 18:161. https://doi.org/10.1186/s13059-017-1289-9
FAOSTAT (2018) Food and Agriculture Organization of the United Nations Statistical Database. http://www.fao.org/faostat/en/#data/QC. Accessed 14 Oct 2018
Fehr WR (1991) Principles of cultivar development: theory and technique, vol 1. Macmillan, New York
Fehr WR, Caviness CE, Burmood DT, Pennington JS (1971) Stage of development descriptions for soybeans, Glycine max (L.) Merrill1. Crop Sci 11:929. https://doi.org/10.2135/cropsci1971.0011183x001100060051x
Flori L, Fritz S, Jaffrézic F et al (2009) The genome response to artificial selection: a case study in dairy cattle. PLoS ONE 4:e6595. https://doi.org/10.1371/journal.pone.0006595
Fragoso CA, Moreno M, Wang Z et al (2017) Genetic architecture of a rice nested association mapping population. G3 Genes Genomes Genet (Bethesda) 7:1913–1926. https://doi.org/10.1534/g3.117.041608
Funatsuki H, Suzuki M, Hirose A et al (2014) Molecular basis of a shattering resistance boosting global dissemination of soybean. Proc Natl Acad Sci USA 111:17797–17802. https://doi.org/10.1073/pnas.1417282111
Gizlice Z, Carter TE, Burton JW (1994) Genetic base for North American Public soybean cultivars released between 1947 and 1988. Crop Sci 34:1143. https://doi.org/10.2135/cropsci1994.0011183X003400050001x
Ha B-K, Kim H-J, Velusamy V et al (2014) Identification of quantitative trait loci controlling linolenic acid concentration in PI483463 (Glycine soja). Theor Appl Genet 127:1501–1512. https://doi.org/10.1007/s00122-014-2314-y
Holland J (2007) Genetic architecture of complex traits in plants. Curr Opin Plant Biol 10:156–161. https://doi.org/10.1016/j.pbi.2007.01.003
Hu Z, Kan G, Zhang G et al (2014) Genetic diversity analysis using simple sequence repeat markers in soybean. Plant Genet Resour 12:S87–S90. https://doi.org/10.1017/S1479262114000331
Hwang E-Y, Song Q, Jia G et al (2014) A genome-wide association study of seed protein and oil content in soybean. BMC Genom 15:1–12. https://doi.org/10.1186/1471-2164-15-1
Hyten DL, Song Q, Zhu Y et al (2006) Impacts of genetic bottlenecks on soybean genome diversity. Proc Natl Acad Sci USA 103:16666–16671. https://doi.org/10.1073/pnas.0604379103
Hyten DL, Choi IY, Song Q et al (2007) Highly variable patterns of linkage disequilibrium in multiple soybean populations. Genetics 175:1937–1944. https://doi.org/10.1534/genetics.106.069740
Jarquín D, Kocak K, Posadas L et al (2014) Genotyping by sequencing for genomic prediction in a soybean breeding population. BMC Genom 15:740. https://doi.org/10.1186/1471-2164-15-740
Kang S-T, Kwak M, Kim H-K et al (2009) Population-specific QTLs and their different epistatic interactions for pod dehiscence in soybean [Glycine max (L.) Merr.]. Euphytica 166:15–24. https://doi.org/10.1007/s10681-008-9810-6
Kong F, Liu B, Xia Z et al (2010) Two coordinately regulated homologs of FLOWERING LOCUS T are involved in the control of photoperiodic flowering in soybean. Plant Physiol 154:1220–1231. https://doi.org/10.1104/pp.110.160796
La T, Large E, Taliercio E et al (2019) Characterization of select wild soybean accessions in the USDA germplasm collection for seed composition and agronomic traits. Crop Sci 59:1–19. https://doi.org/10.2135/cropsci2017.08.0514
Langewisch T, Lenis J, Jiang GL et al (2017) The development and use of a molecular model for soybean maturity groups. BMC Plant Biol 17:91. https://doi.org/10.1186/s12870-017-1040-4
Leamy LJ, Zhang H, Li C et al (2017) A genome-wide association study of seed composition traits in wild soybean (Glycine soja). BMC Genom 18:18. https://doi.org/10.1186/s12864-016-3397-4
