
Abstract
Low temperature at the booting stage of rice causes male sterility resulting in severe yield loss. Cold tolerance has long been an important objective in rice breeding. We identified a quantitative trait locus (QTL) for cold tolerance on the long arm of chromosome 3 from the cold-tolerant breeding line ‘Ukei 840’ by using F2 and BC1F2 populations from crosses between ‘Ukei 840’ and ‘Hitomebore’. The cold tolerance of ‘Ukei 840’ is derived from the Chinese cultivar ‘Lijiangxintuanheigu’. The effect of this QTL on cold tolerance was confirmed by developing ‘Hitomebore’ chromosome segment substitution lines having ‘Lijiangxintuanheigu’ alleles on chromosome 3. By producing recombinants in chromosome 3, the QTL region for cold tolerance was delimited to the region of about 1.2-Mb region between RM3719 and RM7000. All lines heterozygous for the QTL showed seed fertilities as low as that of ‘Hitomebore’, suggesting that the ‘Lijiangxintuanheigu’ allele for cold tolerance in the QTL region is recessive. Determination of a 1.2-Mb nucleotide sequence of ‘Ukei 840’ and comparison with the published genomic sequence of ‘Nipponbare’ showed 254 SNPs, of which 11 were in coding regions of genes, seven in five genes being non-synonymous. SNPs were detected in the 5-kb upstream regions of 89 genes, but no differences of gene expression levels were detected between alleles of these genes. Although further delimitation is required to identify the gene responsible for cold tolerance of ‘Lijiangxintuanheigu’, SNP markers developed here will be useful for marker-assisted selection in a breeding program using ‘Lijiangxintuanheigu’ as a donor of cold tolerance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
High temperature is required for normal development of pollen grains at the booting stage in rice. Low temperature, lower than 19°C, during the period of microspore development causes male sterility, resulting in severe loss of yield (Satake and Hayase 1970). Such damage to gamete development by low temperature is observed only in male organs, while development of female organs is normal (Hayase et al. 1969). In the northern region of Japan and some countries in the temperate zone where the temperature is not consistently high enough for rice production, rice yield is reduced dramatically once every several years. Thus, rice cultivars with high tolerance to low-temperature stress at the booting stage are required for stable rice production in such regions.
In the development of pollen grains, the callose wall, which covers haploid cells of a tetrad, is digested by callase secreted by the tapetum. The haploid cells are released as microspores, and mitotic divisions occur twice in the microspores during development to mature pollen grains. Simultaneously, tapetal cells are degraded. Under low temperature, the tapetal cells expand abnormally, and anther locules are invaded (Nishiyama 1970). A decrease of inorganic phosphate, an increase of nonreducing sugar (Ito 1978), and a decrease of acid phosphatase activity (Nishiyama 1978) have been observed in anthers of rice under low temperature, suggesting that abnormal sugar metabolism causes expansion of the tapetal cells due to a change of osmotic pressure and results in abnormality of pollen development.
Rice breeding for cold tolerance has long been performed in Japan. A deep-water irrigation system using cold water with controlled temperature has been developed as a reliable screening method for cold tolerance and is widely used for development of cold-tolerant cultivars (Matsunaga 2005). ‘Koshihikari’ has been identified as a highly cold-tolerant cultivar, but its flowering time is too late for cultivation in the northern region of Japan. Therefore, ‘Hitomebore’, which has high tolerance and an early flowering trait, has been developed by crossing ‘Koshihikari’ with ‘Hatsuboshi’ (Sasaki 2005). ‘Hitomebore’ shows high seed fertility, more than 70%, after cultivation in the deep-water irrigation system with cold water controlled at 19°C. However, even these cold-tolerant cultivars exhibit low seed fertilities, less than 50%, under cold-deep-water irrigation of 18.5°C. Thus, further improvement of cold tolerance is required.
Cold tolerance of rice at the booting stage is a quantitative trait controlled by multiple genes. Since it is difficult to combine many genes responsible for cold tolerance by investigating plant phenotypes, identifying each gene, and combining the genes by marker-assisted selection are considered to be effective means of developing cold-tolerant cultivars. Since there is a large variation of cold tolerance in rice cultivars (Toriyama and Futsuhara 1960), quantitative trait locus (QTL) analyses using various cultivars have been carried out. ‘Silewah’ and ‘Padi Labou Alumbis’, which have been identified as highly cold-tolerant cultivars by a Japan–China cooperative project for rice genetic resources in Yunnan Province of China (International Rice Research Institute 1977), have been used as parents for developing ‘Norin PL8’ and ‘Norin PL11’, respectively. QTLs for cold tolerance of ‘Norin PL8’ have been detected on chromosome 3 and 4 (Saito et al. 1995, 2001, 2004), and that of ‘Norin PL11’ has been mapped on chromosome 8 (Kuroki et al. 2007). The QTL on chromosome 4 derived from ‘Silewah’ has been delimited to a 56-kb region, and a gene encoding F-box protein among seven genes in this region has been suggested to be the gene responsible for cold tolerance (Saito et al. 2004, 2010). QTLs for cold tolerance of the Japanese leading cultivar ‘Koshihikari’ have been found on chromosomes 1, 7, and 11 (Takeuchi et al. 2001), and those of ‘Kunmingxiaobaigu’ have been detected on chromosomes 3, 6, and 7 (Dai et al. 2004). The presence of many different QTLs in different rice cultivars suggests a complicated mechanism of cold tolerance controlled by many genes.
