
Abstract
Annual wild soybean (Glycine soja Sieb. et Zucc.) is believed to be a potential gene source for future soybean improvement in coping with the world climate change for food security. To evaluate the wild soybean genetic diversity and differentiation, we analyzed allelic profiles at 60 simple-sequence repeat (SSR) loci and variation of eight morph-biological traits of a representative sample with 196 accessions from the natural growing area in China. For comparison, a representative sample with 200 landraces of Chinese cultivated soybean was included in this study. The SSR loci produced 1,067 alleles (17.8 per locus) with a mean gene diversity of 0.857 in the wild sample, which indicated the genetic diversity of G. soja was much higher than that of its cultivated counterpart (total 826 alleles, 13.7 per locus, mean gene diversity 0.727). After domestication, the genetic diversity of the cultigens decreased, with its 65.5% alleles inherited from the wild soybean, while 34.5% alleles newly emerged. AMOVA analysis showed that significant variance did exist among Northeast China, Huang-Huai-Hai Valleys and Southern China subpopulations. UPGMA cluster analysis indicated very significant association between the geographic grouping and genetic clustering, which demonstrated the geographic differentiation of the wild population had its relevant genetic bases. In comparison with the other two subpopulations, the Southern China subpopulation showed the highest allelic richness, diversity index and largest number of specific-present alleles, which suggests Southern China should be the major center of diversity for annual wild soybean.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
It is well known that the cultivated soybean, Glycine max (L.) Merr., was domesticated from its annual wild relative Glycine soja Sieb. et Zucc. in China. The wild soybean has its widest geographical distribution from 53° to 24° north latitude and from 134° to 97° east longitude in China (Li 1993). During the long term of evolution, the species has accumulated a wide range of genetic variation adapted to the geographic, abiotic and biotic environmental conditions. Therefore, the wild soybean germplasm should be a potential genetic reservoir in the improvement of cultivated soybean, especially for coping with today’s worldwide climate change. Since there was no crossability barrier observed between the two species, G. soja might be easily used in soybean breeding programs for the improvement of phenological, morphological and agronomic traits, including resistances and tolerances to biotic and abiotic stresses. Currently, there are about 10,000 accessions of G. soja in global ex situ collections and perhaps no more than 8,500 unique accessions worldwide. Wild soybean collections are considerably smaller than cultivated soybean collections of 23,000 accessions in Chinese gene bank. Since it has been noted that the annual wild soybean habitats are confronting disturbance and fragmentation along with the human population increase and intensive land reclamation, the public society and the government in China have called attention to further collection and conservation of native wild soybeans. For a rational collection strategy as well as a sustainable utilization of the resources, a thorough evaluation of the collected wild accessions for their genetic diversity and differentiation should be a basic step.
Most of the previous studies on G. soja were based on morphological traits and isozymes (Zhuang et al. 1996; Kiang et al. 1992; Li et al. 1995; Pei et al. 1996). Dong et al. (2001) analyzed the phenotypic diversity for the entire collection of G. soja in China and recognized three major centers of diversity in China: the Northeast, the Yellow River Valley and the Southeast Coasts. They indicated that morphological differences among G. soja populations might relate to adaptation to specific environments. Pei et al. (1996) studied four natural populations each with 120 individuals from North China using seven isozyme loci and indicated that a high degree of intra-population variation and inter-population diversification existed in the four G. soja populations. Li et al. (1995) obtained a similar result that both intra-sample and inter-sample differentiation existed in and among 14 samples collected on a hillside in Jinghua, Zhejiang, China by using six isozyme loci data. Because of the limited information provided by isozyme analysis and the influence of growing environment on morphological evaluation, these methods of assessing genetic diversity have been replaced primarily with DNA marker analysis. Li and Nelson (2002) studied 40 accessions of G. max and 40 accessions of G. soja from four provinces in China using randomly amplified polymorphic DNA (RAPD). They indicated that the genetic distance within the G. soja group was larger than that within the G. max group and that the groups formed with cluster analysis generally coincided with their geographic regions of