
Abstract
Glycine soja, the wild progenitor of soybean, is a potential source of useful genetic variation in soybean improvement. The objective of our study was to map quantitative trait loci (QTL) from G. soja that could improve the crop. Five populations of BC2F4-derived lines were developed using the Glycine max cultivar IA2008 as a recurrent parent and the G. soja plant introduction (PI) 468916 as a donor parent. There were between 57 and 112 BC2F4-derived lines in each population and a total of 468 lines for the five populations. The lines were evaluated with simple sequence repeat markers and in field tests for yield, maturity, plant height, and lodging. The field testing was done over 2 years and at two locations each year. Marker data were analyzed for linkage and combined with field data to identify QTL. Using an experimentwise significance threshold of P=0.05, four yield QTL were identified across environments on linkage groups C2, E, K, and M. For these yield QTL, the IA2008 marker allele was associated with significantly greater yield than the marker allele from G. soja. In addition, one lodging QTL, four maturity QTL, and five QTL for plant height were identified across environments. Of the 14 QTL identified, eight mapped to regions where QTL with similar effects were previously mapped. Many regions carrying the yield QTL were also significant for other traits, such as plant height and lodging. When the significance threshold was reduced and the data were analyzed with simple linear regression, four QTL with a positive allele for yield from G. soja were mapped. One epistatic interaction between two genetic regions was identified for yield using an experimentwise significance threshold of P=0.05. Additional research is needed to establish whether multiple trait associations are the result of pleiotropy or genetic linkage and to retest QTL with a positive effect from G. soja.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Improvement of elite soybean [Glycine max (L.) Merr.] germplasm in North America may be limited by genetic bottlenecks during its development. Pedigree analysis shows that 80% of the ancestry of this germplasm can be traced to only 13 ancestral cultivars (Gizlice et al. 1994). In addition, northern U.S. cultivars can trace 50% of their ancestry to only three ancestral lines (Gizlice et al. 1994). Recent estimates show that yield improvements in soybean continue at a modest rate of approximately 30 kg ha-1 year-1 or about 1% year-1 (Specht et al. 1999; Wilcox 2001). These yield improvements are sustained, however, by breeding efforts that continue to increase in size.
The primary gene pool that can be used to improve North American soybean germplasm includes elite soybean germplasm, exotic soybean lines and Glycine soja Sieb. and Zucc., the wild ancestor of soybean (Hymowitz and Singh 1987). The limited diversity in elite soybean germplasm has resulted in an interest in evaluating G. soja as a new source of genetic diversity for improving the crop. There is ample evidence that within G. soja, there is greater genetic diversity than in G. max (Maughan et al. 1995). Introgression of genes from wild species through genetic mapping with molecular markers has been employed successfully in the identification of agronomically important genes in a number of crop species (Tanksley et al. 1996; Tanksley and Nelson 1996; Tanksley and McCouch 1997; Xiao et al. 1998). Using the advanced backcross method, deVicente and Tanksley (1993) identified and transferred quantitative trait loci (QTL) for increased yield and soluble solids from Lycopersicon pennellii, a wild relative of tomato (Lycopersicon esculentum), into cultivated tomato. Further research has resulted in the identification of QTL that agronomically improve tomato from L. peruvianum (Fulton et al. 1997), L. hirsutum (Bernacchi et al. 1998) and L. pimpinellifolium (Tanksley et al. 1996). The advanced backcross method was also used to map genes that enhance rice yield from Oryza rufipogon, a weedy relative of cultivated rice (Xiao et al. 1996, 1998; Moncada et al. 2001).
A number of researchers have evaluated G. soja as a source of useful genes. These researchers were unsuccessful in identifying positive transgressive segregates or in mapping useful QTL from G. soja (Ertl and Fehr 1985; Graef et al. 1989; Suarez et al. 1991) until the employment of genetic markers became widespread. Useful QTL from G. soja were discovered in a population of F2-derived lines from a cross between the G. max experimental line A81–356022 and the G. soja accession PI 468916. In this population, QTL controlling hard seededness (Keim et al. 1990b), plant morphology, maturity (Keim et al. 1990a), and protein and oil concentration (Diers et al. 1992) were mapped. The G. soja alleles for the protein QTL were associated with over 20 g kg-1 greater protein concentration than the G. max alleles (Diers et al. 1992). These G. soja alleles have been backcrossed into the background of the G. max parent and one region continued to increase protein concentration in a BC3 population, although it was associated with reduced yield (Sebolt et al. 2000).
