
Abstract
Non-alcoholic fatty liver disease (NAFLD) is currently the most common liver disease. Non-alcoholic steatohepatitis (NASH) is the advanced form of NAFLD to which a subpopulation of NAFLD patients progress. While NAFLD is manifested by hepatic steatosis, NASH has the additional features of inflammation, cell injury and ballooning, and mitochondrial changes and/or fibrosis. The understanding of the pathogenesis of NAFLD has been evolving but is still not complete. Fat accumulation in the liver is the first step in the disease process. It occurs when there is increased caloric intake and de novo lipogenesis. Peroxisome proliferator-activated receptors (PPARs), Farnesoid X receptor (FXR) and liver X receptor (LXR) have been shown to play a role in pathogenesis. Many mechanisms are known to be involved in the progress of NAFLD to NASH, including oxidative stress, endotoxins, cytokines, mitochondrial dysfunction and induction of the cytochrome P450 system. More recently discovered possible mechanisms include gut microbiota, dietary fructose, toll-like receptors (TLRs), nucleotide-binding oligomerization domain receptors (NOD-like receptors), and the hedgehog signaling pathway.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Diagnosis is performed by ruling out other causes of elevation of liver enzymes and performing imaging studies. Liver biopsy is still the gold standard to differentiate NAFLD (or simple steatosis) from NASH and to stage the disease. New magnetic resonance imaging (MRI) techniques have been shown to be very promising in quantifying fat (MR Proton Density Fat Fraction) (MR-PDFF) and in detect fibrosis (MRE elastography). There is currently no Food and Drug Administration-approved treatment. Weight loss and exercise are generally the first recommended approach. Vitamin E and pioglitazone have been shown to improve liver enzymes and histology; however, the long-term effects are unknown. Finally, statins have been shown to be safe and helpful in NAFLD and NASH patients. Statins are recommended for the treatment of dyslipidemia in these patients. Randomized controlled trials are needed to assess the effects of statins on NAFLD/NASH.
Introduction
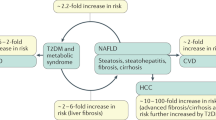
Non-alcoholic fatty liver disease (NAFLD) has emerged as a major health problem in the last decade in parallel with the increasing epidemic of obesity [1]. It has been recognized as the hepatic manifestation of metabolic syndrome and is the most common reason for referral to hepatologists today [2]. The disease has a variable histological course, with some patients only accumulating fat in the liver and not progressing beyond simple steatosis while a subpopulation of patients progress to the more advanced stage of nonalcoholic steatohepatitis (NASH) in which inflammation and cell injury occur [3]. NASH can lead to liver cirrhosis and hepatocellular carcinoma (HCC) and is expected to be the leading cause of liver transplant and liver-associated mortality in the coming decade [4]. It is not surprising that the liver is the major affected organ with the epidemic of obesity as it plays a central role in lipid metabolism through the synthesis of apoproteins and lipoproteins as well as de novo lipogenesis [5]. Since the liver regulates lipid metabolism and secretion, the disruption of normal physiologic lipid regulation can lead to fat accumulation in the liver and subsequently liver injury. In this chapter we highlight the different aspects of the disease, including natural history, epidemiology, pathophysiology, diagnosis and treatment. In addition, we discuss the role of statins and whether they may be harmful or beneficial in NAFLD patients.
Epidemiology and Natural History
NAFLD has become the most common cause of asymptomatic elevation of liver enzymes and the most common reason for referral to liver clinics [1, 6]. While initially thought to be exclusively a disease of adults, it has become the most common liver disease among adolescents in the United States, with older age often being predictive of more advanced disease [7–9]. It is estimated that one in three adult Americans is afflicted with NAFLD, with a higher prevalence in Hispanic populations likely due to the higher prevalence of obesity and insulin resistance in this ethnic group [1]. The importance of genetic and epigenetic changes in the etiology and pathogenesis of NAFLD has been increasingly recognized. Genome-wide association studies have led to increased understanding of genomic variations of NAFLD. Patatin-like phospholipase domain containing family member A3 (PNPLA3, SNP rs738409, encoding I148M), also termed adiponutrin, may be of particular importance [10]. A series of studies has validated that PNPLA3 is associated with increased hepatic fat levels and hepatic inflammation [11]. This allele is most common in Hispanics, with hepatic fat content being more than two-fold higher in G homozygous subjects than in non-carriers. G allele frequency is lower in people of European descent and is lowest in African Americans who constitute the population least likely to have hepatic fat accumulation [11] .
Around 10–20 % of patients with simple steatosis progress to NASH; of those, 10–20 % progress to cirrhosis over 10–20 years [12, 13]. HCC may develop in those who progress to cirrhosis; however, the incidence rate is still unknown [14]. In addition, many reports have described cases of HCC in NASH patients that have developed without underlying cirrhosis [14]. Epidemiologic risk factors associated with NAFLD and NASH include obesity, type 2 diabetes and hyperlipidemia [15]. Metabolic syndrome has been shown to increase the risk of NASH and advanced fibrosis, in particular if there is coexisting diabetes [15]. Cardiovascular diseases have been shown to play a significant role in the natural history, morbidity and mortality of NAFLD [16, 17]. Indeed, cardiovascular events have been thought to be the leading cause of death in NAFLD [18, 19].
Pathogenesis of NAFLD/NASH
A two-hit model has been proposed to explain the progression of NAFLD. The role of hyperinsulinemia and insulin resistance in lipid accumulation in the liver is essential in the disease process [20]. The disease develops with the abnormal hepatic accumulation of triglycerides (TG), which can progress to NASH in some patients. Factors that promote the progression from steatosis to NASH in humans are incompletely understood but include genetic and behavioral factors [21].
Development of Hepatic Steatosis
Increased Fatty Acid Synthesis, Increased Triglyceride Storage and Impaired Secretion
Lipid metabolism is imbalanced in the liver in the setting of obesity and insulin resistance, leading to accumulation of triglycerides in the liver. This process is usually due to increased free fatty acid (FFA) flux from adipose tissue to the liver, increased caloric intake, and increased de novo lipogenesis in the liver [22]. As the adipose tissue is increased with obesity , there is increased hormone-sensitive lipase (HSL) activity and accelerated release of FFA from adipose cells into circulation. FFA uptake by the liver is increased proportionally to the increase in FFA in the blood circulation [22, 23]. The fate of FFA in the liver is either metabolism via oxidation to generate ATP through β-oxidation in the mitochondria or esterification to produce triglycerides. These triglycerides (TG) are either packaged into very-low-density lipoproteins (VLDL) for export or are used for the production of lipids such as phospholipids [22]. These processes have been shown to be impaired in NAFLD, leading to imbalance between the uptake and metabolism of FFA which, in turn, leads to TG accumulation in the liver [22]. Furthermore, when there is increased caloric intake, glucose gets converted to pyruvate which enters the Krebs cycle in the mitochondria. Acetyl-CoA is formed from pyruvate by pyruvate dehydrogenase in the mitochondria. Acetyl-CoA produced in the mitochondria is condensed with oxaloacetate by citrate synthase to form citrate. In the presence of ATP and Coenzyme A, citrate lyase catalyzes the cleavage of citrate to yield acetyl CoA, oxaloacetate, ADP, and orthophosphate. Acetyl-CoA carboxylase (ACC), a biotin-dependent enzyme, catalyzes the irreversible carboxylation of acetyl-CoA to produce malonyl-CoA through its two catalytic activities, biotin carboxylase (BC) and carboxyltransferase. Malonyl-CoA is utilized in fatty acid biosynthesis by the enzyme malonyl coenzyme A:acyl carrier protein transacylase (MCAT). MCAT serves to transfer malonate from malonyl-CoA to the terminal thiol of holo-acyl carrier protein (ACP). Malonyl-CoA also converts to palmitic acid via fatty acid synthase (FAS) [21, 24]. Subsequently, the enzymes stearyl-CoA desaturase (SCD) and long chain fatty acid elongase are used to create other fatty acids such as palmitoleic acid (C16:1), stearic acid (C18:0), or oleic acid (C18:1) [21, 24]. Ultimately these fatty acids form triglycerides.
