
Abstract
Overweight and obesity have been identified as the most important risk factors for many diseases, including cardiovascular disease, type 2 diabetes and lipid disorders, such as non-alcoholic fatty liver disease (NAFLD). The metabolic changes associated with obesity are grouped to define metabolic syndrome, which is one of the main causes of morbidity and mortality in industrialized countries. NAFLD is considered to be the hepatic manifestation of metabolic syndrome and is one of the most prevalent liver diseases worldwide. Inflammation plays an important role in the development of numerous liver diseases, contributing to the progression to more severe stages, such as non-alcoholic steatohepatitis and hepatocellular carcinoma. Peroxisome proliferator-activated receptors (PPARs) are binder-activated nuclear receptors that are involved in the transcriptional regulation of lipid metabolism, energy balance, inflammation and atherosclerosis. Three isotypes are known: PPAR-α, PPARδ/β and PPAR-γ. These isotypes play different roles in diverse tissues and cells, including the inflammatory process. In this review, we discuss current knowledge on the role PPARs in the hepatic inflammatory process involved in NAFLD as well as new pharmacological strategies that target PPARs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
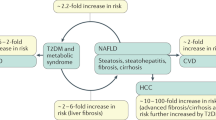
Non-alcoholic fatty liver disease (NAFLD) is characterized by significant lipid deposition in hepatocytes with persistent changes in liver enzymes, such as AST and ALT [1]. Overeating is the most common cause of the accumulation of excess lipids in the liver and it is estimated that over six hundred million overweight people worldwide will develop fatty liver disease [2]. In clinical practice, the initial diagnosis of hepatic steatosis is usually established using radiological imaging techniques and is based on the presence of liver fat accumulation ≥ 5% in the absence of other recognized causes, such as alcohol, virus and medication [3].
NAFLD comprises a broad spectrum of liver damage, ranging from simple macrovesicular steatosis to non-alcoholic steatohepatitis (NASH), advanced fibrosis and cirrhosis [4, 5]. NAFLD is recognized as a cause of end-stage liver disease and is associated with increased rates of hepatocellular carcinoma, liver transplantation and death [6,7,8]. Although not yet well understood, it is recognized that insulin resistance plays an important role in the pathogenesis of NAFLD. In healthy subjects, insulin stimulates hepatic and peripheral glucose absorption and suppresses hepatic glucose production [9]. In the fasting state, the liver becomes the main site of glucose production mediated by glycogenolysis and gluconeogenesis [10, 11]. In patients with insulin resistance, hepatic autoregulation is altered and both gluconeogenesis and glycogenolysis are increased, resulting in the development of hyperglycemia [12].
According to the “multiple hit” theory, hyperinsulinemia in the context of insulin resistance first leads to an increase in the release of free fatty acids from adipocytes, which then accumulate in the liver, resulting in steatosis. This initial step is followed by a series of complex interactions among hepatocytes, Kupffer cells, adipocytes, inflammatory mediators and oxygen radicals, the result of which is non-alcoholic steatohepatitis.
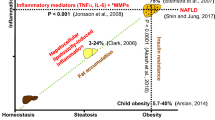
Inflammation plays a central role in several acute and chronic liver diseases, contributing to the progression of liver damage to more severe stages, such as fibrosis, cirrhosis and hepatocellular carcinoma [13, 14]. In addition to hepatocytes, the liver also contains a complex repertoire of lymphoid and non-lymphoid cells that play key roles in hepatic and defense immunoregulation [15]. The cells that are part of innate immunity in the liver include Kupffer cells, monocytes, neutrophils, dendritic cells, natural killer cells and natural killer T cells, which initiate and maintain hepatic inflammation through the production of cytokines [16]. The dysregulated release of cytokines following liver damage may result in the excessive death of hepatocytes, which is a key event in several acute and chronic liver diseases [17].
Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors that are involved in the transcriptional regulation of lipid metabolism, energy balance, inflammation and atherosclerosis. The three known isotypes are PPAR-α, PPAR-δ/β and PPAR-γ [18, 19]. PPAR-α is the most commonly expressed in the liver and expressed in lower levels in the heart, skeletal muscle, intestine and kidneys, where it plays an important role in controlling the oxidation of fatty acids [20]. The activation of PPAR-α is related to the transcription of 80–100 genes involved in fatty acid oxidation, lipid metabolism and inflammation [18]. Fibrates are the synthetic ligands of PPAR-α and are used to treat dyslipidemias in humans [21]. PPAR-δ/β is present in a wide variety of cells, but its greatest expression occurs in tissues that have high metabolic activity, including hepatocytes, macrophages and adipocytes. When activated, PPAR-δ/β primarily regulates lipid metabolism, glucose homeostasis and inflammation [22, 23]. PPAR-γ is a transcription factor found in adipocytes, macrophages, monocytes, hepatocytes, muscle and endothelial cells and is the most studied of the three isotypes. It belongs to the superfamily of nuclear receptors that bind to specific agonists, also known as ligands or activators of PPAR-γ [24]. Thiazolidinediones (TZDs) are synthetic PPAR-γ agonists that act as insulin sensitizers and are used clinically to treat type 2 diabetes [25]. The three isotypes are involved in the inflammatory process in different tissues and cells. In this review, we focus on the role of PPARs in the inflammatory process related to NAFLD and discuss new pharmacological strategies that target PPARs.
PPAR-α
PPAR-α is expressed in many mammalian cells and tissues (Bishop-Bailey 2000; Braissant et al. 1996), including immune cells (e.g., macrophages). Consistent with this broad distribution, PPAR-α plays multiple regulatory functions, including the control of macrophage activity and inflammation [26, 27]. Specifically in rodents, the mRNA expression of PPAR-α is highest in tissues characterized by a high rate of fatty acid oxidation, including brown adipose tissue, liver, kidneys and heart [28]. Considerable evidence indicates that PPAR-α serves as the master regulator of lipid metabolism in the liver, especially during fasting [29].
The activation of PPAR-α occurs after dimerization with the retinoid X receptor, forming a multi-protein complex with a variable set of protein coactivators. In its active form, PPAR-α binds to responsive elements in DNA, enhancing the transcription of various anti-inflammatory proteins, such as the kB-α inhibitor [30]. PPAR-α negatively regulates the gene expression of pro-inflammatory proteins by antagonizing the activities of other transcription factors, including nuclear factor kB (NF-kB), activator protein-1 (AP-1), the nuclear factor of activated T cells and signal transducers and activators of transcription proteins through direct protein–protein interactions in a process known as transrepression, which does not involve binding to typical receptor-specific PPAR response elements (PPRE), as occurs in transactivation [19, 31, 32]. Another PPRE-dependent model of transcriptional regulation has been proposed. Upon ligand activation, PPRE-bound PPAR-α directly interacts with p65 to abolish its binding to an NFκB response element in the complement C3 promoter [33]. The loss of PPAR-α-mediated gene transcription in PPAR-α null macrophages results in enhanced MAPK phosphorylation, leading to increased NF-κB [26]. PPAR-α ligands significantly reduce levels of pro-inflammatory cytokines, such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), cyclooxygenase-2 (COX-2), and inducible nitric oxide synthase (iNOS) through the interference with AP-1 and NF-kB signaling pathways [34].
