
Abstract
Melatonin is involved in many physiological functions and it plays an important role in many pathological processes as well. Melatonin has been shown to reduce the incidence of experimentally induced cancers and can significantly inhibit the growth of some human tumors, namely hormone-dependent cancers. The anticancer effects of melatonin have been observed in breast cancer, both in in vivo with models of chemically induced rat mammary tumors, and in vitro studies on human breast cancer cell lines. Melatonin acts at different physiological levels and its antitumoral properties are supported by a set of complex, different mechanisms of action, involving apoptosis activation, inhibition of proliferation, and cell differentiation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the last two decades, a compelling body of evidence has outlined the relevance of melatonin to human physiology and pathology. Melatonin displays important roles in several biologic functions, among which are circadian rhythms, sleep, mood, reproductive physiology, and diseases of aging [1–8]. Additionally, in the event of elevated oxidative stress, melatonin functions as a highly efficient antioxidant [9–12]. Numerous studies, based on animal as well as on clinical data, have provided evidence that melatonin reduces the incidence of experimentally induced cancers [13–15] and may significantly inhibit the growth of some human tumors [16–18]. The general conclusion is that melatonin inhibits cell proliferation and induces apoptosis in tumors (especially hormone-sensitive cancers), and decreases the incidence and the proliferation rate of chemically induced murine neoplasias. Interestingly, there is no consensus regarding the major mechanisms by which melatonin reduces tumor growth although numerous well-supported explanations have been proposed [19–24].
The first suggestion concerning a potential relationship between the pineal gland and cancer was made more than 80 years ago [25]. Soon after the discovery of melatonin, its role in the control of neoplastic growth quickly was often investigated [26, 27]. In 1978, the seminal paper by Cohen et al. [28] first proposed that the pineal gland and its main secretory product are likely to play an important role in the pathogenesis of breast cancer. These authors suggested that a reduction in pineal function, whatever its cause, and the consequent loss in melatonin secretion, may induce a state of relative hyperestrogenism, thus leading to a prolonged exposure of breast tissue to estrogens and eventually ending in cancer induction. Studies performed on the field in recent decades have highlighted that the anticancer effects exerted by melatonin on breast tumors are complex and can be recognized at different physiological levels (from the molecular to the endocrine level) [21].
Breast cancer is one of the most frequently occurring cancers, and one of the leading causes of death among women aged 40–55 years [29]. Many factors, such as genetics, hormonal environment, age, diet, alcohol consumption, and cigarette smoking, have been hypothesized as contributors to the development of breast cancer [30–32]. However, a major consequence of a modern lifestyle is the disruption of circadian rhythms, a condition that leads to several pathological conditions, including sleep disturbances and depression [20, 33]. The alternation of the day and night circadian cycle is indeed a very important regulator of a wide variety of physiological biorhythms in organisms, including humans. In particular, accumulating evidence shows that alteration of circadian rhythms might lead to increased susceptibility to cancer in humans. Epidemiological studies have revealed the risk for breast cancer to be significantly higher in industrialized societies, and that the risk increases in women who work night shifts, and in individuals who spend more hours working at night [34]. Recently, experimental data has provided compelling evidence in support of such a hypothesis [35].
Growing in parallel with industrialization, the use of artificial light prolonged the “day,” permitting employers to extend their work schedules well into the night and in many cases throughout the 24-h period. Because of this temporal coincidence, light at night suppresses the synthesis and release of melatonin; this drop has been incriminated as a plausible contributor to the elevated cancer risk [26, 36]. What this means is that humans in modern societies are rendering themselves progressively more melatonin-deficient by shortening their daily dark period, which also reduces the total amount of melatonin produced [24]. Kerenyi et al. [37] were early advocates of the idea that “light pollution” might be a potentially important etiologic influence on the genesis of other human cancers. Epidemiologic studies [38, 39] have shown that women working night shifts have a significantly elevated risk of breast cancer, which is likely related to circadian disruption, sleep deprivation, and melatonin suppression [33]. In 1981, Bartsch et al. [40] published an early study demonstrating that plasma concentrations are diminished in patients with breast cancer. Since then, other reports have confirmed that patients with established breast cancer have measurably lower levels of melatonin [41, 42]. Overall, these studies highlight how the nocturnal melatonin rhythm may represent a critically important chronobiotic signal, which not only directly inhibits human breast cancer signal transduction, but temporally organizes cancer metabolism and growth to help maintain the host–cancer balance. Disruption of tumor circadian organization shifts the host–cancer balance in favor of constantly up-regulated tumor metabolism and fuels “runaway” cancer cell proliferation and survival.
Melatonin’s effects on breast cancer: animal studies and clinical trials
Insights into the relationship between melatonin and breast cancer have been provided by studies performed on chemically induced mammary cancer in animals. Reducing circulating melatonin levels in rats (through pinealectomy, or by exposure to different photoperiods), generally leads to increased spontaneous tumor induction or enhanced growth of implanted cancers [43]. Conversely, restoring melatonin levels generally prevents or restrains the development of breast cancer [44, 45].
Melatonin significantly reduces the incidence and tumor size of rat mammary cancers induced by 7,12-dimethylbenz[a]anthracene (DMBA) or N-nitrosomethylurea (NMU) [43, 46]. In DMBA-exposed rats, long-term daily administration of melatonin inhibited tumorigenesis, whereas pinealectomy increased the incidence of breast tumors [47]. Similar results have been reported by several authors [48]. Moreover, constant light (known to suppress melatonin release) reduces the latency and increases the number of DMBA-induced mammary tumors in rats; it also increases the incidence of different spontaneous cancers in female CBA mice [49]. Moreover, in NMU-treated rats, melatonin’s cytostatic effects are similar to those exerted by tamoxifen, i.e., melatonin increases tumor latency, reduces cancer incidence (% of animals developing tumors) and also reduces the number and size of tumors [46]. Furthermore, melatonin retards the rate of tumor-growth and enhances spontaneous tumor regression [50, 51]. Collectively, these data show that melatonin inhibits both cancer initiation and progression, through several mechanisms, including estrogen-pathway modulation, receptor-mediated and receptor-independent effects on different enzymatic processes, as well as anti-oxidant effects [21]. It is well documented that reactive oxygen species (ROS) participate in a variety of processes regulating cell growth, gene transcription, differentiation, and apoptosis [52]. In cancer cells, free radicals and ROS can act as tumor promoters, leading to cancer initiation or to the growth enhancement of already-transformed cells. Therefore, free radical scavengers and antioxidant molecules like melatonin can display a significant role in preventing cancer and/or in hindering its progression [53, 54].
Some results obtained from research carried out on animals have been confirmed in studies performed in humans. In addition to some preliminary anecdotic reports [55, 56], limited clinical trials carried out by Lissoni and colleagues [57–63] have provided evidence of benefits in treating cancer-bearing patients with melatonin. In a pilot study, 14 women with metastatic breast cancer who had no clinical response to tamoxifen alone were given 20 mg of tamoxifen plus 20 mg of melatonin in the evening. A partial response, defined as radiographic-confirmed reduction of lesions by greater than 50 %, was observed in four out of 14 patients (28.5 %) with a median duration of 8 months [57]. In two randomized clinical trials [58, 59] (including several types cancer, including breast tumors), melatonin had beneficial effects among patients with metastatic cancer.
Panzer and Viljoen [64] have reviewed clinical studies with melatonin on patients with different types of cancer, providing evidence about melatonin therapy benefits, and showing that the indoleamine, may: (a) increase survival in a few metastatic patients; (b) retard cancer progression; (c) improve quality of life and performance status; (d) decrease the incidence and severity of some side-effects linked to conventional treatments (hypotension, thrombocytopenia, myelodysplastic syndrome, lymphocytopenia) [61–63, 65]. In the evaluated reports, several types of advanced cancer patients are covered and only a few cases of mammary tumors; thus, little information on the potential efficacy of melatonin treatment in breast cancer patients is currently available. Additionally, these findings require verification by independent and controlled replication studies to overcome statistical bias and methodological deficiencies due to the limited number of patients under study [66].
Melatonin’s effects on breast cancer: in vitro studies
Inhibition of breast cancer cell growth
The anticancer effects attributed to melatonin have often been observed in in vitro studies carried out on estrogen-responsive human breast cancer cell lines. The first such experiments using human MCF-7 cells demonstrated that melatonin, even at physiological concentrations, directly suppresses cancer cell growth [67]. Melatonin appears to exert an inhibitory effect by causing an accumulation of cells in the G0/G1 phase of the cell cycle [68] or, otherwise, by delaying the progression of MCF-7 cells from the G1 phase to the S phase of the cell cycle [69, 70] allowing the cells to achieve a greater differentiation. A similar pattern was observed for other estrogen-sensitive cancer cell lines (T47D and ZR75-1) [71–73]. Growth-inhibition is accompanied by a significant reduction in DNA content and thymidine incorporation [74, 75]. These effects seem to be related to both cancer cell characteristics and culture conditions.
Melatonin receptors and estrogen receptors
Melatonin significantly inhibits cell growth in breast cancer cells expressing estrogen receptors (ERα) [76, 77]. Melatonin does not inhibit the proliferation of MDA-MB-231, MDA-MB-330, or BT-20 ERα-negative human breast tumor cells lines. However, the indoleamine has been demonstrated to reduce growth proliferation on ERα-negative breast cancer and progesterone receptor–negative human breast tumor xenografts growing in nude rats [78]. Moreover, a significant oncostatic action has been observed in ER-negative, non-breast tumors treated with melatonin [79]. These data suggest that some non-estrogen receptor-mediated effects are likely to be elicited by the complex interplay between melatonin and cellular molecules not related to estrogen receptors.
Certainly, some of the effects of the indoleamine are mediated by the interaction with a specific membrane-bound melatonin receptor. Numerous reports have demonstrated that melatonin binds and activates the G protein-coupled membrane receptors 1 (MT1) and 2 (MT2) in a variety of tissues [80, 81].
The oncostatic effects of melatonin on ER-positive breast cancer cells seem to be, at least in a major part, dependent on the presence of the MT1, which has been found in human breast cancer tissues [82]. The MT1 receptor is differentially expressed in ERα-positive and ERα-negative breast cancer cells, with the higher MT1 levels found in the former cell lines [83]. The MT1 receptor couples with different Gαi proteins in multiple cell types, while also coupling with the Gq and G11 proteins in other cell types [84]. Selective MT1 antagonists (i.e., luzindole) suppress melatonin-induced anticancer effects [84, 85] while, overexpression of MT1 receptor in MCF-7 cells significantly enhances the response of these cells to the growth-inhibitory actions of melatonin, both in vitro and in vivo [86, 87]. Similar results have been observed when treating MCF-7 cells with valproic acid, a MT1 receptor inducer [88]. The sensitivity of different MCF-7 strains to melatonin is strongly dependent on MT1 expression [89]. Significantly diminished night-time and early morning levels of MT1 receptors were observed in uteri from old rats compared to adult and young animals; in association with this reduction, the growth-suppressive action of exogenous melatonin was found to be diminished in old rats [90]. The MT2 receptor seems not to be involved in oncostatic effects triggered by melatonin, in that MT2 activation is incapable of mediating the antiproliferative effects of melatonin on breast tumors [91]. Recent findings also demonstrated that the MT1 receptor co-localizes with the Cav-1 antibody, indicating the MT1 receptor resides in the caveola, a key membrane-signaling platform [92].
MT1 and MT2 receptors are G-protein-coupled receptors, which are expressed in various parts of the central nervous system and in numerous peripheral organs (blood vessels, mammary gland, gastrointestinal tract, liver, kidney and bladder, ovary, testis, prostate, skin, and the immune system) [93]. Indeed, melatonin’s receptor may exist in every cell in the body. Melatonin receptors mediate a plethora of intracellular events depending on the cellular milieu. These effects include changes in intracellular cyclic nucleotides (cAMP, cGMP) and calcium levels, activation of certain protein kinase C subtypes, intracellular localization of steroid hormone receptors, and regulation of G protein signaling proteins [81, 94].