Concibido et al. (2003) mapped a QTL allele that was associated with increased seed yield from G. soja PI 407305. The initial mapping was done in a BC2 population developed according to the advanced backcross method (Tanksley and Nelson 1996). In the BC2 population, the QTL allele was associated with a 12% yield increase across testing environments. The QTL was then backcrossed into seven genetic backgrounds and retested. They observed a significant (P<0.05) positive effect on yield for the QTL allele from G. soja in three of the seven backgrounds.
It is difficult to map QTL controlling yield in populations developed directly from crosses between soybean and G. soja, because of the poor general agronomic performance of lines. Agronomic problems such as premature pod dehiscence (seed shattering) highlight the need to study yield in backcross populations that have improved agronomic performance. Unfortunately, advanced backcross populations (Tanksley and Nelson 1996) are difficult to develop in species such as soybean where it is problematic to produce the hundreds of hybrid seed needed for the experiments. We have developed backcross populations for mapping QTL from wild species using a novel approach that does not require the large amount of hybrid seed production required for the advanced backcross method. The objective of our study was to map QTL for yield and other agronomic traits in a series of backcross populations developed using a G. soja line as a donor parent and a soybean cultivar as a recurrent parent.
Materials and methods
Plant material and trait evaluations
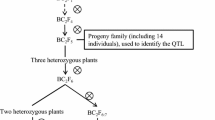
The mapping populations consisted of five BC2 populations developed using the G. max cultivar IA2008 as the recurrent parent and the G. soja plant introduction (PI) 468916 as the donor parent. The cultivar IA2008 is a high yielding, maturity group II public cultivar developed at Iowa State University. PI 468916 is a G. soja plant introduction that was collected from Liaoning Province, People's Republic of China in 1982 (Bernard et al. 1989). PI 468916 is also the G. soja parent of the population used in developing the Iowa State University-USDA genetic map (Keim et al. 1990a; Cregan et al. 1999).
IA2008 and PI 468916 were crossed and a population of F2:3 lines were produced. Five random F2:3 lines were crossed with IA2008 to produce BC1F1 plants. These BC1F1 plants were crossed again with IA2008 to produce BC2F1 seed. Five BC2F1 plants that each trace to a different F2 plant were grown to initiate the development of five backcross populations. The populations were inbred to the F4 generation through single-seed descent. Each F4 plant was then threshed individually to produce BC2F4-derived lines. There were a total of 468 BC2F4-derived lines tested with 110 lines each in populations 324B and 330A, and 79, 57 and 112 lines in populations 326, 334A and 338B, respectively.
The BC2F4-derived lines from the five populations and the IA2008 parent were evaluated in field tests during the summers of 1999 and 2000. Two replicates of the lines were grown in Lincoln, Nebraska and Urbana, Illinois each year. The plots were arranged in a randomized complete block design with each population grown as a separate set. The lines were grown in 4-row plots with a 76-cm row spacing and a seeding rate of 30 seeds m-1. The length of the plots was 3.2 m in Illinois and 3.0 m in Nebraska. The two center rows of each plot were harvested for seed yield measurement. Populations 326 and 334A were not harvested in Nebraska during 2000 because of plot damage in that area of the field. Seed moisture was measured for seed harvested from each plot and was used to calculate seed yield on a moisture-free basis. In addition to yield, the lines were rated for date of maturity, plant height, and lodging. Date of maturity was the day when 95% of the pods had changed to their mature pod color. Plant height was the average length in centimeters of mature plants from the ground to the top node of the main stem. Lodging was rated at maturity with a visual score ranging from 1 to 5, with 1 representing all plants erect and 5 all plants prostrate.
Pearson product-movement correlations among all traits were calculated with PROC CORR (SAS 1988). Estimates of variance components and broad-sense heritabilities were calculated for each population from mean squares (Fehr 1987) obtained from PROC GLM of SAS. Genotypes within populations, locations, and years were analyzed as random effects.
Molecular marker and QTL analysis
The lines in the populations were evaluated with simple sequence repeat (SSR) markers that were mapped previously in soybean (Cregan et al. 1999). Genomic DNA was extracted from 10 bulked seedlings per line using a modified CTAB method (Kisha et al. 1997). Polymerase chain reactions (PCR) were carried out according to Cregan and Quigley (1997). The PCR products were analyzed by electrophoresis in 6% non-denaturing polyacrylamide gels (Sambrook et al. 1989; Wang et al. 2003) and stained with 0.1 μg ml-1 ethidium bromide. DNA from the original parents of the populations (PI 468916 and IA2008) and a bulk of DNA from each population were screened with 606 SSR markers to identify markers polymorphic between the parents and those segregating in the populations. Each bulk consisted of an equal amount of DNA from all lines in the population. The polymorphic markers were then used to evaluate each line in the populations for which they were segregating.
