
Abstract
Alzheimer’s disease is a progressive and fatal neurodegenerative disease affecting the elderly population accompanied by a decrease in cholinergic transmission, impairing cognitive functions. Acetylcholine deficiency is important in the development of disease symptoms. Inhibition of acetylcholinesterase, an important enzyme in acetylcholine hydrolysis, is one of the important drug targets to increase acetylcholine levels. In this study, we aimed to develop acetylcholinesterase inhibitor compounds. For this, we synthesized compounds 6(a–e) bearing 3(2H)-pyridazinone and 1,2,4-triazole ring structures. We determined the IC50, Ki and inhibition types of N-substituted-(p-methoxyphenyl)pyridazin-3(2H)-one derivatives that we synthesized and elucidated their structures. The compound with the best AChE activity was compound 6b (Ki = 3.73 ± 0.9 nM) with the p-methylphenyl group it carried and showed competitive inhibition. Kinetic study was also performed for the compounds with the highest BChE 6a (Ki = 0.95 ± 0.16 nM) inhibitory activities. Molecular docking studies have shown that the p-methylphenyl group is indeed active in the hinge region of the AChE crystal structure as a result of experimental activity. In addition, the best free binding energy (ΔGBind), docking score and Glide score values were determined by examining the interactions with AChE crystal structure for compound 6b and with BChE crystal structure for 6a in silico approaches.

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a fatal neurodegenerative disease associated with the decreased cholinergic transmission, affecting many elderly individuals worldwide, and is the leading cause of dementia [1,2,3]. It is a multifaceted disease in which cholinergic dysfunction, epigenetic abnormalities, genetic defects and environmental risk factors cause neuron death. When the pathology of AD is examined, many factors such as oxidative stress, amyloid β protein (Aβ) accumulation and inflammation play a role [4,5,6]. All these factors and the high prevalence of the disease due to these factors have increased the importance of treatment.
Between 1993 and 2003, the available drugs donepezil, tacrine, galantamine and rivastigmine were introduced to the market (Fig. 1). Recently, the Food and Drug Administration gave rapid approval to aducanumab (Aduhelm) to treat patients with AD, and aducanumab became the first antibody therapy for the presence of Aβ plaques in the brain [7, 8]. However, all these treatments are symptomatic and do not completely cure the disease [9].

The available drugs for Alzheimer’s disease
Biochemical studies for the pathophysiology of AD have shown that the levels of some neuromediators (acetylcholine) are decreased in the cerebral cortex. This decrease in neurotransmitter levels makes it difficult to maintain neurotransmission. The “cholinergic hypothesis” that emphasizes the importance of acetylcholine deficiency in the progression of the disease has gained importance [10]. According to this hypothesis, acetylcholinesterase’s (AChEs’), which perform inhibition of acetylcholine hydrolysis, was developed to increase the decreased acetylcholine level. AChE and butyrylcholinesterase (BChE) enzymes are commonly known as cholinesterase’s. Despite the full knowledge of AChE in the cholinergic transition, the effects of BChE have not been adequately clarified [11]. For this purpose, rivastigmine, donepezil and galantamine are used as AChE inhibitors [12, 13].
Due to the side effects and some pharmacokinetic properties of existing ChEIs, it is aimed to improve treatments by developing new ChEIs. The search for more effective drugs for the treatment of AD has become one of the most targeted pharmacological targets [14,15,16].
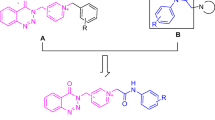
The pyridazinone ring has a broad spectrum of pharmacological activity. It has been reported that compounds containing the pyridazinone ring as the main structure exhibit ChE inhibitory activity both in the literature and in our previous studies [17,18,19,20]. However, when we examined the literature, compounds bearing a triazole ring showed anticholinesterase activity [21,22,23,24]. This study, it was aimed to examine the inhibition of two basic rings showing anticholinesterase activity by combining them. In this direction, newly designed pyridazinone compounds in which the triazole ring-substituted were synthesized (Fig. 2) and in vitro and in silico studies were performed.

Newly designed pyridazinone compounds 6(a–e)
A series of N-substituted-(p-methoxy)pyridazin-3(2H)-derivatives have been developed by synthesis, biological evaluation and molecular docking studies. In addition, compounds 6a and 6b were evaluated in silico studies as a potential lead compound candidate for further optimization.