‘Lijiangxintuanheigu’ (‘LTH’ hereafter), a local variety of Yunnan Province in China, has been reported to be one of the most cold-tolerant cultivars among 148 cultivars (Horisue et al. 1988). Recently, Ye et al. (2010) have detected one QTL for cold tolerance at the booting stage of ‘Lijiangheigu’, which has a name similar to ‘LTH’, on the short arm of chromosome 10. Investigating the effects of low temperature at the booting stage on pollen development in a leading cultivar in Australia, ‘Doongara’, and the highly cold-tolerant cultivar ‘R31’, Oliver et al. (2005) have revealed that sucrose content increases in the anthers and that starch does not accumulate in the pollen grains of ‘Doongara’, while sucrose does not accumulate in anthers having fertile pollen grains in ‘R31’ and that expression of a gene encoding cell-wall-bound acid invertase, which has an important role in transport of sucrose to sink tissues, is suppressed in ‘Doongara’ but is high in ‘R31’. An increase in the level of abscisic acid (ABA) has been observed in ‘Doongara’ treated by low temperature, while not in similarly treated ‘R31’ (Oliver et al. 2007). Treatment of ‘Doongara’ with ABA has been found to result in a change of expression of an invertase gene and in male sterility similar to that caused by low temperature. ABA has been suggested to function as a signaling molecule participating in male sterility due to low-temperature stress by controlling the transport pathway of sugar (Oliver et al. 2007).
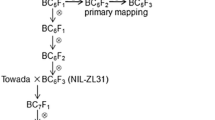
‘LTH’ is a useful breeding material for further improvement of cold tolerance in the present cold-tolerant cultivars such as ‘Hitomebore’. Analysis using progeny plants from a cross between ‘LTH’ and ‘Hitomebore’ may enable identification of genes for cold tolerance. However, ‘LTH’ is tall, which makes it difficult to treat plants with cold water by the deep-water irrigation system. The height of ‘Ukei 840’, which is a cold-tolerant line selected from BC1F6 progeny plants between ‘LTH’ as a donor parent and ‘Hitomebore’ as a recurrent parent (Fig. 1), is as great as that of ‘Hitomebore’. In the present study, we analyzed QTLs for cold tolerance using progeny from a cross between ‘Ukei 840’ and ‘Hitomebore’, and delimited the QTL region for identification of a candidate gene responsible for cold tolerance.

Lineage of ‘Ukei 840’ and progeny population used for analysis of a QTL for cold tolerance
Materials and methods
Plant materials and isolation of DNA
‘Lijiangxintuanheigu’ (‘LTH’), ‘Hitomebore’, and ‘Ukei 840’, which was later renamed ‘Ouu PL4’, were used for developing DNA markers and evaluation of seed fertility after low-temperature treatment. The F2 population of 192 individuals between ‘Ukei 840’ and ‘Hitomebore’, which are BC2F2 plants from a cross between ‘LTH’ as a donor parent and ‘Hitomebore’ as a recurrent parent (Fig. 1), were used for analysis of QTL for cold tolerance at the booting stage in 2005. For reevaluation of QTL for cold tolerance, 192 BC1F2 (BC3F2 between ‘LTH’ and ‘Hitomebore’) individuals were used in 2006. Genomic DNAs were extracted from leaves of ‘LTH’, ‘Ukei 840’ and ‘Hitomebore’ by the CTAB method (Murray and Thompson 1980) and leaves of populations for QTL analyses by the method of Edwards et al. (1991).
Examination of cold tolerance of plants
Populations for QTL analysis and parental lines were grown in a paddy field (Daisen Research Station, National Agricultural Research Center for Tohoku Region, Akita, Japan) and treated by the cold-deep-water irrigation method (Matsunaga 2005) for about 2 months from the panicle initiation stage to full heading. The cold-deep-water irrigation method was developed as a method for examining cold tolerance of rice about 30 years ago, and is widely used for selecting cold-tolerant lines because of its high reliability (Matsunaga 2005; Suh et al. 2010). In the cold-deep-water irrigation method for low-temperature treatment at the booting stage, the water temperature was controlled at 18.4–18.5°C and its depth was 20–25 cm. Seed fertility, which is the percentage of the number of fertile seeds in the number of florets, of plants grown under low-temperature stress was used as an index of cold tolerance.