Along with increased FFA flux into the liver and increased fatty acid formation due to increased caloric intake, de novo lipogenesis is augmented [22, 25]. In the normal state, de novo lipogenesis contributes to less than 5 % of fatty acid, TG, and VLDL synthesis [26]. However, in NAFLD patients this process is upregulated, contributing to synthesis of up to 26 % of fatty acids, TG, and VLDL [25, 27]. Hyperglycemia stimulates carbohydrate response element-binding protein (ChREBP), which transcriptionally stimulates the liver-type pyruvate kinase (L-PK), a key enzyme in glycolysis. LPK stimulates the entry of pyruvate into the mitochondria and its conversion into citrate, which forms acetyl-CoA and hence increases fatty acid synthesis [28, 29]. On the other hand, hyperinsulinemia leads to activation of a membrane-bound transcription factor, sterol regulatory element-binding protein-1c (SREBP-1c), which triggers all lipogenesis genes and thus increases de novo fatty acid synthesis [30]. One of the important effects of increased fatty acid synthesis is increased malonyl-CoA which inhibits carnitine palmitoyl transferase-1 (CPT-1), the protein responsible for fatty acid transport into the mitochondria [31]. TG synthesis has also been shown to be affected by increasing levels of glycerol-3-phosphate acyltransferase (GPAT); and VLDL secretion is impaired by decreasing expression of microsomal transfer protein [32]. Gluconeogenesis is also diminished secondary to SREBP-1c inhibition of phosphoenolpyruvate carboxykinase (PEPCK) [33]. The net result is increased de novo lipogenesis and impaired VLDL packaging and secretion [34].
The Role of PPAR, LXR and FXR Receptors
Peroxisome proliferator-activated receptors (PPARs) are a group of nuclear receptor proteins that function as transcription factors, regulating the expression of genes and playing essential roles in lipid metabolism and hepatic steatosis . There are three PPAR isotypes, including PPARα, PPARβ, and PPARγ. PPARα is mainly expressed in the liver where it increases the use of fatty acids [35]. It induces the transcription of genes for movement of fatty acids into the cell and mitochondria including fatty acid transport protein and CPT1. The result of PPARα activation is increased fatty acid uptake and oxidation, lipolysis, and clearance of ApoB-containing lipoprotein [36]. The role of PPARβ in hepatic steatosis is not completely understood but it is thought to play a role in fatty acid transportation and oxidation. PPARγ is mainly located in adipose tissue but it is also formed to a lesser extent in skeletal muscle, liver, pancreatic beta cells, myeloid dendritic cells, and macrophages. PPARγ agonists (thiazolidinediones and others now being studied or in development) have been shown to act on adipocyte tissue in a way that increases fatty acid uptake and storage and increases insulin sensitivity [36]. This results in redistribution of fat from the liver into the subcutaneous fat. PPARγ also increases production of adiponectin which has significant effects on fatty acid oxidation and insulin sensitivity [37].
A recent role for the liver X receptor (LXR), a member of the nuclear receptor family of transcription factors, has been postulated [38]. The LXR has many similarities to PPARα as both are transcription factors that belong to class II nuclear receptors [39]. LXRα is found mostly in hepatocytes, adipose tissue, and macrophages, whereas LXRβ is more widespread [40]. LXRs induce the key enzymes in the de novo lipogenesis pathway including acetyl-CoA carboxylase 1 (ACC1), FAS, and stearoyl CoA desaturase 1 (SCD1) [41, 42]. Both SREBP -1c and ChREBP have been shown to be target genes of LXRs. LXRs mainly increase hepatic lipogenesis by upregulating the expression of SREBP-1c and to a lesser extent by activating ChREBP [42]. A recent study has shown the role of the LXR-lysophosphatidyl acyltransferase 3 (Lpcat3) pathway in modulating phospholipid metabolism, ER stress and inflammation [38]. Lpcat3 catalyzes the formation of phosphatidylcholine (PC) from saturated lysophosphatidylcholines (LysoPC) and unsaturated fatty acyl-CoAs, with PC containing unsaturated fatty acids preferentially synthesized by this enzyme [43, 44]. It has been shown that increased levels of saturated fatty acids lead to changes in ER membrane composition and induce ER stress [45]. LXR activates Lpcat3 leading to formation of polyunsaturated phospholipids, which decreases membrane saturation. This membrane remodeling leads to decreased ER stress in liver cells. Moreover, the LXR-Lpcat3 pathway decreases hepatic inflammation through a c-Jun NH2-terminal kinase (JNK) pathway-mediated mechanism.
The farnesoid X receptor (FXR) is expressed mainly in the liver, intestine, adrenal glands and kidneys. It has also been shown to be expressed in lower levels in the heart, adipose tissue and vasculature [46, 47]. FXR inhibits SREBP-1c and FAS leading to reduced lipogenesis [48]. It also affects glucose metabolism in the liver by reducing gluconeogenesis via the downregulation of PEPCK and glucose-6-phosphatase (G6Pase). FXR reduces conversion of cholesterol to bile acids by inhibiting enzymes involved in bile acid synthesis such as cytochrome P450 7A1 (CYP7A1) and CYP8B1 [49]. Prior to secretion into the bile, bile acids are conjugated to either glycine or taurine. FXR enhances bile acid conjugation and stimulates the transport of bile acids to the gallbladder [50]. It also decreases bile acid absorption in the small intestine and stimulates reabsorption recycling of bile acids to the liver. FXR reduces hepatic uptake of bile acids and promotes the release of fibroblast growth factor 15 (FGF15) and FGF19 from the intestine [51]. FGF15 and FGF19 circulate to the liver and reduce CYP7A1 expression, thus repressing bile acid synthesis [51]. Recently, bile acids have been shown to play a significant role in glucose homeostasis. They regulate cholesterol, glucose, and metabolic homeostasis in addition to regulating their own synthesis [50]. FXR knockout mice have been shown to have elevated plasma triglycerides and cholesterol levels, impaired glucose hemostasis and decreased insulin sensitivity [52]. FXR agonists inhibit hepatic gluconeogenesis and stimulate glycogen synthesis and storage, resulting in an overall low glucose level [53]. A recent trial in NAFLD patients has shown that an FXR agonist reduces liver inflammation and fibrosis markers in addition to improving insulin sensitivity [54].
Another mediator that has been shown to play a role in metabolism is adenosine monophosphate-activated protein kinase (AMPK). AMPK stimulates fatty acid oxidation and glucose transport. In the liver, it augments fatty acid oxidation and decreases glucose output and cholesterol and triglyceride synthesis, metabolic effects that result in lowered blood glucose levels in hyperglycemic individuals [55]. Two types of oral antihyperglycemic drugs, the biguanidines and thiazolidinediones, have been shown to work in part by directly or indirectly activating AMPK [55]. For example, metformin is known to activate AMPK [56]. Once energy in increased AMP accumulates, it stimulates AMPK and leads to formation of adenosine triphosphate. AMPK inhibits ACC, decreases expression of SREBP-1 and stimulates deactivation of ChREBP. It also increases β-oxidation [57].
The Emerging Role of Gut Microbiota
A relationship between gut microbiota, the collective term for the 100 trillion bacteria that inhabit the GI tract. and the development of NAFLD has been demonstrated in mice and humans. Transplantation of normal cecal microbiota into germ-free mice induced a 60 % increase in body fat and a twofold increase in hepatic fat [58]. One of the first observations of the relationship between gut microbiota and hepatic steatosis was in the 1980s when steatosis, NASH and bacterial overgrowth were seen to develop after intestinal bypass [59]. Interestingly, steatosis was reversed by metronidazole, suggesting a causative role of the microbiota in fatty liver disease and antibiotics as potential candidates for treatment [59].
