
Abstract
Monoamine oxidases (MAOs) are flavoproteins of the outer mitochondrial membrane that catalyze the oxidative deamination of biogenic and xenobiotic amines. In mammals there are two isoforms (MAO-A and MAO-B) that can be distinguished on the basis of their substrate specificity and their sensitivity towards specific inhibitors. Both isoforms are expressed in most tissues, but their expression in the central nervous system and their ability to metabolize monoaminergic neurotransmitters have focused MAO research on the functionality of the mature brain. MAO activities have been related to neurodegenerative diseases as well as to neurological and psychiatric disorders. More recently evidence has been accumulating indicating that MAO isoforms are expressed not only in adult mammals, but also before birth, and that defective MAO expression induces developmental abnormalities in particular of the brain. This review is aimed at summarizing and critically evaluating the new findings on the developmental functions of MAO isoforms during embryogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Monoamine oxidases (MAO, E.C. 1.4.3.4.) are flavoproteins that catalyze the oxidative deamination of a number of biogenic and dietary monoamines forming the corresponding aldehydes, hydrogen peroxide and ammonia (Fig. 1). In humans there are two separate MAO isoforms (MAO-A, MAO-B), which exhibit different but overlapping substrate and inhibitor specificities [1, 2]. MAO-A, which preferentially oxidizes serotonin (5-hydroxytryptamine, 5-HT), norepinephrine (NE) and epinephrine (EN), is irreversibly inhibited by low doses of clorgyline. In contrast, MAO-B prefers phenylethylamine (PEA) as a substrate and is irreversibly inactivated by low doses of deprenyl (selegiline). Dopamine (DA) and tyramine are common substrates for both MAO-A and MAO-B. Because of their capability of metabolizing monoamines that function as neurotransmitters, research on MAO isoforms in the past has been focused on the cerebral role of the two enzymes. However, more recent expression studies have indicated abundant expression of MAO isoforms in peripheral tissues, which clearly exceeds expression in the brain [3]. For instance, human MAO-A is dominantly expressed in placenta, adipose tissue, the thyroid gland and the lung, whereas expression in various parts of the brain is rather low. In contrast, MAO-B is dominantly expressed in various parts of the central nervous system (hypothalamus, prefrontal cortex, amygdala, spinal cord), but uterus, kidney, liver and heart are also rich sources for MAO-B. In the central nervous system MAO-A is predominantly found in catecholaminergic neurons, whereas MAO-B is more abundant in serotonergic and histaminergic neurons as well as in glial cells [2]. Abnormal MAO activity has been implicated in a number of neurological and psychiatric disorders, such as depression and social anxiety [4].

Schematic representation of the oxidative deamination of monoamines to the corresponding aldehydes by MAO. The aldehydes generated in the MAO reaction are further metabolized by aldehyde dehydrogenase and aldehyde reductase
Monoamine oxidase inhibitors have long been developed and are widely used in clinics for the treatment of many neuropsychiatric and neurodegenerative disorders. MAO-A inhibitors have been shown to be effective antidepressant drugs [4]. In contrast, MAO-B inhibitors, such as selegiline and rasagiline, have been used for the treatment of Parkinson’s disease [5], but it remains a matter of discussion whether the MAO inhibitory activities of these compounds are the basis for the therapeutic effect. The recent advances in the development of clinically relevant MAO inhibitors and the improvements in our understanding of the molecular mechanisms of enzyme-inhibitor interactions have not only provided an excellent basis for clinical treatment of neuropsychiatric and neurodegenerative disorders, but also demonstrated the important role of MAO in the brain.
Research on monoamine oxidases has a long history since the discovery of these enzymes by Mary Hare back in 1928 [6]. During all these years MAO research has been largely focused on the functionality of these enzymes in the adult organism. However, over the last couple of years increasing experimental evidence has indicated that MAO enzymes are already present in developing embryos. Moreover, more sensitive analytical techniques (e.g., in vitro embryogenesis, quantitative PCR) and optimized research tools (e.g., antibodies, specific DNA and RNA probes) have recently become available allowing researchers to explore the role of MAO during mammalian embryogenesis [3, 7]. These new developments have generated a growing number of publications that have not yet been discussed in detail. This review is thus intended to summarize and critically discuss the recent advances in our knowledge on the role MAO isoforms in embryogenesis and during the early post-natal developmental period. Consequently, the behavioral abnormalities, which have been related to MAO-A deficiency in mice and men, will not be addressed in detail in this review. The interested reader is referred to other review articles covering these clinically more relevant topics [2, 4]. The first section summarizes the mechanisms of MAO catalysis and the structural biology of the two MAO isoforms, which are the chemical basis for their biologic activity. “Enzymology of MAO isoforms” discusses the diverse effects of MAO activity on cellular signaling and cellular functions, while the third section will introduce the reader to the various amine systems that are available in the developing embryo; this is followed by a critical discussion of the recent publications on the roles of MAO during embryo development in “Monoamine systems during normal embryo development”. The final two sections conclude by respectively focusing on the clinical implications of monoamines in the fetus and a discussion of the perspectives of MAO research in embryogenesis.
Enzymology of MAO isoforms
Reaction mechanism of MAO catalysis
As indicated above, monoamine oxidases (MAOs) are catalytically active flavoproteins that catalyze the oxidation of amines to the corresponding aldehydes. This reaction requires molecular dioxygen and produces stoichiometric amounts of hydrogen peroxide and ammonia [8, 9]. Since hydrogen peroxide is a product of the MAO reaction, the two MAO isoforms have been implicated in cellular redox regulation. Although monoamine oxidases have been studied extensively, their detailed catalytic mechanisms have not been clarified completely. Our current knowledge on MAO catalysis is summarized in Fig. 2. After binding of the amine substrate at the active site, the oxidized FAD cofactor is reduced, and an enzyme-bound imine complex (MAO-FADred-imine) is formed. For most substrates, this complex (MAO-FADred-imine) then reacts with atmospheric oxygen to reoxidize the FAD (MAO-FADox-imine) and to liberate hydrogen peroxide. Next, the MAO-FADox-imine complex dissociates, liberating the imine that hydrolyzes non-enzymatically to yield the aldehyde product and ammonia. Alternatively (not shown), the reduced enzyme-imine complex (MAO-FADred-imine) may first dissociate, liberating the imine, which hydrolyzes to yield the products aldehyde and ammonia. Finally, the reduced enzyme (MAO-FADred) is reoxidized by atmospheric oxygen to form hydrogen peroxide. Thus, the MAO reaction consists of a reductive and an oxidative half reaction, in which the oxidation state of the enzyme-bound FAD cycles between FADox and FADred (Fig. 2). The reductive half-reaction is initiated by a cleavage at the C–H bond of the pro-chiral carbon atom of the amine moiety. The stereochemistry of hydrogen abstraction (pro-R hydrogen removal) is well defined [10], and experiments with bovine MAO-B suggested that hydrogen tunneling may be involved [11]. MAO-catalyzed oxidation of stereospecifically labeled substrates revealed pronounced kinetic isotope effects [12, 13], suggesting that C–H bond cleavage might constitute the rate-limiting step of the catalytic cycle. In principle, initial hydrogen abstraction may occur via three different mechanisms: (1) heterolytic hydride transfer, (2) heterolytic H+ abstraction and (3) homolytic removal of a hydrogen radical (H•). There are pros and cons for each of these mechanisms, and thus, the detailed chemistry of the different elementary reactions remains to be worked out.

Kinetic scheme of MAO reaction indicating reductive and oxidative half-reactions
Another unsolved problem of the reaction mechanism of MAO isoforms is the question of the mode of interaction between MAO substrates and the flavin cofactor. Most experts agree that the deprotonated amine moiety of MAO substrates binds to the active site. The deprotonated amines are then oxidized to the protonated imine, and the oxidized FAD cofactor is reduced to its hydroquinone form. Since deprotonated amines do not appear to be sufficiently nucleophilic to readily add to FAD in model systems, there appear to be factors in the protein facilitating this type of reaction. Structural data of various MAO isoforms suggest that the flavin cofactor at the active site of the enzymes may not be present as a planar moiety. Instead, the isoalloxazine ring exhibits a bent conformation (Fig. 3c) in the oxidized form of the enzyme, to which the substrate is bound. Such bending is likely to induce rearrangements in the electron density distribution of the alloxazine ring [14]. In addition, tyrosyl residues in the proximity of the substrate-binding site (Tyr435 for MAO-B, Tyr444 for MAO-A) have been suggested to increase the nucleophilic character of the amine nitrogen of the substrates, and these two factors may contribute to enabling a nucleophilic attack of the deprotonated amine substrate on the covalently linked FAD cofactor [14].

Structural aspects of human MAO isoforms. a Chromosomal localization of human MOA isoforms. b Amino acid sequence alignment of human MAO isoforms. Differing amino acid residues are highlighted by cyan boxes. c Overlay of the 3D structure of human MAO isoforms. MAO-A is shown in green, and MAO-B is shown in cyan. The FAD group is highlighted in orange. d Comparison of the active site cavities of the human MAO isoforms. MAO-A is shown in green, and MAO-B is shown in cyan. The cavities of MAO-A and MAO-B are represented by blue or green dots, respectively. The FAD group is shown in orange
Structural biology of MAO isoforms
The two MAO isoforms are encoded for by separate genes (Fig. 3a), which are head-to-tail tandemly arranged on the X chromosome (Xp11.4-p11.3). On the protein level, the two isoforms share a high degree (73 %) of amino acid conservation (Fig. 3b). MAO-A contains an extra stretch of nine amino acids close to its N terminus, which is lacking in MAO-B. The three-dimensional structures of the two human isoforms and rat MAO-A have been solved [15–20]; as expected from the high degree of amino acid conservation, their 3D-structures are almost super imposable (Fig. 3c). Since MAO isoforms are anchored in the outer mitochondrial membrane, they contain a membrane-binding motif, and this transmembrane helix is located in the C-terminus of the proteins. In human MAO-B, this region folds into an R-helix protruding perpendicularly from the main globular body of the protein. It should, however, be stressed that the last 20 residues of this motif are too disordered to provide definitive electron density [15]. The C-terminal part of human MAO-A also folds into a transmembrane R-helix [17]. Despite these structural similarities, mutagenesis studies interchanging the membrane binding motifs between MAO-A and MAO-B lead to inactive enzyme species [21, 22], suggesting that there may be subtle differences in the membrane binding structures between the two isozymes.
The protein structures of the active site where the FAD coenzyme is covalently attached are quite similar among the three known MAO structures. The most prominent structural difference between human MAO isoforms is that the X-ray coordinates indicate a MAO-A monomer [16], whereas MAO-B is present as a homodimer [15]. However, rat MAO-A has also been detected as a homodimer [19]. Many membrane proteins form homodimers when arrested in a lipid bilayer, and thus the question arose of whether the monomeric character of human MAO-A might constitute a crystallization artifact. To explore whether MAO isoforms are present as homodimers in their natural environment, pulsed EPR experiments with nitroxide spin-labeled pargyline analogous covalently attached to the FAD coenzyme were carried out. With this technique one can measure distances between the two paramagnetic species in membrane preparations. These data suggest that both human and rat MAO-B are found in their native environment as homodimers, whereas a 1:1 monomer–dimer equilibrium appears to exist for human and rat MAO-A [23].
The crystal structures of both human and rat MAOs revealed that substrate binding and oxidation occur in a more compact cavity in the case of MAO-A and a more bipartite cavity in MAO-B. The respective cavities extend from the flavin binding site at the core of the enzyme to the protein surface. The volume of the reaction cavity of human MAO-A is about 400 Å3. The volume of the corresponding cavity in human MAO-B is about 700 Å3 [14]. The MAO-B cavity consists of two subcavities: the substrate-binding cavity (400 Å3) and the entrance cavity (300 Å3). Whether the active site of MAO-B constitutes a large single space or a bipartite cavity depends on the conformational state of Ile199 (Fig. 3d). The side chain of Ile199 adopts two different conformations [20, 24] depending on the nature of the bound ligand. The corresponding residue in human MAO-A (Phe208) does not appear to function as a gating residue. Exchange of Ile199Phe in human MAO-B suggests that the bulky Phe side chain prevents conformational flexibility, reducing the space of the entrance subcavity and impairing the binding of MAO B-specific inhibitors [25].
Rat MAO isoforms have frequently been used as tools for screening for potential inhibitors of the human enzyme. Despite their high levels of sequence homology (>90 %), there have been reports in the literature indicating that rat enzymes exhibit different affinities towards various compounds relative to the human enzymes. The 3D structural data of human and rat MAO isoforms [16] and the possibility of motional differences between the two proteins either in their soluble or membrane-bound forms have suggested subtle interspecies differences [14]. Thus, care should be taken if data collected with rodent enzymes are translated to the human situation.
Reaction kinetics and specificity
The affinity of the two MAO isoforms towards their biologically most important substrates (serotonin for MAO-A, dopamine for MAO-B) is not particularly impressive, as indicated by the high K M values ranging in the three-digit micromolar range (Table 1). For yeast d-amino acid oxidase, which also contains FAD as a catalytically important co-factor at the active site, the substrate affinity is even lower (K M = 2.6 mM). These data are in sharp contrast to the substrate affinity of oxygenases (cyclooxygenase and lipoxygenase isoforms), which do not contain FAD as a redox mediator (Table 1). The structural basis for the low substrate affinity of the two MAO isoforms remains to be clarified, but it can be speculated that it might be related to the inefficient interaction of the deprotonated substrate with the FAD moiety. On the other hand, the low substrate affinity of the two MOA isoforms might be of biological relevance, since biogenic amines are usually present in biological systems at nanomolar concentrations [26, 27]. These data suggest that the in vivo catalytic activity of MAO-A/B is mainly controlled by substrate availability. It is of course always possible that enzyme substrates are enriched in certain cellular compartments, such as intracellular vesicles, but for the time being there is no indication for such enrichment of MAO substrates at the outer mitochondrial membrane, where the two isoforms are located.
As indicated earlier, MAO isoforms exhibit different substrate and inhibitor specificities. Structure–activity studies on isoform-specific MAO inhibitors suggest that planar compounds with a high degree of π-electron conjugation exhibit high specificities for reversible inhibition of human MAO-B, but they hardly affect the catalytic activity of MAO-A [25, 28]. Detailed investigations into the 3D structures of the two human MAO isoforms provide a plausible explanation for this behavior. As indicated above, MAO-B contains a large bipartite active site, which consists of a central substrate binding cavity and a peripheral entrance cavity intersecting with the protein surface (Fig. 3d). In contrast, the substrate-binding cleft of MAO-A is smaller and only monopartite [25]. Binding of small inhibitors at the active site of MAO-B induces rotation of Ile199, which closes the internal substrate-binding cavity and separates it from the entrance cavity. Permanent opening of the gate, which is achieved by an Ile199Ala exchange, results in an impaired binding affinity of smaller inhibitors that only bind in the internal substrate cavity. MAO-B-specific inhibitors that traverse both the entrance cavity and the substrate cavity force an opposite rotation of Ile199, which leads to fusion of the two subcavities. Thus, the Ile199Ala mutation has a positive effect on the binding of more space-filling inhibitors, which is reflected by an increased binding affinity [24].
Among the naturally occurring amines, the preferred substrate for MAO-A is 5-hydroxytryptamine (5-HT, serotonin). In contrast, β-phenylethylamine (PEA) is frequently used as a MAO-B-specific substrate for isoform-specific activity assays [27, 29]. Since the biological relevance of PEA is still unclear and it does not occur in vivo in substantial concentrations, DA is considered the biologically most relevant endogenous substrate for MAO-B. This is mirrored inter alia by the fact that endogenous DA deficiency (Parkinson's disease) can be treated with selective MAO-B inhibitors [30]. It should, however, be stressed that the isoform specificity of MAO substrates is relative. As indicated in Table 1, MAO-A prefers 5-HT (catalytic efficiency of 11 l/mol s) over DA (catalytic efficiency of 5 l/mol s). In other words, MAO-B also oxidizes 5-HT, and the isoform-specificity coefficient (catalytic efficiency of MAO-A vs. catalytic efficiency of MAO-B) is only about 2. On the other hand, MAO-B prefers DA (catalytic efficiency of 36 l/mol s) over 5-HT (catalytic efficiency of 0.4 l/mol s). Here the isoform-specificity coefficient of 90 is much more pronounced. It should, however, be stressed that these calculations are based on in vitro activity assays obtained with purified human enzyme species under defined reaction conditions employing fixed substrate concentrations. It remains unclear whether the enzymes in their native environments may exhibit similar kinetic properties when substrate concentrations are regulated.
The oxidative half-reaction of MAO catalysis involves reduction of atmospheric dioxygen by the hydroquinone form of the FAD. A key difference between MAO-A on one hand and MAO-B on the other is that the K M for O2 with MAO-B is about 240 μM (57), while the corresponding value for MAO-A is 12 μM (78). Accordingly, MAO-A is operating at saturating oxygen concentrations under normoxic cellular conditions (about 250 μM), whereas MAO-B is less than half saturated. Because of this kinetic peculiarity, MAO-B may function as an oxygen sensor. Depending on the local oxygen concentration, the enzymatic activity is up- or downregulated, and thus the cellular H2O2 concentration is regulated. In other words, depending on the local oxygen concentration, MAO-B modifies the cellular redox state, which impacts the functional phenotype of the cell. Adaption of cellular metabolism to variable oxygen concentrations is essential for embryo development, and hypoxia [31] is a major risk factor for embryonic lethality and teratogenesis [32]. The relative importance of oxygen at different stages of embryogenesis is variable and depends on the anatomic environment the embryo is exposed to. Thus, the developing embryo must be able to cope with these challenges by adapting its metabolism to the variable oxygen concentrations [33]. Oviductal oxygen levels are lower than atmospheric oxygen concentrations [34], and in the uterus, where implantation takes place, even lower oxygen concentrations have been determined [35]. In fact, following fertilization in the oviduct, the embryo encounters a decreasing oxygen gradient when moving down the reproductive tract, and at early implantation the embryo is confronted with an almost anoxic environment [34]. These differences in oxygen concentrations clearly alter the energy metabolism of the developing embryos. But the regulatory events involved in these adaptive responses are not completely understood. For the switch from aerobic to anaerobic energy supply, oxygen sensing might be important and, in addition to HIF-related regulatory mechanisms [36], MAO-B may play a role as oxygen sensor.