The observed segregation ratios of SSR markers were tested for goodness-of-fit to the expected ratio using χ2 tests with a significance level of α=0.05. Conservative estimates of the proportion of the genome that was segregating in each population were calculated by totaling the lengths of genomic regions flanked by segregating markers. The cM lengths of the segregating regions were determined by the map positions of the flanking markers on the composite maps in Cregan et al. (1999) and the SoyBase website (http://soybase.org).
Linkage and map distances among the polymorphic markers were determined using the computer program JoinMap (Stam 1993) with the Kosambi mapping function (Kosambi 1944) and a minimum LOD score of 3.0. The marker orders from the composite maps on SoyBase were used as fixed orders in the linkage analysis. The names of linkage groups (LGs) on SoyBase were used for presenting the results in this paper.
QTL controlling traits were mapped by composite interval mapping (CIM) and simple linear regression with the computer program package QTL-CARTOGRAPHER (Basten et al. 1999). Marker and trait data from each population were analyzed independently in both linkage and QTL analyses. Trait values obtained from each environment and the averages across environments were treated as independent traits in the QTL analysis. The threshold of LOD scores for declaring a putative QTL as significant was determined by 1,000 permutations using the Zmapqtl program in QTL-CARTOGRAPHER. A LOD value corresponding to an experimentwise threshold of α=0.05 was used to declare a QTL as significant. The estimate of the QTL position is the point of maximum LOD score in the region under consideration. Only the results from QTL analyses of the averages across environments are reported in the tables.
A search for epistatic interactions between identified yield QTL and markers not linked to the QTL was performed using the conditional search function of the Epistat program (Chase et al. 1997). The Epistat program examines pairs of markers for additive by additive epistatic interactions (Chase et al. 1997). For each pair of markers examined, at least one marker had a significant additive effect on yield with a log likelihood ratio (LLR) greater than 5 and the other marker was on a different linkage group to avoid the confounding effects of linkage. The LLR threshold for identifying preliminary significant interactions was set to 6. The MNTECRLO program within the Epistat package was then used to determine the significance of the preliminary significant interactions. The MNTECRLO program estimates type I errors and significance thresholds using Monte Carlo methods. One million Monte Carlo simulations were carried out for each preliminary significant interaction. The P values from the output of MNTECRLO program were corrected for the number of interactions analyzed to produce experimentwise P values with the equation P corr =1–(1–P)x, where P corr is the corrected P value, P is the P value from the output of the MNTECRLO program, and x is the number of interactions analyzed. If the corrected P value is less than 0.05, the interaction between the two markers was declared significant. For significant interactions, Tukey tests were carried out to determine the significance of differences between the means of different interacting genotypes using the SAS GLM procedure.
Results
Molecular marker analysis
Of the 606 SSR markers tested using the parental and pooled progeny DNA, 302 were polymorphic between the two parents. One hundred and fifty nine (52%) of these markers segregated in at least one population and 77 (26%) markers segregated in at least two populations. Assuming that no selection occurred during the development of the BC2 populations, each population should segregate 25% of its genome while the remaining 75% should be fixed for the soybean parent. Of the markers that were polymorphic between the two parents, 24.1% segregated in population 326 and 22.7% segregated in population 330A. These values are not significantly (P=0.05) different from the expectation of 25% of the markers segregating based on Chi-square tests. The percentage of markers that segregated in populations 334A, 338B, and 324B were 13.5%, 15.9%, and 18.9%, respectively, and these values were significantly (P<0.05) lower than the expected. The lower than expected percentage of markers segregating in three populations was likely caused by inadvertent selection during population development, since conscious selection was not perfomed. The genomic regions that were verified as segregating in populations with SSR markers are listed in Table 1. These estimates are conservative because additional chromosomal regions adjacent to these flanking regions would also segregate. In addition, other unlinked regions likely went undetected due to a lack of polymorphic markers.
The grouping of markers was generally consistent with Cregan et al. (1999) and composite maps at the SoyBase website. There were two exceptions where markers belonged to different LGs on the composite map, but were grouped together in our analyses. Marker Satt352 from LG G on the composite maps linked close to Satt133 from LG A2 in three of our populations. In addition, marker Satt317 from LG H on the composite map was grouped with markers on LG G in our analysis. As expected, the map distances were not always consistent among populations and with the composite maps. Nevertheless, the relative distances among markers were generally consistent among maps.
