
Abstract
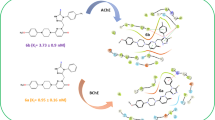
Alzheimer's disease is a complex and progressive form of dementia. Its treatment relies on the behavioral and cognitive symptoms of an individual. Inhibiting acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) is a primary strategy used for treating Alzheimer's disease. Pyrazoline is a well-recognized nitrogen-containing five-membered heterocyclic skeleton. The unique structure of pyrazoline gives it a spatial configuration that leads to the multiple substitution pattern and advanced pharmacological properties. In an attempt to identify potent acetylcholine (AChE) and butyrylcholinesterase (BChE) inhibitors, new pyrazoline derivatives (IIIa–IIId) were synthesized using conventional method. The synthesised compounds were purified by column chromatography and were analysed using LCMS, 1HNMR, 13CNMR, and Mass Spectroscopic HR-MS techniques. The derivatives underwent initial screening for in-vitro antioxidant potential. The most potent compound, IIIa, showed IC50 values of 22.15 and 25.09 nM against AChE and BChE, respectively. Additionally, in-silico screening with AutoDock tools indicated that IIIa had promising binding affinities to the targets, with a binding energy (− 8.9 kcal/mol) comparable to donepezil (− 10.6 kcal/mol). The binding pattern of IIIa to AChE's active site justified its in-vitro inhibitory activity. These findings suggest that compound IIIa has potential as a new therapeutic agent for Alzheimer’s disease and is suitable for pre-clinical evaluation.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer's disease (AD) is one of the major types of dementia predominant in old population (Amin et al. 2021). Primarily it is a neurodegenerative disease in which there is a loss of cognitive abilities along with tremendous behavioral change, loss of memory and finally, death (Li et al. 2017). It is reported that almost 50 million world population is suffering from AD which doubles every 5 years and is expected to reach 152 million by 2050 (Breijyeh and Karaman 2020; Malik, et al. 2023). The pathophysiology of Alzheimer's disease is quite complicated. There are two major hypotheses regarding AD disease; the amyloid hypothesis and the cholinergic hypothesis. Amongst the identified factors which contribute to the development of the disease are a lower level of acetylcholine (AChE), an imbalance of biomarkers, and oxidative stress (Gawad, et al. 2010). The cholinergic hypothesis suggests that due to the loss of cholinergic function in CNS, there is a decrease of cognitive behaviour which ultimately leads to memory disturbance. By inhibiting the acetylcholinesterase enzyme (AChE), the cholinergic function could be restored which then results in the elevated level of acetylcholine (ACh) (Bohnen, et al. 2018). Two different types of cholinesterase enzymes are involved in the degradation of ACh, namely acetylcholine esterase (AChE) and butyrylcholinesterase (BChE). Although acetylcholine is a major enzyme which garnered great attention as a major target for AD, however, butyrylcholine esterase (BChE) is also under consideration due to its main role in the regulation of AChE levels in AD patients (Adeowo et al. 2022).
Pyrazoline is an important nucleus in the field of organic and medicinal chemistry, containing a five-membered nitrogen ring (Kanwal, et al. 2022; Mathew, et al. 2013). Pyrazoline is also emerging as a potent entity in the discipline of drug design and development (Song et al. 2020). Pyrazoline has one endocyclic double bonds and two adjacent nitrogen atoms. Among its chemical forms include (Fig. 1), 1-pyrazoline, 2-pyrazoline, and 3-pyrazoline, the most studied being 2-pyrazoline owing to its stability compared to the other forms (Haider, et al. 2022). The 2-pyrazoline nucleus is the part of many active drugs including, antipyrine, ramifenazone, ibipinabant, and axitinib (Wang, et al. 2013). Pyrazolines are used as an isostere of heterocyclic rings including imidazole, thiazole, isoxazole, tetrazole, oxazole, triazole and more due to their strength to alter the physicochemical properties and pharmacological profile (Yang, et al. 2013; Rathore et al. 2014). First Pyrazoline was synthesized by Knoevenagel and Fisher reactions by α, β-unsaturated ketones and hydrazine cyclization reaction followed by other improved methods (Farooq and Ngaini 2020; Ozgun et al. 2019; James, et al. 2018; Yamali, et al. 2020).