origin. Xu et al. (2002) reported that simple sequence repeat (SSR) marker analysis of chloroplast DNA also showed wild soybean genotypes exhibited regional distributions, which indicated the extensive geographic differentiation at the level of chloroplast DNA. Chen and Nelson (2004) studied the genetic relationship among 92 G. soja, G. max and semi-wild accessions using 137 RAPD markers. They found the soja group had the largest within-cluster genetic distance, whereas the semi-wild group had the smallest within-cluster genetic distance and the latter had a closer relationship to max than to soja. They also found that the soja accessions collected from the same pasture (field) could be similar or genetically quite distinct and, therefore, suggested that multiple samples may be needed from within soja populations to completely capture the genetic diversity of the population. It seems that most of the previous studies have their limitations either in that the sample size of plant materials used was relatively small and mostly from limited regions rather than from the entire country, or in that the data were often incongruent due to different materials or environmental conditions involved. However, Gai et al. (2000) used 194 accessions of G. soja from the whole growing areas, including three eco-regions, i.e. Northeast China, Huang-Huai-Hai Valleys and Southern China, and evaluated the chloroplast and mitochondria DNA variation among accessions using RFLP markers. They concluded that the wild soybean differed remarkably from the cultivated ones in allele frequency, and the Southern China accessions showed the highest cytoplasmic genetic diversity. From the above, although genetic diversity studies on wild soybean have been recognized by some researchers, few studies on genetic differentiation and peculiarity of countrywide wild soybeans in China have been reported.
Simple sequence repeats (SSRs) are tandem repeats of nucleotide units, commonly di-nucleotide or tri-nucleotide repeats, but also compound and imperfect or interrupted motifs. The inheritance of microsatellite alleles from one generation to another follows the co-dominant Mendelian pattern, thus allowing the reconstruction of crosses and the understanding of genealogical histories. Uses of SSR loci as polymorphic DNA markers have become widely accepted for many areas of G. max genome analysis, including genetic diversity, population structure and genetic mapping (Powell et al. 1996; Narvel et al. 2000; Diwan and Cregan 1997; Abe et al. 2003; Wang et al. 2006; Wang and Takahata 2007; Cregan et al. 1999; Song et al. 2004). More than 1,000 SSR markers have been explored and mapped in G. soja involved mapping populations (Song et al. 2004). Therefore, microsatellite marker can be used not only to assess population diversity but also to detect gene flow between wild and cultivated soybean.
Definitude of genetic diversity and peculiarity of wild soybean is invaluable for its efficient utilization and conservation. In order to obtain a better understanding of genetic differentiation and peculiarity of the Chinese wild soybean population, we evaluated the genetic diversity and genetic variation pattern by examining the length polymorphism of alleles of 60 SSR loci on different linkage groups performed in a representative sample of country-wide wild soybeans in China.
Materials and methods
Plant materials and field experiments
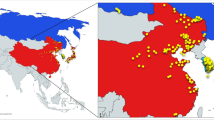
One hundred ninety-six accessions representing the full geographic range of wild soybean from Southern China to Northeast China were sampled and used in this study (Fig. 1, supplementary material 1). In order to investigate the phenotypic diversity, the accessions were evaluated in a randomized blocks design with 0.8 × 0.8 m2 hill-plots, two replications at Jiangpu Experimental Station of Nanjing Agricultural University in the summers of 2004 and 2005. A total of eight morpho-biological traits, including four quantitative traits (days to maturity, days to flowering, 100-seed weight and leaf area) and four qualitative traits (seed sootiness, flower color, leaf shape and seed color) were used to analyze morpho-biological diversity. Classification of the quantitative traits was defined as follows: denoting \( < \overline{x} - 2s,\overline{x} - 2s\sim \,\overline{x} - 1. 7 5s,\overline{x} - 1. 7 5s\,\sim \,\overline{x} \, - 1. 50s, \ldots \,{\text{and}}\, \ge \overline{x} + 2s \) as 1, 2, 3,… and 18, total 18 classes, with an interval of 0.25s (s = standard deviation) in each class. For the qualitative traits, seed sootiness was categorized as smooth and sooty; flower color as white and purple; seed color as black, brown, yellow, green and double color; leaf shape as linear, lanceolate, ellipse, ovate ellipse, ovate round and round according to Dong et al. (2001). For comparison, the data from 200 landraces of cultivated soybean, which covered the entire soybean production area in China, were obtained and treated in the same way here.

