
Abstract
Alzheimer’s disease (AD) is a neurodegenerative condition that affects elderly persons around the world, impairing cognitive function, due to a decrease in cholinergic transmission. Thus, developing drugs with better acetylcholinesterase (AChE) inhibitory characteristics will improve cholinergic transmission. This work aimed to synthesize isoindolone derivatives and test their AChE inhibitory activity both in silico and in vitro, and to compare them with their precursors. Seven racemic mixtures (isoindolones) and four precursors were synthesized in good yields. Their interaction with AChE is mainly mediated by the aromatic ring and the ester group, according to molecular docking. The in vitro experiments revealed competitive inhibition for all tested molecules. The structural analysis showed better Ki values for structures with an extra double bond, and the two ester groups. Racemic mixture rac-6 showed the lowest Ki (55.4 µM) of all compounds. Both aromatic ring moieties and two ester groups in the isoindolone derivatives play an important role in recognition with AChE.

Graphical abstract
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Dementia is the acquired progressive cognitive impairment sufficient to impact activities of daily living [1]. Dementia has physical, psychological, social, and economic impacts; it is a major cause of disability and dependency in older people. In 2015, dementia affected 47 million persons worldwide, but an increase is a forecast for 75 million in 2030 and 132 million by 2050. Although there are several forms of dementia, Alzheimer’s disease (AD) contributes to 60–70% of cases [2]. AD dementia refers to a particular onset and course of cognitive and functional decline associated with age that ultimately results in death [3]. To date, there is no single treatment that can stop or reverse the progression of AD. AD is a neurodegenerative disease primarily caused by inefficient processing in the polymerization of proteins; i.e., detrimental changes in the brain as protein structure and function, promote protein misfolding, aggregation, and deposition in capillaries walls, arteries, and arterioles, causing cerebral angiopathy and degeneration of vascular wall components and blood flow, leading to intraparenchymal hemorrhages [4,5,6]. The pathophysiology of AD continues to be the subject of some controversy, but the prevailing hypothesis places the accumulation of β-amyloid at the center of the process and is referred to as the “amyloid-cascade hypothesis”. The amyloid precursor protein (APP) can undergo sequential cleavage through two different pathways: non-amyloidogenic and amyloidogenic, in the latter, APP is first cleaved by β-secretase producing the extracellular product sAPPβ and C99, the membrane-bound 99 amino acid C-terminal fragment; γ-secretase process C99 producing Αβ peptide [7]. The overproduction of Αβ induces it to self-assemble into oligomers and ultimately into highly regular amyloid fibrils forming plaques, which are toxic and impair cellular function, protein expression, modify signal transduction pathways, and produce neurofibrillary tangles and selective neuron death with consequently neurotransmitter deficits as acetylcholine (ACh). These events cause reduction in synaptic strength, synaptic loss, and neurodegeneration. Metabolic, vascular, and inflammatory changes, as well as comorbid pathologies, are key components of the disease process [7, 8]. The cholinergic neurons are widely distributed in the central nervous system where they regulate several physiological functions including memory, learning, attention, and sensory information, among others [9]. The deterioration of the cholinergic system, especially in the nucleus basalis of Meynert, contributes to memory loss as in AD [10, 11]. In this regard, avoiding the hydrolysis of ACh is fundamental to delaying cognitive impairment. Cholinesterase inhibitors enhance cholinergic transmission, inhibiting the enzyme acetylcholinesterase (AChE), avoiding the hydrolysis of ACh, as well as decreasing the Αβ deposition [12], ameliorating the symptoms of AD. Despite the large amount of evidence that supports the pathogenesis of AD, the inhibition of AChE is the most effective strategy for treating AD; however, AChE inhibitors currently on the market have several non-desirable effects [13]. Therefore, the search for new, more effective drugs with fewer adverse effects is required [14, 15]. Isoindolone moiety is present in several natural products with bioactivities such as the inhibition of the α-glucosidase [16] and topoisomerase inhibitors [17], and urotensin-II receptor antagonists [18], among others. Additionally, isoindolone derivatives have exhibited effects on molecules involved in neuropsychiatric disorders such as schizophrenia, anxiety, and depression through binding the 5-HT2C receptor [19], and also produced the inhibition of AchE, ameliorating the impairment of memory in scopolamine-induced mice [20]. Thus, the aim of the present contribution was the design, synthesis, and evaluation of isoindolone derivatives as potential inhibitors of AChE with a possible therapeutic effect in AD.
Results and discussion
Chemistry
N-phthaloyl phenylalanine compound (S)-1 was obtained in 97% (ee = 93%) overall yield by condensation of L-phenylalanine and phthalic anhydride by heating at 135–140 °C, according to the synthetic route depicted in Scheme 1. Compound (R)-1 was synthesized analogously to (S)-1 from D-phenylalanine in 95% yield (ee = 94.4%). The partial racemization observed in obtaining the compounds is due to the high temperature used during the reaction and confirmed by chiral HPLC. In this regard, in the synthesis of N-phthaloyl amino acids when employing heating conditions similar to those utilized in the present work, racemization has been observed; thus, complete racemization in N-phthaloyl phenylglycine was described in a previous report [21]. Both (S)-1 and (R)-1, employing a Fischer esterification reaction, by treatment with MeOH and a catalytic amount of H2SO4 gave rise to compounds (S)-2 in a 93% (ee = 97.2%) yield and (R)-2 in 93% (ee = 94.4%), respectively (Scheme 1).

Synthesis of N-substituted phthalimides
Based on our previously reported method [21], the cascade reaction between phthalimide (S)-2 and ethyl acrylate was carried out in the presence of lithium bis(trimethylsilyl)amide (LHMDS) at a low temperature to give a 69:31 diastereomeric mixture and a 45% yield corresponding to the rac-3a and rac-3b diastereoisomers in racemic form. In addition, the compound rac-2 was also recovered in racemic form with 42%, as confirmed by chiral HPLC (Scheme 2a). The loss of chirality in the reaction is due to the basic treatment of (S)-2 with the LHMDS to give rise to the anion A. Ideally, the conjugated addition of the (achiral) anion A to the ethyl acrylate to form the (racemic) intermediate B, which, by intramolecular addition to one of the carbonyl groups of the phthalimide fragment, gives rise to the observed products rac-3a and rac-3b. Finally, it should be noted that on increasing the reaction time or the temperature decomposition products were observed whose structure could not be established.

a) Synthesis of rac-3a and rac-3b. b) Two-step cyclization supported with plausible mechanistic insight for the Michael-type addition followed by intramolecular nucleophilic addition via Zimmerman-Traxler-type transition state (TS)
The relative stereochemistry observed in compounds rac-3a and rac-3b can be explained by the general reaction involving a Michael-type addition in the first step followed by an intramolecular nucleophilic addition through the “Zimmerman-Traxler-type transition state (TS)” at the second step. Furthermore, both rac-3a and rac-3b differ only in terms of configuration at the C-3 carbon, representing strong evidence for the independence of both individual steps, as depicted in Scheme 2b [22].
The rac-3a and rac-3b stereoisomers were separated by column chromatography and were easily distinguishable by 1H NMR, revealing similar coupling patterns and chemical shifts for the Ha-1, Hb-2, and Hc-2 protons of the pyrrolidine ring and similat to those reported in previous works [21, 22]. In particular, the 1H NMR spectrum of rac-3a shows a doublet of doublets at 2.48 ppm with geminal and vicinal coupling constants 2J = 13.6 Hz and 3J = 6.8 Hz, respectively, which was assigned to Hc-2, a triplet to 3.04 ppm with 3J = 13.1 Hz that was assigned to Hb-2, and a doublet of doublets at 3.31 ppm with 3J = 12.5 Hz and 3J = 6.7 Hz that was assigned to Ha-1. 1H NMR of rac-3b shows a doublet of doublets at 2.84 ppm with geminal and vicinal coupling constants 2J = 13.4 Hz and 3J = 7.4 Hz, respectively, which was assigned to Hc-2, a triplet at 3.22 ppm with 3J = 12.7 Hz that was assigned to Hb-2, and a doublet of doublets at 1.84 ppm with 3J = 12.1 Hz and 3J = 7.4 Hz that was assigned to Ha-1.