Results and discussion
Chemistry
By our goal, starting from the aromatic heterocyclic dichloro structure, N-substituted-(4-methoxyphenyl)piperazinyl-pyridazin-3(2H)-one derivatives were synthesized as a result of a six-step reaction (Scheme 1). It was started the nucleophilic substitution reaction of 3,6-dichloropyridazine and 1-(4-methoxyphenyl)piperazine to reach the main substrate. In the next step, the oxidation reaction was performed with glacial acetic acid and the compound 2 was obtained. In the other step of the synthesis, the unpaired electrons of the nitrogen atom(2nd) of the pyridazinone group attacked the bromine-bonded carbon of ethyl 2-bromoacetate by the nucleophilic substitution SN2 reaction mechanism to obtain the ethyl 2-(3-(4-(4-methoxyphenyl)piperazin-1-yl)-6-oxopyridazin-1(6H)-yl)acetate (3). The acetohydrazide compound (4) was synthesized by the reaction of the compound 3 and hydrazine hydrate. The final products 6(a–e) were synthesized by the intramolecular condensation reaction of the structure that was obtained as a result of the reaction of aromatic isothiocyanates and the compound 5.

Synthesis of compounds 6(a–e)
The melting points of the white-colored compounds 6(a–e), which were synthesized in yields ranging from 63-39% in general, were 245–269 °C. In the 1H NMR spectrum, while the triazole-NH peak for compound 6a showed at 13.88 ppm, it was not observed in the others. Bridge of triazole and pyridazinone rings –CH2- the peak was observed near 5.07–4.91 ppm in proton NMR. In the 13C NMR spectrum, the pyridazinone ring carbonyl carbon-supported structure accuracy in the range of 157.38–156.86 ppm. The specific thiocarbonyl group of the triazole ring was observed in the range of 168.04–167.46 ppm.
Biological evaluation and structure-activity relationship
In this study, the inhibitory effects of N-substituted-(p-methoxyphenyl)pyridazin-3(2H)-one derivatives 6(a–e) for AChE/BChE was evaluated. The synthesized compounds had inhibitory effect with Ki values in the range of 3.73 ± 0.9–12.8 ± 4.0 nM and IC50 values in the range of 22.9–26.1 nM for AChE. The BChE inhibitory effects of the compounds were found between 0.95 ± 0.16–3.32 ± 1.99 nM Ki values and 18.36–26.96 nM IC50 values. Besides, inhibition types of newly synthesized compounds were determined. These values of the compounds are given in Tables 1 and 2, and Lineweaver-Burk graph of 6(a–e) and Tacrine for AChE and BChE (Figs. 3 and 4).

IC50 graph (A) and Lineweaver-Burk graph (B) of 6b and Tacrine (TAC) for AChE

IC50 graph (A) and Lineweaver-Burk graph (B) of 6a and Tacrine (TAC) for BChE
Its contribution to the activity was evaluated by using phenyl ring and substituted derivatives (-methylphenyl, -bromophenyl, -fluorophenyl, -trifluoromethoxyphenyl). In addition to the substitutions, the inhibitory effects of different groups from the money position were investigated for AChE. Non-substituted compound 6a (-phenyl) had Ki value of 7.0 ± 2.6 nM. In the N-substituted-(p-methoxyphenyl)pyridazine-3(2H)-one derivatives the inhibitory strength order was 6b (4-methyl derivative) >6a (non-derivative) >6d (4-fluoro derivative) >6c (4-bromo derivative) >12.8 (trifluoromethoxy derivative). Compared to the reference compound Tacrine (Ki = 4.96 ± 1.76 nM), the compound with the best activity is the methyl derivative substituted 6b (Ki = 3.73 ± 0.9 nM) with the electron donating group. Also, compound 6b showed a stronger inhibitory effect than Tacrine. Non-substituted derivative 6a (Ki = 7.0 ± 2.6 nM) showed moderate inhibitory effect compared to Tacrine. Among the halogen-bearing compounds, the compound with strong inhibitory effect was the 4-fluorophenyl derivative 6d compound with a Ki value of 10.1 ± 2.9 nM. 6b, 6d and 6e showed competitive inhibition like Tacrine, while 6a and 6c showed non-competitive inhibition.
Compounds 6a, 6c and 6d showed stronger inhibitory effect than Tacrine (Ki = 1.28 ± 0.66 mM). Compound 6a (non-derivative) with the highest BChE inhibition (Ki = 0.95 ± 0.16 mM) were selected as the hit compounds in this series. The best activity was seen in the non-substituted derivative. Substitution of -Br and -F halogens from the para position positively affected the inhibitory effect. All compounds showed competitive inhibition type such as Tacrine.