The map showing locations of 196 wild accessions used in the study
Simple sequence repeat genotyping
Genomic DNA was extracted from young leaves of a single plant per accession according to the method of Doyle and Doyle (1990). Based on their position on the soybean linkage group map (Song et al. 2004), 400 SSR markers, polymorphic in cultivated soybean according to our historical record, were chosen and checked for their polymorphism in a random sample of eight wild accessions. The SSR loci were excluded from the study if they did not show polymorphism. A final set of 60 pairs of SSR primers, evenly distributed on the whole genome with 2–3 loci on each of the 20 linkage groups (LG), were used in genotyping both of the cultivated and wild soybean accessions (Table 1). The primer sequences with their linkage group locations are available at http://bldg6.arsusda.gov/cregan/soymap. The 10 μl PCR reaction buffer consisted of 20 ng total DNA, 0.4 μM forward and reverse primers, 200 μM of each dNTPs, 1 × PCR buffer (10 mM of Tris–HCl, pH 8.3, 50 mM of KCl), 2 mM of MgCl2 and 0.5U of Taq DNA polymerase. PCR was programmed with an initial denaturing at 94°C for 3 min, followed by 30 cycles of 94°C for 30 s, 55°C for 60 s and 72°C for 1 min, with a final extension at 72°C for 8 min. PCR reactions were performed with an MJ Research PTC 225 DNA engine thermal cycler (Bio-RAD, USA). Amplified products were fractionated by electrophoresis through 8% non-denaturing polyacrylamide gels and stained with silver staining, the size of the stained band was analyzed based on its migration distance relative to the pBR322 DNA Marker (MBI Fermentas) using Quantity One software (Version 4.4.0).
Data analysis
Genetic diversity was expressed in both genetic richness A = ∑A i and genetic dispersion (Simpson genetic diversity index H i ), \( H_{i} = 1 - \sum\limits_{i = 1}^{n} {p_{ij}^{2} }. \) For SSR loci, A i is the number of alleles of the ith locus in a population or subpopulation; A is the total number of alleles of all loci in a population or subpopulation; p ij is the frequency of jth allele at ith locus. For phenotypic traits, A i is the number of categories of the ith trait in a population or subpopulation; A is total number of categories of all traits in a population or subpopulation; p ij is the frequency of jth category of the ith trait. Average diversity index (H) was computed with \( H = \sum\limits_{i = 1}^{k} {H_{i} } /k\,, \)where k means the total number of loci or total number of traits tested in the study. Proportion of shared alleles distance (D sa) was used to calculate estimates of genetic distances for all pairs of accessions according to Chakrabortry and Jin’s statistics (1993):
where X ij and Y ij are the frequencies of jth allele at ith locus in accessions (or populations) Xs and Y, respectively, while a i is the number of alleles at ith locus, and m is the number of loci examined. The genetic distance matrix of D sa was used in cluster analysis with the unweighted pair-group method of the arithmetic average (UPGMA) under PowerMarker version 3.25 (Liu and Muse 2005). An analysis of molecular variance (AMOVA, Michalakis and Excoffier 1996) was used to detect the population differentiation and was calculated under the Arlequin software version 3.0 (Excoffier and Schneider 2005). To detect the genetic differentiation among subpopulations for each microsatellite locus, AMOVAs were performed for each locus separately. AMOVA creates a matrix of genetic distances (Φ) between samples in order to measure the genetic structure of the population from which the samples are drawn. F-statistics were tested by 1,000 permutations, and significant differences between populations declared if measured variance is lower than 95% of the variance in the null distribution (Excoffier et al. 1992).
Results
Genetic diversity of Chinese wild soybean population
Genetic diversity of the entire population
The 60 SSR loci were successfully amplified in both G. soja and G. max samples. A total of 1,067 SSR alleles were detected among the 196 wild soybean accessions with a mean of 17.8 per locus and a range from 7 per locus at Satt209 on LG A2 to 47 at Sat_219 on LG I. The mean diversity index H was 0.857 and ranged from 0.29 for Satt163 on LG G to 0.97 for Sat_219 on LG I (Table 1). About 14% of the alleles were unique and detected on only one accession. A total of 118 accessions showed unique alleles. This means that 60.2% (118 out of 196) of the wild soybean studied could be uniquely genotyped. In addition, 15 accessions (7.6%) had heterozygous alleles that implied natural hybridization happened in an earlier generation.