With the isoindolone rac-3a in hand, several derivatives were prepared that incorporate the required isoindolone scaffold. Thus, under conditions of the hydrolysis of rac-3a with one equivalent of LiOH.H2O, the racemic compound rac-4 was regioselectively obtained with a yield of 80%. The selective hydrolysis of the ethyl ester group can be explained by the formation of a coordinated intermediate among the Li+ cation, the HO-group bound to C-9b, and an oxygen atom of the ester group, this to favor the nucleophilic attack of the hydroxyl ion on that ester group [23], while for the complete hydrolysis of rac-3a, four equivalents of LiOH.H2O were necessary to obtain the racemic compound rac-5 with a yield of 93% (Scheme 3).

Synthesis of substituted pyrrolo-isoindolones rac-4 - rac-9. a) 1 eq LiOH, THF/H2O, rt 5 h. b) 4 eq LiOH, THF/H2O, 50 °C, 48 h. c) H2SO4, CH2Cl2, rt, 30 min. d) 5 eq LiOH, THF/H2O, 50 °C, 48 h. e) 4 eq NaOH, MeOH/H2O, 58 °C, 24 h. f) H2/Pd(OH)2, AcOEt, 689.4 kPa, 60 °C, 24 h
The compound rac-3a has a highly reactive hydroxyl group attached to C-9b because it is benzylic and beta-positioned to the ethyl ester group, which allows easy and fast dehydration by acid catalysis. Thus, the rac-6 compound was obtained by dehydration at room temperature, using CH2Cl2 as a solvent and a catalytic amount of H2SO4 with a yield of 98% for only 30 min, without observing possible hydrolysis products of the ester groups (Scheme 2). The racemic compound rac-6 was hydrolyzed by treatment with four equivalents of LiOH.H2O to obtain the racemic compound rac-7 at an 87% yield, four 4 equivalents of NaOH were necessary to obtain the racemic compound rac-8 at a 90% yield. Finally, compound rac-9 at a 52% yield was obtained in its racemic form by the catalytic reduction of compound rac-6 using 20% Pd(OH)2/C in AcOEt at 60 °C (Scheme 2). The structures of the synthetic derivatives were clarified based on 1D, 2D-NMR, and by the analogy of the 1H and 13C NMR spectroscopic data with the structure of similar compounds reported in the literature [22].
Anticholinesterase activity assay
In previous works, phenylethyl phthalimide derivatives exhibited inhibitory activity on EeAChE with good results (Fig. 1a), thus, in this study these leading molecules were used for the design of novel molecules as indicate in color red (Fig. 1b), along with the new ring substituted with different functional groups, that have been described to improve the affinity for the AChE [24, 25]. We describe the synthesis and effect as acetylcholinesterase inhibitors (in vitro and in silico) of four dioxoisoindolines, (S)-1, (R)-1, (S)-2, (R)-2, and seven fused isoindolones (Table 1) that present the tricyclic systems pyrrolo[2,1-a]isoindolone (rac-3a - rac-9). The latter are new chemical entities with multiple functional groups that permit modifications to determine the influence of the nature and location of substituents along with the core of phenylethyl-phthalimides on the inhibition effect of AChE (Fig. 1b). The phenylethyl-phthalimide analogs (S)-1, (R)-1, (S)-2, and (R)-2 contain an ethylene bridge between the phthalimide and the aromatic ring, the carboxylic acid, or a methyl ester of different polarity. They also integrate a stereogenic center; the differences among these compounds will make it possible to determine the influence between the configuration of the stereogenic center and both substituents on the inhibition effect of AChE. On conducting a more detailed analysis of these structures, we can see that the compound with the (R) configuration has better Ki values than those with an (S) configuration. In relation to the substituents, the methyl ester group appears to improve the inhibitory activity, as it is observed for (R)-2 compound, which exhibited the best Ki (270.6 μM) value in comparison with their phenylethyl-phthalimide analogs and those reported in the literature (610 μM) [25]. On the other hand, tricyclic pyrrolo[2,1-a]isoindolone (rac-3a - rac-9) systems, which include a pyrrolidine ring fused to the phenylethylphthalimide core with different substituents, enhanced AChE inhibition up to 10-fold compared to their precursors (S)-1, (R)-1, (S)-2 and (R)-2 (Table 1), suggesting that larger molecules are more effective than smaller ones. In addition, due to the tricyclic compounds were obtained as racemic mixtures was not possible analyze the contribution of the stereogenic centers to AChE inhibiton. Among the structural changes that favored the inhibition of AChE, the double ester group is an important one because the best compounds for inhibiting the enzyme exhibited this pattern compared to acid derivatives. The presence of the double bond in the newly formed ring of isoindolones contributed to the inhibitory activity to a higher degree than the isoindolone with the hydroxyl group or the hydrogenation of the double bond of rac-6. The molecule that reflected all of these aforementioned characteristics was racemic mixture rac-6, which has a double ester group and the presence of the double bond, resulting in a Ki of 55.47 µM, the latter the best in the series of isoindolone derivatives. Likewise, this molecule can be taken as a lead-compound and can generate from this a new family with better inhibitory characteristics. The second-best racemic mixture was rac-8, which was the fully hydrolyzed version of rac-6, obtaining a Ki of 67.16 µM, and in the third-place we find rac-3a and rac-7, which appear to have no significant difference in their Ki (163.4 and 168.7 µM, respectively). On performing a structural analysis, the racemic mixture rac-3a does not have the extra double bond in its structure, but this is compensated for the double ester group, quite the opposite with the racemic mixture rac-7, which has the extra double bond but lacks the double ester group. All of this reinforces the importance of these two parameters in the improvement of the inhibition of the enzyme AChE. All isoindolone derivatives as well as their respective precursors (phenylethyl-phthalimide) demonstrated a competitive inhibition pattern, as we are able to observe in the Lineweaver-Burk plots (Fig. 2). The inhibitory activity of phenylethyl-phthalimide is well known, as we have described in previous works [24, 25], but before this, to our knowledge, the activity of isoindolones had not been explored. Thus, this contribution revealed that isoindolones possess promising activity. Separating the enantiomeric mixtures would allow us to better understand the compounds with higher AChE inhibitory capacity.

Design strategy for the molecules evaluated in this work. a) lead-compound obtained in previous work b) in red is represented the structure of the lead-compound included in the new isoindolone derivatives

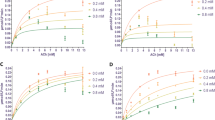
The top three best inhibitors of Electrophorus electricus AChE in the form of racemic mixture. Michaelis-Menten plot for: A) (R)-6/(S)-6; B) (R)-8/(S)-8; C) (1R,3R,9bS)-3a/(1S,3S,9bR)-3a, and Lineweaver-Burk plot for: A’) (R)-6/(S)-6; B’) (R)-8/(S)-8; C’) (1R,3R,9bS)-3a /(1S,3S,9bR)-3a. Non-linear regression analysis was carried out with 95% confidence intervals
Molecular docking and theoretical calculations
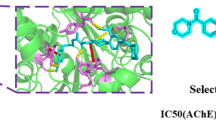
The molecular docking between the ligands and the Electrophorus electricus AChE enzyme was carried out for a better understanding of the phenomenon observed in in vitro experiments. Because the kinetic experiments occurred at a pH = 8 higher than the physiological one, we extrapolated the in silico results to an in vivo condition considering a pH =7.4. The results presented in Table 2 indicate an exergonic interaction for all ligands. Interestingly, both in silico and in vitro experiments demonstrated that the best ligand was the racemic mixture rac-6. The conformational analysis (Table 3) was performed for the ligands that obtained the best inhibition constants in enzyme kinetics. The advantage of in silico experiments lies in that we can study the molecules individually despite being a racemic mixture. The analysis indicated that the ligands are interacting with the amino acid residues located in the gorge formed by the peripheral anionic site, the anionic site, the catalytic active site, the acyl pocket, and the oxyanion hole of the enzyme. As depicted in Table 3 and Fig. 3, some of the aminoacid residues with which the ligands interact are key pieces in recognition, such Trp86, Tyr124, Ser203, His447, Gly121, etc. The numbers of the interactions found in the in silico study for the isoindolones were around 10 interactions on average, while for the reference molecules, these were lower, with eight interactions on average. On the other hand, Table 2 shows that isoindolones and reference molecules possess a similar strength (measure by Gibbs free energy) when binding with the AChE (−8.74 and −8.76, respectively). The type of interactions that occurred were mainly the hydrophobic type, including the π-π type; the majority of the interactions were promoted by the aromatic rings of the ligands, it is worth mentioning that, in the analysis, a clear difference was not observed between the two (R) and (S) configurations. On the other hand, the type of inhibition cannot be predicted by in silico experiments. Nonetheless, the conformational analysis performed by comparison with known references drugs and those that we know to present a competitive inhibition with the enzyme, the position found, and the amino acid residues with which the different compounds interact, suggested that the inhibition could be of a competitive type (Fig. 3d and Table 3), this result correlates with that obtained in the in vitro experiments. It is noteworthy that the in silico experiments were validated with well-known ligands such donepezil, galantamine, rivastigmine, and tacrine, which reproduced the binding modes with the EeAChE enzyme (RMSD 0.9744, 0.0780, 0.1002 and 0.0487 respectively) [24, 26].