The molecular basis for the difference in oxygen affinity of the two MAO isoforms remains unclear but may be related to a steric hindrance of intra-enzyme oxygen diffusion in MAO-B. Molecular dynamics simulations and implicit ligand sampling have recently identified sites of high affinity for molecular dioxygen in D-amino-acid oxidase (DAAO). According to these data [37], a dynamic channel for oxygen diffusion leads from the periphery of the protein to the flavin-binding site. Based on modeling data, amino acids flanking the putative oxygen access channel have been exchanged with bulky residues to introduce steric constraints. In the G52V mutant, the valine side chain occupies the high oxygen affinity region. In this enzyme variant, the reactivity of the reduced enzyme with oxygen is impaired more than 100-fold, and the turnover number is down 1,000-fold, proving the correctness of this concept. Similar data have previously been published for rabbit arachidonate 15-lipoxygenase (ALOX15), a non-flavin oxygenase, suggesting the existence of an oxygen access channel in other types of oxygenases [38]. If steric hindrance of intraenzyme oxygen diffusion is the structural basis for the different oxygen K M values of the two MAO isoforms, similar modeling investigations should prove or disprove the concept. On the other hand, X-ray crystallographic data on MAO-B in the presence of xenon (X-ray oxygen probe) suggested that oxygen may bind inside the substrate-binding cavity and at the cavity entrance [14]. These data suggest that oxygen employs the same path for penetrating to the active site as the amine substrates. If this is the case, the differences in oxygen affinity of the two MAO isoforms is difficult to explain on the basis of steric hindrance of intraenzyme oxygen movement, since the substrate-binding pocket of the two isoforms is big enough to allow unhindered oxygen diffusion.
Cellular and physiological functions of MAO enzymes
The biological function of monoamine oxidases has been mainly defined by their enzymatic activities on biogenic amines and the concomitant production of hydrogen peroxide. Thus, the impact of MAO enzymes has been discussed dominantly with respect to amine metabolism on one hand and to the production of hydrogen peroxide on the other. Little attention, however, has been paid to the other products of the MAO reaction, namely ammonia and aldehydes. Both, ammonia as well as the corresponding aldehydes that are produced by the oxidative deamination may affect cellular metabolism and cellular functions. The two MAO isoforms (MAO-A and MAO-B) that are known in mammals are each characterized by unique enzymatic properties as outlined in the previous section. In addition, each isoform seems to serve specific functions in the organism as indicated by their distinct isoform-specific expression profiles. However, whereas the role of MAO-A as a control enzyme of neurotransmitter levels in particular of 5-HT is well established, the biological role of MAO-B is much less well understood. The following paragraphs will illuminate the various perspectives of MAO research on the biological implications of MAO activities.
MAO enzymes are ubiquitously expressed to maintain physiological amine levels
Research on MAOs has been focused in the past on their roles in the central nervous system, since both isoenzymes are expressed in various cell populations of the brain [39]. Whereas the major sites of MAO-A expression are adrenergic and noradrenergic neurons of the locus coeruleus, MAO-B is dominantly expressed in serotonergic neurons of the raphe and in histaminergic neurons of the posterior hypothalamus as well as in astrocytes [39–41]. The expression of both isoforms in the brain has highlighted their importance in the regulation of neurotransmitter homeostasis in particular of 5-HT and DA [26, 27]. Hence, MAOs have been recognized as important pharmacological targets in the treatment of neurological disorders and neurodegenerative diseases that are characterized by abnormal amine metabolism [4, 30].
In addition to the central nervous system, most peripheral tissues have been found to contain both MAO enzymes [42]. Both isoenzymes are highly expressed in the gastrointestinal tract and in the liver, where they are thought to be vital for the degradation of ingested alimentary amines that might otherwise exhibit deleterious effects on the organism [43]. In other words, MAO enzymes are believed to function as a firewall against alimentary amine bombardment. Pharmacological inhibition of MAO-A activity may result in a fatal hypertensive crisis when the sympathomimetic amine tyramine is ingested [44]. On the other hand, 5-HT, which is degraded by MAO-A, is vital for the regulation of intestinal mucosal function [45]. Despite the fact that expression levels of MAOs in the gastrointestinal tract exceed those in the brain, surprisingly little is known about the roles of MAOs in this organ.
Both MAO isoforms are also found in the kidney where they are involved in the regulation of blood pressure [46]. Expression of MAO-A in the syncytiotrophoblast of the placenta and in interleukin-4-stimulated monocytes, and the expression of MAO-B in platelets and lymphocytes are not entirely understood but have been related to the clearing of endogenous and xenobiotic vasoactive amines [42, 47–50]. Indeed, impaired MAO-A expression in placenta correlates with the occurrence of pre-eclampsia, which is characterized by hypertension [49], but the detailed pathomechanisms have not been completely clarified.
Expression regulation of MAO enzymes
As indicated by their differential expression patterns, both MAO isoforms are likely to share regulatory mechanisms as well as possess distinct pathways that control isoform-specific MAO expression. The two MAO genes have been mapped to a chromosomal region that sometimes is included in the chromosomal aberrations found in patients suffering from Norrie’s disease [51]. In addition, the two MAO genes have a very similar structure of 15 exons and 14 introns, with exon 12 coding for the highly conserved flavin-binding domain. The putative promoter regions of the MAO genes are both characterized by GC-rich sequences that have been shown to interact with proteins of the transcription factor Sp1 (stimulating protein 1) protein family [52], in general, GC-rich promoter sequences (CpG islands) and transcriptional control by the ubiquitous transcription factor Sp1 hallmark promoters of housekeeping genes [53, 54], which is consistent with the ubiquitous expression of the MAO enzymes. Epigenetic modification of the CpG island of the MAO-B promoter by methylation was shown to inhibit MAO-B expression [55], and similar regulatory mechanisms are expected for the MAO-A gene [56]. The Sp1-binding sites of the MAO promoters also interact with the transcriptional repressor CDCA7L (cell division cycle associated 7-like) [57], and CDCA7L overexpression was shown to repress MAO-A expression [7, 57]. The Sp1-binding sites of the MAO-B promoter also carry potential binding sequences for CDCA7L, but its impact on MAO-B expression has not been reported so far.
In addition, MAO-A expression is known to be positively regulated by the transcription factor SRY (sex-determining region Y) ([58]. SRY expression is important for gonadal development [59], but its impact on MAO-A expression in vivo or an involvement of MAO-A in the development of the male and female reproductive tracts have not yet been demonstrated. Similarly, expression of the two MAO genes is under the influence of hormones and cytokines, such as glucocorticoids, retinoic acid, estrogens and interleukin-4 [48, 60–62]. However, although the cis- and trans-regulatory elements that translate cellular signaling into changes in MAO expression have been explored, the physiological implications of these observations are still elusive. In addition to these well-established cis-regulatory DNA elements, expression of MAO-A is affected by a polymorphism within its proximal promoter region [63]. Whereas the precise molecular mechanisms underlying this observation have not been fully understood, this anomaly has been related to the incidence of mood and panic disorders [64–68].
In conclusion, a series of experimental data exist to describe the regulatory elements in the promoters of the MAO genes that affect gene transcription. However, there is no general concept of expression regulation of either isoform. Moreover, little is known about post-transcriptional regulation of MAO expression, and post-translational modifications of MAO proteins have only recently been revealed [69]. Post-transcriptional mechanisms however have been suggested in a number of conditions, under which MAO mRNA levels do not correspond to expression of the corresponding protein [70–72]. Moreover, given the distinct molecular mechanisms that appear to drive tissue-specific expression of MAO enzymes, care must be taken when extrapolating cerebral MAO expression from expression levels in other cell types such as platelets, which are more easily accessible to analysis. Such an approach has occasionally been applied to predict cerebral MAO-B expression in psychiatric and neurological diseases and personality disorders [73–75].
MAO expression affects cellular redox homeostasis
As outlined above, MAO expression has mainly been related to their ability to deactivate amines in the various tissues. However, MAO activity is always accompanied by the production of hydrogen peroxide (Fig. 1). Hydrogen peroxide together with the superoxide anion (O •−2 ) and the hydroxyl radical (OH•) is classified as reactive oxygen species (ROS). ROS have dual biological functions (Fig. 4). Traditionally, ROS have been considered potent biological hazards that cause oxidative modifications of virtually all macromolecules such as nucleic acids, lipids and proteins [33]. Such oxidative modifications may impact the functionality of these molecules and may eventually induce cell death [76]. Interestingly, MAO proteins themselves have been shown to be targeted by oxidative modifications, which cause their subsequent deactivation [77]. On the other hand, it has recently become apparent that the cellular peroxide tone is an important regulatory element of intracellular signaling cascades and of gene expression [33]. Because of this regulatory function the redox equilibrium of a cell is maintained and precisely adjusted by an array of pro-oxidative and antioxidative mechanisms [33]. Production of hydrogen peroxide by MAO activity has been shown to alter this cellular redox equilibrium in several cellular models including renal epithelium [78], myocytes [62, 79] and neurons [72]. Remarkably, expression of MAO-A itself is induced by peroxides in monocytic cells [80], suggesting the existence of positive loops of regulatory feedback.

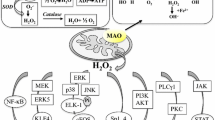
Functional implications of MAO enzymes in cellular signaling pathways. MAO activity results in the production of aldehydes, ammonia and the reactive oxygen species (ROS) hydrogen peroxide (H2O2). ROS can be scavenged by antioxidative enzymes such as glutathione peroxidase (GPx), superoxide dismutase (SOD) and catalase. If the cellular anti-oxidative capacity is exhausted, ROS may cause unordered cell death. On the other hand, ROS second messenger signaling may alter the activity of mitogen-activated protein kinases (MAPK) or of transcription factors. These changes in turn are directly or indirectly via gene expression regulation (dashed arrows) relayed to the proteins that regulate cell proliferation (cyclin D) or apoptotic cell death (Bcl-2 proteins). Further information is found in the main text
Mediators of cell signaling and gene expression regulation that are affected by the cellular peroxide tone include protein kinases such as ERK (extracellular signal-regulated kinase) and JNK (c-Jun N-terminal kinase), and the transcription factors NF-kappaB and AP1 (activator protein 1) [33, 81, 82]. In particular, activation of ERK1/2 through hydrogen peroxide generated by MAO activity has been shown in several reports [72, 78, 83]. Activation of ERK1/2 results in its translocation from the cytoplasm into the nucleus, where it can alter gene expression by phosphorylation of a number of target proteins [84]. Targets of ERK1/2 activation are pro-survival factors such as cell-cycle regulators [85]. Indeed, hydrogen peroxide generated by MAO-A activity was found to stimulate cell proliferation in various cellular models [78, 86]. On the other hand, oxidative stress produced by MAO activity promoted cell death in neuronal and cardiac cells [72, 86]. Thus, the effect of MAO activity on cellular functions depends on the amount of hydrogen peroxide produced and the capacity of the cells to cope with increased levels of hydrogen peroxide [87] (Fig. 4). Dopaminergic neurons in the substantia nigra of Parkinson patients exhibit impaired antioxidative capacities, as indicated by low levels of reduced glutathione [88]. In such cells, a relatively modest increase in MAO activity is expected to lead to pronounced functional alterations. In contrast, cells with a high antioxidative capacity may be less sensitive to redox alterations induced by MAO overexpression. It should be stressed at this point that redox-dependent cell signaling is always coupled with other signaling mechanisms, and in some cases the cellular targets are the same. For instance, in vascular smooth muscle cells hydrogen peroxide acts in concert with 5-HT via activation of 5-HT cell surface receptors to activate ERK1/2 and cause its translocation into the nucleus [79].
In conclusion, MAO activity results in the generation of cellular oxidative stress, which in turn is translated into altered cellular signaling and gene expression (Fig. 4). However, care must be taken when discussing the biological role of this highly reactive product of MAO activity. Depending on the relative hydrogen peroxide concentrations and on the activity of antagonizing pathways, the outcome of signaling induced by hydrogen peroxide can be diametrically opposed (Fig. 4).

Development of monoamine systems in pre- and post-implantation embryo
MAO activity modulates the intrinsic pathway of apoptotic cell death
MAO activity has also been associated with the induction of cell death. Cell death can either occur in a largely unregulated manner (necrosis) or following a highly ordered process (apoptosis) [89]. Signaling pathways of apoptosis can be subdivided into the extrinsic pathway, which relays signals from cell surface receptors to the intracellular apoptotic machinery, and the intrinsic pathway, which depends on mitochondrial integrity. Apoptosis induced by either pathway often results in the activation of the effector caspase-3 [90]. The extrinsic pathway translates signals from cell death receptors at the cell surface via activation of caspase-8 to the effector caspase-3. In contrast, the intrinsic pathway is induced by mitochondrial dysfunction, which leads to the liberation of cytochrome C from the intermembrane space into the cytosol and subsequently to the activation of caspase-9 [91]. Alternatively, a stress response in the endoplasmic reticulum may activate caspase-12, which then activates downstream effector caspases [91, 92].
Their prime position at the outer mitochondrial membrane makes MAO enzymes excellent candidates for regulators of the intrinsic pathway of apoptosis. The key event of the intrinsic pathway—the release of cytochrome c—is regulated by the complex interaction of pro- and anti-apoptotic mitochondrial proteins, in particular of proteins of the B cell lymphoma-2 (Bcl-2) protein family [92]. In addition, the balance between mitochondrial ROS production and the activity of ROS scavenging enzymes including superoxide dismutase (SOD) and glutathione peroxidase 4 (GPx4) modulates this key event of apoptotic signaling [93, 94]. GPx4 activity for instance suppresses apoptotic cell death by reducing the hydroperoxides of cardiolipin, a mitochondria-specific phospholipid [94]. Cytochrome c is retained in the mitochondrial intermembrane space through its association with cardiolipin. This binding is impaired when cardiolipin is oxidized to cardiolipin hydroperoxides. MAO activity on the other hand generates peroxides on the mitochondrial surface, and thus, MAO-A and GPx4 may be considered antagonizing enzymes. Upregulation of MAO-A expression may tip the finely tuned mitochondrial redox equilibrium in favor of pro-oxidative events, which might impair respiratory chain and other mitochondrial functions [95, 96]. In addition, hydrogen peroxide produced by MAO activates protein kinases such as JNK [86]. This kinase in turn initiates signaling events that lead to the induction of pro-apoptotic proteins of the Bcl-2 protein family [97]. Protein kinases of the MAP kinase family also affect MAO gene expression itself. MAP kinase signaling was shown to induce MAO-B expression via transcription factors c-Jun and Egr-1 [98–100], and MAO-A via silencing of the transcriptional repressor CDCA7L [101]. In addition, the MAP kinase p38 was recently shown to inhibit MAO-A activity directly by MAO protein phosphorylation [69].
Taken together, these data implicate MAO at various levels of apoptotic signaling. This includes direct action of ROS produced by MAO on mitochondrial integrity, modulation of MAP kinase signaling and redox sensitive gene expression regulation. In turn, MAO expression and activity are controlled by redox-sensitive MAP kinase signaling and transcription factor activity. These overlapping regulatory circuits make it very difficult to directly translate in vitro phenomena to the in vivo situation.
Monoamine systems during normal embryo development
Monoaminergic transmitter systems are functional in the developing embryo and play an important role in pre- and postnatal development [102]. During embryogenesis monoamines have been detected in pre- and post-implantation stages, and a summary of their activities is given in Fig. 5 and Table 2.
Monoamines in preimplantation embryos
The development of pre-implantation embryos is controlled by an intrinsic genetic program as well as by environmental factors. The roles of monoamines in early embryo development depend on the developmental stage of the embryo and on the physiological state of the maternal organism [103]. Monoamines have been identified in follicular and oviductal fluids [104–106]. NE and DA modulate the biosynthesis of prostaglandins, which are essential for implantation of the embryo and for survival of the blastocyst [107]. 5-HT has been detected in oocytes, cumulus cells, zygotes and blastocysts [108]. In early embryos, 5-HT originates from both endogenous sources of the embryo and active uptake from the surrounding tissues and fluids. Exposure of early embryos to 5-HT or a specific 5-Htr-1d receptor agonist significantly reduces the cell number and increases the incidence of dead cells [109]. Abnormalities in blastocyst formation have been observed when embryonic 5-HT levels are reduced by inhibition of 5-HT biosynthesis [110].
Monoamine receptors are already expressed in embryonic stem cells, and they retain their functionality throughout pre-implantation embryogenesis [111]. Activation of adrenergic and 5-Htr-1d receptors in ovulated oocytes [112], 5-Htr-7 in oocytes and 5-Htr-7, 5-Htr-2a and 5-Htr-2b in ovarian cumulus cells [113] appear to regulate proliferation and survival of early embryonic cells. Activation of α2- and various subtypes of β-AR regulate proliferation and apoptosis in various cell types of pre-implantation embryos, and a variety of signaling pathways involving MAPKs, Akt kinase and Src kinase appear to be involved [114]. β-Adrenergic or α2-adrenergic agonists induce a developmental retardation in early embryos and lower the mean embryonic cell number [115]. Since MAO isoforms are involved in the metabolism of these monoamines up- or downregulation of MAO expression may interfere with these signaling events.
As indicated above, embryonic monoamines originate from embryonic as well as maternal biosynthesis. Immunohistochemical staining of cells expressing biosynthetic enzymes and/or monoamine transporters may help to identify embryonic sources of these mediators. Tryptophane hydroxylases (TPH1 and THP2), which are key enzymes of 5-HT biosynthesis, are expressed in ovarian cumulus cells and oocytes, respectively, and these data suggest autocrine and/or paracrine regulatory mechanisms [116]. Expression of the 5-HT-specific transporter SLC6A4 in oocytes and preimplantation embryos from zygote to blastocyst stage indicates a 5-HT transfer from the environment. Systemic administration of MAO inhibitors during pregnancy elevates maternal NE and 5-HT levels, which leads to developmental retardations of the embryo [117].
Monoamine systems in post-implantation embryos
In various post-implantation stages of embryogenesis monoamines are important for shaping and wiring of the nervous system [118]. They are present during early neuronal differentiation and function as humoral morphogens in the initial stages of neurogenesis. They regulate the time course of neuronal development and program the complex innervation of different organ systems. In later stages of embryogenesis, monoamines become neurotransmitters [119, 120]. This dual function of monoamines and the exact timing of the functional switch of monoamines from humoral morphogens towards neurotransmitters is important for the plasticity of the immature brain and subsequently for memory storage and preservation in the mature brain [121].
Monoamine substrates
As indicated above, monoamines appear in early embryo development before the onset of neurogenesis [122, 123]. This indicates that their developmental roles may not be restricted to the nervous system. In neurons monoamines can be visualized in the cell bodies before axon growth is completed. Within embryonic cerebrogenesis, monoamines appear first in the caudal parts of the brain. Later on, monoamine nerve terminals can be found throughout the entire nervous system, in particular in the regions of the hypothalamus and striatum.