In comparison, the cultivated soybeans showed lower genetic richness in a total of 826 alleles with a mean of 13.7 alleles per locus and in diversity index H = 0.727 than wild soybeans did at the whole genome level. On most of the LGs, such as A2, C2, D1b, F, H, I, J, K, M, N, O, etc., the average number of alleles per LG decreased after the domestication process from wild ancestors to their cultivated offspring (Table 2). This kind of change also existed in the diversity index H i , which indicates that genetic diversity decreased during the domestication process. On the other hand, of the 826 alleles detected in the cultivated soybean, 541 (65.5%) were the same as in G. soja, accounting for only 51% of the total allelic number (1,067) of the wild soybean, and 285 new alleles (34.5%) emerged after domestication in the cultivated soybean.
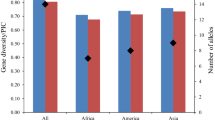
The richness and diversity index at the phenotypic level were not as high as the molecular results for both G. soja and G. max. The average richness (A) and diversity index (H) in wild soybean were 9.1 and 0.649, respectively (Table 3). Generally, qualitative traits revealed less genetic diversity than quantitative traits. Days to flowering had the highest variation among the eight traits followed by days to maturity, leaf area, 100-seed weight, leaf shape, flower color, seed coat color and seed sootiness. In comparing the results of G. soja with those of G. max, the latter showed a higher average phenotypic richness A (10.0) and higher average diversity index H (0.695). It means that the phenotypic diversity in G. soja is smaller than G. max and that only a part of the characteristics of G. max were obtained from G. soja during its domestication and new characteristics appeared after domestication due to artificial selection. It is especially obvious in the observed morphological and agronomic traits. The number of categories for some traits, including days to maturity, seed coat color, flower color, leaf shape and seed sootiness, remained the same in cultivated soybean after domestication, while others, including days to flowering, 100-seed weight and leaf area, increased greatly after domestication (Table 3).
Genetic diversity of the geographic subpopulations
The 196 wild soybean accessions tested were pooled into three geographic subpopulations, namely, those of Northeast China (NEC), Huang-Huai-Hai Valleys (HHH) and Southern China (SOC). The genetic and phenotypic diversity values of the three subpopulations are listed in Tables 2 and 3. The allelic richness and diversity index was the highest in SOC (Total 836 alleles and 13.9 per locus), followed by NEC (783 alleles, 13.5 per locus), and HHH (737 alleles, 12.6 per locus). Furthermore, the SOC subpopulation contained the largest number of alleles in all except four linkage groups (B2, D1b, E and F). The H value followed similar trends (Table 2).
Like the SSR genotyping results, the phenotypic diversity was likewise highest in the SOC subpopulation (A = 56, H = 0.622), followed by HHH (A = 50, H = 0.607), and NEC (A = 50, H = 0.558, Table 3).
Genetic differentiation and peculiarity among geographic subpopulations of G. soja
Geographic differentiation detected with analysis of molecular variance
The analysis of molecular variance (AMOVA) was used to partition the SSR variation into among and within subpopulation components. Most of the variation (97.3%) was detected within subpopulations, while only a small but significant portion of the variation (2.7%, P < 0.001) was attributed to variation among subpopulations, which indicated significant geographic differentiation existed in the Chinese wild soybean population (Table 4).
Geographic differentiation supported by genetic cluster analysis
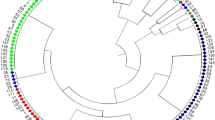
The dendrogram based on the genetic distance (D sa = 0.86) between accessions showed that the 196 accessions formed nine clusters (cluster I to IX, Fig. 2). All the NEC accessions except two were mainly scattered in three clusters (V, VI and VII). About 80% of the HHH accessions were included in one cluster (cluster VI), and the SOC accessions were distributed across eight clusters, but dominated the cluster IV, VIII and IX and fully dominated cluster III. Table 5 shows the two-way classification of the wild soybeans, with SSR data clusters and phenotypic data clusters at the top and the geographic eco-types at the left. It implies that the geographic grouping of the wild soybeans is associated with genetic clustering of SSR data. The χ2-test showed very significant association between the two grouping factors, which meant the geographic grouping into three eco-types was of sound genetic bases.