Binding modes of the best molecules with the amino acid residues of the active site of Electrophorus electricus AChE: A) AChE-(R)-6 complex; A’) AChE-(S)-6 complex; B) AChE-(R)-8 complex; B’) AChE-(S)-8 complex; C) AChE-(1R,3R,9bS)-3a; C’) AChE-(1S,3R,9bR)-3a complex; D) Interaction of isoindolones (yellow) and reference compounds (fuchsia) at the active site of AChE
Conclusions
A series of isoindolone derivatives were synthesized and evaluated as AChE inhibitors. Two families were formed: the precursors (four dioxoisoindolines) and the isoindolones (seven racemic mixtures). These displayed a diverse range in their capacity to inhibit the enzyme, in each case with a competitive pattern supported by in vitro and in silico experiments. According to the in vitro results, those molecules with two ester groups, and a double bond in their structure exhibited the highest affinity for the EeAChE. Among the isoindolone compounds tested herein, rac-6 (a racemic mixture; Ki = 55.47 µM) holds promise as a leading compound for designing new AChE inhibitors, due to the good inhibition it displayed. This structure can be improved, and even separate the enantiomeric mixture to generate new families and reach effectiveness as an inhibitor even better than those in the references. In silico experiments showed that isoindolones were strongly recognized by the gorge of the enzyme, mainly through hydrophobic interactions (π-π). Further research is warranted on isoindolone derivatives focused on molecule rac-6 and related molecules.
Materials and methods
Synthesis and characterization
All reagents purchased for Sigma-Aldrich were used without further purification, except tetrahydrofuran (THF) was distilled with sodium before use. All reactions were performed in an oven-dried flask, while the mixtures were agitated with a magnetic stirring bar and concentrated employing a standard rotary evaporator. The progress of the reaction was routinely monitored by thin-layer chromatography (TLC) on silica gel 60 (precoated F254 Merck plates), under UV lamp (254 nm) irradiation to visualize starting materials and products. Column chromatography was performed using silica gel (230–400 mesh). Melting points were measured on a Melt-Temp “Electrothermal” apparatus and are uncorrected. Infrared spectra (IR) were obtained on a Perkin-Elmer (PC16, Spectrum GX) FT-IR spectrometer with an attenuated total reflectance (ATR) accessory. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on Bruker ASCEND 400 (400 MHz) and on Bruker ASCEND g750 Ultrashield (750 MHz) spectrometers. The data are reported as follows: chemical shift in ppm (δ), integration area, multiplicity (s = singlet, br s = broad singlet, d = doublet, t = triplet, and m = multiplet), and coupling constants in Hz (J) in the solvent indicated. Electrospray ionization high-resolution mass spectrometry (ESI-HRMS) was performed on a Bruker micrOTOF-Q instrument. The HPLC analysis were conducted in a Waters HPLC Alliance 2695 with a UV/Visible detector, with diode array Waters 2996, with the following separation conditions: column Chiralpak AD-H; 92:8 hexane-IPA, and Flux 1.0 mL/min. The catalytic hydrogenation reaction was carried out in a Parr high-pressure hydrogenator (Non-Stirred Pressure Vessels, 4792 HP/HT, 100 mL).
(S)-2-(1,3-dioxoisoindolin-2-yl)-3-phenylpropanoic acid, [(S)-1]
Compound (S)-1 was prepared according to the reported protocol with slight modifications [27]. A 250 mL round-bottomed flask containing a magnetic stirrer and equipped with a reflux condenser was charged with L-phenylalanine (6.7 g, 40.6 mmol) and phthalic anhydride (6.0 g, 40.6 mmol). The resulting mixture was heated to 135–140 °C for 2.5 h and after the completion of the reaction, the reaction mixture was allowed to cool down to room temperature. The crude product was purified by recrystallization from EtOH/H2O (2:1). This product was obtained as a white solid; yield 11.1 g (97%); enantiomeric excess 93.0%; HPLC retention time 19.8 min; mp 181–183 °C (lit. mp 184–186 °C [27]); 1H NMR (Methanol-d4, 400 MHz) δ 7.75 (4H, m, H-Ar), 7.13 (4H, m, H-Ar), 7.07 (1H, m, H-Ar), 5.14 (1H, dd, J = 11.5, 5.3 Hz, CH), 3.50 (2H, m, CHHPh diastereotopic proton); 13C NMR (Methanol-d4, 101 MHz) δ 172.1 (C, HO=O), 168.9 (2 C, NC=O), 138.6 (C, benzyl), 135.6 (2 CH, Phth), 132.7 (2C, Phth), 129.9 (2CHortho, benzyl), 129.5 (2CHmeta, benzyl), 127.8 (CHpara, benzyl), 124.2 (2CH, Phth), 54.5 (CH, CHCO2H), 35.5 (CH2, CH2Ph); HRMS (ESI+) m/z calculated for C17H14NO4 296.09228, found 296.09226 (M + H+).
(R)-2-(1,3-dioxoisoindolin-2-yl)-3-phenylpropanoic acid, [(R)-1]
Compound (R)-1 was synthesized analogously of (S)-1 from D-phenylalanine. The crude product was purified by recrystallization from EtOH/H2O (2:1). It was obtained as a white solid; yield (95%); enantiomeric excess 94.4%; HPLC retention time 33.3 min; mp 180–182 °C (lit. mp 182-183 °C [28]); 1H NMR (Methanol-d4, 301 MHz) δ 7.75 (4H, m, H-Ar), 7.13 (4H, m, H-Ar), 7.08 (1H, m, H-Ar), 5.15 (1H, dd, J = 11.3, 5.6 Hz, CH), 3.50 (2H, m, CHHPh diastereotopic proton); 13C NMR (Methanol-d4, 76 MHz) δ 172.1 (C, HOC=O), 168.9 (2 C, N=O), 138.6 (C, benzyl), 135.6 (2 CH, Phth), 132.8 (2C, Phth), 129.9 (2CHortho, benzyl), 129.5 (2CHmeta, benzyl), 127.8 (CHpara, benzyl), 124.2 (2CH, Phth), 54.5 (CH, CHCO2H), 35.5 (CH2, CH2Ph). HRMS (ESI+) m/z calculated for C17H14NO4 296.09228, found 296.09329 (M + H+).