MT1 expression is regulated by both melatonin and estradiol, as first documented in experiments performed on cells of the pars tuberalis [95]. The steady-state level of MT1 mRNA is significantly enhanced in MCF-7 cells cultured in estradiol-depleted medium. In cancer cells cultured in the presence of fetal bovine serum (FBS), the MT1 receptor steady-state mRNA level is suppressed by the addition of estradiol (1 nM) or significantly diminished by the addition of melatonin, confirming the ability of melatonin to down-regulate the levels of its own receptor, at least at the steady-state mRNA levels [91, 96]. The ability of estradiol to down-regulate MT1 receptors could explain some contradictory results, i.e., the lack of melatonin inhibition on estradiol-induced proliferation of breast cancer cells [97].
While removal of estradiol from the culture media up-regulates MT1 levels, several reports were unable to demonstrate an enhanced growth-inhibitory response to melatonin in MCF-7 cells growing in estradiol-deficient media, as the overall growth of those cells is generally slowed in the absence of estradiol [98]. These results imply that a number of other hormones, cytokines, or growth factor-related signaling pathways modulate MT1 expression, and the hormonal milieu of the tumor at the time of melatonin administration may dramatically impact the responsiveness of the tumor to the anti-proliferative action of melatonin. These actions are generally recognized as hormone-like effects. However, melatonin does not always act in this manner, and several melatonin-induced actions are carried out without the intervention of a receptor [98–100]. Melatonin should be rather considered as a tissue factor, behaving like a paracoid, an autocoid, an antioxidant, or a pro-oxidant factor depending on the physiological context [101].
The oncostatic effects triggered by melatonin are strictly context-dependent. Reducing the FBS concentration abrogates the responsiveness of MCF-7 cells to melatonin, until cells are totally refractory in serum-free medium [102]; on the contrary, melatonin-induced inhibition is enhanced in both human [76] and animal cancer cells [72] cultured in stripped serum supplemented with estradiol. Moreover, differences in MCF-7 cell strains and especially differences in their proliferation rate may account for the different sensitivity to the inhibitory effects induced by melatonin [103]. The effect of melatonin seems specific since melatonin precursors, metabolites, or other pineal methoxyindoles, have not been shown to inhibit breast cancer cell proliferation. Melatonin’s inhibitory activity is dependent on the pattern (continuous or pulsated) of the exposure to the indole hormone in the culture media; the highest antiproliferative effects are obtained when the concentration of melatonin in culture media is changed every 12 h with concentrations ranging from 10−11 to 10−9 M, thus mimicking the physiological day/night oscillation of melatonin in the plasma of most mammals [104].
Culture conditions exert a relevant modulation on cell sensitivity to melatonin. In cells growing in anchorage-dependent monolayer culture with FBS, melatonin inhibits MCF-7 cells according to a bell-shaped curve, showing that the highest cytostatic effect is generally obtained around the physiological range (10−11–10−9 M). Higher or lower concentrations produce little or no inhibition [68]. Growth-inhibition becomes evident after 48–72 h and thereafter increases linearly up to 144 h [70]. However, in an anchorage-independent culture system, the dose–response curve loses its characteristic form and becomes quite linear with increasing melatonin concentrations producing progressively greater inhibition [105]. This result highlights that cellular attachment to a substratum—that is likely to modify both cytoskeleton and cell shape—plays an important role in setting the level of cell sensitivity to melatonin.
Signaling pathways involved: estrogen pathways
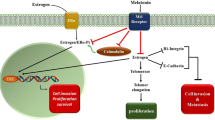
Melatonin influences estrogenic actions on mammary tissue in three different ways: (1) by down-regulating gonadal synthesis of steroids and, consequently, decreasing their circulating levels. Thus melatonin interferes with the systemic effects of estrogens; (2) by interacting with the estrogen-receptor (ER), thus behaving as an anti-estrogen; and (3) by down-regulating the activity of some enzymes, such as aromatase, involved in the synthesis of estrogens from androgens, i.e., behaving as a selective estrogen enzyme modulator [21, 106].
Systemic effects
Melatonin was initially shown to control seasonal reproduction in animals under natural photoperiods [107–109]. In seasonally breeding mammalian species, melatonin controls reproductive function through the activation of receptor sites within the hypothalamic-pituitary axis thus driving the levels of gonadal activity [109, 110]. As part of this action, melatonin down-regulates ovarian estrogen secretion in a variety of mammals. It was initially hypothesized that an impaired pineal secretion leads to reduced circulating melatonin levels, which results in unopposed estrogen secretion and thus to an elevated susceptibility to breast cancer [111]. In turn, normal or high serum melatonin levels, by suppressing of estrogen secretion, or by direct inhibitory effects on breast tissue, might restrain induction of mammary cancer (Fig. 1). Although in humans the role of melatonin on the reproductive physiology is not totally clear [112, 113], an inverse relationship between melatonin and ovarian activity [114] and a role of melatonin in the modulation of neuroendocrine-reproductive axis has been proposed [115, 116]. Indeed, melatonin exerts some modulatory actions on steroidogenesis in human granulosa-luteal cells [117]; moreover, functional melatonin receptors have been identified in cells of antral follicles and corpora lutea of rat ovaries [118]. Together, these data suggest that melatonin may participate in the modulation of ovarian function by down-regulating the production of estrogens, thereby supporting the above-mentioned hypothesis of its role in breast cancer.

Melatonin and estrogens have different actions on breast cancer cells. Estradiol stimulates cell proliferation, while reducing differentiation processes; conversely, melatonin promotes differentiation and reduces breast cancer cell proliferation
Melatonin–ER interactions
Only ERα-positive breast tumor cell lines are growth-inhibited by physiologic concentrations of melatonin, whereas ERα-negative cell lines are unaffected by the indoleamine [77, 119]. Various breast cancer cell lines have been reported to exhibit significant differences in their sensitivity to the antiproliferative action of melatonin. This may correlate with the degree of estrogen responsiveness [120] or the ERα/ERβ ratio [67, 121]; indeed, MCF-7 cell sensitivity to melatonin is abolished by ERβ overexpression.
Since melatonin’s inhibitory activity has been observed principally in estrogen-responsive breast cancer cells, it has been hypothesized that melatonin hinders cancer cell growth by antagonizing the intracellular estrogen-response pathway. Melatonin blocks the mitogenic effects of estradiol as well as counteracting the estradiol-induced invasiveness of MCF-7 cells [122]. Furthermore, the indoleamine augments the sensitivity of MCF-7 cells to anti-estrogens [123] and down-regulates the expression of proteins, growth factors, and proto-oncogenes regulated by estrogens [124]. Moreover, the transfection of MT1 melatonin receptors into MCF-7 cells or MDA-MB-231 cells (ERα negative) significantly enhances the growth-suppressive effects of melatonin exclusively in MCF-7 cells; thus, only cells also expressing an estrogen receptor are inhibited [87]. Indeed, melatonin significantly blunts estrogen-induced ERα transcriptional activity, while the addition of pertussis toxin (a known uncoupler of Gαi2 proteins) suppresses melatonin-induced inhibitory effects [125].
How melatonin interacts with the estrogen pathway remains an open question. Unlike anti-estrogenic drugs, evidence indicates that melatonin does not bind to the ER nor interfere with the binding of estradiol to its receptor [126]. Melatonin reduces the expression of ERα (both at the mRNA and protein level) and inhibits the binding of estradiol–receptor complex to the estrogens response element (ERE) on DNA [47, 127]. These effects likely depend on its binding to a high-affinity membrane-bound receptor coupled to Gi proteins [128] (Fig. 2). Via the activation of Gαi2 protein, melatonin limits the basal phosphorylation level of the ERα. Thus, melatonin behaves as an anti-estrogen, which does not bind to the ER, but to its own membrane receptors; via this binding to its specific receptors, melatonin interacts with the ER signaling pathway. This effect is specific for ERα-mediated effects. One of the desirable properties of a selective estrogen modulator is its ability to specifically block the ERα but not ERβ. Indeed, it was demonstrated [52] that whereas melatonin is a specific inhibitor of estrogen-induced ERα-mediated transcriptional activation, the indoleamine does not inhibit ERβ-mediated transactivation.

Melatonin interacts with the Ca++/calmodulin signaling pathway, either by modifying the intracellular accumulation of Ca++ or by means of a direct interaction with calmodulin. Calmodulin interacts with ER, stimulating the phosphorylation of the receptor and enhancing the binding of the estradiol–ER complex to ERE (estrogen response elements). Estrogens activate adenylate cyclase and increase cAMP; on the contrary, melatonin, after its binding to MT1, inhibits adenylate cyclase and reduces cAMP
Melatonin and nuclear receptors
The ligand-dependent nuclear transcription factors (NRs) play a multitude of essential roles in development, homeostasis, reproduction, and immune function [129]. NRs regulate transcription by several mechanisms and can both activate and inhibit gene expression [130]. The NRs include steroidal transcription factors such as the estrogen (ER), glucocorticoid (GR), thyroid hormone receptor (TR), liver X receptor (LXR), farnesoid X receptor (FXR), vitamin D receptor (VDR), retinoid acid receptor (RAR), retinoid X receptor (RXR), and peroxisome proliferators-activated receptors (PPARs) [131]. Through its role as a required heterodimeric partner, RXRs control the function of many other NRs, thus integrating a unique transcriptional network dependent on RXR responses [132, 133]. RXR forms heterodimers with virtually all NRs including GR, ER, TR, PPAR, VDR, LXR, and FXR. NRs can activate transcription as monomers and/or dimers with the RXR. Once activated, NRs dissociate from co-repressors and recruit co-activator proteins, which promote transcriptional activation [134, 135]. Half of the NRs are so-called “orphan” receptors because the identity of their ligand is unknown. However, some receptors belonging to RXRs are no longer “orphaned”. Evidence of a genomic action of melatonin via nuclear RZR/ROR receptors was initially hypothesized by Becker-Andre in 1994 [136]. Subsequent studies have detected the nuclear melatonin receptor by using in situ hybridization in neuronal tissue [137, 138] including the pineal gland [139] and in non-neuronal other tissues as well [140–142].
Direct evidence of the epigenetic effect of melatonin has been provided by Sharma et al. [143]. In this study, melatonin significantly elevated mRNA expression for various histone deacetylases (HDAC) isoforms and increased histone H3 acetylation in neural stem cell lines. As suggested by Korkmaz and colleagues [144, 145], these effects are indicative of an epigenetic regulation exerted by melatonin at NR/co-regulator level rather than selective enzymatic inhibition or activation. It is not still known if melatonin’s effects are mediated via direct changes in phosphorylation of the nuclear receptor, regulation of coactivator or corepressor phosphorylation, or both [146]. Regardless, these data clearly demonstrate the ability of melatonin, via signal transduction pathways, to influence gene expression in human breast cancer cells.
Melatonin as calmodulin and calcium modulator
Even if the molecular link between melatonin and estrogen pathways has not been fully elucidated, it is likely that cyclic AMP (cAMP) could, at least in part, modulate this function. Estrogens activate adenylate cyclase, thereafter increasing c-AMP levels; in turn, c-AMP synergizes with hormone receptors, enhancing ER-mediated transcription [147]. On the contrary, melatonin, after it binds to MT1, it inhibits adenylate cyclase, reduces c-AMP and in turn protein kinase A (PKA) activity leading to a diminished phosphorylation and activation of ERα and the co-activators CBP/p300, thus blocking the estrogenic effect [148, 149].