a UPGMA dendrogram showing genetic relationships among 196 Chinese wild soybean accessions (Constructed based on the shared allele distances (Dsa) among accessions)
Cluster analyses of phenotypic data based on similarities between accessions showed that the 196 accessions formed six major clusters, and that the clusters likewise largely corresponded to their geographic origin (Table 5). For example, cluster B contained 69 accessions, of which 55 were NEC accessions; cluster F contained 35 accessions, of which 27 were HHH accessions; cluster E contained 18 accessions, of which 17 were SOC accessions. The χ2-test also showed a very significant association between the two grouping factors, which meant the geographic grouping into three eco-types was also of sound phenotypic performance bases.
Geographic differentiation resulted in the differences of specific-present allele and specific-deficit allele among geographic subpopulations
Since cluster analysis showed that accessions formed clusters generally based on their geographic origin, comparisons were made between geographical populations to identify peculiar alleles specific to each of the three subpopulations (Tables 6 and 7). Here we define specific-present allele (SPA) of a subpopulation as an allele present in the subpopulation but not in all others; define specific-deficit allele (SDA) as an allele that is present in all subpopulations but deficit in the specific one; and define complement allele (CA) of one subpopulation for the other as an allele that is specific to its counterpart subpopulation in a pair of comparison. These indicators can mainly reflect the peculiarity of and differentiation among subpopulations (or populations in other cases), and also can provide an estimate of the potential contribution of a subpopulation relative to its counterpart subpopulation. The total number of specific-present alleles was 189 (accounting for 17.9% of the total alleles in the whole population) distributed over all loci. Three geographic subpopulations carried different numbers of SPA. The largest number (83 with an average frequency 0.028) was observed in the SOC subpopulation, followed by the NEC subpopulation (70 with an average frequency 0.031), and the HHH subpopulation (36 with an average frequency 0.030). Table 7 shows SPA with a frequency more than 0.06, where seven alleles from five loci were specific in the SOC subpopulation, six alleles from four loci specific to the HHH subpopulation, and six alleles from five loci specific to the NEC subpopulation. The existence of so many different SPA demonstrated that significant geographic differentiation happened during the long history of the species.
The AMOVA test for each of the SSR loci indicated that, of the total variation, the percentage of variation attributed to differences among three subpopulations ranged from 0.9% at Satt373 to 10.03% at Satt235, with an average over all loci of 2.70%. Here, the percentage of total molecular variance contributed by the SPA of a single locus listed in Table 7 was mostly higher than the average value.
Table 6 shows that the total number of SDA in subpopulations was 278, accounting for 26.3% of the total alleles in the whole population and 91, 110 and 77 for Northeast, Huang-Huai-Hai, and Southern China subpopulations, respectively. Tables 6 and 8 shows that the SOC subpopulation had the smallest number of SDA, but it could provide the largest number of CA for the HHH and NEC subpopulations, followed by the NEC subpopulation and the HHH subpopulation. This means that the SOC subpopulation can provide the most potential contribution to its counterpart subpopulations.
In summary, the present results have provided an outline of the genetic diversity of the Chinese wild soybean population: (1) There is great genetic diversity in the Chinese wild soybean population. This genetic diversity is larger than that of Chinese cultivated soybean population. About two-thirds of the alleles of cultivated soybean were inherited from their wild ancestor, and about one-third new alleles emerged after domestication. (2) The entire population is composed of three geographic subpopulations, and each of them showed a high level of intra-population variation. There exists significant geographic differentiation in the Chinese wild soybean population, and the geographic differentiation has sound genetic bases. Among the geographic subpopulations, the SOC subpopulation is of greatest genetic diversity, therefore, Southern China should be the major center of genetic diversity of the wild soybean among the three regions.