High concentrations of catecholamines have been found in the notochord of mammalian embryos [124]. Catecholaminergic neurons prevail in the developing brainstem and in the hypothalamus. The concentrations of catecholaminergic neurotransmitters increase concomitantly with synapse formation. In late embryogenesis, a surge of catecholamines, particulary of NE, is important for neonatal adaptation at birth [125]. NE neurons develop in the locus coeruleus before 5-HT neurons appear in raphe nuclei [126]. They innervate not only hippocampal pyramidal cells, but also hippocampal polymorph cells and cerebellar Purkinje cells. NE regulates the development of Cajal-Retzius cells, and is important for neuronal migration and laminar formation [127]. Some of the cells in the neuronal crest contain NE during early embryonic development, but become cholinergic later on. These cells dominate in the peripheral nervous system [128]. Depletion of NE from developing embryos during the perinatal developmental period results in subtle changes of dendrite formation and in alterations within cortical differentiation [129]. Dopaminergic neurons appear early on during embryo development [130]. Administration of 6-OH-DA prevents developmental apoptosis of neurons and delays the formation of cortical layers [129].
After midgestation 5-HT-producing cells of the embryo start to synthesize 5-HT. However, cerebral 5-HT concentrations are mainly regulated by the reuptake of maternal 5-HT via 5-HT transporters (5-HTT, see below) and by the degradation of 5-HT (MAO-A, see below) [131]. During early neurulation, 5-HT is mainly found in notochord and the floor plate of the neural tube as well as in adjacent somites and the primitive gut [119]. Within the floor plate, 5-HT is localized predominantly within the mesencephalon and the otic level of the myelencephalon extending caudally into the cervical neural tube. In early embryogenesis, 5-HT regulates cell proliferation, embryonic morphogenesis, gastrulation, neural crest migration, craniofacial and limb development, mesenchymal cell growth and differentiation, specification of neuronal identity and connectivity, bone patterning and generation of body asymmetry [132]. Craniofacial malformations are observed when embryos are cultured in the presence of 5-HT uptake inhibitors or receptor ligands. During mid gestation, 5-HT neurons appear in raphe nuclei later than NE neurons in the locus coeruleus, but earlier than DA neurons in the substantia nigra [126]. In dorsal and medial raphe nuclei, 5-HT neurons innervate neuronal cells in the superior colliculus and stratum griseum superficiale as well as hippocampal pyramidal cells [133]. At birth, 5-HT neurons are present in all cortical layers, but the number of these cells declines significantly after birth. Interference with 5-HT signaling induces variable effects in perinatal brain development. Blocking uptake of 5-HT mediated by 5-HTT during embryogenesis causes wiring defects, which are most prominent in the sensory nervous system. Injection of a 5-HT-specific toxin (5,7-dihydroxytryptamine) into the forebrain bundle of newborn mice produces numerous defects including age- and region-specific increases in the width of the cerebral cortex, impaired social learning and impulse control, increased repetitive behavior and aggression, hyper-responsiveness to environmental changes and impaired fine motor performance [134]. In contrast, depletion of 5-HT after birth seems to have little impact on cortical development. Low maternal plasma 5-HT levels induce autism by retarding embryonic brain development [135].
β-Phenylethylamine (PEA) is a phenylalanine-derived biogenic amine synthesized in the brain, serving as a neuromodulator [136]. It induces biological effects at nanomolar concentrations by binding to trace amine receptors [137]. The origin of PEA and its role in embryogenesis have not been clarified. On the other hand, PEA derivatives are used as psychoactive drugs [138]. PEA has been implicated in the pathophysiology of various neurological disorders such as depression, schizophrenia and hyperactivity disorder [139]. PEA injection in adult mice [140] and guinea pigs [141] induced dyskinesias. This effect has been related to oscillations of the dopaminergic system in neurons [142] and to excessive generation of hydroxyl radicals in the striatum [143]. In adults, long-term consumption of PEA-containing foods, such as chocolate and wine, is considered a risk factor for depression and motoric dysfunction [143]. However, the origin of PEA and its role in embryogenesis has not been clarified, and the impact of alimentary intake of PEA on embryogenesis has not been explored in detail. Since PEA is the preferred in vitro substrate of MAO-B, mice deficient in MAO-B expression have strongly increased PEA levels, which may be the reason for the abnormal behavioral phenotypes in these animals [27].
Monoamine receptors
To induce their biological effects, monoamines bind to cell surface membrane receptors [144]. According to their mode of action there are two principal kinds (metabotropic and ionotropic) of receptors. Metabotropic receptors are expressed at early stages of embryogenesis and play a more modulatory role in the mature brain. Ionotropic monoamine receptors are mainly expressed at later stages of embryogenesis.
5-HT is the native ligand for 20 different receptor subtypes, which are classified in 7 subclasses, 5-Htr-1 to 5-Htr-7 [145]. At gastrulation, 5-Htr-2b is expressed in neural crest progenitor cells, in proliferating cells of the myocardium and in somites of mice [146]. Catecholamines bind to nine different adrenergic receptors, classified into α1, α2, and β subtypes, and to six subtypes of DA receptors, classified into D1-like and D2-like receptors. Blocking NE projections or neurotransmission with receptor antagonist prevents astrogliosis and glial cell proliferation [129]. The extremely high levels of D1 receptors in the pallidum during perinatal developmental period are important for the development of motoric and cognitive functions [147].
Monoamine transporters
Unlike most neurotransmitters, monoamines are frequently recycled by monoamine transporters in addition to being degraded intracellularly by monoamine oxidases [148]. In most cases the affinity of the metabolizing enzymes is lower than that of the vesicular transporters, and thus, at a limiting monoamine concentration the uptake exceeds the extent of intracellular breakdown [149]. Monoamine transporters are localized in the plasma membrane and mediate the re-uptake of monoamine neurotransmitters from the synaptic cleft. There are specific transporters for EN, NE, DA and 5-HT, and each transporter is characterized by its unique substrate specificity, by its ion dependence and its sensitivity towards specific inhibitors [150]. 5-HT transporter (known as SERT or 5-HTT) re-internalizes 5-HT, which was secreted into the synaptic cleft [151]. In mice, expression of the 5-HTT gene starts at midgestation in 5-HT neurons of the raphe nuclei, but expression soon extends to non-serotonergic neurons, including the principal projection neurons of the sensory systems (thalamus, retina, somatosensory cortex) and the corticolimbic centers (hippocampus in the prenatal period, prefrontal/cingulate cortex postnataly). 5-HTT expression in non-serotonergic neurons stops suddenly during the second postnatal week, which coincides with the development of neuronal circuits [152]. In addition to the plasma membrane transporter, there are vesicular monoamine transporters (VMAT1 and VMAT2) inside the neurons. These transporters are responsible for transferring newly synthesized or re-internalized 5-HT into intracellular vesicles. In rats, VMATs are found in the developing neural tube as early as E8 [153].
Monoamine oxidases
Expression of the MAO isoforms has been monitored in different embryonic stages of several mammalian species. In human fetuses the catalytic activities of both isoenzymes were detected [154]. Here, MAO-A appears to be the dominant isoform in most tissues, in particular in the gastrointestinal tract, skin, aorta, lung and the brain. In contrast, MAO-B activity prevails in the heart and liver of human fetuses [154]. After birth, MAO-B expression appears to catch up. In fact, in particular in the brain and in the skin, MAO-B becomes the dominant isoform in adults. Similar data were obtained when the mRNA levels of the two MAO isoforms were profiled. Both mRNA species were detected in most embryonic tissues apart from thymus and adrenal gland, which express neither MAO isoform. In midtrimester human fetuses, very high expressions of both mRNA species were observed in the gastrointestinal tract, kidney, liver, lung and the spinal chord. In human brains, expression of both mRNA species was detected in most parts before birth, but no MAO mRNA expression was detected in the thalamus, hippocampus or striatum [155].
The dominance of MAO-A over MAO-B activity before birth was confirmed in developing rodents [7, 156, 157]. Here again MAO-B becomes the dominant isoform in the organism after birth. Embryonic expression of the two MAO isoforms was detected as early as at gestational day E6.5 (Fig. 7a, b) [7], albeit the levels of expression were higher for MAO-A. In the developing mouse brain mRNA expression of both MAO isoforms was observed from gestational day E10.5 (Fig. 6a, b). However, MAO-A is the dominant isoform in this tissue. In the developing brain expression of both isoforms follows very similar expression kinetics, and expression levels drop below the threshold of detection towards birth. Only immediately around birth MAO mRNA expression becomes detectable again. At E12 MAO-A expression was observed in noradrenergic, adrenergic and dopaminergic cells [158]. Coexpression of both isoforms was observed in serotonergic neurons of the raphe [158]. After birth activities of both isoforms further increase in maturing brains; similar observations are made in both humans and rodents [154, 156]. Surprisingly, in human brain MAO-A mRNA levels decline during aging, and post-transcriptional regulatory mechanisms have been suggested to cause this phenomenon [71].

Expression of MAO-A and MAO-B in the embryonic brain (a, b), lung (c, d) and gastrointestinal tract (e, f). Inbred ICR pregnant mice were obtained from the animal house, and embryos at different developmental stages [gestational day 6.5 (E6.5) to E17.5] were prepared under a stereomicroscope (Olympus, USA). For qRT-PCR, preparations were kept in PBS (0.1 % diethyl pyrocarbonate), and extra-embryonic tissue was removed. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Germany) and was reversely transcribed according to standard protocols with oligo d(T)15 primers and SuperScript III reverse transcriptase (Invitrogen) according to the vendor’s instructions. Quantitative RT-PCR (RT-qPCR) was carried out with a Rotor Gene 3000 system (Corbett Research, Australia) using ImmoMix/SYBR Green (BIOLINE, Germany). Isoform-specific amplification primers were designed for MAO-A (5′-TTC AGC GTC TTC CAA TGG GAG CT-3′/5′-TGC TCC TCA CAC CAG TTC TTC TC-3′), MAO-B (5′-ACT CGT GTG CCT TTG GGT TCA G-3′/5′-TGC TCC TCA CAC CAG TTC TTC TC-3′) and GAPDH (5′-CCA TCA CCA TCT TCC AGG AGC GA-3′/5′-GGA TGA CCT TGC CCA CAG CCT TG-3′). Absence of cross-amplification between the two isoforms was ensured, and the standard protocol was followed as outlined before [296]. RNA preparations were analyzed at least in triplicate, and mean ± SD are given. The experimental raw data were evaluated with the Rotor-Gene Monitor software (version 4.6). To generate standard curves for quantification of expression levels, specific amplicons were used as external standards for each target gene (5 × 103–3 × 106 copy numbers). GAPDH mRNA was used as internal standard to normalize expression of the target transcripts
Despite the focus of research on MAO expression in neuronal structures, MAO expression can also be detected in other parts of the developing embryo. In fact, in contrast to the rather low expression levels of both MAO isoforms in the brain, expression in other tissues was found to be much higher. In the developing lung, for instance, expression of both MAO isoforms was detected, and mRNA levels were about 40-fold higher than those of the brain (Fig. 6c, d). Very high expression of MAO-A and MAO-B was observed in the embryonic gastrointestinal system (Fig. 6e, f). AT E16 expression of MAO-A in the gut was about 50 times higher than in the brain after gestational day E16. MAO-B expression is delayed and started to be detectable after day E17. However, expression of MAO-B, in particular of the embryonic small and large intestine, exceeds its expression in the brain by about three orders of magnitude. The biological relevance of this vast overexpression of MAO-B in the developing intestines is not clear since the gastrointestinal tract of the embryo is not fully functional yet, and there are no dietary amines to be detoxified.
Monoamines in extra-embryonic tissues and in the reproductive tract
Monoamine transporters, in particular NE and 5-HT transporters, are expressed in the placenta to protect the fetus from the detrimental effects of high circulating catecholamine concentrations present in maternal blood [159, 160]. These transporters are localized in the maternal facing microvillous border of the placenta. Circulating catecholamine levels in the fetus are low during embryogenesis but increase suddenly at birth to support the cardiovascular, pulmonary, metabolic and endocrine adaptations in early postnatal life [161]. Placental 5-HTT expression controls 5-HT concentrations and regulates microvascular resistance in the juxta placental myometrium [162]. The placenta also expresses high levels of catechol-O-methyl transferase (COMT) and MAO isoforms (Fig. 7) [49], which limits the exchange on monoamines between fetus and mother [163]. In the placenta, MAO-A expression dominates over MAO-B, which is in line with previous findings [49]. In contrast, in the yolk sac MAO-B mRNA expression dominates considerably over that of MAO-A. When comparing the expression levels of extraembryonic tissues to embryonic MAO expression, a sharp contrast between these tissues becomes apparent. MAO-A (100-fold) and MAO-B (1,000-fold) mRNA levels are much higher in extra-embryonic tissues when compared with embryonic cells (Fig. 7). These findings suggest that maternal MAOs lower monoamine levels in the blood to protect the embryo from their deleterious effects.

Expression of MAO-A and MAO-B in the murine embryo (a, b), yolk sac and placenta (c, d). A detailed description of preparation of whole murine embryos, RNA extraction, reverse transcription and quantitative RT-PCR is given in the legend to Fig. 6
Despite the high clearing capacity of the placenta, embryonic monoamines are mostly of maternal origin. They are essential for normal embryo development in early pregnancy, and thus, excessive clearance of monoamines from maternal blood would be detrimental [131]. Monoamines in the maternal blood originate from various maternal tissues and from the innervation of reproductive organs. Mast cells and nerve fibers are the major sources of 5-HT in the female reproductive tract [164]. Tyrosine hydroxylase and DA beta-hydroxylase activities have been detected in the oviduct [165], and DA, NE and EN were also shown to occur in oviductal tissues [166]. The uterus is innervated extensively by sympathic neurons, and adrenergic nerve fibers are essential for myometric contractility. In contrast, 5-HT plays an important role in endometrial decidualization and implantation during early pregnancy [167, 168]. Selective vascular constriction is induced by 5-HT in pregnant uterine arteries [169]. In fact, elevated 5-HT levels as well as reduced expression of MAO-A are found in cases of pre-eclampsia, a disease characterized by hypertension that endangers survival of both mother and fetus [49]. 5-HTT, COMT and MAO isoforms control the concentrations of monoamines in the uterus and progesterone stimulates the degradation of catecholamines in the endometrium [170].
Animal models of MAO deficiency and their impact on embryogenesis
To study the biological role of MAO enzymes in living organisms several experimental models are available (Table 2). Firstly, MAO-A expression can be interrupted by targeted knockout strategies in embryonic stem cells to generate animals lacking functional MAO gene(s). Secondly, expression of functional MAO isoenzymes can be suppressed at different developmental stages employing specific RNAi strategies. The latter methods are particularly useful when studying small organisms such as developing embryos, in which excessive penetration barriers do not cause major problems. In addition, such experimental setups circumvent genetic drifts or epigenetic phenomena, which frequently hamper the analysis of knockout animals [171–173]. Moreover, targeted knockdown experiments cause a sudden suppression of the gene of interest in embryos, which were allowed to develop normally until the experimental manipulation. Thus, this setup does not facilitate slow adaptive responses, which would ameliorate phenotypes observed in stem cell knockouts. A major drawback of such RNA interference studies is that such strategies can only be applied during certain time windows of development. Finally, a plethora of pharmacologic antagonists are available, which inhibit MAO enzymatic activities. However, the use of pharmacologic inhibitors is limited because of off-target effects and since the inhibitors might not be available to all cells and tissues in sufficient concentrations.
The most obvious biological function of MAO isoforms is to oxidize endogenous or alimentary amines to the corresponding aldehydes. Thus, the major expected defect of any MAO silencing strategy is an impairment of amine degradation, which will consequently result in elevated monoamine levels. However, biological monoamine levels are not only controlled by the rates of their respective catabolizing pathways, but also by the activity of their biosynthesis. Inhibition of monoamine biosynthesis on a background of reduced amine catabolism restores normal amine levels, and rescues biochemical and physiological derailments caused by exposure to excessive amine concentrations.
Moreover, monoamines like 5-HT can be stored in vesicles and can be cleared from their site of action using transporters and membrane channels. Thus, the availability of the respective amines at their site of action needs to be considered when discussing the role of MAOs in amine catabolism. In addition, when only one MAO isoform is lacking, the remaining isoform may take over at least some of the enzymatic workload in amine degradation. However, since the kinetic properties of the two MAO isoforms are different (see “Enzymology of MAO isoforms”), such compensation might be incomplete. Finally, other pathways of amine degradation such as diamine oxidases [174] or catechol-O-methyltransferase [175] may be available to compensate for a specific MAO deficiencies [26].
When discussing the biological role of MAO enzymes in such loss-of-function animal models, other MAO metabolites must be considered: (1) Hydrogen peroxide: H2O2 is formed during the MAO reaction in stoichiometric amounts and might exhibit biological activities. Hydrogen peroxide is a vital signaling molecule but also a hazard for cell survival. Cells contain a number of enzymatic and non-enzymatic mechanisms to maintain a critical peroxide tone. In fact, alterations of the cellular redox state, which result from an imbalance of peroxide-producing and peroxide-degrading reactions, are one of the most important regulatory events in the cellular gene expression regulation [33, 82, 176]. Thus, alterations in the cellular redox state are directly translated into changes of the cellular phenotype. (2) Ammonia: NH4 + is the second side product of the MAO reaction, but its biological relevance in embryogenesis has not been explored in detail. Ammonia is toxic to cells at higher concentrations, and there are several detoxifying mechanisms to protect the organism from the deleterious effects of ammonia. In the liver the majority of ammonia originating from oxidative deamination of glutamate via glutamate dehydrogenase [177] and other deaminating enzymes is used to synthesize carbamoylphosphate [178]. This intermediate is then employed for nitrogen disposal via the urea cycle. In other cells, carbamoylphosphate is used for the biosynthesis of pyrimidines [179]. In the brain, where urea biosynthesis does not occur, ammonia is used to synthesize glutamine from glutamate via glutamine synthase. This enzyme is mainly localized in astrocytes [180]. Glutamine, which is capable of penetrating the blood–brain barrier, is then transported to the kidney where ammonia is liberated via the glutaminase reaction [180] and excreted with the urine. Since urea biosynthesis is restricted to the liver and to a lesser extent to the kidney, glutamine synthase is likely to detoxify ammonia released during the MAO reaction in other cells and tissues. (3) Aldehydes: the highly reactive aldehydes produced by the MAO reaction are quickly transformed into less reactive alcohols or acids by the corresponding reductases or dehydrogenases (Fig. 1) [4]. However, in conditions of high monoamine turnover such as in the substantia nigra of Parkinson’s patients, these aldehydes may overburden the normal degrading pathways. In such conditions, the metabolite of DA degradation 3,4-dihydroxyphenylacetaldehyde (DOPAL) is believed to contribute to the loss of dopaminergic neurons in Parkinson’s disease [181]. In fact, DOPAL was shown to oxidatively modify tyrosine hydroxylase, superoxide dismutase and alpha-synuclein [181–183]. Interestingly, alpha-synuclein is a hallmark of the Lewy body aggregates that are found in the remaining substantia nigra of Parkinson’s patients [181].