Chemical structures of Pyrazoline Scaffold
Pyrazoline derivatives have been reported to exhibit a versatile spectrum of biological activities including, anti-cancer (Rathore et al. 2014), anti-convulsant (Özdemir, et al. 2007), anti-inflammatory (Kharbanda et al. 2014), anti-tubercular, anti-bacterial (Ali et al. 2007; Joshi, et al. 2016), anti-depressant (Prasad, et al. 2005), anti-malarial (Acharya, et al. 2010), anti-fungal (Altıntop, et al. 2015), anti-cholinesterase (Nehra et al. 2020), monoamine oxidase (MAO) (Gökhan-Kelekçi, et al. 2009), carbonic anhydrase (CA) inhibition (Khloya et al. 2015) and neuroprotective activity (Kamogawa et al. 2014). The research described in this paper outlines synthetic strategy, docking analysis, anti-oxidant, acetylcholine esterase (Ach) and butyrylcholinesterase (BChE) inhibitory effect of newly synthesized pyrazoline derivatives.
Experimental
Chemicals and reagents
All building blocks and chemicals were purchased from different suppliers. Di-ketones were purchased from Fluorochem (UK) and nicotinic acid hydrazide (INH) was purchased from Sigma Aldrich (St. Louis, MO, USA). All chemicals were at least 99% pure and of HPLC grade. Reactions were monitored by using Thin Layer Chromatography (TLC) plates (Merck silica gel 60F 254 plates) and were visualized under ultraviolet light (UV) at 254 nm wavelength. Manual column was performed to purify the impure compounds by using silica gel (Merck 9385, 230–400 mesh, ASTM, 40–60 µM). Ethyl acetate was used as a solvent system (mobile phase) in TLC to see the separating profiles of compounds. Bruker AM-300 (Billerica, Massachusetts, UK) was employed to predict the NMR of all synthesized compounds at 400 MHz. These spectra were interpreted by ACD/NMR Processor Academic Edition software. LC–MS was processed on a Water Alliance 2695 HPLC which is coupled with Waters Micro mass ZQ instrument along with a Waters 2996 PDA. Chemical shifts (δH) are expressed in ppm compared to deuterated DMSO, with residual signals δH = 2.5 and 13C δ = 44. Multiplicities of 1HNMR spectra are expressed as s = singlet, d = doublet, t = triplet, m = multiplet. Melting points of all compounds were checked through Digital Kamp apparatus (Sanyo, Osaka, Japan). For the acetylcholine esterase (AChE) and butyrylcholine esterase (BChE) inhibitory assay, an Elisa Colorimetric kit (Catalog # K197-100, Bio Vision) & (Catalog # MBS846801, Bio Vision) was used.
Chemistry
General procedure for the synthesis of (5-substituted -3-methyl-4,5-dihydro-1H-Pyrazol-1-yl) (pyridine-4-yl) methanone (IIIa-IIId)
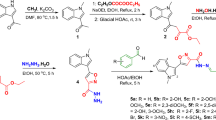
An equimolar quantity of compound I (Isoniazid) (0.1 mol) and 1,3-dicarbonyl (0.1 mol) (II) were taken in a flask and dissolved in ethanol. A few drops of glacial acetic acid (GAA) were used as a catalytic agent to provide an acidic environment. The reaction mixture was refluxed in a water bath. The reaction progress was monitored with the help of TLC (ethyl acetate: pet ether, 1:3 ratio). After the completion of the reaction, the solvent was evaporated using rotary evaporation, and the crude product was purified with the help of column chromatography (dry loading) (Knorr, L.J.B.d.d.c.G. 1883).
(5-ethoxy-5-hydroxy-3-methyl-4,5-dihydro-1Hpyrazol-1-yl)(pyridine-4-yl)methanone (IIIa)
1H NMR: (DMSO-d6, δ ppm): 7.752–8.795 (m, 4H, Ar–H), 1.132 (t, 3H, CH3), 2.229 (s, 3H, CH3) 3.929–4.050(q, 2H, CH2), 3.351–3.429(s, 2H, CH2), 4.683 (s, 1H, OH). 13C NMR (DMSO d6, δ ppm): (14.09, 1C), (25.30, 1C), (50.09, 1C), (61.21, 1C), (121.43, 1C), (121.20, 1C), (123.76, 1C), (149.91, 1C), (149.20, 1C), (151.96, 1C), (162.38, 1C), (169.42, 1C). LC–MS at 5mint run: 3.839 mints, purity: 100%. HR-MS calculated for C12 H15 N3 O3 is 249.1186 [M + H]+, calcd, 250.1181. [M + H]+. (see the supplementary data). COSY NMR (see the supplementary data). Anal. Calculated for C 12 H 15 N 3 O 3: C, 57.82%, H, 6.07%, N, 16.86%, O, 19.26% M.W. 249.27 Yield 55% m.p. 130–140 °C. Rf value 0.91. Colourless needles.