Lee SH, Bailey MA, Mian MAR et al (1996) Molecular markers associated with soybean plant height, lodging, and maturity across locations. Crop Sci 36:728–735. https://doi.org/10.2135/cropsci1996.0011183X003600030035x
Lee J-D, Shannon JG, Vuong TD, Nguyen HT (2009) Inheritance of salt tolerance in wild soybean (Glycine soja Sieb. and Zucc.) Accession PI483463. J Hered 100:798–801. https://doi.org/10.1093/jhered/esp027
Li D, Pfeiffer TW, Cornelius PL (2008) Soybean QTL for yield and yield components associated with alleles. Crop Sci 48:571. https://doi.org/10.2135/cropsci2007.06.0361
Li S, Cao Y, He J et al (2017) Detecting the QTL-allele system conferring flowering date in a nested association mapping population of soybean using a novel procedure. Theor Appl Genet 130:2297–2314. https://doi.org/10.1007/s00122-017-2960-y
Li MW, Liu W, Lam HM, Gendron JM (2019) Characterization of two growth period QTLs reveals modification of PRR3 genes during soybean domestication. Plant Cell Physiol 60:407–420. https://doi.org/10.1093/pcp/pcy215
Liu B, Fujita T, Yan Z-H et al (2007) QTL mapping of domestication-related traits in soybean (Glycine max). Ann Bot 100:1027–1038. https://doi.org/10.1093/aob/mcm149
Liu B, Kanazawa A, Matsumura H et al (2008) Genetic redundancy in soybean photoresponses associated with duplication of the phytochrome A gene. Genetics 180:995–1007. https://doi.org/10.1534/genetics.108.092742
Mao T, Li J, Wen Z et al (2017) Association mapping of loci controlling genetic and environmental interaction of soybean flowering time under various photo-thermal conditions. BMC Genom 18:415. https://doi.org/10.1186/s12864-017-3778-3
Maurer A, Sannemann W, Léon J, Pillen K (2017) Estimating parent-specific QTL effects through cumulating linked identity-by-state SNP effects in multiparental populations. Heredity (Edinb) 118:477–485. https://doi.org/10.1038/hdy.2016.121
Ning W, Zhai H, Yu J et al (2017) Overexpression of Glycine soja WRKY20 enhances drought tolerance and improves plant yields under drought stress in transgenic soybean. Mol Breed 37:19. https://doi.org/10.1007/s11032-016-0614-4
Nisa Z, Mallano A, Yu Y et al (2017) GsSNAP33, a novel Glycine soja SNAP25-type protein gene: improvement of plant salt and drought tolerances in transgenic Arabidopsis thaliana. Plant Physiol Biochem 119:9–20. https://doi.org/10.1016/j.plaphy.2017.07.029
Price AL, Zaitlen NA, Reich D, Patterson N (2010) New approaches to population stratification in genome-wide association studies. Nat Rev Genet 11:459–463. https://doi.org/10.1038/nrg2813
Ray DK, Mueller ND, West PC, Foley JA (2013) Yield trends are insufficient to double global crop production by 2050. PLoS ONE 8:e66428. https://doi.org/10.1371/journal.pone.0066428
Reinprecht Y, Poysa VW, Yu K et al (2006) Seed and agronomic QTL in low linolenic acid, lipoxygenase-free soybean (Glycine max (L.) Merrill) germplasm. Genome 49:1510–1527. https://doi.org/10.1139/g06-112
Rincker K, Nelson R, Specht J et al (2014) Genetic improvement of U.S. soybean in maturity groups II, III, and IV. Crop Sci 54:1419–1432. https://doi.org/10.2135/cropsci2013.10.0665
Rogers J, Chen P, Shi A et al (2015) Agronomic performance and genetic progress of selected historical soybean varieties in the southern USA. Plant Breed 134:85–93. https://doi.org/10.1111/pbr.12222
Rowntree SC, Suhre JJ, Weidenbenner NH et al (2013) Genetic gain × management interactions in soybean: I. Planting date. Crop Sci 53:1128. https://doi.org/10.2135/cropsci2012.03.0157
Sharma R, Draicchio F, Bull H et al (2018) Genome-wide association of yield traits in a nested association mapping population of barley reveals new gene diversity for future breeding. J Exp Bot 69:3811–3822. https://doi.org/10.1093/jxb/ery178