Molecular docking
To investigate the plausible explanation between the target-ligand of the studied compounds, molecular modeling of the representative compound 6b in the active binding pocket was performed using the Schrödinger 2021-2 Glide XP docking protocol. Figure 3A showed the interaction of the most potent compound 6b, and Fig. 3B with tacrine. In Fig. 5A, there is both π–π stacking and π-cation interaction with the 1,2,4-triazole ring and Trp84, an important amino acid residue in the AChE crystal structure (PDB ID:1ACJ), as well as the 4-methylphenyl ring and other important amino acid residues in this crystal structure. The existence of π–π stacking and π-cation interactions with Phe330, which is an amino acid residue, was determined. Also, in Fig. 5A, the existence of π-π stacking between the pyridazinone ring in compound 6b and amino acid Tyr121 is presented.

A 2D interaction diagram of AChE (PDB ID: 1ACJ) and compound 6b. B 2D interaction diagram of AChE (PDB ID: 1ACJ) and compound Tacrine
When the 2D interaction diagram between AChE crystal structure (PDB ID: 1ACJ) and Tacrine was examined in Fig. 5B, it is determined that there are π–π stacking interactions with Trp84 and Phe330, as well as the presence of hydrogen bonding between the –NH2 group in Tacrine and the 1ACJ crystal structure.
Figure 5A, B shows that the AChE crystal structure can be said to have similar binding properties with Tacrine, the reference compound, and compound 6b, which was synthesized by us and had good experimental activities.
Figure 5 shows the binding sites of 6b and Tacrine compounds by molecular docking, which is one of the in silico approach methods with AChE crystal structure. The binding parameter values formed between this crystal structure and ligands are as important as the binding site. Table 3 presents the binding parameters of compound 6a, 6b and Tacrine. By comparing these binding parameter values, docking score, Glide score, free binding energy (ΔGBind), Glide emodel and Glide energy values in the active binding site were determined. The parameters calculated as a result of the interaction of both the synthesized compound 6b with the best activity and the reference compound Tacrine with the 1ACJ crystal structure are shown.
When Table 3 is examined in detail, the docking score value of 6b, one of the binding parameters, was calculated as −13.512 kcal/mol, while the value of Tacrine was calculated as −5.673 kcal/mol. Compound 6b is said to have a better value than the reference compound Tacrine. In addition, this interpretation can be made for the free binding energy. In Table 2, the free binding energy of Tacrine is −32.14 kcal/mol, while the value of compound 6b is −56.89 kcal/mol.
Figure 6 shows the 3D surface model structure of compound 6b docked in the main groove of the 1ACJ crystal structure and its interaction with amino acid residues in the active binding site. Figure 6 shows the 3D interaction of the crystal structure of AChE, 1ACJ and compound 6b through molecular docking.

Representation of 3D amino acid residues of compound 6b interacting with the 1ACJ crystal structure
When the 2D interaction diagram between the BChE crystal structure (PDB ID: 4BDS) and the 6a compound in Fig. 7 was analyzed, it was determined that there was a cation-pi interaction with Hip438, an important amino acid residue.

2D interaction diagram of BChE (PDB ID: 4BDS) and compound 6a
Conclusions
In this study, we studied a number of new pyridazinone-triazole derivative compounds. After elucidating the structure of the compounds, we synthesized with spectral analysis, we examined the AChE inhibition effect and interactions with the enzyme active site by molecular docking studies. Compound 6b showed a stronger AChE inhibitory effect than Tacrine with Ki value of 3.73 ± 0.9 nM, making it the strongest compound in our series. Non-substituted and halogen substituted derivatives were the most potent inhibitor of BChE. It has been shown that these compounds will be developed using various substitutions on phenyl rings to develop better enzyme inhibitors. As a result, the tested compounds will provide new ideas for the development of new drugs. Also, these results showed that the 3(2H)-pyridazinone and 1,2,4-triazole ring was more favorable for both AChE and BChE inhibition. Such biological features highlight 6b as a very interesting prototype in the search for new drugs in the treatment of AD. The interactions between molecular docking and target crystal structure of compounds 6b and 6a, which act on AChE and BChE, respectively, were investigated by in silico approaches. The effect of compound 6b on AChE and the effect of compound 6a on BChE can be said to be promising considering the results obtained in molecular docking and may lead to further studies.