Murine models have demonstrated that PPAR-α expression is also regulated by natural compounds, such as Rutin, which is a plant-derived flavonoid commonly found in fruit, tea, wine [35] curcumin and vitamin E [36] as well as high-protein diets and fish oil [37]. However, the anti-inflammatory mechanisms have not been evaluated. In contrast, hydroxytyrosol, which is a polyphenol with cytoprotective effects, demonstrated anti-inflammatory properties when evaluated in a high-fat diet (HFD) model. HFD-fed mice exhibited inflammation, together with the downregulation of PPAR-α and Nrf2 and the up-regulation of NF-κB. Hydroxytyrosol supplementation attenuated the metabolic changes produced by the high-fat diet, normalizing the activity of Nrf2 and PPAR-α and attenuating the gene expression of the pro-inflammatory mediators NF-κB, TNF-α, IL-6, and IL-1β [38].
Astaxanthin is a natural xanthophyll carotenoid that is abundant in marine organisms, such as microalgae and salmon [39]. It is also a PPAR-α agonist and PPAR-γ antagonist that ameliorates lipid accumulation in HepG2 cells by regulating genes involved in lipid metabolism [40]. Another study by the same group showed that astaxanthin administration reduced the hepatic pro-inflammatory cytokines TNF-α and IL-6 through the activation of PPAR-α in HFD-fed C57BL/6J mice [41].
Fibrates are widely used to ameliorate the macrovascular and microvascular risks associated with metabolic syndrome and are considered PPAR-α agonists [42]. This class includes clofibrate, gemfibrozil, fenofibrate, bezafibrate and ciprofibrate. Clofibrate was developed in the 1960s as the first member of this class and is no longer available due to its adverse effects. Gemfibrozil and fenofibrate are available in the United States, whereas gemfibrozil, fenofibrate, bezafibrate and ciprofibrate are available in Europe. Although the main mechanism of action of fibrates is related to lipid reduction, these substances also have been shown to contribute to a reduction in inflammation. In APOE2-knockout mice fed a western diet containing high levels of sucrose and cholesterol to induce steatohepatitis, treatment with fenofibrate decreased hepatic steatosis, hepatic macrophage accumulation, inflammatory gene expression and the up-regulation of genes involved in beta oxidation [43]. Similar results were found in rats fed a high fat and fructose diet [44, 45].
In a small study involving 16 human patients with biopsy-confirmed NASH, 12 months of clofibrate treatment resulted in no changes in ALT or histological findings in comparison with baseline [46]. Similarly, a small pilot study involving 16 NAFLD patients treated with fenofibrate for 48 weeks showed a lower concentration of plasma ALT concentration, but no significant improvement in histological findings compared to baseline liver biopsies [47]. Studying 90 NAFLD patients for 24 weeks, El-Haggar and Mostafa [48] observed that fenofibrate improved inflammation by reducing plasma levels of TNF-α and acting on fibrotic markers, such as TGF-β. Thus, given the available clinical data, PPAR-α agonists may lower the plasma concentration of ALT and reduce inflammation, but do not appear to produce histological improvements in NASH in humans.
However, fibrates are weak PPAR-α agonists and have limited efficacy due to dose-related adverse events. To address this problem, a new generation of PPAR-α-specific agonists, known as selective PPAR-α modulators (SPPARMα), have been developed to maximize the receptor-mediated effects and diminish the side effects. Pemafibrate is a novel SPPARMα designed to have highly selective and tissue-specific activity without the unwanted side effects of currently used fibrates and was developed for the treatment of dyslipidemia. Recent studies demonstrate that pemafibrate is a more potent PPAR-α-agonist than fenofibrate. In a phase-2 study, pemafibrate reduced plasma concentrations of liver enzymes in patients with dyslipidemia [49]. Moreover, pemafibrate in db/db mice fed an MCD and AMLN diet (exhibiting the three stages of NAFLD: steatosis, steatohepatitis with fibrosis and cirrhosis) inhibited the expression of pro-inflammatory and pro-fibrotic genes (F4/80, TNFα, collagen 1α), indicating that this new drug is a promising therapeutic agent for NAFLD/NASH [50].
Recently, Takei et al. [51] evaluated the effects of K-877 (another selective PPAR-α modulator) on metabolism and inflammation in vitro and in vivo, and compared the results to those achieved with classic PPAR-α agonists. K-877 was found to be associated with beneficial changes in liver disease markers, suggesting the potential of this novel agent in the treatment of NASH/NAFLD, possibly related to PPAR-α pathway activation.
Other PPAR-α agonists have emerged with potential anti-inflammatory activity in the liver. LPSF/GQ-02 is a molecule developed as a thiazolidine derivative. According to da Costa Leite et al. [52], the molecular structure of LPSF/GQ-02 indicates a possible role as a PPAR-γ agonist. However, animal model studies have shown increased PPAR-α expression after LPSF/GQ-02 administration [53]. Soares e Silva et al. [54] found that LPSF/GQ-02 was effective at improving the hepatic architecture, decreasing fat accumulation, reducing the amount of collagen, decreasing inflammation by reducing IL-6, iNOS, COX-2 and F4/80 as well as increasing the protein expression of IκBα, cytoplasmic NFκB-65, eNOS and IRS-1 in LDLR−/− mice fed a high-fat diet. These results suggest direct action on factors that affect inflammation, insulin resistance and fat accumulation. More studies are needed to characterize this receptor better and further clarify the possible mechanisms of action related to the inflammatory process in the liver so that more selective and effective novel molecules can be developed (Fig. 1).

A schematic summarizing the anti-inflammatory activity of PPAR-α in the liver. Green arrows indicate activation, and red bar-headed line indicates inhibition (see text for details)
PPAR-β/δ
PPAR-β/δ is the least studied among the PPAR isotypes, although it has significant expression in tissues responsible for controlling lipid metabolism, such as adipocytes, the small intestine, heart, skeletal muscle, liver and macrophages [55]. The removal of the gene encoding the PPAR-β/δ isoform is lethal for the initial development of nearly all embryos due to a defect in the formation of the placenta [56]. Therefore, this isoform plays an important role not only in the regulation of metabolism, but also in the development of the organism [57]. There are strong indications that fatty acids, triglycerides and prostacyclin are endogenous PPAR-β/δ activators. Experimental evidence suggests that PPAR-β/δ activation may have therapeutic value in the treatment of metabolic syndrome [58,59,60]. Studies indicate that PPAR-β/δ activation exerts regulatory effects on fatty acid catabolism, reverse cholesterol transport and energy metabolism [61]. Moreover, some PPAR-β/δ agonist substances were able to reduce insulin resistance and plasma glucose in an animal model of type 2 diabetes [62, 63].