Animal models of MAO-A deficiency
As outlined in “Monoamine systems during normal embryo development”, expression of MAO-A can be detected in many embryonic tissues during various stages of embryo development. The early expression of MAO-A and the presence of serotonergic transmitter systems during embryogenesis suggest that MAO-A may play an important role for normal embryo development by maintaining normal 5-HT concentrations in embryonic tissues. To explore the role of enzymes in whole organisms, targeted genetic knockout mice are usually created. Unfortunately, no such targeted MAO-A knockout strains are currently available. Instead, native mouse models exist that have acquired MAO-A deficiency by naturally occurring MAO-A gene mutations. The first MAO-A-deficient mouse strain was produced when an interferon beta (IFN-β) mini gene was accidentally inserted into the MAO-A gene [184]. This mouse strain is commonly referred to as Tg8. The insertion of the IFN-β mini gene caused a disruption of the MAO-A gene by displacing exons 2 and 3 [26]. The resulting MAO-A mRNAs that are generated from this altered gene fail to translate into active MAO-A enzymes [26]. This may in part be due to the fact that exon 2, which is lacking in the Tg8 mouse strain, encodes for a protein domain that is vital for binding of the cofactor FAD [185]. These animals do not express functional MAO-A, but it remains unclear whether or not additional genetic alterations have been induced during the generation of this mouse line. A second MAO-A-deficient mouse model was established when a number of individuals were screened for a specific point mutation in the coding region of the MAO-A gene, which has previously been associated with the Brunner syndrome in humans [186, 187]. This point mutation residing in exon 8 causes the insertion of a premature stop codon, which prevents translation of a complete enzyme. Instead, the mutated MAO-A mRNA is presumably subjected to nonsense-mediated mRNA decay. Here again, these mice do not express functional MAO-A, but it remains unclear whether they carry additional mutations in other regions of the genome, which might affect the resulting phenotype. Several behavioral tests were conducted with these animals [186], but a thorough biochemical analysis of this animal model is still lacking. Both MAO-A-deficient mouse strains are fertile and develop into adulthood without major morphological defects [26, 186]. However, adult MAO-A-deficient mice impress with behavioral alterations such as an increased tendency towards aggressive behavior [26, 186] and enhanced emotional learning [188]. The present review article however is focused on the impact of MAOs before and around birth. Thus, the behavioral abnormalities of MAO-A-deficient mice are not covered here, and the interested reader is referred to other review articles covering these topics [2, 4].
In contrast to the mouse strain carrying the Brunner-like point mutation [186], the discovery of the interruption of MAO-A expression in Tg8 mice has led to many studies aimed at characterizing these animals in an attempt to define the biological role of MAO-A. Most importantly, MAO-A-deficient Tg8 animals are characterized by excessively elevated levels of the MAO-A substrate 5-HT [26]. Similar elevated levels of 5-HT were observed when mouse embryos were exposed to clorgyline, a specific inhibitor of MAO-A activity [7], and when MAO-A expression was suppressed by siRNAs [7]. In the developing forebrain of Tg8 mice, 5-HT levels are elevated about fivefold just before birth and about tenfold 5 days after birth [189]. In the brainstem 5-HT concentration is increased fivefold before birth and eightfold at the 5th postnatal day. In contrast, in the cervical spinal cord only a weak increase in 5-HT levels (<1.5-fold) was observed before birth and about 3.5-fold after birth. Interestingly, the elevated levels of 5-HT in Tg8 mice become less pronounced within the first few months after birth [26]. Corresponding data were obtained when the MAO-A metabolite 5-hydroxyindoleacetic acid (5-HIAA) was quantified. Surprisingly, 5-HIAA is still detectable in mice lacking MAO-A activity, but its perinatal plasma concentrations were markedly decreased when compared with wild-type animals. Here again, the difference becomes less obvious within the first couple of months after birth. On the other hand, DA and NE levels remained largely unaffected by MAO-A depletion until birth. In addition, expression of MAO-B remains normal in these mice [26]. These data indicate that there is no compensatory upregulation of MAO-B expression in these mice and that MAO-B appears to be sufficient to maintain normal levels of DA and NE. Moreover, it is reasonable to assume that MAO-B is capable of metabolizing 5-HT in the embryo as indicated by the presence of 5-HIAA. Later on in postnatal life other compensatory mechanisms may be responsible for the decreasing levels of 5-HT in MAO-A knockout mice. These mechanisms may involve alternative pathways of 5-HT degradation or reduced 5-HT biosynthesis. Here again, the naturally increasing levels of MAO-B in adult mice may contribute to 5-HT breakdown. This hypothesis is further supported by data obtained with MAO-A/B double knockout mice. In these animals no detectable levels of 5-HIAA were observed [190], and these data suggest that in vivo 5-HT appears to be a suitable MAO-B substrate, which is consistent with previous in vitro data. The reason for this unexpected finding has not been clarified, but it can be speculated that the in vitro activity assays are not sensitive enough to pick up 5-HT oxidase activity of MAO-B. Alternatively, in vivo MAO-B might exhibit a different substrate specificity, which may be related to posttranslational modification. It has been reported that the catalytic activity of MAO-A is modified by serine phosphorylation [69], and similar post-translational modifications may impact MAO-B properties. The observation that surprisingly MAO-A in newborn mice is capable of oxidizing the classical MAO-B substrate PEA strongly suggests that alterations in the substrate specificity of MAO isoforms are possible [191].
In conclusion, the metabolic hallmark of MAO-A-deficient mice is reduced 5-HT oxidation, which leads to augmented 5-HT levels and a reduced hydrogen peroxide production. It remains to be explored whether or not the hydrogen peroxide deficiency impacts proper embryogenesis.
The lack of MAO-A activity in the developing embryo induces structural and functional abnormalities
The focus of attention, when analyzing MAO-A-deficient mice, has been on 5-HT and its impact on the development of the central nervous system. In fact, MAO-A deficiency in mice alters a number of developmental processes in the brain. Firstly, MAO-A-deficient Tg8 mice show abnormal development of thalamocortical afferent fibers [192, 193]. This is paralleled by the absence of barrel-like structures in layer 4 of the cortex, which are vital in processing information coming in from the large facial whiskers of the snout [26]. This phenomenon is also observed when wild-type mice are treated with clorgyline, an isoform-specific MAO-A inhibitor [192]. Barrel formation takes place during the 1st days after birth [194]. However, the basis for barrel formation depends on the proper migration and development of thalamocortical axons, which is already initiated around embryonic day E13.5. Secondly, the segregation of ipsilateral and contralateral retinal projections, which takes in place in the 1st days after birth, is interrupted in Tg8 mice [195]. Here again growth of retinal ganglion cells is initiated before birth around embryonic day E12 and enters the peak phase of axon growth between E14 and E17 [196]. Thirdly, phrenic motoneurons that are vital for establishing the respiratory network fail to develop normal dendrites in Tg8 mice, which is paralleled by a failure to develop a stable respiratory rhythm [197]. The neuronal structures of the respiratory system mature postnatally but are already set up prenatally, and the first inspiratory discharge can be observed as early as at E15 in fetal mice [198]. Fourthly, in Tg8 mice maturation of neonatal neuronal locomotor networks is transiently delayed [199]. Neuronal networks underlying rhythmic motor activities in vertebrates are termed central pattern generators [200]. Even though maturation of the locomotor network takes place after birth, newborns already possess functional circuits that can be induced to generate rhythmic bursts well before birth [201, 202]. Fifthly, the lack of MAO-A activity in Tg8 mice coincides with an altered distribution of 5-HT in neuronal structures [203]. This includes transient localization of 5-HT to catecholaminergic cells of the substantia nigra, ventral tegmental area, hypothalamus and locus coeruleus, as well as in various neuronal cells of the telencephalon and diencephalon [203]. Interestingly, all these neuronal structures are not competent for 5-HT biosynthesis and have to take up the excess extracellular 5-HT via monoamine plasma membrane transporters [203]. The functional consequences of the altered distribution pattern of 5-HT have not directly been explored, but it may contribute to the above-mentioned developmental alterations in the brains of Tg8 mice [203].
MAO-A-deficient Tg8 mice have not been created using targeted methods, and it remains unclear whether additional alterations in the genome were induced when accidental incorporation of the IFN-β mini gene took place. In addition, the inserted IFN-β minigene may affect the genetic activity of surrounding sequences [186]. Moreover, embryos that have survived functional silencing of the MAO-A gene might have evolved compensatory mechanisms preventing developmental defects. To circumvent these limitations MAO-A expression was knocked down in developing mouse embryos at midgestation (E7.5-E10.5) employing a siRNA strategy [7]. These mouse embryos showed more severe phenotypic alterations than Tg8 mice as indicated by general growth retardations as well as by developmental abnormalities of hind- and midbrain structures. Similar to observations in Tg8 mice, however, these alterations could be reproduced when wild-type embryos were exposed to the inhibitor of MAO-A activity clorgyline at the same experimental time window. In addition, transgenic overexpression of a transcriptional inhibitor of MAO-A expression induced similar alterations at these embryonic stages.
5-HT appears to be the key effector of MAO-A function
The major substrate of MAO-A activity is 5-HT, and consequently 5-HT is massively enriched in MAO-A-deficient mouse embryos. Thus, it is likely that the major developmental defects in such embryos might be related to the excessively elevated 5-HT levels (Fig. 8). As outlined in “Monoamine systems during normal embryo development”, 5-HT does not only function as a neurotransmitter; it also acts as trophic factor for normal embryonic development. Systemic levels of 5-HT are mainly controlled by the balanced equilibrium between biosynthetic and degrading enzymes. The key enzyme of 5-HT biosynthesis is tryptophane hydroxylase (TPH) [204, 205], and embryonic 5-HT levels are fed by embryonic and maternal sources [206]. TPH activity can be suppressed by the specific inhibitor parachlorophenylamine (PCPA), which impairs 5-HT production [207]. PCPA administration shortly after mating greatly impairs fertility. In contrast, when PCPA is given at later stages of embryo development, normal offspring are produced [208]. Treatment of Tg8 MAO-A-deficient mice with PCPA at birth normalizes 5-HT levels and restored the development of normal barrel structures [26] as well as the normal segregation of retinal projections [195]. In addition, pre- and postnatal treatment with PCPA normalized maturation of respiratory networks [197]. Taken together, these data strongly indicate that a precise balance of 5-HT levels just before birth and during the early postnatal period is crucial for normal maturation of various neuronal structures.

Regulatory circuits of serotonin signaling in the developing embryo are modulated by MAO-A activity. The activity of tryptophan hydroxylase (TPH) is crucial for the biosynthesis of serotonin (5-hydroxytyptamine, 5-HT), which in turn is degraded by monoamine oxidase A (MAO-A). 5-HT produced in the cytoplasm is packed into vesicles via the vesicular monoamine transporter (VMAT), which facilitates serotonin release into the extracellular space. Extracellular serotonin then either binds to specific serotonin receptors (5-Htr) to initiate cellular signaling cascades or is cleared from extracellular space via the serotonin transporter (5-HTT) to subject serotonin to degradation by MAO-A. Both activation of 5-Htr as well as the by-product of MAO-A activity hydrogen peroxide (H2O2) control cellular signaling cascades that affect cell proliferation, differentiation or apoptotic cell death
MAO-A deficiency alters cellular signaling cascades involved in the maturation of neuronal networks
Serotonin (5-HT) mediates its biological function via binding to a number of 5-HT receptors (5Htr). Seven major types of 5-HT receptors (5-Htr) have been classified, and some classes consist of different subtypes [209]. Most 5-Htrs are metabotropic G-protein coupled transmembrane receptors, but 5Htr-3 constitutes a ligand-gated cation channel, which increases membrane permeability for Na+ and K+, leading to membrane depolarization. Intracellularly, the metabotropic 5-Htrs are coupled to various signaling cascades, which involve second messengers such as cAMP, IP3 and DAG [210]. Apart from the metabolic pathways controlling systemic 5-HT levels other mechanisms regulate its targeted distribution. Vesicular monoamine transporters (VMAT) clear monoamines from the cytosol to store them in synaptic vesicles [211]. In addition, 5-HT can be internalized from the extracellular space via a specific plasma membrane transporter named serotonin transporter (SERT) or 5-hydroxytryptamine transporter (5HTT) [212]. These transport mechanisms allow the cell to clear 5-HT from its site of action. Deficiency in these cellular reuptake mechanisms was predicted to lead to similar symptoms as global MAO-A malfunction. In fact, reduced 5-HT breakdown in MAO-A-deficient Tg8 mice as well as abrogated 5-HT clearance from extracellular space in 5HTT−/− mice showed impaired formation of barrel-like structures in layer 4 of the cortex [213]. Similarly, both animal models exhibit alterations in the segregation of retinal projections [213]. These data indicate that altered concentrations of 5-HT in the extracellular space are important for the phenotypic alterations. Interestingly, abnormally elevated extracellular 5-HT levels have been associated with a significant reduction in the expression of 5-HT receptor 1a (5Htr-1a) in the cervical cord and in the medulla [197], and of 5Htr-1b in thalamocortical fibers of newborns [192]. Activation of 5Htr1-a appears to be important for rhythmogenesis [214]. Similar regulatory responses were observed for 5Htr-2a and 5Htr-2c in adult Tg8 mice [215]. These findings suggest that increased ligand concentrations downregulate the expression of corresponding receptors at the cell surface and thereby alter cellular 5-HT signaling. This regulatory response may be considered a protective cellular response against augmented ligand concentrations preventing excessive intracellular metabolic alterations. This protective response was confirmed when MAO-A-deficient Tg8 mice and 5HTT-null mice were crossbred with 5Htr-1b-deficient animals [193, 213]. In both cases, silencing of 5Htr-1b expression rescued the functional phenotypes observed in animals that are deficient for MAO-A or 5HTT [193, 213]. On the other hand, 5Htr-1b ablation alone has no impact on thalamocortical or retinogeniculate projections [213]. These data do not support the suggested importance of 5Htr1b for these developmental phenomena. The reasons for this unexpected finding are largely unclear, but compensatory mechanisms in 5Htr-1b−/− mice have been discussed [213]. Activation of 5Htr-1b is negatively coupled to adenylate cyclase. Interestingly, when adenylate cyclase expression is disrupted a barrelless phenotype is observed, which points to a role of cAMP in barrel formation [216]. Disturbed development of the locomotor as well as the respiratory network has also been reported to involve signaling via 5Htr-1b [217]. MAO-A-deficient Tg8 mice showed significantly enhanced expression of the 5Htr-1b receptor subtype in phrenic motoneurons of the cervical cord [217]. This effect was mimicked by prenatal application of a 5Htr-2a receptor agonist to wild-type mice. Similarly, the induction of 5Htr-1b in Tg8 neonates could be reversed by prenatal inhibition of 5Htr-2 receptor subtypes using a pharmacological antagonist against this receptor subclass [217]. In conclusion, these data indicate that MAO-A controls embryonic 5-HT levels and thereby modulates 5-HT receptor signaling. However, 5-HT also affects expression of 5-HT receptors in developing mice. In addition, there appears to be an intense intracellular crosstalk between the signaling pathways initiated by different 5-HT receptor subtypes. In general, the in vivo data obtained in experiments with genetically modified mice as well as the pharmacological intervention studies employing specific receptor antagonists suggest that MAO-A expression is vital for the formation and maturation of neuronal networks that prepare the embryo for extrauterine life.
MAO-A deficiency ameliorates apoptotic signaling in the developing neuroepithelium
The formation of neuronal networks during cerebral embryogenesis requires proper differentiation of individual neurons, and developmental apoptosis is an essential part of this maturation process [218, 219]. In fact, about 50 % of all neurons generated during cortex development are driven into apoptotic cell death [220, 221]. Recently, 5-HT has been shown to act as a modulator of apoptotic cell death [222], and 5-HT signaling proved to be neuroprotective in experimental models of stroke [223]. Moreover, transgenic mice that are devoid of 5-HT transporter (5-HTT) and that are characterized by high 5-HT levels exhibited significantly reduced apoptotic cell death in various regions of the brain around birth [224]. These mice are unable to clear 5-HT from the cell surface to submit it to intracellular breakdown by MAO-A. Similar observations were made when MAO-A expression was knocked down by inhibitory RNAs in developing mouse embryos around midgestation [7]. Here again the number of apoptotic cells in the developing neuroepithelium was greatly reduced. This coincided with a reduction of caspase-9 activation indicating a reduced activity of the intrinsic apoptotic pathway. In contrast, the extrinsic pathway of apoptosis was not affected [7]. In the same experimental model similar results were observed when MAO-A activity was pharmacologically blocked or when its transcription was inhibited by overexpression of a transcriptional inhibitor of MAO-A expression [7]. In accordance with these results, abrogated serotonin signaling by genetic ablation of cerebral VMAT2 results in significantly higher numbers of apoptotic cells in the developing brains of murine neonates [225]. Here again, apoptosis seems to proceed via the intrinsic pathway. Taken together, these data suggest that cytoplasmic enrichment of serotonin, which is either due to the cellular inability to store away the mediator into specialized vesicles or due to impaired intracellular degradation, is a key process for regulation of apoptosis. This conclusion is further supported by experiments in which VMAT2 null mice were crossed with MAO-A-deficient Tg8 animals [225]. The lack of MAO-A activity in the brains of VMAT2 null mice restored normal apoptosis around birth. These data indicate that the catalytic activity of MAO-A regulates apoptotic cell death by modifying the intracellular serotonin concentrations rather than impacting ligand binding to cell surface receptors by altering extracellular ligand concentrations. Although the sequence of events of this pro-apoptotic cascade has not been explored in detail, it may be that the failure to store away synthesized serotonin in vesicles, as is the case in VMAT2 null mice, may favor serotonin breakdown by MAO-A, which in turn increases intracellular hydrogen peroxide concentrations. This oxidative stress may subsequently overcome the neuronal anti-oxidative defense system and cause the induction of neuronal cell death. Thus, inactivation of MAO-A expression on a VMAT2 defective background may reduce the rate of apoptotic cell death.