It has recently been proposed that melatonin may hinder the estrogen pathway through the Ca++/calmodulin signaling pathway, either modifying the intracellular accumulation of Ca++ or by means of a direct interaction with calmodulin (CaM) [150, 151]. Furthermore, melatonin changes CaM subcellular redistribution stimulating its phosphorylation by protein kinase C (PKCα) [152]. It is known that calmodulin interacts with ER, stimulating the phosphorylation of the receptor and thus enhancing the binding of the estradiol–ER complex to the ERE [153]. Conversely, anti-calmodulin compounds inhibit breast cancer growth, probably by interfering with the CaM-ER interplay [154]. Since both melatonin and calmodulin are phylogenetically well preserved, calmodulin–melatonin interaction probably represents a major mechanism for regulation and synchronization of cell physiology and it is likely that melatonin interference with calmodulin functions could contribute to modulate estrogen receptor activation. Moreover, the melatonin-induced rise in both intracellular Ca++ and membrane-bound calmodulin could enhance apoptosis and E-cadherin mediated cell–cell adhesion [155]. Consistent with this, Blask et al. [156] reported that melatonin inhibits Ca++-stimulated MCF-7 cell growth via a glutathione-dependent mechanism. Indeed, once glutathione synthesis is inhibited using buthionine sulfoximine (an inhibiter of γ-glutamylcysteine synthetase) the oncostatic action of 1 nM melatonin was blocked, indicating that glutathione is required for melatonin action [157]. In addition, glutathione depletion has been shown to cause a reduction in microtubule polymerization in cells that may relate to the oxidation of sulfhydryl groups [158]. In contrast, physiological concentrations of melatonin are known to stabilize microtubules by inhibiting Ca++/CaM depolymerization, which is itself a mitogenic signal transduction mechanism [159]. Thus, adequate levels of glutathione may be required to maintain the sulfhydryl groups of microtubule-associated proteins in a reduced state in order for melatonin to suppress Ca++/CaM-mediated depolymerization of the cytoskeleton and thus cell proliferation. These effects are seemingly to be ER-independent, keeping in mind that they have been recorded also in ER-negative breast cancer cells, as well as in non-breast cancer cell lines devoid of the ER; these data therefore could provide a reasonable hypothesis about how melatonin inhibits cell proliferation [44, 160, 161] (Fig. 2). Clearly, further studies are warranted in order to verify and better understand the physiological meaning of melatonin’s modulation of calmodulin and intracellular calcium in breast cancer cells.
Aromatase pathways
In MCF-7 breast cancer cells, as well as in adipose tissue of tumor-bearing breasts, expression of the CYP19 gene, which encodes aromatase P450, the enzyme responsible for estrogen biosynthesis, is regulated by two proximal promoters, i.e., I.3 and II [162], mainly modulated by intracellular cAMP [163]. Therefore, molecules or drugs able to modulate cAMP levels could also influence aromatase expression in breast cancer cells. This is the case with prostaglandin E2 (PGE2) that increases intracellular cAMP levels and stimulates aromatase and estrogen biosynthesis [164]. Estrogens also increase cAMP, as previously mentioned. Thus, in breast cancer cells, but not in normal epithelial cells with different CYP19 promoters, estrogens may induce, through a paracrine loop, the local biosynthesis of estrogens via the increase of cAMP and expression of aromatase. On the other hand, melatonin, after its binding to MT1 membrane receptor linked to Gi proteins, decreases in a dose- and time-dependent manner the activity of adenylate cyclase and subsequently reduces cAMP synthesis thus leading to elevated cGMP levels and a reduced aromatase concentration [165]. Indeed, it has been observed that melatonin, at both physiological (10−9 M) and pharmacological (10−4 M) concentrations, reduces the synthesis of estrogen in MCF-7 cells, through aromatase inhibition [166]. Furthermore, transfection of the MT1 melatonin receptor in MCF-7 cells significantly reduced aromatase activity and MT1-transfected cells showed a level of aromatase activity that was 50 % of that of the control cancer cells when both are treated with melatonin [167]. Moreover, melatonin enhances the inhibitory effect of aminoglutethimide on aromatase activity in breast cancer cells, through a significant reduction in aromatase mRNA expression [168]. Additionally in MCF-7 cells, aromatase activity is stimulated by epidermal growth factor and transforming growth factor-α [169], both of which are down-regulated by melatonin [170]; melatonin-dependent aromatase inhibition could be further achieved through suppression of cyclooxygenase activity and reduced prostaglandin E2 synthesis [171].
As breast cancer occurs in regions of the mammary gland with the highest levels of aromatase expression, the inhibition of aromatase activity by melatonin may be an important mechanism in the ability of this indoleamine to control tumor growth. Other studies confirmed that melatonin efficiently inhibits local, tissue-based biosynthesis of estrogen. It is well recognized that mammary cancer tissue contains all the enzymatic machinery for the local biosynthesis of estrogens [172]. The presence of the type 1 (17β-HSD1) isoform of 17β-hydroxysteroid dehydrogenases, which catalyze the conversion of the relatively weak estrone (E1), androstenedione and 5-androstenedione to the more potent estradiol, has been documented in several human breast cancer cell lines, including T47D and MCF-7 [172]. Estrogen production in normal mammary tissue is displaced toward the production of hormones with low activity (like estrone), whereas in breast adenocarcinomas what predominates is the formation of the active estradiol [173]. This effect is likely to be due to the different enzyme composition of normal and cancerous tissues. The former exhibits a higher activity of both 17β-HSD type 2, which converts estradiol to estrone, and estrogen sulfotransferase, which inactivates both estrone and estradiol; on the other hand, opposite effects are mediated by aromatase and type 2 isoform of 17β-hydroxysteroid dehydrogenase that are largely represented in cancer tissues. It has been demonstrated that melatonin reduces the synthesis of biologically active estrogens in MCF-7 cells, through the contemporary inhibition of sulfatases and 17β-HSD1 and the stimulation of estrogen sulfotransferase, the enzyme responsible for the formation of the biologically inactive estrogen sulphates. As a result, the production of estradiol from estrone in MCF-7 cells decreases two- to three-fold in the presence of melatonin 1 nM [174].
The transcription factor nuclear factor κB (NF-κB) is involved in CYP19 activation by inducing several pro-inflammatory molecules [tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), cyclooxygenase (COX-2), and PGE2] [175]. Over-expression of TNF-α, COX-2, and PGE2 has been demonstrated to induce elevated aromatase expression in both human and mice breast cancer tissue [72, 176]. It is noteworthy that melatonin inhibits every molecule in this pathway, mainly through its nuclear actions [177, 178]. Moreover, melatonin inhibits p300 HAT activity, thus leading to a reduced COX-2 and iNOS synthase expression [179]. This effect is likely to be mediated by inhibition of p52 acetylation and binding of DNA. As expected, melatonin also suppresses NF-κB binding to DNA [180–182], thereby decreasing TNF-α, iNOS, COX-2 and PGE2 levels. Furthermore, melatonin interaction with PPARs and RXR hinders NF-κB transcription, leading to cancer cell growth inhibition [183, 184]. In turn, melatonin-induced activation of both PPARs and RXR receptor inhibits aromatase transcription via NF-κB. Melatonin also inhibits the expression of other estrogen-regulated genes, like pS2 or cathepsin [185]. These findings are important, as pS2 has been demonstrated to be a differentiation factor in the gastrointestinal tract, as well as an inhibitor of adenocarcinoma cell proliferation [186, 187].
Growth-inhibitory mechanisms
Melatonin significantly limits cancer proliferation in vitro. For melatonin to achieve this effect, it seems several mechanisms are involved, including actions on the expression of some proteins involved in the control of the G1–S transition, through the inhibition of cyclin D1 expression and the increase in p53 release (Fig. 3). Cyclin D1 is a key protein of G1 to S transition and seems to mediate the steroid-dependent growth of both normal and malignant mammary epithelial cells [188]; moreover, down-regulation of cyclin D1 expression may be sufficient to drive the inhibitory effects displayed by anti-estrogenic drugs [189]. Cyclin D1 interacts with several transcription factors as well as with nuclear receptors (including GR, ERα, and PPARs). NRs directly bind to cyclin D1 and their ligand-dependent transactivation is modulated by cyclin D1 [190]. It is noteworthy that cyclin D1 participates in the activation process of ERα transcription and cooperates in the down-regulation of both GR and PPARs [191]. Melatonin induces a significant transcriptional down-regulation of the cyclin D1 gene through the inhibition of c-jun and ATF-2 proteins [192]. Both c-jun and ATF-2 proteins are known to transactivate the cAMP-responsive element present in the cyclin D1 promoter element [193].

Melatonin significantly inhibits cancer proliferation by increasing the p53/MDM2 and Akt/Akt-P ratios. The p53 gene activates the expression of p21, which inhibits cyclin-dependent kinases, thus leading to cell cycle arrest. In addition, melatonin induces apoptosis in MCF-7 cancer cells. Melatonin-mediated early apoptosis is a caspase-independent process, involving the apoptosis-inducing factor (AIF). Melatonin-induced late apoptosis is TGFβ-1 and caspase-dependent process. During late apoptosis, activated caspase-9, -7, and cleaved-PARP increase significantly, concomitant with a down-regulation of the Bcl/Bax ratio. By adding anti-TGFβ-1 neutralizing antibodies, growth inhibition and late apoptosis triggered by melatonin are inhibited
The p53 gene is involved in both growth suppression and apoptosis pathways [193]. The p53 gene activates the expression of the WAF1 gene (also known as p21), which inhibits cyclin-dependent kinases thus leading to a failure of the phosphorylation of the retinoblastoma protein and the subsequent cell cycle arrest [194].
In breast cancer cells treated with physiological doses of melatonin, both p53 and p21 expression is significantly augmented [195]. It is likely that up-regulation of p53 occurs downstream to enhanced release of TGFβ-1 induced by melatonin. Indeed, melatonin can up-regulate TGFβ-1 mRNA expression in prostate [196] and breast cancer cells [197]. Moreover, melatonin-inhibitory effect on breast cancer growth should be viewed as a TGFβ-1 dependent process as it can be completely prevented in several breast cancer cell lines by adding anti-TGFβ-1 antibodies [70, 74, 198]. A significant rise in TGFβ-1 levels in MCF-7 cells treated with melatonin are measured after 72 h and an evident growth-inhibitory action is documented only after this period. After adding anti-TGFβ-1 antibodies, the growth-inhibition induced by melatonin was completely prevented. These data point out that melatonin-induced cell-growth inhibition in MCF-7 breast cancer cells is largely mediated through the involvement of the TGFβ-1 pathway.
Other mechanisms are also involved in melatonin-mediated inhibition of cancer cells. The decrease in cAMP production caused by melatonin via MT1 and MT2 receptor interaction reportedly slows down the uptake of linoleic acid, an essential fatty acid, by specific fatty acid transporters [199]. Linoleic acid can be oxidized to 13-hydroxyoctadecadienoic acid by 15-lipoxygenase, serving as an energy source for tumor growth and tumor growth signaling molecules. Inhibition of linoleic acid uptake by melatonin is regarded as one mechanism of its antiproliferative effects.
Melatonin hinders telomerase activity, induced by estrogens or cadmium, both in vitro and in vivo [200, 201]. Melatonin-treated cells display a significant dose-dependent decrement in telomerase reverse transcriptase mRNA expression as well as the mRNA of telomerase-reverse, the RNA telomerase subunit. Similar results have been obtained with GP 52608—an agonist of melatonin nuclear receptors—while treatment with an agonist of melatonin membrane receptors did not produce any effect, thus highlighting the relevance of epigenetic mechanisms triggered by melatonin [202]. Telomerase is a specialized ribonucleoprotein DNA polymerase that extends telomeres of eukaryotic chromosomes and its activity is under control of epigenetic regulation [203]. Telomerase is activated in most human cancers and its inhibition leads to cancer cell death. Therefore, it is tempting to speculate that such a mechanism could also be involved in melatonin-dependent cancer apoptosis [204].
Apoptosis pathways
In contrast to the well-studied inhibition of apoptotic processes by melatonin in normal cells [205, 206], it is well known that in cancer cells, melatonin actually promotes apoptosis. Melatonin induces programmed cell death in colon cancer cells [207–209], hepatocarcinoma cells [201, 210], neuroblastoma [211], Ehrlich ascites carcinoma cells [212], myeloid [213], lymphoma [214], pancreatic cancer [215, 216], renal cancer [217], and leukemia cells [218–220]. However, although inhibitory effects of melatonin in MCF-7 have been well documented, the mechanism by which the apoptotic effects are executed are still a matter of investigation. Cos and coworkers [221] claimed no apoptotic effects on MCF-7 cells treated with different concentrations of melatonin, while other reports [222, 223] documented a significant rise in MCF-7 cells apoptotic rate when melatonin was administered together with retinoids. In the latter studies, melatonin was able to enhance apoptosis by modulating the transcriptional activity of the retinoic acid receptors [224]. A recent study documented a significant increase in caspase-3 activity and DNA fragmentation in tumor tissues obtained from breast cancer-bearing rats treated with melatonin, therefore providing at least an indirect proof of the apoptotic activity exerted by melatonin on breast cancer [225].
These apoptotic actions of melatonin have been confirmed in vitro, where treating MCF-7 cells with nanomolar concentrations of melatonin caused cell death [70]. Both flow cytometry and DNA fragmentation-based techniques documented an early (at 24 h) and a late (at 96 h) apoptosis in melatonin-treated MCF-7 cells. Early apoptosis is a caspase-independent process, and it is likely to be triggered by the apoptosis-inducing factor (AIF). In contrast, a more complex pathway underlies late apoptosis, involving both TGFβ-1 and terminal caspase effectors (caspases-7). Indeed, adding anti-TGFβ-1 antibodies, melatonin-induced late-apoptosis is almost completely suppressed, while early apoptosis remains unaffected. During late apoptosis, activated caspase-9 and -7 and cleaved-PARP increased significantly, concomitantly with a down-regulation of the Bcl-2/Bax ratio. It is noteworthy that melatonin-triggered apoptosis involves both p53 and p73 release. In fact, melatonin-treated MCF-7 cells showed a significant rise in both p73 and p53, but only the p73 protein, the homologue of p53 protein, increased at 96 h; concurrently, MDM2 levels were significantly reduced. These data suggest that p53 is likely activated during early programmed cell death, while only p73 is involved in caspase-dependent late-apoptosis.
It is worth reiterating that MDM2 is decreased as a consequence of melatonin treatment. MDM2 inhibits the transcriptional activity of p53 and promotes its degradation by the proteasome, thus representing the major physiological antagonist of p53 [226]. An autoregulatory negative feedback loop controls the MDM2 expression, where p53 induces MDM2 expression, whereas MDM2 represses p53 activity. Abrogation of the MDM2 expression allows the p53 to escape from the autoregulatory loop, becoming lethally active [227, 228]. Thus, melatonin induces an early down-regulation of MDM2 expression concomitant with a p53 rise, causing a significant rise in the p53/MDM2 ratio (Fig. 4). It is likely that the modified p53/MDM2 ratio could trigger the apoptotic cascade involving both caspase-dependent and caspase-independent pathways.

After binding to the MT1 receptor, melatonin reduces MDM2 levels, thus allowing p53 to escape from the autoregulatory loop. The modified p53/MDM2 ratio triggers the apoptosis cascade involving both caspases-dependent and caspases-independent pathways. In addition, melatonin increases mitochondrial membrane depolarization, releasing cytochrome C and apoptosis inducing factor (AIF). Melatonin is likely to inhibit AKT phosphorylation and subsequently the MAPK-related pathways
We have recently found (unpublished results) that melatonin enhances the depolarization of the mitochondrial membrane, while inhibiting Akt-phosphorylation; these effects are probably involved in melatonin-dependent oncostatic effects and they participate in triggering the complex apoptotic cascade. Similar results have been obtained by adding melatonin together with vitamin D3 [229]. Melatonin and vitamin D3 induced in MCF-7 cells a synergistic proliferative inhibition, with an almost complete cell growth arrest at 144 h. Cell growth blockade is associated to an activation of the TGFβ-1 pathway, leading to increased TGFβ-1, Smad4, and phosphorylated-Smad3 levels. Concomitantly, melatonin and D3, alone or in combination, caused a significant reduction in Akt phosphorylation and MDM2 values, with a consequent initial elevation of p53/MDM2 ratio [229]. These effects were completely suppressed by adding a monoclonal anti-TGFβ-1 antibody to the culture medium.
As Sainz and colleagues [205] reported, “melatonin involvement in apoptotic processes is a new and relevant field of investigation. The results obtained to date appear promising, and if in fact melatonin uniformly induces apoptosis in cancer cells, the findings could have important clinical utility. Many tumors show resistance to drug treatment mainly due to their resistance to undergo apoptosis. Identifying agents which potentiate apoptosis in cancer cells is clearly of great interest”. It may seem paradoxical that a substance could induce apoptosis in cancer cells, while preventing this process in normal cells. However, melatonin shares this unusual behavior with other known anti-oxidant compounds, including epigallocatechins and procyanidins [230]. Clearly melatonin’s ability to trigger apoptotic or anti-apoptotic pathways is largely context-dependent.
Malignant behavior
Some preliminary observations suggest that melatonin can efficiently reduce the metastatic ability of MCF-7 cells. Mao et al. [231] have evaluated the potential anti-invasive actions of melatonin, employing three clones of MCF-7 cells with high metastatic potential including the MCF-7/6 clone derived by serial passages in nude mice, MCF-7Her2.1 cells stably transformed with and overexpressing the Her2-neu/c-erbB2 construct, and MCF-7CXCR4 cells stably transformed with and overexpressing the CXCR4 cytokine G protein-coupled receptor. The invasive capacity of these clones was significantly reduced when they were treated with melatonin (10−8 or 10−9 M). Melatonin treatment resulted in marked suppression (60 to 85 % decrease) of cell invasion using a Transwell assay system and Matrigel-covered inserts.
In an in vitro study, Cos et al. [232] demonstrated that 1 nM melatonin reduced the invasiveness of cancer cells measured in Falcon invasion chambers and also blocked estradiol-induced invasion [123]; both sub-physiological (0.1 pmol) and pharmacological concentrations (10 μM) of melatonin failed to inhibit cell invasion. It is likely that such effects could be attributed to an overall effect of melatonin on cell morphology, which are mediated, at least in part, by a melatonin-induced elevation in the expression of two cell-surface adhesion proteins, E-cadherin and β1-integrin, as well as by an increased gap junctional intercellular communication between adjacent epithelial cells also induced by melatonin [232]. It has been hypothesized that if a cell would be unable to perform gap junctional intercellular communication, normal growth control and cell differentiation would not be possible, thereby favoring the development of malignant neoplasia [233]. Since there is an inverse correlation between the ability of a cell to exhibit gap junctional communication and its ability to metastasize [234], it is likely that melatonin may reduce metastatic behavior through inducing local gap junctional intercellular communication and reshaping the relationships between cancer cells and their microenvironment.
Estrogen treatment is known to induce a marked rearrangement of the cytoskeleton and adhesion structures and enhances the attachment of MCF-7 cells to laminin (a basement membrane component); this action is completely abolished by melatonin and shifts cancer cells to a lower invasive status [235]. These findings indicate that melatonin could exert an additional anti-tumor action by modulating the cross-talk between cells and stroma components (stromal cells, laminin, collagen), and by increasing regulatory signals shared between adjacent cells through intercellular junctions. Also, cancer cell invasiveness is enhanced by the elevated activities of matrix metallo-proteinases which break down interstitial tissue thereby aiding cancer cell movement and access to blood vessels. Since melatonin inhibits the activities of the proteinases that cause the dissolution of the connective tissue elements [236], the indole may also impede cancer cell invasiveness by this means.
Melatonin, cytoskeleton, and cell morphology
From the initial studies carried out by Hill et al. [67], it was already apparent that melatonin significantly modifies breast cancer cell morphology and cytoskeleton architecture. Likewise, melatonin also changes the histomorphology of prostate cancer cells and renders them more sensitive to cytokine-mediated apoptosis [237, 238]. The significance of these findings has been underestimated since only recently compelling evidence has been provided documenting the relevance of melatonin’s influence on the cytoskeleton.
The cytoskeleton is an important group of cellular structures, composed of an intricate fibrous network including microtubules, microfilaments, and intermediate filaments as well as their associated proteins [239]. Dynamic and differential changes in cytoskeletal organization occur during different cellular processes according to the cell type and the specific function. Moreover, the cytoskeleton, together with integrins and other related adhesion proteins, orients much of the metabolic and signal transduction machinery of the cell [240]. Indeed, cells are hard-wired to respond immediately to mechanical stresses transmitted over cell surface receptors that physically couple the cytoskeleton to the extracellular matrix or to other cells [241]. The shape of cells and the internal structure are consequences of physical forces generated in the cytoskeleton as well as in extracellular matrix. Shape, in reflecting cytoskeleton organization [242], is linked to a repertoire of metabolic events, which result from the right ordering in space of the enzymes catalyzing specific pathways. Physical forces (e.g., microgravity) induce dramatic changes in gene expression and alter cellular shape [243, 244]. In particular, distortions in cell shape can switch between distinct cell phenotypes, and this process is viewed as a biological phase transition [245, 246]. Moreover, tumor phenotype reversion is primarily associated with relevant shape modifications, preceding molecular and metabolic “normalization” [247].
The cytoskeleton is a phylogenetically well-preserved structure allowing the cell to have a well-organized structure, a specific shape-associated phenotype, and an optimal functionality. By contrast, cancer is characterized by an abnormal cytoskeletal rearrangement with poor organization and structure [248]. Therefore, the ability of melatonin to preserve normal microfilament distribution and its influence on cytoskeleton rearrangement and cell shape in cancerous tissues deserves further investigation [249].
Cancer cells show an abnormal microfilament organization, reduced stress fiber production, and loosened focal contact adhesion. These changes enhance cell proliferation, cell migration [250], and foster resistance to apoptosis [251]. Highly malignant metastatic cancer cells present poorly structured microfilaments and scarce anchorage to their substratum. These cells have microfilaments and microtubules arranged in membrane ruffles, lamellipodia, and filopodial formations at the leading edge; at the cell rear, a retraction of these cytoskeletal structures occurs [252].
A bimodal effect of melatonin on microtubule organization was first described in 1994 in both in vitro polymerization assays and in a preparation of cytoskeleton material in situ [253]. Melatonin, in the presence of Ca++, augmented tubulin polymerization, causing microtubule enlargement, while melatonin without Ca++ inhibited tubulin polymerization and caused microtubule disruption. Interestingly, in kidney epithelial cells (MDCK), melatonin’s effects on cytoskeletal organization are not mediated by membrane melatonin receptors, while in breast cancer cells, luzindole, an MT1 and MT2 antagonist, completely prevented melatonin-induced effects on microfilaments. Furthermore, studies performed by Benitez-King et al. [254] have demonstrated the complex interaction between melatonin and the cytoskeleton. In experimental conditions designed to measure cell anchorage, melatonin increases the number of focal adhesion contacts by MCF-7 cells, and microfilaments are arranged in thicker bundles of stress fibers assembled with phospho-vinculin to form focal adhesion contacts [255]. These results strongly suggest that melatonin inhibits cancer cell invasion and metastasis formation by changing microfilament phenotypes of migratory cells (ruffles and lamellipodia) to stress fibers that are microfilament phenotypes of attached cells.
Melatonin-induced effects on stress fibers involve protein kinase C (PKC) and the Rho-associated protein kinase (ROCK), downstream of the PKC pathway [256]. In MDCK and MCF-7 cells treated with melatonin, the addition of the PKC inhibitor abolished the augmented number and thickening of stress fibers, as well as the elevated number of focal adhesion contacts elicited by the indoleamine in both cell lines [257]. It is noteworthy that calmodulin also participates in this process [258] and that melatonin modulates stress fiber formation by involving ROCK and Ca++/CaM balance. In MCF-7 cells, melatonin causes a redistribution of CaM and phosphorylated-CaM, recruiting them to specific subcellular compartments, one of which is the cytoskeleton [259]. CaM is redistributed to the membrane cytoskeletal fraction where it becomes associated to myosin phosphorylation, through the myosin light-chain kinase [159].
Conclusions
Numerous studies have documented the oncostatic properties of melatonin both in vivo, with models of chemically induced rat mammary tumors, as well as in vitro using MCF-7 human breast cancer cells. Melatonin exerts both inhibitory as well pro-apoptotic effects, interacting with several molecular pathways. Generally, melatonin’s cytostatic actions seem to be mediated by the interaction of the indoleamine with both the estrogen receptors and the melatonin receptors. However, recently, some receptor-independent and estrogen-independent signaling pathways activated by melatonin have been uncovered. In particular, increasing attention should perhaps be directed to melatonin’s effects on the cytoskeleton and cell shape, as well as identifying how melatonin inhibits both Akt activation and MAPK-related pathways. A recent paper illustrates how melatonin exerts an inhibitory effect on breast cancer cell invasion through down-regulation of the p38 pathway and inhibition of MMP-2 and MMP-9 expression and activity [236]. The anticancer effects triggered by melatonin could be at least in part mediated by a selective genetic modulation, as it was shown that a set of microRNAs are differentially up- or down-regulated by melatonin in MCF-7 cells [260].
Additional research is required to clarify if melatonin administration constitutes, either alone or in combination with chemo-radiotherapy [261], a potentially new anticancer treatment. Given its widespread actions on breast cancer, its virtual absence of toxicity and its low cost, it seems reasonable to strongly recommend more thorough trials as to its usefulness as a preventive or treatment of breast cancer.
References
Hardeland R, Madrid JA, Tal DX et al (2012) Melatonin, the circadian multioscillator system and health: the need for detailed analyses of peripheral melatonin signaling. J Pineal Res 52:139–166
Cardinali DP, Srinivasan V, Brzezinski A et al (2012) Melatonin and its analogues in insomnia and depression. J Pineal Res 52:167–202
Reiter RJ, Tan DX, Manchester LC et al (2009) Melatonin and reproduction revisited. Biol Reprod 81:445–456
Rosales-Corral SA, Acuña-Castroviejo D, Coto-Montes A et al (2012) Alzheimer’s disease: pathological mechanisms and the beneficial role of melatonin. J Pineal Res 52:167–202
Tan DX, Manchester LC, Fuentes-Broto L et al (2011) Significance and application of melatonin in the regulation of brown adipose tissue metabolism: relation to human obesity. Obes Rev 12:167–188
Dominguez-Rodriguez A, Abreu-Gonzalez P, Reiter RJ (2012) Melatonin and cardiovascular disease: myth or reality? Rev Esp Cardiol 65:215–218
Korkmaz A, Ma S, Topal T et al (2012) Glucose: a vital toxin and potential utility of melatonin in protecting against the diabetic state. Mol Cell Endocrinol 349(2):128–137
Sanchez-Barcelo EJ, Mediavilla MD, Tan RJ et al (2010) Scientific basis for the potential use of melatonin in bone diseases: osteoporosis and adolescent idiopathic scoliosis. J Osteoporos ID 830231
Dx Tan, Chen LD, Poeggeler B et al (1993) Melatonin: a potent endogenous hydroxyl radical scavenger. Endocr J 1:57–60
Hardeland R, Poeggeler B (2008) Melatonin beyond its classical functions. Open Physiol J 1:1–23
Reiter RJ, Paredes SD, Manchester LC et al (2009) Reducing oxidative/nitrosative stress: a newly discovered genre for melatonin. Crit Rev Biochem Mol Biol 44:175–200
Galano A, Tan DX, Reiter RJ (2011) Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res 51:1–16
Sharman EH, Sharman KG, Bondy SC (2011) Extended exposure to dietary melatonin reduces tumor number and size in aged male mice. Exp Gerontol 46:18–22
Cutando A, Aneiros-Fernández J, Aneiros-Cachaza J, Arias-Santiago S (2011) Melatonin and cancer: current knowledge and its application to oral cavity tumours. J Oral Pathol Med 40:593–597
Sanchez-Barcelo EJ, Mediavilla MD, Alonso-Gonzalez C, Reiter RJ (2012) Melatonin uses in oncology: breast cancer prevention and reduction of the side effects of chemotherapy and radiation. Expert Opin Investig Drugs 21:819–831
Hill SM, Blask DE, Xiang S et al (2011) Melatonin and associated signaling pathways that control normal breast epithelium and breast cancer. J Mammary Gland Biol Neoplasia. 16:235–245
Lissoni P, Rovelli F (2012) Principles of psychoneuroendocrinoimmunotherapy of cancer. Immunotherapy 4:77–86
Zha L, Fan L, Sun G et al (2012) Melatonin sensitizes human hepatoma cells to endoplasmic reticulum stress-induced apoptosis. J Pineal Res 52:322–331
Blask DE (2009) Melatonin, sleep disturbance and cancer risk. Sleep Med Rev 13:252–264
Jung-Hynes B, Reiter RJ, Ahmad N (2010) Sirtuins, melatonin and circadian rhythms: building a bridge between aging and cancer. J Pineal Res 48:9–19
Mediavilla MD, Sanchez-Barcelo EJ, Tan DX (2010) Basic mechanisms involved in the anti-cancer effects of melatonin. Curr Med Chem 17:4462–4481
Greene MW (2012) Circadian rhythms and tumor growth. Cancer Lett 318:115–123
Blask DE, Dauchy RT, Sauer LA (2005) Putting cancer to sleep at night. Endocrine 27(2):179–188
Russel JR, Tan DX, Korkmaz A (2007) Light at night, chronodisruption, melatonin suppression, and cancer risk: a review. Crit Rev Oncog 13:303–328
Georgiou E (1929) Über die Natur und die Pathogenese der Krebstumoren, Radikale Heilung des Krebses bei weißen Mäusen. J Cancer Res Clin Oncol (Zeitschrift für Kregsforshung) 28(1):562–572
Lapin V, Ebels I (1976) Effects of some low molecular weight sheep pineal fractions and melatonin on different tumours in rats and mice. Oncology 33:110–113
Gupta D, Attanasio A, Reiter RJ (1988) The pineal gland and cancer brain research promotion. Tübingen, Germany, pp 1–383
Cohen M, Lippman M, Chabner B (1978) Role of pineal gland in aetiology and treatment of breast cancer. Lancet 2:814–816
Srinivasan V, Spence DW, Pandi-Perumal SR et al (2008) Therapeutic actions of melatonin in cancer: possible mechanisms. Integr Cancer Ther 7:189–203
Villarini A, Pasanisi P, Traina A (2012) Lifestyle and breast cancer recurrences: the DIANA-5 trial. Tumori 98:1–18
Adams SV, Newcomb PA, White E (2012) Dietary cadmium and risk of invasive postmenopausal breast cancer in the VITAL cohort. Cancer Causes Control 23:845–854
DeVita VT Jr, Rosenberg SA (2012) Two hundred years of cancer research. N Engl J Med 366:2207–2214
Reiter RJ, Tan DX, Erren TC et al (2009) Light-mediated perturbations of circadian timing and cancer risk: a mechanistic analysis. Integr Cancer Ther 8:354–360
Hansen J, Stevens RG (2011) Night shiftwork and breast cancer risk: overall evidence. Occup Environ Med 68:236–240
Blask DE, Dauchy RT, Brainard GC et al (2009) Circadian stage-dependent inhibition of human breast cancer metabolism and growth by the nocturnal melatonin signal: consequences of its disruption by light at night in rats and women. Integr Cancer Ther 8:347–353
Bartsch C, Bartsch H (2006) The anti-tumor activity of pineal melatonin and cancer enhancing life styles in industrialized societies. Cancer Causes Control 17:559–571
Kerenyi NA, Pandula E, Feuer G (1990) Why the incidence of cancer is increasing: the role of "light pollution". Med Hypotheses 33:75–78
Davis S, Mirick DK, Stevens RG (2001) Night shift work, light at night, and risk of breast cancer. J Natl Cancer Inst 93:1557–1562
Hansen J (2001) Increased breast cancer risk among women who work predominantly at night. Epidemiology 12:74–77
Bartsch C, Bartsch H, Jain AK et al (1981) Urinary melatonin levels in human breast cancer patients. J Neural Transm 52:281–294
Bartsch C, Bartsch H, Bellmann O et al (1991) Depression of serum melatonin in patients with primary breast cancer is not due to an increased peripheral metabolism. Cancer 67:1681–1684
Falkson G, Falkson HC, Steyn ME et al (1990) Plasma melatonin in patients with breast cancer. Oncology 47:401–405
Tamarkin L, Cohen M, Roselle D et al (1981) Melatonin inhibition and pinealectomy enhancement of 7–12 dimethylbenz(a)anthracene-induced mammary tumors in the rat. Cancer Res 41:4432–4436
Blask DE (1984) The pineal: an oncostatic gland? In: Reiter RJ (ed) The pineal gland. Raven Press, New York, pp 253–284
Saez MC, Barriga C, Garcia JJ et al (2005) Melatonin increases the survival time of animals with untreated mammary tumours: neuroendocrine stabilization. Mol Cell Biochem 278:15–20
Sanchez-Barcelo EJ, Cos S, Fernandez R et al (2003) Melatonin and mammary cancer: a short review. Endocr Relat Cancer 10:153–159
Kubatka P, Bojkova B, Kalicka K et al (2001) Preventive effects of raloxifene and melatonin in N-methyl-N-nitrosourea-induced mammary carcinogenesis in female rats. Neoplasma 48:313–319
Pawlikowski M, Winczyk K, Karasek M (2002) Oncostatic action of melatonin: facts and question marks. Neuro Endocrinol Lett 23:24–29
Cos S, Mediavilla D, Martinez-Campa C et al (2006) Exposure to light-at-night increases the growth of DMBA-induced mammary adenocarcinomas in rats. Cancer Lett 235:266–271
Anisimov VN (2003) The role of pineal gland in breast cancer development. Crit Rev Oncol Hematol 46:221–234
Blask DE, Pellettier DB, Hill SM et al (1991) Pineal melatonin inhibition of tumor promotion in the N-nitroso-N-methylurea model of mammary carcinogenesis: potential involvement of antiestrogenic mechanisms in vivo. J Cancer Res Clin Oncol 117:526–532
Palmer HJ, Paulson KE (1997) Reactive oxygen species and antioxidants in signal transduction and gene expression. Nutr Rev 55:353–361
Cerutti PA (1985) Prooxidant states and tumor promotion. Science 227:375–381
Reiter RJ, Tan DX, Terron M et al (2007) Melatonin and its metabolites: new findings regarding their production and their radical scavenging actions. Acta Biochim Pol 54:1–9
Burns JK (1973) Administration of melatonin to non-human primates and to women with breast carcinoma. J Physiol 229:38–39
Di Bella L, Scalera G, Rossi MT (1979) Perspectives in pineal function. Prog Brain Res 52:475–478
Lissoni P, Barni S, Meregalli S et al (1995) Modulation of cancer endocrine therapy by melatonin: a phase II study of tamoxifen plus melatonin in metastatic breast cancer patients progressing under tamoxifen alone. Br J Cancer 71:854–856
Lissoni P, Barni S, Cattaneo G et al (1991) Clinical results with the pineal hormone melatonin in advanced cancer resistant to standard antitumor therapies. Oncology 48:448–450
Lissoni P, Barni S, Tancini G et al (1994) A randomised study with subcutaneous low-dose interleukin 2 alone vs interleukin 2 plus the pineal neurohormone melatonin in advanced solid neoplasms other than renal cancer and melanoma. Br J Cancer 69:196–199
Lissoni P, Barni S, Ardizzoia A et al (1994) A randomized study with the pineal hormone melatonin versus supportive care alone in patients with brain metastases due to solid neoplasms. Cancer 73:699–701
Lissoni P, Barni S, Ardizzoia A et al (1992) Randomized study with the pineal hormone melatonin versus supportive care alone in advanced non small cell lung cancer resistant to a first-line chemotherapy containing cisplatin. Oncology 49:336–339
Lissoni P, Barni S, Crispino S et al (1989) Endocrine and immune effects of melatonin therapy in metastatic cancer patients. Eur J Cancer Clin Oncol 25:789–795
Lissoni P, Barni S, Tancini G et al (1987) Clinical study of melatonin in untreatable advanced cancer patients. Tumori 73:475–480
Panzer A, Viljioen M (1997) The validity of melatonin as an oncostatic agent. J Pineal Res 22:184–207
Mills E, Wu P, Seely D, Guyatt G (2005) Melatonin in the treatment of cancer: a systematic review of randomized controlled trials and meta-analysis. J Pineal Res 39:360–366
Bartsch C, Bartsch H, Karasek M (2002) Melatonin in clinical oncology. Neuro Endocrinol Lett 23(1):30–38
Hill SM, Blask DE (1988) Effects of the pineal hormone melatonin on the proliferation and morphological characteristics of human breast cancer cells (MCF-7) in culture. Cancer Res 48:6121–6126
Cos S, Blask DE, Lemus-Wilson A et al (1991) Effects of melatonin on the cell cycle kinetics and oestrogen rescue of MCF-7 human breast cancer cells in culture. J Pineal Res 10:36–42
Cos S, Recio J, Sanchez-Barcelo EJ (1996) Modulation of the cell cycle time of MCF-7 human breast cancer cells by melatonin. Life Sci 58:811–816