Specifically in the liver, PPAR-β/δ expression has been identified in many cell types, such as hepatocytes, Kupffer cells and hepatic stellate cells. Although the main effects of PPAR-β/δ in the liver are related to fatty acid and glucose metabolism, evidence suggests that PPAR-β/δ also controls anti-inflammatory mechanisms in the liver. More specifically, PPARβ/δ is a key regulator of the alternative activation of Kupffer cells towards anti-inflammatory activity in the presence of IL4 and IL13 stimulation [64]. PPAR-β/δ activation in HepG2 cells attenuates interleukin (IL)-6-induced inflammation and insulin resistance. These effects were mediated via the PPARβ/δ inhibition of IL-6-induced signal transducer and activator of transcription (STAT)-3, resulting in the restoration of normal insulin signaling [65]. Kupffer cell-specific deletion of PPAR-β/δ in mice resulted in impaired hepatic Akt phosphorylation coupled with increased hepatic inflammation [64]. Together, these studies demonstrate that the activation of hepatic PPAR-β/δ attenuates inflammation and contributes to improved hepatic insulin sensitivity, since the increase in inflammation is directly related to hepatic insulin resistance.
Many unsaturated fatty acids can bind to PPARβ/δ in a pattern closely resembling the binding to PPAR-α. In relation to the synthetic agonists, the compound GW501516 [66], which is the most potent and specific, has become the reference PPARβ/δ agonist [67] Despite promising early results, however, the further investigation and development of GW501516 was discontinued after observations in animal studies of its association with the rapid induction of cancer in several organs (liver, stomach, tongue, skin, bladder, ovaries, womb and testes). Nonetheless, it is commercially available for non-human research purposes, together with GW0742, which is also a highly selective PPARβ/δ agonist [66]. Lee et al. [68] concluded that GW501516 was able to suppress the activation of inflammasome and reduce IL-1β levels, possibly through the modulation of AMPK phosphorylation, and decreased the production of radical oxygen species in both in vivo and in vitro studies, associating the anti-inflammatory effect with the improvement of hepatic steatosis in mice. In a mouse model of NASH, PPARβ/δ improved hepatic steatosis and inflammation by the regulation of lipid metabolism and the inhibition of the inflammatory response activation of AMPK [69].
Using a type 2 diabetic rat model (in vivo study) as well as HepG2 and Raw264.7 cells (in vitro study), Lee et al. [70] demonstrated that the PPARβ/δ agonist GW0742 attenuated hyperglycemia and fat accumulation in the liver. These changes were possibly due to the suppression of inflammatory cytokines, such as TNF-α and MCP-1, suggesting that this PPAR-β/δ agonist has beneficial effects against NAFLD. In another study using GW0742, the activation of PPAR-β/δ was found to inhibit CCl4-induced liver toxicity through a mechanism involving the PPAR-β/δ-dependent downregulation of pro-inflammatory signaling through interactions between PPAR-β/δ and NF-κB [71].
In several respects, PPAR-β/δ is comparable to PPAR-α, since both are important regulators of the beta oxidation of fatty acids. Sanderson et al. [72] used microarray analysis to compare the hepatic transcriptome of PPAR-α-null mice and PPARβ/δ-null mice, and found little overlap between the PPAR-α-dependent and PPAR-β/δ-dependent regulation of genes. The authors also strengthened observations that PPAR-β/δ governs glucose use and the metabolism of lipoproteins and plays an important anti-inflammatory role in the liver.
The ability of PPAR-β/δ to regulate several important pathways in the liver, such as lipid homeostasis, inflammation and insulin resistance, demonstrates that this transcription factor plays an important role in metabolic regulation (Fig. 2).

A schematic summarizing the anti-inflammatory activity of PPAR-β/δ in the liver. Green arrows indicate activation, and red bar-headed line indicates inhibition (see text for details)
PPAR-γ
PPAR-γ is more highly expressed in adipose tissue and plays a crucial role in the differentiation of adipocytes, inducing the expression of important markers involved in lipid metabolism, such as fatty acid-binding protein (aP2) [73], phosphoenolpyruvate carboxykinase [74] and lipoprotein lipase [75]. It also controls the expression of fatty acid transporter protein 1 and CD-36 [76], both of which are involved in lipid uptake by adipocytes. The importance of PPAR-γ in adipogenesis has been demonstrated in several studies. For example, PPAR-γ is induced during the differentiation of pre-adipocytes in vitro and its ectopic expression in non-adipogenic fibroblasts stimulates adipogenesis in the presence of PPAR-γ ligands [77].
It should also be stressed that increased PPAR-γ expression is a feature of the steatotic liver and several studies attribute a causal role to PPAR-γ in the development of steatosis through mechanisms involving the activation of lipogenic genes and de novo lipogenesis [78, 79]. Accordingly, the targeted deletion of PPAR-γ from hepatocytes and macrophages protects mice against diet-induced hepatic steatosis [80]. Moreover, hepatic PPAR-γ expression is robustly induced in NAFLD patients and experimental models [79, 81,82,83], suggesting a possible pro-steatotic role of PPAR-γ in both parenchymal and non-parenchymal cells.
Since it is a transcription factor, PPAR-γ also affects other important pathways in the organism. It has potent anti-inflammatory properties that modulate the immune inflammatory response [84]. Similarly to PPAR-α, PPAR-γ activation alleviates the inflammatory response through the negative interference in the transcriptional repression of genes, including NF-κB and STAT [85, 86]. PPAR-γ also regulates genes related to inflammation, especially in macrophages [87].
Macrophages are essential components of innate immunity and play a central role in inflammation and host defense [88, 89]. Kupffer cells, which are hepatic resident macrophages, represent the largest group of fixed macrophages in the body and account for 20–25% of non-parenchymal cells in the liver. Moreover, there is increasing evidence that Kupffer cells critically contribute to the pathogenesis of NAFLD [90]. Macrophage polarization is an important mechanism for regulating the inflammatory response and is classically characterized by two subtypes: M1 macrophages, which are pro-inflammatory and induced by TLR ligands, such as LPS and IFNγ, and M2 macrophages, which are anti-inflammatory and activated by IL-4/IL-13 [91]. PPAR-γ activation has been found to play an important role in macrophage polarization [87, 92]. In one study, the disruption of PPAR-γ impaired alternative M2 macrophage activation and predisposed mice to obesity and insulin resistance [64]. Using animal model of NASH, Zhong and Liu [93] confirmed that the activation of PPAR-γ regulates the polarization of the macrophages to M2, leading to the prevention of the development of NAFLD.
A number of natural ligands may activate PPAR-γ, including unsaturated fatty acids, eicosanoids and components of oxidized LDLs. However, the receptor affinity for many of these ligands is low and, in some cases, the physiological relevance of the ligand has not yet been determined [94].