There are several potential mechanisms through which gut microbiota may cause hepatic steatosis and NASH. These may include stimulation of obesity , increased gut permeability, inflammation and altered immune balance, modulation of dietary choline metabolism increasing ethanol production by the bacteria, and regulation of bile acid metabolism [60]. Bile acids damage bacterial cell membranes by interacting with membrane phospholipids which results in bactericidal activity. Conversely, the gut microbiota modulates bile acid metabolism through FXR stimulation. Bile acids are ligands for a G-protein coupled receptor (TGR5/Gpbar-1) and activate FXR. Therefore, through bile acid metabolism and FXR/TGR5 signaling, gut flora could contribute indirectly to the development of NAFLD [61].
Progression from Simple Steatosis to NASH
While increased storage of circulating FFA, increased de novo lipogenesis and impaired β-oxidation and TG secretion may explain the significant triglyceride accumulation in simple steatosis, a “second hit” or more precisely “multiple hits” are thought to be required to promote inflammation, cell death, and fibrosis and the resultant progression to NASH. There are many potential candidates for the additional hits which may play a role in the shift from steatosis to NASH, including oxidative stress, iron, endotoxins , cytokines, mitochondrial dysfunction and induction of the cytochrome P450 system . Lipotoxicity and oxidative stress are key drivers of disease progression. Increased reactive oxygen species (ROS) production has been shown to play a major role in progression to NASH [22]. Sources of increased ROS production include proinflammatory cytokines (such as TNF-α and IL6), iron overload, overburdened and dysfunctional mitochondria, CYPs, and peroxisomes [21]. The role of mitochondria in NASH development has been shown to be essential. In normal conditions fatty acids get oxidized mainly by the mitochondria via β-oxidation and then get transported to the mitochondrial respiratory chain (MRC), leading to production of ATP and generation of CO2 and water. A small portion of oxygen is not utilized, leading to formation of ROS including superoxide, hydrogen peroxide and the hydroxyl radical species [34, 62]. In the setting of increased free fatty acids flux the mitochondria exhaust and fatty acids are then metabolized at other sites in hepatocytes including peroxisomes (β-oxidation) and the CYP enzymes of the smooth endoplasmic reticulum (ω-oxidation) [62, 63]. In the mitochondria, long-chain fatty acids are oxidized and transported using the carnitine shuttle enzymes carnitine palmitoyltransferase I (CPT-I) and carnitine palmitoyltransferase II (CPT-II). This leads to formation of shorter acyl-CoA moieties, acetyl-CoA. This oxidation process is associated with the reduction of oxidized NAD+ and FAD to NADH and FADH2, which produces electrons that transfer to the MRC. These partially reduced oxygen molecules (ROS) lead to oxidant stress as the mitochondria are overwhelmed [62, 64]. CYP2E1 then oxidizes the rest of the excess free fatty acids which further increases ROS production within hepatocytes. Using immunostaining, CYP2E1 has been shown to be increased in NASH patients [65, 66]. Other excess free fatty acids undergo oxidation in the peroxisomes in which electrons from FADH2 and NADH are transferred directly to oxygen leading to further formation of ROS. These overwhelming processes in the mitochondria result in mitochondrial dysfunction manifested by depletion of ATP, and decreased mitochondrial DNA levels and proteins produced by mitochondrial genes [62]. Crystalline inclusions within the mitochondrial matrix seen by electron microscopy and megamitochondria detected by microscopy have been observed in NASH patients [62, 64, 67].
FFAs can also lead to lipotoxicity in an apoptosis process due to translocation into lysosomes resulting in release of lysosomal enzymes and subsequently activation of nuclear factor (NF)-κB activation and TNF-α overexpression in the liver. TNF-α activates two pathways including (NF)-κB and JNK [68]. JNK leads to insulin resistance by phosphorylation of insulin receptor substrate 1 (IRS-1). The (NF)-κB pathway leads to production of proinflammatory cytokines. These pathways have been shown to be activated in NASH patients [68, 69]. The results of the previous process with a central role of the mitochondria collectively lead to NASH progression [70]. Other possible etiologies have been considered in the last few years including the roles of dietary fructose, toll-like receptors (TLRs), nucleotide-binding oligomerization domain receptors (NOD-like receptors), and the hedgehog signaling pathway [71–84].
Fructose consumption has gained significant attention as a possible cause of NAFLD. High-fructose corn syrup has been shown to increase endoplasmic reticulum stress, activate JNK, induce mitochondrial dysfunction, and increase apoptotis in hepatocytes [80, 85, 86]. In addition, dietary fructose intake has been found to have close association with gut-derived endotoxemia, toll-like receptor 4 and NAFLD [87]. Human studies have shown correlation between high fructose consumption and NAFLD [83, 88].
TLRs and NOD-like receptors (NLRs) are pattern recognition signal receptors involved in activation of the innate immune system [89]. In general, activation of NLRs and TLRs induces pro-inflammatory cytokine production, as well as recruitment in the liver of immune cells, including macrophages and T cells, resulting in chronic low-grade inflammation that promotes insulin resistance and contributes to development of fatty liver [90]. In response to pathogens, TLR signaling induces proinflammatory cytokines in immune cells [91]. With the increased intestinal permeability in NASH patients, intestine-derived pathogen-associated molecular patterns (PAMPs), including lipopolysaccharide (LPS), translocate to the liver and activate TLR signaling cascades [92]. The activation of TLR2, TLR4 and TLR9 induce production of various cytokines, including transforming growth factor-beta (TGF-β), interleukin 1 beta (IL-1β), and tumor necrosis factor-alpha (TNF-α), which in turn stimulate hepatic stellate cells (HSC), leading to lipid accumulation and apoptosis in liver cells [91, 93–95]. Moreover, apoptotic hepatocytes activate Kuppfer cells via TLRs and produce inflammatory cytokines such as interleukin 6 (IL-6). TLR2, TLR4, and TLR9 have been reported to be associated with steatohepatitis [94, 96, 97]. In experimental NASH, TLR4 and TLR9 have been shown to promote hepatic inflammation and fibrosis [94, 95], while inactivation of TLR4 has been shown to lead to attenuation of steatosis and NASH [87, 98]. It has also been reported that TLR2 and palmitic acid cooperatively contribute to the development of NASH through inflammasome activation [91]. NLR activation leads to assembly of the caspase 1-containing inflammasome, resulting in inflammation and apoptosis [90]. NOD1 and NOD2 have both been associated with many inflammatory diseases, and both NOD1 and NOD2 mRNA and protein have been shown to be highly expressed in hepatocytes [99]
The Hedgehog pathway is one of the complex signaling cascades that are important for the immune response [71]. Studies in mice have shown that the development of fibrosis and steatohepatitis correlate with the intensity and duration of Hedgehog pathway activation that develops during fatty liver injury [100]. This pathway is essential in embryogenesis and can be triggered in adult life in the setting of tissue regeneration [101]. It has been shown that hepatocyte injury in an environment of lipotoxicity can produce Hedgehog pathway activation which in turn stimulates inflammatory cells and, in particular, natural killer T (NKT) cells. It also promotes growth and hepatocyte differentiation but at the same time activates stellate cells leading to fibrosis [72, 102, 103]. It has been hypothesized that differences in Hedgehog pathway activity may contribute to the varying outcomes of fatty liver injury in NAFLD patients. In a study of a large cohort of NAFLD patients, it was found that the level of Hedgehog activity paralleled the severity of liver damage (hepatocyte ballooning, portal inflammation and liver fibrosis) [71]. The researchers suggested that development of non-invasive tests that quantify Hedgehog pathway activity might help identify patients developing tissue damage related to metabolic syndrome before irreparable end-organ damage occurs.