MAO-A activity alters cellular signaling of apoptotic cell death and cell proliferation—the precise mechanisms by which MAO-A activity and serotonin turnover affect apoptotic signaling remain largely elusive, and more work is needed to elucidate the molecular basis for this effect. However, for neuronal cells some data became currently available [7, 225, 226]. As outlined above, the intrinsic pathway of apoptosis is controlled by proteins of the Bcl-2 family. The Bcl-2 protein family includes anti-apoptotic proteins such as Bcl-2 and Bcl-XL as well as pro-apoptotic proteins such as Bax and Bad [227]. MAO-A activity is thought to negatively affect expression of the anti-apoptotic protein Bcl-2, and thus shifts the equilibrium towards apoptosis in neuronal and mensenchymal stem cells [72, 226]. Inhibition of MAO-A activity restores normal Bcl-2 levels [72]. Similarly, genetic ablation of MAO-A (Tg8 mice) on a VMAT2 defective background enhanced the expression of the anti-apoptotic protein Bcl-XL [225]. However, the molecular mechanisms involved in the repression of these anti-apoptotic proteins by MAO-A activity remains a matter of speculation. Expression of Bcl-XL is promoted by the transcription factor NF-kappaB [228]. Interestingly, the NF-kappaB subunit p50 contains a cysteine residue, which is sensitive to oxidative modification [229], and oxidation of this cysteine impairs binding of NF-kappaB to its cognate cis-regulatory motif in the promoters of target genes. It can thus be speculated that MAO-A activity, which augments the cellular oxidative potential by generating hydrogen peroxide, impairs NF-kappaB driven expression of Bcl-XL. In addition to NF-kappaB, a large variety of other proteins involved in the regulation of gene expression is sensitive to alterations in the cellular redox state [33, 82]. Thus, there is no reason to assume that the regulatory impact of MAO-A-derived hydrogen peroxide is unique for NF-kappaB. However, the hypothesis that MAO-A induced cellular oxidative stress affects the activity of genes that regulate gene expression remains to be tested.
In addition to its impact on apoptotic cell death, MAO-A also appears to modulate the regulation of cell proliferation. Propagation of the cell cycle depends on the expression of regulatory proteins such as cyclins and cyclin-dependent kinases. Cyclin D1 is vital for induction of cell proliferation by mitogenic stimuli from the extracellular matrix. Suppression of MAO-A expression and/or activity in developing embryos reduces cellular cyclin D1 levels [7] and thus renders cells less sensitive to mitogens. Similar data were obtained in MAO-A-deficient mouse embryos carrying a spontaneous point mutation in the MAO-A gene [230]. During late gestation these animals exhibit strongly reduced proliferative activity within neuronal stem cell populations of the developing telencephalon [230]. In addition to stem cell proliferation, neuronal differentiation of stem cells is impaired when MAO-A expression is suppressed by the presence of a neomycin cassette in the MAO-A gene [7]. Pharmacological inhibition of serotonin biosynthesis improved stem cell proliferation in these animals, and these data pointed to an involvement of serotonin in neuronal stem cell proliferation [230]. It should be stressed at this point that these conclusions are solely based on the observations that reduction of proliferation and cyclin D1 expression coincides in time with impairment of cellular MAO-A activity. Unfortunately, the mechanistic basis for a direct connection between MAO-A activity and cell proliferation has not yet been established. Similar to proteins that regulate apoptotic cell death the cellular redox state might be of importance [33]. In fact, moderate levels of hydrogen peroxide have been shown to enhance cyclin D1 expression. In turn it can be speculated that decreased levels of hydrogen peroxide as caused by MAO-A deficiency reduce expression of cyclin D and thus reduce cell cycle progression.
A second mechanistic link between MAO-A activity and cell proliferation is protein kinases, such as ERK1/2, JNK and p38 [72, 231]. These enzymes modulate cellular phenotypes in response to various stimuli including oxidative stress by hydrogen peroxide and are already active in developing embryos [232–234]. However, hydrogen peroxide not only initiates cellular signaling but is also able to modulate virtually all downstream events of most signaling cascades [33]. It is thus a task for investigators to elicit the role of hydrogen peroxide generated by MAO-A in the regulation of cell death and cell proliferation in general and during embryo development in particular.
Animal models of MAO-B deficiency
In contrast to MAO-A, the gene coding for MAO-B has been inactivated in mice by a conventional stem cell knockout strategy [27]. MAO-B-deficient offspring develop normally and do not show major developmental abnormalities as adults. Similar to MAO-A-deficient animals, the lack of MAO-B expression in these knockout mice does not cause a compensatory upregulation of MAO-A expression. However, these mice exhibited massively elevated levels of the typical MAO-B substrate β-phenylethylamine (PEA) while, surprisingly, DA levels remained unaffected. Pharmacological intervention studies using the MAO-B inhibitor selegiline are normally associated with increased levels of DA [235]. Unfortunately, the results of such intervention studies are difficult to interpret because of a potential lack of isoform specificity and owing to potential off-target effects of such inhibitors. On the other hand, the elevated levels of PEA in the brains of MAO-B−/− mice are consistent with corresponding findings in patients suffering from Norrie’s disease [236] and with studies in rodents using pharmacological MAO inhibitors [237, 238]. PEA is a trace neurotransmitter that is synthesized from phenylalanine via primary decarboxylation and is thought to modulate the activity of NE and DA in the brain [239]. In rats, elevated levels of PEA induce downregulation of dopamine D1 receptors [238]. Hence, elevated levels of PEA in MAO-B−/− mice may interfere with the dopaminergic system. High levels of PEA have been associated with altered behavior and bipolar disease and schizo-affective disorders [240–242]. In the brain PEA modulates the cortical blood flow via a yet unknown mechanism, and in MAO-B−/− mice elevated PEA levels correlate with decreased cortical blood flow [243]. In addition, adult MAO-B-deficient mice show increased stress behavior when subjected to the forced swim test [27]. PEA is thought to modulate the stress response modulated by DA and noradrenaline, and this may be the underlying reason for the enhanced stress response in MAO-B−/− mice [244]. In contrast to the effects seen in adult MAO-B-deficient mice, no abnormalities were detected in developing embryos. This finding is not surprising considering the low expression levels of this isoform in embryonic tissues during embryogenesis when compared with MAO-A. Similar observations were made when embryonic MAO-B expression was suppressed employing the siRNA strategy during in vitro culture of mouse embryos [7]. Indeed, isoform-specific knockdown embryos exhibited strongly suppressed MAO-B expression levels but showed no significant developmental abnormalities. Taken together, the data from targeted knockout and knockdown models do not provide experimental evidence for an essential role of MAO-B in mouse embryo development. The results obtained in MAO-B knockout mice indicate that metabolism of all major endogenous amines remains unaffected. However, it remains unclear whether MAO-B might play a role in protecting the developing embryo from the deleterious effects of maternal circulating amines. The high expression of MAO isoforms in tissues that protect the embryo during intrauterine development such as the yolk sac (Fig. 7) and the placenta [49] may support this hypothesis. The kinetic properties of MAO-B, in particular its low affinity for atmospheric oxygen (see “Enzymology of MAO isoforms”), suggest that MAO-B may function as an oxygen sensor during embryo development. In case of oxygen deficiency the enzyme activity is kinetically impaired. Thus, less hydrogen peroxide is produced, which subsequently alters the cellular redox state and gene expression profiles. For the time being, the putative oxygen sensor function of MAO-B is rather speculative and needs to be confirmed by experimental data.
Developmental implications of drugs and stress targeting the monoamine system
Maternal conditions have significant impact on prenatal and postnatal development [245]. As neurotransmitters and neuromodulators, monoamines are not only important for neurogenesis in early embryos, but also for the development of neuronal circuits at later stages [118]. Prenatal or neonatal stress, as well as maternal medications, may disturb neuronal wiring and thus may cause long-term behavioral defects.
Psychoactive drugs
Serotonergic neurotransmission in the brain is the target of a number of psychoactive drugs. When taken by pregnant females these compounds may cross the placenta to enter the fetal bloodstream. In addition, they may alter placental blood flow and biological functions, and thus may indirectly disturb the normal interaction between mothers and fetuses. Prenatal exposure to these substances can interfere with the ontogeny of the developing neuronal circuitry and may induce long-lasting metabolic dysfunctions [246].
Psychostimulants
The cellular uptake of biogenic amines is directly blocked by psychostimulants such as cocaine and amphetamines.
Cocaine: Cocaine inhibits the cellular uptake of 5-HT, NE and DA, and thus controls the monoamine concentrations in the synaptic cleft of the corresponding neurons. Because of its physico-chemical properties cocaine can cross the blood-brain barrier [247]. It is the most common recreational drug and interacts with the catecholaminergic systems in the brain [248]. According to the pregnancy category of drugs in assessment of the risk for fetal injury, cocaine is classified in category C. Animal reproduction studies have shown adverse effects of cocaine on fetal development. Not surprisingly, there are no large-scale clinical studies in humans, but the potential benefits may warrant the use of the drug in pregnant women despite potential risks. Cocaine inhibits the catecholaminergic system by preventing presynaptic transport mechanisms, which remove DA and EN from the synaptic cleft. Acute fetal cocaine administration causes an abrupt increase in circulating catecholamines and an associated increase in fetal arterial pressure [249]. Chronic fetal cocaine administration is associated with an increase in plasma catecholamine levels and a significant reduction in intrauterine catecholamine clearance [250]. Redistribution of blood flow during exposure to cocaine results from exaggerated elevations in plasma catecholamine levels contributing the adverse effects on the fetus [251]. In rats prenatal cocaine exposure leads to dysregulation of neuronal migration and consequently to severe neurobehavioral disturbances. Prenatal cocaine exposure in humans causes atypic motoric activity immediately after birth and abnormal behavior in later life [147].
Amphetamine: Amphetamines belong to the class of phenethylamine-like psychostimulants that increased wakefulness and focus. Amphetamines exert their behavioral effects by modulating the activity of NE, DA and 5-HT throughout the brain [252]. Similarly to cocaine, amphetamine is classified as pregnancy category C.
Antidepressants
Antidepressants are another group of psychoactive drugs to which a fetus is exposed when taken by the mother. Inadequate levels of cerebral 5-HT are a possible cause of depression, and thus, many antidepressants affect 5-HT metabolisms: (1) selective 5-HT reuptake inhibitors (SSRIs), (2) non-selective 5-HT-NE reuptake inhibitors (SNRIs), (3) noradrenergic and specific serotonergic antidepressants (NaSSAs), (4) selective 5-HT reuptake enhancers, (5) NE-DA disinhibitors (NDDIs), (6) tricyclic antidepressants and (7) MAO inhibitor (MAOIs).
Selective serotonin reuptake inhibitors (SSRIs)
SSRIs are widely used for the treatment of depression and anxiety-related disorders. SSRIs target the 5HTTs and prevent reuptake of 5-HT by presynaptic neurons. Thus, they maintain higher concentrations of 5-HT in the synaptic cleft. SSRIs have fewer adverse effects than tricyclics and MAO inhibitors (see below) [253]. Among the side effects of SSRI treatment are drowsiness, dry mouth, nervousness, anxiety, insomnia, decreased appetite, long-term weight gain and sexual disturbance.
Most SSRIs can penetrate the placental barrier or are passed on to the newborns via breastfeeding [254, 255]. There are particular concerns about employing SSRIs as drug of choice for the treatment of depression in women during pregnancy. Maternal exposure of SSRIs has been associated with congenital cardiac malformations and neonatal pulmonary hypertension [256, 257]. Severe dilative cardiomyopathy with decreased survival rates was identified in SSRI treated mice [258]. Adverse pregnancy outcomes in humans included spontaneous abortions, low birth weight, preterm birth and fetal death [259, 260]. However, it remains unclear whether these adverse outcomes are solely related to the side effects of the drugs since contributions of the underlying depression on the pregnancy cannot be excluded [261]. 5-HT has important roles in the establishment of neural plasticity and on long-term synaptic facilitation [262]. The behavioral outcome of SSRI exposure during embryo development is paradoxical [263]. They include increased anxiety/depression, increased REM sleep, blunted pain responses, reduced gap crossing, improved spatial learning and increased cocaine-induced conditioned place preference. Animal studies have revealed that administration of SSRIs results in abnormal brain circuitry and maladaptive behaviors that persist into adulthood [264, 265]. On the other hand, in large cohort trials of maternal SSRI exposure throughout pregnancy no adverse effects were observed on fetal growth and development, behavior and teratogenicity [266, 267].
Monoamine oxidase inhibitors (MAOIs)
First generation MAO inhibitors block the catalytic activity of MAO irreversibly and thus thus strongly inhibit the breakdown of monoaminergic neurotransmitters such as DA, 5-HT, and NE. MAOIs can be as effective as tricyclics, but have a higher incidence of unwanted side effects as well as drug and food interactions. MAOIs of the second generation are as effective as SSRIs and tricyclics [268]. For instance, reversible inhibitors of monoamine oxidase A (RIMA, e.g., moclobemide) acts in a short-lived and selective manner without major interactions with the diet [269]. Side effects of RIMA treatment, which include hepatitis, heart attack, stroke and seizures, are rare. However, in patients receiving multiple medications including MAOIs, the 5-HT syndrome, a life-threatening adverse drug reaction with excess serotonergic activity, may develop [270]. There is evidence that both, MAO-B inhibitors employed for treatment of psychiatric and neurological disorders, as well as clorgyline, a MAO-A inhibitor formerly used as an anti-depressant, have negative impacts on early embryo development [117, 271].
Other drugs
Neuroleptic drugs administrated during pregnancy can block DA receptors and cause long-lasting side effects [147]. Tricyclics are still prescribed in severe depression or when the patients are SSRI intolerant. Tricyclics block the reuptake of both NE and 5-HT, but are less selective and have smaller safety margins than other antidepressants. Side effects include increased heart rate, drowsiness, dry mouth, constipation, urinary retention, blurred vision, dizziness, confusion as well as sexual dysfunction. 5-HT-NE inhibitors (SNRIs) impact the cellular reuptake of NE and 5-HT, and have similar side effects as SSRIs. NE- and specific 5-HT antidepressants (NaSSAs) block presynaptic alpha-2 adrenergic receptors and various 5-HT receptors to enhance NE and 5-HT neurotransmission [272]. Side effects of these drugs include drowsiness, increased appetite and weight gain. NE-DA inhibitors (NDDIs) are 5-Htr-2c receptor antagonists, which inhibit NE and DA release and promote the outflow of these neurotransmitters.
A number of drugs impacting the serotonergic system affect the development of preimplantation embryos. For instance anti-migraine and anti-headache drugs, such as triptans, which are 5-Htr-1d/5-Htr-1b agonists, retard preimplantation development [112]. Potential risks of triptan treatment during pregnancy were reported, but for the time being no large-scale in vivo studies have been performed to quantify the developmental effects of these drugs on preimplantation embryos [273]. Triptan treatment impairs expression of placental NE transporters and was inversely related to umbilical cord NE concentration and to fetal condition at birth [274].
Prenatal stress
Prenatal maternal stress significantly affects pregnancy outcome including reduction in litter size, increased early fetal loss [275] and spontaneous abortion [276]. The fetus is adapted to low oxygen levels. The sympathic tone is low and respiratory movements are suppressed. If fetuses are challenged by birth asphyxia, unlike adults, they becomes hypotonic, hypoventilated and bradycardic owing to inhibition of neurotransmission and/or a lack of neuronal excitatory activities [102]. Chronic prenatal hypoxia alters the monoamine turnover in the locus coeruleus and the nucleus tractus solitarius in adolescent rats leading to disturbed control of respiratory behavior [277]. Clinically, prenatal stress is associated with attention deficit disorders in children because of the disturbance of monoaminergic turnover, subsequent abnormal neurotransmission and receptor reprogramming [278], which may lead to long-term behavioral defects.
Outlook
During embryogenesis monoamines function as neuromodulators and neurotransmitters, and substances interfering with monoamine signaling have become effective drugs used for treatment of neurological and psychiatric disorders. However, in the past few years it became apparent that MAO expression in peripheral tissues exceeds that in the brain, but our knowledge on the biological roles of MAOs in these cells has remained fragmentary. More work is needed to shed light on the biological function of MAO isoforms in the placenta, in the gastrointestinal tract, and in resting and stimulated immune cells, such as alternatively stimulated macrophages (M2). M2 macrophages have been implicated in the resolution phase of acute inflammation, but it remains to be established what function MAO-A may have in this process.
In addition, it has become increasingly evident that MAO activity is not only associated with amine turn-over. Instead, since the biological importance of hydrogen peroxide as a second messenger has emerged, MAO research has paid more attention to this previously neglected side product of the MAO enzymatic reaction.
Monoamine oxidase research has gained further progress following the discovery of mouse strains with defective MAO expression as well as the generation of mouse models with more targeted strategy of gene silencing. Such animal models have subsequently allowed researches to investigate the biological implications of MAO expression in prenatal stages of development. Indeed, MAO activity appears to be involved in perinatal maturation of neuronal structures. Moreover, in particular MAO-A, which in contrast to MAO-B is expressed in significant amounts in the developing embryo, is a vital player early on during embryogenesis. The impact of MAO activity on the developing embryo involves the regulation of cell cycle control, differentiation and apoptotic cell death. The underlying molecular mechanisms are only poorly understood. Both the control of the potent morphogen serotonin and the generation of oxidative stress via the generation of hydrogen peroxide are involved. Whereas the signaling events that are associated to serotonin action have been described in great detail, we are only beginning to understand the means by which hydrogen peroxide modulates cellular signaling cascades. This is further complicated by the fact that hydrogen peroxide can be either toxic or stimulatory depending on the ability of the cell to cope with it. Moreover, the impact of such a highly reactive and ephemeral molecule is further complicated by the fast sequence of spatio-temporal events that take place in the developing embryo in a tightly controlled manner. Furthermore, the various biological functions of 5-HT that have been described in mammalian adults and embryos have to be re-considered, since transgenic mice that are unable to synthesize 5-HT develop normally and show no major behavioral abnormalities [279]. Is serotonin really dispensable for survival or are the compensatory mechanisms resulting from the loss of such an ancient molecule so ingenious that life without serotonin is possible? Thus, new experimental strategies such as inducible or tissue-specific targeted knockout strategies should be developed to further characterize the roles of MAO during embryogenesis. Defining the functions of MAO enzymes in the developing embryo will also help to understand the effects of alimentary amines and of pharmacological molecules interfering with amine metabolism on the unborn child.