Cucina A, Proietti S, D’Anselmi F et al (2009) Evidence for a biphasic apoptotic pathway induced by melatonin in MCF-7 breast cancer cells. J Pineal Res 46:172–180
L’hermite-Baleriaux M, L’hermite M, Pasteels JM et al (1990) Effect of melatonin on the proliferation of human mammary cancer cell lines. Endocr Soc 140:59
Molis T, Muraoka HG, Castles C et al (1991) Growth regulatory effects of melatonin are linked to the oestrogen response pathway of human breast cancer cells. Endocr Soc 588:177
Shellard SA, Whelan RDH, Hill BT (1989) Growth inhibitory and cytotoxic effects of melatonin and its metabolites on human tumour cell lines in vitro. Br J Cancer 60:288–290
Bizzarri M, Cucina A, Valente MG et al (2003) Melatonin and vitamin D3 increase TGF-beta1 release and induce growth inhibition in breast cancer cell cultures. J Surg Res 110:332–337
Cos S, Fernandez F, Sanchez-Barcelo EJ (1996) Melatonin inhibits DNA synthesis in MCF-7 human breast cancer cells in vitro. Life Sci 58:2447–2453
Cos S, Sanchez-Barcelo EJ (2000) Melatonin and mammary pathological growth. Front Neuroendocrinol 21:133–170
Hill SM, Spriggs LL, Simon MA et al (1992) The growth inhibitory action of melatonin on human breast cancer cells is linked to the estrogen response system. Cancer Lett 64:249–256
Dauchy RT, Dauchy EM, Sauer LA et al (2004) Differential inhibition of fatty acid transport in tissue-isolated steroid receptor negative human breast cancer xenografts perfused in situ with isomers of conjugated linoleic acid. Cancer Lett 209:7–15
Blask DE (1993) Melatonin in oncology. In: Yu HS, Reiter RJ (eds) Melatonin. Biosynthesis, physiological effects, and clinical applications. CRC Press, Boca Raton, pp 447–475
Brydon L, Roka F, Petit L (1999) Dual signaling of human Me11a melatonin receptors via Gi2, Gi3 and Gq/11 proteins. Mol Endocrinol 13:2025–2038
Dubocovich ML, Markowska M (2005) Functional MT1 and MT2 melatonin receptors in mammals. Endocrine 2:101–110
Ram PT, Dai J, Yuan L et al (2002) Involvement of the mt1 melatonin receptor in human breast cancer. Cancer Lett 179:141–150
Roka F, Brydon L, Waldhoer M (1999) Tight association of the human Mel (1a)-melatonin receptor and G (i): precoupling and constitutive activity. Mol Pharmacol 56:1014–1024
Dubocovich ML (1988) Luzindole (N-0774): a novel melatonin receptor antagonist. J Pharmacol Exp Ther 246:902–910
Dubocovich ML, Masana MI, Iacob S et al (1997) Melatonin receptor antagonists that differentiate between the human Mel1a and Mel1b recombinant subtypes are used to assess the pharmacological profile of the rabbit retina ML1 presynaptic heteroreceptor. Naunyn Schmiedebergs Arch Pharmacol 355:365–375
Collins A, Yuan L, Kiefer TL et al (2003) Overexpression of the MT1 melatonin receptor in MCF-7 human breast cancer cells inhibits mammary tumour formation in nude mice. Cancer Lett 189:49–57
Yuan L, Collins AR, Dai J et al (2002) MT1 melatonin receptor overexpression enhances the growth suppressive effects of melatonin in human breast cancer cells. Mol Cell Endocrinol 192:147–156
Jawed S, Kim B, Ottenhof T et al (2007) Human melatonin MT1 receptor induction by valproic acid and its effects in combination with melatonin on MCF-7 breast cancer cell proliferation. Eur J Pharmacol 560:17–22
Bahia H, Ashman JN, Cawkwell L et al (2002) Karyotypic variation between independently cultured strains of the cell line MCF-7 identified by multicolour fluorescence in situ hybridization. Int J Oncol 20:489–494
Hill SM, Cheng C, Yuan L et al (2011) Declining melatonin levels and MT1 receptor expression in aging rats is associated with enhanced mammary tumor growth and decreased sensitivity to melatonin. Breast Cancer Res Treat 127:91–98
Lai L, Yuan L, Chen Q et al (2008) The Gαi and Gαq proteins mediate the effects of melatonin on steroid/thyroid hormone receptor transcriptional activity and breast cancer cell proliferation. J Pineal Res 45:476–488
Lai L, Yuan L, Cheng Q et al (2009) Alteration of the MT1 melatonin receptor gene and its expression in primary human breast tumors and breast cancer cell lines. Breast Cancer Res Treat 118:293–305
Slominski RM, Reiter RJ, Schlabritz-Loutsevitch N et al (2012) Melatonin membrane receptors in peripheral tissues: distribution and functions. Mol Cell Endocrinol 351:152–166
Pandi-Perumal SR, Trakht I, Srinivasan V et al (2008) Physiological effects of melatonin: role of melatonin receptors and signal transduction pathways. Prog Neurobiol 85:335–353
Guerrero HY, Gauer F, Schuster C et al (2000) Melatonin regulates the mRNA expression of the mt1 melatonin receptor in the rat pars tuberalis. Neuroendocrinology 71:163–169
Gerdin MJ, Masana MI, Ren D et al (2003) Short-term exposure to melatonin differentially affects the functional sensitivity and trafficking of the hMT1 and hMT2 melatonin receptors. J Pharmacol Exp Ther 304:931–939
Baldwin SW, Travlos GS, Risinger JI et al (1998) Melatonin does not inhibit estradiol-stimulated proliferation in MCF-7 and BG-1 cells. Carcinogenesis 19:1895–1900
Reiter RJ, Tan DX, Manchester LC et al (2007) Medical implications of melatonin: receptor-mediated and receptor-independent actions. Adv Med Sci 52:11–28
Bonnefont-Rousselot D, Collin F, Jore D, Gardès-Albert M (2011) Reaction mechanism of melatonin oxidation by reactive oxygen species in vitro. J Pineal Res 50:328–335
Kilic U, Yilmaz B, Ugur M et al (2012) Evidence that membrane-bound G protein-coupled melatonin receptors MT1 and MT2 are not involved in the neuroprotective effects of melatonin in focal cerebral ischemia. J Pineal Res 52:228–235
Tan DX, Manchester LC, Hardeland R et al (2003) Melatonin: a hormone, a tissue factor, an autocoid, a paracoid, and an antioxidant vitamin. J Pineal Res 34:75–78
Hill SM, Blask DE (1986) Melatonin inhibition of MCF-7 breast cancer cell proliferation: influence of serum factors prolactin and oestradiol. Abstr Endocr Soc 863:246
Cos S, Sanchez-Barcelo EJ (1995) Melatonin inhibition of MCF-7 human breast cancer cells: influence of cell proliferation rate. Cancer Lett 93:207–212
Cos S, Sanchez-Barcelo EJ (1994) Differences between pulsatile or continuous exposure to melatonin on MCF-7 human breast cancer cell proliferation. Cancer Lett 85:105–109
Cos S, Blask DE (1990) Effects of melatonin on the anchorage-independent growth of human breast cancer cells (MCF-7) in a clonogenic culture system. Cancer Lett 50:115–119
Sanchez-Barcelo EJ, Mediavilla MD, Alonso-Gonzalez C (2012) Breast cancer therapy based on melatonin. Recent Pat Endocr Metab Immune Drug Discov 1(6):108–116
Reiter RJ, Fraschini F (1969) Endocrine aspects of the mammalian pineal gland: a review. Neuroendocrinology 5:219–255
Reiter RJ (1980) The pineal and its hormones in the control of reproduction in mammals. Endocr Rev 1:109–131
Barrett P, Bolborea M (2012) Molecular pathways involved in seasonal body weight and reproductive responses governed by melatonin. J Pineal Res 52:376–388
Dubocovich ML, Rivera-Bermudez MA, Gerdin MJ et al (2003) Molecular pharmacology, regulation and function of mammalian melatonin receptors. Front Biosci 8:1093–1108
Cohen M, Lippman M, Chabner B (1978) Role of pineal gland in aetiology and treatment of breast cancer. Lancet 2:814–816
Tamura H, Nakamura Y, Terron MP (2008) Melatonin and pregnancy in the human. Reprod Toxicol 25:291–303
Tamura H, Nakamura Y, Korkmaz A et al (2009) Melatonin and the ovary: physiological and pathophysiological implications. Fertil Steril 92:328–343
Kauppila A, Kivela A, Pakarinen A et al (1987) Inverse seasonal relationship between melatonin and ovarian activity in humans in a region with a strong seasonal contrast in luminosity. J Clin Endocrinol Metab 65:823–828
Aleandri V, Spina V, Morini A (1996) The pineal gland and reproduction. Hum Reprod Update 2:225–235
Luboshitzky R, Lavie P (1999) Melatonin and sex hormone interrelationships: a review. J Pediatr Endocrinol Metab 12:355–362
Woo MM, Tai CJ, Kang SK et al (2001) Direct action of melatonin in human granulosa-luteal cells. J Clin Endocrinol Metab 86:4789–4797
Soares JM, Masana MI, Ersahin C et al (2003) Functional melatonin receptors in rat ovaries at various stages of the estrous cycle. J Pharmacol Exp Ther 306:694–702
Sanchez-Barcelo EJ, Cos S, Mediavilla D et al (2005) Melatonin–estrogen interactions in breast cancer. J Pineal Res 38:217–222
Ram PT, Yuan L, Dai J et al (2000) Differential responsiveness of MCF-7 human breast cancer cell line stocks to the pineal hormone melatonin. J Pineal Res 28:210–218
Del Rio B, Garcia Pedrero JM, Martinez-Campa C et al (2004) Melatonin an endogenous-specific inhibitor of estrogen receptor alpha via calmodulin. J Biol Chem 279:38294–38302
Cos S, Fernandez R, Guezmes A et al (1998) Influence of melatonin on invasive and metastatic properties of MCF-7 human breast cancer cells. Cancer Res 58:4383–4390
Wilson ST, Blask DE, Lemus-Wilson AM (1992) Melatonin augments the sensitivity of MCF-7 human breast cancer cells to tamoxifen in vitro. J Clin Endocrinol Metab 75:669–670
Mediavilla MD, Guezmez A, Ramos S et al (1997) Effects of melatonin on mammary gland lesions in transgenic mice overexpressing N-ras proto-oncogene. J Pineal Res 22:86–94
Kiefer TL, Lai L, Yuan L et al (2005) Differential regulation of estrogen receptor alpha, glucocorticoid, receptor and retinoic acid receptor alpha transcriptional activity by melatonin is mediated via different G proteins. J Pineal Res 38:231–239
Garcia-Rato A, Garcia-Pedrero JM, Martinez MA et al (1999) Melatonin blocks the activation of estrogen receptor for DNA binding. FASEB J 13:857–868
Lawson NO, Wee BE, Blask DE et al (1992) Melatonin decreases estrogen receptor expression in the medial preoptic area of inbred (LSH/SsLak) golden hamsters. Biol Reprod 47:1082–1090
Ram PT, Dai J, Yuan L et al (2002) Involvement of the mt1 melatonin receptor in human breast cancer. Cancer Lett 179:141–150
Mangelsdorf DJ, Thummel C, Beato M et al (1995) The nuclear receptor superfamily: the second decade. Cell 83:835–839
Glass CK, Rosenfeld MG (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14:121–141
Benoit G, Cooney A, Giguere V et al (2006) International Union of Pharmacology LXVI. Orphan nuclear receptors. Pharmacol Rev 58:798–836
Mukherjee R, Davies PJ, Crombie DL et al (1997) Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature 386:407–410
Mukherjee R, Jow L, Croston GE et al (1997) Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARgamma2 versus PPARgamma1 and activation with retinoid X receptor agonists and antagonists. J Biol Chem 272:8071–8076
Mckenna NJ, O’Malley BW (2002) Minireview: nuclear receptor coactivators—an update. Endocrinology 143:2461–2465
Mckenna NJ, O’Malley BW (2002) Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108:465–474
Becker-Andre M, Wiesemberg I, Schaeren-Wiemers N et al. (1994) Pineal gland hormone melatonin binds and activates an orphan of the nuclear receptor superfamily. J Biol Chem 269:28531–28534
Agez L, LaurentV Pevet P et al (2007) Melatonin affects nuclear orphan receptors mRNA in the rat suprachiasmatic nuclei. Neuroscience 144:522–530
Park HT, Baek SY, Kim BS et al (1996) Developmental expression of ‘RZR beta, a putative nuclear-melatonin receptor’ mRNA in the suprachiasmatic nucleus of the rat. Neurosci Lett 217:17–20
Baler R, Coon S, Klein DC (1996) Orphan nuclear receptor RZRbeta: cyclic AMP regulates expression in the pineal gland. Biochem Biophys Res Commun 220:975–978
Naji L, Carillo-Vico A, Guerrero JM et al (2004) Expression of membrane and nuclear melatonin receptors in mouse peripheral organs. Life Sci 74:2227–2236
Bordji K, Grillasca JP, Gouze JN et al (2000) Evidence for the presence of peroxisome proliferator-activated receptor (PPAR) alpha and gamma and retinoid Z receptor in cartilage. PPARgamma activation modulates the effects of interleukin-1beta on rat chondrocytes. J Biol Chem 275:12243–12250
Smirnov AN (2001) Nuclear melatonin receptors. Biochemistry 66:19–26
Sharma R, Ottenhof T, Rzeczkowska PA et al (2008) Epigenetic targets for melatonin: induction of histone H3 hyperacetylation and gene expression in C17.2 neural stem cells. J Pineal Res 45:277–284
Korkmaz A, Sanchez-Barcelo EJ, Tan DX et al (2009) Role of melatonin in the epigenetic regulation of breast cancer. Breast Cancer Res Treat 115:13–27
Korkmaz A (2009) Epigenetic actions of melatonin. J Pineal Res 46:117–118
Korkmaz A, Tamura H, Manchester LC et al (2009) Combination of melatonin and a peroxisome proliferator-activated receptor-gamma agonist induces apoptosis in a breast cancer cell line. J Pineal Res 46:115–116
Aronika SM, Kraus WL, Katzenellenbogen BS (1994) Oestrogen action via the cAMP signalling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc Natl Acad Sci USA 91:8517–8521
Kiefer T, Ram PT, Yuan L, Hill SM (2002) Melatonin inhibits estrogen receptor transactivation and cAMP levels in breast cancer cells. Breast Cancer Res Treat 71:37–45
Ram PT, Kiefer T, Silverman M et al (1998) Estrogen receptor transactivation in MCF-7 breast cancer cells by melatonin and growth factors. Mol Cell Endocrinol 141:53–64
Benítez-King G, Ríos A, Martínez A et al (1996) In vitro inhibition of Ca2+/calmodulin-dependent kinase II activity by melatonin. Biochim Biophys Acta 1290:191–196
Pozo D, Reiter RJ, Calvo JR et al (1997) Inhibition of cerebellar nitric oxide synthase and cyclic GMP production by melatonin via complex formation with calmodulin. J Cell Biochem 1(65):430–442
Soto-Vega E, Ramírez-Rodríguez G, Benitez-King G (2004) Melatonin stimulates calmodulin phosphorylation by protein kinase C. J Pineal Res 37:98–106
Garcia-Pedrero JM, Martinez MA, Del Rio B et al (2002) Calmodulin is a selective modulator of estrogen receptors. Mol Endocrinol 16:947–960
Dai J, Inscho EW, Yuan L et al (2002) Modulation of intracellular calcium and calmodulin by melatonin in MCF-7 human breast cancer cells. J Pineal Res 32:112–119
Li Z, Kim SH, Higgins JMG et al (1999) IQGAP1 and calmodulin modulate E-cadherin function. J Biol Chem 274:37885–37892
Blask DE (1997) Systemic, cellular and molecular aspects of melatonin action on experimental breast carcinogenesis. In: Stevens RG, Wilson BW, Anderson LE (eds) The melatonin hypothesis-breast cancer and use of electric power. Battel Press, Columbus, p 189–230
Blask DE, Wilson ST, Zalatan F (1997) Physiological melatonin inhibition of human breast cancer cell growth in vitro: evidence for a glutathione-mediated Pathway. Cancer Res 57:1909–1914
Leung MF, Chov IN (1989) Relationship between l-chloro-2,4-diitrobenzene induced cytoskeletal perturbations and cellular glutathione. Cell Biol Toxicol 5:51–99
Benitez-King G, Anton-Tay F (1993) Calmodulin mediates melatonin cytoskeletal effects. Experientia 49:635–641
Roth JA, Rabin R, Agnello K (1997) Melatonin suppression of PC12 cell growth and death. Brain Res 768:63–70
Yang QH, Xu YN, Xu RK et al (2007) Antiproliferative effects of melatonin on the growth of rat pituitary prolactin-secreting tumor cells in vitro. J Pineal Res 42:172–179
Zhou D, Clarke P, Wang J et al (1996) Identification of a promoter that controls aromatase expression in human breast cancer and adipose stromal cells. J Biol Chem 271:15194–15202
Michael MD, Michael LF, Simpson ER (1997) A CRE-like sequence that binds CREB and contributes to cAMP-dependent regulation of the proximal promoter of the human aromatase P450 (CYP19) gene. Mol Cell Endocrinol 134:147–156
Zhao Y, Aagarwal VR, Mendelson CR et al (1996) Estrogen biosynthesis proximal to a breast tumor is stimulated by PGE2 via cyclic AMP, leading to activation of promoter II of the CYP19 (aromatase) gene. Endocrinology 137:5739–5742
Cardinali DP, Bonanni Rey RA et al (1992) Diurnal changes in cyclic nucleotide response to pineal indoles in murine mammary glands. J Pineal Res 13:111–116
Cos S, Martinez-Campa C, Mediavilla MD et al (2005) Melatonin modulates aromatase activity in MCF-7 human breast cancer cells. J Pineal Res 38:136–142
Gonzalez A, Martinez-Campa C, Mediavilla MD et al (2007) Effects of MT1 melatonin receptor overexpression on the aromatase-suppressive effect of melatonin in MCF-7 human breast cancer cells. Oncol Rep 17:947–953
Martinez- Campa C, Gonzalez A, Mediavilla MD et al (2005) Melatonin enhances the inhibitory effect of aminoglutethimide on aromatase activity in MCF-7 human breast cancer cells. Breast Cancer Res Treat 94:249–254
Ryde CM, Nicholls JE, Dowsett M (1992) Steroid and growth factor modulation of aromatase activity in MCF-7 and T47D breast carcinoma cell lines. Cancer Res 52:1411–1415
Cos S, Blask DE (1994) Melatonin modulates growth factor activity in MCF-7 human breast cancer cells. J Pineal Res 17:25–32
Martinez-Campa C, Gonzalez A, Mediavilla MD et al (2009) Melatonin inhibits aromatase promoter expression by regulating cyclooxygenases expression and activity in breast cancer cells. Br J Cancer 101:1613–1619
Suzuki T, Miki Y, Nakamura Y et al (2005) Sex steroid-producing enzymes in human breast cancer. Endocr Relat Cancer 12:701–720
Suzuki T, Miki Y, Nakata T et al (2003) Steroid sulfatase and estrogen sulfotransferase in normal human tissue and breast cancer. J Steroid Biochem Mol Biol 86:449–454
Gonzalez A, Cos S, Martinez-Campa C et al (2008) Selective estrogen enzyme modulator actions of melatonin in human breast cancer cells. J Pineal Res 45:86–92
Cai Z, Kwintkiewicz J, Young ME et al (2007) Prostaglandin E2 increases cyp19 expression in rat granulosa cells: implication of GATA-4. Mol Cell Endocrinol 263:181–189
Subbaramaiah K, Howe LR, Port ER et al (2006) HER-2/neu status is a determinant of mammary aromatase activity in vivo: evidence for a cyclooxygenase-2-dependent mechanism. Cancer Res 66:5504–5511
Dong WG, Mei Q, Yu JP et al (2003) Effects of melatonin on the expression of iNOS and COX-2 in rat models of colitis. World J Gastroenterol 9:1307–1311
Mrnka L, Hock M, Rybova M et al (2008) Melatonin inhibits prostaglandin E2 and sodium nitroprusside-induced ion secretion in rat distal colon. Eur J Pharmacol 581:164–170
Deng WG, Tang ST, Tseng HP et al (2006) Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 108:518–5124
Mohan N, Sadeghi K, Reiter RJ et al (1995) The neurohormone melatonin inhibits cytokine, mitogen and ionizing radiation induced NF-kappa B. Biochem Mol Biol Int 37:1063–1070
Chuang JI, Mohan N, Meltz ML et al (1996) Effect of melatonin on NF-kappa-B DNA-binding activity in the rat spleen. Cell Biol Int 20:687–692
Esposito E, Iacono A, Muia C et al (2008) Signal transduction pathways involved in protective effects of melatonin in C6 glioma cells. J Pineal Res 44:78–87
Crowe DL, Chandraratna RA (2004) A retinoid X receptor (RXR)-selective retinoid reveals that RXR-alpha is potentially a therapeutic target in breast cancer cell lines, and that it potentiates antiproliferative and apoptotic responses to peroxisome proliferator-activated receptor ligands. Breast Cancer Res 6:R546–R555
Fan W, Yanase T, Morinaga H et al (2005) Activation of peroxisome proliferator-activated receptor-gamma and retinoid X receptor inhibits aromatase transcription via nuclear factor kappa B. Endocrinology 146:85–92
Molis TM, Spriggs LL, Jupiter Y (1995) Melatonin modulation of estrogen-regulated proteins, growth factors, and proto-oncogenes in human breast cancer. J Pineal Res 18:93–103
Calnaan DPK, Westley BR, May FEB et al (1999) The trefoil peptide TFF1 inhibits the growth of the human gastric adenocarcinoma cell line AGS. J Pathol 188:312–317
Henry JA, Piggott NH, Mallick UK et al (1990) PNR-2/pS2 immunohistochemical staining in breast cancer: correlation with prognostic factors and endocrine response. Br J Cancer 4:615–622
Barnes DM, Gillett CE (1998) Cyclin D1 in breast cancer. Breast Cancer Res Treat 52:1–15
Cicatiello L, Addeo R, Sasso A (2004) Oestrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/oestrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol Cell Biol 24:7260–7274
Fu M, Wang C, Li Z, Sakamaky T et al (2004) Minireview: cyclin D1: normal and abnormal functions. Endocrinology 145:5439–5447
Fu M, Rao M, Bouras T et al (2005) Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. J Biol Chem 280:16934–16941
Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88:323–331
Cini G, Neri B, Pacini A et al (2005) Antiproliferative activity of melatonin by transcriptional inhibition of cyclin D1 expression: a molecular basis for melatonin-induced oncostatic effects. J Pineal Res 39:12–20
Abbas T, Dutta A (2009) p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9:400–414
Mediavilla MD, Cos S, Sanchez-Barcelo EJ (1999) Melatonin increases p53 and p21WAF1 expression in MCF-7 human breast cancer cells in vitro. Life Sci 65:415–420
Rimler A, Matzkin H, Zisapel N (1999) Cross talk between melatonin and TGF-1 in human benign prostate epithelial cells. Prostate 40:211
Molis TM, Priggs LL, Hill SM (1994) Modulation of estrogen receptor mRNA expression by melatonin in MCF-7 human breast cancer cells. Mol Endocrinol 8:1683–1690
Czeczuga-Semeniuk E, Anchim T, Dziecioł J et al (2004) Can transforming growth factor-β1 and retinoids modify the activity of estradiol and antiestrogens in MCF-7 breast cancer cells? Acta Biochim Pol 51:733–745
Blask DE, Sauer LA, Dauchy RT (2002) Melatonin as a chronobiotic/anticanceragent: cellular, biochemical, and molecular mechanisms of action and their implications for circadian-based cancer therapy. Curr Top Med Chem 2:113–132
Leon-Blanco MM, Guerrero JM, Reiter JR et al (2003) Melatonin inhibits telomerase activity in the MCF-7 tumour cell line both in vitro and in vivo. J Pineal Res 35:204–211
Martinez-Campa CM, Alonso-Gonzalez C, Mediavilla MD et al (2008) Melatonin down-regulates hTERT expression induced by either natural estrogens (17b-estradiol) or metalloestrogens (cadmium) in MCF-7 human breast cancer cells. Cancer Lett 268:272–277