Song Q, Hyten DL, Jia G et al (2013) Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS ONE 8:e54985. https://doi.org/10.1371/journal.pone.0054985
Song Q, Hyten DL, Jia G et al (2015) Fingerprinting soybean germplasm and its utility in genomic research. G3 Genes Genomes Genet (Bethesda) 5:1999–2006. https://doi.org/10.1534/g3.115.019000
Song Q, Jenkins J, Jia G et al (2016) Construction of high resolution genetic linkage maps to improve the soybean genome sequence assembly Glyma1.01. BMC Genomics 17:33. https://doi.org/10.1186/s12864-015-2344-0
Specht JE, Chase K, Macrander M et al (2001) Soybean response to water: a QTL analysis of drought tolerance. Crop Sci 41:493–509. https://doi.org/10.2135/cropsci2001.412493x
Tan B, Grattapaglia D, Martins GS et al (2017) Evaluating the accuracy of genomic prediction of growth and wood traits in two Eucalyptus species and their F1 hybrids. BMC Plant Biol. https://doi.org/10.1186/s12870-017-1059-6
United Department of Agriculture (USDA) (2018) National agriculture statistics. https://quickstats.nass.usda.gov/results. Accessed 23 Sept 2018
Vatter T, Maurer A, Kopahnke D et al (2017) A nested association mapping population identifies multiple small effect QTL conferring resistance against net blotch (Pyrenophora teres f. teres) in wild barley. PLoS ONE. https://doi.org/10.1371/journal.pone.0186803
Wang D, Diers BW, Arelli PR, Shoemaker RC (2001) Loci underlying resistance to Race 3 of soybean cyst nematode in Glycine soja plant introduction 468916. Theor Appl Genet 103:561–566. https://doi.org/10.1007/PL00002910
Wang D, Procopiuk AM, Diers BW, Graef GL (2004) Identification of putative QTL that underlie yield in interspecific soybean backcross populations. TAG Theor Appl Genet 108:458–467. https://doi.org/10.1007/s00122-003-1449-z
Wang X, Jiang G-L, Green M et al (2014) Identification and validation of quantitative trait loci for seed yield, oil and protein contents in two recombinant inbred line populations of soybean. Mol Genet Genomics 289:935–949. https://doi.org/10.1007/s00438-014-0865-x
Wei J, Xu S (2016) A random-model approach to QTL mapping in multiparent advanced generation intercross (MAGIC) populations. Genetics 202:471–486. https://doi.org/10.1534/genetics.115.179945
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution (N Y) 38:1358. https://doi.org/10.2307/2408641
Wen Z, Ding Y, Zhao T-J, Gai J (2009) Genetic diversity and peculiarity of annual wild soybean (G. soja Sieb. et Zucc.) from various eco-regions in China. Theor Appl Genet 119:371–381. https://doi.org/10.1007/s00122-009-1045-y
Winter SMJ, Shelp BJ, Anderson TR et al (2007) QTL associated with horizontal resistance to soybean cyst nematode in Glycine soja PI464925B. Theor Appl Genet 114:461–472. https://doi.org/10.1007/s00122-006-0446-4
Wolfgang G, An YC (2017) Genetic separation of southern and northern soybean breeding programs in North America and their associated allelic variation at four maturity loci. Mol Breed 37:8. https://doi.org/10.1007/s11032-016-0611-7
Xavier A, Xu S, Muir WM, Rainey KM (2015) NAM: association studies in multiple populations. Bioinformatics 31:btv448. https://doi.org/10.1093/bioinformatics/btv448
Xavier A, Hall B, Hearst AA et al (2017) Genetic architecture of phenomic-enabled canopy coverage in Glycine max. Genetics 206:1081–1089. https://doi.org/10.1534/genetics.116.198713
Xavier A, Jarquin D, Howard R et al (2018) Genome-wide analysis of grain yield stability and environmental interactions in a multiparental soybean population. G3 Genes Genomes Genet (Bethesda) 8:519–529. https://doi.org/10.1534/g3.117.300300
Xia Z, Watanabe S, Yamada T et al (2012) Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc Natl Acad Sci 109:E2155–E2164. https://doi.org/10.1073/pnas.1117982109