Diagnosis
For diagnosis of NAFLD, clinical history and laboratory and radiological investigations are the first step to exclude other causes of liver disease [104]. History of alcohol intake should be taken carefully to rule out alcoholic liver disease which shares many common findings with NAFLD [104]. Imaging studies are needed to assess hepatic steatosis, with ultrasound being the most widely used method [104]. However, ultrasound and computed tomography (CT) scan lack sensitivity and specificity to detect steatosis [105]. MRI techniques have been shown to be highly accurate in detecting liver fat [106]. MR spectroscopy (MRS) has been shown to be highly accurate in detecting liver fat and in quantifying it. MRS has been used for longitudinal follow up in clinical trials in NASH [106] and has become a reference standard. However, it has been mainly used as a research tool since it requires a special coil and special software and is time consuming. New MRI techniques such as MRI-Proton Density fat fraction have been shown to be highly precise in quantifying liver fat and are easier to use than MRS [107]. While fat can be detected by imaging, liver biopsy remains the gold standard for the accurate diagnosis of NASH and can differentiate simple fatty liver without inflammation , cell injury or fibrosis from NASH [104, 108, 109]. Metabolic syndrome is a strong predictor for the presence of steatohepatitis in NAFLD patients [15, 110, 111]. Histological scoring systems have been proposed to stage and grade the disease. The most widely used scoring system was described by Kleiner et al. from the NASH Clinical Research Network (CRN) established by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) [108, 112, 113]. This scoring system created a numeric score called NAFLD activity score (NAS) for grading activity and for use in clinical trials [108]. NAS consists of three key histological elements in NASH: steatosis, lobular inflammation and ballooning. Validation studies showed that an NAS score of 5–8 correlates with definitive NASH while a score of 1–2 correlates with definitive exclusion of NASH [114, 115]. However, other important histological findings that are seen in NASH such as portal inflammation and megamitochondria suggest that this score can be improved. Indeed, portal inflammation was later found to be associated with clinically and histologically advanced NAFLD in children and adults [116]. Children have two types of histological presentation. One of these types resembles adults where there is zone 3 prominence of steatosis. On the other hand, the most common type consists of either zone 1 prominence of steatosis, or panacinar steatosis [117]. Ballooning has been found to be uncommon in both types. More recently, a study from the NASH CRN has shown that the elderly (defined as > 65 years of age) have more azonal distribution of steatosis and are more likely to have NASH and advanced fibrosis [7].
Non-invasive serum and imaging markers as well as predictive scores of NASH and advanced fibrosis have emerged but are not yet widely utilized. Some of the biomarkers that have been investigated include C-reactive protein, hyaluronic acid (HA), tumor necrosis factor-α, leptin, interleukin-6, ferritin, resistin and adiponectin [118, 119]. Blood levels of cytokeratin 18 have been shown to be promising in predicting NASH but this method is not yet commercially available [120]. The NAFLD fibrosis score has been shown to be a good predictor of fibrosis and cirrhosis [121]. Other scores that have been used include ELF score, modified ELF score, BARD (body mass index, alanine aminotransferase/aspartate aminotransferase ratio, and presence of diabetes) and BAAT (body mass index, alanine aminotransferase, and triglycerides) [122]. Many of these scores have achieved an excellent area under the receiver operating characteristic curve (AUROC). The details of these biomarkers are beyond the scope of this chapter but can be found in reviews [122]. Other evolving imaging techniques such as transient elastography and MR elastography are now being investigated in assessing fibrosis in NASH patients [123–125].
Treatment
Weight loss and exercise are the recommended treatments for NAFLD and NASH by the Food and Drug Administration (FDA). It is thought that at least 7 % reduction of body weight is needed for histological improvement in NASH patients [126]. Because it is often difficult for patients to maintain these lifestyle changes over the long term therapeutic agents have been investigated. However, no pharmaceutical agent has yet been approved [126, 127]. The current focus is to find treatments for NASH. Treatment for simple steatosis has not been a priority since the long term outcome is unknown. Small, mostly uncontrolled studies have been conducted showing limited benefit, if any, of the use of ursodeoxycholic acid, metformin, betaine, N-acetyl cysteine, and orlistat [128–135]. In a trial that randomized 166 NASH patients to ursodeoxycholic acid (13–15 mg/kg daily) or placebo, liver biopsies were performed before and after 2 years of treatment [128]. There was no difference in liver enzymes or histological changes between the two groups. Other clinical trials including high dose ursodeoxycholic acid were unsuccessful in showing a significant effect on NASH and in particular on histology [136, 137].
Metformin has been studied as potential agent for NASH treatment. Pilot trials have shown limited improvement in liver enzymes and less in histology; the beneficial effect was thought to be due to the weight loss effect of metformin [131, 138]. Metformin has also been shown to improve insulin resistance , prevent diabetic complications, and play a role in hepatocellular carcinoma chemoprevention, all of which coexist in NASH [139]. Therefore, although metformin has minimal effect on NASH itself, long-term studies with either metformin monotherapy or with combination therapies that include metformin are needed. In a pilot study of 10 NASH patients, betaine was shown to be a promising agent for treatment of NASH as patients had improvement in aminotransferases and histology [132].
Betaine is required for the generation of methionine from homocysteine, a reaction that is central to the recycling of S-adenosyl-L-methionine (SAMe). Betaine has been shown to increase SAMe levels and protect against steatosis in animal models [140], while alteration of enzymes in the SAMe cycle has been shown to lead to NASH and hepatocellular carcinoma [141]. More recently, a human study suggested a role of methionine adenosyl methyltransferase 1 A (MAT1 A), one of the enzymes in the SAMe cycle in NASH [142]. Although a randomized clinical trial showed that betaine may protect against worsening of steatosis but may not play a role in improvement of the other histological features of NASH, of the study’s initial 55 patients, only 34 patients were available for the exit biopsy [132]; additional research is needed.
Pentoxifylline has been shown to have a possible beneficial effect in improving serum aminotransferase and histology in NASH patients. In one study in which 55 patients were randomized to either pentoxifylline (400 mg, three times daily) or placebo for 1 year, pentoxifylline led to histological improvement of the NAFLD activity score (NAS) in 38.5 % of patients compared to 13.8 % in those given placebo [143]. Larger randomized controlled trials are needed to assess the effect of pentoxifylline.
Thiazolidinediones have been studied extensively in NASH patients. The two most commonly used agents have been rosiglitazone [144] and pioglitazone [145]. Rosiglitazone has led to improvement in steatosis and liver enzymes but not other histological parameters [144]. On the other hand, pioglitazone has been shown to be beneficial in improving liver enzymes and histology [145]. However, weight gain led to less enthusiasm by patients and hepatologists for its use [145–147]. The American Association for the Study of Liver Diseases (AASLD) guidelines state that pioglitazone can be used in biopsy-proved NASH. However, the guidelines highlight the fact that most trials have been carried out with non-diabetics and that the long term effects are unknown [104].
The PIVEN and TONIC trials using vitamin E have shown benefits in the treatment of NASH in adults and children [147, 148]. Although vitamin E has not yet been widely used in clinical practice, the AASLD has recommended vitamin E (d-alpha-tocopherol) administered at a daily dose of 800 IU for non-diabetic adults with biopsy-proven NASH as a first-line pharmacotherapy [104]. The long-term effects of vitamin E therapy in NASH patients have not been determined. Further therapies for NAFLD and NASH are still under investigation. The FXR agonist obeticholic acid is under investigation. Obeticholic acid has been shown to increase insulin sensitivity and decrease markers of inflammation and fibrosis [54]. Bariatric surgery has been shown to improve histology including fibrosis in NASH patients. However randomized clinical trials are needed to confirm benefits and for now a surgical approach is not recommended [149]. Many trials have looked at statins in NAFLD/NASH patients and shown some benefits.