5-ethoxy-4-fluoro-5-hydroxy-3-methyl-4,5-dihydro-1H-pyrazol-1-yl)(pyridine-4-yl)methanone (IIIb)
1H NMR: (DMSO-d6, δ ppm): 6.182–9.193 (m, 4H, Ar–H), 1.661–1.694 (t, 3H, CH3), 2.944 (s, 3H, CH3), 3.780(s, 1H, CH), 4.669–4.693 (q, 2H, CH2), 11.506 (s, 1H, OH). 13C NMR (DMSO d6, δ ppm): (14.09,1C), (15.50, 1C), (50.09, 1C), (108.30,1C), (121.43,1C), (121.20,1C), (123.76, 1C), (149.91,1C), (149.20,1C), (151.96,1C), (162.38,1C), (169.42,1C). LC–MS at 5mint run: 4.314 mints, purity: 100%. HR-MS calculated for C12 H14 F N3 O3 is 267.102 [M + H]+, calcd, 268.109. [M + H]+. (see the supplementary data). COSY NMR (see the supplementary data). Anal. Calculated for C 12 H 14 F N 3 O 3: C, 53.93%, H, 5.82%, F, 7.11, N, 15.72%, O, 17.96% M.W. 267.26 Yield 52% m.p. 132–145 °C. Rf value 0.98. Colourless needles.
3-ethyl-4-fluoro-5-hydroxy-5-methoxy-4,5-dihydro-1H-pyrazol-1-yl)(pyridine-4-yl)methanone (IIIc)
1H NMR: (DMSO-d6, δ ppm): 7.813–8.816 (m, 4H, Ar–H), 1.066 (t, 3H, CH3), 3.360 (s, 3H, CH3), 3.789(s, 1H, CH), 1.124–1.171 (q, 2H, CH2), 10.964 (s, 1H, OH). 13C NMR (DMSO d6, δ ppm): (14.09, 1C), (18.20, 1C), (50.09, 1C), (105.43, 1C), (121.43, 1C), (121.20, 1C), (123.76, 1C), (149.91, 1C), (149.20,1C), (151.96, 1C), (162.38, 1C), (169.42, 1C). LC–MS at 5mint run: 4.156 mints, purity: 100%. HR-MS calculated for C12 H14 F N3 O3 is 267.102 [M + H]+, calcd, 268.109. [M + H]+. (see the supplementary data). COSY NMR (see the supplementary data). Anal. Calculated for C 12 H 14 F N 3 O 3: C, 53.93%, H, 5.28%, F, 7.11, N, 15.72%, O, 17.96% M.W. 267.26 Yield 51% m.p. 132–145 °C. Rf value 0.97. light brown needles.
(4-flouro-5-hydroxy-3,5-dimethoxy-4,5-dihydro-1-hydropyrazol-1-yl)(pyridine-4-yl)methanone (IIId)
1H NMR: (DMSO-d6, δ ppm): 8.191–9.004 (m, 4H, Ar–H), 1.225 (s, 3H, CH3), 4.244(s, 1H, CH), 5.595 (s, 1H, OH). 13C NMR (DMSO d6, δ ppm): (62.48, 1C), (84.83, 1C), (86.80, 1C), (123.90, 1C), (141.59, 1C), (147.49, 1C), (163.49, 1C), (164.90, 1C), (166.14, 1C), (166.38, 1C), (165.62, 1C). LC–MS at 5 mint run: 4.2 mints, purity: 100%. COSY NMR (see the supplementary data). Anal. Calculated for C 11 H 12 F N 3 O 4: C, 49.07%, H, 4.49%, F, 7.06, N, 15.61%, O, 23.77% M.W. 269.229 Yield 42% m.p. 130–147 °C. Rf value 0.93. light brown needles.