Materials and methods/experimental
Chemistry
Unless otherwise noted, all the reagents were commercial quality and were used without purification. The progress of reactions and the purity of the compounds were monitored by TLC using silica gel plates (250 μm, F254) under UV light. Melting points were measured on a hot stage microscope (Stuart SMP30) and are uncorrected. NMR spectra were recorded on a Bruker Avance Neo 500 MHz (1H, 500 MHz; 13C, 125 MHz), in DMSO-d6 (internal standard tetramethylsilane (TMS)). Chemical shifts (δ) are expressed as parts per million (ppm) downfield from TMS and the coupling constants (J) quoted in Hertz. Splitting patterns have been designated as follows: s (singlet), d (doublet), t (triplet) and m (multiplet). High-resolution mass spectra data were collected in sing a Waters LCT Premier XE Mass Spectrometer (high sensitivity orthogonal acceleration time flight instrument) operating in the ESI (+) method, also coupled to an AQUITY Ultra Performance Liquid Chromatography system (Waters Corporation, Milford, MA, USA).
Synthesis of 3-chloro-6-(4-(4-chlorophenyl)piperazin-1-yl)pyridazine (1)
3,6-dichloropyridazine (1 equiv., 10 mmol) and 1-(4-chlorophenyl)piperazine (1 equiv., 10 mmol) were stirred in 15 ml of ethanol by heating under reflux. The reaction medium was poured into ice water; the precipitate was filtered and purified using suitable solvents for example EtOH, MeOH, etc. [17].
Synthesis of 6-(4-(4-chlorophenyl)piperazin-1-yl)pyridazin-3(2H)-one (2)
The compound 1 (5 equiv., 50 mmol) were stirred in 30 ml of glacial acetic acid with heating under reflux for 6 h [25].
Synthesis of ethyl 2-(3-(4-(4-chlorophenyl)piperazin-1-yl)-6-oxopyridazin-1(6H)-yl)acetate (3)
The compound 2 (1 equiv., 10 mmol), ethyl bromoacetate (2 equiv., 20 mmol) and potassium carbonate (2 equiv., 20 mmol) were mixed in 40 ml of acetone by heating under reflux for 24 h. The mixture was cooled, the precipitated salt was filtered off and the acetone was evaporated [26].
Synthesis of 2-(3-(4-(4-chlorophenyl)piperazin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (4)
The compound 3 (1 equiv., 10 mmol) was dissolved in 25 ml of ethanol and hydrazine hydrate (3 ml) was added. The reaction medium was stirred at room temperature for 3 h. The precipitate formed was filtered, washed with water, dried and purified by crystallization from EtOH and water [26].
General procedure for the synthesis of 6(a–e)
The compound 4 (1 equiv., 10 mmol) and substituted aryl isothiocyanate (1 equiv., 10 mmol) were mixed in 25 ml of anhydrous ethanol by heating under reflux for varying times. The reaction medium was cooled; the precipitate was filtered off, washed with ether and dried. Thiosemicarbazide derivatives (1 equiv., 10 mmol) and 2% aqueous NaOH solution (20 ml) were refluxed for 2–3 h. The reaction was cooled and neutralized with dilute HCl. The precipitate was filtered and then crystallized from ethanol [27].
6-(4-(4-Methoxyphenyl)piperazin-1-yl)-2-((4-phenyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)methyl)pyridazin-3(2H)-one (6a)
Yellow solid (45% yield), Rf 0.61 (CH3OH-CHCl3 9:1), mp 248–250 °C, 1H NMR (500 MHz, DMSO-d6) δH 13.88 (s, 1H, -NH), 7.29 (d, 1H, J = 12 Hz, 5-H), 7.46–7.43 (m, 3H, 3′-4′- & 5′- Ph-H), 7.28 (d, 2H, J = 4 Hz, 2′- & 6′- Ph-H), 6.95 (d, 2H, J = 11 Hz, 2″- & 6″-H), 6.83 (d, 2H, J = 11 Hz, 3″- & 5″-H), 6.64 (d, 1H, J = 12 Hz, 4-H), 5.07 (s, 2H, -CH2-), 3.35 (s, 3H, -OCH3), 3.24 (t, 4H, J = 5 Hz, piperazine 2′- & 6′-H), 3.05 (t, 4H, J = 5 Hz, piperazine 1′- & 5′-H), 13C NMR (125 MHz, DMSO-d6) δC 168.04 (5″‘-C), 156.86 (3-C), 153.23 (3″‘-C), 148.58, 148.19, 145.14, 133.17, 130.24, 129.32, 129.18, 127.59, 126.50, 117.87, 114.24, 55.17 (-OCH3), 49.20 (-CH2-), 46.90 (piperazine 2′- & 6′-C), 45.36 (piperazine 1′- & 5′–C), MS m/z (ESI) calcd for C24H25N7O2S (M + H+) 476.1869, found 476.1862.
