
Abstract
Background
Soybean (Glycine max (L.) Merr.) is an economically important crop for vegetable oil and protein production, and yield is a critical trait for grain/vegetable uses of soybean. However, our knowledge of the genes controlling the vegetable soybean yield remains limited.
Objective
To better understand the genetic basis of the vegetable soybean yield.
Methods
The 100-pod fresh weight (PFW), 100-seed fresh weight (SFW), kernel percent (KP) and moisture content of fresh seeds (MCFS) at the R6 stage are four yield-related traits for vegetable soybean. We investigated a soybean mini core collection composed of 224 germplasm accessions for four yield-related traits in two consecutive years. Based on 1514 single nucleotide polymorphisms (SNPs), genome-wide association studies (GWAS) were conducted using a mixed linear model (MLM).
Results
Extensive phenotypic variation existed in the soybean mini core collection and significant positive correlations were shown among most of traits. A total of 16 SNP markers for PFW, SFW, KP and MCFS were detected in all environments via GWAS. Nine SNP markers were repeatedly identified in two environments. Among these markers, eight were located in or near regions where yield-related QTLs have been reported in previous studies, and one was a novel genetic locus identified in this study. In addition, we conducted candidate gene analysis to the large-effect SNP markers, a total of twelve genes were proposed as potential candidate genes of soybean yield at the R6 stage.
Conclusion
These results will be beneficial for understanding the genetic basis of soybean yield at the R6 stage and facilitating the pyramiding of favourable alleles for future high-yield breeding by marker-assisted selection in vegetable soybean.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soybean [Glycine max (L.) Merr.] is an important crop that is grown worldwide as a source of edible oil and high-quality protein (Graham and Vance 2003). Soybean provides approximately 68% of protein meal and 57% of vegetable oil production worldwide (USDA-FAS, http://www.fas.usda.gov/). In accordance with the use of soybean cultivars, it can be divided into grain soybean and vegetable soybean. Grain soybean is harvested in the late maturation stage, when seeds are dehydrated, and is mainly employed for the production of oil, soybean milk, tofu and other bean products. Vegetable soybean is harvested at the R6 stage, when the pods are green and fully filled with the seeds, and is mainly used as a bean vegetable (Fehr and Caviness 1977; Young et al. 2000). With a long soybean breeding history, yield has been considered as one of the preferable traits. It is a very complex trait and depends upon a combination of different plant traits. Seed weight, the seed number per pod and the pod number per plant are three important yield component traits that directly determine yield (Zhou et al. 2009; Hao et al. 2012). Seed shape, plant architecture, the growing period and biomass yield are important yield-related traits that have been proven to influence yield indirectly in several crops (Li et al. 2019a, b; Zhang et al. 2018; Salas Fernandez et al. 2009; Zhang et al. 2015; Raboanatahiry et al. 2018). In addition, vegetable soybean has a high moisture content of approximately 57.0–79.0% in fresh seeds, and a significant positive correlation between moisture content and yield has been observed in soybean landrace populations (Li et al. 2019a, b). Thus, moisture content is also an important yield-related trait in vegetable soybean. In maize, high heritability (h2) of moisture content has been shown, and several quantitative trait loci (QTLs) that are simultaneously related to yield and moisture content have been detected (Austin et al. 2000). Many yield-related QTLs have been shown to be clustered with yield QTLs in several crops (Chen et al. 2007; Shi et al. 2009; Cai et al. 2014; Zhang et al. 2017). Therefore, understanding the genetic basis of yield-related traits will help us to improve yield in vegetable soybean.
Yield-related traits are complex traits that are controlled by multiple genes and influenced by environmental factors. In soybean, QTL analyses have been conducted to elucidate the genetic bases of yield-related traits in segregating populations derived from biparental crosses. More than 300 QTLs have been detected across the whole genome of soybean, most of which have been repeatedly identified in different genetic backgrounds (Kim et al. 2010; Liu et al. 2011; Han et al. 2012; Sun et al. 2012). These studies demonstrated that linkage mapping is an efficient approach for revealing the genetic architecture underlying complex quantitative traits. Linkage mapping has been conducted in cross-populations such as recombinant inbred lines (RILs). In these mapping populations, homozygous alternative alleles are segregated, and each allele is present in 50% of the recombinant lines; thus, high power to detect QTLs can be observed (Huang and Han 2014). However, the establishment of the RIL population is time consuming and labour intensive.