Abbreviations
- MAO:
-
Monoamine oxidase
- LOX:
-
Lipoxygenase
- COX:
-
Cyclooxygenase
- DAAO:
-
d-Amino acid oxidase
- TPH:
-
Tryptophan hydroxylase
- EN:
-
Epinephrine (adrenalin)
- NE:
-
Norepinephrin (noradrenalin)
- DA:
-
Dopamine
- 5-HT:
-
5-Hydroxytryptamine (serotonin)
- 5-Htr:
-
Serotonin receptor
References
Gaweska H, Fitzpatrick PF (2011) Structures and mechanism of the monoamine oxidase family. Biomol Concepts 2(5):365–377. doi:10.1515/BMC.2011.030
Shih JC, Chen K, Ridd MJ (1999) Monoamine oxidase: from genes to behavior. Annu Rev Neurosci 22:197–217. doi:10.1146/annurev.neuro.22.1.197
Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 101(16):6062–6067. doi:10.1073/pnas.0400782101.0400782101
Bortolato M, Chen K, Shih JC (2008) Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev 60(13–14):1527–1533. doi:10.1016/j.addr.2008.06.002
Mandel S, Grunblatt E, Riederer P, Gerlach M, Levites Y, Youdim MB (2003) Neuroprotective strategies in Parkinson’s disease : an update on progress. CNS Drugs 17(10):729–762. pii: 17104
Blaschko H, Richter D, Schlossmann H (1937) The oxidation of adrenaline and other amines. Biochem J 31(12):2187–2196
Wang CC, Borchert A, Ugun-Klusek A, Tang LY, Lui WT, Chu CY, Billett E, Kuhn H, Ufer C (2011) Monoamine oxidase a expression is vital for embryonic brain development by modulating developmental apoptosis. J Biol Chem 286(32):28322–28330. doi:10.1074/jbc.M111.241422
Edmondson DE, Mattevi A, Binda C, Li M, Hubalek F (2004) Structure and mechanism of monoamine oxidase. Curr Med Chem 11(15):1983–1993
Scrutton NS (2004) Chemical aspects of amine oxidation by flavoprotein enzymes. Nat Prod Rep 21(6):722–730. doi:10.1039/b306788m
Yu PH, Bailey BA, Durden DA, Boulton AA (1986) Stereospecific deuterium substitution at the alpha-carbon position of dopamine and its effect on oxidative deamination catalyzed by MAO-A and MAO-B from different tissues. Biochem Pharmacol 35(6):1027–1036. pii: 0006-2952(86)90094-8
Jonsson T, Edmondson DE, Klinman JP (1994) Hydrogen tunneling in the flavoenzyme monoamine oxidase B. Biochemistry 33(49):14871–14878
Nandigama RK, Edmondson DE (2000) Structure–activity relations in the oxidation of phenethylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry 39 (49):15258–15265. pii: bi001957h
Walker MC, Edmondson DE (1994) Structure–activity relationships in the oxidation of benzylamine analogues by bovine liver mitochondrial monoamine oxidase B. Biochemistry 33(23):7088–7098
Edmondson DE, Binda C, Wang J, Upadhyay AK, Mattevi A (2009) Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry 48(20):4220–4230. doi:10.1021/bi900413g
Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A (2002) Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol 9(1):22–26. doi:10.1038/nsb732
De Colibus L, Li M, Binda C, Lustig A, Edmondson DE, Mattevi A (2005) Three-dimensional structure of human monoamine oxidase A (MAO A): relation to the structures of rat MAO A and human MAO B. Proc Natl Acad Sci USA 102(36):12684–12689. doi:10.1073/pnas.0505975102
Son SY, Ma J, Kondou Y, Yoshimura M, Yamashita E, Tsukihara T (2008) Structure of human monoamine oxidase A at 2.2-A resolution: the control of opening the entry for substrates/inhibitors. Proc Natl Acad Sci USA 105(15):5739–5744. doi:10.1073/pnas.0710626105
Binda C, Wang J, Pisani L, Caccia C, Carotti A, Salvati P, Edmondson DE, Mattevi A (2007) Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J Med Chem 50(23):5848–5852. doi:10.1021/jm070677y
Ma J, Yoshimura M, Yamashita E, Nakagawa A, Ito A, Tsukihara T (2004) Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. J Mol Biol 338(1):103–114. doi:10.1016/j.jmb.2004.02.032
Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A (2003) Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc Natl Acad Sci USA 100(17):9750–9755. doi:10.1073/pnas.1633804100
Chen K, Wu HF, Shih JC (1996) Influence of C terminus on monoamine oxidase A and B catalytic activity. J Neurochem 66(2):797–803
Gottowik J, Malherbe P, Lang G, Da Prada M, Cesura AM (1995) Structure/function relationships of mitochondrial monoamine oxidase A and B chimeric forms. Eur J Biochem 230(3):934–942
Upadhyay AK, Borbat PP, Wang J, Freed JH, Edmondson DE (2008) Determination of the oligomeric states of human and rat monoamine oxidases in the outer mitochondrial membrane and octyl beta-D-glucopyranoside micelles using pulsed dipolar electron spin resonance spectroscopy. Biochemistry 47(6):1554–1566. doi:10.1021/bi7021377
Milczek EM, Binda C, Rovida S, Mattevi A, Edmondson DE (2011) The ‘gating’ residues Ile199 and Tyr326 in human monoamine oxidase B function in substrate and inhibitor recognition. FEBS J 278(24):4860–4869. doi:10.1111/j.1742-4658.2011.08386.x
Hubalek F, Binda C, Khalil A, Li M, Mattevi A, Castagnoli N, Edmondson DE (2005) Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J Biol Chem 280(16):15761–15766. doi:10.1074/jbc.M500949200
Cases O, Seif I, Grimsby J, Gaspar P, Chen K, Pournin S, Muller U, Aguet M, Babinet C, Shih JC et al (1995) Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science 268(5218):1763–1766
Grimsby J, Toth M, Chen K, Kumazawa T, Klaidman L, Adams JD, Karoum F, Gal J, Shih JC (1997) Increased stress response and beta-phenylethylamine in MAOB-deficient mice. Nat Genet 17(2):206–210. doi:10.1038/ng1097-206
Chen JF, Steyn S, Staal R, Petzer JP, Xu K, Van Der Schyf CJ, Castagnoli K, Sonsalla PK, Castagnoli N Jr, Schwarzschild MA (2002) 8-(3-Chlorostyryl)caffeine may attenuate MPTP neurotoxicity through dual actions of monoamine oxidase inhibition and A2A receptor antagonism. J Biol Chem 277(39):36040–36044. doi:10.1074/jbc.M206830200
Broadley KJ (2010) The vascular effects of trace amines and amphetamines. Pharmacol Ther 125(3):363–375. doi:10.1016/j.pharmthera.2009.11.005
Henchcliffe C, Severt WL (2011) Disease modification in Parkinson’s disease. Drugs Aging 28(8):605–615. doi:10.2165/11591320-000000000-00000
Chen EY, Fujinaga M, Giaccia AJ (1999) Hypoxic microenvironment within an embryo induces apoptosis and is essential for proper morphological development. Teratology 60(4):215–225. doi:10.1002/(SICI)1096-9926(199910)60:4<215:AID-TERA6>3.0.CO;2-2
Hansen JM (2006) Oxidative stress as a mechanism of teratogenesis. Birth Defects Res C Embryo Today 78(4):293–307. doi:10.1002/bdrc.20085
Ufer C, Wang CC, Borchert A, Heydeck D, Kuhn H (2010) Redox control in mammalian embryo development. Antioxid Redox Signal 13(6):833–875. doi:10.1089/ars.2009.3044
Leese HJ (1995) Metabolic control during preimplantation mammalian development. Hum Reprod Update 1(1):63–72
Fischer B, Bavister BD (1993) Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J Reprod Fertil 99(2):673–679
Webster WS, Abela D (2007) The effect of hypoxia in development. Birth Defects Res C Embryo Today 81(3):215–228. doi:10.1002/bdrc.20102
Saam J, Rosini E, Molla G, Schulten K, Pollegioni L, Ghisla S (2010) O2 reactivity of flavoproteins: dynamic access of dioxygen to the active site and role of a H+ relay system in d-amino acid oxidase. J Biol Chem 285(32):24439–24446. doi:10.1074/jbc.M110.131193
Saam J, Ivanov I, Walther M, Holzhutter HG, Kuhn H (2007) Molecular dioxygen enters the active site of 12/15-lipoxygenase via dynamic oxygen access channels. Proc Natl Acad Sci USA 104(33):13319–13324. doi:10.1073/pnas.0702401104
Jahng JW, Houpt TA, Wessel TC, Chen K, Shih JC, Joh TH (1997) Localization of monoamine oxidase A and B mRNA in the rat brain by in situ hybridization. Synapse 25(1):30–36. doi:10.1002/(SICI)1098-2396(199701)25:1<30:AID-SYN4>3.0.CO;2-G
Westlund KN, Denney RM, Rose RM, Abell CW (1988) Localization of distinct monoamine oxidase A and monoamine oxidase B cell populations in human brainstem. Neuroscience 25(2):439–456 0306-4522(88)90250-3 [pii]
Saura J, Bleuel Z, Ulrich J, Mendelowitsch A, Chen K, Shih JC, Malherbe P, Da Prada M, Richards JG (1996) Molecular neuroanatomy of human monoamine oxidases A and B revealed by quantitative enzyme radioautography and in situ hybridization histochemistry. Neuroscience 70(3):755–774. pii: S0306-4522(96)83013-2
Billett EE (2004) Monoamine oxidase (MAO) in human peripheral tissues. Neurotoxicology 25(1–2):139–148. doi:10.1016/S0161-813X(03)00094-9
Yamada M, Yasuhara H (2004) Clinical pharmacology of MAO inhibitors: safety and future. Neurotoxicology 25(1–2):215–221. doi:10.1016/S0161-813X(03)00097-4
Youdim MB, Finberg JP (1987) Monoamine oxidase B inhibition and the “cheese effect”. J Neural Transm Suppl 25:27–33
Hansen MB, Witte AB (2008) The role of serotonin in intestinal luminal sensing and secretion. Acta Physiol (Oxf) 193(4):311–323. doi:10.1111/j.1748-1716.2008.01870.x
Li G, Xu J, Wang P, Velazquez H, Li Y, Wu Y, Desir GV (2008) Catecholamines regulate the activity, secretion, and synthesis of renalase. Circulation 117(10):1277–1282. doi:10.1161/CIRCULATIONAHA.107.732032
Ben-Harari RR, Youdim MB (1981) Ontogenesis of uptake and deamination of 5-hydroxytryptamine, dopamine and beta-phenylethylamine in isolated perfused lung and lung homogenates from rats. Br J Pharmacol 72(4):731–737
Chaitidis P, Billett EE, O’Donnell VB, Fajardo AB, Fitzgerald J, Kuban RJ, Ungethuem U, Kuhn H (2004) Th2 response of human peripheral monocytes involves isoform-specific induction of monoamine oxidase-A. J Immunol 173(8):4821–4827. pii: 173/8/4821
Sivasubramaniam SD, Finch CC, Billett MA, Baker PN, Billett EE (2002) Monoamine oxidase expression and activity in human placentae from pre-eclamptic and normotensive pregnancies. Placenta 23(2–3):163–171. doi:10.1053/plac.2001.0770
Bond PA, Cundall RL (1977) Properties of monoamine oxidase (MAO) in human blood platelets, plasma, lymphocytes and granulocytes. Clin Chim Acta 80(2):317–326
Suarez-Merino B, Bye J, McDowall J, Ross M, Craig IW (2001) Sequence analysis and transcript identification within 1.5 MB of DNA deleted together with the NDP and MAO genes in atypical Norrie disease patients presenting with a profound phenotype. Hum Mutat 17(6):523. doi:10.1002/humu.1140
Chen ZY, Hotamisligil GS, Huang JK, Wen L, Ezzeddine D, Aydin-Muderrisoglu N, Powell JF, Huang RH, Breakefield XO, Craig I et al (1991) Structure of the human gene for monoamine oxidase type A. Nucleic Acids Res 19(16):4537–4541
Kundu TK, Rao MR (1999) CpG islands in chromatin organization and gene expression. J Biochem 125(2):217–222
Wierstra I (2008) Sp1: emerging roles–beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun 372(1):1–13. doi:10.1016/j.bbrc.2008.03.074
Wong WK, Chen K, Shih JC (2003) Decreased methylation and transcription repressor Sp3 up-regulated human monoamine oxidase (MAO) B expression during Caco-2 differentiation. J Biol Chem 278(38):36227–36235. doi:10.1074/jbc.M305549200
Shumay E, Fowler JS (2010) Identification and characterization of putative methylation targets in the MAOA locus using bioinformatic approaches. Epigenetics 5(4):325–342. pii: 11719
Chen K, Ou XM, Chen G, Choi SH, Shih JC (2005) R1, a novel repressor of the human monoamine oxidase A. J Biol Chem 280(12):11552–11559. doi:10.1074/jbc.M410033200
Wu JB, Chen K, Li Y, Lau YF, Shih JC (2009) Regulation of monoamine oxidase A by the SRY gene on the Y chromosome. FASEB J 23(11):4029–4038. doi:10.1096/fj.09-139097
Wilhelm D, Palmer S, Koopman P (2007) Sex determination and gonadal development in mammals. Physiol Rev 87(1):1–28. doi:10.1152/physrev.00009.2006
Wu JB, Chen K, Ou XM, Shih JC (2009) Retinoic acid activates monoamine oxidase B promoter in human neuronal cells. J Biol Chem 284(25):16723–16735. doi:10.1074/jbc.M901779200
Shih JC, Wu JB, Chen K (2011) Transcriptional regulation and multiple functions of MAO genes. J Neural Transm 118(7):979–986. doi:10.1007/s00702-010-0562-9
Manoli I, Le H, Alesci S, McFann KK, Su YA, Kino T, Chrousos GP, Blackman MR (2005) Monoamine oxidase-A is a major target gene for glucocorticoids in human skeletal muscle cells. FASEB J 19(10):1359–1361. doi:10.1096/fj.04-3660fje
Sabol SZ, Hu S, Hamer D (1998) A functional polymorphism in the monoamine oxidase A gene promoter. Hum Genet 103(3):273–279
Syagailo YV, Stober G, Grassle M, Reimer E, Knapp M, Jungkunz G, Okladnova O, Meyer J, Lesch KP (2001) Association analysis of the functional monoamine oxidase A gene promoter polymorphism in psychiatric disorders. Am J Med Genet 105(2):168–171 10.1002/ajmg.1193
Gutierrez B, Arias B, Gasto C, Catalan R, Papiol S, Pintor L, Fananas L (2004) Association analysis between a functional polymorphism in the monoamine oxidase A gene promoter and severe mood disorders. Psychiatr Genet 14 (4):203–208. pii: 00041444-200412000-00007
Jorm AF, Henderson AS, Jacomb PA, Christensen H, Korten AE, Rodgers B, Tan X, Easteal S (2000) Association of a functional polymorphism of the monoamine oxidase A gene promoter with personality and psychiatric symptoms. Psychiatr Genet 10(2):87–90
Kirov G, Norton N, Jones I, McCandless F, Craddock N, Owen MJ (1999) A functional polymorphism in the promoter of monoamine oxidase A gene and bipolar affective disorder. Int J Neuropsychopharmacol 2(4):293–298. doi:10.1017/S1461145799001601
Kunugi H, Ishida S, Kato T, Tatsumi M, Sakai T, Hattori M, Hirose T, Nanko S (1999) A functional polymorphism in the promoter region of monoamine oxidase-A gene and mood disorders. Mol Psychiatry 4(4):393–395
Cao X, Rui L, Pennington PR, Chlan-Fourney J, Jiang Z, Wei Z, Li XM, Edmondson DE, Mousseau DD (2009) Serine 209 resides within a putative p38(MAPK) consensus motif and regulates monoamine oxidase-A activity. J Neurochem 111(1):101–110. doi:10.1111/j.1471-4159.2009.06300.x
Morishima M, Harada N, Hara S, Sano A, Seno H, Takahashi A, Morita Y, Nakaya Y (2006) Monoamine oxidase A activity and norepinephrine level in hippocampus determine hyperwheel running in SPORTS rats. Neuropsychopharmacology 31(12):2627–2638. doi:10.1038/sj.npp.1301028
Rothmond DA, Weickert CS, Webster MJ (2012) Developmental changes in human dopamine neurotransmission: cortical receptors and terminators. BMC Neurosci 13:18. doi:10.1186/1471-2202-13-18
Fitzgerald JC, Ufer C, De Girolamo LA, Kuhn H, Billett EE (2007) Monoamine oxidase-A modulates apoptotic cell death induced by staurosporine in human neuroblastoma cells. J Neurochem 103(6):2189–2199. doi:10.1111/j.1471-4159.2007.04921.x
Chen K, Wu HF, Shih JC (1993) The deduced amino acid sequences of human platelet and frontal cortex monoamine oxidase B are identical. J Neurochem 61(1):187–190
Youdim MB (1988) Platelet monoamine oxidase B: use and misuse. Experientia 44(2):137–141
Klaiber EL, Broverman DM, Vogel W, Peterson LG, Snyder MB (1996) Individual differences in changes in mood and platelet monoamine oxidase (MAO) activity during hormonal replacement therapy in menopausal women. Psychoneuroendocrinology 21(7):575–592. pii: S0306453096000236
Magder S (2006) Reactive oxygen species: toxic molecules or spark of life? Crit Care 10(1):208. doi:10.1186/cc3992
Wiesner R, Kasuschke A, Kuhn H, Anton M, Schewe T (1989) Oxygenation of mitochondrial membranes by the reticulocyte lipoxygenase. Action on monoamine oxidase activities A and B. Biochim Biophys Acta 986(1):11–17. pii: 0005-2736(89)90266-6
Vindis C, Seguelas MH, Lanier S, Parini A, Cambon C (2001) Dopamine induces ERK activation in renal epithelial cells through H2O2 produced by monoamine oxidase. Kidney Int 59(1):76–86. doi:10.1046/j.1523-1755.2001.00468.x
Mialet-Perez J, Bianchi P, Kunduzova O, Parini A (2007) New insights on receptor-dependent and monoamine oxidase-dependent effects of serotonin in the heart. J Neural Transm 114(6):823–827. doi:10.1007/s00702-007-0695-7
Chaitidis P, Billett E, Kuban RJ, Ungethuem U, Kuhn H (2005) Expression regulation of MAO isoforms in monocytic cells in response to Th2 cytokines. Med Sci Monit 11(8):BR259–BR265. pii: 6629
Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ (1996) Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem 271(8):4138–4142
Brigelius-Flohe R, Flohe L (2011) Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal 15(8):2335–2381. doi:10.1089/ars.2010.3534