Leon-Blanco MM, Guerrero JM, Reiter RJ et al (2004) RNA expression of human telomerase subunits TR and TERT is differentially affected by melatonin receptor agonists in the MCF-7 tumor cell line. Cancer Lett 216:73–80
Blasco MA (2007) The epigenetic regulation of mammalian telomeres. Nat Rev Genet 8:299–309
Reiter RJ (2004) Mechanisms of cancer inhibition by melatonin. J Pineal Res 37:213–214
Sainz RM, Mayo JC, Rodriguez C et al (2003) Melatonin and cell death: differential actions on apoptosis in normal and cancer cells. Cell Mol Life Sci 60:1407–1426
Sánchez-Hidalgo M, Guerrero JM, Villegas I (2012) Melatonin, a natural programmed cell death inducer in cancer. Curr Med Chem (in press)
Karasek M, Winczyk K, Kunert-Radk J et al (1998) Antiproliferative effects of melatonin and CGP52608 on the murine colon 38 adenocarcinoma in vitro and in vivo. Neuroendocrinol Lett 19:71–78
Winczyk K, Pawlikowski M, Lawnicka H et al (2002) Effects of melatonin and melatonin receptors ligand N-[(4-methoxy-1H-indol-2yl) methyl] propanamide on murine Colon 38 cancer growth in vitro and in vivo. Neuroendocrinol Lett 1:50–54
Motilva V, García-Mauriño S, Talero E et al (2011) New paradigms in chronic intestinal inflammation and colon cancer: role of melatonin. J Pineal Res 51:44–60
Carbajo-Pescador S, García-Palomo A, Martín-Renedo J et al (2011) Melatonin modulation of intracellular signaling pathways in hepatocarcinoma HepG2 cell line: role of the MT1 receptor. J Pineal Res 51:463–471
Garcia-Santos G, Antolin I, Herrera F et al (2006) Melatonin induces apoptosis in human neuroblastoma cancer cells. J Pineal Res 41:130–135
El-Missiry MA, Abd EL-Aziz AF (2000) Influence of melatonin on proliferation and antioxidant system in Ehrlich ascites carcinoma cells. Cancer Lett 151:119–125
Rubio S, Estevez F, Cabrera J et al (2007) Inhibition of proliferation and induction of apoptosis by melatonin in human myeloid HL-60 cells. J Pineal Res 42:131–138
Trubiani O, Recchioni R, Moroni F et al (2005) Melatonin provokes cell death in human B-lymphoma cells by mitochondrial-dependent apoptotic pathway activation. J Pineal Res 39:425–431
Leja-Szpak A, Jaworek J, Pierzchalski P et al (2010) Melatonin induces pro-apoptotic signaling pathway in human pancreatic carcinoma cells (PANC-1). J Pineal Res 49:248–255
Gonzalez A, del Castillo-Vaquero A, Miro-Moran A et al (2011) Melatonin reduces pancreatic tumor cell viability by altering mitochondrial physiology. J Pineal Res 50:250–260
Um HJ, Park JW, Kwon TK (2011) Melatonin sensitizes Caki renal cancer cells to kahweol-induced apoptosis through CHOP-mediated up-regulation of PUMA. J Pineal Res 50:359–366
Wolfler A, Caluba HC, Abuja PM et al (2001) Prooxidant activity of melatonin promotes fas-induced cell death in human leukemic Jurkat cells. FEBS Lett 502:127–131
Bejarano I, Espino J, Barriga C et al (2011) Pro-oxidant effect of melatonin in tumour leucocytes: relation with its cytotoxic and pro-apoptotic effects. Basic Clin Pharmacol Toxicol 108:14–20
Casado-Zapico S, Martín V, García-Santos G et al (2011) Regulation of the expression of death receptors and their ligands by melatonin in haematological cancer cell lines and in leukaemia cells from patients. J Pineal Res 50:345–355
Cos S, Mediavilla MD, Fernandez R et al (2002) Does melatonin induce apoptosis in MCF-7 human breast cancer cells in vitro? J Pineal Res 32:90–96
Czeczuga-Semeniuk E, Wołczyński S, Anchim T et al (2002) Effect of melatonin and all-trans retinoic acid on the proliferation and induction of the apoptotic pathway in the culture of human breast cancer cell line MCF-7. Pol J Pathol 53:59–65
Eck-Enriquez KM, Yuan L, Duffy L et al (1998) A sequential treatment regimen with melatonin and all-trans retinoic acid induces apoptosis in MCF-7 tumour cells. Br J Cancer 77:2129–2137
Eck-Enriquez KM, Kiefer TL, Spriggs LL et al (2000) Pathways through which a regimen of melatonin and retinoic acid induces apoptosis in MCF-7 human breast cancer cells. Breast Cancer Res Treat 61:229–239
Abd El-Aziz MA, Hassan HA, Mohamed MH et al (2005) The biochemical and morphological alterations following administration of melatonin, retinoic acid and Nigella sativa in mammary carcinoma: an animal model. Int J Exp Pathol 86:383–396
Momand J, Wu HH, Dasgupta G (2000) MDM2–master regulator of the p53 tumor suppressor protein. Gene 242:15–29
RozieresS DE, Maya R, Oren M et al (2000) The loss of MDM2 induces p53-mediated apoptosis. Oncogene 19:1691–1697
Inoue T, Geyer RK, Yu ZK et al (2001) Downregulation of MDM2 stabilizes p53 by inhibiting p53 ubiquitination in response to specific alkylating agents. FEBS Lett 490:196–201
Proietti S, Cucina A, D’Anselmi F et al (2011) Melatonin and vitamin D3 synergistically down-regulate Akt and MDM2 leading to TGFβ-1-dependent growth inhibition of breast cancer cells. J Pineal Res 50:150–158
Lopez-Burillo S, Tan DX, Mayo JC et al (2003) Melatonin, xanthurenic acid, resveratrol, EGCG, vitamin C and a-lipoic acid differentially reduce oxidative DNA damage induced by Fenton reagents: a study of their individual and synergistic actions. J Pineal Res 34:269–277
Mao L, Yuan L, Hill SM. Inhibition of cell proliferation and blockade of cell invasion by melatonin in human breast cancer cells mediated through multiple signaling pathways. Paper presented at: 97th annual meeting of the American Association for Cancer Research 2006 Washington, DC. Abstract 495
Cos S, Fernandez R (2000) Melatonin effects on intercellular junctional communication in MCF-7 human breast cancer cells. J Pineal Res 29:166–171
Trosko JE, Chang CC, Madhukar BV et al (1990) Chemical, oncogene and growth factor inhibition of gap junctional intercellular communication: an integrative hypothesis of carcinogenesis. Pathobiology 58:265–278
Hamada J, Takeichi N, Kobayashi H (1987) Inverse correlation between the metastatic capacity of cell clones derived from a rat mammary carcinoma and their intercellular communication with normal fibroblasts. Gann 78:1175–1178
Crespo D, Fernandez-Viadero C, Verdura R et al (1994) Interaction between melatonin and estradiol on morphological and morphometric features of MCF-7 human breast cancer cells. J Pineal Res 16:215–222
Swarnakar R, Paul S, Singh LP et al (2011) Matrix metalloproteinases in health and disease: regulation by melatonin. J Pineal Res 50:8–20
Sainz RM, Mayo JC, Tan DX et al (2005) Melatonin reduces prostate cancer cell growth leading to neuroendocrine differentiation via a receptor and PKA independent mechanism. Prostate 63:29–43
Rodriguez-Garcia A, Mayo JC, Heria D et al (2012) Phenotypic changes caused by melatonin increased sensitivity of prostate cancer cells to cytokine-induced apoptosis. J Pineal Res (in press)
Roberts K (1974) Cytoplasmic microtubules and their functions. Prog Biophys Mol Biol 28:371–420
Ingber DE (2003) Tensegrity, II. How structural networks influence cellular information processing networks. J Cell Sci 116:1397–1408
Wang N, Tytell JD, Ingber DE (2009) Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nature Rev Mol Cell Biol 10:75–82
Pourati J, Maniotis A, Speigel DM et al (1998) Is cytoskeletal tension a major determinant of cell deformability in adherent endothelial cells? Am J Physiol 274:C1283–C1289
Hammond TG, Lewis FC, Goodwin TJ et al (1999) Gene expression in space. Nat Med 5:359
Stein GS, Van Wijnen AJ, Stein JL et al (1999) Implications for interrelationships between nuclear architecture and control of gene expression under microgravity conditions. FASEB 13:S157–S166
Chen CS, Mrksich M, Huang S et al (1997) Geometric control of cell life and death. Science 276:1425–1428
Huang S, Ingber DE (2000) Shape-dependent control of cell growth, differentiation, and apoptosis: switching between attractors in cell regulatory networks. Exp Cell Res 261:91–103
D’Anselmi F, Valerio MC, Cucina A et al (2011) Metabolism and cell shape in cancer: a fractal analysis. Int J Biochem Cell Biol 43:1052–1058
Raz A, Geiger B (1982) Altered organization of cell-substrate contacts and membrane-associated cytoskeleton in tumor cell variants exhibiting different metastatic capabilities. Cancer Res 42:5183–5190
Benitez-King G, Tunez I, Bellon A et al (2003) Melatonin prevents cytoskeletal alterations and oxidative stress induced by okadaic acid in N1E−115 cells. Exp Neurol 182:151–159
Wang LH (2004) Molecular signaling regulating anchorage independent growth of cancer cells. Mt Sinai J Med 71:361–367
Bharadwaj S, Thanawala R, Bon G et al (2005) Resensitization of breast cancer cells to anoikis by tropomyosin-1: role of Rho kinase-dependent cytoskeleton and adhesion. Oncogene 24:8291–8303
Raftopoulou M, Hall A (2004) Cell migration: Rho GTPases lead the way. Dev Biol 265:23–32
Huerto-Delgadillo L, Anton-Tay F, Benitez-King G (1994) Effects of melatonin on microtubule assembly depend on hormone concentration: role of melatonin as a calmodulin antagonist. J Pineal Res 17:55–62
Benitez-King G, Soto-Vega E, Ramirez-Rodriguez G (2009) Melatonin modulates microfilament phenotypes in cancer cells: implications for adhesion and inhibition of cancer cell migration. Histol Histopathol 24:789–799
Ortiz-Lopez L, Morales-Mulia S, Ramiraz-Rodriguez G et al (2009) ROCK-regulated cytoskeletal dynamics participate in the inhibitory effect of melatonin on cancer cell migration. J Pineal Res 46:15–21
Ramirez-Rodriguez G, Meza I, Hernandez ME et al (2003) Melatonin induced cyclic modulation of vectorial water transport in kidney-derived MDCK cells. Kidney Int 63:1356–1364
Ramirez-Rodriguez G, Ortiz-Lopez L, Benitez-King G (2007) Melatonin increases stress fibers and focal adhesions in MDCK cells: participation of Rho-associated kinase and protein kinase C. J Pineal Res 42:180–190
Yuan J, Shi GX, Shao Y et al (2008) Calmodulin bound to stress fibers but not microtubules involves regulation of cell morphology and motility. Int J Biochem Cell Biol 40:284–293
Benitez-King G (2006) Melatonin as a cytoskeletal modulator: implications for cell physiology and disease. J Pineal Res 40:1–9
Lee SE, Kim SJ, Youn JP et al (2011) MicroRNA and gene expression analysis of melatonin-exposed human breast cancer cell lines indicating involvement of the anticancer effect. J Pineal Res 51:345–352
Sanchez-Barcelo EJ, Mediavilla MD, Alonso-Gonzalez C et al (2012) Melatonin uses in oncology: breast cancer prevention and reduction of the side effects of chemotherapy and radiation. Expert Opin Investig Drugs 21:819–831
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Proietti, S., Cucina, A., Reiter, R.J. et al. Molecular mechanisms of melatonin’s inhibitory actions on breast cancers. Cell. Mol. Life Sci. 70, 2139–2157 (2013). https://doi.org/10.1007/s00018-012-1161-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1161-8