Genome-wide association studies (GWAS), also referred to as linkage disequilibrium (LD) mapping, are an effective method for studying the genetic architecture of complex traits. In contrast to linkage mapping, GWAS is a time- and labour-saving approach with a high resolution. It can be performed in natural populations with complex genetic backgrounds that have accumulated a large number of historic recombination events and can couple events with linked DNA polymorphisms to reveal associations with phenotypic diversity within a relatively small region of the genome (Rafalski 2010; Zhao et al. 2015). In addition, GWAS has the ability to reveal elite alleles at genetic loci in germplasm. GWAS has been successfully used in soybean. For example, 118 SNPs and 11 SSRs were identified for soybean flowering time under various photothermal conditions in 91 soybean cultivars from different maturity groups, four of which were large-effect markers located on Gm 11, Gm 16 and Gm 20 and were detected in eight environments (Mao et al. 2017). Based on SNP markers and SNP-based haplotype analysis, a total of 11, 17 and 59 SNP-based haplotypes have been shown to be associated with seed yield, 100-seed weight and plant height, respectively (Contreras et al. 2017). For seed size and shape, a total of 21 significant loci were identified in a soybean landrace population, which explained 8.12–14.25% of the observed phenotypic variance. The new locus SW9-1, on chromosome 9, is a reliable locus related to seed size traits that exerts a relatively high phenotypic effect (13.67) on 100-seed weight (Li et al. 2019a, b). For the vegetable soybean yield, Li et al. (2019a, b) detected 35 significant SNPs and proposed five potential candidate genes for soybean yield at the R6 stage (Li et al. 2019a, b). However, the genetic basis of yield and yield-related traits in soybean remains largely ambiguous.
In this study, we genotyped a soybean mini core collection composed of 224 germplasm accessions using 1514 SNP markers and surveyed four yield-related traits in two consecutive years. The objectives of this study were to (1) reveal the genetic basis of yield-related traits in soybean at the R6 stage and (2) evaluate marker-based allelic effects for application in vegetable soybean breeding programmes.
Materials and methods
Plant materials
Two hundred and twenty-four accessions obtained from the Chinese soybean mini core collection were selected from 23,587 germplasm accessions conserved in the Chinese National Soybean GenBank (CNSGB). The 224 accessions consisted of 196 landraces and 28 cultivars which had been reported by Huang (Table S1) (Huang et al. 2016). The population preserved approximately 63.5% of the soybean genetic diversity present in China and is an excellent population for identifying QTLs and revealing the genetic bases of target traits by association mapping (Huang et al. 2016; Qiu et al. 2009).
Field trials and trait measurement
The 224 accessions were planted in double-row plots in a randomized complete block design with three replications on the Experimental Farms of Nanjing Agricultural University located in two environments: Jiangpu (32.04° N 118.63° E, 2015 and 2016) in Nanjing, China. The rows were 4 m in length and 0.5 m in width, and there were 40 plants in a row. At the R6 stage, fifty fresh pods from normally developed plants were harvested for trait investigation in the autumn of 2015 and 2016.
Four yield-related traits, 100-pod fresh weight (PFW), 100-seed fresh weight (SFW), the kernel percent (KP) and the moisture content of fresh seeds (MCFS), were investigated. First, PFW (g) could be calculated by weighing the fresh pods with an electronic balance. Second, SFW (g) was measured in fresh seeds whose pod husks were stripped. Third, KP (%) was the ratio of the fresh seed weight to the fresh pod weight. Finally, the fresh seeds were killed at 110 °C for 30 min and dried at 65 °C in a dryer until a constant weight was reached. Then, the MCFS (%) could be calculated.
Statistical analysis
Analysis of variance (ANOVA) of the phenotype data across multiple environments was performed via the PROC GLM process in SAS version 9.4 (SAS Institute, Inc., Cary, NC, USA). The model for the phenotypic traits was as follows: \({y}_{ijk}=\mu +{\alpha }_{i}+{\beta }_{j}+{\gamma }_{kj}+{(\alpha \beta )}_{ij}+{\varepsilon }_{ijk}\), where µ is the overall mean; αi is the genetic effect of the ith genotype; βj is the effect of the jth environment; γkj is the block effect within the jth environment; (αβ)ij is the interaction effect between the ith genotype and the jth environment; and εijk is the random error following N(0, α2ε). Broad-sense heritability was calculated as follows:\({h}^{2}={\alpha }_{g}^{2}/({\alpha }_{g}^{2}+{\alpha }_{ge}^{2}/n+{\alpha }_{\varepsilon }^{2}/rn)\), where σ2g is the genotypic variance; σ2ge is the genotype by environment interaction variance; α2ε is the error variance; n is the number of environments; and r is the number of replications. All of the above variance values can be calculated by using the REML method via the SAS VARCOMP procedure.
The best linear unbiased prediction (BLUP) is the best statistical method for predicting breeding values of target traits under multi-environments. The BLUPs for PFW, SFW, KP and MCFS in each accession were calculated using PROC MIXED in SAS. Then the breeding values from the mixed BLUP model were used as phenotypic data for association mapping.
Genotyping and quality control
1645 SNPs were used to genotype the population and had been described by Huang et al. (2016). After excluding SNP markers with > 15% missing data, a minor allele frequency (MAF) < 5% and heterozygosity > 15% (Liu and Muse 2005), a total of 1514 high-quality SNPs were selected for association mapping (Table S2).
Population structure and linkage disequilibrium
Population structure analysis was performed using the Bayesian model-based clustering method in the program STRUCTURE v2.2 (Pritchard et al. 2000).Pairwise kinship (K) was calculated in TASSEL 5. Based on the Q and K, the Chinese soybean mini core collection was divided into two subgroups (Huang et al. 2016). The pairwise LD between SNPs was calculated as the squared correlation coefficient (r2) of alleles using TASSEL 5 software (Bradbury et al. 2007). The LD estimation had been reported by Huang and the average LD of the population was 544.01 kb (Huang et al. 2016).