Field data analysis
There was significant (P<0.01) genetic variance among lines for yield, plant height, maturity, and lodging in each population. Location was significant for all traits in all populations except for plant height and lodging in population 334A. Year had a significant effect on yield in all populations except population 334A. Year was not significant for plant height in three of the five populations but was significant for maturity and lodging in all populations. The interactions of genotype-by-year and genotype-by-location were not significant for all traits. However, the interaction of genotype-by-year-by-location was significant for all four traits.
The population means for yield ranged from 2,263 kg ha-1 for population 330A to 2,697 kg ha-1 for population 334A (Table 2, Fig. 1). In no population were lines identified that had significantly greater yield than IA2008, the recurrent parent. The heritabilities for yield varied from 0.29 in population 326 to 0.85 in population 324B.

Yield distributions of lines over all environments for each population
Analysis of QTL
Four significant (experimentwise P<0.05) yield QTL were identified with CIM on LGs C2, E, K and M (Table 3). The QTL on LG C2 was significant for population 334A in all three environments where data were collected. The QTL explained 40% of the phenotypic variance for yield in this population. The QTL on LGs E and M were identified in population 338B. The LG E QTL explained 28% of the phenotypic variance for yield and the LG M QTL explained 29% of the phenotypic variance for yield. The QTL on LG E was significant in three of the four environments and the QTL on LG M was significant in all environments. The significant QTL on LG K was identified in populations 324B and 330A and was significant in all environments that the populations were tested. This QTL explained 38–40% of the total phenotypic variance for yield in the two populations. For all four significant yield QTL, the marker alleles from IA2008, the G. max parent, were associated with greater yield than the G. soja alleles.
Three of the four yield QTL that we mapped were located in the same region or near previously mapped yield QTL. The QTL for yield on LG C2 mapped to the same location as yield QTL that were mapped in three previous studies. Two of these populations were from crosses between 'Minsoy' and 'Noir 1' (Mansur et al. 1993a, 1993b, Mansur et al. 1996) and the third was from a cross between 'Noir 1' and 'Archer' (Orf et al. 1999a, 1999b). The QTL for yield on LG M also was identified in the two populations developed by crossing 'Minsoy' and 'Noir 1' (Mansur et al. 1993a, 1996). Yuan et al. (2002) found evidence of a yield QTL on LG K in populations developed from crossing 'Flyer' with 'Hartwig' and 'Essex' with 'Forrest'. The yield QTL on LG K in both populations of Yuan et al. (2002) mapped near Satt337, which is 22 cM from our LOD peak according to the composite map. Additional work is needed to verify whether the QTL in our study and the study of Yuan et al. (2002) map to the same location or if they are different QTL. The QTL for yield on LG E has not been identified in earlier mapping studies, suggesting the possibility of this being a new yield QTL.
Five QTL for plant height were identified on five LGs (Table 3). Four of the five QTL mapped to the same locations as the yield QTL on LGs C2, E, K and M. The QTL on LG K was identified with significant LOD scores in population 324B in three of the four test environments and in all four environments for population 330A. The QTL on LGs C2 and E were significant in three of the four test environments and the QTL on LGs O and M were significant in all four environments. The height QTL on LG C2 (Mansur et al. 1996, Orf et al. 1999b) and M (Specht et al. 2001) mapped to regions where plant height QTL were previously identified in G. max populations.
Four QTL for maturity were identified on LGs C2, L, M, and O (Table 3). The four QTL were identified in four different populations and were identified with significant LOD scores in all test environments. The maturity QTL on LGs C2, and M, mapped to the same regions as the QTL for plant height and yield in these populations. In addition, the maturity QTL on LG O mapped to the same position as a height QTL. Other researchers mapped QTL for maturity to the same regions on LG C2 (Orf et al. 1999b), L (Orf et al. 1999b), and M (Mansur et al. 1993a, 1993b, 1996; Lark et al. 1994; Orf et al. 1999b) in G. max populations.
Only one QTL was identified for lodging in this study and it has not been identified in earlier studies. This QTL was identified in population 330A with significant LOD scores in all four environments. This QTL maps to the same region as a QTL for plant height and yield. Surprisingly, the region from IA2008 is associated with greater plant height but less lodging than the region from G. soja. This contradicts the normal association that is observed between greater plant height and increased lodging. One explanation for this contradiction is that in this region IA2008 carries a QTL allele, that confers less plant lodging and a second QTL that confers taller plant height. Alternatively, there could be a single QTL with a pleiotropic effect of increasing both plant height and stem strength.