(3,5-diethoxy-4-fluoro-5-hydroxy-4,5-dihydro-1H-pyrazole-1-yl) (pyridine-4-yl) methanone (IIIe)
1H NMR: (DMSO-d6, δ ppm): 8.051–8.960 (m, 4H, Ar–H), 1.211–1.247 (t, 3H, CH3), 1.339–1.373 (t, 3H, CH3), 5.588 (s, 1H, OH), 5.705 (s, 1H, CH), 4.214–4.265 (q, 3H, –CH2), 4.369–4.422 (q, 2H, –CH2). 13C NMR (DMSO d6, δ ppm): (14.29, 2C), (62.48, 1C), (84.83, 1C), (86.80, 1C), (123.90, 1C), (141.59, 1C), (147.49, 1C), (163.49, 1C), (164.90, 1C), (166.14, 1C), (166.38, 1C), (165.62, 1C). LC–MS at 5mint run: 4.9 mints, purity: 100%. COSY NMR (see the supplementary data). Anal. Calculated for C 13 H 16 F N 3 O 4: C, 52.52%, H, 5.42%, F, 6.39, N, 14.13%, O, 21.53% M.W. 297.282 Yield 41% m.p. 133–145 °C. Rf value 0.96. dark brown needles.
Biological study
diphenylpicrylhydrazide (DPPH) free radical scavenging activity
This assay was performed by using the published protocol with minimal changes (Bibi et al. 2011). Stock solution of PDDH was prepared in 100 ml of methanol by dissolving 9.2 mg of DPPH. Vitamin C (ascorbic acid) was used as standard drug and its stock was prepared by mixing 1 mg/ml in DMSO solvent. 4 mg from each compound was dissolved in 1 ml of DMSO to prepare final stock solution. All samples were tested for DPPH potential and it was confirmed through the change of purple colour into yellow in DMSO solution. In a 96-well microplate, 10 µl of the test sample and 190 µl of DPPH reagent were added to prepare the final volume of 200 µg/ml of the sample. The sample plate was incubated in darkness for 60 min at 37 °C. DMSO and ascorbic acid were used as negative and positive control respectively. Microplate reader was used to measure the absorbance of the samples at 517 nm (Miana, et al. 2022). A decrease in absorbance indicated the presence of Free Radical Scavenging potential of the test compounds. All samples and standards were diluted three folds to perform this assay. The percentage of radical scavenging activity was determined using the following formula.
%age scavenging activity = (1-Abs/Abc) × 100).
Abs = absorbance of sample.
Abc = absorbance of negative control.
The serial dilutions were as: 40, 20 and 10 µg/ml.
In-vitro acetylcholinesterase (AChE) inhibitory activity
Based on the antioxidant results, the most active compound (IIIa) was evaluated for its ability to inhibit the acetylcholine esterase enzyme. This test was performed using the AChE screening colorimetric kit (catalogue # K197-100, BioVision). This kit is in a 96-well form and used for rapid screening of AChE inhibitors. The inhibitor was diluted 100X or higher concentration and diluted to 20X using AChE assay buffer. From the dilution of 20X, 10µL of test inhibitor was added into the designated 96-well plate. 412 nm was employed to measure the absorbance (OD) for 40mints under kinetic mode. DMSO was used as control. GraphPad Prism program package (Graph Pad Software) was used to calculate the non-linear regression analysis of the response-concentration (log) curve. This method was employed to finally calculate the IC50 values (Eissa, et al. 2022).
In-vitro butyrylcholinesterase (BChE) inhibitory activity
The most active analogue (IIIa) was further investigated for its in-vitro butyrylcholinesterase inhibitory effect using the BChE activity colorimetric kit (Catalogue # MBS846801, Biovision). 96-well plate assay kit was used for this test. 120-fold dilution for BChE was prepared and it was kept on ice. In each well containing test samples, 100 µL of diluted BChE substrate was merged. BChE was used as positive control. In each well BChE assay buffer of 100 µL was added. Multiwall spectrophotometer was used to measure the absorbance at 412 nm for 30 mints in kinetic mode. IC50 values were calculated using Graph Pad Prism program package (Graph Pad Software) through non-linear regression analysis (Eissa, et al. 2022).