PPAR-γ is the molecular target of a class of synthetic linkers known as thiazolidinediones (TZDs). The main characteristic of these molecules is the presence of a dione ring. Troglitazone was the first PPAR-γ agonist approved by the US Food and Drug Administration (FDA) for the treatment of type 2 diabetes in 1997, but was withdrawn from the market after confirmation of severe hepatotoxicity and death [95]. Rosiglitazone and pioglitazone, which were approved for use by the FDA in 1999, are considered the second generation of PPAR-γ agonists.
TZDs primarily sensitize the action of insulin directly on adipocytes and indirectly on the release of adipocytokines. In the direct effect, TZDs promote the uptake and storage of fatty acids in adipose tissue, increasing the mass of this tissue and sparing other insulin-sensitive tissues, such as skeletal muscle and the liver [96]. In other words, TZDs promote the distribution of fat from liver and skeletal muscle cells to adipocytes.
Several studies in the literature point out that the pharmacological activation of PPAR-γ has favorable effects on the liver. The adenovirus-mediated overexpression of PPAR-γ reduced hepatic steatosis, inflammation and fibrosis in a model of steatohepatitis [97]. Similarly, the use of rosiglitazone prevented the development of NASH in an animal model induced with a choline–methionine deficient diet [98]. Recently, Deng et al. [99] demonstrated that the PPAR-γ agonist pioglitazone exerts its anti-inflammatory and anti-fibrotic effects by repressing the expression level of platelet-derived growth factor and tissue inhibitor of metalloproteinase-2 in an animal model of NAFLD. van der Veen et al. [100] found that inflammatory and fibrotic markers were reduced after the use of pioglitazone in a model of phosphatidylethanolamine N-methyltransferase-deficient mice, even without altering steatosis levels. In a model of acute hepatic injury in rats, rosiglitazone was able to reduce levels of inflammatory markers, such as TNF-α, IL-6 and COX-2, through the downregulation of NFκB [101].
In humans, pioglitazone has been shown to improve hepatic steatosis, inflammation and fibrosis in a meta-analysis including both diabetic and non-diabetic patients with NASH [102]. In the PIVENS trial involving 247 non-diabetic adults with NASH randomized to receive pioglitazone, vitamin E or a placebo for 96 weeks, pioglitazone did not meet its primary endpoint, although it demonstrated a reduction in hepatic steatosis, lobular inflammation as well as serum alanine and aspartate aminotransferase levels. However, subjects receiving pioglitazone gained more weight than those who received vitamin E or the placebo [103], which is a side effect seen in several other studies. Although many clinical studies report the risk of adverse effects with regard to congestive cardiac failure, bladder cancer and osteoporosis [104,105,106], the European and American Associations for the Study of the Liver recommend pioglitazone for the treatment of NASH, but with important considerations regarding the long-term risks [107, 108].
Controversially, some experimental studies indicate that increased PPAR-γ expression is a characteristic of the steatotic liver and several studies attribute a causal role of PPAR-γ in the development of steatosis through mechanisms involving the activation of genes from the pathways of lipogenesis and de novo lipogenesis [80, 100, 109]. PPAR-γ deletion in hepatocytes and macrophages protected mice against diet-induced hepatic steatosis [80], suggesting a pro-steatotic role of PPAR-γ in both parenchymal and non-parenchymal cells. Moreover, treatment of ob/ob mice with rosiglitazone did not reverse the histological changes of NAFLD, but increased oxidative stress and steatosis [109]. Consistent with these findings, Soares e Silva et al. [54] found that pioglitazone increased levels of hepatic triglycerides in a mouse model of NAFLD, with a consequent increase in steatosis associated with elevated inflammatory markers (IL-6, COX-2, F4/80 and iNOS).
Based on all of these findings, it is difficult to confirm whether PPAR-γ is the causal factor or a consequence of the accumulation of fat in the liver. These limitations highlight the need for novel approaches, such as more selective PPAR-γ agonists or drugs that effectively activate downstream targets (Fig. 3).

A schematic summarizing the anti-inflammatory activity of PPAR-γ in the liver. Green arrows indicate activation, and red bar-headed line indicates inhibition (see text for details)
Dual and pan agonists
The involvement of PPARs in various biological processes, especially in the modulation of lipid metabolism and inflammatory response, makes these nuclear receptors an important target for the development of new molecules. In recent years, agonists of PPARs have arisen with affinity for binding to multiple isoforms, known as dual agonists or PAN agonists.
The first developed dual agonists, called glitazars (PPARα/γ), have shown interesting results by reducing important inflammatory markers in mice [110,111,112], but these studies were later abandoned because of serious adverse effects [113, 114]. Recently, a new dual agonist PPARα/γ, saroglitazar, has presented interesting and promising results in murine model [115] and there was an approval for its use in India in a prospective, randomized, and registered clinical trial comparing saroglitazone with pioglitazone in patients with NAFLD (GLAZED) (https://clinicaltrials.gov/ct2/show/NCT02265276). This study began in October 2014 and was scheduled to end in September 2015, but no published results are currently available [42].
The GFT505, also known as Elafibranor, is PPAR-α/δ agonist and it has presented interesting results in reducing parameters of the metabolic syndrome that are related to the development of NAFLD. Short-term phase II studies demonstrated that GFT505 improves several metabolic parameters in patients with dyslipidemia and/or prediabetes [116, 117]. Staels and cols [118] evaluated the action of GTF505 on animal models of NAFLD (WD in hApoE2 KI mice, MCD diet in db/db mice and fibrosis in rat induced for CCl4), and observed that the dual agonist reduced inflammatory parameters (IL-1β, TNF-α and F4/80) associated with decreased hepatic lipid accumulation and fibrosis. More recent, Ratziu and cols [119], evaluated the action of Elafibranor in an international, randomized, double-blind placebo-controlled trial of patients with non-alcoholic steatohepatitis (NASH). The patients received Elafibranor 80 mg or 120 mg each day for 52 weeks. Liver enzymes, lipids, glucose profiles, and markers of systemic inflammation were significantly reduced including improvement of the cardiometabolic risk profile.
In this context, IVA337 is classified as a new agonist generation of PPARs, having the ability to activate in a moderate and balanced way the three isoforms of PPAR (α, β/δ and γ). In a recent study, the authors investigated the effects of IVA337 on several preclinical models reproducing the main metabolic and hepatic features associated with NASH. As a result, they found that this novel compound was able to reduce inflammasome genes (NLRP3, ASC, Caspase-1, IL-1β and IL-18) and important inflammatory cascade genes (CCL5, CCR2, NF-κB) as well as important markers of lipid and fibrotic metabolism, representing a potential future drug for the treatment of patients with NASH [120].