Association mapping
To minimize the effects of environmental variation, the best linear unbiased predictions (BLUPs) of individual lines were calculated for each trait using the R package lme4 (Bates et al. 2012). A total of 1514 SNPs (MAF > 0.05) and four yield-related traits (PFW, SFW, KP and MCFS) for all genotypes from the association panel across two years were used to perform marker-trait association analysis. GWAS was performed with a mixed linear model (MLM) using TASSEL 5, which takes both familial relatedness and population structure into account. The significance threshold of marker-trait association was set to a P value < 10−3 (− Log10 P > 3.00).
Prediction of candidate genes
To identify candidate genes underlying the association signals, we selected significant SNP markers which identified in multiple environments and associated with large-effect QTLs to search candidate genes in their candidate regions (LD ± 544. 01 kb). The functional annotations and tissue expression of genes located in the candidate regions were obtained from Soybase (https://www.soybase.org/) and Phytozome (http://phytozome.net). The soybean reference genome was Wm82.a1.v1.1. Based on the soybean genomic annotations and expression data, potential candidate genes were predicted. The heatmap profile of the candidate genes were drawn with R software.
Results
Phenotypic analysis of four yield-related traits
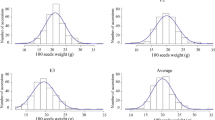
A total of 224 soybean accessions were planted in two consecutive years from 2015 to 2016, and four yield-related traits were investigated. Abundant phenotypic variation was observed for all of these traits in the 224 soybean accessions (Table 1). The 100-pod fresh weight (PFW) showed 9.26 or 7.81 fold variation, ranging from 35.85 to 332.08 g and 41.71 to 326.04 g during 2015 and 2016, respectively. The 100-seed fresh weight (SFW) showed an 8.35 or 7.17 fold variation, ranging from 8.66 to 72.35 g and 8.24 to 59.13 g in 2015 and 2016, respectively. The remaining two traits of the kernel percent (KP) and the moisture content of fresh seeds (MCFS) displayed 1.46 to 1.64 or 1.26 to 1.39 fold variation, respectively. The analysis of the coefficient of variation (CV) for these traits showed similar results, suggesting that this population presented a high latent capacity for increases in yield. The skewness ranged from − 0.18 to 1.56, and kurtosis ranged from − 0.50 to 7.29 for the four traits in 2015 and 2016, respectively. Following normalization, the phenotypic data of KP (2015 and 2016) and MCFS (2016) were normally distributed, and the other two traits did not fit a normal distribution (Fig. 1; Table 1).

Frequency distributions of 100-pod fresh weight (PFW), 100-seed fresh weight (SFW), kernel percent (KP) and moisture content of fresh seed (MCFS) in 2015 and 2016
Variance analysis of the four yield-related traits showed that the effects of the genotype (G), the environment (E) and the interaction between the genotype and environment (G × E) were all at very significant level (P < 0.0001) (Table 2). The broad-sense heritability (h2) of the four traits was calculated (Table 1). PFW, SFW and MCFS showed a high heritability (≥ 80%), indicating that genetic effects play a predominant role in the phenotype variation of these traits. KP showed a low heritability (54%) and had a great phenotype variation between years. In addition, there was a significant positive correlation between PFW, SFW and MCFS (rp 0.198–0.946, P < 0.01), suggesting that MCFS is also an important factor influencing soybean yield development at the R6 stage (Table 3).
Association mapping and candidate genes for soybean yield-related traits
Through GWAS, a total of 45 associations for the four yield-related traits (− Log10(P) > 3) were identified under the MLM conditions, which accounted for both population structure and familial relatedness. The 45 associations included 16 SNPs located on 10 of 20 soybean chromosomes (Chr) (Figs. 2, 3, 4, 5). Six, five and seven SNPs associated with PFW were identified in 2015, 2016 and the BLUP across 2 years, respectively, representing 8 genomic regions covering six soybean chromosomes. The contribution of individual SNPs to the observed phenotypic variation was 5.13–11.42%. Four SNP markers, Map-0676 on Chr 04, Q-07-0082152 on Chr 07, and Map-2049 and Map-2055 on Chr 11, were repeatedly detected in all environments (Table 4). For SFW, four, six and six significant SNPs were detected in 2015, 2016 and the BLUP across 2 years, respectively, which represented 8 genomic regions covering seven chromosomes. Each SNP could explain 5.25–9.78% of the observed phenotypic variation. Two SNP markers, Map-2049 and Map-2055, were repeatedly detected in all environments (Table 5). Four, one and two SNP markers were detected for KP using the BLUP across two years and within the individual years, respectively. These SNPs were located in 5 genomic regions across the four chromosomes and explained 3.05–3.68% of the phenotypic variation for KP. No SNPs were repeatedly identified in all environments. Only the SNPs Map-2976 and Q-05-0211601 were repeatedly detected in individual year and the BLUP (Table 6). For MCFS, two, one and one SNP markers were detected in 2015, 2016 and the BLUP across 2 years, respectively, which were located in the same genomic region on chromosome 5. The phenotypic variation interpretation rate of a single SNP was 5.79–8.69%. There was only one SNP marker (Q-05-0194474 on Chr 5) that was repeatedly detected in all environments (Table 7).