Statins in NAFLD/NASH
A growing body of evidence is beginning to elucidate the extent to which alcoholic liver disease , chronic hepatitis C, and NAFLD raise a patient’s risk of a significant cardiovascular event. Many of the factors mediating this increased cardiovascular risk include disruption of lipid metabolism resulting in unfavorable lipid profiles, insulin resistance , and features of metabolic syndrome. Thus, the use of lipid-regulating agents such as HMG-CoA reductase inhibitors (statins) may play an important role to help mitigate this pro-atherogenic profile seen in liver disease [150]. However, statins have been previously thought to be a common cause of abnormal liver enzymes, a major concern in the setting of already present liver disease. Of note, liver disease from chronic hepatitis B infection is associated with a far more favorable lipid profile, including decreased total cholesterol and decreased triglyceride levels, less steatosis, far less association with insulin resistance and type 2 diabetes, and in keeping with these factors lower cardiovascular risk [151]. Chronic HCV on the other hand has a unique constellation of findings. On the one hand, it is associated with decreased levels of total cholesterol, LDL, and triglycerides. Yet it has been demonstrated to have a significant association with hepatic steatosis , increased visceral fat and insulin resistance , and excess type 2 diabetes risk. HCV infection has been shown to be an independent predictor of angiographically detected coronary artery stenosis as well as increased carotid intimal thickness [152, 153]. While there has been an argument that statins may have limited value in the setting of decreased cholesterol and LDL, they have been shown to independently lower AST and ALT levels [154]. In conjunction with interferon alpha and ribavirin, statins may also increase rates of rapid virologic response (RVR), early virological response, and sustained virological response (SVR) [155]. Significant alcohol intake has been demonstrated to be a common cause of hyperlipidemia [156]. Patients with chronic alcoholic liver diseases have been demonstrated to have elevations in serum levels of triglycerides, chylomicrons , and VLDL. These lipid derangements in the setting of alcohol-induced pro-inflammatory and pro-thrombotic changes, insulin resistance, and features of metabolic syndrome yield a definite increase in cardiovascular risk for patients with alcoholic liver disease [5]. Patients with NAFLD/NASH arguably have the worst lipid profile with a combination of elevated triglyceride levels along with a significantly decreased HDL. LDL is not different in NAFLD patients; however, higher levels of small, dense LDL particles (nontype A), which are more atherogenic than type A LDL particles, are seen in these patients [16, 157]. The mechanisms for these changes are not completely understood but involve overproduction of the very-low-density lipoprotein (VLDL) particles and abnormal clearance of various lipoproteins. Thus it comes as no surprise that there is a significant increase in cardiovascular biomarkers (such as coronary calcium score or carotid artery intima-media thickness) in NAFLD patients. Indeed, cardiovascular events have been proved to be the leading cause of death in NAFLD patients [18, 19, 158].
Statins have been demonstrated to significantly mitigate the risk of cardiovascular events and provide a benefit to patients with these underlying causes of liver disease . However, because statins are cleared by the liver and are known to cause elevations of liver enzymes, there was much concern that patients with underlying liver disease may be at increased risk for statin-induced hepatotoxicity. This, however, has proven not to be the case. Two retrospective cohort studies have served to alleviate this concern. In patients with underlying liver disease and abnormal liver biochemistries who were treated for 6–12 months with statins there was no significant increase in the frequency of liver biochemistry abnormalities nor in severe liver disease when compared with patients who had normal liver biochemistries at baseline and received the same treatment [159, 160].
Are Statins Harmful in NAFLD/NASH?
The current evidence points toward no harmful effect of statins on NAFLD/NASH patients and a possible beneficial effect. In a long-term study of 86 patients who were followed for up to 16 years, 17 patients were on statins. Patients on statins had higher BMI and more sever hepatic steatosis at baseline [161]. Patients who were on statins had a decrease in their histological steatosis. However, there was a slight increase in fibrosis progression in the statin group. This was attributed to possibly more severe lipotoxicity ay baseline in the statins subgroup and more rapid fibrosis progression despite therapeutic measures such as statins [161]. In a prospective study of high-dose pravastatin therapy of patients with chronic liver diseases, including 64 % with NAFLD, there was a reduction in LDL cholesterol, without a significant change in aminotransferase elevation [162]. There is growing evidence that statins are safe in patients with NAFLD/NASH and may have histological benefit.
Are Statins Useful in NAFLD/NASH?
Treatment modalities for the spectrum of NAFLD remain controversial. However, one common cause of morbidity and mortality that is of great concern in these patients is the significantly atherogenic lipid profile that may play a role in the progression of the disease as well as contribute to the increased cardiovascular disease risk. One possible NAFLD treatment that is being explored is the use of statins. In Table 9.2, eight studies are outlined which assessed the effect of statin therapy in patients with NAFLD. While the dosing and length of treatment varied, all of the studies demonstrated a significant and persistent improvement of aminotransferase levels after the treatment period. Athyros et al demonstrated complete normalization of aminotransferases in all of their patients using atorvastatin; other smaller studies confirmed this improvement in liver enzymes [163–166]. In a non-randomized trial in which rosuvastatin was used for approximately 8 months in NAFLD patients, there was complete resolution of biochemical and ultasonographic evidence of NAFLD in 67 % of patients [163]. Multiple retrospective studies which have looked at the effects of simvastatin and pravastatin in NAFLD patients have shown improvement in aminotransferases [161, 167, 168]. A post-hoc analysis of the GREek Atorvastatin and Coronary-heart-disease Evaluation (GREACE) study demonstrated that atorvastatin improves liver enzymes and cardiac outcomes in patients with increased liver enzymes most likely due to NAFLD [169]. Further studies demonstrating histological improvement of NAFLD with statin treatment may bolster their use in treating fatty liver, especially in those with abnormal lipid panels (Table 9.1).
With the evidence of benefit seen with statin use in NAFLD, it would stand to reason that there may also be benefits from their use in NASH. The majority of studies of statin use in biopsy-proven NASH echo the findings of improved aminotransferases seen with NAFLD (Table 9.3). The first evidence came from a pilot study of 7 biopsy-proven NASH patients. Although there was no statistically significant improvement of aminotransferase after 12 months of atorvastatin, there were improvements in both steatosis and inflammation. In a prospective non-randomized trial, a total of 44 biopsy-proven NASH patients were enrolled in the study. Patients without dyslipidemia (n = 17) were given ursodeoxycholic acid (UDCA) while patients with dyslipidemia (n = 27) were given atorvastatin 10 mg daily for 6 months [170]. There was more significant improvement in liver enzymes in the atorvastatin group compared to the UDCA group. There was also improvement in steatosis measured by CT in the atorvastatin group which was not seen in the UDCA group. Other small studies with different durations of treatment have confirmed the beneficial effect of atorvastatin on histology in NASH patients.
Pitvastatin and simvastatin have been studied less extensively, with a beneficial effect on aminotransferases shown with pitvastatin but no such effect shown with simvastatin [171, 172]. Both medications failed to show a significant effect on improving histology in NASH patients. Because randomized clinical trials haven’t been performed to examine the effect of statins in NASH patients, they have not been recommended by the AASLD as a treatment for NASH. However, statins are recommended to address the dyslipidemia that is estimated to occur in from 20–80 % of NAFLD/NASH patients [121, 173–175]. Further studies may advance our understanding of the possible value of statins in the slowing the progression of NASH to end stage liver disease.
Summary
NAFLD is the most common liver disease in western countries and is very prevalent today. It is thought to be benign unless it progresses to NASH. NASH can lead to cirrhosis and liver morbidity and mortality. Metabolic syndrome and, in particular, type 2 diabetes are thought to be risk factors for developing NASH. Thus, special attention should be paid to NAFLD patients with diabetes. Liver biopsy should be considered for staging. New imaging techniques have evolved to quantify liver fat and to assess fibrosis, including MRI-PDFF and MR elastography. There is currently no FDA-approved treatment for NASH but vitamin E and pioglitazone have been shown to be helpful; the long-term effects for these are unknown. Statins have not been shown to be harmful in NAFLD and NASH patients and may be beneficial. Larger studies and randomized trials are needed to explore the effect of statins on NAFLD/NASH patients, especially in those with dyslipidemia.
Financial Support
USC Research Center of Liver Diseases. P30 DK48522
Conflict of Interest
No conflicts of interest exist
References
Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–31.
Pais R, Ratziu V. [Epidemiology and natural history of nonalcoholic fatty liver disease]. Rev Prat. 2012;62:1416–8.