The yield and marker data were analyzed with the simple linear regression option of QTL-CARTOGRAPHER and a significance threshold of P=0.05 for each marker association to determine if any positive yield QTL alleles from G. soja could be identified at this lower threshold. These analyses led to the identification of four QTL where the G. soja alleles were significantly associated with improved yield across all environments tested (Table 4). In addition, the LG D1b QTL was significantly associated with maturity and the LG L QTL was significantly associated with lodging. According to Soybase, the regions where these loci mapped have not been previously identified as containing significant yield QTL. However, PI468916 was previously found to carry a QTL allele for resistance to soybean cyst nematode (SCN, Heterodera glycines Ichinohe) near Satt491 on LG E (Wang et al. 2001). Although high SCN infestations were not found in the field tests, there may have been sufficient pressure to cause the SCN resistance QTL to have a significant effect on yield. Because these yield QTL were not identified with a high significance threshold through CIM, they will need to be retested in additional studies to confirm their effect.
Significant epistatic interactions for yield across environments were found with the computer program Epistat in only population 324B. The significant interactions in this population were between Satt349 and Satt417 in the region of the yield QTL on LG K, and Satt189 on LG D1b. On the Soybase composite map, Satt349 and Satt417 are positioned at 65 and 71 cM, respectively, on LG K and Satt189 is located at 113 cM on LG D1b. In the significant interactions between the two markers on LG K and the marker on LG D1b, the yield of genotypes that are homozygous for G. max alleles on LG K and G. soja alleles on LG D1b is higher than genotypes homozygous for G. max alleles on both LG K and LG D1b. However, this difference is not statistically significant at P=0.05. These two interacting regions also segregate in population 330A, but no significant interaction was detected between them in this population. There are two possible explanations for these inconsistent results. The first is that the significant interaction between the two regions identified in population 324B is actually a type I error and the second is that at least one additional region in the genome is needed for the interaction to occur.
Discussion
A major goal of this research effort was to map the locations of favorable yield QTL alleles from G. soja that could be useful in soybean improvement. We were not successful in finding these QTL using the CIM method with a LOD threshold corresponding to an experimentwise P=0.05. This lack of success stands in contrast to reports of the identification of positive alleles for yield from wild accessions in other species (Tanksley et al. 1996, Xiao et al. 1998) and soybean (Concibido et al. 2003). There are a number of explanations for our lack of success in this study. The first is simply that PI 468916, the G. soja parent of the population, lacks positive yield QTL that could improve IA2008, the G. max parent. Another possibility is that the G. soja parent did have one or more positive QTL, but they were not detected in this study because they were not segregating in any population. This is possible because only 52% of the markers that were polymorphic between the parents segregated in at least one population. In addition, positive QTL from G. soja may have segregated in one or more populations, but were not detected. This may have occurred if there were no segregating markers near the QTL or if polymorphic markers were present but the populations used in this study were of insufficient size to detect the QTL. Larger populations were not used in this study, in part, because of the difficultly in field testing the G. soja introgressed lines, which tend to be viny and prone to pod dehiscence (shattering). Many plots had to be hand-harvested prior to machine harvesting to avoid pod dehiscence affecting yield results. In addition, our goal was to identify relatively large, positive QTL from the G. soja parent, which should be identifiable in relatively small populations.
In this study, we used a series of backcross populations instead of the advanced backcross method, which has been successfully used to map positive QTL from exotic sources (Tanksley and Nelson 1996). In the advanced backcross method, populations are advanced typically to the backcross two generation before trait evaluations are initiated. This backcrossing is done by producing hundreds of F1 seed. Producing these F1 seed is not difficult in crops such as maize and tomato, which can be crossed easily, but is a problem in crops such as soybean where the production of F1 seed is tedious and time consuming. In response to the difficulty in producing the large number of F1 seed needed in the advanced backcross method, we developed a series of backcross populations that were used in our study.