6-(4-(4-Methoxyphenyl)piperazin-1-yl)-2-((5-thioxo-4-(p-tolyl)-4,5-dihydro-1H-1,2,4-triazol-3-yl)methyl)pyridazin-3(2H)-one (6b)
White solid (63% yield), Rf 0.58 (CH3OH-CHCl3 9:1), mp 267–269 °C, 1H NMR (500 MHz, DMSO-d6) δH 7.4 (d, 1H, J = 12 Hz, 5-H), 7.15 (d, 2H, J = 10 Hz, 2′- & 6′- Ph-H), 7.04 (d, 2H, J = 10 Hz, 3′- & 5′- Ph-H), 6.94 (d, 2H, J = 11 Hz, 2″- & 6″-H), 6.83 (d, 2H, J = 11 Hz, 3″- & 5″-H), 6.67 (d, 1H, J = 12 Hz, 4-H), 4.91 (s, 2H, -CH2-), 3.69 (s, 3H, -OCH3), 3.22 (t, 4H, J = 6 Hz, piperazine 2′- & 6′-H), 3.05 (t, 4H, J = 5 Hz, piperazine 1′- & 5′-H), 2.31 (s, 3H, Ph-CH3), 13C NMR (125 MHz, DMSO-d6) δC 167.58 (5″‘-C), 157.01 (3-C), 153.22 (3″‘-C), 148.28, 146.13, 136.33, 134.23, 130.33, 128.75, 127.72, 125.96, 117.87, 114.27, 55.18 (-OCH3), 49.34 (-CH2-), 46.09 (piperazine 2′- & 6′-C), 45.57 (piperazine 1′- & 5′-C), 20.73 (Ph-CH3), MS m/z (ESI) calcd for C25H27N7O2S (M + H+) 490.2025, found 490.2028.
2-((4-(4-Bromophenyl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)methyl)-6-(4-(4-methoxyphenyl)piperazin-1-yl)pyridazin-3(2H)-one (6c)
White solid (53% yield), Rf 0.63 (CH3OH-CHCl3 9:1), mp 245–246 °C, 1H NMR (500 MHz, DMSO-d6) δH 7.5 (d, 2H, J = 11 Hz, 2′- & 6′- Ph-H), 7.43 (d, 1H, J = 13 Hz, 5-H), 7.13 (d, 2H, J = 11 Hz, 3′- & 5′- Ph-H), 6.94 (d, 2H, J = 12 Hz, 2″- & 6″-H), 6.83 (d, 2H, J = 12 Hz, 3″- & 5″-H), 6.67 (d, 1H, J = 13 Hz, 4-H), 4.91 (s, 2H, -CH2-), 3.69 (s, 3H, -OCH3), 3.2 (t, 4H, J = 5 Hz, piperazine 2′- & 6′-H), 3.05 (t, 4H, J = 5 Hz, piperazine 1′- & 5′-H), 13C NMR (125 MHz, DMSO-d6) δC 167.46 (5″‘-C), 156.97 (3-C), 153.21 (3″‘-C), 148.31, 145.98, 136.22, 131.15, 130.31, 130.11, 125.95, 120.11, 117.85, 114.28, 55.19 (-OCH3), 49.32 (-CH2−), 46.01 (piperazine 2′- & 6′-C), 45.46 (piperazine 1′- & 5′-C), MS m/z (ESI) calcd for C24H24N7BrO2S (M + H+) 554.0974, found 554.0974.
2-((4-(4-Fluorophenyl)-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)methyl)-6-(4-(4-methoxyphenyl)piperazin-1-yl)pyridazin-3(2H)-one (6d)
Light yellow solid (57% yield), Rf 0.48 (CH3OH-CHCl3 9:1), mp 286–288 °C, 1H NMR (500 MHz, DMSO-d6) δH 7.52 (d, 2H, J = 11 Hz, 2′- & 6′- Ph-H), 7.46 (d, 1H, J = 13 Hz, 5-H), 7.14 (d, 2H, J = 11 Hz, 3′- & 5′- Ph-H), 6.98 (d, 2H, J = 12 Hz, 2″- & 6″-H), 6.86 (d, 2H, J = 12 Hz, 3″- & 5″-H), 6. 7 (d, 1H, J = 13 Hz, 4-H), 4.92 (s, 2H, -CH2−), 3.73 (s, 3H, -OCH3), 3.22 (t, 4H, J = 5 Hz, piperazine 2′- & 6′-H), 3.09 (t, 4H, J = 5 Hz, piperazine 1′- & 5′-H), 13C NMR (125 MHz, DMSO-d6) δC 167.58 (5″‘-C), 157.03 (3-C), 153.46 (3″‘-C), 148.57, 148.11, 136.4, 131.29, 130.49, 130.31, 126.01, 120.33, 117.95, 114.37, 55.27 (-OCH3), 49.56 (-CH2−), 46.19 (piperazine 2′- & 6′-C), 45.57 (piperazine 1′- & 5′-C), MS m/z (ESI) calcd for C24H24N7FO2S (M + H+) 494.1591, found 494.1696.