Harrison SA, Torgerson S, Hayashi PH. The natural history of nonalcoholic fatty liver disease: a clinical histopathological study. Am J Gastroenterol. 2003;98:2042–7.
Charlton MR, Burns JM, Pedersen RA, et al. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology. 2011;141:1249–53.
Loria P, Marchesini G, Nascimbeni F, et al. Cardiovascular risk, lipidemic phenotype and steatosis. A comparative analysis of cirrhotic and non-cirrhotic liver disease due to varying etiology. Atherosclerosis. 2014;232:99–109.
Kim WR, Brown RS Jr, Terrault NA, et al. Burden of liver disease in the United States: summary of a workshop. Hepatology. 2002;36:227–42.
Noureddin M, Yates KP, Vaughn IA, et al. Clinical and histological determinants of nonalcoholic steatohepatitis and advanced fibrosis in elderly patients. Hepatology. 2013;58:1644–54.
Corey KE, Stanley TL, Misdraji J, et al. Prevalence and outcome of non-alcoholic fatty liver disease in adolescents and young adults undergoing weight loss surgery. Pediatr Obes. 2014;9:e91–3.
Schwimmer JB, Deutsch R, Kahen T, et al. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388–93.
Li YY. Genetic and epigenetic variants influencing the development of nonalcoholic fatty liver disease. World J Gastroenterol. 2012;18:6546–51.
Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–5.
Day CP. Natural history of NAFLD: remarkably benign in the absence of cirrhosis. Gastroenterology. 2005;129:375–8.
Caldwell S, Argo C. The natural history of non-alcoholic fatty liver disease. Dig Dis. 2010;28:162–8.
Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–32.
Loomba R, Abraham M, Unalp A, et al. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology. 2012;56:943–51.
Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363:1341–50.
Vanwagner LB, Bhave M, Te HS, et al. Patients transplanted for nonalcoholic steatohepatitis are at increased risk for postoperative cardiovascular events. Hepatology. 2012;56:1741–50.
Misra VL, Khashab M, Chalasani N. Nonalcoholic fatty liver disease and cardiovascular risk. Curr Gastroenterol Rep. 2009;11:50–5.
Rafiq N, Bai C, Fang Y, et al. Long-term follow-up of patients with nonalcoholic fatty liver. Clin Gastroenterol Hepatol. 2009;7:234–8.
Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–5.
Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–52.
Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–23.
Marchesini G, Brizi M, Bianchi G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–50.
Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–38.
Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003;29:478–85.
Diraison F, Beylot M. Role of human liver lipogenesis and reesterification in triglycerides secretion and in FFA reesterification. Am J Physiol. 1998;274:E321–7.
Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–51.
Yamashita H, Takenoshita M, Sakurai M, et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U S A. 2001;98:9116–21.
Kawaguchi T, Takenoshita M, Kabashima T, et al. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc Natl Acad Sci U S A. 2001;98:13710–5.
Horton JD, Shah NA, Warrington JA, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–32.
McGarry JD, Mannaerts GP, Foster DW. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest. 1977;60:265–70.
Gonzalez-Baro MR, Lewin TM, Coleman RA. Regulation of Triglyceride Metabolism. II. Function of mitochondrial GPAT1 in the regulation of triacylglycerol biosynthesis and insulin action. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1195–9.
Chakravarty K, Leahy P, Becard D, et al. Sterol regulatory element-binding protein-1c mimics the negative effect of insulin on phosphoenolpyruvate carboxykinase (GTP) gene transcription. J Biol Chem. 2001;276:34816–23.
Fabbrini E, Mohammed BS, Magkos F, et al. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134:424–31.
Christodoulides C, Vidal-Puig A. PPARs and adipocyte function. Mol Cell Endocrinol. 2010;318:61–8.
Wang YX. PPARs: diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010;20:124–37.
Maeda N, Takahashi M, Funahashi T, et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50:2094–9.
Rong X, Albert CJ, Hong C, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013;18:685–97.
Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol. 2012;13:213–24.
Viscarra JA, Ortiz RM. Cellular mechanisms regulating fuel metabolism in mammals: role of adipose tissue and lipids during prolonged food deprivation. Metabolism. 2013;62:889–97.
Vacca M, Degirolamo C, Mariani-Costantini R, et al. Lipid-sensing nuclear receptors in the pathophysiology and treatment of the metabolic syndrome. Wiley Interdiscip Rev Syst Biol Med. 2011;3:562–87.
Mitro N, Mak PA, Vargas L, et al. The nuclear receptor LXR is a glucose sensor. Nature. 2007;445:219–23.
Hishikawa D, Shindou H, Kobayashi S, et al. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc Natl Acad Sci U S A. 2008;105:2830–5.
Li Z, Ding T, Pan X, et al. Lysophosphatidylcholine acyltransferase 3 knockdown-mediated liver lysophosphatidylcholine accumulation promotes very low density lipoprotein production by enhancing microsomal triglyceride transfer protein expression. J Biol Chem. 2012;287:20122–31.
Borradaile NM, Han X, Harp JD, et al. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–37.
Halilbasic E, Baghdasaryan A, Trauner M. Nuclear receptors as drug targets in cholestatic liver diseases. Clin Liver Dis. 2013;17:161–89.
Zhang Y, Lee FY, Barrera G, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A. 2006;103:1006–11.
Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–18.
Eloranta JJ, Kullak-Ublick GA. Coordinate transcriptional regulation of bile acid homeostasis and drug metabolism. Arch Biochem Biophys. 2005;433:397–412.
Lefebvre P, Cariou B, Lien F, et al. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–91.
Kim I, Ahn SH, Inagaki T, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48:2664–72.
Kong B, Luyendyk JP, Tawfik O, et al. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther. 2009;28:116–22.
Knop FK. Bile-induced secretion of glucagon-like peptide-1: pathophysiological implications in type 2 diabetes? Am J Physiol Endocrinol Metab. 2010;299:E10–3.
Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145(574–582):e571.
Gruzman A, Babai G, Sasson S. Adenosine Monophosphate-Activated Protein Kinase (AMPK) as a new target for antidiabetic drugs: a review on metabolic, pharmacological and chemical considerations. Rev Diabet Stud. 2009;6:13–36.
Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74.
Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–83.
Backhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–23.
Drenick EJ, Fisler J, Johnson D. Hepatic steatosis after intestinal bypass-prevention and reversal by metronidazole, irrespective of protein-calorie malnutrition. Gastroenterology. 1982;82:535–48.
Miele L, Marrone G, Lauritano C, et al. Gut-liver axis and microbiota in NAFLD: insight pathophysiology for novel therapeutic target. Curr Pharm Des. 2013;19:5314–24.
Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–9.
Fromenty B, Robin MA, Igoudjil A, et al. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004;30:121–38.
Sunny NE, Parks EJ, Browning JD, et al. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–10.
Begriche K, Igoudjil A, Pessayre D, et al. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28.
Emery MG, Fisher JM, Chien JY, et al. CYP2E1 activity before and after weight loss in morbidly obese subjects with nonalcoholic fatty liver disease. Hepatology. 2003;38:428–35.
Weltman MD, Farrell GC, Hall P, et al. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. 1998;27:128–33.
Caldwell SH, Chang CY, Nakamoto RK, et al. Mitochondria in nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:595–617.
Hui JM, Hodge A, Farrell GC, et al. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46–54.
Puri P, Mirshahi F, Cheung O, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–76.
Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–88.
Guy CD, Suzuki A, Zdanowicz M, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology. 2012;55:1711–21.
Syn WK, Choi SS, Liaskou E, et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology. 2011;53:106–15.
Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–87.
Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–9.
Cario E. Bacterial interactions with cells of the intestinal mucosa: toll-like receptors and NOD2. Gut. 2005;54:1182–93.
Schultz A, Neil D, Aguila MB, et al. Hepatic adverse effects of fructose consumption independent of overweight/obesity. Int J Mol Sci. 2013;14:21873–86.
Basaranoglu M, Basaranoglu G, Sabuncu T, et al. Fructose as a key player in the development of fatty liver disease. World J Gastroenterol. 2013;19:1166–72.