Discussion
Changes of genetic diversity from wild to cultivated soybean
The present analysis detected a high level of polymorphism at 60 SSR loci in 196 Chinese annual wild soybean accessions tested. The genetic diversity observed among the wild soybean accessions was much higher than the values detected in 200 widely grown Chinese landraces of cultivated soybean. Moreover, the cultivated soybean only inherited half the alleles of the wild soybean. Accordingly, our results support the point that the cultivated soybean was domesticated from a part or parts of the wild soybean gene pool (Kuroda et al. 2006). The higher variation in wild soybean than that of cultivated soybean is consistent with earlier studies (Maughan et al. 1995; Xu et al. 2002; Gai et al. 2000). Kuroda et al. (2006) analyzed the microsatellite variation of 616 Japanese wild soybean and 53 cultivated soybean using 20 pairs of microsatellite primers and found that cultivated soybeans had only 57% of the Nei’s diversity value of wild soybeans. It is a common phenomenon that domesticated relatives have lower genetic diversity than wild relatives do in most cereals, and in general, about two-thirds of the genetic diversity in wild relatives (Buckler et al. 2001). However, although genetic variation in cultivated soybean was lower than that for wild soybean, some new alleles have emerged and existed only in cultivated soybean. In this study, there were 285 microsatellite alleles found only in cultivars, and the majority of them (84.3%) were at low frequencies (<0.1). The 285 new alleles is a primary estimate obtained from the two tested samples and the exact number should be estimated from further larger samples. However, it can be supposed that the causation of the new alleles in cultivated soybean might be new mutations after domestication combined with both artificial and natural selection in addition to sampling fluctuation. Here both artificial selection and natural evolution processes might be involved, which should be an interesting subject for our further study.
On the other hand, despite the drop of SSR alleles during domestication, cultivated soybean still showed higher diversity than wild soybean at the phenotypic level in the present study. A possible explanation is that the ancient Chinese farmers played key roles in diversification selection. Some of mutant alleles with agronomic profits were kept and multiplied while a great number of old and mutant alleles without agronomic importance, including some alleles neutral to artificial selection, were discarded during the history. Since the mutated SSR alleles are usually not a part of the DNA coding region and are mostly neutral to artificial selection, they might not be reserved during the artificial selection. An alternate possible explanation is that alleles with potential to affect agronomic productivity resided in G. soja, but had no opportunity to express, phenotypically, because of the diminutive G.soja plant type (Carter et al. 2004). These could partially explain the reduced variability in SSR markers and increased variability in phenotypic traits in G. max in comparison with G. soja.
Genetic diversity and peculiarity related to geographic differentiation
Along with the migration of wild soybean from one place to another, it evolved and became adapted to the local environment. In this way, new geographic ecotypes formed. Among the ecotypes, there should be obvious genetic differentiation. The present results detected a number of specific-present alleles, specific-deficit alleles, and complement alleles between ecotypes, as well as a number of accession-unique alleles, which caused the cluster analysis clearly allocated the observed diversity into geographic-associated clusters. The results demonstrated the real existence of genetic differentiation among geographic ecotypes. Since all the alleles were formed and accumulated during the long systematic development of the species, the number of mutant alleles due to geographic differentiation should be relatively small. Therefore, there exists difference among subpopulations but most of the allele variation is still left within subpopulations. Chen and Nelson (2005) found that the SSR variation among individuals of cultivated soybean within the four Chinese provinces accounted for 90% of the total. The variation among provinces accounted for only 10% of the total but was highly significant. This is the similar situation as in our wild soybean study. Here the inter-subpopulation variation in cultivated soybean is larger than what we obtained in wild soybean (10.0% vs. 2.7%). We think the reason for that is basically due to more artificial selection difference among provinces in cultivated soybean in addition to natural selection. It might explain why relatively higher intra-population (97.3%) portion of the variation did exist among the three subpopulations. Such kind of geographic-related variation was observed also in Japanese wild soybean by Kiang et al. (1992) and Fujita et al. (1997). Kuroda et al. (2006) indicated that the nuclear microsatellite variation of wild soybeans in northern Japan is distinct from southern Japan, and wild soybeans of central Japan had variation from both regions based on the graphical ordination analysis. Tozuka et al. (1998) analyzed RFLP (restriction fragment length polymorphism) patterns of mitochondrial DNA using 1,097 individuals of wild soybean collected from throughout Japan and found genetic differentiation between northern and southern parts of Japan. Wang and Takahata (2007) demonstrated the Japanese and Chinese wild soybean germplasm pools differed from each other according to their data of 10 SSR markers. Based on the present results, further studies are required for explaining the genetic mechanism of geographic differentiation in detail.