6-(4-(4-Methoxyphenyl)piperazin-1-yl)-2-((5-thioxo-4-(4-(trifluoromethoxy)phenyl)-4,5-dihydro-1H-1,2,4-triazol-3-yl)methyl)pyridazin-3(2H)-one (6e)
White solid (39% yield), Rf 0.59 (CH3OH-CHCl3 9:1), mp 253–255 °C, 1H NMR (500 MHz, DMSO-d6) δH 7.4 (d, 1H, J = 10 Hz, 5-H), 7.33 (d, 2H, J = 9 Hz, 2′- & 6′- Ph-H), 7.30 (d, 2H, J = 9 Hz, 3′- & 5′- Ph-H), 6.95 (d, 2H, J = 9 Hz, 2″- & 6″-H), 6.84 (d, 2H, J = 9 Hz, 3″- & 5″-H), 6.61 (d, 1H, J = 10 Hz, 4-H), 5.01 (s, 2H, -CH2-), 3.7 (s, 3H, -OCH3), 3.22 (t, 4H, J = 5 Hz, piperazine 2′- & 4′-H), 3.06 (t, 4H, J = 5 Hz, piperazine 1′- & 5′-H), 13C NMR (125 MHz, DMSO-d6) δC 167.95 (5″‘-C), 157.38 (3-C), 153.70 (3″‘-C), 153.70 (4″-C), 148.81 (6-C), 147.11 (Ph-4C), 146.57, 145.69, 136.40, 130.68, 130.20, 126.29, 121.25, 118.33, 114.75, 55.66 (-OCH3), 49.77 (-CH2−), 46.49 (piperazine 2′- & 6′-C), 45.56 (piperazine 1′- & 5′-C), MS m/z (ESI) calcd for C25H24F3N7O3S (M + H+) 560.1692, found 560.1692.
Cholinesterase (ChE) enzyme inhibition studies
AChE and BChE enzyme were supplied ready. ChE enzymes activity was determined according to the method performed by Ellman et al. [28]. The ChE enzymes has two substrates, DTNB [(Ellman’s Reagent) 5,5-dithio-bis-(2-nitrobenzoic acid)] and acetyltiyocholiniodate and butyryltiyocholiniodate. Thiocholine is formed because of the hydrolysis of the substrates. The thiocholine formed reacts with DTNB and forms the yellow 5-thio-2-nitrobenzoate anion. This molecule gives maximum absorbance at 412 nm wavelength [29]. A percent activity versus inhibitor concentration graph was drawn for the designation of the inhibition efficacy of each the new N-substituted-(p-tolyl)pyridazin-3(2H)-one 6(a–e) on the ChE enzymes. The IC50 values were obtained from these graphs. For the calculation of Ki values, three different of these compounds concentrations and five substrate concentrations were used. The study also has included inhibition graph of the most effective compound which was drawn. The same procedures were performed for Tacrine, the standard inhibitor of AChE and BChE enzymes, and both IC50 and Ki values were calculated.
Molecular docking
Molecular docking studies were applied to determine the amino acid residues in the active site of compounds 6b and 6a, which were interacted with AChE and BChE enzymes in silico, respectively, and to calculate the binding parameters. To investigate the binding mode of compounds 6a and 6b was carried out using Schrödinger 2021-2 software [30,31,32,33].
The possible conformations of the studied compound were optimized using the “Ligand preparation wizard” [34] program of Schrödinger 2021-2. Possible tautomeric states of pH 7.0 ± 2.0 in the Epic ligand preparation portion were used to generate a net negative substitution change that varied in each case.