Kunduzova OR, Bianchi P, Pizzinat N, Escourrou G, Seguelas MH, Parini A, Cambon C (2002) Regulation of JNK/ERK activation, cell apoptosis, and tissue regeneration by monoamine oxidases after renal ischemia-reperfusion. FASEB J 16(9):1129–1131. doi:10.1096/fj.01-1008fje
Yoon S, Seger R (2006) The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24(1):21–44. doi:10.1080/02699050500284218
Molnar A, Theodoras AM, Zon LI, Kyriakis JM (1997) Cdc42Hs, but not Rac1, inhibits serum-stimulated cell cycle progression at G1/S through a mechanism requiring p38/RK. J Biol Chem 272(20):13229–13235
Kaludercic N, Carpi A, Menabo R, Di Lisa F (1813) Paolocci N (2011) Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim Biophys Acta 7:1323–1332. doi:10.1016/j.bbamcr.2010.09.010
Giorgio M, Trinei M, Migliaccio E, Pelicci PG (2007) Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 8(9):722–728. doi:10.1038/nrm2240
Pearce RK, Owen A, Daniel S, Jenner P, Marsden CD (1997) Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J Neural Transm 104(6–7):661–677
Kanduc D, Mittelman A, Serpico R, Sinigaglia E, Sinha AA, Natale C, Santacroce R, Di Corcia MG, Lucchese A, Dini L, Pani P, Santacroce S, Simone S, Bucci R, Farber E (2002) Cell death: apoptosis versus necrosis (review). Int J Oncol 21(1):165–170
Porter AG, Janicke RU (1999) Emerging roles of caspase-3 in apoptosis. Cell Death Differ 6(2):99–104. doi:10.1038/sj.cdd.4400476
Franklin JL (2011) Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid Redox Signal 14(8):1437–1448. doi:10.1089/ars.2010.3596
Martinou JC, Youle RJ (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 21(1):92–101. doi:10.1016/j.devcel.2011.06.017
Holley AK, Dhar SK, St Clair DK (2010) Manganese superoxide dismutase versus p53: the mitochondrial center. Ann N Y Acad Sci 1201:72–78. doi:10.1111/j.1749-6632.2010.05612.x
Imai H, Nakagawa Y (2003) Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med 34(2):145–169. pii: S0891584902011978
Cohen G, Farooqui R, Kesler N (1997) Parkinson disease: a new link between monoamine oxidase and mitochondrial electron flow. Proc Natl Acad Sci USA 94(10):4890–4894
Cadenas E, Davies KJ (2000) Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 29 (3–4):222–230. pii: S0891-5849(00)00317-8
Del Rio D, Serafini M, Pellegrini N (2002) Selected methodologies to assess oxidative/antioxidant status in vivo: a critical review. Nutr Metab Cardiovasc Dis 12(6):343–351
Cao X, Li XM, Mousseau DD (2009) Calcium alters monoamine oxidase-A parameters in human cerebellar and rat glial C6 cell extracts: possible influence by distinct signalling pathways. Life Sci 85(5–6):262–268. doi:10.1016/j.lfs.2009.06.004
Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ (2002) Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci USA 99(23):14783–14788. doi:10.1073/pnas.192569699
Wong WK, Ou XM, Chen K, Shih JC (2002) Activation of human monoamine oxidase B gene expression by a protein kinase C MAPK signal transduction pathway involves c-Jun and Egr-1. J Biol Chem 277(25):22222–22230. doi:10.1074/jbc.M202844200
Ou X-M, Chen K, Shih JC (2006) Monoamine oxidase A and repressor R1 are involved in apoptotic signaling pathway. Proc Nat Acad Sci 103(29):10923–10928. doi:10.1073/pnas.0601515103
Herlenius E, Lagercrantz H (2004) Development of neurotransmitter systems during critical periods. Exp Neurol 190(Suppl 1):S8–21. doi:10.1016/j.expneurol.2004.03.027
Cikos S, Fabian D, Makarevich AV, Chrenek P, Koppel J (2011) Biogenic monoamines in preimplantation development. Hum Reprod 26(9):2296–2305. doi:10.1093/humrep/der233
Fernandez-Pardal J, Gimeno MF, Gimeno AL (1986) Catecholamines in sow graafian follicles at proestrus and at diestrus. Biol Reprod 34(3):439–445
Khatchadourian C, Menezo Y, Gerard M, Thibault C (1987) Catecholamines within the rabbit oviduct at fertilization time. Hum Reprod 2(1):1–5
Way AL, Barbato GF, Killian GJ (2001) Identification of norepinephrine in bovine oviductal fluid by high performance liquid chromatography. Life Sci 70(5):567–576
Skarzynski DJ, Uenoyama Y, Kotwica J, Okuda K (1999) Noradrenaline stimulates the production of prostaglandin f2alpha in cultured bovine endometrial cells. Biol Reprod 60(2):277–282
Basu B, Desai R, Balaji J, Chaerkady R, Sriram V, Maiti S, Panicker MM (2008) Serotonin in pre-implantation mouse embryos is localized to the mitochondria and can modulate mitochondrial potential. Reproduction 135(5):657–669. doi:10.1530/REP-07-0577
Il’kova G, Rehak P, Vesela J, Cikos S, Fabian D, Czikkova S, Koppel J (2004) Serotonin localization and its functional significance during mouse preimplantation embryo development. Zygote 12(3):205–213
Khozhai LI, Puchkov VF, Otellin VA (1995) The effect of a serotonin deficiency on mammalian embryonic development. Ontogenez 26(5):350–355
Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA (2002) “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science 298(5593):597–600. doi:10.1126/science.1072530
Vesela J, Rehak P, Mihalik J, Czikkova S, Pokorny J, Koppel J (2003) Expression of serotonin receptors in mouse oocytes and preimplantation embryos. Physiol Res 52(2):223–228
Amireault P, Dube F (2005) Intracellular cAMP and calcium signaling by serotonin in mouse cumulus-oocyte complexes. Mol Pharmacol 68(6):1678–1687. doi:10.1124/mol.104.010124
Cikos S, Vesela J, Il’kova G, Rehak P, Czikkova S, Koppel J (2005) Expression of beta adrenergic receptors in mouse oocytes and preimplantation embryos. Mol Reprod Dev 71(2):145–153. doi:10.1002/mrd.20256
Cikos S, Rehak P, Czikkova S, Vesela J, Koppel J (2007) Expression of adrenergic receptors in mouse preimplantation embryos and ovulated oocytes. Reproduction 133(6):1139–1147. doi:10.1530/REP-07-0006
Amireault P, Dube F (2005) Serotonin and its antidepressant-sensitive transport in mouse cumulus-oocyte complexes and early embryos. Biol Reprod 73(2):358–365. doi:10.1095/biolreprod.104.039313
Mihalik J, Maslankova J, Spakovska T, Marekova M, Hodorova I, Kusnir J, Rybarova S, Ferenc P, Schmidtova K (2010) Impact of 2 doses of clorgyline on the rat preimplantation embryo development and the monoamine levels in urine. Reprod Sci 17(8):734–741. doi:10.1177/1933719110369181
Herlenius E, Lagercrantz H (2001) Neurotransmitters and neuromodulators during early human development. Early Hum Dev 65(1):21–37. pii: S037837820100189X
Lauder JM, Wallace JA, Krebs H (1981) Roles for serotonin in neuroembryogenesis. Adv Exp Med Biol 133:477–506
Golden GS (1982) A review of the neuroembryology of monoamine systems. Brain Res Bull 9(1–6):553–558
Fox K, Henley J, Isaac J (1999) Experience-dependent development of NMDA receptor transmission. Nat Neurosci 2(4):297–299. doi:10.1038/7203
Lauder JM (1993) Neurotransmitters as growth regulatory signals: role of receptors and second messengers. Trends Neurosci 16(6):233–240
Buznikov GA, Lambert HW, Lauder JM (2001) Serotonin and serotonin-like substances as regulators of early embryogenesis and morphogenesis. Cell Tissue Res 305(2):177–186
Pendleton RG, Rasheed A, Roychowdhury R, Hillman R (1998) A new role for catecholamines: ontogenesis. Trends Pharmacol Sci 19(7):248–251. pii: S0165614798012188
Ringstedt T, Tang LQ, Persson H, Lendahl U, Lagercrantz H (1995) Expression of c-fos, tyrosine hydroxylase, and neuropeptide mRNA in the rat brain around birth: effects of hypoxia and hypothermia. Pediatr Res 37(1):15–20
Lauder JM, Bloom FE (1974) Ontogeny of monoamine neurons in the locus coeruleus, Raphe nuclei and substantia nigra of the rat. I. Cell differentiation. J Comp Neurol 155(4):469–481. doi:10.1002/cne.901550407
Naqui SZ, Harris BS, Thomaidou D, Parnavelas JG (1999) The noradrenergic system influences the fate of Cajal-Retzius cells in the developing cerebral cortex. Brain Res Dev Brain Res 113(1–2):75–82
Patterson PH, Chun LL (1977) The induction of acetylcholine synthesis in primary cultures of dissociated rat sympathetic neurons. I. Effects of conditioned medium. Dev Biol 56(2):263–280. pii: 0012-1606(77)90269-X
Berger-Sweeney J, Hohmann CF (1997) Behavioral consequences of abnormal cortical development: insights into developmental disabilities. Behav Brain Res 86(2):121–142. pii: S0166-4328(96)02251-6
Sundstrom E, Kolare S, Souverbie F, Samuelsson EB, Pschera H, Lunell NO, Seiger A (1993) Neurochemical differentiation of human bulbospinal monoaminergic neurons during the first trimester. Brain Res Dev Brain Res 75(1):1–12
Yavarone MS, Shuey DL, Tamir H, Sadler TW, Lauder JM (1993) Serotonin and cardiac morphogenesis in the mouse embryo. Teratology 47(6):573–584. doi:10.1002/tera.1420470609
Levin M, Buznikov GA, Lauder JM (2006) Of minds and embryos: left-right asymmetry and the serotonergic controls of pre-neural morphogenesis. Dev Neurosci 28(3):171–185. doi:10.1159/000091915
Lauder JM, Krebs H (1978) Serotonin as a differentiation signal in early neurogenesis. Dev Neurosci 1(1):15–30
Hohmann CF, Walker EM, Boylan CB, Blue ME (2007) Neonatal serotonin depletion alters behavioral responses to spatial change and novelty. Brain Res 1139:163–177. doi:10.1016/j.brainres.2006.12.095
Connors SL, Matteson KJ, Sega GA, Lozzio CB, Carroll RC, Zimmerman AW (2006) Plasma serotonin in autism. Pediatr Neurol 35(3):182–186. doi:10.1016/j.pediatrneurol.2006.02.010
Janssen PA, Leysen JE, Megens AA, Awouters FH (1999) Does phenylethylamine act as an endogenous amphetamine in some patients? Int J Neuropsychopharmacol 2(3):229–240. doi:10.1017/S1461145799001522
Kitamura T, Munakata M, Haginoya K, Tsuchiya S, Iinuma K (2008) Beta-phenylethylamine inhibits K+ currents in neocortical neurons of the rat: a possible mechanism of beta-phenylethylamine-induced seizures. Tohoku J Exp Med 215(4):333–340. pii: JST.JSTAGE/tjem/215.333
Hirano M, Uchimura H, Shiraishi A, Kuroki T, Matsumoto T, Tsutsumi T (1989) Beta-phenylethylamine and amphetamine: similar aspects in their behavioropharmacological and neurochemical characteristics. Yakubutsu Seishin Kodo 9(4):335–348
Branchek TA, Blackburn TP (2003) Trace amine receptors as targets for novel therapeutics: legend, myth and fact. Curr Opin Pharmacol 3(1):90–97. pii: S1471489202000280
Jackson DM (1975) Beta-phenylethylamine and locomotor activity in mice. Interaction with catecholaminergic neurones and receptors. Arzneimittelforschung 25(4):622–626
Costall B, Naylor RJ, Pinder RM (1975) Dyskinetic phenomena caused by the intrastriatal injection of phenylethylamine, phenylpiperazine, tetrahydroisoquinoline and tetrahydronaphthalene derivatives in the guinea pig. Eur J Pharmacol 31(1):94–109
Zharikov SI, Zharikova AD, Budantsev A (1979) Effect of beta-phenylethylamine on the dopaminergic system of rat brain. Neirofiziologiia 11(6):578–584
Sengupta T, Mohanakumar KP (2010) 2-Phenylethylamine, a constituent of chocolate and wine, causes mitochondrial complex-I inhibition, generation of hydroxyl radicals and depletion of striatal biogenic amines leading to psycho-motor dysfunctions in Balb/c mice. Neurochem Int 57(6):637–646. doi:10.1016/j.neuint.2010.07.013
Lopez-Gil X, Artigas F, Adell A (2010) Unraveling monoamine receptors involved in the action of typical and atypical antipsychotics on glutamatergic and serotonergic transmission in prefrontal cortex. Curr Pharm Des 16(5):502–515. pii: CPD-Albert Adell (Albert Adell)
Zifa E, Fillion G (1992) 5-Hydroxytryptamine receptors. Pharmacol Rev 44(3):401–458
Choi DS, Maroteaux L (1996) Immunohistochemical localisation of the serotonin 5-HT2B receptor in mouse gut, cardiovascular system, and brain. FEBS Lett 391(1–2):45–51. pii: 0014-5793(96)00695-3
Boyson SJ, Adams CE (1997) D1 and D2 dopamine receptors in perinatal and adult basal ganglia. Pediatr Res 41(6):822–831
Eisenhofer G (2001) The role of neuronal and extraneuronal plasma membrane transporters in the inactivation of peripheral catecholamines. Pharmacol Ther 91(1):35–62. pii: S0163-7258(01)00144-9
Nirenberg MJ, Liu Y, Peter D, Edwards RH, Pickel VM (1995) The vesicular monoamine transporter 2 is present in small synaptic vesicles and preferentially localizes to large dense core vesicles in rat solitary tract nuclei. Proc Natl Acad Sci USA 92(19):8773–8777
Bjork K, Svenningsson P (2011) Modulation of monoamine receptors by adaptor proteins and lipid rafts: role in some effects of centrally acting drugs and therapeutic agents. Annu Rev Pharmacol Toxicol 51:211–242. doi:10.1146/annurev-pharmtox-010510-100520
Murphy DL, Lesch KP (2008) Targeting the murine serotonin transporter: insights into human neurobiology. Nat Rev Neurosci 9(2):85–96. doi:10.1038/nrn2284
Berbel P, Auso E, Garcia-Velasco JV, Molina ML, Camacho M (2001) Role of thyroid hormones in the maturation and organisation of rat barrel cortex. Neuroscience 107(3):383–394. pii: S0306-4522(01)00368-2
Hansson SR, Mezey E, Hoffman BJ (1998) Ontogeny of vesicular monoamine transporter mRNAs VMAT1 and VMAT2. II. Expression in neural crest derivatives and their target sites in the rat. Brain Res Dev Brain Res 110(1):159–174. pii: S0165380698001035
Lewinsohn R, Glover V, Sandler M (1980) Development of benzylamine oxidase and monoamine oxidase A and B in man. Biochem Pharmacol 29(9):1221–1230
Grimsby J, Lan NC, Neve R, Chen K, Shih JC (1990) Tissue distribution of human monoamine oxidase A and B mRNA. J Neurochem 55(4):1166–1169
Rao K, Nagendra SN, Subhash MN (1995) Monoamine oxidase isoenzymes in rat brain: differential changes during postnatal development but not aging. Neurobiol Aging 16(5):833–836. pii: 019745809500061I
Nicotra A, Pierucci F, Parvez H, Senatori O (2004) Monoamine oxidase expression during development and aging. Neurotoxicology 25(1–2):155–165. doi:10.1016/S0161-813X(03)00095-0
Vitalis T, Fouquet C, Alvarez C, Seif I, Price D, Gaspar P, Cases O (2002) Developmental expression of monoamine oxidases A and B in the central and peripheral nervous systems of the mouse. J Comp Neurol 442(4):331–347. doi:10.1002/cne.10093
Nguyen TT, Tseng YT, McGonnigal B, Stabila JP, Worrell LA, Saha S, Padbury JF (1999) Placental biogenic amine transporters: in vivo function, regulation and pathobiological significance. Placenta 20(1):3–11. doi:10.1053/plac.1998.0348
Ramamoorthy S, Prasad PD, Kulanthaivel P, Leibach FH, Blakely RD, Ganapathy V (1993) Expression of a cocaine-sensitive norepinephrine transporter in the human placental syncytiotrophoblast. Biochemistry 32(5):1346–1353
Padbury JF (1989) Functional maturation of the adrenal medulla and peripheral sympathetic nervous system. Baillieres Clin Endocrinol Metab 3(3):689–705
Prasad PD, Hoffmans BJ, Moe AJ, Smith CH, Leibach FH, Ganapathy V (1996) Functional expression of the plasma membrane serotonin transporter but not the vesicular monoamine transporter in human placental trophoblasts and choriocarcinoma cells. Placenta 17(4):201–207
Bzoskie L, Blount L, Kashiwai K, Tseng YT, Hay WW Jr, Padbury JF (1995) Placental norepinephrine clearance: in vivo measurement and physiological role. Am J Physiol 269(1 Pt 1):E145–E149
Juorio AV, Chedrese PJ, Li XM (1989) The influence of ovarian hormones on the rat oviductal and uterine concentration of noradrenaline and 5-hydroxytryptamine. Neurochem Res 14(9):821–827
Belmar J, Lara H, Galleguillos X (1983) Changes in noradrenergic vesicle markers of rabbit oviducts during progesterone treatment. Biol Reprod 29(3):594–604
Helm G, Owman C, Rosengren E, Sjoberg NO (1982) Regional and cyclic variations in catecholamine concentration of the human fallopian tube. Biol Reprod 26(4):553–558
Hansson SR, Bottalico B, Noskova V, Casslen B (2009) Monoamine transporters in human endometrium and decidua. Hum Reprod Update 15(2):249–260. doi:10.1093/humupd/dmn048
Mitchell JA, Hammer RE, Goldman H (1983) Serotonin-induced disruption of implantation in the rat: II. Suppression of decidualization. Biol Reprod 29(1):151–156
Lang U, Prada J, Clark KE (1993) Systemic and uterine vascular response to serotonin in third trimester pregnant ewes. Eur J Obstet Gynecol Reprod Biol 51(2):131–138