Manhattan plots and QQ plots of estimated − log10(P) from association analysis of PFW. a 2015 PFW: 100-pod fresh weight in 2015, b 2016 PFW:100-pod fresh weight in 2016; c BLUP PFW: 100-pod fresh weight in the BLUP across 2 years

Manhattan plots and QQ plots of estimated − log10(P) from association analysis of SFW. a 2015 SFW:100-seed fresh weight in 2015; b 2016 SFW:100-seed fresh weight in 2016; c BLUP SFW: 100-seed fresh weight in the BLUP across 2 years

Manhattan plots and QQ plots of estimated − log10(P) from association analysis of KP. a 2015 KP: kernel percent in 2015; b 2016 KP: kernel percent in 2016; c BLUP KP: kernel percent in the BLUP across 2 years

Manhattan plots and QQ plots of estimated − log10(P) from association analysis of MCFS. a 2015 MCFS: moisture content of fresh seed in 2015; b 2016 MCFS: moisture content of fresh seed in 2016; c BLUP MCFS: moisture content of fresh seed in the BLUP across 2 years
To identify the beneficial allele of each SNP associated with the target trait, the average phenotypic difference was calculated from the soybean accessions that possessed different SNP alleles. On average, the cultivars carrying the favorable allele (allele 1) exhibited higher phenotypic values than those with the unfavorable allele (allele 2) and the entire association panel. For PFW, the phenotypic difference between the cultivars with allele 1 and the cultivars with allele 2 reached 7.10–25.84 g. The phenotypic difference between the cultivars with allele 1 and the entire association panel was 0.71–5.83 g (Table 4). For SFW, the phenotypic values of the cultivars with allele 1 were 1.33–4.05 g higher than those of the cultivars with allele 2 and were 0.27–1.53 g higher than those of the entire association panel (Table 5). The difference in KP between the cultivars carrying allele 1 and allele 2 was 0.45–1.38%. The mean KP of the cultivars with allele 1 was 0.13–1.22% higher than that of the entire association panel (Table 6). With respect to MCFS, the cultivars with allele 1 presented approximately 2.13–2.87% and 0.58–1.34% higher phenotypic values than those with allele 2 and the entire association panel (Table 7).
In the current study, we identified a series of pleiotropic SNPs associated with multiple traits. Six common SNP markers (Map-0676, Q-07-0082152, Map-2049, Map-2055, Map-2565 and Q-18-0006557) were associated with both PFW and SFW, and one SNP marker, Q-05-0194474, was associated with both SFW and MCFS (Table 8). This was consistent with the significant positive correlations among PFW, SFW and MCFS. This result suggested that the positive correlations between PFW and SFW, SFW and MCFS mainly occurred at the molecular level via coincident quantitative trait nucleotides (QTNs). Thus, these coincident QTNs are critical for breeding vegetable soybean cultivars with a high yield.
A total of nine significant SNP markers were repeatedly detected in at least two environments (2015, 2016, and the BLUP across the two years) (Table 8). Among these markers, eight SNP markers were located in or near regions where QTLs have been previously mapped by linkage analysis. These reported QTLs were mainly related to seed yield, seed weight, seed shape, pod wall width and pod maturity. In this study, only one SNP marker (Map-2565) was identified as a new locus associated with soybean yield at the R6 stage.
To shed light on the potential candidate genes involved in the development of soybean yield at the R6 stage, two SNP markers, Map-0676 and Q-05-0194474, which were repeatedly detected in multiple environments and associated with large-effect QTLs were selected to conduct the candidate gene analysis. The PFW and SFW marker Map-0676 (R2 7.64–11.42%) was located on chromosome 4, the PFW and SFW of the materials carrying the favorable allele (AA) at Map-0676 were 6.77 g and 1.63 g higher than the materials carrying the unfavorable allele (GG), respectively (Tables 4, 5). The MCFS marker Q-05-0194474 (R2 5.79–8.69%) located on chromosome 5, the MCFS of the materials carrying the favorable allele (AA) at Q-05-0194474 was 2.13% higher than the materials carrying the unfavorable allele (GG) (Table 7). Based on gene annotation and tissue expression, we selected 12 potential candidate genes for soybean yield at the R6 stage in the candidate regions of Map-0676 (42369904–43457924) and Q-05-0194474 (37953252–39041272) (Table 9). The candidate genes encode a plant intracellular ras-group-related leucine-rich repeat protein (PIRL9), a RAB GTPase homolog E1B (RABE1b), a FUS3-complementing gene protein (FC2), a seed imbibition protein (SIP2), a protein-l-isoaspartate methyltransferase (PIMT1), a BTB and TAZ domain protein (BT2), a pectin lyase-like superfamily protein (PPME1), a retinoblastoma-related protein (RBR1), a HCP-like superfamily protein, a phosphatidylethanolamine-binding protein (MFT) and two relative of early flowering proteins (REF6), respectively. The 12 genes showed significant difference in expression during the development of soybean pod (Fig. 6). The combined results suggested that these genes would be as the potential candidate genes, which might participate in the development of soybean yield at the R6 stage.