Southern China as the major center of genetic diversity of wild soybean
From the above, Southern China is believed the major center of genetic diversity of wild soybean in China. The evidences are that the SOC subpopulation possessed the greatest allelic richness, had the highest genetic diversity index, contained the largest number of regional special alleles, held smallest number of special deficit alleles, had largest number of complementary alleles for its counterparts, and was distributed across eight out of nine UPGMA clusters and six of six phenotypic clusters. This concept is consistent with our previous study (Xu et al. 1999) in which morphological characters, isozymes and RFLPs of cytoplamic DNA were examined for more than 200 accessions of annual wild soybean from various ecological regions in China. However, our conclusion is somewhat different from Dong et al. (2001) and Wang and Takahata (2007). The former suggested that there were three centers of genetic diversity for annual wild soybean in China, namely the Northeast, the Yellow River Valley and the Southeast Coasts, and the center of Northeast China was the most genetically diverse region (Southeast Coasts is the east part of Southern China). In their study, the materials were obviously biased among regions, and the phenotypic traits were recorded in respective eco-regions without uniform experiment. Therefore, strictly, those data were not exactly comparable and only phenotypic diversity rather than genetic diversity was involved. The latter (Wang and Takahata 2007) suggested the region between Changjiang River and the Yellow River (the same as Huang-Huai-Hai region) as the most genetically diverse region by using length polymorphism of alleles found in 10 SSR loci. However, their study did not include a wide range of accessions and enough alleles, and thus in fact could not authoritatively make reasonable inference on the center of genetic diversity of wild soybean. Consequently, we believe the rationality of our conclusion, even further evidence to support our conclusion is to be pursued.
References
Abe J, Xu DH, Suzuki Y, Kanazawa A (2003) Soybean germplasm pools in Asia revealed by nuclear SSR. Theor Appl Genet 106:445–453
Buckler ESIV, Thornsberry JM, Kresovich S (2001) Molecular diversity and domestication of grasses. Genet Res 77:213–218
Carter TE, Nelson RL, Sheller CH, Cui ZL (2004) Genetic diversity in soybean. In: Boerma HR, James ES (eds) Soybeans: improvement, production, and uses, 3rd edn. ASA,CSSA and SSSA, Madison, pp 309–310
Chakrabortry R, Jin L (1993) A unified approach to study hypervariable polymorphisms: statistical considerations of determining relatedness and population distances. In: Pena SDJ, Chakraborjt R, Epplen JT, Jeffreys AJ (eds) DNA fingerprinting: state of the science. Birkhäuser Verlag, Basel, pp 153–175
Chen YW, Nelson RL (2004) Genetic variation and relationships among cultivated, wild, and semiwild soybean. Crop Sci 44:316–325
Chen YW, Nelson RL (2005) Relationship between origin and genetic diversity in Chinese soybean germplasm. Crop Sci 45:1645–1652
Cregan PB, Jarvik T, Bush AL, Shoemaker RC, Lark KG, Kahler AL, Kaya N, VanToai TT, Lohnes DG, Chung J, Especht J (1999) An intergrated genetic linkage map of the soybean genome. Crop Sci 39:1464–1490
Diwan N, Cregan PB (1997) Automated sizing of fluorescent-labeled simple sequence repeat (SSR) markers to assay genetic variation in soybean. Theor Appl Genet 95:723–733
Dong YS, Zhuang BC, Zhao LM, Sun H, He MY (2001) The genetic diversity of annual wild soybeans grown in China. Theor Appl Genet 103:98–1030
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Excoffier L, Schneider S (2005) Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolut Bioinform Online 1:47–50
Excoffier L, Smouse P, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes:application to human mitochondrial DNA restriction data. Genetics 131:479–491