The crystal structures used were obtained from the protein database (https://www.rcsb.org/). In molecular docking studies, PDB ID: 1ACJ [35] crystal structure for AChE and PDB ID: 4BDS [36] crystal structure for BChE were used. Crystal structures were prepared with the “Protein Preparation Wizard” [37] interface of Schrödinger 2021-2 software. It was prepared by sequential processes such as deletion of water molecules, addition of missing side chains and hydrogen atoms, protonation states, assignment of partial charges, optimization and minimization using the OPLS-2005 force field.
Prime MM/GBSA [38] analysis was used to calculate ligand binding energies using the OPLS_2005 force field and the VSGB solvent model. MM/GBSA analysis was applied to determine the free binding energies of 6a on BChE and 6b on AChE via molecular docking.
References
Andrade-Jorge E, Rivera-Sánchez F, Rodríguez JE, Lagos-Cruz JA, Reyes-Vallejo N, Villalobos-Molina R, et al. Isoindolone derivatives as novel potential anti-Alzheimer’s candidates: synthesis, in silico, and AChE inhibitory activity evaluation. Med Chem Res. 2022;31:851–66.
Bhilare NV, Marulkar VS, Kumar D, Chatap VK, Patil KS, Shirote PJ. An insight into prodrug strategy for the treatment of Alzheimer’s disease. Med Chem Res. 2022;31:383–99.
Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377:1019–31.
Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–55.
Jagust W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci. 2018;19:687–700.
Kumar N, Kumar V, Anand P, Kumar V, Ranjan Dwivedi A, Kumar V. Advancements in the development of multi-target directed ligands for the treatment of Alzheimer’s disease. Bioorg Med Chem. 2022;61:116742.
Ricciarelli R, Fedele E. The amyloid cascade hypothesis in Alzheimer’s disease: it’s time to change our mind. Curr Neuropharmacol. 2017;15:926–35.
Townsend KP, Praticò D. Novel therapeutic opportunities for Alzheimer’s disease: focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005;19:1592–601.
Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–98.
Özdemir Z, Ozcelik AB, Uysal M. Approaches based on cholinergic hypothesis and cholinesterase inhibitors in the treatment of Alzheimer’s disease. In: Atta-ur-Rahman FRS, editor. Frontiers in Clinical Drug Research-Alzheimer Disorders. Cambridge, UK; 2019. pp. 154–90.
Campanella L, Achilli M, Sammartino MP, Tomassetti M. Butyrylcholine enzyme sensor for determining organophosphorus inhibitors. Bioelectrochem Bioenerg. 1991;26:237–49.
Atanasova M, Stavrakov G, Philipova I, Zheleva D, Yordanov N, Doytchinova I. Galantamine derivatives with indole moiety: docking, design, synthesis and acetylcholinesterase inhibitory activity. Bioorg Med Chem. 2015;23:5382–9.
Wang L, Wang Y, Tian Y, Shang J, Sun X, Chen H, et al. Design, synthesis, biological evaluation, and molecular modeling studies of chalcone-rivastigmine hybrids as cholinesterase inhibitors. Bioorg Med Chem. 2017;25:360–71.
Anand P, Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res. 2013;36:375–99.
Mehta M, Adem A, Sabbagh M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int J Alzheimers Dis. 2012;2012:728983.
David B, Schneider P, Schäfer P, Pietruszka J, Gohlke H. Discovery of new acetylcholinesterase inhibitors for Alzheimer’s disease: virtual screening and in vitro characterisation. J Enzym Inhib Med Chem. 2021;36:491–6.
Özdemir Z, Alagöz MA, Uslu H, Karakurt A, Erikci A, Ucar G, et al. Synthesis, molecular modelling and biological activity of some pyridazinone derivatives as selective human monoamine oxidase-B inhibitors. Pharmacol Rep. 2020;72:692–704.
Bozbey İ, Özdemir Z, Uslu H, Özçelik AB, Şenol FS, Orhan İE, et al. A series of new hydrazone derivatives: synthesis, molecular docking and anticholinesterase activity studies. Mini Rev Med Chem. 2020;20:1042–60.
Dubey S, Bhosle PA. Pyridazinone: an important element of pharmacophore possessing broad spectrum of activity. Med Chem Res. 2015;24:3579–98.
Çöl ÖF, Bozbey İ, Türkmenoğlu B, Uysal M. 3(2H)-pyridazinone derivatives: synthesis, in-silico studies, structure-activity relationship and in-vitro evaluation for acetylcholinesterase enzyme inhibition. J Mol Struct. 2022;1261:132970.
Krasiński A, Radić Z, Manetsch R, Raushel J, Taylor P, Sharpless KB, et al. In situ selection of lead compounds by click chemistry: target-guided optimization of acetylcholinesterase inhibitors. J Am Chem Soc. 2005;127:6686–92.