Methyl (S)-2-(1,3-dioxoisoindolin-2-yl)-3-phenylpropanoate, [(S)-2]
A 100 mL round-bottomed flask containing a magnetic stirrer and equipped with a reflux condenser was charged with (S)-1 (12.0 g, 40.6 mmol, 93.0% ee) and was dissolved in methanol (50 mL) prior to the addition of H2SO4 (0.9 mL). The reaction mixture was heated to reflux for 3 h and then allowed to cool to room temperature. The solvent was removed at reduced pressure before the addition of water (100 mL) and the acid-reaction mixture was neutralized with K2CO3 powder. The residue was dissolved in water (200 mL) and EtOAc (100 mL), and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine (30 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. This was obtained as a white solid; yield 10.9 g (93%); enantiomeric excess 97.2%; HPLC retention time 14.7 min; mp 124-126 °C (lit. mp 124.8-126.8 °C [29]); TLC: Rf (hexane/AcOEt, 7:3) = 0.53; 1H NMR (CDCl3, 400 MHz) δ 7.77 (2H, dd, J = 5.5, 3.1 Hz, H-Ar), 7.68 (2H, dd, J = 5.5, 3.0 Hz, H-Ar), 7.17 (4H, m, H-Ar), 7.12 (1H, m, H-Ar), 5.16 (1H, dd, J = 11.2, 5.4 Hz, CH), 3.78 (3H, s, OCH3), 3.55 (2H, m, CHHPh diastereotopic proton); 13C NMR (CDCl3, 101 MHz) δ 169.3 (C, O=O), 167.4 (2 C, NC=O), 136.7 (C, benzyl), 134.1 (2 CH, Phth), 131.5 (2C, Phth), 128.8 (2CHortho, benzyl), 128.5 (2CHmeta, benzyl), 126.8 (CHpara, benzyl), 123.4 (2CH, Phth), 53.2 (CH, CHCO2CH3), 52.8 (CH3, OCH3), 34.6 (CH2, CH2Ph); HRMS (ESI+) m/z calculated for C18H16NO4 310.10793, found 310.10670 (M + H+).
Methyl (R)-2-(1,3-dioxoisoindolin-2-yl)-3-phenylpropanoate, [(R)-2]
Compound (R)-2 was synthesized analogously of (S)-2 from (R)-1 (94.4% ee). It was obtained as a white solid; yield (93%); enantiomeric excess 94.4%; HPLC retention time 18.8 min; mp 119-120 °C (lit. mp 124.8–126.8 °C [29]); 1H NMR (CDCl3, 301 MHz) δ 7.77 (2H, m, H-Ar), 7.68 (2H, m, H-Ar), 7.17 (4H, m, H-Ar), 7.11 (1H, m, H-Ar), 5.16 (1H, dd, J = 11.0, 5.5 Hz, CH), 3.78 (3H, s, OCH3), 3.56 (2H, m, CHHPh diastereotopic proton); 13C NMR (CDCl3, 76 MHz) δ 169.3 (C, O=O), 167.4 (2 C, N=O), 136.7 (C, benzyl), 134.1 (2 CH, Phth), 131.6 (2C, Phth), 128.8 (2CHortho, benzyl), 128.5 (2CHmeta, benzyl), 126.8 (CHpara, benzyl), 123.5 (2CH, Phth), 53.2 (CH, CHCO2CH3), 52.9 (CH3, OCH3), 34.6 (CH2, CH2Ph); HRMS (ESI+) m/z calculated for C18H16NO4 310.10793, found 310.10820 (M + H+).
1-Ethyl 3-methyl (1R,3R,9bS)- and (1S,3S,9bR)-3-benzyl-9b-hydroxy-5-oxo-2,3,5,9b-tetrahydro-1H-pyrrolo[2,1-a]isoindole-1,3-dicarboxylate, [rac-3a] and 1-ethyl 3-methyl (1R,3S,9bS)- and (1S,3R,9bR)-3-benzyl-9b-hydroxy-5-oxo-2,3,5,9b-tetrahydro-1H-pyrrolo[2,1-a]isoindole-1,3-dicarboxylate, [rac-3b]
This compound was prepared according to the protocol reported with slight modifications [22]. In a 100 mL round bottom flask containing a magnetic stirrer, (S)-2 (5.0 g, 16.2 mmol) was placed before sealing with a rubber septum. To maintain an inert atmosphere, nitrogen was passed through syringe needles that were inserted through the rubber septum to allow nitrogen in and out. After 30 min, dry THF (50 ml) was added via cannula, ethyl acrylate (3.6 ml, 34.0 mmol) was added with a syringe and the reaction mixture was stirred for a few minutes. Subsequently, the mixture was cooled in a dry ice-acetone bath, and lithium bis (trimethylsilyl) amide (1 M in THF, LHMDS, 24.3 ml, 24.3 mmol) was added dropwise using a syringe in order to not to expose them to the atmosphere. The resulting mixture was stirred for 1 h at the same temperature prior to the addition of aq. NH4Cl (30 mL) and the reaction mixture were extracted with EtOAc (3 × 30 mL). The organic extracts were combined, washed with brine (30 mL), and dried over anhydrous Na2SO4. Subsequently, the solvent was removed under reduced pressure and the crude product was purified by column chromatography (hexane/EtOAc, 7:3) and yielded gave rac-3a (2.0 g, 31%) and rac-3b (0.90 g, 14%).
Data for the rac-3a: white solid; HPLC retention time 25.9 and 27.6 min; mp 178–180 °C; TLC: Rf = 0.26 (hexane/EtOAc 7:3); ATR-FTIR νmax 3479 (O-H), 1733 (C=O ester), 1706 (C=O lactam) cm-1; 1H NMR (CDCl3, 400 MHz) δ 7.70 (2H, m, H-6 and H-9), 7.54 (1H, td, J = 7.5, 1.2 Hz, H-8), 7.47 (1H, td, J = 7.5, 1.1 Hz, H-7), 7.27 (4H, m, H-Ar), 7.21 (1H, dd, J = 9.3, 4.8 Hz, H-Ar), 4.20 (2H, m, OCH2CH3), 3.70 (3H, s, OCH3), 3.61 (1H, d, J = 13.9 Hz, PhCHH diastereotopic proton), 3.31 (1H, dd, J = 12.5, 6.7 Hz, Ha-1), 3.21 (1H, d, J = 14.0 Hz, PhCHH diastereotopic proton), 3.04 (1H, t, J = 13.1 Hz, Hb-2), 2.48 (1H, dd, J = 13.6, 6.8 Hz, Hc-2), 1.52 (1H, m, OH), 1.28 (3H, t, J = 7.2 Hz, OCH2CH3); 13C NMR (CDCl3, 101 MHz) δ 172.6 (C, MeOC=O), 169.3 (C, NC=O), 169.1 (C, EtOC=O), 145.9 (C, C-9a), 135.7 (C, benzyl), 133.1 (CH, C-8), 131.7 (C, C-5a), 131.5 (2CHortho, benzyl), 130.0 (CH, C-7), 128.2 (2CHmeta, benzyl), 127.2 (CHpara, benzyl), 124.0 (CH, C-6), 123.9 (CH, C-9), 94.8 (C, C-9b), 66.0 (C, C-3), 61.3 (CH2, OCH2CH3), 52.8 (CH3, OCH3), 48.9 (CH, C-1), 39.8 (CH2, C-2), 38.3 (CH2, CH2Ph), 14.2 (CH3, OCH2CH3); HRMS (ESI+) m/z calculated for C23H24NO6 410.1604, found 410.1598 (M + H+).
Data for rac-3b: white solid; HPLC retention time 26.7 and 28.7 min; mp 180–182 °C; TLC: Rf = 0.14 (hexane/EtOAc 7:3); ATR-FTIR νmax 3174 (O-H), 1747 (C=O ester), 1727 (C=O lactam), 1680, 1666 cm-1. 1H NMR (CDCl3, 750 MHz) δ 7.81 (1H, dd, J = 6.6, 1.7 Hz, H-Ar), 7.70 (1H, dd, J = 6.7, 1.5 Hz, H-Ar), 7.53 (2H, m, H-Ar), 7.15 (2H, d, J = 7.9 Hz, H-Ar), 7.12 (3H, dd, J = 10.4, 6.9 Hz, H-Ar), 4.20 (2H, m, OCH2CH3), 4.16 (1H, d, J = 14.1 Hz, PhCHH diastereotopic proton), 3.85 (3H, s, OCH3), 3.28 (1H, d, J = 14.1 Hz, PhCHH diastereotopic proton), 3.22 (1H, t, J = 12.7 Hz, Hb-2), 2.84 (1H, dd, J = 13.4, 7.4 Hz, Hc-2), 1.84 (1H, dd, J = 12.1, 7.4 Hz, Ha-1), 1.31 (3H, t, J = 7.2 Hz, OCH2CH3), 1.26 (1H, br s, OH); 13C NMR (CDCl3, 189 MHz) δ 173.9 (C, MeOC=O), 169.2 (C, N=O), 166.4 (C, EtOC=O), 144.3 (C, C-9a), 134.9 (C, benzyl), 133.4 (C, C-5a), 132.8 (CH, C-Ar), 130.7 (2CHortho, benzyl), 130.1 (CH, C-7), 128.5 (2CHmeta, benzyl), 127.2 (CHpara, benzyl), 123.6 (CH, C-6), 123.5 (CH, C-9), 95.6 (C, C-9b), 66.2 (C, C-3), 61.5 (CH2, OCH2CH3), 53.3 (CH, C-1), 48.3 (CH3, OCH3), 39.1 (CH2, CH2Ph), 36.5 (CH2, C-2), 14.1 (CH3, OCH2CH3); HRMS (ESI+) m/z calculated for C23H24NO6 410.1604, found 410.1598 (M + H+).
(1R,3R,9bS)- and (1S,3S,9bR)-3-benzyl-9b-hydroxy-3-(methoxycarbonyl)-5-oxo-2,3,5,9b-tetrahydro-1H-pyrrolo[2,1-a]isoindole-1-carboxylic acid, [rac-4]
A 100 mL round-bottomed flask containing a magnetic stirrer, rac-3 (491 mg, 1.2 mmol) and LiOH.H2O (51.2 mg, 1.2 mmol) were dissolved in dry THF (25 mL) prior to the addition of water (1.7 mL) and the reaction mixture was stirred at room temperature for 5 h. Subsequently, the solvent was evaporated, water (10 mL) and EtOAc (10 mL) were added to the obtained mixture. After that, the phases were separated, EtOAc (5 mL) was added to the aqueous phase, and the mixture was carefully acidified to pH 1 with aqueous 1 N HCl, then gently extracted with more EtOAc (3 × 5 mL), and the organic extracts were combined, washed with brine (10 mL) and dried over anhydrous Na2SO4. Subsequently, the solvent was removed under reduced pressure and the crude product was purified by recrystallization from hexane/EtOAc (7:3). It was obtained as a white solid; yield: 390 mg (80%); HPLC retention time 53.0 and 70.0 min; mp 155–157 °C; TLC: Rf = 0.31 (hexane/EtOAc, 6:4); ATR-FTIR νmax 1738 (C=O ester), 1700 (C=O lactam), 1265, 1204, 1176, 770, 696 cm−1; 1H NMR (CDCl3, 750 MHz) δ 7.80 (1H, d, J = 7.5 Hz, H-6), 7.76 (1H, d, J = 7.7 Hz, H-9), 7.59 (1H, dt, J = 7.5, 1.2 Hz, H-7), 7.53 (1H, td, J = 7.5, 1.0 Hz, H-8), 7.30 (4H, m, H-Ar), 7.25 (1H, m, H-Ar), 3.75 (3H, s, OCH3), 3.66 (1H, d, J = 14.1 Hz, CHHPh diastereotopic proton), 3.41 (1H, dd, J = 12.5, 6.7 Hz, Ha-1), 3.25 (1H, d, J = 14.1 Hz, CHHPh diastereotopic proton), 3.08 (1H, t, J = 13.0 Hz, Hb-2), 2.53 (1H, dd, J = 13.4, 6.7 Hz, Hc-2), 1.50 (1H, br s, OH); 13C NMR (CDCl3, 188.6 MHz) δ 173.5 (C, HOC=O), 172.6 (C, MeOC=O), 169.4 (C, NC=O), 145.5 (C, C-9a), 135.8 (C, benzyl), 133.4 (CH, C-8), 131.6 (C, C-5a), 131.5 (2CHortho, benzyl), 130.2 (CH, C-7), 128.3 (2CHmeta, benzyl), 127.3 (CHpara, benzyl), 124.3 (CH, C-6), 123.9 (CH, C-9), 94.7 (C, C-9b), 66.1 (C, C-3), 53.0 (CH3, OCH3), 48.8 (CH, C-1), 39.7 (CH2, C-2), 38.1 (CH2, CH2Ph); HRMS (ESI+) m/z calculated for C21H20NO6 382.1291, found 382.1276 (M + H+).
(1R,3R,9bS)- and (1S,3S,9bR)-3-benzyl-9b-hydroxy-5-oxo-2,3,5,9b-tetrahydro-1H-pyrrolo[2,1-a]isoindole-1,3-dicarboxylic acid, [rac-5]
In a 100 mL round-bottomed flask equipped with a reflux condenser, rac-3 (286 mg, 0.7 mmol) and LiOH.H2O (122.5 mg, 2.9 mmol) were dissolved in dry THF (15 mL) before the addition of water (0.7 mL) and the reaction mixture was stirred for 48 h at 50 °C. Subsequently, the solvent was evaporated, and water (10 mL) and EtOAc (10 mL) were added to the obtained mixture. After that, the phases were separated, EtOAc (5 mL) was added to the aqueous phase and the mixture was carefully acidified to pH 1 with aqueous 1 N HCl and then gently extracted with more EtOAc (3 x 5 mL), the organic extracts were combined, washed with brine (10 mL), and dried over anhydrous Na2SO4. Subsequently, the solvent was removed under reduced pressure and the crude product was purified by recrystallization from hexane/EtOAc (7:3). It was obtained as a white solid; yield 240 mg (93%); HPLC: retention time 31.8 min; mp 184-186 °C; TLC: Rf = 0.15 (hexane/EtOAc, 6:4); ATR-FTIR νmax 3054 (O–H), 1760 (C=O carboxylic acid), 1710 (C=O lactam), 1690, 1644, 1404, 767, 700 cm−1; 1H NMR (DMSO-D6, 750 MHz) δ 12.79 (2H, br s, 2CO2H), 7.67 (2H, dd, J = 12.1, 7.4 Hz, H-6 and H-9), 7.57 (1H, td, J = 7.5, 1.3 Hz, H-7), 7.52 (1H, td, J = 7.5, 1.1 Hz, H-8), 7.17 (2H, m, H-Ar), 7.13 (3H, m, H-Ar), 6.70 (1H, br s, OH), 3.72 (1H, d, J = 13.5 Hz, CHHPh diastereotopic proton), 3.44 (1H, d, J = 13.5 Hz, CHHPh diastereotopic proton), 3.10 (1H, t, J = 12.8 Hz, Hb-2), 2.53 (1H, dd, J = 13.2, 7.3 Hz, Hc-1), 1.92 (1H, dd, J = 12.4, 7.3 Hz, Ha-2); 13C NMR (DMSO-D6, 188.6 MHz) δ 174.2 (C, HO=O), 170.3 (C, HO=O), 166.0 (C, NC=O), 145.3 (C, C-9a), 136.0 (C, benzyl), 133.8 (CH, C-8), 132.4 (C, C-5a), 130.7 (2CHortho, benzyl), 129.8 (CH, C-7), 128.3 (2CHmeta, benzyl), 127.1 (CHpara, benzyl), 124.3 (CH, C-6), 122.5 (CH, C-9), 95.8 (C, C-9b), 65.8 (C, C-3), 48.3 (CH, C-1), 37.8 (CH2, CH2Ph), 36.6 (CH2, C-2); HRMS (ESI+) m/z calculated for C20H17NO6Na 390.0954, found 390.0948 (M + Na+).
1-Ethyl 3-methyl (RS)-3-benzyl-5-oxo-2,5-dihydro-3H-pyrrolo[2,1-a]isoindole-1,3-dicarboxylate, [rac-6]
In a 100 mL round-bottomed flask containing a magnetic stirrer, rac-3 (4.1 g, 10 mmol) was dissolved in DCM (40 mL) before the addition of H2SO4 (1.3 mL) and the reaction mixture was stirred at room temperature for 30 min. Subsequently, water (25 mL) was added, and the product was extracted with DCM (3 × 15 mL), and the organic extracts were combined, washed with brine (15 mL), and dried over anhydrous Na2SO4. Subsequently, the solvent was removed under reduced pressure and the crude product was purified by recrystallization from methanol. It was obtained as a white solid; yield 3.8 g (98%); HPLC retention time 11.9 and 20.1 min; mp 127-128 °C; TLC: Rf = 0.46 (hexane/EtOAc, 7:3); ATR-FTIR νmax 1748 (C=O ester), 1699 (C=O lactam), 1650 (C=C), 1229, 1211, 1168, 1103 cm−1; 1H NMR (CDCl3, 400 MHz) δ 8.40 (1H, m, H-9), 7.91 (1H, m, H-6), 7.61 (2H, dd, J = 5.7, 3.1 Hz, H-7 and C-8), 7.17 (5H, m, H-Ar), 4.23 (2H, m, Hz, OCH2CH3), 3.96 (1H, d, J = 14.2 Hz, CHHPh diastereotopic proton), 3.82 (3H, s, OCH3), 3.53 (1H, d, J = 17.7 Hz, CHH-2 diastereotopic proton), 3.43 (1H, d, J = 17.7 Hz, CHH-2 diastereotopic proton), 3.34 (1H, d, J = 14.1 Hz, CHHPh diastereotopic proton), 1.33 (3H, t, J = 7.1 Hz, OCH2CH3); 13C NMR (CDCl3, 101 MHz) δ 172.2 (C, MeOC=O), 164.2 (C, EtOC=O), 163.9 (C, NC=O), 148.6 (C, C-9b), 135.8 (C, benzyl), 134.5 (C, C-9a), 132.4 (CH, C-8), 131.4 (CH, C-7), 130.6 (2CHortho, benzyl), 129.3 (C, C-5a), 128.5 (2CHmeta, benzyl), 127.2 (CHpara, benzyl), 126.7 (CH, C-9), 123.7 (CH, C-6), 107.5 (CH, C-1), 66.7 (C, C-3), 60.8 (CH2, OCH2CH3), 53.3 (CH3, OCH3), 43.7 (CH2, C-2), 38.3 (CH2, CH2Ph), 14.4 (CH3, OCH2CH3); HRMS (ESI+) m/z calculated for C23H22NO5 392.1498, found 392.1492 (M + H+).
(RS)-3-benzyl-1-(ethoxycarbonyl)-5-oxo-2,5-dihydro-3H-pyrrolo[2,1-a]isoindole-3-carboxylic acid, [rac-7]
A 100 mL round-bottomed flask equipped with a reflux condenser, rac-6 (1.0 g, 2.5 mmol) and LiOH.H2O (427.9 mg, 10.2 mmol) were dissolved in dry THF (30 mL) before the addition of water (0.7 mL) and the reaction mixture was stirred for 48 h at 50 °C. Subsequently, the solvent was evaporated, and water (10 mL) and EtOAc (10 mL) were added to the obtained mixture. After that, the phases were separated, t EtOAc (5 mL) was addedo the aqueous phase and the mixture was carefully acidified to pH 1 with aqueous 1 N HCl and then gently extracted with more EtOAc (3 x 5 mL), and the organic extracts were combined, washed with brine (10 mL), and dried over anhydrous Na2SO4. Subsequently, the solvent was removed under reduced pressure and the crude product was purified by recrystallization from hexane/EtOAc (7:3). It was obtained as a white solid; yield 249 mg (87%); HPLC retention time 14.8 and 22.9 min; mp 160–162 °C; TLC: Rf = 0.46 (hexane/EtOAc, 6:4); ATR-FTIR νmax 1745(C=O ester), 1674, 1645 (C=C), 1257, 1126, 780, 747, 701 cm-1; 1H NMR (CDCl3, 750 MHz) δ 8.40 (1H, m, H-9), 7.90 (1H, m, H-6), 7.62 (2H, pq, J = 7.4, 1.4 Hz, H-7 and H-8), 7.16 (5H, m, H-Ar), 5.12 (1H, br s, COOH), 4.25 (2H, m, OCH2CH3), 3.82 (1H, dd, J = 14.2, 3.0 Hz, CHHPh diastereotopic proton), 3.71 (1H, dd, J = 17.9, 2.9 Hz, CHH-2 diastereotopic proton), 3.52 (1H, dd, J = 17.9, 2.5 Hz, CHH-2 diastereotopic proton), 3.32 (1H, d, J = 14.1 Hz, CHHPh diastereotopic proton), 1.33 (3H, t, J = 7.1 Hz, OCH2CH3); 13C NMR (CDCl3, 188.6 MHz) δ 173.4 (C, HO=O), 165.8 (C, EtOC=O), 163.6 (C, N=O), 147.3 (C, C-9b), 135.0 (C, benzyl), 133.7 (C, C-9a), 132.9 (CH, C-5a), 131.5 (CH, C-8), 130.2 (2CHortho, benzyl), 129.1 (CH, C-6), 128.5 (2CHmeta, benzyl), 127.5 (CHpara, benzyl), 126.9 (CH-7), 123.9 (CH, C-9), 109.8 (C, C-1), 68.0 (C, C-3), 61.0 (CH2, OCH2CH3), 43.4 (CH2, C-2), 39.4 (CH2, CH2Ph), 14.3 (CH3, OCH2CH3); HRMS (ESI+) m/z calculated for C22H20NO5 378.1341, found 378.1336 (M + H+).
(RS)-3-benzyl-5-oxo-2,5-dihydro-3H-pyrrolo[2,1-a]isoindole-1,3-dicarboxylic acid [rac-8]
A 250 mL round-bottomed flask equipped with a reflux condenser, rac-6 (300 mg, 0.7 mmol) was dissolved in methanol (30 mL), and NaOH (121.6 mg, 3.0 mmol) was added before the addition of water (3 mL), and the reaction mixture was stirred for 24 h at 58 °C. Subsequently, the solvent was evaporated, water (10 mL) was added, the obtained mixture was carefully acidified to pH 1 with aqueous 1 N HCl and extracted with EtOAc (3 × 5 mL), and the organic extracts were combined, washed with brine (10 mL) and dried over anhydrous Na2SO4. Subsequently, the solvent was removed under reduced pressure and the crude product was purified by recrystallization from methanol/water (5:1). It was obtained as a white solid; yield 229 mg (90%); HPLC retention time 28.7 and 37.7 min; mp 231–233 °C (dec.); TLC: Rf = 0.21 (hexane/EtOAc, 6:4); ATR-FTIR νmax 1726 (C=O carboxylic acid), 1693 (C=O lactam), 1643 (C=C), 1260, 1211, 1181, 748, 699 cm−1; 1H NMR (DMSO-D6, 750 MHz) δ 13.25 (2H, br s, 2CO2H), 8.32 (1H, d, J = 7.5 Hz, H-9), 7.85 (1H, d, J = 7.4 Hz, H-6), 7.71 (2H, m, H-7 and H-8), 7.16 (4H, m, H-Ar), 7.14 (1H, dd, J = 5.6, 2.9 Hz, H-Ar), 3.68 (1H, d, J = 14.1 Hz, CHHPh diastereotopic proton), 3.43 (1H, d, J = 17.8 Hz, CHH-2 diastereotopic proton), 3.33 (1H, d, J = 17.8 Hz, CHH-2 diastereotopic proton), 3.28 (1H, d, J = 14.1 Hz, CHHPh diastereotopic proton); 13C NMR (DMSO-D6, 188.6 MHz) δ 172.7 (C, HOC=O), 165.0 (C, HOC=O), 163.3 (C, NC=O), 147.3 (C, C-9b), 135.4 (C, benzyl), 135.2 (CH, C-9a), 133.0 (CH, C-8), 132.0 (C, C-5a), 130.4 (2CHortho, benzyl), 128.8 (CH, C-6), 128.5 (2CHmeta, benzyl), 127.2 (CHpara, bencyl), 126.5 (CH, C-7), 123.6 (CH, C-9), 108.8 (CH, C-1), 65.9 (C, C-3), 43.9 (CH2, C-2), 37.6 (CH2, CH2Ph); HRMS (ESI+) m/z calculated for C20H16NO5 350.1028, found 350.1023 (M + H+).
1-Ethyl 3-methyl (1R,3R,9bS)- and (1S,3S,9bR)-3-benzyl-5-oxo-2,3,5,9b-tetrahydro-1H-pyrrolo[2,1-a]isoindole-1,3-dicarboxylate [rac-9]
The Parr hydrogenator was rinsed with EtOAc (10 mL), (rac-6) (300 mg, 0.7 mmol), and Pearlman’s catalyst (33 mg, 20% Pd(OH)2/C) was added. The hydrogenation reactor was purged three times with nitrogen (to 689.4 kPa), and then three times with hydrogen (to 689.4 kPa). The hydrogenation reactor was finally pressurized to 689.4 kPa of hydrogen and heated with stirring at 60 °C for 24 h. Completion of the reaction was confirmed by TLC and the catalyst was filtered. Subsequently, the solvent was removed under reduced pressure and the crude product was purified by column chromatography (hexane/EtOAc, 7:3). It was obtained as a white solid; yield 156 mg (52%); HPLC retention time 42.9 and 44.6 min; mp 124–126 °C; TLC: Rf = 0.19 (hexane/EtOAc, 7:3); ATR-FTIR νmax 1727 (C=O ester), 1691 (C=O lactam), 1175, 742 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.84 (1H, m, H-6), 7.50 (1H, td, J = 7.4, 1.4 Hz, H-8), 7.45 (1H, td, J = 7.4, 1.3 Hz, H-7), 7.33 (1H, dd, J = 7.1, 1.2 Hz, H-9), 7.20 (5H, m, H-Ar), 4.16 (1H, d, J = 6.7 Hz, Hd-9b), 3.8 (2H, m, OCH2CH3), 3.70 (1H, d, J = 14.0 Hz, CHHPh), 3.70 (3H, s, OCH3), 3.22 (1H, d, J = 13.9 Hz, CHHPh), 3.06 (2H, m, Ha-1 and Hb-2), 2.70 (1H, dd, J = 14.1, 7.9 Hz, Hc-2), 0.79 (3H, t, J = 7.2 Hz, OCH2CH3); 13C NMR (CDCl3, 100.5 MHz) δ 172.0 (C, MeOC=O), 170.0 (C, EtOC=O), 167.7 (C, NC=O), 143.9 (C, C-9a), 135.6 (C, benzyl), 134.6 (C, C-5a), 131.3 (2CHortho, benzyl), 131.1 (CH, C-8), 128.3 (2CHmeta, benzyl), 127.9 (CH, C-7), 126.9 (CHpara, benzyl), 124.0 (CH, C-6), 123.7 (CH, C-9), 66.3 (CH, C-9b), 64.4 (C, C-3), 60.7 (CH2, OCH2CH3), 52.6 (CH3, OCH3), 42.8 (CH, C-1), 40.7 (CH2, C-2), 40.2 (CH2, CH2Ph), 13.4 (CH3, OCH2CH3); HRMS (ESI+) m/z calculated for C23H24NO5 394.1654, found 394.1649 (M + H+).
Solutions
Solutions were prepared as follows: (1) 0.1 M phosphate buffer solution (PBS), pH 8.0 (dissolve 7.48 g of K2HPO4 + 0.916 g of KH2PO4 in distilled water, adjust pH to 8.0, and reach a final volume of 500 mL); (2) 4.2 M sodium hydroxide solution (dissolve 16.96 g NaOH in distilled water, final volume 100 mL); (3) 2.4 M hydroxylamine hydrochloride solution (dissolve 16.84 g NH2OH·HCl in distilled water, final volume 100 mL); (4) alkaline hydroxylamine solution (mix equal volumes of 2 and 3 solutions, prepare it immediately before use); (5) 0.75 M ferric chloride solution (dissolve 20.31 g FeCl3·6H2O in HCl solution 7.5 M, final volume 100 mL); 6) 256 mM acetylcholine stock solution (dissolve 50 mg ACh iodide in 0.695 mL of solution 1) and prepare dilutions for the standard curve of ACh (48, 32, 24, 16, 12, 8, 6, and 4 mM); 7) 1 U/mL stock solution of Electrophorus electricus AChE dissolved according to the supplier’s instructions (Sigma Chemical C3389). Test compounds and reference drugs (Neostigmine and Galantamine) were prepared at appropriate concentrations and dissolved in DMSO (<1%).
Acetylcholinesterase activity assay
In a 96-well microplate in different lines, the reactions were set in place for the activity assay as follows: (1) Blank: 160 μL of PBS; (2) standard curve of ACh: 20 μL of each dilution of ACh (48, 32, 24, 16, 12, 8, 6, and 4 mM) were placed in different wells and 140 μL of PBS was added; 3) AChE curve: 20 μL of ACh + 20 μL of AChE + 120 μL of PBS; 4) Inhibition curve: 20 μL of ACh + 20 μL of AChE + 20 μL of test compound or reference drugs + 100 μL of PBS. The microplate was incubated at 37 °C for 20 min in a water bath. After that time, the reaction was stopped by adding 40 μL of alkaline hydroxylamine solution. The colorimetric reaction was developed through the addition of 100 μL of FeCl3 and mixed for 30 s; then, the optical density (OD) was read in a wavelength of 540 nm (Accuris MR9600) [25, 30]. The enzymatic assays were developed in triplicate.
Molecular docking
The molecular approach experiment was designed to provide insights into the in vitro results. Electrophorus electricus Acetylcholinesterase (EeAChE; Protein Data Bank code:1C2O) was used as target [31], as well as the proposed molecules. Gaussian 16 and GaussView 6 were used for determining the lower energy conformation [32], with the semi-empirical PM6 method, CCPM solvation in water, and the ionization state of molecules at physiological pH (7.4). Then, the number of rotational bonds, the degree of torsional freedom, and Gasteiger charges for each ligand were established. In addition, Kollman partial charges and polar hydrogens were assigned for the receptor by using AutoDock tools 1.5.6 and Raccoon [33, 34]. The amino acids of the active sites of the EeAChE were obtained from a previous report [26]. The latter aided us to design the grid box: the grid center was established at X = 42.27, Y = 66.809, and Z = −81.47, with a size of 60 Å x 60 Å x 60 Å, and a mesh separation of 0.375 Å. Docking conditions were established using a hybrid Lamarckian genetic algorithm, with 100 randomly placed individuals as the initial population. Finally, the calculations were carried out in Linux Fedora 22 as the operating system and AutoDock4 [35]. Gibbs free energy (ΔG), dissociation constant (Kd), the negative logarithm of the dissociation constant (pKd), interactions, distance, and binding type were obtained with AutoDock Tools 1.5.6 and BIOVIA Discovery Studio 2020 [34]. The validation of these experiments was performed by comparing the results with other well-known AChE inhibitors (Donepezil, Galantamine, Rivastigmine, and Tacrine), as reported in previous works of the group [25].
Statistical analysis
The data are expressed as the mean ± 95% confidence intervals for all of the assays. Non-linear regression analysis and Lineweaver–Burk plot were used to obtain Ki and the type of inhibition (GraphPad Prism statistical software). The kinetic module was employed to obtain the corresponding values.
References
Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol 2018;25:59–70. https://doi.org/10.1111/ene.13439
World Health Organization Global Action Plan on the Public Health Response to Dementia 2017–2025. Geneva World Heal. Organ. 2017, License: C, 52
Association A. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019;2019:321–87. https://doi.org/10.1016/j.jalz.2019.01.010
Silva MVF, Loures CDMG, Alves LCV, De Souza LC, Borges KBG, Carvalho MDG. Alzheimer’s disease: risk factors and potentially protective measures. J Biomed Sci 2019;26:33 https://doi.org/10.1186/s12929-019-0524-y
Cortes-Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, et al. Fibrinogen and β-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron. 2010;66:695–709. https://doi.org/10.1016/j.neuron.2010.05.014
Adav SS, Sze SK. Insight of brain degenerative protein modifications in the pathology of neurodegeneration and dementia by proteomic profiling. Mol Brain. 2016;9:1–22. https://doi.org/10.1186/s13041-016-0272-9
Soria Lopez, JA; González, HM; Léger, GC Alzheimer’s disease. In Handbook of Clinical Neurology; Elsevier B.V., 2019; Vol. 167, pp. 231-55. https://doi.org/10.1016/B978-0-12-804766-8.00013-3
Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet 2010;19:R12–R20. https://doi.org/10.1093/hmg/ddq160
Voytko M, Lou, Olton DS, Richardson RT, Gorman LK, Tobin JR, et al. Erratum for Voytko et Al., Basal Forebrain Lesions in Monkeys Disrupt Attention but Not Learning and Memory. J Neurosci. 1995;15:np.2-np https://doi.org/10.1523/JNEUROSCI.15-03-j0001.1995
H. Ferreira-Vieira T, M. Guimaraes I, R. Silva F, M. Ribeiro F. Alzheimer’s disease: targeting the cholinergic system. Curr Neuropharmacol 2016;14:101–15. https://doi.org/10.2174/1570159x13666150716165726
Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol 1981;10:122–6. https://doi.org/10.1002/ana.410100203
Anand P, Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res 2013;36:375–99. https://doi.org/10.1007/s12272-013-0036-3
Hitzeman N. Cholinesterase Inhibitors for Alzheimer’s Disease. Am Fam Physician. 2006;74:747–9. https://doi.org/10.1093/med/9780199377527.003.0001
T M, S E, P A. Behavioral side effects in rats treated with acetylcholinesterase inhibitors suggested used as prophylactics against nerve agents. Pharmacol Biochem Behav. 2010;95:338–43. https://doi.org/10.1016/J.PBB.2010.02.010
Campbell NL, Perkins AJ, Gao S, Skaar TC, Li L, Hendrie HC, et al. Adherence and tolerability of Alzheimer’s disease medications: a pragmatic randomized trial. J Am Geriatr Soc 2017;65:1497–504. https://doi.org/10.1111/jgs.14827
Miyazawa M, Takahashi T, Horibe I, Ishikawa R. Two new aromatic compounds and a new D-arabinitol ester from the mushroom hericium erinaceum. Tetrahedron. 2012;68:2007–10. https://doi.org/10.1016/j.tet.2011.11.068
Guillaumel J, Léonce S, Pierré A, Renard P, Pfeiffer B, Arimondo PB, et al. Synthesis and biological activity of 6H-Isoindolo[2,1-a]Indol-6-Ones, analogues of batracylin, and related compounds. Eur J Med Chem 2006;41:379–86. https://doi.org/10.1016/j.ejmech.2005.10.008
Luci DK, Lawson EC, Ghosh S, Kinney WA, Smith CE, Qi J, et al. Generation of novel, potent urotensin-II receptor antagonists by alkylation-cyclization of isoindolinone C3-carbanions. Tetrahedron Lett. 2009;50:4958–61. https://doi.org/10.1016/j.tetlet.2009.06.025
Chagraoui A, Thibaut F, Skiba M, Thuillez C, Bourin M. 5-HT2C receptors in psychiatric disorders: a review. Prog Neuro-Psychopharmacol Biol Psychiatry. 2016;66:120–35. https://doi.org/10.1016/j.pnpbp.2015.12.006
Gong YX, Zhu QF, Zhong JQ, Liu LF, Li XF, Zheng XH, et al. Design, synthesis and biological evaluation of Novel [1,3] Dioxolo[4,5-Fjisoindolone derivatives. Yaoxue Xuebao. 2015;50:191–8.
Reyes A, Regla I, Fragoso MC, Vallejo LA, Demare P, Jiménez-Vázquez HA, et al. Stereoselective tandem michael-intramolecular cyclization approach to functionalized pyrroloisoindolones. Tetrahedron. 1999;55:11187–202. https://doi.org/10.1016/S0040-4020(99)00645-6
Sánchez-Antonio O, González-Olvera R, Aguilera-Cruz A, Reyes-Ramírez A, Juaristi E. Synthesis of novel isoindolone derivatives via cascade reactions. contrasting diastereoselectivity under solution-phase vis-a-vis solvent-free ball-milling reaction conditions. Tetrahedron. 2019;75:130594 https://doi.org/10.1016/j.tet.2019.130594
Li Z, Yang X, Tsumori N, Liu Z, Himeda Y, Autrey T, et al. Tandem nitrogen functionalization of porous carbon: toward immobilizing highly active palladium nanoclusters for dehydrogenation of formic acid. ACS Catal. 2017;7:2720–4. https://doi.org/10.1021/acscatal.7b00053
Ruiz-Maciel O, Padilla-Martínez II, Sánchez-Labastida LA, Soriano-Ursúa MA, Andrade-Jorge E, Trujillo-Ferrara JG. Inhibitory activity on cholinesterases produced by aryl-phthalimide derivatives: green synthesis, in silico and in vitro evaluation. Med Chem Res 2020;29:1030–40. https://doi.org/10.1007/s00044-020-02543-2
Andrade-Jorge E, Sánchez-Labastida LA, Soriano-Ursúa MA, Guevara-Salazar JA, Trujillo-Ferrara JG. Isoindolines/Isoindoline-1,3-diones as AChE inhibitors against alzheimer’s disease, evaluated by an improved ultra-micro assay. Med Chem Res 2018;27:2187–98. https://doi.org/10.1007/s00044-018-2226-5
Altamirano‐Espino JA, Sánchez‐Labastida LA, Martínez‐Archundia M, Andrade‐Jorge E, Trujillo‐Ferrara JG. Acetylcholinesterase inhibition (potential anti‐Alzheimer effects) by aminobenzoic acid derivatives: synthesis, in vitro and in silico evaluation. ChemistrySelect. 2020;5:14177–82. https://doi.org/10.1002/slct.202003471
Zeng Q, Liu Z, Li B, Wang F. Mild and effective n-phthaloylation of amino acids. Amino Acids. 2004;27:183–6. https://doi.org/10.1007/s00726-004-0109-1
Bose AK, Greer F, Price CC. A procedure for phthaloylation under mild conditions. J Org Chem 1958;23:1335–8. https://doi.org/10.1021/jo01103a025
Peterson PE, Niemann C. Some reactions of α-phthalimidonitriles including those leading to the synthesis of α-aminoamidoximes and α-aminothioamides 1. J Am Chem Soc 1957;79:1389–95. https://doi.org/10.1021/ja01563a034
Bonting SL, Featherstone RM. Ultramicro assay of the cholinesterases. Arch Biochem Biophys 1956;61:89–98. https://doi.org/10.1016/0003-9861(56)90319-8
Bourne Y, Grassi J, Bougis PE, Marchot P. Conformational flexibility of the acetylcholinesterase tetramer suggested by X-ray crystallography. J Biol Chem 1999;274:30370–6. https://doi.org/10.1074/jbc.274.43.30370
Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; et al. Gaussian 09, Revision A. 1; Gaussian; 2009
Morris GM, Ruth H, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91. https://doi.org/10.1002/jcc.21256
Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, Olson AJ. Virtual screening with autodock: theory and practice. Expert Opin Drug Disco. 2010;5:597–607. https://doi.org/10.1517/17460441.2010.484460
Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; et al. Gaussian 16, Revision A.03, 2016
Acknowledgements
E.A.J. received a postdoctoral fellowship from DGAPA of the Universidad Nacional Autónoma de México (UNAM). This work was supported by PAPIIT IN218721 (to AR-R), PAPIIT IN223519 (to RV-M), and PAPIIT IN226819 (to IAGO) DGAPA from the UNAM. Additionally, this work was supported by CONACYT-México, SIP-projects (20210712 and 20212107) from the Instituto Politécnico Nacional IPN-ESM.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
About this article

Cite this article
Andrade-Jorge, E., Rivera-Sánchez, F., Rodríguez, J.E. et al. Isoindolone derivatives as novel potential anti-Alzheimer’s candidates: synthesis, in silico, and AChE inhibitory activity evaluation. Med Chem Res 31, 851–866 (2022). https://doi.org/10.1007/s00044-022-02884-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-022-02884-0