Fujita R, Ohara M, Okazaki K, Shimamoto Y (1997) The extent of natural cross-pollination in wild soybean (Glycine soja). J Hered 88:124–128
Gai JY, Xu DH, Gao Z, Shimamoto Y, Abe J, Fukushi H, Kitajima S (2000) Studies on the evolutionary relationship among eco-types of G. max and G. soja in China. Acta Agron Sin 26(5):513–520
Kiang YT, Chiang YC, Kaizuma N (1992) Genetic diversity in natural populations of wild soybean in Iwate prefecture, Japan. J Hered 83:325–329
Kuroda Y, Kaga A, Tomooka N, Vaughan AD (2006) Population genetic structure of Japanese wild soybean(Glycine soja) based on microsatellite variation. Mol Ecol 15:959–974
Li FS (1993) Studies on the ecological geographical distribution of the Chinese resources of wild soybean. Sci Agr Sin 26:47–55
Li Zl, Nelson RL (2002) RAPD marker diversity among cultivated and wild soybean accessions from four Chinese Provinces. Crop Sci 42:1737–1744
Li J, Tao Y, Zheng SZ, Zhou JL (1995) Isozymatic differentiation in local population of Glycine soja sieb & zucc. Acta Bot Sin 37:669–676
Liu K, Muse SV (2005) PowerMarker: integrated analysis environment for genetic marker data. Bioinformatics 21:2128–2129
Maughan PJ, Saghai MA, Buss GR (1995) Microsatellite and amplified sequence length polymorphisms in cultivated and wild soybean. Genome 38:715–723
Michalakis Y, Excoffier L (1996) A generic estimation of population subdivision using distances between alleles with special reference to microsatellite loci. Genetics 142:1061–1064
Narvel JM, Fehr WR, Chu WC, Grant D, Shoemaker RC (2000) Simple sequence repeat diversity among soybean plant introductions and elite genotypes. Crop Sci 40:1452–1458
Pei YL, Wang L, Ge S, Wang LZ (1996) Studies on genetic diversity of Glycine soja isozyme variation in four populations. Soybean Sci 15:302–309
Powell W, Morgante M, Doyle JJ, McNicol JW, Tingey SV, Rafalski AJ (1996) Genepool variation in Genus Glycine subgenus Soja revealed by polymorphic nuclear and chloroplast microsatellites. Genetics 144:791–803
Song QJ, Marek LF, Shoemaker RC, Lark KG, Concibido VC, Delannay X, Specht JE, Cregan PB (2004) A new integrated genetic linkage map of the soybean. Theor Appl Genet 109:122–128
Tozuka A, Fukushi H, Hirata T, Ohara M, Kanazawa A, Mikaml T, Abe J, Shimamoto Y (1998) Composite and clinal distribution of Glycine soja Japan revealed by RFLP analysis of mitochondrial DNA. Theor Appl Genet 96:170–176
Wang KJ, Takahata Y (2007) A preliminary comparative evaluation of genetic diversity between Chinese and Japanese wild soybean (Glycine soja) germplasm pools using SSR markers. Genet Resour Crop Evol 54:157–165
Wang LX, Guan RX, Liu ZX, Chang RZ, Qiu LJ (2006) Genetic diversity of chinese cultivated soybean revealed by SSR markers. Crop Sci 46:1032–1038
Xu DH, Gao Z, Tian QZ, Gai JY, Fukushi H, Kitajma S, Abe J, Shimamoto Y (1999) Genetic diversity of the annual soybean (Glycine soja) in China. Appl Environ Biol 5:439–443
Xu DH, Abe J, Gai JY, Shimamoto Y (2002) Diversity of chloroplast DNA SSRs in wild and cultivated soybeans: evidence for multiple origins of cultivated soybean. Theor Appl Genet 105:645–653
Zhuang BC, Xu H, Wang YM (1996) Polymorphism and geographical distribution of the stem and leaf characters of wild soybean (Glycine soja) in China. Acta Agron Sin 22:583–586
Acknowledgments
The project was supported by the Natural Science Foundation of China (30671266), the National Key Basic Research Program (2006CB101708, 2009CB118404), the National “863” Program (2006AA100104) and the MOE 111 Project (B08025). The authors would thank the editor and reviewers for their relevant comments on the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by A. Schulman.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Wen, Z., Ding, Y., Zhao, T. et al. Genetic diversity and peculiarity of annual wild soybean (G. soja Sieb. et Zucc.) from various eco-regions in China. Theor Appl Genet 119, 371–381 (2009). https://doi.org/10.1007/s00122-009-1045-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-009-1045-y