Vos MB, Lavine JE. Dietary fructose in nonalcoholic fatty liver disease. Hepatology. 2013;57:2525–31.
Perito ER, Rodriguez LA, Lustig RH. Dietary treatment of nonalcoholic steatohepatitis. Curr Opin Gastroenterol. 2013;29:170–6.
Lustig RH. Fructose: it’s “alcohol without the buzz”. Adv Nutr. 2013;4:226–35.
Assy N, Nasser G, Kamayse I, et al. Soft drink consumption linked with fatty liver in the absence of traditional risk factors. Can J Gastroenterol. 2008;22:811–6.
Abid A, Taha O, Nseir W, et al. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J Hepatol. 2009;51:918–24.
Abdelmalek MF, Suzuki A, Guy C, et al. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1961–71.
Lim JS, Mietus-Snyder M, Valente A, et al. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–64.
Aguirre V, Uchida T, Yenush L, et al. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem. 2000;275:9047–54.
McCarthy EM, Rinella ME. The role of diet and nutrient composition in nonalcoholic fatty liver disease. J Acad Nutr Diet. 2012;112:401–9.
Spruss A, Kanuri G, Wagnerberger S, et al. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology. 2009;50:1094–104.
Abdelmalek MF, Lazo M, Horska A, et al. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology. 2012;56:952–60.
Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32.
Meli R, Mattace Raso G, Calignano A. Role of innate immune response in non-alcoholic fatty liver disease: metabolic complications and therapeutic tools. Front Immunol. 2014;5:177.
Miura K, Yang L, van Rooijen N, et al. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. 2013;57:577–89.
Farhadi A, Gundlapalli S, Shaikh M, et al. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008;28:1026–33.
Csak T, Ganz M, Pespisa J, et al. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–44.
Rivera CA, Adegboyega P, van Rooijen N, et al. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47:571–9.
Miura K, Kodama Y, Inokuchi S, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–34 e327.
Miura K, Seki E, Ohnishi H, et al. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic fatty liver disease. Gastroenterol Res Pract. 2010;2010:362847.
Szabo G, Velayudham A, Romics L Jr, et al. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: the role of toll-like receptors 2 and 4. Alcohol Clin Exp Res. 2005;29:140S–5S.
Csak T, Velayudham A, Hritz I, et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G433–41.
Scott MJ, Chen C, Sun Q, et al. Hepatocytes express functional NOD1 and NOD2 receptors: a role for NOD1 in hepatocyte CC and CXC chemokine production. J Hepatol. 2010;53:693–701.
Syn WK, Jung Y, Omenetti A, et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology. 2009;137:1478–1488 e1478.
Omenetti A, Choi S, Michelotti G, et al. Hedgehog signaling in the liver. J Hepatol. 2011;54:366–73.
Rangwala F, Guy CD, Lu J, et al. Increased production of sonic hedgehog by ballooned hepatocytes. J Pathol. 2011;224:401–10.
Syn WK, Witek RP, Curbishley SM, et al. Role for hedgehog pathway in regulating growth and function of invariant NKT cells. Eur J Immunol. 2009;39:1879–92.
Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–23.
van Werven JR,M, Nederveen AJ, et al. Assessment of hepatic steatosis in patients undergoing liver resection: comparison of US, CT, T1-weighted dual-echo MR imaging, and point-resolved 1H MR spectroscopy. Radiology. 2010;256:159–68.
Reeder SB. Emerging quantitative magnetic resonance imaging biomarkers of hepatic steatosis. Hepatology. 2013;58:1877–80.
Noureddin M, Lam J, Peterson MR, et al. Utility of magnetic resonance imaging versus histology for quantifying changes in liver fat in nonalcoholic fatty liver disease trials. Hepatology. 2013;58:1930–40.
Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21.
Brunt EM. Nonalcoholic steatohepatitis: definition and pathology. Semin Liver Dis. 2001;21:3–16.
Kang H, Greenson JK, Omo JT, et al. Metabolic syndrome is associated with greater histologic severity, higher carbohydrate, and lower fat diet in patients with NAFLD. Am J Gastroenterol. 2006;101:2247–53.
Musso G, Gambino R, Cassader M, et al. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011;43:617–49.
Mendler MH, Kanel G, Govindarajan S. Proposal for a histological scoring and grading system for non-alcoholic fatty liver disease. Liver Int. 2005;25:294–304.
Brunt EM, Janney CG, Di Bisceglie AM, et al. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–74.
Lee KH, Park SH, Kim YJ, et al. [Validity and reliability of the nonalcoholic fatty liver diseases activity score (NAS) in Korean NAFLD patients and its correlation with clinical factors]. Korean J Hepatol. 2010;16:29–37.
Hjelkrem M, Stauch C, Shaw J, et al. Validation of the non-alcoholic fatty liver disease activity score. Aliment Pharmacol Ther. 2011;34:214–8.
Brunt EM, Kleiner DE, Wilson LA, et al. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): a histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology. 2009;49:809–20.
Schwimmer JB, Behling C, Newbury R, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology. 2005;42:641–9.
Dowman JK, Tomlinson JW, Newsome PN. Systematic review: the diagnosis and staging of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2011;33:525–40.
Adams LA, Feldstein AE. Non-invasive diagnosis of nonalcoholic fatty liver and nonalcoholic steatohepatitis. J Dig Dis. 2011;12:10–6.
Wieckowska A, Zein NN, Yerian LM, et al. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44:27–33.
Angulo P, Hui JM, Marchesini G, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 2007;45:846–54.
Noureddin M, Loomba R. Nonalcoholic fatty liver disease: indications for liver biopsy and noninvasive biomarkers. Clin Liver Dis. 2012;1:104–7.
Kwok R, Tse YK, Wong GL, et al. Systematic review with meta-analysis: non-invasive assessment of non-alcoholic fatty liver disease-the role of transient elastography and plasma cytokeratin-18 fragments. Aliment Pharmacol Ther. 2014;39:254–69.
Chen J, Talwalkar JA, Yin M, et al. Early detection of nonalcoholic steatohepatitis in patients with nonalcoholic fatty liver disease by using MR elastography. Radiology. 2011;259:749–56.
Kim D, Kim WR, Talwalkar JA, et al. Advanced fibrosis in nonalcoholic fatty liver disease: noninvasive assessment with MR elastography. Radiology. 2013;268:411–9.
Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–9.
Pearlman M, Loomba R. State of the art: treatment of nonalcoholic steatohepatitis. Curr Opin Gastroenterol. 2014;30:223–37.
Lindor KD, Kowdley KV, Heathcote EJ, et al. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology. 2004;39:770–8.
Torres DM, Jones FJ, Shaw JC, et al. Rosiglitazone versus rosiglitazone and metformin versus rosiglitazone and losartan in the treatment of nonalcoholic steatohepatitis in humans: a 12-month randomized, prospective, open-label trial. Hepatology. 2011;54:1631–9.
Krakoff J, Clark JM, Crandall JP, et al. Effects of metformin and weight loss on serum alanine aminotransferase activity in the diabetes prevention program. Obesity. 2010;18:1762–7.
Loomba R, Lutchman G, Kleiner DE, et al. Clinical trial: pilot study of metformin for the treatment of non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2009;29:172–82.
Abdelmalek MF, Sanderson SO, Angulo P, et al. Betaine for nonalcoholic fatty liver disease: results of a randomized placebo-controlled trial. Hepatology. 2009;50:1818–26.
Baumgardner JN, Shankar K, Hennings L, et al. N-acetylcysteine attenuates progression of liver pathology in a rat model of nonalcoholic steatohepatitis. J Nutr. 2008;138:1872–9.
Harrison SA, Fecht W, Brunt EM, et al. Orlistat for overweight subjects with nonalcoholic steatohepatitis: a randomized, prospective trial. Hepatology. 2009;49:80–6.
Zelber-Sagi S, Kessler A, Brazowsky E, et al. A double-blind randomized placebo-controlled trial of orlistat for the treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4:639–44.