Graphical genotyping of ‘Ukei 840’
We used 684 DNA markers including 251 SSR (simple sequence repeat) (McCouch et al. 2002), 12 SCAR (sequence-characterized amplified region), 19 CAPS (cleaved amplified polymorphic region) (Shirasawa et al. 2004a), 306 PCR-RF-SSCP (PCR-restriction fragment-single strand conformation polymorphism) (Shirasawa et al. 2004b), and 96 dot-blot-SNP (single nucleotide polymorphism) markers (Shirasawa et al. 2006) for polymorphism analysis between ‘LTH’ and ‘Hitomebore’. The dot-blot-SNP markers were primer pairs for specific amplification of a single DNA fragment and allele-specific oligonucleotide probes, which are hybridized to dot-blotted PCR products on nylon membrane together with competitive oligonucleotides having sequences of the other alleles. Eighty SNP markers were also developed by genome-wide sequencing of ‘LTH’ using a 454 sequencer (Genome Sequencer FLX System, Roche, USA) and by comparison of sequence data with the published genome sequence of ‘Nippponbare’ (International Rice Genome Sequencing Project 2005). These 80 SNP markers were dot-blot-SNP markers using the bridge hybridization method (Shiokai et al. 2010). A total of 275 markers (Supplementary Table 1), which can detect polymorphisms between ‘LTH’ and ‘Hitomebore’, were used for graphical genotyping of ‘Ukei 840’.
QTL analysis
Forty-eight markers, which were mapped on chromosome segments derived from ‘LTH’ in ‘Ukei 840’, were used for genotyping of the 192 F2 (BC2F2 between ‘LTH’ and ‘Hitomebore’) plants (Fig. 1). A linkage map was constructed using Mapmaker 3.0 (http://www.broad.mit.edu/ftp/distribution/software/mapmaker3/). Analysis of QTL for cold tolerance was performed by composite interval mapping (CIM) (Jansen and Stam 1994; Zeng 1994) using Windows QTL cartographer 2.5 (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm). For determining threshold value, a permutation test was carried out 1,000 times.
Delimitation of a region containing a QTL for cold tolerance
Genotypes of 1,504 BC1F3 individuals (BC3F3 between ‘LTH’ and ‘Hitomebore’) were determined using seven makers within a QTL region (Fig. 1). For preparation of PCR templates for a large number of plants, the leaf-punch method (Shiokai et al. 2009) was used. Ten individuals having recombination in the QTL region were selected. Six types of recombinant lines were selfed, and genotypes of DNA markers in the QTL region of 64 BC1F4 progenies (BC3F4 for ‘LTH’) of each recombinant line were determined. Eighteen individuals homozygous for the QTL region in each recombinant line were selected and seed fertility was evaluated for delimiting the QTL region.
Sequencing of the QTL region for cold tolerance
The QTL region for cold tolerance of ‘Ukei 840’ was amplified by PCR using 152 pairs of primers, which were designed for amplifying each 5 kb of the candidate region using published sequence data of ‘Nipponbare’. A 20 μl PCR mixture consisting of 20 ng template genomic DNA, 20 pmol primers, 1× PCR buffer, 0.4 mM dNTPs, and 0.4 Units DNA polymerase (KOD-FX: TOYOBO, Japan) was used. PCR was carried out as follows: 2-min denaturation at 94°C, 40 cycles of 10-s denaturation at 98°C and 6-min extension at 68°C. After isolation by agarose gel electrophoresis, PCR products were purified using UltraClean 15 DNA Purification Kit (MO BIO, USA). All purified products were mixed and concentrated by ethanol precipitation. Nucleotide sequences of the PCR products were determined by Genome Analyzer IIx (Illumina, USA). Sequence data of ‘Ukei 840’ were compared with the published nucleotide sequence of ‘Nipponbare’ in the corresponding region. SNPs between ‘Ukei 840’ and ‘Nipponbare’ were detected.
Gene expression analysis
Total RNAs were extracted using the SV Total RNA Isolation System (Promega, USA) from 0.03 g spikelets of young panicles at the booting stage of ‘Hitomebore’ and ‘HCL3-homo’ grown in the cold-deep-water paddy field under normal growing conditions. First-strand cDNA was synthesized using First-Strand cDNA Synthesis Kit (GE Healthcare, USA). Genes having SNPs between ‘Ukei 840’ and ‘Nipponbare’ within 5 kb upstream regions of translation initiation sites were analyzed by RT-PCR using the first-strand cDNA as a template. Sequences of primer pairs are shown in Supplementary Table 2. The rice actin gene was used as a control. PCR was performed under the following conditions: 1 min denaturation at 94°C, 30 cycles of 30 s denaturation at 94°C, 30 s annealing at 58°C, and 30 s extension at 72°C, and 1 min extension at 72°C.