Xu M, Xu Z, Liu B et al (2013) Genetic variation in four maturity genes affects photoperiod insensitivity and PHYA-regulated post-flowering responses of soybean. BMC Plant Biol 13:91. https://doi.org/10.1186/1471-2229-13-91
Xu Y, Li P, Yang Z, Xu C (2017) Genetic mapping of quantitative trait loci in crops. Crop J 5:175–184. https://doi.org/10.1016/j.cj.2016.06.003
Yan L, Xing L-L, Yang C-Y et al (2014) Identification of quantitative trait loci associated with soybean seed protein content using two populations derived from crosses between Glycine max and Glycine soja. Plant Genet Resour 12:S104–S108. https://doi.org/10.1017/S1479262114000379
Yan L, Hofmann N, Li S et al (2017) Identification of QTL with large effect on seed weight in a selective population of soybean with genome-wide association and fixation index analyses. BMC Genom 18:1–11. https://doi.org/10.1186/s12864-017-3922-0
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178:539–551. https://doi.org/10.1534/genetics.107.074245
Zhang J, Song Q, Cregan PB et al (2015) Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genom 16:217. https://doi.org/10.1186/s12864-015-1441-4
Zhang H, Li C, Davis EL et al (2016) Genome-wide association study of resistance to soybean cyst nematode (Heterodera glycines) HG type 2.5.7 in wild soybean (Glycine soja). Front Plant Sci 7:1–11. https://doi.org/10.3389/fpls.2016.01214
Zhang H, Mittal N, Leamy LJ et al (2017a) Back into the wild—apply untapped genetic diversity of wild relatives for crop improvement. Evol Appl 10:5–24
Zhang H, Song Q, Griffin JD, Song B-H (2017b) Genetic architecture of wild soybean (Glycine soja) response to soybean cyst nematode (Heterodera glycines). Mol Genet Genomics 292:1257–1265. https://doi.org/10.1007/s00438-017-1345-x
Zhang S, Zhang Z, Bales C et al (2017c) Mapping novel aphid resistance QTL from wild soybean, Glycine soja 85-32. Theor Appl Genet 130:1941–1952. https://doi.org/10.1007/s00122-017-2935-z
Zhang X, Wang W, Guo N et al (2018) Combining QTL-seq and linkage mapping to fine map a wild soybean allele characteristic of greater plant height. BMC Genom 19:226. https://doi.org/10.1186/s12864-018-4582-4
Zhou Z, Jiang Y, Wang Z et al (2015) Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol 33:408–414. https://doi.org/10.1038/nbt.3096
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome J 1:5. https://doi.org/10.3835/plantgenome2008.02.0089
Acknowledgements
The authors would like to acknowledge funding from the Missouri Soybean Merchandising Council and the United Soybean Board. We also thank personnel from the soybean breeding program at the University of Missouri for their time and effort in preparing and conducting the experiments in 2016 and 2017.
Author information
Authors and Affiliations
Contributions
EB conducted field evaluations and data analysis; AMS acquired funding and supervised the work; QS performed the genotyping; RN developed the initial populations; GS revised the manuscript; JDG, TB, and JD provided statistical expertise and revised the manuscript; EB, AMS, and JG wrote the paper. All authors read the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Albrecht E. Melchinger.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Randall Nelson was retired from USDA-Agricultural Research Service.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Beche, E., Gillman, J.D., Song, Q. et al. Nested association mapping of important agronomic traits in three interspecific soybean populations. Theor Appl Genet 133, 1039–1054 (2020). https://doi.org/10.1007/s00122-019-03529-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03529-4