In addition to reducing the number of F1 seed needed, there are other advantages to using the series backcross method compared to the advanced backcross method. One advantage is a reduction in marker data collection. After an initial marker screen of the parents and a separate bulk of lines from each population, the populations are tested with only those markers that segregate in the population. With the advanced backcross method, the entire population needs to be tested with all polymorphic markers. Another advantage of our series backcross populations is that although all of our populations are smaller than would be used in the advanced backcross method, only a small proportion of the genome segregates in each population. In addition, there should be an equal frequency for the alleles from the wild and domestic parent for segregating markers in the series backcross method. In the advanced backcross method, the marker frequency will be skewed in the population. Both the small proportion of the genome segregating in each population and the equal frequency of alleles will be advantages compared to the advanced backcross method. A disadvantage of the series backcross method is the inability to assay the entire G. soja genome for QTL, because many regions will not segregate in any population. Of the markers that were polymorphic between the parents, we found that only 52% segregated in at least one population. This is significantly lower than the expectation of 76% of the G. soja genome segregating in at least one population. Because we did not conduct QTL mapping with both our series backcross method and with the advanced backcross method, it is impossible to know whether we would have had greater success using the advanced backcross method.
Another difference between our procedures and the advanced backcross method procedures is whether selection is done during population development. Tanksley and Nelson (1996) recommend selection for good agronomic type during population development with the advanced backcross method. We avoided conscious selection because we were concerned that it would result in the segregation of even less of the G. soja genome in populations which may result in our missing positive QTL from G. soja. For example, selection for good plant type would result in missing an allele that increases yield that happens to be in coupling linkage with an allele for viny growth. In hindsight, there would have been advantages for selection against premature pod dehiscence during population development. This trait made yield testing difficult, resulting in the need for hand harvesting of many plots. We could have started with a larger initial population and selected for favorable agronomic traits during population development. This would have allowed us to evaluate the selected lines in more replications and environments using the same total resources during the field evaluation phase.
Many regions contained QTL that were significant for more than one trait (Table 3). In fact, of the four yield QTL identified, all were also significant for plant height and two were significant for maturity. There was no clear pattern in the relationship between yield and these other traits. For example, the marker alleles from IA2008 on LG C2 were significant for greater yield, shorter plants, and earlier maturity, whereas the IA2008 alleles on LG M were significant for greater yield, taller plants and later maturity. More research is needed to dissect whether these multiple trait associations are the result of pleiotropy or genetic linkage.
Of the four significant yield QTL identified through CIM (Table 3), only the QTL on LG E was not identified in previous studies. There was also a similar trend for associations with plant height, maturity and lodging with five of the ten QTL for these traits identified in other studies. The consistency of our results with previous studies suggests that although our population sizes were small, we have mapped real QTL. However, our relatively small populations likely caused an overestimation of the effects of the QTL we mapped (Beavis 1994).
Most previous QTL mapping in soybean was done in populations developed from intraspecific soybean crosses. Because our populations were interspecific, we expected to assay allelic variation from G. soja that was not present in soybean, and therefore did not expect so many of our QTL to have been identified previously. Our finding of consistency in the locations of QTL across different species is consistent with results in tomato. Chen et al. (1999) compared QTL identified for fruit weight and soluble solid content in seven mapping studies conducted by crossing tomato with four related wild species. They found as much consistency in the locations where QTL mapped when comparing two populations developed by crossing tomato with the same wild species as they did when comparing these populations to a population developed by crossing tomato to another species.
Results from our study and those reported in the literature suggest that it will be difficult to unlock positive allelic diversity from G. soja. There are no documented cases in the North American literature of soybean being improved through recent introgressions from G. soja. There are examples of the mapping of useful QTL from G. soja, but in most cases, evidence has later shown that these QTL were already present in G. max germplasm. This includes the high protein QTL identified by Diers et al. (1992) which later evidence suggested was already in high protein G. max germplasm (Sebolt et al. 2000). The yield QTL identified by Concibido et al. (2003) had a positive effect in some genetic backgrounds but not others, which suggests either the G. soja allele is already present in some genetic backgrounds or that the allele requires additional gene(s) in epistatic interactions. There is also new evidence that one of the two SCN resistance QTL from G. soja identified by Wang et al. (2001) also exists in G. max germplasm (Yue et al. 2001). These results, together with our finding that many of the yield and agronomic QTL we identified were also mapped in G. max populations, shows the difficulty in identifying new useful genetic diversity from G. soja.