Hosseini S, Pourmousavi SA, Mahdavi M, Taslimi P. Synthesis, and in vitro biological evaluations of novel naphthoquinone conjugated to aryl triazole acetamide derivatives as potential anti-Alzheimer agents. J Mol Struct. 2022;1255:132229.
Saeedi M, Maleki A, Iraji A, Hariri R, Akbarzadeh T, Edraki N, et al. Synthesis and bio-evaluation of new multifunctional methylindolinone-1,2,3-triazole hybrids as anti-Alzheimer’s agents. J Mol Struct. 2021;1229:129828.
Saeedi M, Safavi M, Karimpour-Razkenari E, Mahdavi M, Edraki N, Moghadam FH, et al. Synthesis of novel chromenones linked to 1,2,3-triazole ring system: Investigation of biological activities against Alzheimer’s disease. Bioorg Chem. 2017;70:86–93.
Çeçen M, Oh JM, Özdemir Z, Büyüktuncel SE, Uysal M, Abdelgawad MA, et al. Design, synthesis, and biological evaluation of pyridazinones containing the (2-Fluorophenyl) piperazine moiety as selective MAO-B inhibitors. Molecules. 2020;25:5371.
Bozbey İ, Özdemir Z, Uslu H, Özçelik BA, Şenol SF, Orhan Eİ, et al. A series of new hydrazone derivatives: synthesis, molecular docking and anticholinesterase activity studies. Mini Rev Med Chem. 2020;20:1042–60.
Wujec M, Pachuta-Stec A, Stefańska J, Kuśmierz E, Siwek A. Synthesis and antibacterial activity of some new derivatives of thiosemicarbazide and 1,2,4-triazole. Phosphorus Sulfur Silicon Relat Elem. 2013;188:1661–9.
Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95.
Shirinzadeh H, Dilek E, Alım Z. Evaluation of naphthalenylmethylen hydrazine derivatives as potent inhibitors on, antiatherogenic enzymes, paraoxonase I and acetylcholinesterase activities. ChemistrySelect. 2022;7:e202104489.
Schrödinger Release 2021-2: Glide S, LLC, New York, NY; 2021.
Dadou S, Altay A, Koudad M, Türkmenoğlu B, Yeniçeri E, Çağlar S, et al. Design, synthesis, anticancer evaluation and molecular docking studies of new imidazo [2, 1-b] thiazole-based chalcones. Med Chem Res. 2022;31:1369–83.
ÇÖL ÖF, Bozbey İ, Türkmenoğlu B, Uysal M. 3 (2H)-pyridazinone derivatives: synthesis, in-silico studies, structure-activity relationship and in-vitro evaluation for acetylcholinesterase enzyme i̇nhibition. J Mol Struct. 2022;1261:132970.
Anil DA, Aydin BO, Demir Y, Turkmenoglu B. Design, synthesis, biological evaluation and molecular docking studies of novel 1H-1, 2, 3-Triazole derivatives as potent inhibitors of carbonic anhydrase, acetylcholinesterase and aldose reductase. J Mol Struct. 2022;1257:132613.
Schrödinger Release 2021-2: LigPrep S, LLC, New York, NY; 2021.
Harel M, Schalk I, Ehret-Sabatier L, Bouet F, Goeldner M, Hirth C, et al. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc Natl Acad Sci USA. 1993;90:9031–5.
Nachon F, Carletti E, Ronco C, Trovaslet M, Nicolet Y, Jean L, et al. Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer’s drugs targeting acetyl-and butyryl-cholinesterase. Biochem J. 2013;453:393–9.
Schrödinger Release 2021-2: Protein Preparation Wizard; Epik S, LLC, New York, NY; 2021; Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY; 2021.
Schrödinger Release 2021-2: Prime S, LLC, New York, NY; 2021.
Acknowledgements
The authors would like to thank Erzincan Binali Yıldırım University, Basic Sciences Application and Research Center (EBYU-EUTAM) for the Schrödinger Maestro 2021-2 program. The authors would like to thank Assoc. Prof. Azime Berna Özçelik for HRMS analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Merde, İ.B., Önel, G.T., Türkmenoğlu, B. et al. Pyridazinones containing the (4-methoxyphenyl)piperazine moiety as AChE/BChE inhibitors: design, synthesis, in silico and biological evaluation. Med Chem Res 31, 2021–2031 (2022). https://doi.org/10.1007/s00044-022-02968-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-022-02968-x