In-silico analysis
To further understand the inhibitory effects of all compounds on AChE enzyme, all compounds were docked against the AChE enzyme using AutoDock Vina version 4.2.6 to assess their potential binding energies at the binding site of the targeted protein in San Diego, CA, USA (Miana, et al. 2022; Herowati and G.P.J.Q.S.-a.R. Widodo 2017). The X-ray crystal structure of the AChE enzyme (PDB ID: 4PQE) was downloaded from the protein data bank (http://www.rcsb.org/pdb) (Volkamer et al. 2012). The DoGi Site Scorer (http://bio.tools/dogisitescorer) was used to locate the active binding pockets of the targeted protein. For the purpose of docking, ligand–protein complexes were formulated, and the targeted proteins were cleaned using Biovia Discovery Visualizer (DSV) to remove water and co-crystallized ligands prior to being stored in PDB format. The structures of the synthesized compounds were drawn in ChemSketch, and their mol files were generated. All files were converted into PDB format using Open Babel. AutoDock Tools of version 1.5.6 was also used to create PDBQT files of all ligands and the targeted protein. Furthermore, PyRx, a digital molecular docking software, was employed. The conformational poses and ligand-target interactions were interpreted using Biovia DSV. The best poses of the co-crystallized ligands were compared with the best poses of the re-docked confirmation to validate the docking.
Results and discussion
Chemistry
In this study we synthesized series of new pyrazoline derivatives (IIIa, IIIb, IIIc, IIId, IIIe) by coupling reaction of Isonicotinic hydrazide (INH) using ethanol as solvent with reflux for 7–8 h (method reported in experimental section) (Scheme 1). Pyrazoline based compounds are reported for a wide range of pharmacological actions including antiepileptic, antidepressant, and anti-neurodegenerative effects (Sapnakumari, et al. 2015; Özdemir, et al. 2018). These effects can be justified, thanks to the lipophilic nature of these compounds, enabling them to cross the blood brain barrier (BBB). The characterization of all the compounds was done by 1HNMR, 13C NMR, LC–MS and HR-MS analysis. In the 1HNMR data of compounds (IIIa-IIIe), the multiplets of aromatic protons, were observed in the range of aromatic region i.e., 6.18–9.19 ppm. The quartets of methylene group (–CH2) were observed at 4.66–4.69 ppm. The singlet of hydroxyl group –OH was seen at 4.68 ppm and 10.96–11.50 ppm. The triplets of methyl group –CH3 were also observed in their expected area. The other spectroscopic data and elemental analysis were also in agreement with the proposed structures of the compounds Table 1.

The schematic path and reagents used for the synthesis of Pyrazoline derivatives (IIIa-IIId)
Biological results
Anti-oxidant effect of Pyrazoline derivatives
The DPPH assay was performed to assess the antioxidant potential of all newly synthesized derivatives using a 96-well plate through microplate reader. The results of the antioxidant assay for all synthesized compounds are summarized in Table 2 in terms of mean percentage inhibition by the DPPH assay. The IC50 values were calculated by non-linear regression analysis of the response-concentration log curve using the GraphPad Prism program package (GraphPad Software). Multiple dilutions (2.5, 5 and 10 µL) were used. The compound IIIa showed best antioxidant potential (IC50 = 17.88 ± 10.60 nM), closely followed by IIIb (IC50 = 22.52 ± 6.11 nM) as given in Table 2. It seems that the addition of electronegative atom (Fluorine) effected the antioxidant potential (IIIb) in a negative way while the removal of ‘OET’ group and adding F′ atom as side chain diminished the activity (IIIc, IIId & IIIe). Consequently, by changing the R′ groups on final derivative and adding electronegative atoms the biological activity was badly affected. As the results depicted that the first compound IIIa has prominent biological effect, by adding electronegative fluorine (F) atom at 4th position the activity gradually reduced. By altering R′ groups at 3rd and 5th position along with the addition of F’ atom the biological potential predominantly decreased (IIIb, IIIc & IIId). The best results of the compound IIIa encouraged us to further screen it using in-silico and in-vivo models.