Conclusion
Non-alcoholic fatty liver disease (NAFLD) is the most common pathology of liver and is considered part of metabolic syndrome. The evolution of NAFLD is related to the development and progression of inflammation and many studies show that the reduction of inflammation is a key point for the improvement of this pathology. The action potential of PPARs in NAFLD has been evaluated for several years and it is undeniable that these transcription factors regulate many inflammatory processes in the liver. The bulk of these studies demonstrate the considerable anti-inflammatory potential of PPARs. However, since these receptors are transcription factors and are involved in a complex metabolic network, further studies must be conducted to explore and clarify the signaling pathways involved in the hepatic anti-inflammatory process.
References
Sonsuz A, Basaranoglu M, Ozbay G (2000) Relationship between aminotransferase levels and histopathological findings in patients with nonalcoholic steatohepatitis inducible nitric oxide synthase activity is expressed not only in inflamed but also in normal colonic mucosa in patients with ulcerat. Am J Gastroenterol 95:1370–1371
Stephen S, Baranova A, Younossi ZM (2012) Nonalcoholic fatty liver disease and bariatric surgery. Expert Rev Gastroenterol Hepatol 6:163–171. https://doi.org/10.1586/egh.11.97
Byrne CD, Targher G (2015) NAFLD: a multisystem disease. J Hepatol 62:S47–S64. https://doi.org/10.1016/j.jhep.2014.12.012
Angulo P (2002) Nonalcoholic fatty liver disease. N Engl J Med 346:1221–1231. https://doi.org/10.1056/NEJMra011775
Musso G, Gambino R, Cassader M, Pagano G (2011) Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med 43:617–649. https://doi.org/10.3109/07853890.2010.518623
Charlton MR, Kondo M, Roberts SK et al (1997) Liver transplantation for cryptogenic cirrhosis. Liver Transpl Surg 3:359–364. https://doi.org/10.1002/lt.500030402
Mccullough AJ (2002) Update on nonalcoholic fatty liver disease. J Clin Gastroenterol 34:255–262
Sass DA, Chang P, Chopra KB (2005) Nonalcoholic fatty liver disease: a clinical review. Dig Dis Sci 50:171–180. https://doi.org/10.1007/s10620-005-1267-z
Edens MA, Kuipers F, Stolk RP (2009) Non-alcoholic fatty liver disease is associated with cardiovascular disease risk markers. Obes Rev 10:412–419. https://doi.org/10.1111/j.1467-789X.2009.00594.x
Dowman JK, Tomlinson JW, Newsome PN (2010) Pathogenesis of non-alcoholic fatty liver disease. QJM 103:71–83. https://doi.org/10.1093/qjmed/hcp158
Targher G, Day CP, Bonora E (2010) Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med 363:1341–1350. https://doi.org/10.1056/NEJMra0912063
Gaggini M, Morelli M, Buzzigoli E et al (2013) Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 5:1544–1560. https://doi.org/10.3390/nu5051544
Szabo G, Mandrekar P, Golganiuc A (2007) Innate immune response and hepatic inflammation. Semin Liver Dis 27:339–350. https://doi.org/10.1055/s-2007-991511
Weiß J, Rau M, Geier A (2014) Non-alcoholic fatty liver disease: epidemiology, clinical course, investigation, and treatment. Dtsch Arztebl Int 111:447–452. https://doi.org/10.3238/arztebl.2014.0447
Bogdanos DP, Gao B, Gershwin ME (2013) Liver Immunology. In: Comprehensive physiology. Wiley, Hoboken
Serino M, Menghini R, Fiorentino L et al (2007) Mice heterozygous for tumor necrosis factor-a converting enzyme are protected from obesity-induced insulin resistance and diabetes. Diabetes 56:2541–2546. https://doi.org/10.2337/db07-0360
Wunderlich FT, Ströhle P, Könner AC et al (2010) Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab 12:237–249. https://doi.org/10.1016/j.cmet.2010.06.011
Ahmed W, Ziouzenkova O, Brown J et al (2007) PPARs and their metabolic modulation: new mechanisms for transcriptional regulation? J Intern Med 262:184–198. https://doi.org/10.1111/j.1365-2796.2007.01825.x
Poulsen LLC, Siersbæk M, Mandrup S (2012) PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol 23:631–639. https://doi.org/10.1016/j.semcdb.2012.01.003
Lefebvre P, Chinetti G, Fruchart JC, Staels B (2006) Sorting out the roles of PPARα in energy metabolism and vascular homeostasis. J Clin Invest 116:571–580. https://doi.org/10.1172/JCI27989
Forman BM, Chen J, Evans RM (1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci USA 94:4312–4317. https://doi.org/10.1073/pnas.94.9.4312
Giordano Mp G, Attianese Desvergne B (2015) Integrative and systemic approaches for evaluating PPARβ/δ (PPARD) function. Nucl Recept Signal 13:1–32. https://doi.org/10.1621/nrs.13001
Neels JG, Grimaldi PA (2014) Physiological functions of peroxisome proliferator-activated receptor. Physiol Rev 94:795–858. https://doi.org/10.1152/physrev.00027.2013
Heikkinen S, Auwerx J, Argmann CA (2007) PPARgamma in human and mouse physiology. Biochim Biophys Acta Lipids Lipid Metab 1771:999–1013. https://doi.org/10.1016/j.bbalip.2007.03.006
Day C (1999) Thiazolidinediones: a new class of antidiabetic drugs. Diabet Med 16:179–192. https://doi.org/10.1046/j.1464-5491.1999.00023.x
Crisafulli C, Cuzzocrea S (2008) The role endogenous and exogenous ligands for the peroxisome proliferator-activated receptor alpha (PPAR-alpha) in the regulation of inflammation in macrophAGES. Shock 32:62–73. https://doi.org/10.1097/SHK.0b013e31818bbad6
Rigamonti E, Chinetti-Gbaguidi G, Staels B (2008) Regulation of macrophage functions by PPAR-α, PPAR-γ, and LXRs in mice and men. Arterioscler Thromb Vasc Biol 28:1050–1059. https://doi.org/10.1161/ATVBAHA.107.158998
Escher P, Michalik L, Wahli W (2001) Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology 142:4195–4202
Hashimoto T, Cook WS, Qi C et al (2000) Defect in peroxisome proliferator-activated receptor ??-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem 275:28918–28928. https://doi.org/10.1074/jbc.M910350199
Delerive P, Gervois P, Fruchart JC, Staels B (2000) Induction of IκBα expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-α activators. J Biol Chem 275:36703–36707. https://doi.org/10.1074/jbc.M004045200
Ricote M, Glass C (2007) PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta Mol Cell Biol Lipids 1771:926–935. https://doi.org/10.1016/j.bbalip.2007.02.013
Glass CK, Saijo K (2010) Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol 10:365–376. https://doi.org/10.1038/nri2748