Leuschner UF, Lindenthal B, Herrmann G, et al. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52:472–9.
Ratziu V, de Ledinghen V, Oberti F, et al. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol. 2011;54:1011–9.
Idilman R, Mizrak D, Corapcioglu D, et al. Clinical trial: insulin-sensitizing agents may reduce consequences of insulin resistance in individuals with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2008;28:200–8.
Doycheva I, Loomba R. Effect of metformin on ballooning degeneration in nonalcoholic steatohepatitis (NASH): when to use metformin in nonalcoholic fatty liver disease (NAFLD). Adv Ther. 2014;31:30–43.
Barak AJ, Beckenhauer HC, Junnila M, et al. Dietary betaine promotes generation of hepatic S-adenosylmethionine and protects the liver from ethanol-induced fatty infiltration. Alcohol Clin Exp Res. 1993;17:552–5.
Mato JM, Martinez-Chantar ML, Lu SC. S-adenosylmethionine metabolism and liver disease. Ann Hepatol. 2013;12:183–9.
Murphy SK, Yang H, Moylan CA, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. 2013;145:1076–87.
Zein CO, Yerian LM, Gogate P, et al. Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial. Hepatology. 2011;54:1610–9.
Ratziu V, Giral P, Jacqueminet S, et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled fatty liver improvement with rosiglitazone therapy (FLIRT) trial. Gastroenterology. 2008;135:100–10.
Belfort R, Harrison SA, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–307.
Aithal GP, Thomas JA, Kaye PV, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–84.
Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85.
Lavine JE, Schwimmer JB, Van Natta ML, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. 2011;305:1659–68.
Mathurin P, Hollebecque A, Arnalsteen L, et al. Prospective study of the long-term effects of bariatric surgery on liver injury in patients without advanced disease. Gastroenterology. 2009;137:532–40.
Corey KE, Chalasani N. Management of dyslipidemia as a cardiovascular risk factor in individuals with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2014;12:1077–84.
Wang CH, Chen CJ, Lee MH, et al. Chronic hepatitis B infection and risk of atherosclerosis-related mortality: a 17-year follow-up study based on 22,472 residents in Taiwan. Atherosclerosis. 2010;211:624–9.
Alyan O, Kacmaz F, Ozdemir O, et al. Hepatitis C infection is associated with increased coronary artery atherosclerosis defined by modified reardon severity score system. Circ J. 2008;72:1960–5.
Petta S, Torres D, Fazio G, et al. Carotid atherosclerosis and chronic hepatitis C: a prospective study of risk associations. Hepatology. 2012;55:1317–23.
Henderson LM, Patel S, Giordano TP, et al. Statin therapy and serum transaminases among a cohort of HCV-infected veterans. Dig Dis Sci. 2010;55:190–5.
Zhu Q, Li N, Han Q, et al. Statin therapy improves response to interferon alfa and ribavirin in chronic hepatitis C: a systematic review and meta-analysis. Antiviral Res. 2013;98:373–9.
Chait A, Mancini M, February AW, et al. Clinical and metabolic study of alcoholic hyperlipidaemia. Lancet. 1972;2:62–4.
Speliotes EK, Massaro JM, Hoffmann U, et al. Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: the Framingham Heart Study. Hepatology. 2010;51:1979–87.
Nseir W, Shalata A, Marmor A, et al. Mechanisms linking nonalcoholic fatty liver disease with coronary artery disease. Dig Dis Sci. 2011;56:3439–49.
Chalasani N, Aljadhey H, Kesterson J, et al. Patients with elevated liver enzymes are not at higher risk for statin hepatotoxicity. Gastroenterology. 2004;126:1287–92.
Vuppalanchi R, Teal E, Chalasani N. Patients with elevated baseline liver enzymes do not have higher frequency of hepatotoxicity from lovastatin than those with normal baseline liver enzymes. Am J Med Sci. 2005;329:62–5.
Ekstedt M, Franzen LE, Mathiesen UL, et al. Statins in non-alcoholic fatty liver disease and chronically elevated liver enzymes: a histopathological follow-up study. J Hepatol. 2007;47:135–41.
Lewis JH, Mortensen ME, Zweig S, et al. Efficacy and safety of high-dose pravastatin in hypercholesterolemic patients with well-compensated chronic liver disease: results of a prospective, randomized, double-blind, placebo-controlled, multicenter trial. Hepatology. 2007;46:1453–63.
Antonopoulos S, Mikros S, Mylonopoulou M, et al. Rosuvastatin as a novel treatment of non-alcoholic fatty liver disease in hyperlipidemic patients. Atherosclerosis. 2006;184:233–4.
Athyros VG, Mikhailidis DP, Didangelos TP, et al. Effect of multifactorial treatment on non-alcoholic fatty liver disease in metabolic syndrome: a randomised study. Curr Med Res Opin. 2006;22:873–83.
Hatzitolios A, Savopoulos C, Lazaraki G, et al. Efficacy of omega-3 fatty acids, atorvastatin and orlistat in non-alcoholic fatty liver disease with dyslipidemia. Indian J Gastroenterol. 2004;23:131–4.
Gomez-Dominguez E, Gisbert JP, Moreno-Monteagudo JA, et al. A pilot study of atorvastatin treatment in dyslipemid, non-alcoholic fatty liver patients. Aliment Pharmacol Ther. 2006;23:1643–7.
Abel T, Feher J, Dinya E, et al. Safety and efficacy of combined ezetimibe/simvastatin treatment and simvastatin monotherapy in patients with non-alcoholic fatty liver disease. Med Sci Monit. 2009;15:MS6–11.
Riley P, Sudarshi D, Johal M, et al. Weight loss, dietary advice and statin therapy in non-alcoholic fatty liver disease: a retrospective study. Int J Clin Pract. 2008;62:374–81.
Athyros VG, Tziomalos K, Gossios TD, et al. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the greek atorvastatin and coronary heart disease evaluation (GREACE) study: a post-hoc analysis. Lancet. 2010;376:1916–22.
Kiyici M, Gulten M, Gurel S, et al. Ursodeoxycholic acid and atorvastatin in the treatment of nonalcoholic steatohepatitis. Can J Gastroenterol. 2003;17:713–8.
Nelson A, Torres DM, Morgan AE, et al. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis: a randomized placebo-controlled trial. J Clin Gastroenterol. 2009;43:990–4.
Hyogo H, Tazuma S, Arihiro K, et al. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism. 2008;57:1711–8.
Matteoni CA, Younossi ZM, Gramlich T, et al. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–9.
Angulo P, Keach JC, Batts KP, et al. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356–62.
Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–23.
Rallidis LS, Drakoulis CK, Parasi AS. Pravastatin in patients with nonalcoholic steatohepatitis: results of a pilot study. Atherosclerosis. 2004;174:193–6.
Georgescu EF, Georgescu M. Therapeutic options in non-alcoholic steatohepatitis (NASH). Are all agents alike? Results of a preliminary study. J Gastrointestin Liver Dis. 2007;16:39–46.
Kimura Y, Hyogo H, Yamagishi S, et al. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J Gastroenterol. 2010;45:750–7.
Hyogo H, Ikegami T, Tokushige K, et al. Efficacy of pitavastatin for the treatment of non-alcoholic steatohepatitis with dyslipidemia: an open-label, pilot study. Hepatol Res. 2011;41:1057–65.
Harlander JC, Kwo PY, Cummings OW. Atorvastatin for the treatment of NASH. Gastroenterol. 2001;120A:544.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Noureddin, M., Alexanian, D., Kaplowitz, N. (2015). Non-Alcoholic Fatty Liver Disease (NAFLD): The Lipid Disease of the Liver and the Effect of Statins. In: Yassine, H. (eds) Lipid Management. Springer, Cham. https://doi.org/10.1007/978-3-319-11161-2_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-11161-2_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-11160-5
Online ISBN: 978-3-319-11161-2
eBook Packages: MedicineMedicine (R0)