
Abstract
Skeletal muscle has a robust capacity for regeneration following injury. However, few if any effective therapeutic options for volumetric muscle loss are available. Autologous muscle grafts or muscle transposition represent possible salvage procedures for the restoration of mass and function but these approaches have limited success and are plagued by associated donor site morbidity. Cell-based therapies are in their infancy and, to date, have largely focused on hereditary disorders such as Duchenne muscular dystrophy. An unequivocal need exists for regenerative medicine strategies that can enhance or induce de novo formation of functional skeletal muscle as a treatment for congenital absence or traumatic loss of tissue. In this review, the three stages of skeletal muscle regeneration and the potential pitfalls in the development of regenerative medicine strategies for the restoration of functional skeletal muscle in situ are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Skeletal muscle comprises 40%-50% of the human body mass (Huard et al. 2002a) and injuries to the musculoskeletal system are common. In sports, muscle injuries account for 10%–55% of all sustained injuries with over 90% of these being either contusions or strains (Beiner and Jokl 2001; Counsel and Breidahl 2010; Garrett 1996; Jarvinen et al. 2000, 2005; Lehto and Jarvinen 1991). Traumatic injuries to the extremities from motor vehicle accidents and increased survival following limb salvage for extremity tumors are contributing to an increasing need for better treatment options for volumetric muscle loss. Despite the clinical importance, relatively few clinical studies are available on the treatment of volumetric muscle loss, because of the heterogeneity in the severity of injuries and the varied anatomic locations of such injuries. Accordingly, the current treatment principles for muscle injuries and volumetric muscle loss have been derived either from experimental studies or from empirical observations.
The most common skeletal muscle injuries such as tears, lacerations and contusions result in physical trauma without significant loss of muscle tissue (Beiner and Jokl 2001; Garrett 1996; Jarvinen et al. 2000, 2005; Lehto and Jarvinen 1991). In these cases, skeletal muscle has a robust capacity for regeneration relying in a large part upon the presence of mononuclear myogenic satellite cells. Indeed, skeletal muscle can be completely removed in an animal model, minced and placed back into its compartment and will subsequently regenerate enough to contract and produce force (Carlson and Gutmann 1972). However, the ability of a muscle to repair itself following damage is dependent on the type and severity of the injury and even in less severe injuries, the repair process is not 100% efficient. When loss of skeletal muscle is associated with traumatic injury, the repair capacity of the muscle diminishes and if more than 20% of the muscle is lost, the natural repair process will fail to repair the defect and result in an accumulation of scar tissue, dennervation of muscle distal to the defect and a loss of function (Aarimaa et al. 2004; Crow et al. 2007; Garrett et al. 1984; Menetrey et al. 1999; Terada et al. 2001). In such cases, the current standard of care is surgical intervention with flaps and vascularized tissue transfer to cover defects and hopefully to return partial function. In cases of extensive traumatic injury, amputation represents the standard of care. Surgical intervention, while restoring function, does not regenerate the lost muscle tissue and typically results in alterations of the anatomy and biomechanics for both the recipient and the donor sites (Tu et al. 2008). Individuals can expect to live the rest of their lives with a physical handicap. As a result, a clear need exists for therapeutic strategies that can enhance the innate ability of skeletal muscle to regenerate following severe local trauma and/or to induce de novo formation of functional muscle.
Tissue engineering and regenerative medicine might offer a partial solution to this need. Traditionally, tissue engineering has employed progenitor cells and/or scaffolds to replace loss or damaged tissue (Koning et al. 2009; Liao and Zhou 2009). Significant advances in the in vitro culture and formation of skeletal muscle have been made in recent years (Borschel et al. 2004; Huang et al. 2005; Larkin et al. 2006; Levenberg et al. 2005; du Moon et al. 2008). Nevertheless, muscle tissue engineering in vitro has limitations. Functional skeletal muscle requires the parallel alignment of muscle fibers, the formation of functional vascular beds and the innervation of the tissue to produce directed forces. In addition, the implantation of muscle tissue created outside the body presents problems of biocompatibility and integration with the host tissue. Recently, as knowledge of developmental biology and the cellular and biochemical cues associated with muscle repair has increased, the focus has shifted away from in vitro tissue synthesis to an in vivo regenerative medicine strategy that provides the biochemical cues to modify the in vivo microenvironment and facilitate rapid restoration of lost tissue with minimal scarring. Such an in vivo approach is also not without its potential pitfalls. In this review, the pathobiology of skeletal muscle repair and some of the recent advances in regenerative medicine that target key stages of muscle repair in order to promote the restoration of functional skeletal muscle in situ, particularly following volumetric muscle loss, are described.
Pathobiology of skeletal muscle repair
Irrespective of the type or severity of the injury, the repair of skeletal muscle follows a set pattern, which can be divided into three phases. These are:
-
1.
The destruction/inflammatory phase characterized by the rupture and necrosis of myofibers and an inflammatory cell reaction
-
2.
The repair phase characterized by the phagocytosis of the necrotic muscle fibers, the generation of new muscle fibers and the production of a tissue scar
-
3.
The remodeling phase characterized by the reorganization of the muscle fibers, the remodeling of the scar tissue and the restoration of muscle function
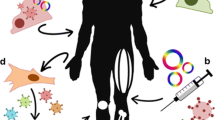
This repair process has been described in detail in a number of articles (Huard et al. 2002a; Jarvinen et al. 2005; Lehto and Jarvinen 1991; Garrett et al. 1984) and so only a brief overview of the process will be presented here (Fig.1).

Repair of a large muscle defect with volumetric loss. Following injury to the muscle with volumetric loss (a), damaged and necrotic myofibers are degraded by invading inflammatory cells, which attract satellite cells and remove cellular debris (b). Satellite cells begin to differentiate into myoblasts, while fibroblasts begin to deposit scar tissue (c). The satellite cells begin forming myotubes that begin to fuse with existing myofibers to form new muscle tissue (d). In cases of volumetric muscle loss, the formation of scar tissue proceeds more rapidly than myogenesis, with the production of a dense cap that prevents myofibers from bridging the wound (e) and that splits the muscle. As a consequence, the distal tissue (right) that does not contain any neuromuscular junctions becomes dennervated (f)
Following muscle injury, myofibers are sheared or torn exposing the intracellular contents to the extracellular environment. Activation of calcium-dependent proteases leads to the rapid disintegration of the myofibrils, with a contraction band of cytoskeletal proteins forming to prevent complete destruction of the myofiber. Activation of the complement cascade induces chemotactic recruitment of neutrophils and then later macrophages. These cells begin the process of digestion of the necrotic myofibers and cellular debris by phagocytosis. The neutrophils and macrophages release cytokines that amplify the inflammatory response and recruit muscle satellite cells to the injury site. Two distinct macrophage populations sequentially invade the injured muscle tissue. First, pro-inflammatory M1 phenotype macrophages phagocytose the necrotic tissue and promote the proliferation of satellite cells, followed by tissue remodeling M2 macrophages, which promote myoblast proliferation, growth and differentiation (Tidball and Villalta 2010).
During the repair process, nerves, blood vessels and muscle cells infiltrate the wound area. Satellite cells migrate and differentiate, becoming myoblasts that fuse with other myoblasts or with existing myofibers to form new skeletal muscle. However, satellite cells are not the only group of progenitor cells capable of contributing to skeletal muscle repair. There is intense interest and controversy, in the contribution of other muscle-derived stem cell groups and circulating progenitor cells, such as bone-marrow-derived cells, to new muscle formation either through myogenic differentiation or by secretion of paracrine factors that affect the surrounding cells (Quintero et al. 2009; Sun et al. 2009; Tedesco et al. 2010; Ten Broek et al. 2010). Concurrent with the formation of new muscle, the formation of scar tissue bridges the gap between the remaining functional muscle fibers and preserves the transduction of force along the muscle. With less severe injuries, such as contusions or sprains, this tissue scar also acts as a conduit to promote myofiber formation. In severe injuries with volumetric tissue loss, fibroblasts quickly deposit scar tissue forming a dense cap that blocks regenerating muscle fibers from bridging the wound, thereby effectively splitting the original muscle and creating two muscles in series (Fig. 1).
The remodeling phase is a continuation of the repair process in which the new myofibers formed by invading progenitor cells subsequently mature and form attachments to the surrounding extracellular matrix (ECM). However, this remodeling process can often be associated with marked reorganization of the muscle tissue including the formation of forked fibers, satellite myofibers, or orphan myofibers that form outside the basal lamina (Schmalbruch 1976). Ultimately, the ends of the damaged muscle fibers might not reunite but, instead, form a new myotendinous junction with the interposed scar tissue. Regenerative medicine techniques are now being explored specifically to target and augment the immune response and the individual processes of progenitor cell proliferation, mobilization and differentiation.
Controlling the immune response
Of interest for any regenerative medicine strategy is the relationship between skeletal muscle repair and the innate immune response. Unlike embryonic muscle development, the microenvironment of the myogenic response to injury in adults is rich with immune effector cells, the concentration of which can exceed 100,000 cells/mm3 of muscle tissue (Abood and Jones 1991; Brunelli and Rovere-Querini 2008; Wehling et al. 2001). This immune response can either help or hinder regenerative medicine therapies. Techniques that can proactively manipulate this response could have profound effects in promoting endogenous skeletal muscle repair.
Within 2 h of muscle damage, neutrophils begin to invade the wound site, peaking between 6 and 24 h post injury before rapidly declining in number. Phagocytic (M1) macrophages then invade the wound site ~24 h post injury, peak at about day 2, before declining in number. This M1 macrophage response phase is followed by an increase in nonphagocytic (M2) macrophages that peak in numbers at ~4 days following injury and persist until well into the remodeling phase of skeletal muscle repair (Brunelli and Rovere-Querini 2008; Frenette et al. 2000; St Pierre and Tidball 1994a; Tidball 1995; Chazaud et al. 2009). The influx of M1 macrophages at 24 h coincides with the activation and recruitment of muscle progenitor cells to the wound site and the increased expression of myogenic transcription factors such as MyoD and myogenin by muscle satellite cells (Launay et al. 2001; Yablonka-Reuveni and Rivera 1994). In vitro studies have shown that, whereas the expression of these transcription factors is not dependent on macrophages, secreted products from macrophages significantly increase transcription factor expression levels and promote satellite cell proliferation (Tidball and Villalta 2010; Arnold et al. 2007; Cantini and Carraro 1995; Malerba et al. 2010).
M1 macrophages release a complex milieu of prostaglandins, cytokines and chemokines, which have been implicated in promoting muscle precursor proliferation and transition to the differentiation stage. Chen et al. (2005) and Warren et al. (2002) have demonstrated the importance of tumor necrosis factor alpha (TNFα) in promoting satellite cell proliferation during the early stages of muscle repair (Chen et al. 2005; Warren et al. 2002), whereas others have demonstrated its function as a chemoattractant for myoblasts and satellite cells in vitro (Al-Shanti et al. 2008; Lolmede et al. 2009; Torrente et al. 2003). Interleukin-6 (IL-6) has also been revealed to play a role in progenitor cell recruitment (Al-Shanti et al. 2008; Wang et al. 2008) with Serrano et al. (2008) showing that elimination of IL-6 greatly diminishes muscle growth. Conversely, both of these molecules have been shown to inhibit the differentiation and maturation of skeletal muscle myoblasts suggesting that the transition from a pro-inflammatory M1 response to a tissue remodeling M2 response is essential for the progression of myogenic differentiation and muscle repair (Szalay et al. 1997; Tsujinaka et al. 1996).
The shift in phenotype from M1 to M2 macrophages coincides with the beginning of myogenic differentiation in muscle progenitor cells (St Pierre and Tidball 1994b). Tidball and Wehling-Henricks (2007) have shown that the depletion of M2 macrophages severely disrupts myoblast differentiation and fusion resulting in decreased muscle fiber diameter (Tidball and Wehling-Henricks 2007). The release of interleukin-10 (IL-10), a characteristic marker of M2 macrophages, is thought to play a key role in promoting the fusion and maturation of myotubes (Arnold et al. 2007; Strle et al. 2007). Although this sequence of inflammatory cell infiltration and macrophage transition has obvious benefits to skeletal muscle repair in relatively minor injuries, the inflammatory response can be severe and prolonged following volumetric muscle loss or severe injury. The administration of anti-inflammatory medication following acute injury, particularly cyclo-oxygenase-2 (COX-2) inhibitors, can markedly delay the muscle repair process (Mackey et al. 2007; Mishra et al. 1995). A careful balance and control of the macrophage phenotype, particularly the promotion of an M2 phenotype, has been suggested as a potential therapeutic strategy to promote in situ muscle repair.
A group from the University of Padova, Italy has recently demonstrated, in a series of studies, the importance of macrophage-secreted products in controlling skeletal muscle repair, both in vitro and in vivo (Cantini and Carraro 1995; Malerba et al. 2010; Malerba et al. 2009; Vitello et al. 2004; Cantini et al. 2002). They have shown that, in vitro, macrophages can secrete factors that increase myoblast/myotube differentiation and enhance the proliferation of neonatal myoblasts from mouse, rat, human and chicken (Cantini and Carraro 1995; Cantini et al. 2002). In addition, they have reported that a macrophage-conditioned culture medium, unlike M1 macrophage products in vivo, does not inhibit myoblast differentiation inducing an increase in contractile myotube formation (Cantini and Carraro 1995; Malerba et al. 2009). Following injection of macrophage conditioned media in a rat model of severe muscle trauma with 80% tissue loss, a significant increase occurs in the number and diameter of regenerating muscle fibers, with twice as much new muscle forming in treated animals compared with controls by the end of the study (Cantini et al. 2002). More recent studies of myogenic progenitor cells isolated from patients with Duchenne muscular dystrophy have shown that macrophage-conditioned media can enhance cell proliferation while maintaining myogenicity resulting in over a 7000-fold increase in cell number after 38 days in culture (Malerba et al. 2010).
An alternative approach by which the macrophage phenotype can be influenced might be through the use of scaffolds derived from decellularized tissues. Biologic scaffolds composed of ECM are commonly used for the reconstruction of musculotendinous, dermal, cardiovascular and gastrointestinal tissues (Badylak 2007). Numerous commercial products are now approved for such use and are readily available (Table 1). Whereas these products differ in their tissue source, species of origin and methods of preparation, such biologic scaffold materials are typically xenogeneic in origin. As such, we might anticipate that these products will elicit a typical foreign-body reaction when implanted. However, if processed correctly, ECM scaffolds induce a macrophage response that rapidly transitions to a predominately M2 phenotype promoting constructive tissue remodeling with minimal scar tissue formation (Ariganello et al. 2011; Badylak et al. 2008; Brown et al. 2009; Valentin et al. 2009).
Of note, processing methods, particularly the use of chemical crosslinking agents, play an essential role in controlling the host response to these ECM materials (Valentin et al. 2009). Typically, only products that are non-crosslinked elicit this M2 macrophage polarization response with associated tissue remodeling. Chemically crosslinked products or poorly-decellularized tissues elicit a chronic M1 pro-inflammatory response and fibrosis (Badylak et al. 2008; Brown et al. 2009). The mechanisms underlying these different responses to ECM scaffolds are largely unknown, although recent studies are beginning to provide clues. Ariganello et al. (2010) have shown that, in vitro, exposure of a macrophage cell line to decellularized tissue elicits lower esterase and phophatase activity consistent with a subdued inflammatory response. Similarly, Rieder et al. (2005) have demonstrated that decellularization can reduce the chemotactic potential of heart valve tissue for macrophages but does not inhibit the activation of macrophages.
Work within the authors’ laboratory has shown that non-crosslinked ECM scaffold materials, such as those derived from porcine small intestinal submucosa (SIS), elicit a notably different immune response compared with the host response to either the cross-linked version of the same material or, perhaps more surprisingly, autologous tissue grafts when used in a model of muscle regeneration (Badylak et al. 2008; Valentin et al. 2009). All three materials elicit an intense macrophage response within the first 14 days. However, the non-crosslinked SIS-ECM elicits a predominantly M2 macrophage response, whereas the response to crosslinked SIS-ECM polarizes toward the M1 macrophage phenotype. Interestingly, autologous tissue elicits a response that shows no preference for M1 or M2, with equal numbers of each phenotype being present (Valentin et al. 2009). Therefore, since SIS-ECM has the ability to promote an increase in M2 polarization over autologous tissue, implantation of this material might result in improved myoblast differentiation and fusion and an increase in the amount of functional muscle restored following injury (Turner et al. 2010; Valentin et al. 2010). An additional benefit of ECM scaffolds, as will be discussed later, is their ability to modify the wound microenvironment through the release of latent growth factors and/or degradation products that can act as chemoattractants for myogenic precursor cells.
Recruitment of myogenic progenitor cells
If therapeutic techniques for in situ skeletal muscle reconstruction in cases of volumetric muscle loss are to be successful, such regenerative therapies must involve myogenic progenitor cells. The involvement of these cells can occur either through the isolation, expansion and injection of autologous cells to the wound site or by the recruitment of tissue resident or circulating progenitor cells in situ. Nevertheless, perhaps a more important question is which progenitor cell population should these strategies target?
Muscle regeneration requires a cell population that is capable of sustained proliferation, self-renewal, myogenesis and resistance to oxidative or hypoxic stress. A number of potential myogenic progenitor cell populations have been identified that might have therapeutic potential. However, a lack of standardized assays to characterize progenitor cell populations and assess myogenic potential means that the true potential of these cells to form new muscle tissue in vivo is largely unknown and controversy exists over their contribution to the repair process.
The most obvious cell population to target is probably represented by the muscle satellite cell that resides between the plasma membrane and basement membrane of the individual muscle fibers (Cossu and Biressi 2005). Characterized by their expression of Pax-7 (paired box protein-7), these cells remain quiescent until activated by muscle injury, at which point they are capable of migration within and between myofibers to contribute to muscle repair (Jarvinen et al. 2005). Such satellite cells have the capacity for self-renewal either through cell division or through withdrawal from the differentiation pathway back to a quiescent state. However, these satellite cells are relatively scarce within skeletal muscle comprising ~1%-5% of total muscle nuclei (Alameddine et al. 1989). Although as few as seven satellite cells have been shown to be capable of repopulating irradiated muscle and generating over 100 new muscle fibers (Collins et al. 2005) in large skeletal muscle defects with loss of muscle tissue, they might not be able to be recruited in high enough numbers to facilitate in situ repair. Strong evidence also exists that the satellite cell pool is actually a heterogeneous population (Biressi and Rando 2010; Harel et al. 2009; Rouger et al. 2004). Furthermore, whereas these cells can be isolated from muscle tissue and expanded in culture, satellite cells appear to show predetermination, despite their stem-cell-like qualities, i.e., the source of the muscle fiber type predisposes satellite cell differentiation. Huang et al. (2006) have shown that muscle fibers derived from soleus muscle satellite cells in vitro have different contractile abilities from muscle fibers derived from anterior tibialis satellite cells. McLoon et al. (2007) have also suggested that the source muscle may impart unique abilities to the satellite cells that it contains, such as increased resistance to apoptosis, indicative of their different developmental origins (Harel et al. 2009). Moreover, evidence suggests that the procedures used for isolation and in vitro expansion can cause satellite cells to lose their regenerative capacity (Renault et al. 2000; Sherwood and Wagers 2006; Montarras et al. 2005), with some suggesting the use of muscle grafts to provide a satellite cell population rather than enzymatic isolation techniques (Montarras et al. 2005; Collins and Zammit 2009; Mong 1988; Schultz et al. 1988). Despite these problems, a number of examples of satellite cell transplantation have led to the successful restoration of muscle injuries (Alameddine et al. 1989; Alameddine et al. 1994; el Andalousi Boubaker et al. 2002; Li et al. 2010; Marzaro et al. 2002; Vindigni et al. 2004).
In addition to satellite cells, a number of muscle-derived stem cell (MDSC) populations have been identified, although the precise origin, identity and location of these cells remain speculative. These include myogenic progenitor cells characterized as CD56+, CD34-, CD144-, CD45-, and CD146-; myo-endothelial cells characterized as CD56+, CD34+, CD144+, CD45-, and CD146-; perivascular progenitor cells characterized as CD56-, CD34-, CD144-, CD45- and CD146+; and a muscle-derived side population that has similar properties to hematopoietic stem cells in the bone marrow (Quintero et al. 2009; Ten Broek et al. 2010; Crisan et al. 2009; Deasy et al. 2004; Huard 2008; Jankowski et al. 2002; Kallestad and McLoon 2010; Lecourt et al. 2010; Peault et al. 2007; Qu-Petersen et al. 2002; Usas and Huard 2007; Wu et al. 2010). MDSCs, once activated, are capable of differentiating along myogenic, osteogenic, chondrogenic and adipogenic lineages in vitro similar to other mesenchymal progenitor cells (Gates et al. 2008). The precise location of these progenitor cells within the muscle is unknown, although increasing evidence suggests that they are located outside the basal lamina of the muscle fibers in close proximity to blood vessels (Crisan et al. 2009; Abou-Khalil et al. 2010; Corselli et al. 2010; Dellavalle et al. 2007). Indeed, myogenic progenitor cells, myoendothelial cells and perivascular cells have been suggested to be an interrelated cell population (Peault et al. 2007; Abou-Khalil et al. 2010). Interestingly, following muscle trauma, the numbers of MDSCs increase rapidly, although whether this increase is attributable to rapid proliferation or recruitment from other sites is uncertain (Jackson et al. 2010; Nesti et al. 2008). The majority of reports describing the use of MDSCs to treat muscle injury are limited to animal models. Successful results have been reported in the treatment of Duchenne muscular dystrophy with MDSCs successfully restoring dytrophin following the implantation or systemic delivery of MDSCs (Bachrach et al. 2006; Ikezawa et al. 2003; Payne et al. 2005; Galvez et al. 2006). Dellavalle et al. (2007) have shown the ability of perivascular MDSCs to colonize dystrophic muscle and to become localized to the satellite cell niche suggesting that these cells contribute to the satellite cell pool. The translation of in vitro studies of MDSCs to in vivo therapeutic treatments has been led by the treatment of stress urinary incontinence (Furuta et al. 2008; Huard et al. 2002b; Kim et al. 2007; Smaldone and Chancellor 2008; Yokoyama et al. 2001). An early clinical trial has demonstrated the successful restoration of detrusor muscle function for over 1 year following MDSC injection, with no adverse outcomes (Carr et al. 2008).
Similar to MDSCs, adipose-derived stem cells reside in proximity to the capillaries within adipose tissue (Bailey et al. 2010; Lin et al. 2010). Although these cells appear to be a more heterogeneous population than MDSCs, containing smooth muscle cells among their numbers, they demonstrate the same myogenic differentiation capacity as other mesenchymal progenitor cells (Pettersson et al. 1984; Zuk et al. 2001; Gimble and Guilak 2003). Adipose-derived stem cells could prove useful as a source of injectable progenitor cells, since the progenitor cells are present at high density, allowing the possibility of direct injection without the need for prolonged culture in vitro (Padoin et al. 2008). In addition, bone marrow is a rich source of myogenic mesenchymal stem cells (MSCs) that are known to contribute to the repair process following muscluloskeletal injuries (Sun et al. 2009; Abedi et al. 2007; Drapeau et al. 2010; Rosu-Myles et al. 2005).
In studies of the MDX mouse model of muscular dystrophy, bone-marrow-derived progenitor cells became incorporated into skeletal muscle and were found occupying the satellite cell niche (Bittner et al. 1999; Corti et al. 2002; Fukada et al. 2002; Saito et al. 1995; Quintero et al. 2009). LaBarge and Blau (2002) showed that, in response to injury, bone marrow cells differentiated into functioning muscle satellite cells prior to becoming differentiated myofibers (LaBarge and Blau 2002). Bone-marrow-derived cells have also been demonstrated to respond to eccentric exercise or muscle overload by mobilization from the bone marrow to engraft within the stimulated muscle (Palermo et al. 2005). An additional benefit of bone-marrow- and adipose-derived MSCs is that, in cases of significant muscle loss, these mutlipotent cells can contribute to the restoration of other muscle components such as blood vessels and nerves, which will also have been lost (Ladak et al. 2011; Guiducci et al. 2010; Tille and Pepper 2002). Evidence suggests that the use of MSCs can improve muscle regeneration without the stem cells even being incorporated into myofibers, through their release of paracrine growth factors and other compounds that might facilitate the regeneration of existing fibers and satellite cells (Kagiwada et al. 2008; Natsu et al. 2004; Hocking and Gibran 2010). Injection of bone-marrow-derived cells restores contractile force more effectively following crush injury compared with normal repair process (Matziolis et al. 2006). Experimental models have shown that injected bone-marrow-derived cells are incorporated into newly formed myofibers of regenerating skeletal muscles and contribute to the satellite cell pool in a similar way to endogenous cells (Saito et al. 1995; LaBarge and Blau 2002; Dezawa et al. 2005; Ferrari et al. 1998; Muguruma et al. 2003). Conversely, in vitro studies have revealed that not all MSCs respond to differentiation signals, with as few as 35%-40% of cells in some cultures exhibiting a morphology consistent with their intended phenotype (Izadpanah et al. 2006; Phinney 2007).
However, given the multipotency of all of these mesenchymal progenitor cell populations, controversy exists as to whether these cells can adopt and maintain a myogenic phenotype when implanted at a site of muscle injury or recruited there via chemotactic factors. Indeed, evidence suggests that, in severely traumatized muscle, the dominant phenotype of these cells is osteogenic rather than myogenic (Jackson et al. 2009).
In addition to tissue resident or bone-marrow-derived MSCs, another population of circulating CD133+ progenitor cells has been identified with myogenic potential. Initially identified as circulating endothelial progenitor cells (Urbich and Dimmeler 2004), numerous groups have demonstrated the ability of these cells to form new muscle tissue in vitro (Alessandri et al. 2004; Torrente et al. 2004; Shmelkov et al. 2005). Being a circulating cell population, these CD133+ cells present an interesting cell pool that could be targeted to home to the site of injury and contribute to muscle repair. The ability of these cells to home and respond to injury has been reported in a number of studies, particularly in ischemic injury of the heart (Kania et al. 2009; Kubo et al. 2008; Voo et al. 2008; Navarro-Sobrino et al. 2010). Torrente et al. (2004) have shown that the direct injection of CD133+ cells into dystrophic muscle improves skeletal muscle structure and replenishes the satellite cell pool, leading to a successful phase I clinical trial of autologous CD133+ cell injection in eight individuals with Duchenne muscular dystrophy (Torrente et al. 2007). Similarly, Negroni et al. (2009) have demonstrated that, compared with human myoblasts, the direct injection of CD133+ cells increases the stem cell pool and results in greater muscle regeneration. Turner et al. (2010) have also recently reported that, following severe muscle trauma with massive tissue loss, replacement of the defect with an ECM-scaffold results in the recruitment of CD133+ cells to the site of injury and is associated with the formation of new skeletal muscle and blood vessels in the first 4 months following injury (Turner et al. 2010).
A variety of potential cell populations clearly exist that can contribute to endogenous skeletal muscle repair. However, each of these populations equally clearly varies in its abundance within the body and in its ability to form and maintain a myogenic phenotype. Whereas intramuscular injection of these cells might prove beneficial for some muscle injuries or congenital muscle disorders, the endogenous repair of severe muscle trauma associated with tissue loss might prove difficult without reliable techniques to attract these cells in high numbers to the site of injury and to trigger their proliferation and differentiation.
Control of the regenerative microenvironment
In addition to the secreted cytokines and chemokines released by macrophages, skeletal muscle injury triggers the release of a complex mix of growth factors and cellular and extracellular proteins that together define the regenerative microenvironment responsible for stimulating and coordinating skeletal muscle repair. Whereas the exact composition of this biochemical milieu remains largely unknown, a number of key regulators have been identified. Controlling this regenerative microenvironment might represent an effective approach to endogenous skeletal muscle repair by allowing the body to function as its own bioreactor to restore skeletal muscle in a site-specific manner and by eliminating the problems associated with the exogenous fabrication of skeletal muscle constructs.
Growth factors are essential regulators of the muscle repair process, controlling the proliferation and differentiation of muscle progenitor cells (Ten Broek et al. 2010; Charge and Rudnicki 2004). Such factors are secreted by active immune cells and by muscle cells following injury and from the vasculature, progenitor cells and neurons (Cantini et al. 2002; Hawke and Garry 2001; Cannon and St Pierre 1998). The sequence of their release appears to be important for the control of muscle repair (Hayashi et al. 2004). Key growth factors include the hepatocyte growth factor (HGF), basic fibroblast growth factor (FGF), insulin-like growth factor-1 (IGF-1), vascular endothelial growth factor (VEGF) and platelet-derived growth factors–AA and –BB (Table 2). In particular, IGF-1 is critical for muscle growth and myoblast proliferation. In vitro studies have shown the ability of IGF-1 to promote the proliferation of satellite cells and to alter the expression of myogenic factors (Charge and Rudnicki 2004; Chakravarthy et al. 2001; Hsu et al. 1997; Philippou et al. 2007). In vivo systemic injection of IGF-1 results in skeletal muscle hypertrophy, whereas direct injection of IGF-1 into muscle enhances muscle regeneration (Hsu et al. 1997; Kasemkijwattana et al. 1998; Menetrey et al. 2000; Sato et al. 2003). HGF is the primary regulator of satellite cell proliferation, with HGF expression increasing in proportion to the degree of injury (Suzuki et al. 2002; Tatsumi 2010; Tatsumi et al. 2002). Bischoff (1997) has shown that HGF is essential for the migration of muscle progenitor cells to the site of injury, whereas others have demonstrated the importance of HGF in satellite cell activation and regulation of proliferation and differentation (Allen et al. 1995; Gal-Levi et al. 1998; Li et al. 2009; Sheehan et al. 2000). Conversely, HGF also inhibits myotube formation and does not promote regeneration if injected into the muscle during the differentiation stage of muscle repair thereby demonstrating a pleiotrophic effect (Miller et al. 2000; Tatsumi et al. 1998). HGF is also known to have synergistic effects with FGF-2 and −6, which in combination significantly increases satellite cell proliferation compared with the individual growth factors alone (Sheehan and Allen 1999).
Overall, it appears that IGF-1 and HGF are the main growth factors involved in regulating skeletal muscle repair but as the pleiotrophic effects of HGF show, improving skeletal muscle repair particularly in severe trauma cases might not be as simple as a single injection of the growth factor. Whereas the growth factors above have been extensively studied, the complex interaction that occurs in the wound microenvironment is still largely uncharacterized. In addition to growth factors that stimulate repair, some growth factors inhibit this process.
Transforming growth factor β1 (TGFβ1) has perhaps the greatest inhibitory effect on muscle progenitor cells, decreasing cell proliferation and differentiation and inhibiting myoblast recruitment (Florini and Magri 1989; Kollias and McDermott 2008; Liu et al. 2001; Massague et al. 1986). In addition, TGFβ1 induces the formation of scar tissue that limits muscle regeneration (Jarvinen et al. 2005; O'Kane and Ferguson 1997; Shah et al. 1999). TGFβ1 can be bound to the ECM in a latent form (Lawrence 2001; Wipff and Hinz 2008) and released during muscle injury at which time it acts as a chemoattractant for macrophages (Shen et al. 2008; Smith et al. 2007). It is also synthesized by macrophages and acts in an autocrine manner to stimulate its own synthesis. Thus, the inhibition of TGFβ1 might decrease muscle fibrosis and control the inflammatory response promoting greater muscle healing. Sato et al. (2003) have shown that decorin can prevent TGFβ1-induced muscle fibrosis but perhaps the most successful mediator of the effects of TGFβ1 has been the application of TGFβ3 (Ghosh et al. 2006; Hosokawa et al. 2003; Occleston et al. 2009; Wu et al. 1997). While TGFβ3 has not been shown to have direct effects on muscle repair, extensive research has shown TGFβ3 to be a potent inhibitor of scar tissue formation reducing the influx of inflammatory cells to the wound site without delaying wound healing (Wu et al. 1997). The indirect effects of inhibiting TGFβ1 could plausibly result in enhanced progenitor cell proliferation, differentiation and recruitment, while decreasing scar tissue.
In addition to active growth factors, many cells essential for skeletal muscle repair release latent growth factors that are sequestered by the ECM and that only become active following the degradation of this ECM during muscle repair (Schultz and Wysocki 2009; Kresse and Schonherr 2001; Miura et al. 2006). Many research groups are now focusing on the ECM as a regulator and stimulator of tissue repair in many body sites including muscle (Borschel et al. 2004; Badylak 2007; Turner et al. 2010; Jensen and Host 1997; Owen and Shoichet 2010; Robinson et al. 2005). The main constituents of skeletal muscle ECM are type IV collagen, laminin and heparan sulfate proteoglycans (HSPGs). Cells attach to these ECM components via integrins, the differential expression of which regulates the migration and activation of progenitor cells (Brzoska et al. 2006; Grabowska et al. 2010; Velleman and McFarland 2004). The ECM acts as a reservoir for growth factors. HSPGs, for example, sequester HGF thereby preventing satellite cell proliferation in healthy muscle (Tatsumi and Allen 2004). Indeed, in vitro experiments have shown that HSPGs are essential for proper signaling by FGF and HGF in muscle (Cornelison et al. 2001; Miller et al. 1991). Following skeletal muscle injury, invading macrophages and satellite cells release matrixmetalloproteinases (MMP) such as MMP-2 and MMP-9 (Lolmede et al. 2009; Tatsumi 2010). These enzymes liberate the bound growth factors, which subsequently stimulate myoblast and progenitor cell migration, proliferation and differentiation.
Laminin-2 is unique to skeletal muscle and is found in the skeletal muscle basement membrane (Shibuya et al. 2003). Knockout studies have shown that a lack of laminin-2 results in an almost complete absence of a basement membrane and decreased proliferation with increased apoptosis of satellite cells in response to injury (Laprise et al. 2002; Vachon et al. 1996; Miyagoe et al. 1997). Therefore, a site specificity of ECM exists that might be targeted to enhance skeletal muscle repair.
ECM has been commonly used as a delivery vehicle. The seeding of an ECM scaffold with myogenic progenitor cells, followed by transfer to an in vivo site, allows the full development of myotubes and functional muscle tissue to occur (Liao and Zhou 2009; Borschel et al. 2004; du Moon et al. 2008; Vindigni et al. 2004; Beier et al. 2009; Merritt et al. 2010). However, these in vitro techniques generally produce muscle with small contractile forces, since the amount of scaffold material is high and can inhibit myoblast fusion. In addition, the ultrastructure of the ECM controls the attachment, alignment and differentiation of myoblasts (Altomare et al. 2010; Grenier et al. 2005; Huang et al. 2010; Matsumoto et al. 2007). Many studies involve the use of gel-based constructs of mainly collagen, laminin, or fibrin that do not necessarily replicate the complex ultrastructure of native tissue (Huang et al. 2005; Beier et al. 2009; Matsumoto et al. 2007; Rowlands et al. 2007; Vandenburgh et al. 1988). Acellular implants are now being developed through the decellularization of tissues and organs in an attempt to retain the ultrastructure (Gilbert et al. 2006). These acellular matrices, typically xenogeneic in origin, have several advantages over other materials. First, the process of removing cells eliminates adverse immune response and, as was discussed earlier, if processed correctly, acellular matrices can actually promote a constructive remodeling phenotype in macrophages (Badylak et al. 2008; Valentin et al. 2009). Second, the complex three-dimensional ultrastructure of the starting tissue is largely retained. For acellular skeletal muscle, this retained ultrastructure might include residual endomysial tubes and remnant neural and vascular pathways that can promote myoblast alignment and fusion and stimulate neurovascular growth (Borschel et al. 2004; Di Benedetto et al. 1994; Gillies et al. 2011).
Biologic scaffolds composed of ECM have been successfully used to repair or replace a variety of damaged or diseased tissues, including cardiac (Akhyari et al. 2008; Badylak et al. 2006; Kochupura et al. 2005), esophageal, (Nieponice et al. 2006, 2008), dermal (Brigido et al. 2004) and musculotendinous (Aurora et al. 2007; De Deyne and Kladakis 2005; Dejardin et al. 2001; Snyder et al. 2009) tissues. Studies have shown that these materials are rich in site-specific glycosaminoglycans and a wide variety of growth factors including FGF, VEGF and epidermal growth factor. The biomechanical properties of ECM scaffolds have also been extensively characterized (Derwin et al. 2006; Sacks and Gloeckner 1999; Freytes et al. 2008a, 2008b; Gilbert et al. 2008) but these properties, as with any implanted material, inevitably and rapidly change in vivo as a result of the concurrent processes of scaffold degradation, neomatrix deposition and host cellular remodeling events (Badylak et al. 2001a; Ko et al. 2006; Liang et al. 2006). The remodeling process is characterized by an immediate intense mononuclear cell infiltrate, followed by a period of intense vascularization and a progressive degradation of the ECM material between days 3 and 14. A deposition of a host-derived neomatrix and the appearance of site-specific parenchymal cells such as skeletal muscle myoblasts and circulating progenitor cells occurs, which in turn repopulate the injury site to form functional tissue (Turner et al. 2010; Badylak and Gilbert 2008; Badylak et al. 2001b). In terms of skeletal muscle repair, Conconi et al. (2005) have found benefits by using decellularized abdominal muscle seeded with cultured myoblasts; this group has shown that, when implanted in the abdominal wall, these constructs generate a complex skeletal muscle tissue that is contractile and vascularized after only 9 days.
Work within our own group has demonstrated that, in many cases, the acellular ECM is capable of restoring functional tissue without any additional cellular component. Valentin et al. (2010) report that, in a rat abdominal wall defect model, acellular ECM derived from SIS achieves almost complete replacement of the defect site with islands and sheets of functional skeletal muscle after 6 months (Valentin et al. 2010). This new muscle generates ~80% of the contractile force of native muscle. Similarly, in a model of severe muscle trauma with massive tissue loss, implantation of SIS-ECM promotes the formation of vascularized skeletal muscle that is functionally innervated and almost indistinguishable from native muscle by 6 months (Turner et al. 2010). Interestingly, the same proteases that degrade a surgically placed ECM scaffold are also responsible for degrading the remnant ECM of native tissue following injury. The degradation products of this surgically placed ECM have potent biologic effects including mitogenic and chemotactic effects on muscle progenitor cells (Crisan et al. 2009; Tottey et al. 2011; Vorotnikova et al. 2010). In related work, Calve et al. (2010) have demonstrated that ECM deposited during amphibian limb regeneration provides instructional cues for both myoblasts and satellite cells; these cues are vital for muscle repair.
Together, these findings have translated into the successful restoration of skeletal muscle in human patients that have suffered volumetric muscle loss. Early results have shown that acellular ECM scaffolds have restored significant amounts of new skeletal muscle with significant improvements in function and isokinetic performance (Mase et al. 2010).
Importance of vascularization, innervation and mechanical loading
Three additional factors play a critical role in determining the regeneration potential of injured muscle: the reinnervation of the regenerating muscle fibers, the formation of new blood vessels within the repaired muscle and mechanical loading and the generation of mechanical force.
As discussed earlier, muscle injuries with volumetric loss typically will not heal without scar tissue formation, which might be associated with the muscle distal to the site of injury becoming denervated and non-functional. Therefore, an essential step in any regenerative medicine approach is the reestablishment of nerves to the regenerating muscle and the formation of new motor endplates. These motor endplates not only confer functional control to the new muscle but their chemotropic and stimulatory effects also influence muscle fiber type, alignment and size (Donghui et al. 2010; Grubic et al. 1995; Lefeuvre et al. 1996; Washabaugh et al. 1998). If neuromuscular connections are not reestablished then regenerating muscle will become atrophic (Jarvinen et al. 2005, 2007). The importance of nerve activity in muscle regeneration has been clearly demonstrated following the use of bupivacaine to induce muscle injury. Bupivacaine induces necrosis of all muscle fibers but leaves satellite cells, nerves and blood vessels intact (Benoit and Belt 1970; Foster and Carlson 1980). In this model, new myotubes begin to form after ~3 days, with new motor endplates forming soon thereafter resulting in rapid maturation of the muscle tissue and restoration of function (Jirmanova and Thesleff 1972; Grubb et al. 1991). Conversely, if the muscle tissue is treated with bupivacaine and also dennervated, although new myotubes still form, no rapid growth or restoration of function occurs (Carlson and Faulkner 1996, 1998). In the rat soleus muscle, this rapid maturation of myotubes coincides with a switch from expression of embryonic to neonatal myosin heavy chains to adult myosin (Whalen et al. 1990). While this change is independent of nerve function, Mendler et al. (2008) have shown that, following dennervation and reinnervation, muscle fibers switch from slow-type fibers to fast-type fibers following reinnervation of the muscle fibers (Mendler et al. 2008). Furthermore, Gregorevic et al. (2004) have demonstrated that these regenerating fibers adopt their fast and slow twitch properties as they regenerate (Gregorevic et al. 2004). Studies investigating the innervation of skeletal muscle have, to date, been limited to in vitro co-cultures of myoblasts, satellite cells and neurons (Larkin et al. 2006; Das et al. 2007; Mars and Martincic 2001; Mehrke et al. 1984; Wagner et al. 2003). These studies have revealed that nerve-muscle constructs generate greater contractile force compared with muscle-only constructs. Dhawan et al. (2007) have found that in vivo innvervation of a muscle construct results in increased contractility compared with non-innvervated constructs (Dhawan et al. 2007). However, returning functional innervation in severe muscle injuries has proven difficult. One successful example is represented by the SIS-ECM-mediated gastrocnemius muscle repair in the model of Turner et al. (2010) in which new nerves invade the site of injury within 2 months of implantation of the SIS-ECM scaffold. Furthermore, these new nerves form new motor endplates with the new muscle and make a functional reconnection with the sciatic nerve as demonstrated by the recorded action potentials in the muscle following nerve stimulation. This re-innervation also coincides with a switch in muscle fiber type from predominately slow fiber types to a 50:50 mix of fast and slow muscle approximating the composition of the uninjured gastrocnemius muscle (Turner et al. 2010).
In contrast to the limited research into improving the innervation of regenerating muscle in situ, extensive research has been directed to the enhancement of the vascularization of the site of muscle injury. In particular, the promotion of angiogenesis can activate resident satellite cells and provide a conduit through which myogenic progenitor cells can travel. One of the major limiting factors in the repair of skeletal muscle is the requirement for nutrients and oxygen by myoblasts. Myoblasts are unable to proliferate or differentiate further than ~150 μm from a nutrient source and oxygen supply (Dennis and Kosnik 2000). Promotion of angiogenesis might therefore be one of the key mediators for stimulating the repair of volumetric muscle injuries in situ.
Techniques for the successful promotion of vascularization in vivo have achieved mixed results. Direct injection of VEGF has been shown to stimulate muscle healing by promoting angiogenesis and increasing the nutrient and oxygen supply (Deasy et al. 2009; Gowdak et al. 2000; Messina et al. 2007; Springer et al. 1998). However, the effects of VEGF may be time-dependent, requiring early injection in order to stimulate the activation of MDSCs derived from the vasculature and satellite cell activating growth factors. In addition, VEGF tends to promote a random angiogenic response rather than a directed response, thus potentially limiting the muscle repair process. Other tissue engineering approaches, particularly those utilizing cellular constructs or the direct injection of cells have sought to develop a vascular bed that would integrate with the host system once implanted into the muscle. For example, a number of studies have utilized the foreign-body response to promote the formation of vascularized tissue surrounding the muscle construct (Saxena et al. 2001; Beier et al. 2006; Tanaka et al. 2000). Others have employed the co-culture of myoblasts and endothelial cells to create a vascularized muscle construct in vitro (Levenberg et al. 2005; Borschel et al. 2006). Interestingly, acellular ECM scaffolds have been shown to promote angiogenesis. Valentin et al. (2010) report that, in the rat abdominal wall, SIS-ECM promotes increased angiogenesis during muscle repair, which is comparable with autologous tissue repair, with the number of new blood vessels almost doubling compared with uninjured tissue (Valentin et al. 2010).
The final consideration and perhaps the most overlooked is the importance of mechanical loading and exercise on the promotion of muscle repair. If the goal of a regenerative medicine approach to muscle repair is to promote the rapid repair of skeletal muscle, a concomitant aim must be the rapid restoration of functional use. Currently, the most commonly prescribed treatments for muscle injury are rest, ice application, compression and elevation, i.e., immobilization of the muscle. This “RICE” therapy is typically followed by an extensive period of physiotherapy to address the muscle atrophy that results from prolonged immobilization (Jarvinen et al. 2000, 2007). As is well known, cells sense their environment through anchoring and pulling on the ECM in which they reside. Similarly, tension within the ECM as a result of mechanical load translates to the resident cells and in turn influences their phenotype (Bischoff 1997; Xu et al. 2009). As has previously been mentioned, the formation of functional skeletal muscle depends in part on the alignment of new muscle fibers in order to direct the appropriate contractile forces without further muscle damage, whereas the formation of an organized neurovascular bed is essential for muscle fiber maturation. A key approach to developing differentiated and functional skeletal muscle tissue is electrical stimulation during myogenesis, either through innervation or by artificial methods. The expression of desmin, myosin heavy chain and other myogenic factors can be increased significantly by electrical stimulation of the repairing muscle (Flaibani et al. 2009; Nicolaidis and Williams 2001; Park et al. 2008; Sasaki et al. 2007; Serena et al. 2008). This type of stimulation is thought to mimic neuronal activity and has achieved success, both in vitro and in vivo, for enhancing myotube formation and contractile force. Although little evidence exists to show that electrical stimulation promotes the migration or differentiation of myogenic progenitor cells, it has been demonstrated markedly to increase angiogenesis thereby resulting in decreased fibrosis (Dobsak et al. 2006; Ljubicic et al. 2005; Nagasaka et al. 2006). Similarly, in healthy muscle, voluntary exercise promotes myogenesis and muscle hypertrophy and has marked effects on the release of MMPs and the secretion of growth factors, such as IGF-1, which might stimulate the recruitment of myogenic progenitor cells (Mackey et al. 2007; Frystyk 2010; Kivela et al. 2008; Wilborn et al. 2009). This information indicates that, at least for some forms of skeletal muscle injury, prolonged rest and immobilization might be detrimental to healing and that a controlled exercise regimen would enhance skeletal muscle healing.
Concluding remarks
Development of a therapy that avoids the collection, isolation and purification of autologous stem cells or the construction of muscle tissue by using bioreactor systems with subsequent re-introduction to the patient would almost certainly reduce the regulatory hurdles for clinical translation, reduce cost of treatment and avoid the risks associated with ex-vivo approaches. This is not to say that these new strategies that stimulate the endogenous repair of skeletal muscle do not have their own unique challenges. Whereas skeletal muscle possesses a robust capacity to repair itself, the ability to augment and enhance this process would significantly advance the treatment of congenital muscle disorders and severe muscle trauma for which, even with the best of present-day treatments, physical handicap or amputation are the most likely outcomes. However, for these endogenous repair therapies to advance, it is essential that an understanding exists of the biochemical, cellular and mechanical cues that promote skeletal muscle repair.
References
Aarimaa V, Kaariainen M, Vaittinen S, Tanner J, Jarvinen T, Best T, Kalimo H (2004) Neuromuscul Disord 14:421–428
Abedi M, Foster BM, Wood KD, Colvin GA, McLean SD, Johnson KW, Greer DA (2007) Br J Haematol 138:792–801
Abood EA, Jones MM (1991) Acta Anat (Basel) 140:201–212
Abou-Khalil R, Mounier R, Chazaud B (2010) Cell Cycle 9:892–896
Akhyari P, Kamiya H, Haverich A, Karck M, Lichtenberg A (2008) Eur J Cardiothorac Surg 34:229–241
Alameddine HS, Dehaupas M, Fardeau M (1989) Muscle Nerve 12:544–555
Alameddine HS, Louboutin JP, Dehaupas M, Sebille A, Fardeau M (1994) Cell Transplant 3:3–14
Alessandri G, Pagano S, Bez A, Benetti A, Pozzi S, Iannolo G, Baronio M, Invernici G, Caruso A, Muneretto C et al (2004) Lancet 364:1872–1883
Allen RE, Sheehan SM, Taylor RG, Kendall TL, Rice GM (1995) J Cell Physiol 165:307–312
Al-Shanti N, Saini A, Faulkner SH, Stewart CE (2008) Growth Factors 26:61–73
Altomare L, Riehle M, Gadegaard N, Tanzi MC, Fare S (2010) Int J Artif Organs 33:535–543
Andalousi Boubaker R el, Daussin PA, Micallef JP, Roux C, Nougues J, Chammas M, Reyne Y, Bacou F (2002) Cell Transplant 11:169–180
Ariganello MB, Labow RS, Lee JM (2010) J Biomed Mater Res A 93:280–288
Ariganello MB, Simionescu DT, Labow RS, Lee JM (2011) Biomaterials 32:439–449
Arnold L, Henry A, Poron F, Baba-Amer Y, Rooijen N van, Plonquet A, Gherardi RK, Chazaud B (2007) J Exp Med 204:1057–1069
Aurora A, McCarron J, Iannotti JP, Derwin K (2007) J Shoulder Elbow Surg 16:S171–178
Bachrach E, Perez AL, Choi YH, Illigens BM, Jun SJ, Nido P del, McGowan FX, Li S, Flint A, Chamberlain J et al (2006) Muscle Nerve 34:44–52
Badylak SF (2007) Biomaterials 28:3587–3593
Badylak SF, Gilbert TW (2008) Semin Immunol 20:109–116
Badylak S, Kokini K, Tullius B, Whitson B (2001a) J Surg Res 99:282–287
Badylak SF, Park K, Peppas N, McCabe G, Yoder M (2001b) Exp Hematol 29:1310–1318
Badylak SF, Kochupura PV, Cohen IS, Doronin SV, Saltman AE, Gilbert TW, Kelly DJ, Ignotz RA, Gaudette GR (2006) Cell Transplant 15 (Suppl 1):S29–S40
Badylak SF, Valentin JE, Ravindra AK, McCabe GP, Stewart-Akers AM (2008) Tissue Eng A 14:1835–1842
Bailey AM, Kapur S, Katz AJ (2010) Curr Stem Cell Res Ther 5:95–102
Beier JP, Stern-Straeter J, Foerster VT, Kneser U, Stark GB, Bach AD (2006) Plast Reconstr Surg 118:1113–1124
Beier JP, Klumpp D, Rudisile M, Dersch R, Wendorff JH, Bleiziffer O, Arkudas A, Polykandriotis E, Horch RE, Kneser U (2009) BMC Biotechnol 9:34
Beiner JM, Jokl P (2001) J Am Acad Orthop Surg 9:227–237
Benoit PW, Belt WD (1970) J Anat 107:547–556
Biressi S, Rando TA (2010) Semin Cell Dev Biol 21:845–854
Bischoff R (1997) Dev Dyn 208:505–515
Bittner RE, Schofer C, Weipoltshammer K, Ivanova S, Streubel B, Hauser E, Freilinger M, Hoger H, Elbe-Burger A, Wachtler F (1999) Anat Embryol (Berl) 199:391–396
Borschel GH, Dennis RG, Kuzon WM Jr (2004)Contractile skeletal muscle tissue-engineered on an acellular scaffold. Plast Reconstr Surg 113:595–604
Borschel GH, Dow DE, Dennis RG, Brown DL (2006) Plast Reconstr Surg 117:2235–2242
Brigido SA, Boc SF, Lopez RC (2004) Orthopedics 27:s145–s149
Brown BN, Valentin JE, Stewart-Akers AM, McCabe GP, Badylak SF (2009) Biomaterials 30:1482–1491
Brunelli S, Rovere-Querini P (2008) Pharmacol Res 58:117–121
Brzoska E, Bello V, Darribere T, Moraczewski J (2006) Differentiation 74:105–118
Calve S, Odelberg SJ, Simon HG (2010) Dev Biol 344:259–271
Cannon JG, St Pierre BA (1998) Mol Cell Biochem 179:159–167
Cantini M, Carraro U (1995) J Neuropathol Exp Neurol 54:121–128
Cantini M, Giurisato E, Radu C, Tiozzo S, Pampinella F, Senigaglia D, Zaniolo G, Mazzoleni F, Vitiello L (2002) Neurol Sci 23:189–194
Carlson BM, Faulkner JA (1996) J Gerontol A Biol Sci Med Sci 51:B43–B49
Carlson BM, Faulkner JA (1998) J Gerontol A Biol Sci Med Sci 53:B52–B57
Carlson BM, Gutmann E (1972) Exp Neurol 36:239–249
Carr LK, Steele D, Steele S, Wagner D, Pruchnic R, Jankowski R, Erickson J, Huard J, Chancellor MB (2008) Int Urogynecol J Pelvic Floor Dysfunct 19:881–883
Chakravarthy MV, Booth FW, Spangenburg EE (2001) Int J Sport Nutr Exerc Metab 11 (Suppl):S44–S48
Charge SB, Rudnicki MA (2004) Physiol Rev 84:209–238
Chazaud B, Brigitte M, Yacoub-Youssef H, Arnold L, Gherardi R, Sonnet C, Lafuste P, Chretien F (2009) Exerc Sport Sci Rev 37:18–22
Chen SE, Gerken E, Zhang Y, Zhan M, Mohan RK, Li AS, Reid MB, Li YP (2005) Am J Physiol Cell Physiol 289:C1179–C1187
Collins CA, Zammit PS (2009) Meth Mol Biol 482:319–330
Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA, Morgan JE (2005) Cell 122:289–301
Conconi MT, De Coppi P, Bellini S, Zara G, Sabatti M, Marzaro M, Zanon GF, Gamba PG, Parnigotto PP, Nussdorfer GG (2005) Biomaterials 26:2567–2574
Cornelison DD, Filla MS, Stanley HM, Rapraeger AC, Olwin BB (2001) Dev Biol 239:79–94
Corselli M, Chen CW, Crisan M, Lazzari L, Peault B (2010) Arterioscler Thromb Vasc Biol 30:1104–1109
Corti S, Strazzer S, Del Bo R, Salani S, Bossolasco P, Fortunato F, Locatelli F, Soligo D, Moggio M, Ciscato P et al (2002) Exp Cell Res 277:74–85
Cossu G, Biressi S (2005) Semin Cell Dev Biol 16:623–631
Counsel P, Breidahl W (2010) Semin Musculoskelet Radiol 14:162–175
Crisan M, Chen CW, Corselli M, Andriolo G, Lazzari L, Peault B (2009) Ann NY Acad Sci 1176:118–123
Crow BD, Haltom JD, Carson WL, Greene WB, Cook JL (2007) J Orthop Res 25:396–403
Das M, Rumsey JW, Gregory CA, Bhargava N, Kang JF, Molnar P, Riedel L, Guo X, Hickman JJ (2007) Neuroscience 146:481–488
De Deyne PG, Kladakis SM (2005) Clin Podiatr Med Surg 22:521–532
Deasy BM, Li Y, Huard J (2004) Curr Opin Biotechnol 15:419–423
Deasy BM, Feduska JM, Payne TR, Li Y, Ambrosio F, Huard J (2009) Mol Ther 17:1788–1798
Dejardin LM, Arnoczky SP, Ewers BJ, Haut RC, Clarke RB (2001) Am J Sports Med 29:175–184
Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, Innocenzi A, Galvez BG, Messina G, Morosetti R et al (2007) Nat Cell Biol 9:255–267
Dennis RG, Kosnik PE 2nd (2000) In Vitro Cell Dev Biol Anim 36:327–335
Derwin KA, Baker AR, Spragg RK, Leigh DR, Iannotti JP (2006) J Bone Joint Surg Am 88:2665–2672
Dezawa M, Ishikawa H, Itokazu Y, Yoshihara T, Hoshino M, Takeda S, Ide C, Nabeshima Y (2005) Science 309:314–317
Dhawan V, Lytle IF, Dow DE, Huang YC, Brown DL (2007) Tissue Eng 13:2813–2821
Di Benedetto G, Lassner F, Riccio M, Pugnaloni A, Tappa MM, Biagini G, Berger A, Bertani A (1994) Clin Mater 16:137–143
Dobsak P, Novakova M, Siegelova J, Fiser B, Vitovec J, Nagasaka M, Kohzuki M, Yambe T, Nitta S, Eicher JC et al (2006) Circ J 70:75–82
Donghui C, Shicai C, Wei W, Fei L, Jianjun J, Gang C, Hongliang Z (2010) Laryngoscope 120:353–358
Drapeau C, Antarr D, Ma H, Yang Z, Tang L, Hoffman RM, Schaeffer DJ (2010) Cell Cycle 9:1819–1823
Ferrari G, Cusella-De Angelis G, Coletta M, Paolucci E, Stornaiuolo A, Cossu G, Mavilio F (1998) Science 279:1528–1530
Flaibani M, Boldrin L, Cimetta E, Piccoli M, De Coppi P, Elvassore N (2009) Tissue Eng A 15:2447–2457
Florini JR, Magri KA (1989) Am J Physiol 256:C701–C711
Foster AH, Carlson BM (1980) Anesth Analg 59:727–736
Frenette J, Cai B, Tidball JG (2000) Am J Pathol 156:2103–2110
Freytes DO, Stoner RM, Badylak SF (2008a) J Biomed Mater Res B Appl Biomater 84:408–414
Freytes DO, Tullius RS, Valentin JE, Stewart-Akers AM, Badylak SF (2008b) J Biomed Mater Res A 87:862–872
Frystyk J (2010) Med Sci Sports Exerc 42:58–66
Fukada S, Miyagoe-Suzuki Y, Tsukihara H, Yuasa K, Higuchi S, Ono S, Tsujikawa K, Takeda S, Yamamoto H (2002) J Cell Sci 115:1285–1293
Furuta A, Jankowski RJ, Pruchnic R, Egawa S, Yoshimura N, Chancellor MB (2008) Int Urogynecol J Pelvic Floor Dysfunct 19:1229–1234
Gal-Levi R, Leshem Y, Aoki S, Nakamura T, Halevy O (1998) Biochim Biophys Acta 1402:39–51
Galvez BG, Sampaolesi M, Brunelli S, Covarello D, Gavina M, Rossi B, Constantin G, Torrente Y, Cossu G (2006) J Cell Biol 174:231–243
Garrett WE Jr (1996) Am J Sports Med 24:S2–8
Garrett WE Jr, Seaber AV, Boswick J, Urbaniak JR, Goldner JL (1984) J Hand Surg Am 9:683–692
Gates CB, Karthikeyan T, Fu F, Huard J (2008) J Am Acad Orthop Surg 16:68–76
Ghosh J, Baguneid M, Khwaja N, Murphy MO, Turner N, Halka A, Ferguson MW, Kielty CM, Walker MG (2006) J Vasc Surg 43:142–149
Gilbert TW, Sellaro TL, Badylak SF (2006) Biomaterials 27:3675–3683
Gilbert TW, Wognum S, Joyce EM, Freytes DO, Sacks MS, Badylak SF (2008) Biomaterials 29:4775–4782
Gillies AR, Smith LR, Lieber RL, Varghese S (2011) Method for decellularizing skeletal muscle without detergents or proteolytic enzymes. Tissue Eng Part C Methods 17:383–389
Gimble J, Guilak F (2003) Cytotherapy 5:362–369
Gowdak LH, Poliakova L, Wang X, Kovesdi I, Fishbein KW, Zacheo A, Palumbo R, Straino S, Emanueli C, Marrocco-Trischitta M et al (2000) Circulation 102:565–571
Grabowska I, Szeliga A, Moraczewski J, Czaplicka I, Brzoska E (2010) Cell Biol Int 35:125–133
Gregorevic P, Plant DR, Stupka N, Lynch GS (2004) J Physiol (Lond) 558:549–560
Grenier G, Remy-Zolghadri M, Larouche D, Gauvin R, Baker K, Bergeron F, Dupuis D, Langelier E, Rancourt D, Auger FA et al (2005) Tissue Eng 11:90–100
Grubb BD, Harris JB, Schofield IS (1991) J Physiol (Lond) 441:405–421
Grubic Z, Komel R, Walker WF, Miranda AF (1995) Neuron 14:317–327
Guiducci S, Porta F, Saccardi R, Guidi S, Ibba-Manneschi L, Manetti M, Mazzanti B, Dal Pozzo S, Milia AF, Bellando-Randone S et al (2010) Ann Intern Med 153:650–654
Harel I, Nathan E, Tirosh-Finkel L, Zigdon H, Guimaraes-Camboa N, Evans SM, Tzahor E (2009) Dev Cell 16:822–832
Hawke TJ, Garry DJ (2001) J Appl Physiol 91:534–551
Hayashi S, Aso H, Watanabe K, Nara H, Rose MT, Ohwada S, Yamaguchi T (2004) Histochem Cell Biol 122:427–434
Hocking AM, Gibran NS (2010) Exp Cell Res 316:2213–2219
Hosokawa R, Nonaka K, Morifuji M, Shum L, Ohishi M (2003) J Dent Res 82:558–564
Hsu HH, Zdanowicz MM, Agarwal VR, Speiser PW (1997) Biochem Mol Med 60:142–148
Huang NF, Lee RJ, Li S (2010) Am J Transl Res 2:43–55
Huang YC, Dennis RG, Larkin L, Baar K (2005) J Appl Physiol 98:706–713
Huang YC, Dennis RG, Baar K (2006) Am J Physiol Cell Physiol 291:C11–C17
Huard J (2008) J Musculoskelet Neuronal Interact 8:337
Huard J, Li Y, Fu FH (2002a) J Bone Joint Surg Am 84A:822–832
Huard J, Yokoyama T, Pruchnic R, Qu Z, Li Y, Lee JY, Somogyi GT, Groat WC de, Chancellor MB (2002b) Gene Ther 9:1617–1626
Ikezawa M, Cao B, Qu Z, Peng H, Xiao X, Pruchnic R, Kimura S, Miike T, Huard J (2003) Hum Gene Ther 14:1535–1546
Izadpanah R, Trygg C, Patel B, Kriedt C, Dufour J, Gimble JM, Bunnell BA (2006) J Cell Biochem 99:1285–1297
Jackson WM, Aragon AB, Bulken-Hoover JD, Nesti LJ, Tuan RS (2009) J Orthop Res 27:1645–1651
Jackson WM, Lozito T, Djouad F, Kuhn NZ, Nesti LJ, Tuan RS (2010) Differentiation and regeneration potential of mesenchymal progenitor cells derived from traumatized muscle tissue. J Cell Mol Med (in press)
Jankowski RJ, Deasy BM, Huard J (2002) Gene Ther 9:642–647
Jarvinen TA, Kaariainen M, Jarvinen M, Kalimo H (2000) Curr Opin Rheumatol 12:155–161
Jarvinen TA, Jarvinen TL, Kaariainen M, Kalimo H, Jarvinen M (2005) Am J Sports Med 33:745–764
Jarvinen TA, Jarvinen TL, Kaariainen M, Aarimaa V, Vaittinen S, Kalimo H, Jarvinen M (2007) Best Pract Res Clin Rheumatol 21:317–331
Jensen LT, Host NB (1997) Cardiovasc Res 33:535–539
Jirmanova I, Thesleff S (1972) Z Zellforsch Mikrosk Anat 131:77–97
Kagiwada H, Yashiki T, Ohshima A, Tadokoro M, Nagaya N, Ohgushi H (2008) J Tissue Eng Regen Med 2:184–189
Kallestad KM, McLoon LK (2010) J Cell Physiol 222:676–684
Kania G, Blyszczuk P, Stein S, Valaperti A, Germano D, Dirnhofer S, Hunziker L, Matter CM, Eriksson U (2009) Circ Res 105:462–470
Kasemkijwattana C, Menetrey J, Somogyl G, Moreland MS, Fu FH, Buranapanitkit B, Watkins SC, Huard J (1998) Cell Transplant 7:585–598
Kim YT, Kim DK, Jankowski RJ, Pruchnic R, Usiene I, de Miguel F, Chancellor MB (2007) Muscle Nerve 36:391–393
Kivela R, Silvennoinen M, Lehti M, Jalava S, Vihko V, Kainulainen H (2008) Cardiovasc Diabetol 7:13
Ko R, Kazacos EA, Snyder S, Ernst DM, Lantz GC (2006) J Surg Res 135:9–17
Kochupura PV, Azeloglu EU, Kelly DJ, Doronin SV, Badylak SF, Krukenkamp IB, Cohen IS, Gaudette GR (2005) Circulation 112:I144–I149
Kollias HD, McDermott JC (2008) J Appl Physiol 104:579–587
Koning M, Harmsen MC, van Luyn MJ, Werker PM (2009) J Tissue Eng Regen Med 3:407–415
Kresse H, Schonherr E (2001) J Cell Physiol 189:266–274
Kubo H, Jaleel N, Kumarapeli A, Berretta RM, Bratinov G, Shan X, Wang H, Houser SR, Margulies KB (2008) Circulation 118:649–657
LaBarge MA, Blau HM (2002) Cell 111:589–601
Ladak A, Olson J, Tredget EE, Gordon T (2011) Differentiation of mesenchymal stem cells to support peripheral nerve regeneration in a rat model. Exp Neurol 228:242–252
Laprise P, Poirier EM, Vezina A, Rivard N, Vachon PH (2002) J Cell Physiol 191:69–81
Larkin LM, Van der Meulen JH, Dennis RG, Kennedy JB (2006) In Vitro Cell Dev Biol Anim 42:75–82
Launay T, Armand AS, Charbonnier F, Mira JC, Donsez E, Gallien CL, Chanoine C (2001) J Histochem Cytochem 49:887–899
Lawrence DA (2001) Mol Cell Biochem 219:163–170
Lecourt S, Marolleau JP, Fromigue O, Vauchez K, Andriamanalijaona R, Ternaux B, Lacassagne MN, Robert I, Boumediene K, Chereau F et al (2010) Exp Cell Res 316:2513–2526
Lefeuvre B, Crossin F, Fontaine-Perus J, Bandman E, Gardahaut MF (1996) Mech Dev 58:115–127
Lehto MU, Jarvinen MJ (1991) Ann Chir Gynaecol 80:102–108
Levenberg S, Rouwkema J, Macdonald M, Garfein ES, Kohane DS, Darland DC, Marini R, van Blitterswijk CA, Mulligan RC, D'Amore PA et al (2005) Nat Biotechnol 23:879–884
Li J, Reed SA, Johnson SE (2009) Exp Cell Res 315:2284–2292
Li Q, Bai Y, Xu Y, Yu H (2010) Foot Ankle Int 31:706–711
Liang R, Woo SL, Takakura Y, Moon DK, Jia F, Abramowitch SD (2006) J Orthop Res 24:811–819
Liao H, Zhou GQ (2009) Tissue Eng B Rev 15:319–331
Lin CS, Xin ZC, Deng CH, Ning H, Lin G, Lue TF (2010) Histol Histopathol 25:807–815
Liu D, Black BL, Derynck R (2001) Genes Dev 15:2950–2966
Ljubicic V, Adhihetty PJ, Hood DA (2005) Can J Appl Physiol 30:625–643
Lolmede K, Campana L, Vezzoli M, Bosurgi L, Tonlorenzi R, Clementi E, Bianchi ME, Cossu G, Manfredi AA, Brunelli S et al (2009) J Leukoc Biol 85:779–787
Mackey AL, Kjaer M, Dandanell S, Mikkelsen KH, Holm L, Dossing S, Kadi F, Koskinen SO, Jensen CH, Schroder HD et al (2007) J Appl Physiol 103:425–431
Malerba A, Vitiello L, Segat D, Dazzo E, Frigo M, Scambi I, De Coppi P, Boldrin L, Martelli L, Pasut A et al (2009) Exp Cell Res 315:915–927
Malerba A, Pasut A, Frigo M, De Coppi P, Baroni MD, Vitiello L (2010) Neurol Res 32:55–62
Mars T, Martincic DS (2001) Pflugers Arch 442:R177–R178
Marzaro M, Conconi MT, Perin L, Giuliani S, Gamba P, De Coppi P, Perrino GP, Parnigotto PP, Nussdorfer GG (2002) Int J Mol Med 10:177–182
Mase VJ Jr, Hsu JR, Wolf SE, Wenke JC, Baer DG, Owens J, Badylak SF, Walters TJ (2010) Orthopedics 33:511
Massague J, Cheifetz S, Endo T, Nadal-Ginard B (1986) Proc Natl Acad Sci USA 83:8206–8210
Matsumoto T, Sasaki J, Alsberg E, Egusa H, Yatani H, Sohmura T (2007) PLoS ONE 2:e1211
Matziolis G, Winkler T, Schaser K, Wiemann M, Krocker D, Tuischer J, Perka C, Duda GN (2006) Tissue Eng 12:361–367
McLoon LK, Thorstenson KM, Solomon A, Lewis MP (2007) Oral Dis 13:134–140
Mehrke G, Jockusch H, Faissner A, Schachner M (1984) Neurosci Lett 44:235–239
Mendler L, Pinter S, Kiricsi M, Baka Z, Dux L (2008) J Histochem Cytochem 56:111–123
Menetrey J, Kasemkijwattana C, Fu FH, Moreland MS, Huard J (1999) Am J Sports Med 27:222–229
Menetrey J, Kasemkijwattana C, Day CS, Bosch P, Vogt M, Fu FH, Moreland MS, Huard J (2000) J Bone Joint Surg Br 82:131–137
Merritt EK, Cannon MV, Hammers DW, Le LN, Gokhale R, Sarathy A, Song TJ, Tierney MT, Suggs LJ, Walters TJ et al (2010) Tissue Eng A 16:2871–2881
Messina S, Mazzeo A, Bitto A, Aguennouz M, Migliorato A, De Pasquale MG, Minutoli L, Altavilla D, Zentilin L, Giacca M et al (2007) FASEB J 21:3737–3746
Miller RR, Rao JS, Burton WV, Festoff BW (1991) Int J Dev Neurosci 9:259–267
Miller KJ, Thaloor D, Matteson S, Pavlath GK (2000) Am J Physiol Cell Physiol 278:C174–C181
Mishra DK, Friden J, Schmitz MC, Lieber RL (1995) J Bone Joint Surg Am 77:1510–1519
Miura T, Kishioka Y, Wakamatsu J, Hattori A, Hennebry A, Berry CJ, Sharma M, Kambadur R, Nishimura T (2006) Biochem Biophys Res Commun 340:675–680
Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, Takeda S (1997) FEBS Lett 415:33–39
Mong FS (1988) Experientia 44:601–603
Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M (2005) Science 309:2064–2067
Moon G du, Christ G, Stitzel JD, Atala A, Yoo JJ (2008) Tissue Eng A 14:473–482
Muguruma Y, Reyes M, Nakamura Y, Sato T, Matsuzawa H, Miyatake H, Akatsuka A, Itoh J, Yahata T, Ando K et al (2003) Exp Hematol 31:1323–1330
Nagasaka M, Kohzuki M, Fujii T, Kanno S, Kawamura T, Onodera H, Itoyama Y, Ichie M, Sato Y (2006) Clin Exp Pharmacol Physiol 33:623–627
Natsu K, Ochi M, Mochizuki Y, Hachisuka H, Yanada S, Yasunaga Y (2004) Tissue Eng 10:1093–1112
Navarro-Sobrino M, Rosell A, Hernandez-Guillamon M, Penalba A, Ribo M, Alvarez-Sabin J, Montaner J (2010) Microvasc Res 80:317–323
Negroni E, Riederer I, Chaouch S, Belicchi M, Razini P, Di Santo J, Torrente Y, Butler-Browne GS, Mouly V (2009) Mol Ther 17:1771–1778
Nesti LJ, Jackson WM, Shanti RM, Koehler SM, Aragon AB, Bailey JR, Sracic MK, Freedman BA, Giuliani JR, Tuan RS (2008) J Bone Joint Surg Am 90:2390–2398
Nicolaidis SC, Williams HB (2001) Microsurgery 21:241–247
Nieponice A, Gilbert TW, Badylak SF (2006) Ann Thorac Surg 82:2050–2058
Nieponice A, McGrath K, Qureshi I, Beckman EJ, Luketich JD, Gilbert TW, Badylak SF (2008) Gastrointest Endosc 69:289–296
Occleston NL, Fairlamb D, Hutchison J, O'Kane S, Ferguson MW (2009) Expert Opin Investig Drugs 18:1231–1239
O'Kane S, Ferguson MW (1997) Int J Biochem Cell Biol 29:63–78
Owen SC, Shoichet MS (2010) J Biomed Mater Res A 94:1321–1331
Padoin AV, Braga-Silva J, Martins P, Rezende K, Rezende AR, Grechi B, Gehlen D, Machado DC (2008) Plast Reconstr Surg 122:614–618
Palermo AT, Labarge MA, Doyonnas R, Pomerantz J, Blau HM (2005) Dev Biol 279:336–344
Park H, Bhalla R, Saigal R, Radisic M, Watson N, Langer R, Vunjak-Novakovic G (2008) J Tissue Eng Regen Med 2:279–287
Payne TR, Oshima H, Sakai T, Ling Y, Gharaibeh B, Cummins J, Huard J (2005) Gene Ther 12:1264–1274
Peault B, Rudnicki M, Torrente Y, Cossu G, Tremblay JP, Partridge T, Gussoni E, Kunkel LM, Huard J (2007) Mol Ther 15:867–877
Pettersson P, Cigolini M, Sjostrom L, Smith U, Bjorntorp P (1984) Acta Med Scand 215:447–451
Philippou A, Maridaki M, Halapas A, Koutsilieris M (2007) In Vivo 21:45–54
Phinney DG (2007) Cell Cycle 6:2884–2889
Quintero AJ, Wright VJ, Fu FH, Huard J (2009) Clin Sports Med 28:1–11
Qu-Petersen Z, Deasy B, Jankowski R, Ikezawa M, Cummins J, Pruchnic R, Mytinger J, Cao B, Gates C, Wernig A et al (2002) J Cell Biol 157:851–864
Renault V, Piron-Hamelin G, Forestier C, DiDonna S, Decary S, Hentati F, Saillant G, Butler-Browne GS, Mouly V (2000) Exp Gerontol 35:711–719
Rieder E, Seebacher G, Kasimir MT, Eichmair E, Winter B, Dekan B, Wolner E, Simon P, Weigel G (2005) Circulation 111:2792–2797
Robinson KA, Li J, Mathison M, Redkar A, Cui J, Chronos NA, Matheny RG, Badylak SF (2005) Circulation 112:I135–I143
Rosu-Myles M, Stewart E, Trowbridge J, Ito CY, Zandstra P, Bhatia M (2005) J Cell Sci 118:4343–4352
Rouger K, Brault M, Daval N, Leroux I, Guigand L, Lesoeur J, Fernandez B, Cherel Y (2004) Cell Tissue Res 317:319–326
Rowlands AS, Hudson JE, Cooper-White JJ (2007) Expert Rev Med Devices 4:709–728
Sacks MS, Gloeckner DC (1999) J Biomed Mater Res 46:1–10
Saito T, Dennis JE, Lennon DP, Young RG, Caplan AI (1995) Tissue Eng 1:327–343
Sasaki A, Aizawa T, Tomiya A, Matsubara Y, Kokubun S, Itoi E (2007) Ups J Med Sci 112:175–181
Sato K, Li Y, Foster W, Fukushima K, Badlani N, Adachi N, Usas A, Fu FH, Huard J (2003) Muscle Nerve 28:365–372
Saxena AK, Willital GH, Vacanti JP (2001) Biomed Mater Eng 11:275–281
Schmalbruch H (1976) Tissue Cell 8:673–692
Schultz E, Albright DJ, Jaryszak DL, David TL (1988) Anat Rec 222:12–17
Schultz GS, Wysocki A (2009) Wound Repair Regen 17:153–162
Serena E, Flaibani M, Carnio S, Boldrin L, Vitiello L, De Coppi P, Elvassore N (2008) Neurol Res 30:207–214
Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Canoves P (2008) Cell Metab 7:33–44
Shah M, Revis D, Herrick S, Baillie R, Thorgeirson S, Ferguson M, Roberts A (1999) Am J Pathol 154:1115–1124
Sheehan SM, Allen RE (1999) J Cell Physiol 181:499–506
Sheehan SM, Tatsumi R, Temm-Grove CJ, Allen RE (2000) Muscle Nerve 23:239–245
Shen W, Li Y, Zhu J, Schwendener R, Huard J (2008) J Cell Physiol 214:405–412
Sherwood RI, Wagers AJ (2006) Trends Mol Med 12:189–192
Shibuya S, Wakayama Y, Inoue M, Kojima H, Oniki H (2003) Med Electron Microsc 36:213–220
Shmelkov SV, Meeus S, Moussazadeh N, Kermani P, Rashbaum WK, Rabbany SY, Hanson MA, Lane WJ, St Clair R, Walsh KA et al (2005) Circulation 111:1175–1183
Smaldone MC, Chancellor MB (2008) World J Urol 26:327–332
Smith CA, Stauber F, Waters C, Alway SE, Stauber WT (2007) J Appl Physiol 102:755–761
Snyder SJ, Arnoczky SP, Bond JL, Dopirak R (2009) Arthroscopy 25:329–333
Springer ML, Chen AS, Kraft PE, Bednarski M, Blau HM (1998) Mol Cell 2:549–558
St Pierre BA, Tidball JG (1994a) Am J Pathol 145:1463–1471
St Pierre BA, Tidball JG (1994b) J Appl Physiol 77:290–297
Strle K, McCusker RH, Tran L, King A, Johnson RW, Freund GG, Dantzer R, Kelley KW (2007) J Neuroimmunol 188:48–55
Sun D, Martinez CO, Ochoa O, Ruiz-Willhite L, Bonilla JR, Centonze VE, Waite LL, Michalek JE, McManus LM, Shireman PK (2009) FASEB J 23:382–395
Suzuki S, Yamanouchi K, Soeta C, Katakai Y, Harada R, Naito K, Tojo H (2002) Biochem Biophys Res Commun 292:709–714
Szalay K, Razga Z, Duda E (1997) Eur J Cell Biol 74:391–398
Tanaka Y, Tsutsumi A, Crowe DM, Tajima S, Morrison WA (2000) Br J Plast Surg 53:51–57
Tatsumi R (2010) Anim Sci J 81:11–20
Tatsumi R, Allen RE (2004) Muscle Nerve 30:654–658
Tatsumi R, Anderson JE, Nevoret CJ, Halevy O, Allen RE (1998) Dev Biol 194:114–128
Tatsumi R, Hattori A, Ikeuchi Y, Anderson JE, Allen RE (2002) Mol Biol Cell 13:2909–2918
Tedesco FS, Dellavalle A, Diaz-Manera J, Messina G, Cossu G (2010) J Clin Invest 120:11–19
Ten Broek RW, Grefte S, Von den Hoff JW (2010) J Cell Physiol 224:7–16
Terada N, Takayama S, Yamada H, Seki T (2001) Scand J Plast Reconstr Surg Hand Surg 35:233–238
Tidball JG (1995) Med Sci Sports Exerc 27:1022–1032
Tidball JG, Villalta SA (2010) Am J Physiol Regul Integr Comp Physiol 298:R1173–R1187
Tidball JG, Wehling-Henricks M (2007) J Physiol (Lond) 578:327–336
Tille JC, Pepper MS (2002) Exp Cell Res 280:179–191
Torrente Y, El Fahime E, Caron NJ, Del Bo R, Belicchi M, Pisati F, Tremblay JP, Bresolin N (2003) Cell Transplant 12:91–100
Torrente Y, Belicchi M, Sampaolesi M, Pisati F, Meregalli M, D'Antona G, Tonlorenzi R, Porretti L, Gavina M, Mamchaoui K et al (2004) J Clin Invest 114:182–195
Torrente Y, Belicchi M, Marchesi C, Dantona G, Cogiamanian F, Pisati F, Gavina M, Giordano R, Tonlorenzi R, Fagiolari G et al (2007) Cell Transplant 16:563–577
Tottey S, Corselli M, Jeffries EM, Londono R, Peault B, Badylak SF (2011) Tissue Eng A 17:37–44
Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, Tanaka K, Katsume A, Ohsugi Y, Shiozaki H et al (1996) J Clin Invest 97:244–249
Tu YK, Yen CY, Ma CH, Yu SW, Chou YC, Lee MS, Ueng SW (2008) Injury 39 (Suppl 4):75–95
Turner NJ, Yates AJ Jr, Weber DJ, Qureshi IR, Stolz DB, Gilbert TW, Badylak SF (2010) Tissue Eng A 16:3309–3317
Urbich C, Dimmeler S (2004) Circ Res 95:343–353
Usas A, Huard J (2007) Biomaterials 28:5401–5406
Vachon PH, Loechel F, Xu H, Wewer UM, Engvall E (1996) J Cell Biol 134:1483–1497
Valentin JE, Stewart-Akers AM, Gilbert TW, Badylak SF (2009) Tissue Eng A 15:1687–1694
Valentin JE, Turner NJ, Gilbert TW, Badylak SF (2010) Biomaterials 31:7475–7484
Vandenburgh HH, Karlisch P, Farr L (1988) In Vitro Cell Dev Biol 24:166–174
Velleman SG, McFarland DC (2004) Poult Sci 83:245–252
Vindigni V, Mazzoleni F, Rossini K, Fabbian M, Zanin ME, Bassetto F, Carraro U (2004) Reconstruction of ablated rat rectus abdominis by muscle regeneration. Plast Reconstr Surg 114:1509–1518
Vitello L, Radu C, Malerba A, Segat D, Cantini M, Carraro U, Baroni MD (2004) Basic Appl Myol 14:45–51
Voo S, Eggermann J, Dunaeva M, Ramakers-van Oosterhoud C, Waltenberger J (2008) Eur Heart J 29:241–250
Vorotnikova E, McIntosh D, Dewilde A, Zhang J, Reing JE, Zhang L, Cordero K, Bedelbaeva K, Gourevitch D, Heber-Katz E et al (2010) Matrix Biol 29:690–700
Wagner S, Dorchies OM, Stoeckel H, Warter JM, Poindron P, Takeda K (2003) Pflugers Arch 447:14–22
Wang X, Wu H, Zhang Z, Liu S, Yang J, Chen X, Fan M (2008) Cell Mol Neurobiol 28:113–124
Warren GL, Hulderman T, Jensen N, McKinstry M, Mishra M, Luster MI, Simeonova PP (2002) FASEB J 16:1630–1632
Washabaugh CH, Ontell MP, Shan Z, Hoffman EP, Ontell M (1998) Dev Dyn 211:177–190
Wehling M, Spencer MJ, Tidball JG (2001) J Cell Biol 155:123–131
Whalen RG, Harris JB, Butler-Browne GS, Sesodia S (1990) Dev Biol 141:24–40
Wilborn CD, Taylor LW, Greenwood M, Kreider RB, Willoughby DS (2009) J Strength Cond Res 23:2179–2187
Wipff PJ, Hinz B (2008) Eur J Cell Biol 87:601–615
Wu L, Siddiqui A, Morris DE, Cox DA, Roth SI, Mustoe TA (1997) Arch Surg 132:753–760
Wu X, Wang S, Chen B, An X (2010) Cell Tissue Res 340:549–567
Xu R, Boudreau A, Bissell MJ (2009) Cancer Metastasis Rev 28:167–176
Yablonka-Reuveni Z, Rivera AJ (1994) Dev Biol 164:588–603
Yokoyama T, Huard J, Pruchnic R, Yoshimura N, Qu Z, Cao B, Groat WC de, Kumon H, Chancellor MB (2001) Urology 57:826–831
Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, Benhaim P, Lorenz HP, Hedrick MH (2001) Tissue Eng 7:211–228
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Turner, N.J., Badylak, S.F. Regeneration of skeletal muscle. Cell Tissue Res 347, 759–774 (2012). https://doi.org/10.1007/s00441-011-1185-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-011-1185-7