Results
Graphical genotyping of ‘Ukei 840’
DNA polymorphism between ‘LTH’ and ‘Hitomebore’ was detected by analysis using 297 DNA markers including 144 SSR, 8 SCAR, 4 CAPS, 100 PCR-RF-SSCP, and 41 dot-blot-SNP markers from among 251 SSR, 12 SCAR, 19 CAPS, 306 PCR-RF-SSCP, and 96 dot-blot-SNP markers tested. To develop SNP markers for the regions in which the distances between markers exceed 3 Mb, nucleotide sequences of genomic DNA of ‘LTH’ were determined using a 454 nucleotide sequencer, and 80 SNPs between ‘LTH’ and ‘Nipponbare’ were selected. Removing closely linked DNA markers, 275 markers were used for genotyping of ‘Ukei 840’. ‘Ukei 840’ was homozygous for ‘LTH’ alleles in 42 markers (ca. 20%), homozygous for ‘Hitomebore’ alleles in 225 markers (ca. 75%), and heterozygous in 8 markers (ca. 5%) (Fig. 2). Regions homozygous for ‘LTH’ alleles were 12 regions in short and long arms of chromosomes 3, 5, and 11 and long arms of chromosomes 4, 8, 9, and 12. Heterozygous regions were in the long arms of chromosomes 1, 2, and 11. Genotypes of all the tested DNA markers on the short arm of chromosome 10 of ‘Ukei 840’, on which a QTL for cold tolerance of ‘LTH’ has been mapped by Ye et al. (2010), were homozygous for ‘Hitomebore’ alleles.

Graphical genotyping of ‘Ukei 840’. Bars indicate DNA markers, black boxes show homozygous regions for ‘LTH’ alleles, white boxes represent homozygous regions for ‘Hitomebore’ alleles, and hatched boxes indicate heterozygous regions. Triangles represent centromeres
QTL analysis of cold tolerance
Seed fertilities of 192 F2 plants derived from a cross between ‘Ukei 840’ and ‘Hitomebore’ grown in a deep-water paddy field with water controlled at 18.5°C showed continuous distribution from 17.4 to 84.2% (Fig. 3). Genotypes of the 192 F2 plants were analyzed using the 48 DNA markers in the regions homozygous for ‘LTH’ alleles and the heterozygous region, and QTLs for the seed fertilities were investigated. One QTL with an LOD score of 7.1, higher than the threshold value of 5% significance represented as LOD score 2.5, was detected at marker C11223 on the long arm of chromosome 3. The phenotypic variance explained by this QTL was 24.4%. The additive effect was 7, indicating that substitution of this region from homozygous ‘Hitomebore’ alleles to homozygous ‘LTH’ alleles increases seed fertility by 14% (Table 1). This QTL was named qLTB3 (QTL for Low-temperature Tolerance at Booting stage on chromosome 3).

QTL analysis using an F2 population. a Distribution of seed fertilities of F2 plants between ‘Ukei 840’ and ‘Hitomebore’ cultivated in the cold-deep-water irrigation field at the booting stage. b Position of a QTL detected by analysis using an F2 population. Black boxes represent homozygous regions for ‘LTH’ alleles, white boxes indicate homozygous regions for ‘Hitomebore’ alleles and gray boxes show heterozygous regions. A circle indicates a QTL region
An F1 plant obtained from a cross between ‘LTH’ and ‘Hitomebore’ was backcrossed with ‘Hitomebore’ to obtain 192 BC1F2 plants, which are BC3F2 plants for ‘LTH’ and ‘Hitomebore’ (Fig. 1). These BC1F2 plants had ‘LTH’ alleles of the DNA markers on the long arms of chromosomes 3 and 8 (Supplementary Fig. 1). Seed fertilities of these BC1F2 plants grown in the cold-deep-water paddy field were distributed continuously from 0 to 60.3% (Supplementary Fig. 1). Analyzing genotypes of these plants using the DNA markers in these regions, QTLs were again investigated. A significant QTL was detected at marker RM7000 in qLTB 3. The LOD score of this QTL was 6.9, and the explained phenotypic variance was 18.1% (Table 1).
Development of chromosome segment substitution lines (CSSLs) and delimitation of the QTL region for cold tolerance
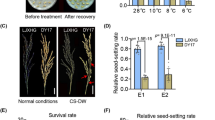
A BC1F2 plant, which is heterozygous for DNA markers in qLTB3 and homozygous for ‘Hitomebore’ alleles in the other genomic regions, named ‘HCL3-hetero’ (‘Hitomebore’ Chromosome segment substitution line with qLTB3-hetero), was selected, and ‘HCL3-homo’ was obtained from its selfed progeny. Six recombinants between RM6970 and RM7389 in qLTB3 were selected from 1,504 selfed progeny of ‘HCL3-hetero’. Determining genotypes of selfed progeny obtained from the six recombinant lines using seven DNA markers in qLTB3, we selected plants homozygous for ‘LTH’ alleles (Fig. 4). These lines, 18 plants for each line, were grown in the cold-deep-water paddy field together with ‘HCL3-homo’ and ‘Hitomebore’, and seed fertility of each plant was investigated in 2008. ‘HCL3-homo’ and ‘Hitomebore’ showed seed fertilities of 31 and 21%, respectively. The difference of seed fertility between the tested lines was significant at the 1% level by ANOVA. Lines with significantly higher seed fertility than that of ‘Hitomebore’ at the 1% level by the Dunnett test were ‘HCL3-3’, ‘HCL3-6’, ‘HCL3-7’, and ‘HCL3-homo’, while seed fertilities of ‘HCL3-5’, ‘HCL3-8’, and ‘HCL3-10’ were not significantly different from that of ‘Hitomebore’. Cold tolerances of these lines were again examined using selfed progeny of these lines in 2009. ‘Hitomebore’ showed 18% seed fertility, while ‘HCL3-homo’ exhibited 32% seed fertility. Seed fertilities of ‘HCL3-3’, ‘HCL3-6’, ‘HCL3-7’, and ‘HCL3’ were significantly higher than that of ‘Hitomebore’ at the 1% level, while those of the other lines were comparable to that of ‘Hitomebore’ (Fig. 4). These results suggest that the QTL for cold tolerance can be delimited to the region from RM3719 to RM7000.