Hobel CJ, Parvez H, Parvez S, Lirette M, Papiernik E (1981) Enzymes for epinephrine synthesis and metabolism in the myometrium, endometrium, red blood cells, and plasma of pregnant human subjects. Am J Obstet Gynecol 141(8):1009–1018
Beglopoulos V, Shen J (2004) Gene-targeting technologies for the study of neurological disorders. Neuromolecular Med 6(1):13–30. doi:10.1385/NMM:6:1:013
Senechal Y, Larmet Y, Dev KK (2006) Unraveling in vivo functions of amyloid precursor protein: insights from knockout and knockdown studies. Neurodegener Dis 3(3):134–147. doi:10.1159/000094772
Wolfer DP, Crusio WE, Lipp HP (2002) Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci 25(7):336–340. pii: S0166223602021926
Ochiai Y, Itoh K, Sakurai E, Adachi M, Tanaka Y (2006) Substrate selectivity of monoamine oxidase A, monoamine oxidase B, diamine oxidase, and semicarbazide-sensitive amine oxidase in COS-1 expression systems. Biol Pharm Bull 29(12):2362–2366. pii: JST.JSTAGE/bpb/29.2362
Napolitano A, Cesura AM, Da Prada M (1995) The role of monoamine oxidase and catechol O-methyltransferase in dopaminergic neurotransmission. J Neural Transm Suppl 45:35–45
Forman HJ, Maiorino M, Ursini F (2010) Signaling functions of reactive oxygen species. Biochemistry 49(5):835–842. doi:10.1021/bi9020378
Oliver R 3rd, Friday E, Turturro F, Welbourne T (2010) Troglitazone regulates anaplerosis via a pull/push affect on glutamate dehydrogenase mediated glutamate deamination in kidney-derived epithelial cells; implications for the Warburg effect. Cell Physiol Biochem 26(4–5):619–628. doi:10.1159/000322329
Rubio V, Grisolia S (1981) Human carbamoylphosphate synthetase I. Enzyme 26(5):233–239
Colombo JP, Beruter J, Bachmann C, Peheim E (1977) Enzymes of ammonia detoxication after portacaval shunt in the rat. I. Carbamylphosphate synthetase and aspartate transcarbamylase. Enzyme 22(6):391–398
Albrecht J, Zielinska M, Norenberg MD (2010) Glutamine as a mediator of ammonia neurotoxicity: A critical appraisal. Biochem Pharmacol 80(9):1303–1308. doi:10.1016/j.bcp.2010.07.024
Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, O’Dell M, Li SW, Pan Y, Chung HD, Galvin JE (2008) Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol 115(2):193–203. doi:10.1007/s00401-007-0303-9
Mexas LM, Florang VR, Doorn JA (2011) Inhibition and covalent modification of tyrosine hydroxylase by 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite. Neurotoxicology 32(4):471–477. doi:10.1016/j.neuro.2011.03.013
Anderson DG, Mariappan SV, Buettner GR, Doorn JA (2011) Oxidation of 3,4-dihydroxyphenylacetaldehyde, a toxic dopaminergic metabolite, to a semiquinone radical and an ortho-quinone. J Biol Chem 286(30):26978–26986. doi:10.1074/jbc.M111.249532
Seif I, De Maeyer E, Riviere I, De Maeyer-Guignard J (1991) Stable antiviral expression in BALB/c 3T3 cells carrying a beta interferon sequence behind a major histocompatibility complex promoter fragment. J Virol 65(2):664–671
Grimsby J, Chen K, Wang LJ, Lan NC, Shih JC (1991) Human monoamine oxidase A and B genes exhibit identical exon-intron organization. Proc Natl Acad Sci USA 88(9):3637–3641
Scott AL, Bortolato M, Chen K, Shih JC (2008) Novel monoamine oxidase A knock out mice with human-like spontaneous mutation. NeuroReport 19(7):739–743. doi:10.1097/WNR.0b013e3282fd6e88
Brunner HG, Nelen M, Breakefield XO, Ropers HH, van Oost BA (1993) Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science 262(5133):578–580
Kim JJ, Shih JC, Chen K, Chen L, Bao S, Maren S, Anagnostaras SG, Fanselow MS, De Maeyer E, Seif I, Thompson RF (1997) Selective enhancement of emotional, but not motor, learning in monoamine oxidase A-deficient mice. Proc Natl Acad Sci USA 94(11):5929–5933
Lajard AM, Bou C, Monteau R, Hilaire G (1999) Serotonin levels are abnormally elevated in the fetus of the monoamine oxidase-A-deficient transgenic mouse. Neurosci Lett 261(1–2):41–44. pii: S0304-3940(98)01012-X
Chen K, Holschneider DP, Wu W, Rebrin I, Shih JC (2004) A spontaneous point mutation produces monoamine oxidase A/B knock-out mice with greatly elevated monoamines and anxiety-like behavior. J Biol Chem 279(38):39645–39652. doi:10.1074/jbc.M405550200
Koide Y, Kobayashi K (1984) Developmental changes in the activity and substrate specificities of mouse brain monoamine oxidase. Neurochem Res 9(5):595–606
Cases O, Vitalis T, Seif I, De Maeyer E, Sotelo C, Gaspar P (1996) Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron 16(2):297–307. pii: S0896-6273(00)80048-3
Rebsam A, Seif I, Gaspar P (2002) Refinement of thalamocortical arbors and emergence of barrel domains in the primary somatosensory cortex: a study of normal and monoamine oxidase a knock-out mice. J Neurosci 22(19):8541–8552. pii: 22/19/8541
Li H, Crair MC (2011) How do barrels form in somatosensory cortex? Ann N Y Acad Sci 1225:119–129. doi:10.1111/j.1749-6632.2011.06024.x
Upton AL, Salichon N, Lebrand C, Ravary A, Blakely R, Seif I, Gaspar P (1999) Excess of serotonin (5-HT) alters the segregation of ispilateral and contralateral retinal projections in monoamine oxidase A knock-out mice: possible role of 5-HT uptake in retinal ganglion cells during development. J Neurosci 19(16):7007–7024
Petros TJ, Rebsam A, Mason CA (2008) Retinal axon growth at the optic chiasm: to cross or not to cross. Annu Rev Neurosci 31:295–315. doi:10.1146/annurev.neuro.31.060407.125609
Bou-Flores C, Lajard AM, Monteau R, De Maeyer E, Seif I, Lanoir J, Hilaire G (2000) Abnormal phrenic motoneuron activity and morphology in neonatal monoamine oxidase A-deficient transgenic mice: possible role of a serotonin excess. J Neurosci 20(12):4646–4656. pii: 20/12/4646
Greer JJ, Funk GD, Ballanyi K (2006) Preparing for the first breath: prenatal maturation of respiratory neural control. J Physiol 570(Pt 3):437–444. doi:10.1113/jphysiol.2005.097238
Cazalets JR, Gardette M, Hilaire G (2000) Locomotor network maturation is transiently delayed in the MAOA-deficient mouse. J Neurophysiol 83(4):2468–2470
Nishimaru H, Kudo N (2000) Formation of the central pattern generator for locomotion in the rat and mouse. Brain Res Bull 53(5):661–669. pii: S0361-9230(00)00399-3
Iizuka M, Nishimaru H, Kudo N (1998) Development of the spatial pattern of 5-HT-induced locomotor rhythm in the lumbar spinal cord of rat fetuses in vitro. Neurosci Res 31(2):107–111. pii: S0168-0102(98)00029-7
Narayanan CH, Fox MW, Hamburger V (1971) Prenatal development of spontaneous and evoked activity in the rat (Rattus norvegicus albinus). Behaviour 40(1):100–134
Cases O, Lebrand C, Giros B, Vitalis T, De Maeyer E, Caron MG, Price DJ, Gaspar P, Seif I (1998) Plasma membrane transporters of serotonin, dopamine, and norepinephrine mediate serotonin accumulation in atypical locations in the developing brain of monoamine oxidase A knock-outs. J Neurosci 18(17):6914–6927
Walther DJ, Bader M (2003) A unique central tryptophan hydroxylase isoform. Biochem Pharmacol 66(9):1673–1680. pii: S0006295203005562
McKinney J, Knappskog PM, Haavik J (2005) Different properties of the central and peripheral forms of human tryptophan hydroxylase. J Neurochem 92(2):311–320. doi:10.1111/j.1471-4159.2004.02850.x
Cote F, Fligny C, Bayard E, Launay JM, Gershon MD, Mallet J, Vodjdani G (2007) Maternal serotonin is crucial for murine embryonic development. Proc Natl Acad Sci USA 104(1):329–334. doi:10.1073/pnas.0606722104
Koe BK, Weissman A (1966) p-Chlorophenylalanine: a specific depletor of brain serotonin. J Pharmacol Exp Ther 154(3):499–516
Acharya SB, Goswami NG, Debnath PK (1989) Uterine and placental 5-HT profile in different gestational period of albino rats. Indian J Exp Biol 27(6):505–509
Pytliak M, Vargova V, Mechirova V, Felsoci M (2011) Serotonin receptors—from molecular biology to clinical applications. Physiol Res 60(1):15–25. pii: 931903
Raymond JR, Mukhin YV, Gelasco A, Turner J, Collinsworth G, Gettys TW, Grewal JS, Garnovskaya MN (2001) Multiplicity of mechanisms of serotonin receptor signal transduction. Pharmacol Ther 92(2–3):179–212. pii: S0163725801001693
Zheng G, Dwoskin LP, Crooks PA (2006) Vesicular monoamine transporter 2: role as a novel target for drug development. AAPS J 8(4):E682–E692. doi:10.1208/aapsj080478
Pratuangdejkul J, Schneider B, Launay JM, Kellermann O, Manivet P (2008) Computational approaches for the study of serotonin and its membrane transporter SERT: implications for drug design in neurological sciences. Curr Med Chem 15(30):3214–3227
Salichon N, Gaspar P, Upton AL, Picaud S, Hanoun N, Hamon M, De Maeyer E, Murphy DL, Mossner R, Lesch KP, Hen R, Seif I (2001) Excessive activation of serotonin (5-HT) 1B receptors disrupts the formation of sensory maps in monoamine oxidase a and 5-ht transporter knock-out mice. J Neurosci 21(3):884–896. pii: 21/3/884
Di Pasquale E, Monteau R, Hilaire G (1994) Involvement of the rostral ventro-lateral medulla in respiratory rhythm genesis during the peri-natal period: an in vitro study in newborn and fetal rats. Brain Res Dev Brain Res 78(2):243–252
Shih JC, Ridd MJ, Chen K, Meehan WP, Kung MP, Seif I, De Maeyer E (1999) Ketanserin and tetrabenazine abolish aggression in mice lacking monoamine oxidase A. Brain Res 835(2):104–112. pii: S0006-8993(99)01478-X
Welker C (1971) Microelectrode delineation of fine grain somatotopic organization of (SmI) cerebral neocortex in albino rat. Brain Res 26(2):259–275. pii: 0006-8993(71)90218-6
Bras H, Gaytan SP, Portalier P, Zanella S, Pasaro R, Coulon P, Hilaire G (2008) Prenatal activation of 5-HT2A receptor induces expression of 5-HT1B receptor in phrenic motoneurons and alters the organization of their premotor network in newborn mice. Eur J Neurosci 28(6):1097–1107. doi:10.1111/j.1460-9568.2008.06407.x
Doseff AI (2004) Apoptosis: the sculptor of development. Stem Cells Dev 13(5):473–483. doi:10.1089/1547328042417255
Greenwood J, Gautier J (2005) From oogenesis through gastrulation: developmental regulation of apoptosis. Semin Cell Dev Biol 16(2):215–224. doi:10.1016/j.semcdb.2004.12.002
Blaschke AJ, Staley K, Chun J (1996) Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 122(4):1165–1174
Ferrer I, Soriano E, del Rio JA, Alcantara S, Auladell C (1992) Cell death and removal in the cerebral cortex during development. Prog Neurobiol 39(1):1–43. pii: 0301-0082(92)90029-E
Gaspar P, Cases O, Maroteaux L (2003) The developmental role of serotonin: news from mouse molecular genetics. Nat Rev Neurosci 4(12):1002–1012. doi:10.1038/nrn1256
Schaper C, Zhu Y, Kouklei M, Culmsee C, Krieglstein J (2000) Stimulation of 5-HT(1A) receptors reduces apoptosis after transient forebrain ischemia in the rat. Brain Res 883(1):41–50. pii: S0006-8993(00)02876-6
Persico AM, Baldi A, Dell’Acqua ML, Moessner R, Murphy DL, Lesch KP, Keller F (2003) Reduced programmed cell death in brains of serotonin transporter knockout mice. NeuroReport 14(3):341–344. doi:10.1097/01.wnr.0000058244.21747.83
Stankovski L, Alvarez C, Ouimet T, Vitalis T, El-Hachimi KH, Price D, Deneris E, Gaspar P, Cases O (2007) Developmental cell death is enhanced in the cerebral cortex of mice lacking the brain vesicular monoamine transporter. J Neurosci 27(6):1315–1324. doi:10.1523/JNEUROSCI.4395-06.2007
Trouche E, Mias C, Seguelas MH, Ordener C, Cussac D, Parini A (2010) Characterization of monoamine oxidases in mesenchymal stem cells: role in hydrogen peroxide generation and serotonin-dependent apoptosis. Stem Cells Dev 19(10):1571–1578. doi:10.1089/scd.2009.0353
Petros AM, Olejniczak ET, Fesik SW (2004) Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta 1644(2–3):83–94. doi:10.1016/j.bbamcr.2003.08.012
Glasgow JN, Qiu J, Rassin D, Grafe M, Wood T, Perez-Pol JR (2001) Transcriptional regulation of the BCL-X gene by NF-kappaB is an element of hypoxic responses in the rat brain. Neurochem Res 26(6):647–659
Mitomo K, Nakayama K, Fujimoto K, Sun X, Seki S, Yamamoto K (1994) Two different cellular redox systems regulate the DNA-binding activity of the p50 subunit of NF-kappa B in vitro. Gene 145(2):197–203
Cheng A, Scott AL, Ladenheim B, Chen K, Ouyang X, Lathia JD, Mughal M, Cadet JL, Mattson MP, Shih JC (2010) Monoamine oxidases regulate telencephalic neural progenitors in late embryonic and early postnatal development. J Neurosci 30(32):10752–10762. doi:10.1523/JNEUROSCI.2037-10.2010
Ou XM, Chen K, Shih JC (2006) Monoamine oxidase A and repressor R1 are involved in apoptotic signaling pathway. Proc Natl Acad Sci USA 103(29):10923–10928. doi:10.1073/pnas.0601515103
Alonso M, Melani M, Converso D, Jaitovich A, Paz C, Carreras MC, Medina JH, Poderoso JJ (2004) Mitochondrial extracellular signal-regulated kinases 1/2 (ERK1/2) are modulated during brain development. J Neurochem 89(1):248–256. doi:10.1111/j.1471-4159.2004.02323.x
Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M (2000) Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol 20(10):2175–2183
Naska S, Cenni MC, Menna E, Maffei L (2004) ERK signaling is required for eye-specific retino-geniculate segregation. Development 131(15):3559–3570. doi:10.1242/dev.01212
Gerlach M, Youdim MB, Riederer P (1996) Pharmacology of selegiline. Neurology 47(6 Suppl 3):S137–S145
Collins FA, Murphy DL, Reiss AL, Sims KB, Lewis JG, Freund L, Karoum F, Zhu D, Maumenee IH, Antonarakis SE (1992) Clinical, biochemical, and neuropsychiatric evaluation of a patient with a contiguous gene syndrome due to a microdeletion Xp11.3 including the Norrie disease locus and monoamine oxidase (MAOA and MAOB) genes. Am J Med Genet 42(1):127–134. doi:10.1002/ajmg.1320420126
Juorio AV, Greenshaw AJ, Wishart TB (1988) Reciprocal changes in striatal dopamine and beta-phenylethylamine induced by reserpine in the presence of monoamine oxidase inhibitors. Naunyn Schmiedebergs Arch Pharmacol 338(6):644–648
Berry MD, Scarr E, Zhu MY, Paterson IA, Juorio AV (1994) The effects of administration of monoamine oxidase-B inhibitors on rat striatal neurone responses to dopamine. Br J Pharmacol 113(4):1159–1166
Boulton AA, Juorio AV, Paterson IA (1990) Phenylethylamine in the CNS: effects of monoamine oxidase inhibiting drugs, deuterium substitution and lesions and its role in the neuromodulation of catecholaminergic neurotransmission. J Neural Transm Suppl 29:119–129
Linnoila M, Karoum F, Cutler NR, Potter WZ (1983) Temporal association between depression-dependent dyskinesias and high urinary phenylethylamine output. Biol Psychiatry 18(4):513–516
Potkin SG, Wyatt RJ, Karoum F (1980) Phenylethylamine (PEA) and phenylacetic acid (PAA) in the urine of chronic schizophrenic patients and controls. Psychopharmacol Bull 16(1):52–54
Sabelli HC, Fawcett J, Gusovsky F, Javaid JI, Wynn P, Edwards J, Jeffriess H, Kravitz H (1986) Clinical studies on the phenylethylamine hypothesis of affective disorder: urine and blood phenylacetic acid and phenylalanine dietary supplements. J Clin Psychiatry 47(2):66–70
Scremin OU, Holschneider DP, Chen K, Li MG, Shih JC (1999) Cerebral cortical blood flow maps are reorganized in MAOB-deficient mice. Brain Res 824(1):36–44. pii: S0006-8993(99)01167-1
Nagatsu T (2004) Progress in monoamine oxidase (MAO) research in relation to genetic engineering. Neurotoxicology 25(1–2):11–20. doi:10.1016/S0161-813X(03)00085-8
Fligny C, Hatia S, Amireault P, Mallet J, Cote F (2009) Mammalian prenatal development: the influence of maternally derived molecules. BioEssays 31(9):935–943. doi:10.1002/bies.200800217
Salisbury AL, Ponder KL, Padbury JF, Lester BM (2009) Fetal effects of psychoactive drugs. Clin Perinatol 36(3):595–619. doi:10.1016/j.clp.2009.06.002
Fattore L, Piras G, Corda MG, Giorgi O (2009) The Roman high- and low-avoidance rat lines differ in the acquisition, maintenance, extinction, and reinstatement of intravenous cocaine self-administration. Neuropsychopharmacology 34(5):1091–1101. doi:10.1038/npp.2008.43
Seidler FJ, Temple SW, McCook EC, Slotkin TA (1995) Cocaine inhibits central noradrenergic and dopaminergic activity during the critical developmental period in which catecholamines influence cell development. Brain Res Dev Brain Res 85(1):48–53. pii: 0165380694001864
Chan K, Dodd PA, Day L, Kullama L, Ervin MG, