
Abstract
As the only striated muscle tissues in the body, skeletal and cardiac muscle share numerous structural and functional characteristics, while exhibiting vastly different size and regenerative potential. Healthy skeletal muscle harbors a robust regenerative response that becomes inadequate after large muscle loss or in degenerative pathologies and aging. In contrast, the mammalian heart loses its regenerative capacity shortly after birth, leaving it susceptible to permanent damage by acute injury or chronic disease. In this review, we compare and contrast the physiology and regenerative potential of native skeletal and cardiac muscles, mechanisms underlying striated muscle dysfunction, and bioengineering strategies to treat muscle disorders. We focus on different sources for cellular therapy, biomaterials to augment the endogenous regenerative response, and progress in engineering and application of mature striated muscle tissues in vitro and in vivo. Finally, we discuss the challenges and perspectives in translating muscle bioengineering strategies to clinical practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The body possesses two types of striated muscle, cardiac and skeletal. Human skeletal muscles can be further categorized into slow-oxidative/Type I, fast-oxidative/Type IIa, and fast-glycolytic/Type IIb types based on their contractile and metabolic phenotypes [1]. Striated muscles are required for whole-body oxygen supply, metabolic balance, and locomotion. While structurally and functionally similar, the two striated muscles have vastly different sizes and regenerative capacities. In adult humans weighing ~70 kg, skeletal muscle tissue weighs ~27 kg comprising 35 % of the total body mass [2], while cardiac muscle is ~100 times smaller weighing in average 270 g [3]. Skeletal muscle can regenerate in response to small muscle tears that occur during exercise or daily activity owing to the abundances of resident muscle stem cells called satellite cells (SCs), which upon injury activate, proliferate, and fuse to repair damaged or form new muscle fibers [4]. In contrast, cardiac muscle does not possess a cardiomyogenic stem cell pool and has little to no regenerative ability, with injury resulting in the formation of a fibrotic scar and, eventually, impaired pump function [5].
Functional deficit in skeletal muscle occurs with age, chronic degenerative diseases such as muscular dystrophy [6], defects in metabolism such as Pompe disease [7], and with large volume muscle loss due to trauma or surgical resection [8]. In these conditions, SC pool is often depleted due to the disruption of SC niche or continuous SC activation resulting in impaired muscle regeneration and chronic fibrosis [9]. Cardiac muscle dysfunction arises largely from narrowing of coronary arteries caused by vascular disease and less frequently from non-ischemic cardiomyopathy, congenital abnormalities, diastolic disease, and certain muscular dystrophies [10, 11].
In this review, we first discuss the structure, function, and regenerative potential of healthy striated muscles, representing the desired outcome of any cell, biomaterial, drug, or gene therapy for muscle disorders. We then review the progress made with cellular therapies, where immature cells are directly delivered in vivo, as well as cell-free therapies where biomaterials are implanted to augment and replicate the natural repair capacity of muscle tissue. Lastly, we review recent developments in engineering mature striated muscle tissues in vitro and highlight the hurdles that need to be overcome to translate these promising approaches to the clinic.
Striated muscle structure and function
Striated muscles are highly organized tissues (Fig. 1) that convert chemical energy to physical work. The primary function of striated muscles is to generate force and contract to support respiration, locomotion, and posture (skeletal muscle) and to pump blood throughout the body (cardiac muscle).

Structure and function of striated muscles. a Adult skeletal muscle contains uniformly aligned, long multinucleated myofibers, blood vessels, and resident satellite cells, with fewer fibroblasts relative to cardiac muscle. b Adult cardiac muscle consists of a branched network of shorter cardiomyocytes connected via intercalated discs and surrounded by blood vessels and extracellular matrix secreted primarily by fibroblasts. c Skeletal muscle excitation–contraction (e–c) coupling begins with a depolarization-induced conformational change in l-type Ca2+ channels (CaV1.1, dihydropyridine receptor, DHPR) that triggers release of Ca2+ from two neighboring SR terminae via opening of the Ryanodine receptor (RyR1) channels, creating a triad (rather than a diad as in cardiac muscle) with the T-tubules. Calcium is pumped back into the SR via the SR-ATP-ase (SERCA1a). d Cardiac E-C coupling occurs through a Ca2+-dependent Ca2+ release wherein T-tubular entry of extracellular Ca2+ through depolarization activated L-type Ca2+ channels (CaV1.2) triggers a release of Ca2+ stored in the sarcoplasmic reticulum (SR) via opening of the RyR2 channels. Calcium is pumped back into the SR via the SR-ATP-ase (SERCA2a). e Tetanic responses of slow and fast-twitch skeletal muscle fibers showing increased ability to recover from fast-paced stimulation in fast-twitch fibers. f Comparison of active and passive tension-length relationships in cardiac and skeletal muscle. Both striated muscles exhibit stronger active (contractile) force with increased muscle length followed by decay at higher levels of stretch (Frank-Starling relationship). While skeletal muscle operates close to the peak of its active force–length curve, cardiac muscle operates at the ascending limb of the curve to allow more forceful contraction at larger diastolic filling. Simultaneously, passive tension of cardiac muscle at its operating length is markedly higher than that of skeletal muscle, primarily due to higher stiffness of titin molecules within the sarcomeres. g Unlike skeletal muscle, cardiac muscle can propagate action potentials (APs) between myocytes that are connected via gap junctions. Schematic depicts an isochrone map showing AP propagation through cardiac muscle, from which conduction velocity can be measured. h Positive force-frequency relationship of cardiac muscle demonstrating increased force production at higher excitation rates
Ultrastructure
Under light microscopy, striated muscles have highly ordered ultrastructure consisting of sarcomeres, which are basic contractile units containing a central myosin-rich dark anisotropic (A) band and two actin-dominated light isotropic (I) bands [12, 13]. While the sarcomeric proteins are conserved among cardiac and slow-twitch and fast-twitch skeletal muscles, the existence of specific isoforms of these proteins contributes to observed differences in rates of muscle contraction and relaxation [14, 15]. All types of striated muscle contain a branched network of membrane invaginations called T-tubules that enable synchronous calcium release throughout the entire cell volume. The T-tubules contact the sarcoplasmic reticulum (SR) between the A and I bands in skeletal muscle and at the Z-disc in cardiac muscle. Dihydropyridine-sensitive voltage-gated Ca2+ channels in the T-tubules associate with the Ryanodine receptors (RyR) on the junctional face of the SR, converting action potentials to calcium release [16].
In skeletal muscle, the Ca2+ channel (CaV1.1) and RyR1 physically interact, producing a fast release of calcium from SR (~2 ms) without the need for extracellular calcium entry into the cell (Fig. 1c). In cardiac muscle, the Ca2+ channel (CaV1.2)-RyR2 complex requires extracellular calcium entry for the calcium release from SR, which significantly slows the maximal frequency at which cardiac muscle can contract and prevent generation of tetanus, a hallmark of skeletal muscle contraction [17] (Fig. 1d). Where the T-tubules and SR meet, the SR enlarges, fuses, and forms expanded chambers called terminal cisternae. In skeletal muscle the T-tubule meets with two terminal cisternae to form a triad, but only with a single terminal cisternae in cardiac muscle to form a diad. The triads enable sufficient supply of calcium from SR to sustain tetanic contractions.
Micro-/macroscopic structure
Despite possessing the same functional units, the microscopic structures of skeletal and cardiac muscle fibers are different. Individual skeletal muscle fibers arise from the fusion of many muscle cells, producing multi-nucleated linear fibers, millimeters to centimeters in length (Fig. 1a). In contrast, cardiac muscle consists of a cellular syncytium wherein individual cells are electromechanically interconnected in a branched pattern via specialized structures known as intercalated discs (Fig. 1b). Within the intercalated disc, gap junctions allow for a rapid propagation of electrical impulses (Fig. 1g), which results in a near-simultaneous depolarization of the entire cardiac syncytium. Unlike the fused skeletal muscle fibers, individual cardiomyocytes are much smaller (~120 μm in length) [20], contain centrally located nuclei, and remain predominantly (~65 %) mononucleated during all stages of human heart development [21], which is in contrast to a maturation-induced shift towards binucleation known to occur in murine hearts [22]. Finally, while skeletal muscle fibers are directly innervated by motor neurons, cardiomyocytes are excited via a conduction cascade that begins with specialized pacemaking cells of the sinoatrial node and terminates at the ventricular cardiomyocytes.
Skeletal muscle fibers are encased in a basement membrane rich in collagen IV, heparin sulfate proteoglycans (HSPGs), and laminin, which plays a key role in force transmission to the outer three connective tissue layers, the endo-, peri-, and epimysium. These layers predominantly consist of Type I, II and III collagens synthesized by fibroblasts. Healthy skeletal muscle has a high volume ratio of muscle cells to fibroblasts, with fibroblast nuclei comprising 8–15 % of all nuclei in the tissue [23, 24]. In the heart, Collagen I is the main ECM protein made by cardiac fibroblasts that until recently were believed to be the most dominant cell type in the heart, with different abundances reported in different species [25–27]. Recent studies, however, identified endothelial cells as the predominant cell type in both mouse and human adult ventricles, representing 45–55 % of the total cell count (compared with ~30 % cardiomyocytes and ~15 % fibroblasts), although, similar to skeletal muscle, taking up only a small fraction of the ventricular volume [28]. While evidence for electrical coupling between fibroblasts and cardiomyocytes is present in vitro [29, 30], the ability of these cell types to form functional heterocellular gap junctions in vivo is still widely debated [31, 32]. Both in skeletal and cardiac muscles, blood is delivered to cells in a hierarchical manner with primary arteries progressively bunching into smaller vessels and capillaries [33, 34]. Greater capillary density is found in slow-oxidative than fast-glycolytic muscles [1, 35] and in epicardium compared to endocardium with an average of 1.3-2.5 capillaries being associated with each cardiomyocyte or myofiber [33, 36, 37].
Contractile function
Skeletal muscle force output is primarily achieved by summation and motor unit recruitment. Slow (10–30 Hz) and fast (50–100 Hz) fibers achieve peak force at different frequencies due to the speed of contraction and relaxation (Fig. 1e), which is dictated by the number of terminal cisternae and the fast or slow isoform dominance of sarcomeric and calcium handling proteins [1]. Although lacking a tetanic response characteristic of skeletal muscle, the human heart still displays a positive force-frequency relationship (Fig. 1h), largely attributed to an increased net uptake of Ca2+ into SR during faster heart rates [38]. Furthermore, force generation in skeletal muscle is typically controlled by motor unit recruitment, in order from smallest to largest, which results in an exponential increase in force and enables a single muscle to produce both delicate and explosive movements [39]. In contrast, cardiac muscle contracts as a syncytium and obeys an “all-or-none” phenomenon.
Both skeletal and cardiac muscles display a biphasic force–length relationship, which is, in part, explained by the sliding filament theory [40]. Lengthening of the muscle increases the overlap of myosin and actin filaments within each sarcomere, which produces a higher contractile force during the power stroke of the contraction cycle. Further increases in muscle length can decrease the myosin/actin overlap, causing a drop in the active tension (Fig. 1f). However, in cardiac muscle the predominant cellular basis for the force–length relationship is the increased affinity of troponin C to calcium, which yields opening of additional sites for binding of myosin to actin and permits greater force generation [41]. In contrast to skeletal muscle, which operates close to the peak of its active force–length curve, cardiac muscle operates on the ascending part of the curve to allow stronger pumping for increased ventricular filling (Fig. 1f). Furthermore, at their respective resting/working lengths, cardiac muscle experiences significantly higher passive tension than skeletal muscle (that has ~0 passive tension; Fig. 1f), reflecting the existence of much stiffer isoforms of titin and collagen in cardiac muscle [42].
Endogenous striated muscle repair
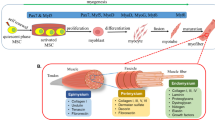
In response to injury, adult skeletal muscle exhibits robust regenerative response that involves a highly orchestrated action of multiple cell types. The most important cells are Pax7+ satellite cells (SCs), which are located between the sarcolemma and the basal lamina [43] and are essential for muscle regeneration [44]. During embryogenesis, myogenic transcription factors Myf5 and Mrf4 are transiently expressed to give rise to a significant number of SCs that persist throughout development [45, 46]. In adult muscle, SCs are a heterogeneous population with the majority of cells expressing Myf5 and being able to directly commit to myogenic differentiation [47]. SCs not expressing Myf5 preferentially self-renew and fill the SC niche but can divide asymmetrically to express Myf5 and support myogenesis [47, 48]. Regardless, the maintenance of SCs in adult muscle is dependent upon Myf5 highlighting the complex non-hierarchical regulatory network that controls SC stemness [49]. In response to injury and exercise, SCs that are typically quiescent become activated and contribute to muscle repair, as reviewed elsewhere [4, 50]. Briefly, upon activation, Pax7+ SCs begin to express MyoD, proliferate and divide asymmetrically to either: (1) commit to muscle differentiation by losing Pax7, and expressing Myf5, MyoD, and ultimately myogenin and then fusing into damaged fibers or forming de novo fibers that express myogenin and Mrf4, or (2) maintain a quiescent state by losing MyoD expression and contributing to future rounds of muscle regeneration (Fig. 2a) [50, 51]. SCs comprise 2-7 % of total nuclear content of skeletal muscle, yet no equivalent cell type has been discovered in cardiac muscle. The c-Kit+ cell niche that emerged over the last decade is likely the closest cardiac relative of the SC niche, although the ability of c-Kit+ cells to generate functional cardiomyocytes has created a wide scientific debate that continues to this day [52, 53]. Regenerative process in skeletal muscle is controlled primarily by the innate immune response (Fig. 2a), with deletion of monocytes completely preventing muscle regeneration [54]. During the first stage of muscle repair, infiltrated neutrophils promote degeneration of muscle fibers while M1 macrophages stimulate a pro-inflammatory cytokine release and muscle cell lysis [55, 56]. Subsequently, the SCs undergo activation and differentiation, which is followed by ECM deposition, angiogenesis to revascularize regenerating tissue, and reinnervation of the new myofibers. Proliferating SCs also migrate bi-directionally along the longitudinal but not the horizontal axis of the degenerating fibers to aid in the redistribution of regenerating muscle progenitors [57]. Conversion to an M2 macrophage phenotype is critical for the repair process as it shifts SCs towards differentiation and away from proliferation seen predominantly with M1 macrophages [58]. Among the other cell types important for successful muscle regeneration, fibroadipogenic progenitors (FAPs) are known to promote myofiber formation through controlled ECM deposition and paracrine factors [59, 60].

Endogenous and exogenous repair of striated muscles. a Damage to skeletal muscle results in proliferation and migration of satellite cells (SCs) along the longitudinal axis of dying fibers (gray) and initial infiltration of pro-inflammatory M1-macrophages and neutrophils which aids in the degeneration of damaged fibers. Conversion to and infiltration of M2-macrophages stimulates SCs to differentiate and eventually fuse into functional myofibers. b Ischemic injury to cardiac muscle results in death of cardiomyocytes (CMs), an initial infiltration of neutrophils and upregulation of matrix metalloproteinases. Release of TGF-β from necrotic CMs induces late migration of macrophages and fibroblasts as well as transformation of fibroblasts into myofibroblasts, which secrete collagen to ultimately produce a fibrotic scar. c Striated muscle repair can be augmented via exogenous delivery of single cells, biomaterials with or without cells, as well as transplantation of in vitro engineered functional muscle tissues
In contrast to skeletal muscle, the adult mammalian heart lacks robust regenerative potential [5, 61]. However, shortly after birth mammalian hearts can still regenerate, as recently shown in the case of a newborn child with myocardial infarction [62]. In neonatal mice, this regenerative process is associated with the activation of epicardial-specific genes, angiogenesis, and a global proliferation of existing cardiomyocytes that restores cardiac function [63] and is dependent upon infiltration of neonatal macrophages [64]. In contrast, cardiac injury in adult heart produces a primarily fibrotic scar that leads to a decline in function. The cellular and molecular events responsible for this process can be broken down into early (<72 h) and late (>72 h) phases [65] (Fig. 2b). The early remodeling phase consists of neutrophil infiltration, activation of matrix metalloproteinasaes and early degradation of extracellular matrix (ECM), which leads to wall thinning and ventricular dilation. During the late phase, initial release of TGF-β1 from necrotic cardiomyocytes induces chemotaxis of fibroblasts and macrophages, proliferation and transformation into myofibroblasts, synthesis of Type I & III collagen, and eventual fibrosis. Pathological hypertrophy also develops as an adaptive response to the decreased contractile function and is largely initiated by the myofiber stretch following chamber dilation and neurohormonal activation, in particular via combined actions of norepinephrine, endothelin-1, and angiotensin II [65].
Striated muscle dysfunction
In contrast to certain amphibians [66] and fish [67], the endogenous repair mechanisms in mammalian skeletal muscle can only regenerate a finite amount of muscle tissue and can thus be overwhelmed by the deposition of fibrotic scar following volumetric muscle loss resulting from trauma or large resection [68]. Furthermore, in sarcopenia, skeletal muscle mass, function, and regenerative ability decrease with age and can cause or exacerbate health problems [69, 70]. Sarcopenia is characterized by a muscular atrophy, loss of muscle fibers, decreased number and size of motor units, and increased fibrosis and fat accumulation [71–74], and can likely be attributed to SC depletion and impaired SC function. In mice, aged satellite cells have reduced regenerative potential [75] due, in part, to defective FGFR1 signaling and consequent p38αβ MAPK activation which promotes SC exhaustion [76]. Inhibition of p38 MAPK restores the regenerative potential of aged SCs, and can restore force generation of muscles in aged mice to levels found in younger animals [75, 76].
Similar to skeletal muscle, impaired function of cardiac muscle can arise from various congenital or acquired diseases. In ischemic heart disease, the leading cause of morbidity and mortality in the world [77], the buildup of atherosclerotic plaques within coronary arteries increases with age and can cause tissue ischemia or necrosis (myocardial infarction) upon rupture, which can precipitate into congestive heart failure over time. Numerous myopathies, including autoimmune and viral myocarditis, can similarly lead to heart failure if left untreated. Diastolic cardiac dysfunction occurs due to pathological hypertrophy and stiffening of the heart walls that impairs chamber relaxation and filling during the cardiac cycle. The long-standing hypertension, ischemic injuries, aging, and aortic stenosis, as wells as certain metabolic diseases such as glycogen storage disease [78] can all cause diastolic dysfunction. Lastly, congenital heart defects (CHDs) are the most common type of birth defect and represent the leading cause of mortality in infants [79, 80]. Three of the top five common types of CHDs—ventricular septal defects, Tetralogy of Fallot, and atrial septal defects—all contain a defect in the heart wall that requires surgical correction when sufficiently large, and often multiple surgeries due to failed or inadequate grafts [81].
Since a large number of proteins are expressed in all striated muscles, the same genetic defect can result in both cardiac and skeletal muscle disease. For example, mutations of proteins in the dystrophin-associated glycoprotein complex (DGC) or associated ECM proteins such as laminin and merosin can lead to a wide range of muscular dystrophies [82]. The DGCs link the sarcomeres to the ECM and are involved in the transmission of force and protection of the membrane from shear stress [83]. Thus, dystrophic muscles are damaged more easily, resulting in a repetitive degeneration/regeneration cycles, eventual exhaustion of the SC pool, muscle loss, and fibrosis [9, 84]. Still, not all skeletal muscles are affected equally by muscular dystrophy, with the ocular muscles being typically spared [85], but the diaphragm which is in constant use being the most severely affected muscle [86] and load-bearing muscles such as the gluteus maximus and the posterior muscles of the lower legs [87, 88] also being severely affected. Furthermore, different diseases can affect cardiac and skeletal muscles differently. Duchenne Muscular dystrophy (DMD), the most severe form of muscular dystrophy characterized by the complete loss of functional dystrophin, typically has a more severe skeletal muscle phenotype and a milder cardiac phenotype [89]. In contrast, Becker’s Muscular Dystrophy (BMD), characterized by a partial loss of functional dystrophin, has a milder skeletal phenotype and a more pronounced cardiac phenotype, potentially due to BMD patients being more physically active than DMD patients and placing more strain on their heart muscle [90, 91].
Therapies for striated muscle disorders
Cell injection therapies
The first cell-based approaches to improve function of injured or diseased striated muscles involved transplantation of stem or progenitor cells into the site of tissue damage. These strategies require expanding a suitable cell population in vitro to generate sufficient cell numbers for transplantation. The desired characteristics of the expanded cells would be to: (1) retain innate stemness or myogenicity during in vitro culture, (2) be immuno-privileged to obviate a need for long-term immunosuppression, (3) survive and robustly engraft upon transplantation, (4) Structurally and functionally integrate with host tissue, (5) repopulate and replenish the tissue-resident stem cell niches and (6) permanently improve muscle function following transplantation. Additionally, having cells that can be delivered throughout the circulatory system and efficiently home to damaged tissues would be desirable to avoid damage due to multiple intramuscular injections and when needed enable the uniform distribution of cells throughout the entire organ.
Skeletal muscle
Primary myoblasts and satellite cells
Given the robust regenerative potential of satellite cells (SCs), it was hypothesized that expanding these cells in vitro and injecting them into damaged muscles may counter loss of muscle mass and function. Initial transplantations of in vitro expanded SCs into skeletal muscle; however, resulted in poor survival, motility, and engraftment/fusion with host myofibers as well as immunorejection [92]. The poor outcomes in these studies have been attributed in part to the in vitro conditions used to expand SCs [93, 94]. Traditional culture for SC expansion is optimized to maximize cell proliferation, which activates SCs and rapidly leads to their commitment and differentiation to myoblasts that can not be readily reverted back to a self-renewing state. Furthermore, committed myoblasts with high proliferation and differentiation potential undergo apoptosis following implantation of whole muscle cultures, with the cells that survive and engraft being the slow-dividing “stem-like” cells [95]. Selection and implantation of these slow-dividing cells promoted greater and longer-lasting muscle regeneration in vivo [96]. Recently, SCs cultured on 12 kPa compliant PEG hydrogels (mimicking stiffness of native skeletal muscle [97]) were shown to engraft more efficiently compared to cells grown on standard tissue culture dishes but still less efficiently than freshly isolated SCs [98], suggesting that mechanical cues of native SC microenvironment are an important but not the only determinant of SC “stemness” to be replicated during SC expansion in vitro.
Of note is that encouraging results have been recently reported in a Phase I/IIa clinical trial of myoblast transplantation in patients with oculopharyngeal muscular dystrophy, where the dystrophic phenotype is limited to pharyngeal muscles. Two years after autologous myoblast transplantation patients showed improved quality of life and no deterioration of swallowing function [99]. This suggests that despite low survival and engraftment, transplanting large numbers of myoblasts could still yield successful repair of small muscles. For larger muscle defects, improved culture conditions to increase the proportion of slow-dividing myogenic cells and/or self-renewing SCs will be required to develop efficient cell-based therapies. On the other hand, myoblasts have low engraftment efficiency when delivered systemically, which prevents their use in treatment of congenital diseases that affect all muscles in the body, thus necessitating an alternative cell source.
Human pluripotent stem cell-derived skeletal muscle cells
Human pluripotent stem cells (hPSCs) have the potential to overcome the issue of in vitro satellite cell expansion due to their unlimited proliferative potential. The generation of muscle progenitors from hPSCs has been achieved by utilizing cell sorting for defined extracellular markers, direct genetic reprogramming, or small molecule inhibitors. After the first published protocol for derivation of muscle progenitors from hESCs by sorting for CD73+/CD56+ cells [100] that has been difficult to reproduce, researchers have overexpressed the myogenic transcription factors Pax7 [101–103] or MyoD [104–106] in hPSCs to generate muscle progenitors in as little as 5 days. These cells efficiently differentiated to myotubes in vitro, fused more efficiently to existing myofibers in vivo than human myoblasts and improved muscle function. Still the risk of undesired genetic recombination or reactivation makes this approach unlikely to be used clinically. Recently small molecules have been used to differentiate hPSCs to muscle progenitors capable of fusing in vitro and with mouse myofibers in vivo [107–110] by protocols that combined GSK3β inhibition and IGF-1, HGF, and/or FGF2 supplementation, with overall efficiency that could be increased via cell sorting [108] or the addition of forskolin [109].
Interstitial cells, fibroadipogenic progenitor cells, pericytes and mesoangioblasts
In addition to SCs, other tissue-resident cells with regenerative potential have been recently identified in rodent and human skeletal muscle. Specifically, interstitial muscle progenitor cells are a heterogeneous cell pool that does not reside under the basal lamina. A fraction of these cells termed Pw1 interstitial cells (PICs) express Pw1/Peg3, an early marker of the myogenic lineage [111], and do not express Pax7 [112]. Developmentally, PICs have distinct origin from SCs but can give rise to SCs indicating that they are upstream of SCs in muscle precursor lineage hierarchy. PICs may also contribute to muscle regeneration through paracrine release of pro-myogenic and differentiation factors. Fibroadipogenic progenitors, which are a subset of PICs, are activated in response to muscle injury and shown to promote murine muscle regeneration via paracrine growth factor release [59]. They are also identified in human skeletal muscle [113]. However, in response to chronic injury or aging, they can contribute to fibrosis and fat accumulation [114, 115]. Further delineation of how exogenous transplantation of PICs regulates muscle regeneration is required but given their greater abundance in extraocular muscles, which are unaffected by sarcopenia and muscular dystrophy [116], PICs are a promising cell source for muscle regeneration.
Other types of PICs include pericytes, which surround endothelial cells in microvessels, and mesoangioblasts, which are thought to be a subset of pericytes or pericyte precursors that exhibit multi-lineage developmental potential [117–122]. Pericytes display greater spontaneous myogenic differentiation in vitro than mesoangioblasts but in contrast to SCs, do not express myogenic regulatory factors until after myotube formation [121]. Pericytes can be further classified into type 1 and type 2 [120]. Type 1 pericytes express the adipogenic marker PDGFRα and contribute to fat accumulation and do not appear to be directly involved in muscle regeneration. They have significant overlap with FAPs in terms of cell marker expression and cell behavior in vivo and in vitro and therefore under certain circumstances may secrete factors that enhance muscle self-repair. Conversely, type 2 pericytes do not express PDGFRα, are highly myogenic both in vitro and in vivo and upon implantation fuse into damaged myofibers. When delivered intra-arterially, pericytes and mesoangioblasts show higher survival, retention and fusion into myofibers compared to satellite cells and distribute throughout all muscles in the body [123]. Furthermore, these cells proliferate extensively in vitro and can be genetically modified making them an attractive cell source for treatment of congenital myopathies such as muscular dystrophy [124–126]. Additionally, iPSCs derived from mesoangioblasts fuse more efficiently with skeletal muscle compared to iPSCs derived from fibroblasts suggesting that the mesoangioblasts may be a more ideal source from which to generate iPSC-derived myogenic progenitors [127]. To date, all published work with mesoangioblasts and pericytes has been preclinical, being performed in mice and dogs, and it is currently unclear if this promising cell therapy approach will translate to humans and be clinically feasible. Interestingly, PW1 is required for mesoangioblasts to cross blood vessel walls and engraft into myofibers and silencing of PW1 inhibits myogenic differentiation potential of mesoangioblasts [128]. Therefore, engineering and/or selecting cells with high PW1 expression may enhance the efficacy of cell transplantation but this has yet to be tested.
Cardiac muscle
Owing to the vast prevalence of heart disease throughout the world, a wide variety of cell types have been used as cardiac cell therapies. Recent reviews have nicely summarized the most important clinical trials using autologous cell types [129, 130], which can be broadly grouped into three main categories: (1) skeletal myoblasts, (2) adipose- and bone marrow-derived cells, and (3) Resident cardiac progenitors and cardiosphere-derived cells.
Myoblasts
Given the limited success of myoblast transplantation into diseased skeletal muscles and the similarities of the two types of striated muscle, early investigations aimed to determine whether myoblasts could also be used for cardiac repair. Initial small non-randomized phase I trials showed a functional benefit following myoblast transplantation but the interpretation of these results is difficult due to the use of confounding treatments such as left ventricular assist device or coronary bypass grafting surgery [131–133]. However, larger Phase II trials found no improvement in LVEF [134, 135] and lack of electromechanical integration between donor myotubes and host myocardium, which even if possible could increase the incidence of arrhythmias [136]. The larger Phase II trials did find an attenuation of LV remodeling and decrease in LV volume, which was attributed to myoblast paracrine action [137, 138]. Overall, the use of myoblasts for cardiac myoplasty is likely to never permit sufficient return of function that would outweigh the incidence and risks of arrhythmia, and thus alternative cell sources have been pursued.
Bone marrow- and adipose-derived cells
Bone marrow-derived cells have been a natural source for cell therapy owing to their relative ease of isolation and the presence of stem cells that have the ability to differentiate into various tissues. These cells are a mixed population of largely undifferentiated cells that consist of early committed cells, hematopoietic and endothelial progenitor cells (~2–4 %), as well as mesenchymal stem cells (MSCs, <0.1 %) [139]. MSCs are a unique multipotent cell group of mesodermal origin characterized by expression of the surface antigens CD73, CD90, CD105, CD44, CD106, and CD166, but not CD34, CD31, CD14, CD45, CD133, or CD105 [140]. Given the relative scarcity of these cells in the bone marrow, MSCs have also been successfully isolated from numerous other organs, including periosteum, synovial membrane, skeletal muscle, skin, pericytes, peripheral blood, deciduous teeth, periodontal ligament, umbilical cord, and adipose tissue, the latter of which holds much promise given the ubiquitous presence of adipose tissues in the body and limited patient morbidity and discomfort upon isolation [141]. To date, two recent meta-analysis studies assessed a combined 82 clinical trials (close to 5000 patients enrolled) using bone marrow-derived cells for cardiac repair and found modest yet significant improvements in LVEF and reduction in scar size (both ~4 %) [142, 143]. However, a 3rd meta-analysis comprising 22 clinical trials failed to identify a significant effect of bone marrow cell treatment on MRI-derived cardiac parameters or clinical outcomes [144]. As typical with cellular therapies, this discrepancy likely stems from differences in the exact cell type, amount of injected cells, route of delivery, and timing and number of injections, suggesting that even larger studies may be needed to establish the utility of bone marrow-derived cells for treatment of heart disease. Similarly, a recent power meta-analysis involving 1225 patients found a statistically significant improvement in LVEF following MSC treatment, with a majority of clinical trials that demonstrated decreased LV dilation and remodeling employing exogenous MSCs [145]. Further trials are ongoing or planned to determine the exact mechanisms of functional improvements and identify MSC subfractions (such as CD133+ cells [146]) with enhanced therapeutic benefits.
Resident cardiac progenitors and cardiosphere-derived cells
Since the ground-breaking discovery of endogenous cardiac progenitors (“cardiac stem cells”, CSCs) over a decade ago [147], numerous pre-clinical studies have assessed whether in vitro expansion followed by administration of these cells into the heart can be a viable therapeutic approach for cardiac repair. CSCs are an exceptionally rare c-Kit+/Lin− population, comprising 0.002–0.005 % of all cells in the heart, and are typically subcultured from cardiac biopsies [130]. Alternatively, cell clusters termed cardiospheres can be cultured from endocardial biopsies and dissociated to obtain cardiosphere-derived cells, a heterogeneous population predominantly containing c-Kit+/Lin− CSCs and CD105+/CD90+/CD45− non-hematopoietic cardiac mesenchymal cells [148]. Recent advancements in endocardial biopsy techniques have significantly improved the ability to isolate CSCs and cardiosphere-derived cells [149]. A recent randomized controlled phase I trial (SCIPIO) demonstrated that intracoronary administration of purified c-Kit+ CSCs significantly improved LV systolic function (up to 12 % increase) and reduced infarct size after 1 year when administered to patients suffering from chronic ischemic cardiomyopathy [150]. Another randomized controlled phase I clinical trial (CADUCEUS) similarly showed that administration of cardiosphere-derived cells (95 % CD105+ without c-Kit+ cell purification) improved regional function of infarcted myocardium and decreased scar size, this time in patients with a more acute LV dysfunction after a myocardial infarction [151]. As such, these studies have paved the way for future phase II clinical trials, which will aim to establish the curative effect of cardiosphere-derived cells and/or CSCs in both acute and chronic settings of ischemic cardiomyopathy.
Human pluripotent stem cell-derived cardiomyocytes
Since the first successful cardiac differentiation of human embryonic stem cells (hESCs, [152]) and induced pluripotent stem cells (hiPSCs, [153]), hPSC-derived cardiomyocytes (hPSC-CMs) have rapidly gained interest for use in heart repair by being the only currently available source of functional human cardiomyocytes. To date, hPSC-CMs have been only tested in infarcted hearts of mice [154], rats [155–158], guinea pigs [159, 160], pigs [161], and most recently, Macaque monkeys [162], but not in humans. A majority of these studies reported some structural and functional improvements (decreased scar size, thicker heart wall, increased LVEF, etc.; for a recent review see [163]), although the Macaque studies have also risen concerns about the potential arrhythmogenicity of the therapy. Despite this limitation, the cryopreservation techniques developed by the Murry’s group that allowed them to transplant 1 billion cardiomyocytes into the non-human primate hearts [162] is an important milestone prior to clinical translation, and it is foreseeable that in the near future hPSC-CM therapies will progress to clinical trials.
Cell-free, biomaterial therapies
An alternative to cell transplantation is the injection of biomaterials that stimulate different aspects of endogenous repair process or provide favorable mechanical environment to enhance survival of muscle cells. This approach decreases the costs and circumvents technical issues associated with expanding large numbers of patient specific cells, thus permitting the treatment to start sooner after injury. Additionally, the ability to deliver biomaterials in a liquid form has potential for repairing injuries of any shape or size.
Skeletal muscle
Promising biomaterials for repair of large skeletal muscle defects are decellularized scaffolds, where native tissue is stripped of cellular material to prevent a host immune response. When implanted, decellularized scaffolds show a progression of events that mimic natural tissue repair [164]. In practice, decellularization protocols are intended to provide a balance between sufficient cellular removal and maintenance of ECM structure. Cellular debris in decellularized scaffolds [165, 166] and excessive modification of native ECM structure with cross-linkers [167] are both associated with poor remodeling outcomes. Despite the clinical use of decellularized scaffolds, the Food and Drug Administration has not established standards for tissue decellularization, resulting in high variability between different manufacturers and variable functional outcomes upon implantation [168].
The gastrointestinal small intestine submucosa (SIS) and bladder are the most commonly used tissues to derive acellular scaffolds for skeletal muscle repair, with SIS showing the same level of muscle regeneration compared to a skeletal muscle derived decellularized ECM [169]. In animal models, acellular scaffolds can restore muscle function though the level of functional repair can vary significantly [164, 170, 171]. Preliminary work in humans has mirrored the findings of animal studies, namely a variable functional recovery following scaffold implantation [164, 172]. It should be noted, however, that in the human studies scaffold implantation occurred months to years after volumetric muscle loss injury, which could preclude more favorable outcomes expected to result from earlier implantations. In addition to technical improvements in acellular scaffolds, combined implantation of cells and scaffolds [171] as well as optimization of physical therapy may further improve functional recovery in these patients [173].
Cardiac muscle
In terms of applicability in the heart, the SIS-ECM decellularized matrix has been demonstrated to be feasible and safe in 40 patients with congenital heart disease [174], while its utility for the repair of ischemic myocardium has been shown in murine [175, 176] and rabbit [177] models of myocardial infarction with moderate improvements in cardiac function. Similar to the SIS-ECM, decellularized matrix from rat [178] or porcine [179, 180] hearts has been used for myocardial repair in rat [178, 179] as well as porcine [180] models of myocardial infarction, the latter being delivered via a percutaneous transendocardial injection, demonstrating potential translatability to human therapies. Outside of decellularized matrices, a variety of scaffold materials have shown promising results in animal models of MI, including natural derivatives such as fibrin, alginate, collagen, chitosan, Matrigel, hyaluronic acid, and Gelfoam/gelatin, as well as synthetic materials such as self-assembling peptides and polymer-based systems (reviewed in detail in [181, 182]). In these studies, biomaterial injections in peri-infarct zone mainly served to reduce the compliance mismatch between the scar and remote myocardium and consequently promote cell survival and favorable remodeling of the heart. Additionally, numerous studies have demonstrated further therapeutic benefits by incorporation of growth factors (bFGF, TGF- β1, IGF-1, VEGF, Neuregulin-1β) and/or cells in the injected biomaterials [183–185]. Many of such studies continue to be performed in small animals; however, studies in larger animal models [186] are required to develop effective treatment strategies with prospect for clinical translation.
Engineered tissue therapies
The inability to engraft sufficient viable cells after transplantation by a bolus injection [187] has prompted development of alternative approaches to pre-engineer a striated muscle tissue in vitro followed by its implantation at the injury site in vivo. In an ideal case, the advantages of this approach are that tissue function is known prior to implantation, tissue implants can be preconditioned for maximized survival and function, patients can expect a more immediate return of function, restoration of large defects can be performed with a single intervention, and any adverse immune response is localized. It is accepted, although debatable, that the key to engineering a functional tissue in vitro is to best replicate the in vivo milieu, which can be achieved by providing cells with an appropriate ECM, biochemical, and mechanical microenvironment to support maximal differentiation and maturation. The ECM should be biocompatible and biodegradable, provide a high surface area for cell adhesion and structural support, and amplify autocrine and paracrine effects by minimizing diffusion distances [188, 189]. Furthermore, cells are exquisitely sensitive to the stiffness to which they are exposed to [190, 191] requiring that the provided ECM should be of similar stiffness to that of native tissue in vivo. The similar ECM protein composition and stiffness of skeletal (12 kPa) [97, 98, 191] and cardiac (8-18 kPa) [192, 193] muscles have resulted in the use of similar biomaterials for their in vitro tissue engineering.
Skeletal muscle
The most abundant ECM protein in adult striated muscles is collagen type I [194], and thus the first engineered skeletal muscle was produced by embedding avian myotubes within a collagen I hydrogel anchored between two fixed points [195]. This was followed by demonstrations that murine C2C12 myoblast cell line can undergo myogenesis (i.e., proliferate, fuse, and differentiate) within a 3D hydrogel environment [196–199]. Mechanical tension between the anchor points within hydrogel promoted myoblast alignment and stimulated muscle growth. Compared to culture in traditional 2D dishes, the 3D environment permitted longer culture times and greater maturation levels. This method has been used to engineer muscle from primary rodent [200–204] myoblasts but active forces were only generated when collagen was mixed with matrigel. The only reported functional engineered muscle using a pure collagen matrix was constructed by alternate layering of rat myotubes and collagen [205] and yielded specific forces of 2 mN/mm2, significantly smaller than 230 mN/mm2 measured in native muscle [206].
More recent studies have utilized fibrin hydrogels that permit significant matrix remodeling and ECM synthesis, and exhibit stiffness resembling that of skeletal muscle [207, 208]. The first fibrin-based engineered skeletal muscle constructs utilized primary neonatal rat myoblasts and generated specific forces (36 mN/mm2) ~6 times lower than native adult rat muscles but greater than collagen-based muscle constructs [209]. Further improvements in specific force to approach values of 100 mN/mm2 [210] have been achieved by including Matrigel [211], a matrix containing laminin and collagen IV proteins found in the basal lamina, dynamic culture and decreased tissue size to maximize nutrient and oxygen delivery [211, 212], and using a highly fusigenic progenitor population [212]. Overall, the absolute and specific forces generated by engineered muscles made of neonatal rat myoblasts have been far higher than those reported when using cells from other species [213–215], including human myoblasts which under similar conditions produced specific forces of 7.2 mN/mm2 [215].
An alternative approach to use of hydrogels/scaffolds to support muscle formation is to allow cells to secrete their own ECM and self-organize into a 3D tissue. Using saran wrap as a cell culture substrate and adding fibroblasts to secrete sufficient ECM enabled the self-assembly of muscle tissue constructs that expressed adult isoforms of myosin heavy chain [216]. This method was improved by replacing saran wrap with laminin, the major component of the basal lamina [217]. While rat skeletal muscle cells in these tissues generated a specific force of 4.1 mN/mm2, primary mouse cells generated 15.6 mN/mm2, possibly due to the greater ECM secretion by fibroblasts to support muscle differentiation and force transmission. The long time to formation (~35 days) and challenges with scale-up have limited utilization of this technique compared to use of natural or synthetic scaffolds. Alternatively, self-assembly of large-area muscle constructs has been achieved using a cell sheet engineering technique in which mixtures of muscle cells and fibroblasts were cultured on dishes coated with thermoresponsive polymer poly(N-isopropylacrylamide) until they generated sufficient ECM and then detached from tissue culture plates by decreasing temperature [218–220]. Stacking of the sheets can allow generation of thicker muscle tissues and incorporation of endothelial and neuronal layers [218, 219]; however, functionality of these muscle constructs has yet to be reported.
While state-of-the-art in vitro engineered skeletal muscle tissues can display postnatal levels of generated contractile force and physiological length-tension and force-frequency relationships [210], their twitch:tetanus ratios mostly resemble embryonic state, although this may be dependent upon the geographical origin of the culture serum [221]. Furthermore, engineered muscles retain the contractile and metabolic phenotype specific to a muscle used for cell isolation [213, 222] and have been recently designed to successfully regenerate in vitro in response to cardiotoxin injury [210], thus exhibiting features characteristics of native mature muscle. Still, the myofiber diameter and functional parameters of engineered muscle remain inferior to those typically found in adult skeletal muscles. Generation of more complex tissue constructs containing additional non-muscle cell types may yield improved properties and utility of engineered muscles. For example, the addition of fibroblasts [223] or their paracrine factors [224] enhanced engineered muscle formation, though excessive fibroblasts can negatively impact force generation. Muscle–tendon units have been engineered by fabricating composite scaffolds to simultaneously form muscle and tendon de novo [225, 226], or by using native tendons [227, 228] or bone-tendon structures [229] as engineered muscle anchors. Addition of motor neurons [230–232] or spinal cord explants [233] resulted in the formation of neuromuscular junctions and promoted force generation of engineered muscle [233]. Whilst engineering a muscle–tendon-bone unit is expected to benefit in vivo implantation, it is unclear if neuron-muscle implants would integrate into the host neuronal system. Alternatively, factors that promote acetylcholine receptor formation and clustering on muscle fiber sarcolemma, such as agrin, may provide more benefit to functional integration of engineered muscle implants in vivo [234–236].
Although soluble factors and co-culturing with other muscle-resident cells are likely to promote myofiber maturation, engineering an adult-like muscle construct in vitro will require additional biophysical and metabolic cues. The continuous passive stretch applied to skeletal muscle by bone growth is critical for muscle development [237] while cyclic mechanical loading is crucial for regulating adult muscle mass [238]. Replicating these distinct loading regimes with ramp or cyclic stretch resulted in improved myotube alignment, hypertrophy, and mass of engineered muscle [239–242]. In addition to mechanical loading, tonic and voluntary muscle contractions induced by functional innervation are required for complete muscle development [243, 244], maintenance of muscle mass [245], and regulation of muscle fiber phenotype [246]. Thus application of electrical stimulation to mimic neuronal input induced rapid increase in force generation [222, 247–251], while mimicking fast and slow neural activation patterns directed engineered muscle contractile and metabolic phenotype and protein expression towards particular fiber type [213, 222, 248]. While the above methodologies will support maturation of engineered muscle in vitro and survival and functional integration in vivo, the scale-up of tissue size along with sufficient vascular supply remain as major roadblocks to clinical translation.
Cardiac muscle
Almost a decade after the emergence of the first tissue-engineered skeletal muscles, embryonic chick cardiomyocytes were embedded in a collagen gel loaded between Velcro-coated glass tubes to create the first tissue-engineered cardiac muscle [252]. These early cardiac tissues generated contractile stresses (specific forces) of 0.2–0.3 mN/mm2 and showed length- and Ca2+-dependent force increase characteristic of native myocardium. Owing to their relative ease of isolation and low cost, neonatal rat ventricular myocytes (NRVMs) have been the first, widely utilized mammalian cell source for cardiac tissue engineering. The first NRVM-based cardiac tissues were engineered by Bursac et al. in 1999 using fibrous polyglycolic scaffolds [253], followed by the first gelatin mesh-based cardiac tissues implanted by Li et al. [254]. Since then, NRVMs were utilized in 3D alginate scaffolds [255], collagen gels [256, 257] and collagen/Matrigel matrices [258], and have progressed towards the use in scaffold-free cell sheets [259] and cylinders [260], fibrin gels [261–263], and fibrin/Matrigel constructs [264, 265], the latter of which have generated contractile stresses of ~9 mN/mm2 [264], still significantly lower than those measured in adult rat myocardium (~70 mN/mm2 [266, 267]).
In addition to force of contraction, which is arguably the most critical functional output of engineered skeletal muscle, the engineered cardiac muscle must also replicate the fast action potential propagation (assessed by measuring conduction velocity, CV) that allows the native heart to rapidly contract in an orderly fashion. In terms of cardiac repair applications, mature electrophysiological properties of engineered cardiac tissues, including high conduction velocity, are necessary to ensure electrical safety of the therapy, while high contractile forces would contribute the therapeutic efficacy. To this end, several studies with NRVMs [264, 268–272] have utilized the technique of optical mapping, whereby incubation of engineered tissues in a voltage-sensitive dye such as Di-4-ANEPPS and stimulation by a point electrode allow CV measurement by analyzing the spatial spread of action potential wavefronts. Measured CVs have ranged from 14.4 cm/s [268] to 36.1 cm/s [272], still inferior to values reported for adult rat ventricular tissues (66–69 cm/s [273, 274]). Beside the evaluation of the basic tissue-scale functional parameters, robust formation of T-tubules (hallmark of excitation–contraction coupling maturation) was reported in NRVMs after 3–4 weeks of culture in fibrin-based constructs [272]. While these results were obtained without any external stimulation, application of chronic cyclic stretch to better mimic the in vivo cardiac mechanics has also yielded the T-tubular formation in NRVMs [258]. Recently, chronic electrical stimulation (4 Hz over 5 days) yielded a positive force-frequency relationship, which despite being at sub-physiological rates and small in magnitude, is still the first demonstration of an important adult-like physiological response [275].
With the advent of stem cell research, experiences with engineering of NRVM constructs have been applied to generate cardiac tissues from mouse [276–280] and, more recently, human embryonic and induced pluripotent stem cell-derived cardiomyocytes, ESC-CMs and iPSC-CMs [281]. In particular, numerous groups have engineered 3D cardiac tissues from hESCs and hiPSCs [163, 282–285] that generated specific forces ranging from 0.08 mN/mm2 [286] to 11.8 mN/mm2 [287] and conduction velocities ranging from 4.9 cm/s [288] to 25.1 cm/s [287, 289]. Thus, further progress in the field will be necessary to bring the state of human cardiac tissue engineering closer to replicating the robust phenotype of the adult human myocardium (specific forces of 25-44 mN/mm2 [290, 291], average conduction velocity of ~50 cm/s [292]). In particular, as hESC- and hiPSC-CMs are structurally and functionally more similar to human fetal than neonatal or adult CMs [293], numerous strategies have been attempted to improve maturation of cardiomyocytes, including long-term culture, electrical and mechanical stimulation, supplementation with growth factors and hormones such as IGF-1 and T3, and fostering heterocellular interactions with non-myocytes [294–296]. For example, 3-4 month 2D culture of hPSC-CMs has produced structurally and electrophysiologically more mature cardiomyocytes [297], while 1-year 2D-cultured hPSC-CMs demonstrated appearance of M-bands (in <10 % of CMs), a hallmark of advanced sarcomeric maturation [298]. Recently, a 4-month long culture within 3D PEG (poly(ethylene glycol))-fibrinogen hydrogels yielded the formation of T-tubules in hPSC-CMs [282]; still the CM volume in all these studies remained significantly smaller than that of the adult CMs.
Since long-term cultures are obviously time and resource intensive, much recent focus has been placed on accelerated maturation of human cardiomyocytes in vitro. Application of chronic electrical stimulation to engineered tissues improved hPSC-CM ultrastructural organization, contractile force generation, electrophysiological properties, and conduction velocity [299, 300], while cyclic stretch improved hPSC-CM alignment and hypertrophy [286]. Treatment of hPSC-derived cardiac tissues with thyroid hormone T3, which normally stimulates cardiac maturation during development, has led to increased cardiomyocyte size, sarcomere length, and contractile force, all indicative of structural and functional maturation [295]. Furthermore, several groups have demonstrated the crucial role that non-myocytes, primarily fibroblasts, smooth muscle cells, and endothelial cells, play in the development of functional 3D cardiac tissues [276, 286–289]. However, there exists a delicate balance in the cardiomyocyte to non-myocyte ratio, as insufficient non-myocytes will inadequately remodel and compact a 3D tissue, while their overabundance can impede the formation of a functional, electromechanically coupled syncytium [253]. Studies of various cell ratios using mESC-derived CMs suggest that as few as 3 % fibroblasts are enough to remodel a tissue matrix without impeding inter-myocyte connections [276], while a moderately high hESC-derived cardiomyocyte fraction (60–80 %) is optimal for generating human 3D tissues with highest forces of contraction [287]. Overall, success of the above individual modifications has been modest, and it is likely that achieving a true, adult cardiomyocyte phenotype in vitro will entail more sophisticated and mechanistically driven approaches to provide the key time-varying environmental cues present in the postnatal cardiac development (e.g. biomechanical, metabolic, endocrine, cell–matrix, cell–cell, etc.).
Engineered tissue vascularization and implantation
While the above strategies are expected to result in successful generation of mature striated muscles suitable for disease modeling and drug development in vitro, one of the main obstacles to successful therapies in vivo will be the ability to rapidly supply implanted muscle cells with oxygen and nutrients from the host circulation. The high metabolic demand of any larger size avascular striated muscle implants would result in rapid consumption of diffused oxygen within the tens of microns from the host capillaries leading to hypoxia and cell death. At least in the case of skeletal muscle implants, some regeneration could occur from resident satellite cells [210, 301] while similar effects in cardiac tissue constructs would only be expected if implanted cardiomyocytes remained proliferative in vivo. Formation of pre-vascularized tissues in vitro prior to implantation and/or conditions to accelerate vascularization in vivo offers avenues to improve implant survival and enhance therapeutic effects.
Skeletal muscle
In early studies, pre-vascularized engineered skeletal muscle tissues formed by tri-culture of myoblasts, endothelial cells (ECs) and fibroblasts in scaffolds composed of 50 % poly-(l-lactic acid) (PLLA) and 50 % polylactic-co-glycolic acid (PLGA) [302] showed improved cell survival and vascularization compared to tissues formed by co-culture of myoblasts and endothelial cells. In these constructs, fibroblasts were necessary to stabilize and increase formation of lumen structures, while increased tri-culture time prior to implantation further enhanced survival and function of muscle implants in vivo [303]. On the other hand, acellular bladder matrix, which naturally supports rapid angiogenesis and vascular infiltration [304], allowed the use of only ECs and muscle progenitor cells yielding vascularization, neuron infiltration, and maturation of engineered muscle tissue in vivo [305]. As skeletal muscle requires an axial vascular pedicle to maintain function and develop adequate vascularization [306], an arteriovenous (AV) loop connecting an artery and vein was used to provide such a pedicle [307–309], and combined with a tri-culture scaffold in vivo allowed the repair of large muscle defects [310]. Alternatively, engineered muscle tissues were placed along the femoral vessels to provide an axial vascular supply leading to the formation of viable myofibers throughout the tissue cross-section [306, 311]. These studies, among others, are highlighted in Table 1, which summarizes the function, vascularization, and in vivo tissue repair of in vitro engineered skeletal muscle implanted into various animal models.
Other approaches have involved incorporation of vascular endothelial growth factor (VEGF) into biodegradable polymer scaffolds or engineering of myoblasts to secrete angiogenic factors [312–314]. The sustained levels of VEGF by these methods result in far greater angiogenesis than bolus VEGF delivery. The results of these studies can be further improved by co-delivery of pro-myogenic growth factors, such as IGF-1, resulting in enhanced muscle regeneration and innervation [315, 316]. Lastly, a recent approach involving the use of human mesangioblasts transduced with the proangiogenic placenta growth factor (PlGF) embedded in fibrinogen-PEG hydrogel restored a full murine tibialis anterior muscle defect yielding vascular and neuronal ingrowth into engineered muscle implants [317]. Mesangioblast implants not expressing PlGF showed decreased vascular infiltration, muscle size and density, suggesting that the proangiogenic effects of PlGF promoted survival of muscle fibers [317, 318]. While single explanted myofibers from the regenerated muscle had specific forces similar to those of native myofibers, it is unclear if they were purely of human origin and if contractile force from the whole regenerated muscle (reflective of functional recovery) was the same as that of the intact TA muscle.
Cardiac muscle
Similar to skeletal muscle, successful pre-vascularization of engineered cardiac tissues has typically required co- and/or tri-culture of cardiomyocytes and vascular cells, most commonly endothelial cells and either fibroblasts or smooth muscle cells [277, 286, 319–322] (reviewed in [323]). Beside adding supporting cell types, engineering cardiac tissues using porous scaffolds [184, 319, 324, 325] or impregnating scaffolds with pro-angiogenic factors such as VEGF [184, 326] or stromal-cell derived factor-1 (SCF-1) [326] have enhanced host neovasculogenesis upon tissue implantation in both normal and infarcted hearts. Hence, numerous studies have assessed the ability of engineered cardiac tissue implants to actively repair infarcted myocardium in small and large animal models (Table 2). While various studies have shown some degree of structural and functional repair, the results differ widely on the extent of functional recovery, graft vascularization, and electrical integration with the host heart. Of note, very few studies assessed in vitro contractile function of engineered tissues prior to their implantation, and no studies reported important measures of electrical function and electrical safety, such as conduction velocity. A recent comparison between injected hPSC-CMs and transplanted hPSC-CM patches showed that injected cells were able to form electrical connections with host myocardium [327] while functional integration of transplanted patches was hampered by the existence of fibrous layer between the patch and host myocardium. Thus, the lack of direct electromechanical integration between implant and host cardiomyocytes remains an important challenge in the field, resolution of which would be expected to further enhance therapeutic effects of engineered cardiac tissues beyond mere paracrine effects. Importantly Menasche et al. has recently presented the first clinical implantation of a hESC-derived cardiac progenitor patch into a human heart [328]. No adverse effects and preliminary evidence of significant improvement in the patient’s heart failure severity offer encouraging prospects for the use of hPSC-derived cardiac tissues in human heart repair, and will undoubtedly be followed up by future clinical trials and in-depth mechanistic studies.
Future considerations
Successful therapies for striated muscle dysfunction will require further technological advances and improved understanding of muscle physiology, development, and disease. Congenital muscle pathologies are most likely to benefit from gene therapies that employ modern genome engineering techniques [342–345], while cell therapies with healthy or genetically corrected cells will be limited to repair of the heart and specific skeletal muscles, or potentially all striated muscles via intra-arterial delivery of mesoangioblasts [124, 126], The use of biomaterials with or without cells will likely be most beneficial for treatment of myocardial infarction [346] and traumatic skeletal muscle injuries such as volumetric muscle loss [168, 172].
One of the important challenges in the cell-based human skeletal muscle repair is the inability to expand satellite cells in sufficient quantities without loss of their myogenic potential [94]. Successful methods to recreate SC niche and enable SC self-renewal in vitro may require the presence of differentiated myofibers and various non-muscle cells (FAPs, ECs, pericytes), manipulation of ECM proteins and substrate mechanics, and use of 3D cell culture [75, 98, 212]. Alternatively, generating functional SCs from hPSCs may overcome this challenge but improved differentiation protocols, via identification of novel small molecules, cell sorting, or genome editing, will be required to increase the efficiency of myogenic hPSC differentiation. Furthermore, while hPSC-derived myogenic precursors have been shown to fuse with mouse myofibers and contribute injury repair in vivo, their ability to form functional human muscle is still unknown. For systemic delivery of myogenic cells it will be also important to promote their passing through capillary walls, which could be achieved by use of gene editing techniques (e.g. CRISPR/Cas9) to safely and stably express proteins such as Pw1/Peg3 [128].
In contrast to skeletal muscle, where implantation of muscle progenitors can contribute to formation of new mature myofibers and muscle repair, injection of immature hPSC-CMs in the heart may not lead to the formation of mature cardiac muscle and may predispose heart to life-threatening arrhythmias [162]. Tissue engineering strategies have emerged as alternative cell delivery methods given their ability to improve cell retention and survival and to promote the maturation of hPSC-CMs, although even state-of-the-art 3D cardiac tissues still lack various properties of adult myocardium. It is possible that full maturation of engineered hPSC-CM tissues can be achieved through a combination of biochemical factors and electromechanical conditioning that closely mimic the complex microenvironment of developing myocardium. Moreover, understanding the complex interactions between hPSC-CMs and non-CMs (fibroblasts, endothelial cells, smooth muscle cells, pericytes, macrophages, etc.) will be likely required for the successful engineering of mature cardiac tissues. Alternatively, combinations of non-physiological stimuli may be identified that jump-start maturation programs in hPSC-CMs to accelerate their metabolic adaptation, hypertrophy, and acquisition of an adult functional phenotype.
For successful tissue engineering therapies, incorporating a vascular and neuronal component will be required to overcome the size and survival limitations. Obtaining robust vascular networks in vitro will likely require the presence of both endothelial and perivascular (pericyte, macrophage, smooth muscle, fibroblast) cells, yet the exact proportions of these cell types remains unclear. Furthermore, in vitro conditions for vascularization need to be identified that do not compromise differentiation, force generation, or electrical conduction (for cardiac) of engineered muscle. Related challenge is the inability to functionally integrate (i.e. enable synchronous activity) of engineered and host muscles in vivo. For engineered skeletal muscle, stable and functional NMJs are required to both promote muscle maturation and enable its innervation upon engraftment. In vitro or in vivo application of synaptogenic factors (e.g. agrin) [234, 236] or intermittent neuromuscular electrical stimulation [347] may be necessary to promote implanted engineered muscle survival, maturation, and formation of stable connections with the host motor neurons. For engineered cardiac muscle, the main contributor to the lack of electrical coupling between the implanted and host cardiac tissue is the fibrous layer that forms between the tissue implant and epicardium [327]. Surgical strategies to better adhere engineered tissues to the heart surface, or genetic engineering strategies to render the fibrous layer electrically conducting [348] may yield functional synchronization between the implanted and host cardiomyocytes.
Conclusions
Current pharmacological therapies show limited capacity to augment the regenerative response of human striated muscles to ischemic injury, excessive trauma, or degenerative disease. Over the past 35 years, a range of alternative cell-, gene-, and material-based strategies have evolved that have shown promising results in both small and large animal studies. Still, a greater understanding of the specific cellular and molecular mechanisms that can benefit striated muscle repair and regeneration is required to improve the efficacy of these approaches. For skeletal muscle therapies, improved cell isolation and culture techniques are needed to expand functional stem cells and maintain their myogenic potential in vitro. For effective cardiac repair, robust methods are required to accelerate in vitro cardiomyocyte maturation and promote transplanted cell survival in vivo. Finally, methods to produce large striated muscle tissues that can be rapidly vascularized and functionally integrated with host muscles need to be developed to enable successful translation of these promising strategies to clinical practice.
Abbreviations
- CM:
-
Cardiomyocyte
- CSC:
-
Cardiac stem cell
- CV:
-
Conduction velocity
- CHD:
-
Congenital heart defect
- DGC:
-
Dystrophin-associated glycoprotein complex
- ECM:
-
Extracellular matrix
- FAP:
-
Fibroadipogenic progenitors
- hESC:
-
Human embryonic stem cell
- hiPSC:
-
Human induced pluripotent stem cell
- mESC-CM:
-
Mouse embryonic stem cell-derived cardiomyocyte
- MHC:
-
Myosin heavy chain
- MMP:
-
Matrix metalloproteinase
- MSC:
-
Mesenchymal stem cells
- NRVM:
-
Neonatal rat ventricular myocyte
- PIC:
-
Pw1 interstitial cell
- RyR:
-
Ryanodine receptor
- SR:
-
Sarcoplasmic reticulum
- SERCA:
-
Sarcoplasmic reticulum Ca2+ ATPase
- SC:
-
Satellite cell
- SIS:
-
Small intestine submucosa
- T-tubule:
-
Transverse tubule
References
Schiaffino S, Reggiani C (2011) Fiber types in mammalian skeletal muscles. Physiol Rev 91:1447–1531
Janssen I, Heymsfield SB, Wang ZM, Ross R (1985) Skeletal muscle mass and distribution in 468 men and women aged 18-88 yr. J Appl Physiol 2000(89):81–88
Arnold HD (1899) Weight of the “Normal” Heart in Adults. J Boston Soc Med Sci 3:174–184
Dumont NA, Wang YX, Rudnicki MA (2015) Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 142:1572–1581
Uygur A, Lee RT (2016) Mechanisms of Cardiac Regeneration. Dev Cell 36:362–374
Wallace GQ, McNally EM (2009) Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu Rev Physiol 71:37–57
DiMauro S, Spiegel R (2011) Progress and problems in muscle glycogenoses. Acta Myol 30:96–102
Grogan BF, Hsu JR (2011) Skeletal trauma research C. Volumetric muscle loss. J Am Acad Orthop Surg 19(Suppl 1):S35–S37
Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL et al (2011) Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle 1:21
Jessup M, Brozena S (2003) Heart failure. N Engl J Med 348:2007–2018
Fahed AC, Gelb BD, Seidman JG, Seidman CE (2013) Genetics of congenital heart disease: the glass half empty. Circ Res 112:707–720
Hanson J, Huxley HE (1953) Structural basis of the cross-striations in muscle. Nature 172:530–532
Huxley HE (1953) Electron microscope studies of the organisation of the filaments in striated muscle. Biochim Biophys Acta 12:387–394
Luther PK (2009) The vertebrate muscle Z-disc: sarcomere anchor for structure and signalling. J Muscle Res Cell Motil 30:171–185
Knöll R, Buyandelger B, Lab M (2011) The sarcomeric Z-disc and Z-discopathies. J Biomed Biotechnol 2011:569628
Al-Qusairi L, Laporte J (2011) T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet Muscle 1:26
Fill M, Copello JA (2002) Ryanodine receptor calcium release channels. Physiol Rev 82:893–922
Schiaffino S, Margreth A (1969) Coordinated development of the sarcoplasmic reticulum and T system during postnatal differentiation of rat skeletal muscle. J Cell Biol 41:855–875
Franzini-Armstrong C, Ferguson DG, Champ C (1988) Discrimination between fast- and slow-twitch fibres of guinea pig skeletal muscle using the relative surface density of junctional transverse tubule membrane. J Muscle Res Cell Motil 9:403–414
Sawada K, Kawamura K (1991) Architecture of myocardial cells in human cardiac ventricles with concentric and eccentric hypertrophy as demonstrated by quantitative scanning electron microscopy. Heart Vessels 6:129–142
Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY et al (2013) Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci USA 110:1446–1451
Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ (1996) Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol 271:H2183–H2189
Venable JH (1966) Constant cell populations in normal, testosterone-deprived and testosterone-stimulated levator ani muscles. Am J Anat 119:263–270
Enesco M, Puddy D (1964) Increase in the Number of Nuclei and Weight in Skeletal Muscle of Rats of Various Ages. Am J Anat 114:235–244
Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA (2007) Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol 293:H1883–H1891
Souders CA, Borg TK, Banerjee I, Baudino TA (2012) Pressure overload induces early morphological changes in the heart. Am J Pathol 181:1226–1235
Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S et al (2015) Dynamics of cell generation and turnover in the human heart. Cell 161:1566–1575
Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R et al (2016) Revisiting cardiac cellular composition. Circ Res 118:400–409
Gaudesius G, Miragoli M, Thomas SP, Rohr S (2003) Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res 93:421–428
McSpadden LC, Kirkton RD, Bursac N (2009) Electrotonic loading of anisotropic cardiac monolayers by unexcitable cells depends on connexin type and expression level. Am J Physiol Cell Physiol 297:C339–C351
Rohr S (2012) Arrhythmogenic implications of fibroblast-myocyte interactions. Circ Arrhyth Electrophysiol 5:442–452
Ongstad E, Kohl P (2016) Fibroblast-myocyte coupling in the heart: Potential relevance for therapeutic interventions. J Mol Cell Cardiol 91:238–246
Bourdeau-Martini J, Odoroff CL, Honig CR (1974) Dual effect of oxygen on magnitude and uniformity of coronary intercapillary distance. Am J Physiol 226:800–810
Korthuis RJ (2011) Skeletal Muscle Circulation. San Rafael
Andersen P, Kroese AJ (1978) Capillary supply in soleus and gastrocnemius muscles of man. Pflugers Arch 375:245–249
Poole DC, Mathieu-Costello O (1989) Skeletal muscle capillary geometry: adaptation to chronic hypoxia. Respir Physiol 77:21–29
Poole DC, Mathieu-Costello O (1990) Analysis of capillary geometry in rat subepicardium and subendocardium. Am J Physiol 259:H204–H210
Layland J, Kentish JC (1999) Positive force- and [Ca2+]i-frequency relationships in rat ventricular trabeculae at physiological frequencies. Am J Physiol 276:H9–H18
Henneman E, Somjen G, Carpenter DO (1965) Excitability and inhibitability of motoneurons of different sizes. J Neurophysiol 28:599–620
Huxley AF (1957) Muscle structure and theories of contraction. Prog Biophys Biophys Chem 7:255–318
Allen DG, Kentish JC (1985) The cellular basis of the length-tension relation in cardiac muscle. J Mol Cell Cardiol 17:821–840
Granzier HL, Irving TC (1995) Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J 68:1027–1044
Mauro A (1961) Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9:493–495
Lepper C, Partridge TA, Fan CM (2011) An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 138:3639–3646
Sambasivan R, Comai G, Le Roux I, Gomes D, Konge J, Dumas G et al (2013) Embryonic founders of adult muscle stem cells are primed by the determination gene Mrf4. Dev Biol 381:241–255
Biressi S, Bjornson CR, Carlig PM, Nishijo K, Keller C, Rando TA (2013) Myf5 expression during fetal myogenesis defines the developmental progenitors of adult satellite cells. Dev Biol 379:195–207
Kuang S, Kuroda K, Le Grand F, Rudnicki MA (2007) Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129:999–1010
Cossu G, Tajbakhsh S (2007) Oriented cell divisions and muscle satellite cell heterogeneity. Cell 129:859–861
Gunther S, Kim J, Kostin S, Lepper C, Fan CM, Braun T (2013) Myf5-positive satellite cells contribute to Pax7-dependent long-term maintenance of adult muscle stem cells. Cell Stem Cell 13:590–601
Charge SB, Rudnicki MA (2004) Cellular and molecular regulation of muscle regeneration. Physiol Rev 84:209–238
Yin H, Price F, Rudnicki MA (2013) Satellite cells and the muscle stem cell niche. Physiol Rev 93:23–67
Leri A, Rota M, Pasqualini FS, Goichberg P, Anversa P (2015) Origin of cardiomyocytes in the adult heart. Circ Res 116:150–166
van Berlo JH, Molkentin JD (2016) Most of the Dust Has Settled: cKit+ Progenitor Cells Are an Irrelevant Source of Cardiac Myocytes In Vivo. Circ Res 118:17–19
Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A et al (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204:1057–1069
Madaro L, Bouche M (2014) From innate to adaptive immune response in muscular dystrophies and skeletal muscle regeneration: the role of lymphocytes. Biomed Res Int 2014:438675
Aurora AB, Olson EN (2014) Immune modulation of stem cells and regeneration. Cell Stem Cell 15:14–25
Webster MT, Manor U, Lippincott-Schwartz J, Fan CM (2016) Intravital Imaging Reveals Ghost Fibers as Architectural Units Guiding Myogenic Progenitors during Regeneration. Cell Stem Cell 18:243–252
Saclier M, Cuvellier S, Magnan M, Mounier R, Chazaud B (2013) Monocyte/macrophage interactions with myogenic precursor cells during skeletal muscle regeneration. FEBS J 280:4118–4130
Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J et al (2010) Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 12:153–163
Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM et al (2013) Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153:376–388
Foglia MJ, Poss KD (2016) Building and re-building the heart by cardiomyocyte proliferation. Development 143:729–740
Haubner BJ, Schneider J, Schweigmann U, Schuetz T, Dichtl W, Velik-Salchner C et al (2016) Functional recovery of a human neonatal heart after severe myocardial infarction. Circ Res 118:216–221
Xin M, Olson EN, Bassel-Duby R (2013) Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol 14:529–541
Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R et al (2014) Macrophages are required for neonatal heart regeneration. J Clin Investig 124:1382–1392
Sutton MG, Sharpe N (2000) Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 101:2981–2988
Morrison JI, Loof S, He P, Simon A (2006) Salamander limb regeneration involves the activation of a multipotent skeletal muscle satellite cell population. J Cell Biol 172:433–440
Poss KD, Keating MT, Nechiporuk A (2003) Tales of regeneration in zebrafish. Dev Dyn 226:202–210
Turner NJ, Badylak SF (2012) Regeneration of skeletal muscle. Cell Tissue Res 347:759–774
Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R (2004) The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc 52:80–85
Berger MJ, Doherty TJ (2010) Sarcopenia: prevalence, mechanisms, and functional consequences. Interdiscip Top Gerontol 37:94–114
Ryall JG, Schertzer JD, Lynch GS (2008) Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology 9:213–228
Morse CI, Thom JM, Reeves ND, Birch KM, Narici MV (1985) In vivo physiological cross-sectional area and specific force are reduced in the gastrocnemius of elderly men. J Appl Physiol 2005(99):1050–1055
Hughes VA, Frontera WR, Wood M, Evans WJ, Dallal GE, Roubenoff R et al (2001) Longitudinal muscle strength changes in older adults: influence of muscle mass, physical activity, and health. J Gerontol A Biol Sci Med Sci 56:B209–B217
Porter MM, Vandervoort AA, Lexell J (1995) Aging of human muscle: structure, function and adaptability. Scand J Med Sci Sports 5:129–142
Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY et al (2014) Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med 20:255–264
Bernet JD, Doles JD, Hall JK (2014) Kelly Tanaka K, Carter TA, Olwin BB. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med 20:265–271
Johnson NB, Hayes LD, Brown K, Hoo EC, Ethier KA, Centers for Disease C et al (2014) CDC national health report: leading causes of morbidity and mortality and associated behavioral risk and protective factors–United States, 2005–2013. Morb Mort Week Rep Surveill Summ 63(Suppl 4):3–27
Satpathy C, Mishra TK, Satpathy R, Satpathy HK, Barone E (2006) Diagnosis and management of diastolic dysfunction and heart failure. Am Fam Physician 73:841–846
Yang Q, Chen H, Correa A, Devine O, Mathews TJ, Honein MA (2006) Racial differences in infant mortality attributable to birth defects in the United States, 1989–2002. Birth Defects Res A 76:706–713
Hoffman JI, Kaplan S (2002) The incidence of congenital heart disease. J Am Coll Cardiol 39:1890–1900
Avolio E, Caputo M, Madeddu P (2015) Stem cell therapy and tissue engineering for correction of congenital heart disease. Front Cell Dev Biol 3
Mercuri E, Muntoni F (2013) Muscular dystrophy: new challenges and review of the current clinical trials. Curr Opin Pediatr 25:701–707
Lapidos KA, Kakkar R, McNally EM (2004) The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res 94:1023–1031
Gaeta M, Messina S, Mileto A, Vita GL, Ascenti G, Vinci S et al (2012) Muscle fat-fraction and mapping in Duchenne muscular dystrophy: evaluation of disease distribution and correlation with clinical assessments. Preliminary experience. Skeletal Radiol 41:955–961
Khurana TS, Prendergast RA, Alameddine HS, Tome FM, Fardeau M, Arahata K et al (1995) Absence of extraocular muscle pathology in Duchenne’s muscular dystrophy: role for calcium homeostasis in extraocular muscle sparing. J Exp Med 182:467–475
Goldstein JA, McNally EM (2010) Mechanisms of muscle weakness in muscular dystrophy. J General Physiol 136:29–34
Kim HK, Laor T, Horn PS, Racadio JM, Wong B, Dardzinski BJ (2010) T2 mapping in Duchenne muscular dystrophy: distribution of disease activity and correlation with clinical assessments. Radiology 255:899–908
Kinali M, Arechavala-Gomeza V, Cirak S, Glover A, Guglieri M, Feng L et al (2011) Muscle histology vs MRI in Duchenne muscular dystrophy. Neurology 76:346–353
McNally EM, Pytel P (2007) Muscle diseases: the muscular dystrophies. Annu Rev Pathol 2:87–109
Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M et al (1996) Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation 94:3168–3175
Verhaert D, Richards K, Rafael-Fortney JA, Raman SV (2011) Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations. Circ Cardiovas Imaging 4:67–76
Mouly V, Aamiri A, Perie S, Mamchaoui K, Barani A, Bigot A et al (2005) Myoblast transfer therapy: is there any light at the end of the tunnel? Acta Myol 24:128–133
Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A et al (2005) Direct isolation of satellite cells for skeletal muscle regeneration. Science 309:2064–2067
Machida S, Spangenburg EE, Booth FW (2004) Primary rat muscle progenitor cells have decreased proliferation and myotube formation during passages. Cell Prolif 37:267–277
Beauchamp JR, Morgan JE, Pagel CN, Partridge TA (1999) Dynamics of myoblast transplantation reveal a discrete minority of precursors with stem cell-like properties as the myogenic source. J Cell Biol 144:1113–1121
Ono Y, Masuda S, Nam HS, Benezra R, Miyagoe-Suzuki Y, Takeda S (2012) Slow-dividing satellite cells retain long-term self-renewal ability in adult muscle. J Cell Sci 125:1309–1317
Engler AJ, Griffin MA, Sen S, Bonnemann CG, Sweeney HL, Discher DE (2004) Myotubes differentiate optimally on substrates with tissue-like stiffness: pathological implications for soft or stiff microenvironments. J Cell Biol 166:877–887
Gilbert PM, Havenstrite KL, Magnusson KE, Sacco A, Leonardi NA, Kraft P et al (2010) Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 329:1078–1081
Périé S, Trollet C, Mouly V, Vanneaux V, Mamchaoui K, Bouazza B et al (2014) Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: a phase I/IIa clinical study. Mol Ther 22:219–225
Barberi T, Bradbury M, Dincer Z, Panagiotakos G, Socci ND, Studer L (2007) Derivation of engraftable skeletal myoblasts from human embryonic stem cells. Nat Med 13:642–648
Darabi R, Arpke RW, Irion S, Dimos JT, Grskovic M, Kyba M et al (2012) Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 10:610–619
Darabi R, Santos FN, Filareto A, Pan W, Koene R, Rudnicki MA et al (2011) Assessment of the myogenic stem cell compartment following transplantation of Pax3/Pax7-induced embryonic stem cell-derived progenitors. Stem Cells 29:777–790
Darabi R, Pan W, Bosnakovski D, Baik J, Kyba M, Perlingeiro RC (2011) Functional Myogenic Engraftment from Mouse iPS Cells. Stem Cell Rev 7:948–957
Abujarour R, Bennett M, Valamehr B, Lee TT, Robinson M, Robbins D et al (2014) Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Trans Med 3:149–160
Albini S, Coutinho P, Malecova B, Giordani L, Savchenko A, Forcales SV et al (2013) Epigenetic reprogramming of human embryonic stem cells into skeletal muscle cells and generation of contractile myospheres. Cell Rep 3:661–670
Goudenege S, Lebel C, Huot NB, Dufour C, Fujii I, Gekas J et al (2012) Myoblasts derived from normal hESCs and dystrophic hiPSCs efficiently fuse with existing muscle fibers following transplantation. Mol Ther 20:2153–2167
Shelton M, Metz J, Liu J, Carpenedo RL, Demers SP, Stanford WL et al (2014) Derivation and expansion of PAX7-positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell Rep 3:516–529
Borchin B, Chen J, Barberi T (2013) Derivation and FACS-mediated purification of PAX3 +/PAX7 + skeletal muscle precursors from human pluripotent stem cells. Stem Cell Reports. 1:620–631
Xu C, Tabebordbar M, Iovino S, Ciarlo C, Liu J, Castiglioni A et al (2013) A zebrafish embryo culture system defines factors that promote vertebrate myogenesis across species. Cell 155:909–921
Chal J, Oginuma M, Al Tanoury Z, Gobert B, Sumara O, Hick A et al (2015) Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat Biotechnol 33:962–969
Relaix F, Weng X, Marazzi G, Yang E, Copeland N, Jenkins N et al (1996) Pw1, a novel zinc finger gene implicated in the myogenic and neuronal lineages. Dev Biol 177:383–396
Mitchell KJ, Pannerec A, Cadot B, Parlakian A, Besson V, Gomes ER et al (2010) Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat Cell Biol 12:257–266
Arrighi N, Moratal C, Clement N, Giorgetti-Peraldi S, Peraldi P, Loubat A et al (2015) Characterization of adipocytes derived from fibro/adipogenic progenitors resident in human skeletal muscle. Cell Death Dis 6:e1733
Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K (2010) Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol 12:143–152
Mozzetta C, Consalvi S, Saccone V, Tierney M, Diamantini A, Mitchell KJ et al (2013) Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Mol Med 5:626–639
Formicola L, Marazzi G, Sassoon DA (2014) The extraocular muscle stem cell niche is resistant to ageing and disease. Front Aging Neurosci 6:328
Tonlorenzi R, Dellavalle A, Schnapp E, Cossu G, Sampaolesi M (2007) Isolation and characterization of mesoangioblasts from mouse, dog, and human tissues. Curr Protoc Stem Cell Biol 2 (Unit 2B 1)
Minasi MG, Riminucci M, De Angelis L, Borello U, Berarducci B, Innocenzi A et al (2002) The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 129:2773–2783
De Angelis L, Berghella L, Coletta M, Lattanzi L, Zanchi M, Cusella-De Angelis MG et al (1999) Skeletal myogenic progenitors originating from embryonic dorsal aorta coexpress endothelial and myogenic markers and contribute to postnatal muscle growth and regeneration. J Cell Biol 147:869–878
Birbrair A, Zhang T, Wang ZM, Messi ML, Enikolopov GN, Mintz A et al (2013) Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Res 10:67–84
Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L et al (2007) Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol 9:255–267
Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS et al (2008) A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3:301–313
Tedesco FS, Cossu G (2012) Stem cell therapies for muscle disorders. Curr Opin Neurol 25:597–603
Tedesco FS, Gerli MF, Perani L, Benedetti S, Ungaro F, Cassano M et al (2012) Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci Trans Med 4:140ra89
Sampaolesi M, Blot S, D’Antona G, Granger N, Tonlorenzi R, Innocenzi A et al (2006) Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 444:574–579
Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D’Antona G, Pellegrino MA et al (2003) Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science 301:487–492
Quattrocelli M, Swinnen M, Giacomazzi G, Camps J, Barthelemy I, Ceccarelli G et al (2015) Mesodermal iPSC-derived progenitor cells functionally regenerate cardiac and skeletal muscle. J Clin Investig 125:4463–4482
Bonfanti C, Rossi G, Tedesco FS, Giannotta M, Benedetti S, Tonlorenzi R et al (2015) PW1/Peg3 expression regulates key properties that determine mesoangioblast stem cell competence. Nat Commun. 6:6364
Alrefai MT, Murali D, Paul A, Ridwan KM, Connell JM, Shum-Tim D (2015) Cardiac tissue engineering and regeneration using cell-based therapy. Stem Cells Cloning Adv Appl 8:81–101
Wang X, Zhang J, Zhang F, Li J, Li Y, Tan Z et al (2015) The clinical status of stem cell therapy for ischemic cardiomyopathy. Stem Cells Int 2015:135023
Menasché P, Hagège AA, Scorsin M, Pouzet B, Desnos M, Duboc D et al (2001) Myoblast transplantation for heart failure. Lancet 357:279–280
Siminiak T, Kalawski R, Fiszer D, Jerzykowska O, Rzeźniczak J, Rozwadowska N et al (2004) Autologous skeletal myoblast transplantation for the treatment of postinfarction myocardial injury: phase I clinical study with 12 months of follow-up. Am Heart J 148:531–537
Hagège AA, Marolleau JP, Vilquin JT, Alhéritière A, Peyrard S, Duboc D et al (2006) Skeletal myoblast transplantation in ischemic heart failure: long-term follow-up of the first phase I cohort of patients. Circulation 114:I108–I113
Menasche P, Alfieri O, Janssens S, McKenna W, Reichenspurner H, Trinquart L et al (2008) The Myoblast Autologous Grafting in Ischemic Cardiomyopathy (MAGIC) Trial. First Randomized Placebo-Controlled Study of Myoblast Transplantation. Circulation
Duckers HJ, Houtgraaf J, Hehrlein C, Schofer J, Waltenberger J, Gershlick A et al (2011) Final results of a phase IIa, randomised, open-label trial to evaluate the percutaneous intramyocardial transplantation of autologous skeletal myoblasts in congestive heart failure patients: the SEISMIC trial. EuroIntervention 6:805–812
Reinecke H, Murry CE (2000) Transmural replacement of myocardium after skeletal myoblast grafting into the heart. Too much of a good thing? Cardiovasc Pathol 9:337–344
Farahmand P, Lai TY, Weisel RD, Fazel S, Yau T, Menasche P et al (2008) Skeletal myoblasts preserve remote matrix architecture and global function when implanted early or late after coronary ligation into infarcted or remote myocardium. Circulation 118:S130–S137
Shintani Y, Fukushima S, Varela-Carver A, Lee J, Coppen SR, Takahashi K et al (2009) Donor cell-type specific paracrine effects of cell transplantation for post-infarction heart failure. J Mol Cell Cardiol 47:288–295
Dimmeler S, Zeiher AM (2009) Cell therapy of acute myocardial infarction: open questions. Cardiology 113:155–160
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317
Mizuno H, Tobita M, Uysal AC (2012) Concise review: Adipose-derived stem cells as a novel tool for future regenerative medicine. Stem Cells 30:804–810
Jeevanantham V, Butler M, Saad A, Abdel-Latif A, Zuba-Surma EK, Dawn B (2012) Adult bone marrow cell therapy improves survival and induces long-term improvement in cardiac parameters: a systematic review and meta-analysis. Circulation 126:551–568
Sadat K, Ather S, Aljaroudi W, Heo J, Iskandrian AE, Hage FG (2014) The effect of bone marrow mononuclear stem cell therapy on left ventricular function and myocardial perfusion. J Nuclear Cardiol Off Publ Am Soc Nuclear Cardiol 21:351–367
de Jong R, Houtgraaf JH, Samiei S, Boersma E, Duckers HJ (2014) Intracoronary stem cell infusion after acute myocardial infarction: a meta-analysis and update on clinical trials. Circ Cardiovas Interven 7:156–167
Fisher SA, Brunskill SJ, Doree C, Mathur A, Taggart DP, Martin-Rendon E (2014) Stem cell therapy for chronic ischaemic heart disease and congestive heart failure. Cochrane Database Syst Rev 4:CD007888
Terzic A, Behfar A (2014) Regenerative heart failure therapy headed for optimization. Eur Heart J
Nadal-Ginard B, Kajstura J, Leri A, Anversa P (2003) Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ Res 92:139–150
Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F et al (2004) Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res 95:911–921
Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E et al (2007) Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 115:896–908
Bolli R, Chugh AR, D’Amario D, Loughran JH, Stoddard MF, Ikram S et al (2011) Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet 378:1847–1857
Malliaras K, Makkar RR, Smith RR, Cheng K, Wu E, Bonow RO et al (2014) Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction). J Am Coll Cardiol 63:110–122
Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A et al (2001) Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Investig 108:407–414
Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP et al (2009) Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res 104:e30–e41
van Laake LW, Passier R, Monshouwer-Kloots J, Verkleij AJ, Lips DJ, Freund C et al (2007) Human embryonic stem cell-derived cardiomyocytes survive and mature in the mouse heart and transiently improve function after myocardial infarction. Stem Cell Res. 1:9–24
Caspi O, Huber I, Kehat I, Habib M, Arbel G, Gepstein A et al (2007) Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J Am Coll Cardiol 50:1884–1893
Laflamme MA, Gold J, Xu C, Hassanipour M, Rosler E, Police S et al (2005) Formation of human myocardium in the rat heart from human embryonic stem cells. Am J Pathol 167:663–671
Fernandes S, Naumova AV, Zhu WZ, Laflamme MA, Gold J, Murry CE (2010) Human embryonic stem cell-derived cardiomyocytes engraft but do not alter cardiac remodeling after chronic infarction in rats. J Mol Cell Cardiol 49:941–949
Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK et al (2007) Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol 25:1015–1024
Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J et al (2012) Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature 489:322–325
Shiba Y, Filice D, Fernandes S, Minami E, Dupras SK, Biber BV et al (2014) Electrical Integration of Human Embryonic Stem Cell-Derived Cardiomyocytes in a Guinea Pig Chronic Infarct Model. J Cardiovas Pharmacol Therap
Ye L, Chang YH, Xiong Q, Zhang P, Zhang L, Somasundaram P et al (2014) Cardiac repair in a porcine model of acute myocardial infarction with human induced pluripotent stem cell-derived cardiovascular cells. Cell Stem Cell 15:750–761
Chong JJ, Yang X, Don CW, Minami E, Liu YW, Weyers JJ et al (2014) Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 510:273–277
Jackman CP, Shadrin IY, Carlson AL, Bursac N (2015) Human Cardiac Tissue Engineering: from Pluripotent Stem Cells to Heart Repair. Curr Opin Chem Eng 7:57–64
Sicari BM, Rubin JP, Dearth CL, Wolf MT, Ambrosio F, Boninger M et al (2014) An acellular biologic scaffold promotes skeletal muscle formation in mice and humans with volumetric muscle loss. Sci Trans Med 6:234ra58
Keane TJ, Londono R, Turner NJ, Badylak SF (2012) Consequences of ineffective decellularization of biologic scaffolds on the host response. Biomaterials 33:1771–1781
Brown BN, Valentin JE, Stewart-Akers AM, McCabe GP, Badylak SF (2009) Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials 30:1482–1491
Valentin JE, Stewart-Akers AM, Gilbert TW, Badylak SF (2009) Macrophage participation in the degradation and remodeling of extracellular matrix scaffolds. Tissue Eng Part A 15:1687–1694
Londono R, Badylak SF (2015) Biologic Scaffolds for Regenerative Medicine: Mechanisms of In vivo Remodeling. Ann Biomed Eng 43:577–592
Wolf MT, Daly KA, Reing JE, Badylak SF (2012) Biologic scaffold composed of skeletal muscle extracellular matrix. Biomaterials 33:2916–2925
Valentin JE, Turner NJ, Gilbert TW, Badylak SF (2010) Functional skeletal muscle formation with a biologic scaffold. Biomaterials 31:7475–7484
Corona BT, Ward CL, Baker HB, Walters TJ, Christ GJ (2014) Implantation of in vitro tissue engineered muscle repair constructs and bladder acellular matrices partially restore in vivo skeletal muscle function in a rat model of volumetric muscle loss injury. Tissue Eng Part A 20:705–715
Mase VJ, Jr., Hsu JR, Wolf SE, Wenke JC, Baer DG, Owens J et al (2010) Clinical application of an acellular biologic scaffold for surgical repair of a large, traumatic quadriceps femoris muscle defect. Orthopedics 33
Aurora A, Garg K, Corona BT, Walters TJ (2014) Physical rehabilitation improves muscle function following volumetric muscle loss injury. BMC Sports Sci Med Rehabil 6:41
Scholl FG, Boucek MM, Chan KC, Valdes-Cruz L, Perryman R (2010) Preliminary experience with cardiac reconstruction using decellularized porcine extracellular matrix scaffold: human applications in congenital heart disease. World J Pediat Congen Heart Surg 1:132–136
Okada M, Payne TR, Oshima H, Momoi N, Tobita K, Huard J (2010) Differential efficacy of gels derived from small intestinal submucosa as an injectable biomaterial for myocardial infarct repair. Biomaterials 31:7678–7683
Toeg HD, Tiwari-Pandey R, Seymour R, Ahmadi A, Crowe S, Vulesevic B et al (2013) Injectable small intestine submucosal extracellular matrix in an acute myocardial infarction model. Ann Thorac Surg 96:1686–1694 (discussion 94)
Tan MY, Zhi W, Wei RQ, Huang YC, Zhou KP, Tan B et al (2009) Repair of infarcted myocardium using mesenchymal stem cell seeded small intestinal submucosa in rabbits. Biomaterials 30:3234–3240
Dai W, Gerczuk P, Zhang Y, Smith L, Kopyov O, Kay GL et al (2013) Intramyocardial injection of heart tissue-derived extracellular matrix improves postinfarction cardiac function in rats. J Cardiovas Pharmacol Therap 18:270–279
Singelyn JM, Sundaramurthy P, Johnson TD, Schup-Magoffin PJ, Hu DP, Faulk DM et al (2012) Catheter-deliverable hydrogel derived from decellularized ventricular extracellular matrix increases endogenous cardiomyocytes and preserves cardiac function post-myocardial infarction. J Am Coll Cardiol 59:751–763
Seif-Naraghi SB, Singelyn JM, Salvatore MA, Osborn KG, Wang JJ, Sampat U et al (2013) Safety and efficacy of an injectable extracellular matrix hydrogel for treating myocardial infarction. Sci Transl Med 5:173ra25
Johnson TD, Christman KL (2013) Injectable hydrogel therapies and their delivery strategies for treating myocardial infarction. Expert Opin Drug Deliv 10:59–72
Burdick JA, Mauck RL, Gorman JH, 3rd, Gorman RC (2013) Acellular biomaterials: an evolving alternative to cell-based therapies. Sci Trans Med 5:176ps4
Cohen JE, Purcell BP, MacArthur JW Jr, Mu A, Shudo Y, Patel JB et al (2014) A bioengineered hydrogel system enables targeted and sustained intramyocardial delivery of neuregulin, activating the cardiomyocyte cell cycle and enhancing ventricular function in a murine model of ischemic cardiomyopathy. Circ Heart Fail 7:619–626
Miyagi Y, Chiu LL, Cimini M, Weisel RD, Radisic M, Li RK (2011) Biodegradable collagen patch with covalently immobilized VEGF for myocardial repair. Biomaterials 32:1280–1290
Miyagi Y, Zeng F, Huang XP, Foltz WD, Wu J, Mihic A et al (2010) Surgical ventricular restoration with a cell- and cytokine-seeded biodegradable scaffold. Biomaterials 31:7684–7694
Ungerleider JL, Christman KL (2014) Concise review: injectable biomaterials for the treatment of myocardial infarction and peripheral artery disease: translational challenges and progress. Stem Cells Trans Med 3:1090–1099
Malliaras K, Marban E (2011) Cardiac cell therapy: where we’ve been, where we are, and where we should be headed. Br Med Bull 98:161–185
Bach AD, Beier JP, Stern-Staeter J, Horch RE (2004) Skeletal muscle tissue engineering. J Cell Mol Med 8:413–422
Rosso F, Giordano A, Barbarisi M, Barbarisi A (2004) From cell-ECM interactions to tissue engineering. J Cell Physiol 199:174–180
Even-Ram S, Artym V, Yamada KM (2006) Matrix control of stem cell fate. Cell 126:645–647
Engler AJ, Sen S, Sweeney HL, Discher DE (2006) Matrix elasticity directs stem cell lineage specification. Cell 126:677–689
Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, Jayasankar V et al (2006) Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol 290:H2196–H2203
Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S et al (2004) Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 110:155–162
Light N, Champion AE (1984) Characterization of muscle epimysium, perimysium and endomysium collagens. Biochem J 219:1017–1026
Vandenburgh HH, Karlisch P, Farr L (1988) Maintenance of highly contractile tissue-cultured avian skeletal myotubes in collagen gel. In Vitro Cell Dev Biol 24:166–174
Shansky J, Del Tatto M, Chromiak J, Vandenburgh H (1997) A simplified method for tissue engineering skeletal muscle organoids in vitro. In Vitro Cell Dev Biol Anim 33:659–661
Okano T, Matsuda T (1998) Tissue engineered skeletal muscle: preparation of highly dense, highly oriented hybrid muscular tissues. Cell Transpl 7:71–82
Okano T, Matsuda T (1997) Hybrid muscular tissues: preparation of skeletal muscle cell-incorporated collagen gels. Cell Transpl 6:109–118
Okano T, Satoh S, Oka T, Matsuda T (1997) Tissue engineering of skeletal muscle. Highly dense, highly oriented hybrid muscular tissues biomimicking native tissues. ASAIO J 43:M749–M753
Lee PH, Vandenburgh HH (2013) Skeletal muscle atrophy in bioengineered skeletal muscle: a new model system. Tissue Eng Part A 19:2147–2155
Vandenburgh H, Shansky J, Benesch-Lee F, Skelly K, Spinazzola JM, Saponjian Y et al (2009) Automated drug screening with contractile muscle tissue engineered from dystrophic myoblasts. FASEB J. 23:3325–3334
Vandenburgh H, Shansky J, Benesch-Lee F, Barbata V, Reid J, Thorrez L et al (2008) Drug-screening platform based on the contractility of tissue-engineered muscle. Muscle Nerve 37:438–447
Mudera V, Smith AS, Brady MA, Lewis MP (2010) The effect of cell density on the maturation and contractile ability of muscle derived cells in a 3D tissue-engineered skeletal muscle model and determination of the cellular and mechanical stimuli required for the synthesis of a postural phenotype. J Cell Physiol 225:646–653
Brady MA, Lewis MP, Mudera V (2008) Synergy between myogenic and non-myogenic cells in a 3D tissue-engineered craniofacial skeletal muscle construct. Journal of tissue engineering and regenerative medicine. 2:408–417
Yan W, George S, Fotadar U, Tyhovych N, Kamer A, Yost MJ et al (2007) Tissue engineering of skeletal muscle. Tissue Eng 13:2781–2790
Close RI (1972) Dynamic properties of mammalian skeletal muscles. Physiol Rev 52:129–197
Collet JP, Shuman H, Ledger RE, Lee S, Weisel JW (2005) The elasticity of an individual fibrin fiber in a clot. Proc Natl Acad Sci USA 102:9133–9137
Chiron S, Tomczak C, Duperray A, Lainé J, Bonne G, Eder A et al (2012) Complex interactions between human myoblasts and the surrounding 3D fibrin-based matrix. PLoS One 7:e36173
Huang YC, Dennis RG, Larkin L, Baar K (2005) Rapid formation of functional muscle in vitro using fibrin gels. J Appl Physiol 98:706–713
Juhas M, Engelmayr GC Jr, Fontanella AN, Palmer GM, Bursac N (2014) Biomimetic engineered muscle with capacity for vascular integration and functional maturation in vivo. Proc Natl Acad Sci USA 111:5508–5513
Hinds S, Bian W, Dennis RG, Bursac N (2011) The role of extracellular matrix composition in structure and function of bioengineered skeletal muscle. Biomaterials 32:3575–3583
Juhas M, Bursac N (2014) Roles of adherent myogenic cells and dynamic culture in engineered muscle function and maintenance of satellite cells. Biomaterials 35:9438–9446
Khodabukus A, Baar K (2015) Contractile and metabolic properties of engineered skeletal muscle derived from slow and fast phenotype mouse muscle. J Cell Physiol 230:1750–1757
Khodabukus A, Baar K (2009) Regulating Fibrinolysis to Engineer Skeletal Muscle from the C2C12 Cell Line. Tissue Eng Part C Methods 15:501–511
Madden L, Juhas M, Kraus WE, Truskey GA, Bursac N (2015) Bioengineered human myobundles mimic clinical responses of skeletal muscle to drugs. Elife. 4:e04885
Strohman RC, Bayne E, Spector D, Obinata T, Micou-Eastwood J, Maniotis A (1990) Myogenesis and histogenesis of skeletal muscle on flexible membranes in vitro. In Vitro Cell Dev Biol 26:201–208
Dennis RG, Kosnik PE 2nd, Gilbert ME, Faulkner JA (2001) Excitability and contractility of skeletal muscle engineered from primary cultures and cell lines. Am J Physiol Cell Physiol 280:C288–C295
Nagamori E, Ngo TX, Takezawa Y, Saito A, Sawa Y, Shimizu T et al (2013) Network formation through active migration of human vascular endothelial cells in a multilayered skeletal myoblast sheet. Biomaterials 34:662–668
Takahashi H, Okano T (2015) Cell sheet-based tissue engineering for organizing anisotropic tissue constructs produced using microfabricated thermoresponsive substrates. Adv Healthc Mater. 4:2388–2407
Takahashi H, Shimizu T, Nakayama M, Yamato M, Okano T (2013) The use of anisotropic cell sheets to control orientation during the self-organization of 3D muscle tissue. Biomaterials 34:7372–7380
Khodabukus A, Baar K (2014) The effect of serum origin on tissue engineered skeletal muscle function. J Cell Biochem 115:2198–2207
Huang YC, Dennis RG, Baar K (2006) Cultured slow vs. fast skeletal muscle cells differ in physiology and responsiveness to stimulation. Am J Physiol Cellphysiol 291:C11–C17
Li M, Dickinson CE, Finkelstein EB, Neville CM, Sundback CA (2011) The role of fibroblasts in self-assembled skeletal muscle. Tissue Eng Part A 17:2641–2650
Hinds S, Tyhovych N, Sistrunk C, Terracio L (2013) Improved tissue culture conditions for engineered skeletal muscle sheets. Sci World J 2013:370151
Merceron TK, Burt M, Seol YJ, Kang HW, Lee SJ, Yoo JJ et al (2015) A 3D bioprinted complex structure for engineering the muscle-tendon unit. Biofabrication 7:035003
Ladd MR, Lee SJ, Stitzel JD, Atala A, Yoo JJ (2011) Co-electrospun dual scaffolding system with potential for muscle-tendon junction tissue engineering. Biomaterials 32:1549–1559
Larkin LM, Calve S, Kostrominova TY, Arruda EM (2006) Structure and functional evaluation of tendon-skeletal muscle constructs engineered in vitro. Tissue Eng 12:3149–3158
Kostrominova TY, Calve S, Arruda EM, Larkin LM (2009) Ultrastructure of myotendinous junctions in tendon-skeletal muscle constructs engineered in vitro. Histol Histopathol 24:541–550
VanDusen KW, Syverud BC, Williams ML, Lee JD, Larkin LM (2014) Engineered Skeletal Muscle Units for Repair of Volumetric Muscle Loss in the Tibialis Anterior Muscle of a Rat. Tissue Eng Part A
Das M, Rumsey JW, Bhargava N, Stancescu M, Hickman JJ (2010) A defined long-term in vitro tissue engineered model of neuromuscular junctions. Biomaterials 31:4880–4888
Guo X, Gonzalez M, Stancescu M, Vandenburgh HH, Hickman JJ (2011) Neuromuscular junction formation between human stem cell-derived motoneurons and human skeletal muscle in a defined system. Biomaterials 32:9602–9611
Morimoto Y, Kato-Negishi M, Onoe H, Takeuchi S (2013) Three-dimensional neuron-muscle constructs with neuromuscular junctions. Biomaterials 34:9413–9419
Larkin LM, Van der Meulen JH, Dennis RG, Kennedy JB (2006) Functional evaluation of nerve-skeletal muscle constructs engineered in vitro. In Vitro Cell Dev Biol Anim 42:75–82
Bian W, Bursac N (2012) Soluble miniagrin enhances contractile function of engineered skeletal muscle. FASEB J 26:955–965
Wang L, Shansky J, Vandenburgh H (2013) Induced formation and maturation of acetylcholine receptor clusters in a defined 3D bio-artificial muscle. Mol Neurobiol 48:397–403
Ko IK, Lee BK, Lee SJ, Andersson KE, Atala A, Yoo JJ (2013) The effect of in vitro formation of acetylcholine receptor (AChR) clusters in engineered muscle fibers on subsequent innervation of constructs in vivo. Biomaterials 34:3246–3255
Olwin BB, Arthur K, Hannon K, Hein P, McFall A, Riley B et al (1994) Role of FGFs in skeletal muscle and limb development. Mol Reprod Dev 39:90–100 (discussion-1)
Tidball JG (2005) Mechanical signal transduction in skeletal muscle growth and adaptation. J Appl Physiol 98:1900–1908
Vandenburgh HH (1982) Dynamic mechanical orientation of skeletal myofibers in vitro. Dev Biol 93:438–443
Vandenburgh HH, Swasdison S, Karlisch P (1991) Computer-aided mechanogenesis of skeletal muscle organs from single cells in vitro. Faseb J. 5:2860–2867
Powell CA, Smiley BL, Mills J, Vandenburgh HH (2002) Mechanical stimulation improves tissue-engineered human skeletal muscle. Am J Physiol Cell Physiol 283:C1557–C1565
du Moon G, Christ G, Stitzel JD, Atala A, Yoo JJ (2008) Cyclic mechanical preconditioning improves engineered muscle contraction. Tissue Eng Part A 14:473–482
Harris AJ (1981) Embryonic growth and innervation of rat skeletal muscles. I. Neural regulation of muscle fibre numbers. Philos Trans R Soc Lond B Biol Sci 293:257–277
Duxson MJ, Sheard PW (1995) Formation of new myotubes occurs exclusively at the multiple innervation zones of an embryonic large muscle. Dev Dyn 204:391–405
Kern H, Boncompagni S, Rossini K, Mayr W, Fanò G, Zanin ME et al (2004) Long-term denervation in humans causes degeneration of both contractile and excitation-contraction coupling apparatus, which is reversible by functional electrical stimulation (FES): a role for myofiber regeneration? J Neuropathol Exp Neurol 63:919–931
Salmons S, Sreter FA (1976) Significance of impulse activity in the transformation of skeletal muscle type. Nature 263:30–34
Khodabukus A, Baar K (2015) Streptomycin decreases the functional shift to a slow phenotype induced by electrical stimulation in engineered muscle. Tissue Eng Part A 21:1003–1012
Khodabukus A, Baehr LM, Bodine SC, Baar K (2015) Role of Contraction Duration in Inducing Fast-To-Slow Contractile and Metabolic Protein and Functional Changes in Engineered Muscle. J Cell Physiol
Khodabukus A, Baar K (2012) Defined electrical stimulation emphasizing excitability for the development and testing of engineered skeletal muscle. Tissue Eng Part C Methods 18:349–357
Donnelly K, Khodabukus A, Philp A, Deldicque L, Dennis RG, Baar K (2010) A novel bioreactor for stimulating skeletal muscle in vitro. Tissue Eng Part C Methods 16:711–718
Ito A, Yamamoto Y, Sato M, Ikeda K, Yamamoto M, Fujita H et al (2014) Induction of functional tissue-engineered skeletal muscle constructs by defined electrical stimulation. Sci Rep 4:4781
Eschenhagen T, Fink C, Remmers U, Scholz H, Wattchow J, Weil J et al (1997) Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: a new heart muscle model system. FASEB J 11:683–694
Bursac N, Papadaki M, Cohen RJ, Schoen FJ, Eisenberg SR, Carrier R et al (1999) Cardiac muscle tissue engineering: toward an in vitro model for electrophysiological studies. Am J Physiol 277:H433–H444
Li RK, Jia ZQ, Weisel RD, Mickle DA, Choi A, Yau TM (1999) Survival and function of bioengineered cardiac grafts. Circulation 100:II63–II69
Leor J, Aboulafia-Etzion S, Dar A, Shapiro L, Barbash IM, Battler A et al (2000) Bioengineered cardiac grafts: A new approach to repair the infarcted myocardium? Circulation 102:III56-II61
Kofidis T, Akhyari P, Wachsmann B, Boublik J, Mueller-Stahl K, Leyh R et al (2002) A novel bioartificial myocardial tissue and its prospective use in cardiac surgery. Eur J Cardiothorac Surg 22:238–243
Zimmermann WH, Fink C, Kralisch D, Remmers U, Weil J, Eschenhagen T (2000) Three-dimensional engineered heart tissue from neonatal rat cardiac myocytes. Biotechnol Bioeng 68:106–114
Zimmermann WH, Schneiderbanger K, Schubert P, Didie M, Munzel F, Heubach JF et al (2002) Tissue engineering of a differentiated cardiac muscle construct. Circ Res 90:223–230
Shimizu T, Yamato M, Isoi Y, Akutsu T, Setomaru T, Abe K et al (2002) Fabrication of pulsatile cardiac tissue grafts using a novel 3-dimensional cell sheet manipulation technique and temperature-responsive cell culture surfaces. Circ Res 90:e40
Baar K, Birla R, Boluyt MO, Borschel GH, Arruda EM, Dennis RG (2005) Self-organization of rat cardiac cells into contractile 3-D cardiac tissue. Faseb J. 19:275–277
Black LD, Meyers JD, Weinbaum JS, Shvelidze YA, Tranquillo RT (2009) Cell-induced alignment augments twitch force in fibrin gel-based engineered myocardium via gap junction modification. Tissue Eng Part A
Hirt MN, Sorensen NA, Bartholdt LM, Boeddinghaus J, Schaaf S, Eder A et al (2012) Increased afterload induces pathological cardiac hypertrophy: a new in vitro model. Basic Res Cardiol 107:307
Tao ZW, Mohamed M, Hogan M, Gutierrez L, Birla RK (2014) Optimizing a spontaneously contracting heart tissue patch with rat neonatal cardiac cells on fibrin gel. J Tissue Eng Regen Med
Bian W, Jackman CP, Bursac N (2014) Controlling the structural and functional anisotropy of engineered cardiac tissues. Biofabrication 6:024109
Bian W, Liau B, Badie N, Bursac N (2009) Mesoscopic hydrogel molding to control the 3D geometry of bioartificial muscle tissues. Nat Protoc 4:1522–1534
Solaro RJ, Lee JA, Kentish JC, Allen DG (1988) Effects of acidosis on ventricular muscle from adult and neonatal rats. Circ Res 63:779–787
Raman S, Kelley MA, Janssen PM (2006) Effect of muscle dimensions on trabecular contractile performance under physiological conditions. Pflugers Arch 451:625–630
Radisic M, Fast VG, Sharifov OF, Iyer RK, Park H, Vunjak-Novakovic G (2009) Optical mapping of impulse propagation in engineered cardiac tissue. Tissue Eng Part A 15:851–860
Sondergaard CS, Mathews G, Wang L, Jeffreys A, Sahota A, Wood M et al (2012) Contractile and electrophysiologic characterization of optimized self-organizing engineered heart tissue. The Annals of thoracic surgery 94:1241–1248 (discussion 9)
Bursac N, Loo Y, Leong K, Tung L (2007) Novel anisotropic engineered cardiac tissues: studies of electrical propagation. Biochem Biophys Res Commun 361:847–853
Shimizu T, Sekine H, Isoi Y, Yamato M, Kikuchi A, Okano T (2006) Long-term survival and growth of pulsatile myocardial tissue grafts engineered by the layering of cardiomyocyte sheets. Tissue Eng 12:499–507
Bian W, Badie N, Himel HDt, Bursac N (2014) Robust T-tubulation and maturation of cardiomyocytes using tissue-engineered epicardial mimetics. Biomaterials 35:3819–3828
Nygren A, Kondo C, Clark RB, Giles WR (2003) Voltage-sensitive dye mapping in Langendorff-perfused rat hearts. Am J Physiol Heart Circ Physiol 284:H892–H902
Zimmermann WH, Melnychenko I, Wasmeier G, Didie M, Naito H, Nixdorff U et al (2006) Engineered heart tissue grafts improve systolic and diastolic function in infarcted rat hearts. Nat Med 12:452–458