Seed fertilities investigated in 2008 (a), in 2009 (b) and graphical genotypes of six CSSLs (HCL3-3, 5, 6, 7, 8 and 10), HCL3-homo (HCL3) and ‘Hitomebore’ in the QTL region (c). Double asterisk indicate that lines showed significant differences in seed fertilities at 1% level against ‘Hitomebore’. Black boxes show homozygous regions for ‘LTH’ alleles and white boxes represent homozygous regions for ‘Hitomebore’ alleles. Gray boxes show regions containing recombination break points
The dominance effect of the QTL was investigated using heterozygous plants of ‘HCL3-3’, ‘HCL3-6’, and ‘HCL3-hetero’. All these heterozygotes showed seed fertilities as low as that of ‘Hitomebore’, suggesting that the ‘LTH’ allele for cold tolerance in qLTB3 is recessive (Fig. 5).

Seed fertilities (a) and graphical genotypes of heterozygous CSSLs of HCL3-3, HCL3-6 and HCL3, ‘Ukei 840′, and ‘Hitomebore’ in the QTL region (b). Double asterisks indicate that lines showed significant differences in seed fertilities at 1% level against ‘Ukei 840’. Black boxes show regions of ‘LTH’ alleles and white boxes represent regions of ‘Hitomebore’ alleles
Identification of SNPs in qLTB3
The ‘Nipponbare’ genome sequence of the region between RM3719 and RM7000 is 1,161,293 bp. Genomic DNA of ‘Ukei 840’ in this region was amplified by long PCR for ca. 5 kb using 216 primer pairs. For amplification of the regions which could not be amplified by these primer pairs, 12 other primer pairs were designed. Genomic DNA fragments of ‘Ukei 840’ covering the whole region between RM3719 and RM7000 were obtained. Nucleotide sequences of these PCR products were determined using an Illumina genome analyzer, and 172 times coverage on average was obtained. The obtained 1,161,293-bp sequence of ‘Ukei 840’ was compared with the ‘Nipponbare’ genome sequence. In this nucleotide sequence analysis, many short sequences of ca. 30 nt were aligned with the published ‘Nipponbare’ genome sequence, and therefore insertions in ‘LTH’ genome could not be detected. However, all the DNA fragments amplified from ‘Ukei 840’ had the same sizes as those from ‘Nipponbare’, indicating that there is no large insertion in this genomic region of ‘Ukei 840’. Deletions of nucleotides in ‘Ukei 840’ were not detected. Detected SNPs were 254, of which 223 were present outside of the assigned gene regions. Among the 31 SNPs identified in the gene regions, seven SNPs detected in five genes were variations causing amino acid changes in encoded proteins. In Os03g0790700, Ile at 728 from the first Met, which is a conserved amino acid residue among genes for aldehyde oxidase-2, was replaced by Asn in ‘Ukei 840’. Leu at 59 and Glu at 439 in Os03g0793700 were replaced by Phe and Gly, respectively (Table 2).
SNPs were detected in the 5-kb upstream regions of 89 genes. In spikelets of young panicles at the booting stage, gene expression of 57 genes was detected, but no differences of gene expression levels were detected in these genes between ‘HCL3-homo’ and ‘Hitomebore’ nor between plants grown in the cold-water paddy field and those under the normal growing conditions.
Seven dot-blot-SNP markers, i.e., qLTB3-1 to -7, were developed with SNPs identified in Os03g0789800, Os03g0790700, Os03g0793700, Os03g0800500, and Os03g0806700 (Table 3). All the markers showed clear dot-blot signals for each allele. In all the markers, ‘Ukei 840’ showed genotypes of ‘LTH’ type, and ‘Hitomebore’ did genotypes of ‘Nipponbare’ type (Fig. 6).

A map of SNPs used for developing dot-blot-SNP markers. Dot-blot signals detected by the SNP markers are shown to the right. qLTB3-n-NP and qLTB3-n-LTH are probes for ‘Nipponbare’ alleles and ‘LTH’ alleles, respectively
Discussion
Repeated QTL analyses using ‘Hitomebore’ and ‘Ukei 840’, which is a cold-tolerant breeding line derived from a crossing of ‘LTH’ with ‘Hitomebore’, revealed the presence of a QTL for cold tolerance at the booting stage on the long arm of chromosome 3. Although only one QTL with a significant effect was detected, phenotypic variance explained by this QTL was only 24.4%. A CSSL having a region of chromosome 3 derived from ‘LTH’ with a genetic background of ‘Hitomebore’ developed by backcrossing showed significantly higher cold tolerance than ‘Hitomebore’, but QTL analysis using a backcrossed population again showed this region to have a small explained phenotypic variance, 18.1%. The presence of many other QTLs with minor effects might be one of the reasons for this low explained phenotypic variance, but backcrossing to remove allelic variations at other QTLs did not increase the phenotypic variance explained by the QTL in chromosome 3, suggesting that most of the remaining phenotypic variance, ca. 80%, is due to environmental factors. Although further improvement is required to develop a more reliable method for testing genetic effects on the cold tolerance of breeding lines in rice, detection of significant QTLs at the same region, i.e., qLTB3, by repeated analysis and significant differences of seed fertilities between the chromosome segment substitution lines having qLTB3, i.e., ‘HCL3’ lines, and ‘Hitomebore’ suggests the reliability of the effect of qLTB3.
Three, four, three, and one QTLs for cold tolerance at the booting stage have been observed by Takeuchi et al. (2001), Dai et al. (2004), Suh et al. (2010), and Ye et al. (2010), respectively, explained phenotypic variance of each QTL being less than 22% with summed explained phenotypic variances of 32, 45, 27, and 20.5%, respectively. Since reevaluations of QTLs using chromosome segment substitution lines or progeny populations were not carried out in these studies, it cannot be speculated whether these low values of summed explained phenotypic variances are due to large environmental effects. Only one QTL for cold tolerance at the booting stage of ‘Hokkai-PL9’ detected on chromosome 8 has explained the higher phenotypic variance, i.e., 26.6% (Kuroki et al. 2007), than the QTL in the present study. In the previous QTL studies, cold-tolerance QTLs have been detected on every chromosome from chromosome 1 to 12, and a few QTLs shared by different cold-tolerant cultivars have been found (Andaya and Mackill 2003; Dai et al. 2004; Takeuchi et al. 2001, Ye et al. 2010). It can be inferred that many genes participate in cold tolerance at the booting stage of rice.
In the chromosomal region of about 1.2 Mb containing a gene for cold tolerance delimited by analysis using CSSLs, 143 genes have been annotated in the ‘Nipponbare’ genome (International Rice Genome Sequencing Project 2005) and full-length cDNA clones of them have been isolated. In the present study, SNPs were detected in 5-kb upstream regions of 89 genes. No difference in gene expression of these genes was detected between the CSSL having qLTB3 and ‘Hitomebore’ nor between plants grown in the cold-water paddy field and those under normal growing condition.
Determining the nucleotide sequence in this region of ‘Ukei 840’, we revealed seven SNPs in five genes causing amino acid substitutions. Among them, one SNP in Os03g0790700 was found to be responsible for alteration of a conserved amino acid residue. Os03g0790700 is a possible candidate of the cold-tolerance gene of ‘LTH’. Os03g0790700 is similar to AAO2 in A. thaliana, which is considered to be a member of aldehyde oxidase functioning in ABA biosynthesis (Koiwai et al. 2004; Seo et al. 2004). Os03g0806700, which has been reported to be expressed in the anther (RiceXPro, Sato et al. 2011), encodes DUF family protein, the function of which is unknown. Proteins having an amino acid sequence similarity to Os03g0806700, e.g., SORBI_01g004400 of Sorghum bicolor, 100381620 of Zea mays, and RCOM_0557310 of Ricinus communis, have serine at 244 aa similar to a protein encoded by the ‘LTH’ allele, not leucine encoded by the ‘Nipponbare’ allele.
Although there is a possibility that the SNPs identified in Os03g0790700 and Os03g0806700 are variations responsible for cold tolerance in ‘Ukei 840’, further analysis is required to demonstrate participation of these SNPs in cold tolerance. Since Tos17 insertion lines of Os03g0790700 and Os03g0806700 are available (http://tos.nias.affrc.go.jp/), we are transforming these Tos17 insertion lines with ‘LTH’ alleles and ‘Hitomebore’ alleles of Os03g0790700 and Os03g0806700 to produce transgenic plants for testing their cold tolerance. However, such transgenic plants should be cultivated in an isolated greenhouse in line with requirements of the Biodiversity Convention. Although many plants per line should be examined for cold tolerance to minimize errors, it is not easy to test many transgenic plants in such an isolated greenhouse. Production of recombinants only in upstream and downstream intergenic regions by meiotic recombination may enable a large-scale evaluation of cold tolerance using a deep-water field with controlled water temperature for demonstrating the function of a candidate gene.
The only gene so far identified as a gene for cold tolerance at the booting stage in rice is the F-box protein gene from ‘Silewah’ (Saito et al. 2010). Although a 30% increase of seed fertility under low temperature has been shown in transgenic plants having the F-box protein gene from ‘Silewah’, there is neither a nucleotide sequence variation nor a difference of gene expression levels between ‘Silewah’ and a cold-sensitive cultivar. Since 687 genes have been reported as F-box protein genes in rice (Jain et al. 2007), it is not easy to elucidate the function of one F-box protein gene.
Further study on delimitation of the cold-tolerance QTL of ‘LTH’ is required for elucidation of the cold-tolerance gene. However, the SNP markers developed in the present study would be useful for marker-assisted selection of cold-tolerant lines in rice breeding programs using ‘LTH’ as a gene source of cold tolerance.
References
Andaya VC, Mackill DJ (2003) QTLs conferring cold tolerance at the booting stage of rice using recombinant inbred lines from a japonica × indica cross. Theor Appl Genet 106:1084–1090
Dai L, Lin X, Ye C, Ise K, Saito K, Kato A, Xu F, Yu T, Zhang D (2004) Identification of quantitative trait loci controlling cold tolerance at the reproductive stage in Yunnan landrace of rice, Kunmingxiaobaigu. Breed Sci 54:253–258
Edwards K, Johnstone C, Thompson C (1991) A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res 19:1349
Hayase H, Satake T, Nishiyama T, Ito N (1969) Male sterility caused by cooling treatment at the meiotic stage in rice plants. II. The most sensitive stage to cooling and the fertilizing ability of pistils. Proc Crop Sci Soc Jpn 38:706–711
Horisue N, Kunihiro Y, Higashi T, Oyamada Z, Huaiyi W, Jianhua X, Suchu Z, Zhiyong L, Yonghua W (1988) Screening for cold tolerance of Chinese and Japanese rice varieties and selection of standard varieties. Trop Agric Res Ser 21:76–92 (in Japanese with English summary)
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
International Rice Research Institute (1977) Screening for cold tolerance. IRRI annual report for 1977, pp 142
Ito N (1978) Male sterility caused by cooling treatment at the young microspore stage in rice plants. XVI. Changes in carbohydrates, nitrogenous and phosphorous compounds in rice anthers after cooling treatment. Jpn J Crop Sci 47:318–323
Jain M, Nijhawan A, Arora R, Agarwal P, Ray S, Sharma P, Kapoor S, Tyagi AK, Khurana JP (2007) F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol 143:1467–1483
Jansen RC, Stam P (1994) High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136:1447–1455
Koiwai H, Nakaminami K, Seo M, Mitsuhashi W, Toyomasu T, Koshiba T (2004) Tissue-specific localization of an abscisic acid biosynthetic enzyme, AAO3, in Arabidopsis. Plant Physiol 134:1697–1707
Kuroki M, Saito K, Matsuba S, Yokogami N, Shimizu H, Ando I, Sato Y (2007) A quantitative trait locus for cold tolerance at the booting stage on rice chromosome 8. Theor Appl Genet 115:593–600
Matsunaga K (2005) Establishment of an evaluation method for cold tolerance at the booting stage of rice using deep water irrigation system and development of highly cold-tolerant rice varieties by combining cold tolerance genes. Bull Miyagi Furukawa Agric Exp Stn 4:1–78 (in Japanese with English summary)
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Cartinhour S, Ware D, Stein L (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Nishiyama I (1970) Male sterility caused by cooling treatment at the young microspore stage in rice plants. VII. Electron microscopical observations on tapetal cells dilated by the cooling treatment. Proc Crop Sci Soc Japan 39:480–486
Nishiyama I (1978) Male sterility caused by cooling treatment at the young microspore stage in rice plants. XVIII. Some enzyme activities in anthers during and after the cooling. Jpn J Crop Sci 47:551–556
Oliver SN, Van Dongen JT, Alfred SC, Mamun EA, Zhao X, Saini HS, Fernandes SF, Blanchard CL, Sutton BG, Geigenberger P, Dennis ES, Dolferus R (2005) Cold-induced repression of the rice anther-specific cell wall invertase gene OSINV4 is correlated with sucrose accumulation and pollen sterility. Plant Cell Environ 28:1534–1551
Oliver SN, Dennis ES, Dolferus R (2007) ABA regulates apoplastic sugar transport and is a potential signal for cold-induced pollen sterility in rice. Plant Cell Physiol 48:1319–1330
Saito K, Miura K, Nagano K, Hayano-Saito Y, Saito A, Araki H, Kato A (1995) Chromosomal location of quantitative trait loci for cool tolerance at the booting stage in rice variety ‘Norin-PL8’. Breed Sci 45:337–340