References
Basten CJ, Weir BS, Zeng Z-B (1999) QTL Cartographer, Version 1.13. Department of Statistics, North Carolina State University, Raleigh, NC
Beavis WD (1994) The power and deceit of QTL experiments: Lessons from comparative QTL studies. In: Proceedings of the Forty-sixth Annual Corn and Sorghum Industry Research Conference ASTA, Washington, DC
Bernacchi D, Beck Bunn T, Eshed Y, Lopez J, Petiard V, Uhlig J, Zamir D, Tanksley S (1998) Advanced backcross QTL analysis in tomato. I. Identification of QTLs for traits of agronomic importance from Lycopersicon hirsutum. Theor Appl Genet 97:381–397
Bernard RL, Juvik GA, Nelson RL (1989) USDA soybean germplasm collection inventory, vol 2(30). University of Illinois at Urbana-Champaign
Chase K, FR Adler, KG Lark (1997) Epistat: a computer program for identifying and testing interactions between pairs of quantitative trait loci. Theor Appl Genet 94:724–730
Chen FQ, Foolad MR, Hyman J, St. Clair DA, Bellaman RB (1999) Mapping of QTLs for lycopene and other fruit traits in a Lycopersicon esculentum × L. pimpinellifolium cross and comparison of QTLs across tomato species. Mol Breeding 5:283–299
Concibido VC, Vallee BL, Mclaird P, Pineda N, Meyer J, Hummel L, Yang J, Wu K, Delannay X (2003) Introgression of a quantitative trait locus for yield from Glycine soja into commercial soybean cultivars. Theor Appl Genet 106:575–582
Cregan PB, Quigley CV (1997) Simple sequence repeat DNA marker analysis. In: Caetano-Anolles G, Gresshoff PM (eds) DNA markers: protocols, applications and overviews. Wiley, New York, pp 173–185
Cregan PB, Jarvik T, Bush AL, Shoemaker RC, Lark KG, Kahler AL, Kaya N, VanToai TT, Lohnes DG, Chung J, Specht JE (1999) An integrated genetic linkage map of the soybean genome. Crop Sci 39:1464–1490
deVicente MC, Tanksley SD (1993) QTL analysis of transgressive segregation in an interspecific tomato cross. Genetics 134:585–596
Diers BW, Keim P, Fehr WR, Shoemaker RC (1992) RFLP analysis of soybean seed protein and oil content. Theor Appl Genet 83:608–612
Ertl DS, Fehr WR (1985) Agronomic performance of soybean genotypes from Glycine max × Glycine soja crosses. Crop Sci 25:589–592
Fehr WR (1987) Principles of cultivar development. Macmillan, New York
Fulton TM, Beck Bunn T, Emmatty D, Eshed Y, Lopez J, Petiard V, Uhlig J, Zamir D, Tanksley SD (1997) QTL analysis of an advanced backcross of Lycopersicon peruvianum to the cultivated tomato and comparisons with QTLs found in other wild species. Theor Appl Genet 95:881–894
Gizlice Z, Carter TE, Burton JW (1994) Genetic base for North American public soybean cultivars released between 1947 and 1988. Crop Sci 34:1143–1151
Graef GL, Fehr WR, Cianzio SR (1989) Relation of isozyme genotypes to quantitative characters in soybean. Crop Sci 29:683–688
Hymowitz T, Singh RJ (1987) Taxonomy and speciation. In: Wilcox JR (ed) Soybean: improvement, production and uses, 2nd edn, vol 16. American Society of Agronomy, Madison, Wis., pp 23–48
Keim P, Diers BW, Olson TC, Shoemaker RC (1990a) RFLP mapping in soybean: association between marker loci and variation in quantitative traits. Genetics 126:735–742
Keim P, Diers BW, Shoemaker RC (1990b) Genetic analysis of soybean hard seededness with molecular markers. Theor Appl Genet 79:465–469
Kisha TJ, Sneller CH, Diers BW (1997) Relationship between genetic distance among parents and genetic variance in populations of soybean. Crop Sci 37:1317–1325
Kosambi DD (1944) The estimation of map distance from recombination values. Ann Eugen 12:185–199
Lark KG, Orf J, Mansur LM (1994) Epistatic expression of quantitative trait loci (QTL) in soybean (Glycine max (L.) Merr.) determined by QTL association with RFLP alleles. Theor Appl Genet 88:486–489
Mansur LM, Lark KG, Kross H, Oliveira A (1993a) Interval mapping of quantitative trait loci for reproductive, morphological, and seed traits of soybean (Glycine max L.). Theor Appl Genet 86:907–913
Mansur LM, Orf J, Lark KG (1993b) Determining the linkage of quantitative trait loci to RFLP markers using extreme phenotypes of recombinant inbreds of soybean (Glycine max L. Merr.). Theor Appl Genet 86:914–918
Mansur LM, Orf JH, Chase K, Jarvik T, Cregan PB, Lark KG (1996) Genetic mapping of agronomic traits using recombinant inbred lines of soybean. Crop Sci 36:1327–1336