Heatmap profiles of the candidate genes in tissues
Discussion
The large phenotype variations observed within the yield-related traits allowed us to identify the best genes with the largest effects (Table 1). The KP (2015 and 2016) and MCFS (2016) traits were normally distributed, but the other two traits were not normally distributed, the different results might be caused by the small sample size used in this study. Heritabilities ranged from 80 to 96% for PFW, SFW and MCFS. Heritability for KP was only 54%. The low heritability of KP might due to the significant genotype-by-environment interaction justified by ANOVA (Table 2). In previous study, a low heritability (51%) for KP and a low correlation coefficient (0.057–0.13) between different environments were observed in maize (Ma et al. 2021), which was similar with the present study.
Soybean yield-related traits constitute complex quantitative traits that are governed by multiple genetic loci, each of which mostly displays minor effects (Zhang et al. 2016). Because of the environmental influence, loci with minor effects are often poor repeatable. Up to date, numerous soybean yield-related trait QTLs have been reported in different independent experiments, but specific and in-depth analyses of these loci are still lacking. Using a mini core collection with abundant genetic variations, a total of 45 significant yield-related associations were identified, which involved 16 SNP markers. Nine SNP markers were repeatedly detected in different environments, indicating that these SNPs were essential for yield-related traits. One of these SNPs was newly associated with soybean yield, and eight were located in or near known QTL regions (Table 8). Five stable SNP markers (Map-0676 on Chr 04, Q-07-0082152 on Chr 07, Map-2049 and Map-2055 on Chr 11, and Q-18-0006557 on Chr 18) that were simultaneously associated with PFW and SFW, overlapped with ten previously reported QTLs, including five seed yield-related QTLs (seed yield 1-2, seed weight 47-3, seed weight 49-15, seed weight 7-2, seed weight 10-8) (Mansur et al. 1993; Li et al. 2010; Teng et al. 2009; Orf et al. 1999; Specht et al. 2001), four seed and pod shape-related QTLs (seed length 1-15, seed length 5-3, seed thickness 1-5, pod wall width 1-7) (Salas et al. 2006; Hu et al. 2013; Tae-Hwan et al. 2014; Guo et al. 2011), and one pod maturity-related QTL (pod maturity 33-1) (Sun et al. 2013). Two stable SNP markers (Q-05-0211601 on Chr 05 and Map-2976 on Chr 16) that associated with KP were located in or near the reported QTL regions containing three yield-related QTLs (seed weight 7-3, seed weight 2-6, seed yield 29-2), one seed shape-related QTL (seed height 1-4) and one pod maturity-related QTL (pod maturity 9-1) (Orf et al. 1999; Salas et al. 2006; Mian et al. 1996; Eskandari et al. 2013). The last stable SNP marker, Q-05-0194474 on Chr 05, was concurrently related to MCFS and SFW; this marker was situated in a yield-related QTL region (seed weight 10-1) located 60 kb away from a seed shape QTL (seed thickness 1-3) (Specht et al. 2001; Jun et al. 2014).
In this study, a total of 12 potential candidate genes associated with soybean yield at the R6 stage were predicted within the candidate regions of two large-effect SNP markers (Table 9 and Fig. 6). Among these 12 genes, 10 genes are proposed as the candidate genes for PFW and SFW. Glyma04g36190 encodes a plant intracellular Ras-group-related leucine-rich repeat protein (PIRL9). In Arabidopsis thaliana, PIRL1 and PIRL9 are essential for differentiation of microspores into pollen, the T-DNA knockout double-mutants stopped growing as microspores, failed to initiate vacuolar fission, aborted, and underwent cytoplasmic degeneration (Forsthoefel et al. 2010). Glyma04g36430 encodes protein-l-isoaspartate methyltransferase 1(PIMT1). In Oryza sativa, PIMT1 is involved in the repair of damaged proteins containing abnormal isoaspartyl (isoAsp) which accumulated as seeds age, the overexpression of OsPIMT1 reduced the accumulation of isoAsp-containing protein in embryos and increased embryo viability (Wei et al. 2015). Glyma04g36500 encodes a BTB and TAZ domain protein (BT2). In Arabidopsis thaliana, BT proteins perform crucial roles in gametophyte development and BT2 seems to be the predominant gene in this process (Robert et al. 2009). The expression levels of Glyma04g36190, Glyma04g36430 and Glyma04g36500 showed significant increase in the late stage of seed development (seed 21–42 days), it suggested that these genes might participate in the soybean seed development, and thereby influence the soybean yield. Glyma04g36250 is a homologous to AT4G20360 which encodes a RAB GTPase homolog E1B (RABE1b). In Arabidopsis thaliana, RABE1b plays an important role in cell division during the embryonic development (Li et al. 2011). Glyma04g36360 is a homologous to AT4G24740, which encodes a FUS3-complementing protein. FUS3 is an important component of the regulatory pathway controlling late embryogenesis and seed maturation (Reidt et al. 2001). Glyma04g36410 encodes a seed imbibition protein (SIP2). SIP2 is an alkaline-galactosidase with a distinct substrate specificity for raffinose (Peters et al. 2010). In plants, the raffinose family of oligosaccharides have been proposed to play roles in the acquisition of desiccation tolerance in seeds (Ooms et al. 1993; Brenac et al. 1997). Glyma04g36520 encodes a pectin lyase-like superfamily protein. In Japanese apricot, the pectin lyase-like superfamily protein (PPME1) have relationships with the pistil abortion which would affect flowers development and seriously decrease the yield in production (Ting et al. 2012). In soybean development, the genes Glyma04g36250, Glyma04g36360, Glyma04g36410 and Glyma04g36520 display a similar expression pattern: they are highly expressed in the early pod development stage. It indicates that they may affect the early pod development by influencing cell division, embryonic development, seed imbibition and pistil fertility. Glyma04g36620 and Glyma04g36630 are homologous to AT3G48430, which encodes a relative of early flowering protein (REF6). In Arabidopsis thaliana, REF6 may participate in the regulation of flowering time and the formation of cotyledon boundary (Noh et al. 2004; Cui et al. 2016). Tissue expression analysis showed the expression of Glyma04g36620 and Glyma04g36630 were highly expressed in early pod and seed development, but were lowly expressed in late pod and seed development. It suggested that the dynamic changes of REF6 genes expression might involved in the regulation of soybean pod and seed development. Glyma04g36700 encodes a retinoblastoma-related protein (RBR1). In Oryza sativa, the RBR1 co-suppression mutants showed low seed setting rate or complete sterility (Zhang 2012). There was a high-level expression of Glyma04g36700 in root, flower, young leaf and pod. It hinted that Glyma04g36700 might play a positive role in soybean yield formation. For MCFS, there were only two genes, Glyma05g33770 and Glyma05g34030, predicted as the potential candidate genes. Glyma05g33770 encodes a HCP-like superfamily protein which might involved in the regulation of cytosolic calcium, and it was upregulated and then downregulated under flooding and drought stress (Xin and Setsuko 2017). The maturity of soybean seed need to go through the seed expanding and dhydration stage. Glyma05g33770 was upregulated in seed expanding stage (seed 10–25 days), and downregulated in seed dhydration stage (seed 28–42 days). It suggested that it might be related to the change of moisture content during seed development. Glyma05g34030 encodes a phosphatidylethanolamine-binding protein which shares 83.2% amino acid sequence identity with MOTHER OF FT AND TFL1 (MFT) in Arabidopsis thaliana, it might regulate seed germination via the ABA and GA signaling pathways (Xi and Yu 2010). In previous study, Glyma05g34030 was predicted as the candidate gene of MCFS and displayed a significantly different expression between varieties with different moisture content (Li et al. 2019a, b). Glyma05g34030 was highly expressed during the seed development, it suggested that Glyma05g34030 might related to the MCFS of soybean seed development. In this study, we postulate that the twelve genes are potential candidate genes for PFW, SFW and MCFS. However, further evidence is needed to functionally validate this hypothesis.
Conclusion
In this study, we identified 45 significant associations for PFW, SFW, KP and MCFS in all environments via GWAS, which included 16 SNP markers. Nine SNP markers were repeatedly detected in different environments, among which eight were located in or near yield-related QTLs that were previously reported, and one was a new locus associated with soybean yield. We were particularly interested in the large-effect markers Map-0676 and Q-05-0194474, which explained 7.64%-11.42% and 5.79%-8.69% of the observed phenotypic variance, respectively. According to genetic annotation and tissue expression analysis, a total of twelve genes, including Glyma04g36190, Glyma04g36250, Glyma04g36360, Glyma04g36410, Glyma04g36430, Glyma04g36500, Glyma04g36520, Glyma04g36620, Glyma04g36630, Glyma04g36700, Glyma05g33770 and Glyma05g34030, were proposed as potential candidate genes of soybean yield at the R6 stage, which requires further investigation for verification. These results provide useful insights for the yield improvement of vegetable soybean.
Availability of data and material
The datasets supporting the results of this article are included within the article and its additional files.
References
Austin D, Lee M, Veldboom L, Hallauer A (2000) Genetic mapping in maize with hybrid progeny across testers and generations: grain yield and grain moisture. Crop Sci 40(1):30–39
Bates D, Maechler M, Bolker B (2012) lme4: linear mixed-effects models using S4 classes. R package version 0.999999-2.
Bradbury P, Zhang Z, Kroon D, Casstevens T, Ramdoss Y, Buckler E (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635
Brenac P, Horbowicz M, Downer S, Dickerman AM et al (1997) Raffinose accumulation related to desic-cation tolerance during maize seed development and maturation. J Plant Physiol 150:481–488
Cai D, Xiao Y, Yang W, Ye W, Wang B, Muhammad Y et al (2014) Association mapping of six yield-related traits in rapeseed (Brassica napus L.). Theor Appl Genet 127:85–96
Chen W, Zhang Y, Liu X, Chen B, Tu J, Fu T (2007) Detection of QTL for six yield-related traits in oil seed rape (Brassica napus) using DH and immortalized F2 populations. Theor Appl Genet 115:849–858
Contreras R, Mora F, Mar O, Higashi W, Schuster I (2017) A genome-wide association study for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis. PLoS ONE 12(2):e0171105
Cui X, Lu F, Qiu Q, Zhou B et al (2016) REF6 recognizes a specific DNA sequence to demethylate H3K27me3 and regulate organ boundary formation in Arabidopsis. Nat Genet 48:694–699
Eskandari M, Cober E, Rajcan I (2013) Genetic control of soybean seed oil: II. QTL and genes that increase oil concentration without decreasing protein or with increased seed yield. Theor Appl Genet 126(6):1677–1687
Fehr W, Caviness C (1977) Stages of soybean development. Spec Rep 87
Forsthoefel N, Dao T, Vernon D (2010) PIRL1 and PIRL9 encoding members of a novel plant-specific family of leucine-rich repeat proteins, are essential for differentiation of microspores into pollen. Planta 232(5):1101–1114
Graham P, Vance C (2003) Legumes: importance and constraints to greater use. Plant Physiol 131(3):872–877
Guo G, Sun R, Hou M, Guo Y et al (2011) Quantitative trait locus (QTL) analysis of pod related traits in different environments in soybean. Afr J Biotechnol 10(56):11848–11854
Han Y, Li D, Zhu D, Li H et al (2012) QTL analysis of soybean seed weight across multi-genetic backgrounds and environments. Theor Appl Genet 125(4):671–683
Hao D, Chen H, Yin Z, Cui S et al (2012) Identification of single nucleotide polymorphisms and haplotypes associated with yield and yield components in soybean (Glycine max) landraces across multiple environments. Theor Appl Genet 124(3):447–458
Hu Z, Zhang H, Kan G, Ma D et al (2013) Determination of the genetic architecture of seed size and shape via linkage and association analysis in soybean (Glycine max L. Merr.). Genetica 141(4–6):247–254
Huang X, Han B (2014) Natural variations and genome-wide association studies in crop plants. Annu Rev Plant Biol 65:531–551
Huang J, Guo N, Li Y, Sun J et al (2016) Phenotypic evaluation and genetic disection of resistance to Phytophthora sojae in the Chinese soybean mini core collection. BMC Genet 17(1):85
Jun T, Freewalt K, Michel A, Mian R (2014) Identification of novel QTL for leaf traits in soybean. Plant Breed 133(1):61–66
Kim H, Kim Y, Kim S, Son B et al (2010) Analysis of quantitative trait loci (QTLs) for seed size and fatty acid composition using recombinant inbred lines in soybean. J Life Sci 20(8):1186–1192
Li D, Sun M, Han Y, Teng W, Li W (2010) Identification of QTL underlying soluble pigment content in soybean stems related to resistance to soybean white mold (Sclerotinia sclerotiorum). Euphytica 172(1):49–57
Li Y, Lu X, Shen S, Zhang C (2011) Study on involvement of the GTPase RABE1b in embryonic development of Arabidopsis thaliana. Agric Sci Technol 12(12):1795–1798
Li J, Zhao J, Li Y, Gao Y et al (2019a) Identification of a novel seed size associated locus SW9-1 in soybean. Crop J 7(04):548–559
Li X, Zhang X, Zhu L, Bu Y et al (2019b) Genome-wide association study of four yield-related traits at the R6 stage in soybean. BMC Genet 20(1):39
Liu K, Muse S (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21(9):2128–2129
Liu W, Kim M, Van K, Lee Y et al (2011) QTL identification of yield-related traits and their association with flowering and maturity in soybean. J Crop Sci Biotechnol 14(1):65–70
Ma J, Wang L, Cao Y, Li H (2021) Genome-wide association studies for kernel ratio in maize. J Plant Genet Resour 22(02):448–454 (in Chinese)
Mansur L, Lark K, Kross H, Oliveira A (1993) Interval mapping of quantitative trait loci for reproductive, morphological, and seed traits of soybean (Glycine max L.). Theor Appl Genet 86(8):907–913
Mao T, Li J, Wen Z, Wu T et al (2017) Association mapping of loci controlling genetic and environmental interaction of soybean flowering time under various photo-thermal conditions. BMC Genom 18(1):415
Mian M, Bailey M, Tamulonis J, Shipe E et al (1996) Molecular markers associated with seed weight in two soybean populations. Theor Appl Genet 93(7):1011–1016
Noh B, Lee S, Kim H et al (2004) Divergent roles of a pair of homologous jumonji/zinc-finger–class transcription factor proteins in the regulation of Arabidopsis flowering time. Plant Cell 16(10):2601–2613
Ooms J, Leon K, Koornneef M, Karssen C (1993) Acquisition of desiccation tolerance and longevity in seeds of Arabidopsis thaliana. Plant Physiol 102:1185–1191
Orf J, Chase K, Jarvik T, Mansur L et al (1999) Genetics of soybean agronomic traits: I. Comparison of three related recombinant inbred populations. Crop Sci 39(6):1642–1651
Peters S, Egert A, Stieger B et al (2010) Functional identification of Arabidopsis ATSIP2 (At3g57520) as an alkaline α-galactosidase with a substrate specificity for raffinose and an apparent sink-specific expression pattern. IEEE Mtt-s International Microwave Symposium Digest