Saito K, Miura K, Nagano K, Hayano-Saito Y, Araki H, Kato A (2001) Identification of two closely linked quantitative trait loci for cold tolerance on chromosome 4 of rice and their association with anther length. Theor Appl Genet 103:862–868
Saito K, Hayano-Saito Y, Maruyama-Funatsuki W, Sato Y, Kato A (2004) Physical mapping and putative candidate gene identification of a quantitative trait locus Ctb1 for cold tolerance at the booting stage of rice. Theor Appl Genet 109:515–522
Saito K, Hayano-Saito Y, Kuroki M, Sato Y (2010) Map-based cloning of the rice cold tolerance gene Ctb1. Plant Sci 179:97–102
Sasaki T (2005) Identification of rice germplasm for cold tolerance at the booting stage and breeding of Hitomebore, a cold-tolerant rice cultivar having excellent grain and eating quality. Bull Miyagi Furukawa Agric Exp Sta 4:79–128 (in Japanese with English summary)
Satake T, Hayase H (1970) Male sterility caused by cooling treatment at the young microspore stage in rice plants. V. Estimation of pollen developmental stage and the most sensitive stage to coolness. Proc Crop Sci Soc Jpn 39:468–473
Sato Y, Antonio BA, Namiki N, Takehisa H, Minami H, Kamatsuki K, Sugimoto K, Shimizu Y, Hirochika H, Nagamura Y (2011) RiceXPro: a platform for monitoring gene expression in japonica rice grown under natural field conditions. Nucleic Acids Res 39:D1141–D1148
Seo M, Aoki H, Koiwai H, Kamiya Y, Nambara E, Koshiba T (2004) Comparative studies on the Arabidopsis aldehyde oxidase (AAO) gene family revealed a major role of AAO3 in ABA biosynthesis in seeds. Plant Cell Physiol 45:1694–1703
Shiokai S, Kitashiba H, Shirasawa K, Nagano K, Nishio T (2009) Leaf-punch method to prepare a large number of PCR templates from plants for SNP analysis. Mol Breed 23:329–336
Shiokai S, Shirasawa K, Sato Y, Nishio T (2010) Improvement of the dot-blot-SNP technique for efficient and cost-effective genotyping. Mol Breed 25:179–185
Shirasawa K, Kishitani S, Nishio T (2004a) Conversion of AFLP markers to sequence-specific markers for closely related lines in rice by use of the rice genome sequence. Mol Breed 14:283–292
Shirasawa K, Monna L, Kishitani S, Nishio T (2004b) Single nucleotide polymorphisms in randomly selected genes among japonica rice (Oryza sativa L.) varieties identified by PCR-RF-SSCP. DNA Res 11:275–283
Shirasawa K, Shiokai S, Yamaguchi M, Kishitani S, Nishio T (2006) Dot-blot-SNP analysis for practical plant breeding and cultivar identification in rice. Theor Appl Genet 113:147–155
Suh JP, Jeung JU, Lee JI, Choi YH, Yea JD, Virk PS, Mackill DJ, Jena KK (2010) Identification and analysis of QTLs controlling cold tolerance at the reproductive stage and validation of effective QTLs in cold-tolerant genotypes of rice (Oryza sativa L.). Theor Appl Genet 120:985–995
Takeuchi Y, Hayasaka H, Chiba B, Tanaka I, Shimano T, Yamagishi M, Nagano K, Sasaki T, Yano M (2001) Mapping quantitative trait loci controlling cool-temperature tolerance at booting stage in temperate Japonica rice. Breed Sci 51:191–197
Toriyama K, Futsuhara Y (1960) Genetic studies on cool tolerance in rice. I. Inheritance of cool tolerance. Jpn J Breed 10:7–16 (in Japanese)
Ye C, Fukai S, Godwin ID, Koh H, Reinke R, Zhou Y, Lambrides C, Jiang W, Snell P, Redona E (2010) A QTL controlling low temperature induced spikelet sterility at booting stage in rice. Euphytica 176:291–301
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgments
This study was partly supported by a Grant-in-Aid for Scientific Research (Research Activity Start-up: 22880005).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. Dubcovsky.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2011_1758_MOESM1_ESM.tif
Supplementary Fig. 1 QTL analysis using a backcrossed population. (a) Seed fertilities of backcross population after cultivation under low temperature conditions at the booting stage. (b) Position of a QTL detected using a backcrossed population. Black boxes indicate regions having segregated genotypes and white boxes show regions having a fixed genotype of homozygous ‘Hitomebore’ alleles. A circle represents a QTL region. A gray bar indicates candidate region of the QTL and a triangle shows the peak of the LOD score. (TIFF 86 kb)
Rights and permissions
About this article
Cite this article
Shirasawa, S., Endo, T., Nakagomi, K. et al. Delimitation of a QTL region controlling cold tolerance at booting stage of a cultivar, ‘Lijiangxintuanheigu’, in rice, Oryza sativa L.. Theor Appl Genet 124, 937–946 (2012). https://doi.org/10.1007/s00122-011-1758-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-011-1758-6