Mogilenko DA, Kudriavtsev IV, Shavva VS et al (2013) Peroxisome proliferator-activated receptor α positively regulates complement C3 expression but inhibits tumor necrosis factor-mediated activation of C3 gene in mammalian hepatic-derived cells. J Biol Chem 288:1726–1738. https://doi.org/10.1074/jbc.M112.437525
Ramanan S, Zhao W, Riddle DR, Robbins ME (2010) Review article: role of PPARs in radiation-induced brain injury. PPAR Res. https://doi.org/10.1155/2010/234975
Liu Q, Pan R, Ding L et al (2017) Rutin exhibits hepatoprotective effects in a mouse model of non-alcoholic fatty liver disease by reducing hepatic lipid levels and mitigating lipid-induced oxidative injuries. Int Immunopharmacol 49:132–141. https://doi.org/10.1016/j.intimp.2017.05.026
Heritage ML, Jaskowski LA, Bridle KR et al (2017) Combination curcumin and vitamin E treatment attenuates diet-induced steatosis in Hfe−/− mice. World J Gastrointest Pathophysiol 8:67. https://doi.org/10.4291/wjgp.v8.i2.67
Adi N, Adi J, Lassance-Soares RM, Kurlansky P, Yu H, Webster KA (2016) High protein/fish oil diet prevents hepatic steatosis in NONcNZO10 mice; association with diet/genetics-regulated micro-RNAs. J Diabetes Metab. https://doi.org/10.4172/2155-6156.1000676
Valenzuela R, Echeverria F, Ortiz M et al (2017) Hydroxytyrosol prevents reduction in liver activity of Δ-5 and Δ-6 desaturases, oxidative stress, and depletion in long chain polyunsaturated fatty acid content in different tissues of high-fat diet fed mice. Lipids Health Dis 16:1–16. https://doi.org/10.1186/s12944-017-0450-5
Hussein G, Goto H, Oda S et al (2006) Antihypertensive potential and mechanism of action of astaxanthin: III. Antioxidant and histopathological effects in spontaneously hypertensive rats. Biol Pharm Bull 29:684–688. https://doi.org/10.1248/bpb.29.684
Jia Y, Kim JY, Jun HJ et al (2012) The natural carotenoid astaxanthin, a PPAR-?? agonist and PPAR-?? antagonist, reduces hepatic lipid accumulation by rewiring the transcriptome in lipid-loaded hepatocytes. Mol Nutr Food Res 56:878–888. https://doi.org/10.1002/mnfr.201100798
Jia Y, Wu C, Kim J et al (2016) Astaxanthin reduces hepatic lipid accumulations in high-fat-fed C57BL/6J mice via activation of peroxisome proliferator-activated receptor (PPAR) alpha and inhibition of PPAR gamma and Akt. J Nutr Biochem 28:9–18. https://doi.org/10.1016/j.jnutbio.2015.09.015
Liss KHH, Finck BN (2017) PPARs and nonalcoholic fatty liver disease. Biochimie 136:65–74. https://doi.org/10.1016/j.biochi.2016.11.009
Shiri-Sverdlov R, Wouters K, Gorp PJV et al (2006) Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J Hepatol 44:732–741. https://doi.org/10.1016/j.jhep.2005.10.033
Zhang N, Lu Y, Shen X et al (2015) Fenofibrate treatment attenuated chronic endoplasmic reticulum stress in the liver of nonalcoholic fatty liver disease mice. Pharmacology 95:173–180. https://doi.org/10.1159/000380952
Abd El-Haleim EA, Bahgat AK, Saleh S (2016) Resveratrol and fenofibrate ameliorate fructose-induced nonalcoholic steatohepatitis by modulation of genes expression. World J Gastroenterol 22:2931–2948. https://doi.org/10.3748/wjg.v22.i10.2931
Laurin J, Lindor KD, Crippin JS et al (1996) Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis : a pilot study. Hepatology 23:1464–1467
Fernández-Miranda C, Pérez-Carreras M, Colina F et al (2008) A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis 40:200–205. https://doi.org/10.1016/j.dld.2007.10.002
El-Haggar SM, Mostafa TM (2015) Comparative clinical study between the effect of fenofibrate alone and its combination with pentoxifylline on biochemical parameters and liver stiffness in patients with non-alcoholic fatty liver disease. Hepatol Int 9:471–479. https://doi.org/10.1007/s12072-015-9633-1
Ishibashi S, Yamashita S, Arai H et al (2016) Effects of K-877, a novel selective PPARα modulator (SPPARMα), in dyslipidaemic patients: a randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis 249:36–43. https://doi.org/10.1016/j.atherosclerosis.2016.02.029
Honda Y, Kessoku T, Ogawa Y et al (2017) Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci Rep 7:1–11. https://doi.org/10.1038/srep42477
Takei K, Han SI, Murayama Y et al (2017) Selective peroxisome proliferator-activated receptor-α modulator K-877 efficiently activates the peroxisome proliferator-activated receptor-α pathway and improves lipid metabolism in mice. J Diabetes Investig 8:446–452. https://doi.org/10.1111/jdi.12621
da Costa Leite LFC, Veras Mourão RH, de Lima Mdo CA et al (2007) Synthesis, biological evaluation and molecular modeling studies of arylidene-thiazolidinediones with potential hypoglycemic and hypolipidemic activities. Eur J Med Chem 42:1263–1271. https://doi.org/10.1016/j.ejmech.2007.02.015
Araújo S, Soares e Silva A, Gomes F, et al (2016) Effects of the new thiazolidine derivative LPSF/GQ-02 on hepatic lipid metabolism pathways in non-alcoholic fatty liver disease (NAFLD). Eur J Pharmacol 788:306–314. https://doi.org/10.1016/j.ejphar.2016.06.043
Soares e Silva AK, de Oliveira Cipriano Torres D, dos Santos Gomes FO et al (2015) LPSF/GQ-02 Inhibits the development of hepatic steatosis and inflammation in a mouse model of non-alcoholic fatty liver disease (NAFLD). PLoS One 10:e0123787. https://doi.org/10.1371/journal.pone.0123787
Gross B, Staels B (2007) PPAR agonists: multimodal drugs for the treatment of type-2 diabetes. Best Pract Res Clin Endocrinol Metab 21:687–710. https://doi.org/10.1016/j.beem.2007.09.004
Nadra K, Anghel SI, Joye E et al (2006) Differentiation of trophoblast giant cells and their metabolic functions are dependent on peroxisome proliferator-activated receptor. Mol Cell Biol 26:3266–3281. https://doi.org/10.1128/MCB.26.8.3266-3281.2006
Grimaldi PA (2007) Regulatory functions of PPARβ in metabolism: implications for the treatment of metabolic syndrome. Biochim Biophys Acta Mol Cell Biol Lipids 1771:983–990. https://doi.org/10.1016/j.bbalip.2007.02.006
Leibowitz MD, Fiévet C, Hennuyer N et al (2000) Activation of PPARdelta alters lipid metabolism in db/db mice. FEBS Lett 473:333–336. https://doi.org/10.1016/S0014-5793(00)01554-4
Oliver WR, Shenk JL, Snaith MR et al (2001) A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci USA 98:5306–5311. https://doi.org/10.1073/pnas.091021198
Graham TL, Mookherjee C, Suckling KE et al (2005) The PPARδ agonist GW0742X reduces atherosclerosis in LDLR−/− mice. Atherosclerosis 181:29–37. https://doi.org/10.1016/j.atherosclerosis.2004.12.028
Stienstra R, Duval C, Müller M, Kersten S (2007) PPARs, obesity, and inflammation. PPAR Res. https://doi.org/10.1155/2007/95974
Shearer BG, Billin AN (2007) The next generation of PPAR drugs: do we have the tools to find them? Biochim Biophys Acta Mol Cell Biol Lipids 1771:1082–1093. https://doi.org/10.1016/j.bbalip.2007.05.005
Michalik L, Auwerx J, Berger JP et al (2006) International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev 58:726–741. https://doi.org/10.1124/pr.58.4.5.(NR1C1)
Odegaard J, Ricardo-Gonzalez R, Goforth MH et al (2007) Macrophage-specific PPAR&ggr; controls alternative activation and improves insulin resistance. Nature 447:1116–1120. https://doi.org/10.1038/nature05894.Macrophage-specific
Serrano-Marco L, Barroso E, El Kochairi I et al (2012) The peroxisome proliferator-activated receptor (PPAR) β/δ agonist GW501516 inhibits IL-6-induced signal transducer and activator of transcription 3 (STAT3) activation and insulin resistance in human liver cells. Diabetologia 55:743–751. https://doi.org/10.1007/s00125-011-2401-4
Sznaidman ML, Haffner CD, Maloney PR et al (2003) Novel selective small molecule agonists for peroxisome proliferator-activated receptor δ (PPARδ)—synthesis and biological activity. Bioorganic Med Chem Lett 13:1517–1521. https://doi.org/10.1016/S0960-894X(03)00207-5
Kostadinova R, Montagner A, Gouranton E et al (2012) GW501516-activated PPARβ/δ promotes liver fibrosis via p38-JNK MAPK-induced hepatic stellate cell proliferation. Cell Biosci 2:1–16. https://doi.org/10.1186/2045-3701-2-34
Lee HJ, Yeon JE, Ko EJ et al (2015) Peroxisome proliferator-activated receptor-delta agonist ameliorated inflammasome activation in nonalcoholic fatty liver disease. World J Gastroenterol 21:12787–12799. https://doi.org/10.3748/wjg.v21.i45.12787
Barroso E, Rodríguez-Calvo R, Serrano-Marco L et al (2011) The PPARβ/δ activator GW501516 prevents the down-regulation of AMPK caused by a high-fat diet in liver and amplifies the PGC-1α-lipin 1-PPARα pathway leading to increased fatty acid oxidation. Endocrinology 152:1848–1859. https://doi.org/10.1210/en.2010-1468
Lee MY, Chung CH, Lee MY et al (2012) Peroxisome proliferator-activated receptor δ agonist attenuates hepatic steatosis by anti-inflammatory mechanism. Exp Mol Med 44:578–585. https://doi.org/10.3858/emm.2012.44.10.066
Shan W, Palkar PS, Murray IA et al (2008) Ligand activation of peroxisome proliferator-activated receptor β/δ (PPARβ/δ) attenuates carbon tetrachloride hepatotoxicity by downregulating proinflammatory gene expression. Toxicol Sci 105:418–428. https://doi.org/10.1093/toxsci/kfn142
Sanderson LM, Boekschoten MV, Desvergne B et al (2010) Transcriptional profiling reveals divergent roles of PPARα and PPARβ/δ in regulation of gene expression in mouse liver. Physiol Genomics 41:42–52. https://doi.org/10.1152/physiolgenomics.00127.2009
Tontonoz P, Hu E, Graves RA et al (1994) mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 8:1224–1234
Tontonoz P, Hu E, Devine J et al (1995) PPAR gamma 2 regulates adipose expression of the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol 15:351–357
Schoonjans K, Peinado-Onsurbe J, Lefebvre AM et al (1996) PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J 15:5336–5348
Sfeir Z, Ibrahimi A, Amri E et al (1997) Regulation of FAT/CD36 gene expression: further evidence in support of a role of the protein in fatty acid binding/transport. Prostaglandins Leukot Essent Fat Acids 57:17–21. https://doi.org/10.1016/S0952-3278(97)90487-7
Tontonoz P, Hu E, Spiegelman BM (1994) Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 79:1147–1156. https://doi.org/10.1016/0092-8674(94)90006-X
Yu S, Matsusue K, Kashireddy P et al (2003) Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor γ1 (PPARγ1) overexpression. J Biol Chem 278:498–505. https://doi.org/10.1074/jbc.M210062200
Gavrilova O, Haluzik M, Matsusue K et al (2003) Liver peroxisome proliferator-activated receptor γ contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem 278:34268–34276. https://doi.org/10.1074/jbc.M300043200
Moran-Salvador E, Lopez-Parra M, Garcia-Alonso V et al (2011) Role for PPAR in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J 25:2538–2550. https://doi.org/10.1096/fj.10-173716
Yu S, Matsusue K, Kashireddy P et al (2003) Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor γ1 (PPARγ1) overexpression. J Biol Chem 278:498–505. https://doi.org/10.1074/jbc.M210062200
Pettinelli P, Videla LA (2011) Up-regulation of PPAR-γ mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab 96:1424–1430. https://doi.org/10.1210/jc.2010-2129
Nakamuta M, Kohjima M, Morizono S et al (2005) Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med 16:631–635
Chinetti G, Fruchart JC, Staels B (2003) Peroxisome proliferator-activated receptors: new targets for the pharmacological modulation of macrophage gene expression and function. Curr Opin Lipidol 14:459–468. https://doi.org/10.1097/01.mol.0000092630.86399.00
Pascual G, Fong AL, Ogawa S et al (2005) A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 437:759–763. https://doi.org/10.1038/nature03988
Konstantinopoulos PA, Vandoros GP, Sotiropoulou-Bonikou G et al (2007) NF-κB/PPARγ and/or AP-1/PPARγ “on/off” switches and induction of CBP in colon adenocarcinomas: correlation with COX-2 expression. Int J Colorectal Dis 22:57–68. https://doi.org/10.1007/s00384-006-0112-y
Villanueva CJ, Tontonoz P (2010) Licensing PPARγ to work in macrophages. Immunity 33:647–649. https://doi.org/10.1016/j.immuni.2010.11.017
Wynn TA, Chawla A, Pollard JW (2013) Origins and hallmarks of macrophages: development, homeostasis, and disease. Nature 496:445–455. https://doi.org/10.1038/nature12034.origins
Ginhoux F, Schultze JL, Murray PJ et al (2016) New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 17:34–40. https://doi.org/10.1038/ni.3324
Baffy G (2009) Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J Hepatol 51:212–223. https://doi.org/10.1016/j.jhep.2009.03.008
Dixon LJ, Barnes M, Tang H et al (2013) Kupffer cells in the liver. In: Comprehensive Physiology. Wiley, Hoboken
Luo W, Xu Q, Wang Q et al (2017) Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep 7:1–13. https://doi.org/10.1038/srep44612
Zhong X, Liu H (2017) Honokiol attenuates diet-induced nonalcoholic steatohepatitis by regulating macrophage polarization through activating PPARgamma. J Gastroenterol Hepatol 33:524–532. https://doi.org/10.1111/jgh.13853
Bensinger SJ, Tontonoz P (2008) Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 454:470–477. https://doi.org/10.1038/nature07202
Isley WL (2003) Hepatotoxicity of thiazolidinediones. Expert Opinion Drug Saf 2:581–586
Yki-Järvinen H (2004) Thiazolidinediones. N Engl J Med 351:1106–1118. https://doi.org/10.1056/NEJMra041001
Nan YM, Han F, Kong LB et al (2011) Adenovirus-mediated peroxisome proliferator activated receptor gamma overexpression prevents nutritional fibrotic steatohepatitis in mice. Scand J Gastroenterol 46:358–369. https://doi.org/10.3109/00365521.2010.525717
Nan YM, Fu N, Wu WJ et al (2009) Rosiglitazone prevents nutritional fibrosis and steatohepatitis in mice. Scand J Gastroenterol 44:358–365. https://doi.org/10.1080/00365520802530861
Deng W, Meng Z, Sun A, Yang Z (2017) Pioglitazone suppresses inflammation and fibrosis in nonalcoholic fatty liver disease by down-regulating PDGF and TIMP-2: evidence from in vitro study. Cancer Biomarkers 20:411–415. https://doi.org/10.3233/CBM-170157
van der Veen JN, Lingrell S, Gao X, et al (2016) Pioglitazone attenuates hepatic inflammation and fibrosis in phosphatidylethanolamine N-methyltransferase-(PEMT) deficient mice. Am J Physiol Gastrointest Liver Physiol 4:ajpgi.00243.2015. https://doi.org/10.1152/ajpgi.00243.2015
Chen W, Lin Y, Zhou X et al (2016) Rosiglitazone protects rat liver against acute liver injury by the NF-kB pathway. Can J Physiol Pharmacol. https://doi.org/10.1139/cjpp-2015-0230
Boettcher E, Csako G, Pucino F et al (2012) Meta-analysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther 35:66–75. https://doi.org/10.1111/j.1365-2036.2011.04912.x
Sanyal AJ, Chalasani N, Kowdley KV et al (2010) Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 362:1675–1685. https://doi.org/10.1056/NEJMoa0907929
Lago RM, Singh PP, Nesto RW (2007) Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidinediones: a meta-analysis of randomised clinical trials. Lancet 370:1129–1136. https://doi.org/10.1016/S0140-6736(07)61514-1
Tuccori M, Filion KB, Yin H et al (2016) Pioglitazone use and risk of bladder cancer: population based cohort study. BMJ. https://doi.org/10.1136/bmj.i1541
Lecka-Czernik B (2010) Bone loss in diabetes: use of antidiabetic thiazolidinediones and secondary osteoporosis. Curr Osteoporos Rep 8:178–184. https://doi.org/10.1007/s11914-010-0027-y
Marchesini G, Day CP, Dufour JF et al (2016) EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. J Hepatol 64:1388–1402. https://doi.org/10.1016/j.jhep.2015.11.004
Chalasani N, Younossi Z, Lavine JE et al (2012) The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 55:2005–2023. https://doi.org/10.1002/hep.25762
García-Ruiz I, Rodríguez-Juan C, Díaz-Sanjuán T et al (2007) Effects of rosiglitazone on the liver histology and mitochondrial function in ob/ob mice. Hepatology 46:414–423. https://doi.org/10.1002/hep.21687
Brand CL, Gotfredsen CF, Fleckner J, Fledelius C, Hansen BF, Andersen B, Ye JM, Sauerberg P, Wassermann KSJ (2002) Dual PPARalpha/gamma activation provides enhanced improvement of insulin sensitivity and glycemic control in ZDF rats. Am J Physiol Endocrinol Metab 284:E841–E854. https://doi.org/10.1152/ajpendo.00348.2002
Harrity T, Farrelly D, Tieman A et al (2006) Preserves-cell function in db/db mice. Diabetes 55:240–248
Ye J-M, Iglesias MA, Watson DG et al (2003) PPARalpha/gamma ragaglitazar eliminates fatty liver and enhances insulin action in fat-fed rats in the absence of hepatomegaly. Am J Physiol Endocrinol Metab 284:531–540. https://doi.org/10.1152/ajpendo.00299.2002
Henry RR, Lincoff AM, Mudaliar S et al (2009) Effect of the dual peroxisome proliferator-activated receptor-α/γ agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (SYNCHRONY): a phase II, randomised, dose-ranging study. Lancet 374:126–135. https://doi.org/10.1016/S0140-6736(09)60870-9
Hamrén B, Öhman KP, Svensson MK, Karlsson MO (2012) Pharmacokinetic-pharmacodynamic assessment of the interrelationships between tesaglitazar exposure and renal function in patients with type 2 diabetes mellitus. J Clin Pharmacol 52:1317–1327. https://doi.org/10.1177/0091270011416937
Jain MR, Giri SR, Bhoi B et al (2017) Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. https://doi.org/10.1111/liv.13634
Cariou B, Hanf R, Lambert-Porcheron S et al (2013) Dual peroxisome proliferator- activated receptor α/δ agonist gft505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 36:2923–2930. https://doi.org/10.2337/dc12-2012
Cariou B, Zaïr Y, Staels B, Bruckert E (2011) Effects of the new dual PPARα/δ agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 34:2008–2014. https://doi.org/10.2337/dc11-0093
Staels B, Rubenstrunk A, Noel B et al (2013) Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 58:1941–1952. https://doi.org/10.1002/hep.26461
Ratziu V, Harrison SA, Francque S et al (2016) Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150:1147–1159e5. https://doi.org/10.1053/j.gastro.2016.01.038
Wettstein G, Luccarini J-M, Poekes L et al (2017) The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol Commun 1:524–537. https://doi.org/10.1002/hep4.1057
Acknowledgements
The authors would like to thank the following Brazilian foundations for financial support: Conselho Nacional de Desenvolvimento Científico e Tecnológico (the Brazilian National Council for Scientific and Technological Development) (CNPq; #301777/2012-8) and CAPES/PNPD Program for Silva AKS post-Doc scholarship.
Author information
Authors and Affiliations
Contributions
The two authors contributed equally in the elaboration of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Rights and permissions
About this article

Cite this article
Silva, A.K.S., Peixoto, C.A. Role of peroxisome proliferator-activated receptors in non-alcoholic fatty liver disease inflammation. Cell. Mol. Life Sci. 75, 2951–2961 (2018). https://doi.org/10.1007/s00018-018-2838-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2838-4