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Qiu L, Li Y, Guan R, Liu Z, Wang L, Chang R (2009) Establishment, representative testing and research progress of soybean core collection and mini core collection. Crop J 35(4):571–579
Raboanatahiry N, Chao H, Dalin H, Pu S et al (2018) QTL alignment for seed yield and yield related traits in brassica napus. Front Plant Sci 9:1127
Rafalski J (2010) Association genetics in crop improvement. Curr Opin Plant Biol 13(2):174–180
Reidt W, Ellerstrm M, Klle K et al (2001) FUS3-dependent gene regulation during late embryogenesis. J Plant Physiol 158(4):411–418
Robert H, Quint A, Brand D, Vivian-Smith A, Offringa R (2009) BTB and TAZ domain scaffold proteins perform a crucial function in Arabidopsis development. Plant J 58(1):109–121
Salas P, Oyarzo-Llaipen J, Wang D, Chase K, Mansur L (2006) Genetic mapping of seed shape in three populations of recombinant inbred lines of soybean (Glycine max L. Merr.). Theor Appl Genet 113(8):1459–1466
Salas Fernandez M, Becraft P, Yin Y, Lübberstedt T (2009) From dwarves to giants? Plant height manipulation for biomass yield. Trends Plant Sci 14:454–461
Shi J, Li R, Qiu D, Jiang C et al (2009) Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182:851–861
Specht J, Chase K, Macrander M, Graef G et al (2001) Soybean response to water: a QTL analysis of drought tolerance. Crop Sci 41(2):493–509
Sun Y, Pan J, Shi X, Du X, Wu Q, Qi Z et al (2012) Multi-environment mapping and meta-analysis of 100-seed weight in soybean. Mol Biol Rep 39(10):9435–9443
Sun S, Kim M, Lee Y, Li B, Lee S (2013) QTLs for resistance to phomopsis seed decay are associated with days to maturity in soybean (Glycine max). Theor Appl Genet 126(8):2029–2038
Tae-Hwan J, Freewalt K, Michal A, Mian R (2014) Identification of novel QTL for leaf traits in soybean. Plant Breed 133(1):61–66
Teng W, Han Y, Du Y, Sun D et al (2009) QTL analyses of seed weight during the development of soybean (Glycine max L. Merr.). Heredity 102(4):372
Ting S, Zhihong G, Liangju W et al (2012) Identification of differentially-expressed genes associated with pistil abortion in Japanese apricot by genome-wide transcriptional analysis. PLoS ONE 7(10):e47810
Wei Y, Xu H, Diao L et al (2015) Protein repair l-isoaspartyl methyltransferase 1 (PIMT1) in rice improves seed longevity by preserving embryo vigor and viability. Plant Mol Biol 89(4–5):475–492
Xi W, Yu H (2010) MOTHER OF FT AND TFL1 regulates seed germination through a negative feedback loop modulating ABA signaling in Arabidopsis. Plant Signal Behav 22(6):1733–1748
Xin W, Setsuko K (2017) Proteomic analysis of calcium effects on soybean root tip under flooding and drought stresses. Plant Cell Physiol 8:8
Young G, Mebrahtu T, Johnson J (2000) Acceptability of green soybeans as a vegetable entity. Plant Foods Hum Nutr 55(4):323–333
Zhang Q (2012) Functional study of the OsRBR1 in rice. Huanan Agricultural University (in Chinese)
Zhang J, Song Q, Cregan P, Nelson R et al (2015) Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genomics 16(1):217
Zhang J, Song Q, Cregan P, Jiang G (2016) Genome-wide association study, genomic prediction and marker-assisted selection for seed weight in soybean (Glycine max). Theor Appl Genet 129(1):117–130
Zhang W, Hu D, Raman R, Guo S et al (2017) Investigation of the genetic diversity and quantitative trait loci accounting for important agronomic and seed quality traits in Brassica carinata. Front Plant Sci 8:615
Zhang X, Wang W, Guo N, Zhang Y, Bu Y, Zhao J (2018) Combining QTL-seq and linkage mapping to fine map a wild soybean allele characteristic of greater plant height. BMC Genom 19(1):226
Zhao X, Han Y, Li Y, Liu D, Sun M, Zhao Y (2015) Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. Plant J Cell Mol Biol 82(2):245–255
Zhou R, Chen H, Wang X, Zhang X et al (2009) QTL analysis of yield, yield components, and lodging in soybean. Acta Agron Sin 35(5):821–830 (in Chinese)
Acknowledgements
This work was supported by the Key R & D project of Jiangsu Province (BE2019376), National Key R&D Program of China (2017YFD0101500), Genetically Modified Organisms Breeding Major Projects (2016ZX08004002), Modern Agro-industry Technology Research System of China (CARS-04-PS10), the Fundamental Research Funds for the Central Universities (KYT201801), Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT_17R55) and Jiangsu Collaborative Innovation Center for Modern Crop Production, Science and Technology Innovative Team in Fujian Academy of Agricultural Sciences (STIT2017-2-6), Cyrus Tang Innovation Center for Seed Industry.
Author information
Authors and Affiliations
Contributions
All authors have read and approved the manuscript. HX and JMZ conceived and coordinated the work. NG and JMZ conducted planting soybean materials. XNL, YZ, YPB and XFW conducted phenotyping experiments. XNL conducted association mapping experiments and wrote the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, X., Zhou, Y., Bu, Y. et al. Genome-wide association analysis for yield-related traits at the R6 stage in a Chinese soybean mini core collection. Genes Genom 43, 897–912 (2021). https://doi.org/10.1007/s13258-021-01109-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-021-01109-9