Godier-Furnemont AF, Tiburcy M, Wagner E, Dewenter M, Lammle S, El-Armouche A et al (2015) Physiologic force-frequency response in engineered heart muscle by electromechanical stimulation. Biomaterials 60:82–91
Liau B, Christoforou N, Leong KW, Bursac N (2011) Pluripotent stem cell-derived cardiac tissue patch with advanced structure and function. Biomaterials 32:9180–9187
Masumoto H, Matsuo T, Yamamizu K, Uosaki H, Narazaki G, Katayama S et al (2012) Pluripotent stem cell-engineered cell sheets reassembled with defined cardiovascular populations ameliorate reduction in infarct heart function through cardiomyocyte-mediated neovascularization. Stem Cells 30:1196–1205
Miki K, Uenaka H, Saito A, Miyagawa S, Sakaguchi T, Higuchi T et al (2012) Bioengineered myocardium derived from induced pluripotent stem cells improves cardiac function and attenuates cardiac remodeling following chronic myocardial infarction in rats. Stem Cells Transl Med 1:430–437
Didie M, Christalla P, Rubart M, Muppala V, Doker S, Unsold B et al (2013) Parthenogenetic stem cells for tissue-engineered heart repair. J Clin Investig 123:1285–1298
Christoforou N, Liau B, Chakraborty S, Chellapan M, Bursac N, Leong KW (2013) Induced pluripotent stem cell-derived cardiac progenitors differentiate to cardiomyocytes and form biosynthetic tissues. PLoS One 8:e65963
Hirt MN, Hansen A, Eschenhagen T (2014) Cardiac tissue engineering: state of the art. Circ Res 114:354–367
Kerscher P, Turnbull IC, Hodge AJ, Kim J, Seliktar D, Easley CJ et al (2015) Direct Hydrogel Encapsulation of Pluripotent Stem Cells Enables Ontomimetic Differentiation and Growth of Engineered Human Heart Tissues. Biomaterials
Riegler J, Tiburcy M, Ebert A, Tzatzalos E, Raaz U, Abilez OJ et al (2015) Human engineered heart muscles engraft and survive long term in a rodent myocardial infarction model. Circ Res 117:720–730
Eng G, Lee BW, Protas L, Gagliardi M, Brown K, Kass RS et al (2016) Autonomous beating rate adaptation in human stem cell-derived cardiomyocytes. Nat Commun 7:10312
Guyette JP, Charest J, Mills RW, Jank B, Moser PT, Gilpin SE et al (2015) Bioengineering Human Myocardium on Native Extracellular Matrix. Circ Res
Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, Hauch KD et al (2011) Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ Res 109:47–59
Zhang D, Shadrin IY, Lam J, Xian HQ, Snodgrass HR, Bursac N (2013) Tissue-engineered cardiac patch for advanced functional maturation of human ESC-derived cardiomyocytes. Biomaterials 34:5813–5820
Kensah G, Roa Lara A, Dahlmann J, Zweigerdt R, Schwanke K, Hegermann J et al (2013) Murine and human pluripotent stem cell-derived cardiac bodies form contractile myocardial tissue in vitro. Eur Heart J 34:1134–1146
Thavandiran N, Dubois N, Mikryukov A, Masse S, Beca B, Simmons CA et al (2013) Design and formulation of functional pluripotent stem cell-derived cardiac microtissues. Proc Natl Acad Sci USA 110:E4698–E4707
Mulieri LA, Hasenfuss G, Leavitt B, Allen PD, Alpert NR (1992) Altered myocardial force-frequency relation in human heart failure. Circulation 85:1743–1750
Hasenfuss G, Mulieri L, Blanchard E, Holubarsch C, Leavitt B, Ittleman F et al (1991) Energetics of isometric force development in control and volume- overload human myocardium. Comparison with animal species. Circ Res 68:836–846
Durrer D, van Dam RT, Freud GE, Janse MJ, Meijler FL, Arzbaecher RC (1970) Total excitation of the isolated human heart. Circulation 41:899–912
Yang X, Pabon L, Murry CE (2014) Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res 114:511–523
Feric NT, Radisic M (2015) Maturing human pluripotent stem cell-derived cardiomyocytes in human engineered cardiac tissues. Adv Drug Deliv Rev
Yang X, Rodriguez M, Pabon L, Fischer KA, Reinecke H, Regnier M et al (2014) Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J Mol Cell Cardiol 72:296–304
Rangarajan S, Madden L, Bursac N (2014) Use of flow, electrical, and mechanical stimulation to promote engineering of striated muscles. Ann Biomed Eng 42:1391–1405
Lundy SD, Zhu W-Z, Regnier M, Laflamme MA (2013) Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev 22:1991–2002
Kamakura T, Makiyama T, Sasaki K, Yoshida Y, Wuriyanghai Y, Chen J et al (2013) Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ J Off J Japn Circ Soc 77:1307–1314
Hirt MN, Boeddinghaus J, Mitchell A, Schaaf S, Börnchen C, Müller C et al (2014) Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. J Mol Cell Cardiol 74:151–161
Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B et al (2013) Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods 10:781–787
Zhang Y, King OD, Rahimov F, Jones TI, Ward CW, Kerr JP et al (2014) Human skeletal muscle xenograft as a new preclinical model for muscle disorders. Hum Mol Genet 23:3180–3188
Levenberg S, Rouwkema J, Macdonald M, Garfein ES, Kohane DS, Darland DC et al (2005) Engineering vascularized skeletal muscle tissue. Nat Biotechnol 23:879–884
Koffler J, Kaufman-Francis K, Shandalov Y, Egozi D, Pavlov DA, Landesberg A et al (2011) Improved vascular organization enhances functional integration of engineered skeletal muscle grafts. Proc Natl Acad Sci USA 108:14789–14794
Kajbafzadeh AM, Payabvash S, Salmasi AH, Sadeghi Z, Elmi A, Vejdani K et al (2007) Time-dependent neovasculogenesis and regeneration of different bladder wall components in the bladder acellular matrix graft in rats. J Surg Res 139:189–202
Criswell TL, Corona BT, Wang Z, Zhou Y, Niu G, Xu Y et al (2013) The role of endothelial cells in myofiber differentiation and the vascularization and innervation of bioengineered muscle tissue in vivo. Biomaterials 34:140–149
Borschel GH, Dow DE, Dennis RG, Brown DL (2006) Tissue-engineered axially vascularized contractile skeletal muscle. Plast Reconstr Surg 117:2235–2242
Mian R, Morrison WA, Hurley JV, Penington AJ, Romeo R, Tanaka Y et al (2000) Formation of new tissue from an arteriovenous loop in the absence of added extracellular matrix. Tissue Eng 6:595–603
Messina A, Bortolotto SK, Cassell OC, Kelly J, Abberton KM, Morrison WA (2005) Generation of a vascularized organoid using skeletal muscle as the inductive source. Faseb J. 19:1570–1572
Bach AD, Arkudas A, Tjiawi J, Polykandriotis E, Kneser U, Horch RE et al (2006) A new approach to tissue engineering of vascularized skeletal muscle. J Cell Mol Med 10:716–726
Shandalov Y, Egozi D, Koffler J, Dado-Rosenfeld D, Ben-Shimol D, Freiman A et al (2014) An engineered muscle flap for reconstruction of large soft tissue defects. Proc Natl Acad Sci USA 111:6010–6015
Dhawan V, Lytle IF, Dow DE, Huang YC, Brown DL (2007) Neurotization improves contractile forces of tissue-engineered skeletal muscle. Tissue Eng 13:2813–2821
Zhou W, He DQ, Liu JY, Feng Y, Zhang XY, Hua CG et al (2013) Angiogenic gene-modified myoblasts promote vascularization during repair of skeletal muscle defects. J Tissue Eng Regen Med
Shvartsman D, Storrie-White H, Lee K, Kearney C, Brudno Y, Ho N et al (2014) Sustained delivery of VEGF maintains innervation and promotes reperfusion in ischemic skeletal muscles via NGF/GDNF signaling. Mol Ther 22:1243–1253
Lu Y, Shansky J, Del Tatto M, Ferland P, Wang X, Vandenburgh H (2001) Recombinant vascular endothelial growth factor secreted from tissue-engineered bioartificial muscles promotes localized angiogenesis. Circulation 104:594–599
Borselli C, Storrie H, Benesch-Lee F, Shvartsman D, Cezar C, Lichtman JW et al (2010) Functional muscle regeneration with combined delivery of angiogenesis and myogenesis factors. Proc Natl Acad Sci USA 107:3287–3292
Borselli C, Cezar CA, Shvartsman D, Vandenburgh HH, Mooney DJ (2011) The role of multifunctional delivery scaffold in the ability of cultured myoblasts to promote muscle regeneration. Biomaterials 32:8905–8914
Fuoco C, Rizzi R, Biondo A, Longa E, Mascaro A, Shapira-Schweitzer K et al (2015) In vivo generation of a mature and functional artificial skeletal muscle. EMBO Mol Med 7:411–422
Gargioli C, Coletta M, De Grandis F, Cannata SM, Cossu G (2008) PlGF-MMP-9-expressing cells restore microcirculation and efficacy of cell therapy in aged dystrophic muscle. Nat Med 14:973–978
Lesman A, Habib M, Caspi O, Gepstein A, Arbel G, Levenberg S et al (2010) Transplantation of a tissue-engineered human vascularized cardiac muscle. Tissue Eng Part A 16:115–125
Sekine H, Shimizu T, Hobo K, Sekiya S, Yang J, Yamato M et al (2008) Endothelial cell coculture within tissue-engineered cardiomyocyte sheets enhances neovascularization and improves cardiac function of ischemic hearts. Circulation 118:S145–S152
Masumoto H, Ikuno T, Takeda M, Fukushima H, Marui A, Katayama S et al (2014) Human iPS cell-engineered cardiac tissue sheets with cardiomyocytes and vascular cells for cardiac regeneration. Sci Rep 4:6716
Stevens KR, Kreutziger KL, Dupras SK, Korte FS, Regnier M, Muskheli V et al (2009) Physiological function and transplantation of scaffold-free and vascularized human cardiac muscle tissue. Proc Natl Acad Sci USA 106:16568–16573
Sun X, Altalhi W, Nunes SS (2015) Vascularization strategies of engineered tissues and their application in cardiac regeneration. Adv Drug Deliv Rev
Xiong Q, Hill KL, Li Q, Suntharalingam P, Mansoor A, Wang X et al (2011) A fibrin patch-based enhanced delivery of human embryonic stem cell-derived vascular cell transplantation in a porcine model of postinfarction left ventricular remodeling. Stem Cells 29:367–375
Thomson KS, Korte FS, Giachelli CM, Ratner BD, Regnier M, Scatena M (2013) Prevascularized microtemplated fibrin scaffolds for cardiac tissue engineering applications. Tissue Eng Part A 19:967–977
Dvir T, Kedem A, Ruvinov E, Levy O, Freeman I, Landa N et al (2009) Prevascularization of cardiac patch on the omentum improves its therapeutic outcome. Proc Natl Acad Sci USA 106:14990–14995
Gerbin KA, Yang X, Murry CE, Coulombe KL (2015) Enhanced Electrical Integration of Engineered Human Myocardium via Intramyocardial versus Epicardial Delivery in Infarcted Rat Hearts. PLoS One 10:e0131446
Menasche P, Vanneaux V, Hagege A, Bel A, Cholley B, Cacciapuoti I et al (2015) Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: first clinical case report. Euro Heart J
Miyagawa S, Sawa Y, Sakakida S, Taketani S, Kondoh H, Memon IA et al (2005) Tissue cardiomyoplasty using bioengineered contractile cardiomyocyte sheets to repair damaged myocardium: their integration with recipient myocardium. Transplantation 80:1586–1595
Sekine H, Shimizu T, Dobashi I, Matsuura K, Hagiwara N, Takahashi M et al (2011) Cardiac cell sheet transplantation improves damaged heart function via superior cell survival in comparison with dissociated cell injection. Tissue Eng Part A
Fujimoto KL, Clause KC, Liu LJ, Tinney JP, Verma S, Wagner WR et al (2011) Engineered fetal cardiac graft preserves its cardiomyocyte proliferation within postinfarcted myocardium and sustains cardiac function. Tissue Eng Part A 17:585–596
Wendel JS, Ye L, Zhang P, Tranquillo RT, Zhang JJ (2014) Functional consequences of a tissue-engineered myocardial patch for cardiac repair in a rat infarct model. Tissue Eng Part A 20:1325–1335
Marsano A, Maidhof R, Luo J, Fujikara K, Konofagou EE, Banfi A et al (2013) The effect of controlled expression of VEGF by transduced myoblasts in a cardiac patch on vascularization in a mouse model of myocardial infarction. Biomaterials 34:393–401
Leontyev S, Schlegel F, Spath C, Schmiedel R, Nichtitz M, Boldt A et al (2013) Transplantation of engineered heart tissue as a biological cardiac assist device for treatment of dilated cardiomyopathy. Eur J Heart Fail 15:23–35
Chen YL, Sun CK, Tsai TH, Chang LT, Leu S, Zhen YY et al (2015) Adipose-derived mesenchymal stem cells embedded in platelet-rich fibrin scaffolds promote angiogenesis, preserve heart function, and reduce left ventricular remodeling in rat acute myocardial infarction. Am J Trans Res 7:781–803
Kawamura M, Miyagawa S, Miki K, Saito A, Fukushima S, Higuchi T et al (2012) Feasibility, safety, and therapeutic efficacy of human induced pluripotent stem cell-derived cardiomyocyte sheets in a porcine ischemic cardiomyopathy model. Circulation 126:S29–S37
Mihic A, Li J, Miyagi Y, Gagliardi M, Li S-H, Zu J et al (2014) The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes. Biomaterials 35:2798–2808
Xiong Q, Ye L, Zhang P, Lepley M, Tian J, Li J et al (2013) Functional consequences of human induced pluripotent stem cell therapy: myocardial ATP turnover rate in the in vivo swine heart with postinfarction remodeling. Circulation 127:997–1008
Simpson DL, Dudley SC Jr (2013) Modulation of human mesenchymal stem cell function in a three-dimensional matrix promotes attenuation of adverse remodelling after myocardial infarction. J Tissue Eng Regen Med 7:192–202
Roura S, Soler-Botija C, Bago JR, Llucia-Valldeperas A, Fernandez MA, Galvez-Monton C et al (2015) Postinfarction Functional recovery driven by a three-dimensional engineered fibrin patch composed of human umbilical cord blood-derived mesenchymal stem cells. Stem Cells Trans Med 4:956–966
VanDusen KW, Syverud BC, Williams ML, Lee JD, Larkin LM (2014) Engineered skeletal muscle units for repair of volumetric muscle loss in the tibialis anterior muscle of a rat. Tissue Eng Part A 20:2920–2930
Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E et al (2016) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351:400–403
Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, Yan WX et al (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351:407–411
Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM et al (2016) In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351:403–407
Wright AV, Nunez JK, Doudna JA (2016) Biology and applications of CRISPR systems: harnessing nature’s toolbox for genome engineering. Cell 164:29–44
Shuman JA, Zurcher JR, Sapp AA, Burdick JA, Gorman RC, Gorman JH 3rd et al (2013) Localized targeting of biomaterials following myocardial infarction: a foundation to build on. Trends Cardiovasc Med 23:301–311
Sakellariou P, O’Neill A, Mueller AL, Stadler G, Wright WE, Roche JA et al (2015) Neuromuscular electrical stimulation promotes development in mice of mature human muscle from immortalized human myoblasts. Skelet Muscle 6:4
Kirkton RD, Bursac N (2011) Engineering biosynthetic excitable tissues from unexcitable cells for electrophysiological and cell therapy studies. Nat Commun 2:300
Acknowledgments
This work was supported by the NIH Grants AR055226 and AR065873 from National Institute of Arthritis and Musculoskeletal and Skin Disease, NIH Grants HL104326 and HL122079 from National Heart, Lung, and Blood Institute, NIH Grant T32 GM007171-Medical Scientist Training Program, UH3TR000505 Grant from the NIH Common Fund for the Microphysiological Systems Initiative, and a Grant from the Fondation Leducq. The content of the manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Author information
Authors and Affiliations
Corresponding author
Additional information
I. Y. Shadrin and A. Khodabukus equally contributing authors.
Rights and permissions
About this article

Cite this article
Shadrin, I.Y., Khodabukus, A. & Bursac, N. Striated muscle function, regeneration, and repair. Cell. Mol. Life Sci. 73, 4175–4202 (2016). https://doi.org/10.1007/s00018-016-2285-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2285-z