In-vitro acetylcholinesterase (AChE) & Butyrylcholinesterase (BChE) inhibitory activity of the compound IIIa
The inhibitory potential of the compound IIIa against AChE and BChE was assessed by using the acetylcholinesterase and butyrylcholinesterase inhibitor assay kit, following the reported protocol (Ellman et al. 1961). This assay is based on the capability of human AChE to hydrolyze the colorimetric substrate, generating the yellow colour which can be detected through absorbance at 412 nm. DNP (reversible AChE inhibitor) is responsible for the suppression of enzyme activity which may lead to the prohibiting the chromophore generation. The results are expressed in the form of standard error of mean ± SEM in Table 3. The potent compound IIIa exhibited a significant ability to inhibit the acetylcholinesterase (AChE) and butyrylcholine esterase (BChE) with IC50 values of 25.15 nM and 22.09 nM respectively compared to Donepezil. The values are expressed as mean ± SEM. Symbols *p < 0.05, **p < 0.01 and ***p < 0.001 show significance level compared to the standard drug (donepezil). Data was analyzed via TWO-WAY ANOVA followed by Bonferroni post-test.
In-silico analysis
To further validate the results, molecular docking study was performed using the protein AChE via AutoDock Vina version 4.2.6. Co-crystallized ligands with INH derivatives (IIIa, IIIb, IIIc) were docked at the active sites of the targeted protein. The X-ray crystal structure of the protein AChE (PDBID: 4PQE) was downloaded from the online Protein Data Bank. Re-docking of the co-crystallized ligand with the same target was done to confirm and validate the docking procedures. Pose analysis confirmed the perfect alignment with the original ligand and exhibited the same binding interactions as depicted by the X-ray crystallographic PDB file. Different 2D binding interactions of compounds IIIa, IIIb, and IIIc with the receptor's active sites, resulting from the re-docked co-crystallized ligand, are shown in Fig. 2A, B. All results in the form of binding energies are summarized in Table 4.

A post-dock analysis performed through Biovia Discovery studio visualizer depicting 2D poses of interactions between compound IIIa, IIIb, IIIc & AChE active binding pockets. B post-dock analysis performed through Biovia Discovery studio visualizer depicting 2D poses of interactions between Donepezil (DNP) & AChE active binding pockets
For compound IIIa, one conventional hydrogen bond was observed between the hydrogen atom of the –OH group and the tyrosine (A: 121) residue, indicating its good interaction with the targeted active site. This interaction also had a distance of less than 2 Å from the molecule. One carbon-hydrogen bond was observed between the glycine (A: 118) moiety and the carbon atom. Additionally, one pi-pi hydrophobic interaction was seen between the tryptamine (A: 84) amino acid and the aromatic ring of compound IIIa. These interactions confirmed its biological potential. For compound IIIb, one conventional hydrogen bond was observed between the tyrosine (A: 121) residue and the –OH group in the compound. This bond lies at a distance of 1.78 Å from the molecule. Two good hydrophobic interactions were observed between the aromatic ring residue and the amino acid residues GLU (A: 199) and tryptamine (A: 84) present at the active site of the protein. A pi-sigma bond was observed between the phenylalanine (A: 330) and the alkyl residue. In the case of the binding interactions of compound IIIc with the targeted protein, one conventional favourable hydrogen bond was observed between the –OH group and the tyrosine (A: 121) amino acid residue. Pi-donor hydrogen bond and one carbon-hydrogen bond were seen between the alkyl residue and SER (A: 200) and oxygen atom and TRP (A: 84) amino acid residues, respectively. Pi-pi stacked and pi-cation bonds were located between the nitrogen atom and PHE (A: 330) and TYR (A: 334) linked through the methyl residue. These two interactions unfavorably affected the biological aspects of compound IIIc.
Conclusion
A new series of pyrazoline derivatives (IIIa-IIId) was designed and synthesized. These compounds were characterized using various spectroscopic techniques. Initially, all derivatives were screened for their antioxidant effects via the DPPH assay. Among them, compound IIIa exhibited the best results, with an IC50 value of 17.88 ± 10.60 nM. This potent compound was further assessed for its inhibitory potential against AChE and BChE enzymes and demonstrated significant inhibition with IC50 values of 22.15 nM and 20.09 nM, respectively. These results were rationalised by molecular docking studies targeting the AChE enzyme, in comparison with donepezil (DNP), a standard drug. The compounds displayed favorable binding energies with the target protein's active pockets (− 8.9, − 8.2, − 8.3 kcal/mol). Collectively, these findings strongly suggest that the synthesized pyrazoline derivative (IIIa), with its notable antioxidant and enzyme inhibitory effects, is a promising candidate for further research as a therapeutic agent in treating Alzheimer’s disease.
Data availability
The data supporting this article are available in the supplementary files.
References
Acharya BN et al (2010) Synthesis and antimalarial evaluation of 1, 3, 5-trisubstituted pyrazolines. Eur j Med Chem 45(2):430–438
Adeowo FY et al (2022) Pharmacophore Mapping of the Crucial Mediators of Acetylcholinesterase and Butyrylcholinesterase Dual Inhibition in Alzheimer’s Disease 26(5):2761–2774
Ali, M.A., M. Shaharyar, and A.A.J.E.j.o.m.c. Siddiqui, Synthesis, structural activity relationship and anti-tubercular activity of novel pyrazoline derivatives. 2007. 42(2): p. 268–275.
Altıntop, M.D., et al., A novel series of thiazolyl–pyrazoline derivatives: Synthesis and evaluation of antifungal activity, cytotoxicity and genotoxicity. 2015. 92: p. 342–352.
Amin KM et al (2021) Design and Synthesis of Novel Coumarin Derivatives as Potential Acetylcholinesterase Inhibitors for Alzheimer’s Disease 110:104792
Bibi G et al (2011) Antitumor, cytotoxic and antioxidant potential of Aster thomsonii extracts. Afr J Pharm Pharmacol 5(2):252–258
Bohnen, N.I., et al., Recent advances in cholinergic imaging and cognitive decline—revisiting the cholinergic hypothesis of dementia. 2018. 7: p. 1–11.
Breijyeh Z, Karaman RJM (2020) Comprehensive Review on Alzheimer’s Disease: Causes and Treatment 25(24):5789
Eissa, K.I., et al., Design, synthesis, and biological evaluation of thienopyrimidine and thienotriazine derivatives as multitarget anti‐Alzheimer agents. 2022. 83(6): p. 1394–1407.
Ellman GL et al (1961) A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity 7(2):88–95
Farooq S, Ngaini ZJTL (2020) One-Pot and Two-Pot Synthesis of Chalcone Based Mono and Bis-Pyrazolines 61(4):151416
Gawad, N.M.A., et al., Synthesis and antitumor activity of some 2, 3-disubstituted quinazolin-4 (3H)-ones and 4, 6-disubstituted-1, 2, 3, 4-tetrahydroquinazolin-2H-ones. 2010. 45(12): p. 6058–6067.
Gökhan-Kelekçi, N., et al., New pyrazoline bearing 4 (3H)-quinazolinone inhibitors of monoamine oxidase: Synthesis, biological evaluation, and structural determinants of MAO-A and MAO-B selectivity. 2009. 17(2): p. 675–689.
Haider, K., et al., A comprehensive review on pyrazoline based heterocyclic hybrids as potent anticancer agents. 2022: p. 100042.
Herowati, R. and G.P.J.Q.S.-a.R. Widodo, Molecular docking analysis: Interaction studies of natural compounds to anti-inflammatory targets. 2017. 63(10.5772).
P. James, J., et al., Design, synthesis, molecular modeling, and ADMET studies of some pyrazoline derivatives as shikimate kinase inhibitors. 2018. 27: p. 546–559.
Joshi, S.D., et al., Synthesis, antimycobacterial screening and ligand-based molecular docking studies on novel pyrrole derivatives bearing pyrazoline, isoxazole and phenyl thiourea moieties. 2016. 107: p. 133–152.
Kamogawa, E., Y.J.B. Sueishi, and M.C. Letters, A multiple free-radical scavenging (MULTIS) study on the antioxidant capacity of a neuroprotective drug, edaravone as compared with uric acid, glutathione, and trolox. 2014. 24(5): p. 1376–1379.
Kanwal, M., et al., New pyrazolone derivatives, synthesis, characterization, and neuroprotective effect against PTZ-induced neuroinflammation in mice. 2022. 25(12): p. 1424.