Maughan PJ, Saghai Maroof MA, Buss GR (1995) Microsatellite and amplified sequence length polymorphisms in cultivated and wild soybean. Genome 38:715–723
Moncada P, Martinez CP, Borrero J, Chatel M, Gauch H, Jr., Guimaraes E, Tohme J, McCouch SR (2001) Quantitative trait loci for yield and yield components in an Oryza sativa × Oryza rufipogon BC2F2 population evaluated in an upland environment. Theor Appl Genet 102:41–52
Orf JH, Chase K, Adler FR, Mansur LM, Lark KG (1999a) Genetics of soybean agronomic traits. II. Interactions between yield quantitative trait loci in soybean. Crop Sci 39:1652–1657
Orf JH, Chase K, Jarvik T, Mansur LM, Cregan PB, Adler FR, Lark KG (1999b) Genetics of soybean agronomic traits: I. Comparison of three related recombinant inbred populations. Crop Sci 39:1642–1651
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Plainview, NY
SAS (1988) SAS/STAT user's guide, version 6.03. SAS Institute, Cary, NC
Sebolt AM, Shoemaker RC, Diers BW (2000) Analysis of a quantitative trait locus allele from wild soybean that increases seed protein concentration in soybean. Crop Sci 40:1438–1444
Specht JE, Hume DJ, Kumudini SV (1999) Soybean yield potential-a genetic and physiological perspective. Crop Sci 39:1560–1570
Specht JE, Chase K, Macrander M, Graef GL, Chung J, Markwell JP, Germann M, Orf JH, Lark KG (2001) Soybean response to water: a QTL analysis of drought tolerance. Crop Sci 41:493–509
Stam P (1993) Construction of integrated genetic linkage maps by means of a new computer package: JoinMap. Plant J 3:739–744
Suarez JC, Graef GL, Fehr WR, Cianzio SR (1991) Association of isozyme genotypes with agronomic and seed composition traits in soybean. Euphytica 52:137–146
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066
Tanksley SD, Nelson JC (1996) Advanced backcross QTL analysis: a method for the simultaneous discovery and transfer of valuable QTLs from unadapted germplasm into elite breeding lines. Theor Appl Genet 92:191–203
Tanksley SD, Grandillo S, Fulton TM, Zamir D, Eshed Y, Petiard V, Lopez J, Beck Bunn T (1996) Advanced backcross QTL analysis in a cross between elite processing line of tomato and its wild relative L. pimpinellifolium. Theor Appl Genet 92:213–224
Wang D, Arelli PR, Shoemaker RC, Diers BW (2001) Loci underlying resistance to race 3 of soybean cyst nematode in Glycine soja plant introduction 468916. Theor Appl Genet 103:561–566
Wang D, Shi J, Carlson SR, Cregan PB, Ward RW, Diers BW (2003) A low-cost, high-throughput polyacrylamide gel electrophoresis system for genotyping with microsatellite DNA markers. Crop Sci 43:1828–1832
Wilcox JR (2001) Sixty years of improvement in publicly developed elite soybean lines. Crop Sci 49:1711–1716
Xiao J, Grandillo S, Ahn SN, McCouch SR, Tanksley SD, Li J, Yuan L, Xiao JH, Li JM, Yuan LP (1996) Genes from wild rice improve yield. Nature 384:223–224
Xiao J, Li J, Grandillo S, Ahn S, Yuan L, Tanksley SD, McCouch SR (1998) Identification of trait-improving quantitative trait loci alleles from a wild rice relative, Oryza rufipogon. Genetics 150:899–909
Yuan J, Njiti VN, Meksem K, Iqbal MJ, Triwitayakorn K, Kassem MA, Davis GT, Schmidt ME, Lightfoot DA (2002) Quantitative trait loci in two soybean recombinant inbred line populations segregating for yield and disease resistance. Crop Sci 42:271–277
Yue P, Arelli PR, Sleper DA (2001) Molecular characterization of resistance to Heterodera glycines in soybean PI 438489B. Theor Appl Genet 102:921–928
Acknowledgements
This material is based upon work supported by USDA-NRI Award No. 99–35300–7820. We appreciate the help of Kevin Chase and Gordon Lark for their assistance in using the program Epistat. This publication is a contribution of the University of Nebraska Agricultural Research Division, Lincoln, NE, 68583, Journal Series No. 13989.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H.C. Becker
Rights and permissions
About this article
Cite this article
Wang, D., Graef, G.L., Procopiuk, A.M. et al. Identification of putative QTL that underlie yield in interspecific soybean backcross populations. Theor Appl Genet 108, 458–467 (2004). https://doi.org/10.1007/s00122-003-1449-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-003-1449-z