Kharbanda C et al (2014) Synthesis and Evaluation of Pyrazolines Bearing Benzothiazole as Anti-Inflammatory Agents 22(21):5804–5812
Khloya P et al (2015) Sulfonamide bearing pyrazolylpyrazolines as potent inhibitors of carbonic anhydrase isoforms I. II, IX and XII 25(16):3208–3212
Knorr, L.J.B.d.d.c.G., Einwirkung von acetessigester auf phenylhydrazin. 1883. 16(2): p. 2597–2599.
Li Q et al (2017) Recent Progress in the Identification of Selective Butyrylcholinesterase Inhibitors for Alzheimer’s Disease 132:294–309
Malik, S., et al., Synthesis, characterization, in-silico, and pharmacological evaluation of new 2-amino-6‑trifluoromethoxy benzothiazole derivatives. 2023. 130: p. 106175.
Mathew, B., et al., Pyrazoline: A promising scaffold for the inhibition of monoamine oxidase. 2013. 13(3): p. 195–206.
Miana, G., et al., In-silico, Antioxidant and Antiepileptic Effect of N (2, 3-methylenedioxy-4benzoyloxy-phenthylamine)-3, 4-dimethyl-1, propanoamide Derivatives. 2022. 44(4).
Nehra B et al (2020) Recent Advancements in the Development of Bioactive Pyrazoline Derivatives 205:112666
Özdemir, Z., et al., Synthesis and studies on antidepressant and anticonvulsant activities of some 3-(2-furyl)-pyrazoline derivatives. 2007. 42(3): p. 373–379.
Özdemir, A., et al., Design, synthesis, and neuroprotective effects of a series of pyrazolines against 6-hydroxydopamine-induced oxidative stress. 2018. 23(9): p. 2151.
Ozgun DO et al (2019) Synthesis and Bioactivities of Pyrazoline Benzensulfonamides as Carbonic Anhydrase and Acetylcholinesterase Inhibitors with Low Cytotoxicity 84:511–517
Prasad, Y.R., et al., Synthesis and antidepressant activity of some 1, 3, 5-triphenyl-2-pyrazolines and 3-(2 ″-hydroxy naphthalen-1 ″-yl)-1, 5-diphenyl-2-pyrazolines. 2005. 15(22): p. 5030–5034.
Rathore P et al (2014) Synthesis and Evaluation of Some New Pyrazoline Substituted Benzenesulfonylureas as Potential Antiproliferative Agents 24(7):1685–1691
Sapnakumari, M., et al., Design, synthesis, and pharmacological evaluation of new pyrazoline derivatives. 2015. 146: p. 1015–1024.
Song Y et al (2020) Synthesis and Biological Evaluation of Novel Pyrazoline Derivatives Containing Indole Skeleton as Anti-Cancer Agents Targeting Topoisomerase II 200:112459
Volkamer A et al (2012) Combining Global and Local Measures for Structure-Based Druggability Predictions 52(2):360–372
Wang, H.-H., et al., Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives containing benzodioxole as potential anticancer agents. 2013. 21(2): p. 448–455.
Yamali, C., et al., Synthesis, structure elucidation, and in vitro pharmacological evaluation of novel polyfluoro substituted pyrazoline type sulfonamides as multi-target agents for inhibition of acetylcholinesterase and carbonic anhydrase I and II enzymes. 2020. 96: p. 103627.
Yang, W., et al., Design, modification and 3D QSAR studies of novel naphthalin-containing pyrazoline derivatives with/without thiourea skeleton as anticancer agents. 2013. 21(5): p. 1050–1063.
Acknowledgements
The authors are highly grateful for the technical support and lab facilities provided by the Institute of Pharmaceutical Sciences at King's College London for conducting chemical synthesis, and the Pharmaceutical Chemistry lab of the Pharmaceutical Sciences department at Riphah International University, Islamabad, Pakistan, for performing in-vitro assays.
Funding
This research has no funding.
Author information
Authors and Affiliations
Contributions
MK: original manuscript writing, KMR: supervisor, review and edit of manuscript, SS & HN: co-supervisor, editorial review, RZ: experimental work.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare no conflict of interest to publish this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kanwal, M., Sarwar, S., Nadeem, H. et al. Synthesis of Pyrazoline derivatives, condensation of β-dicarbonyl compounds with isoniazid (INH), and their biological evaluation as multitarget anti-Alzheimer’ disease agents. Chem. Pap. 78, 2033–2042 (2024). https://doi.org/10.1007/s11696-023-03224-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-023-03224-1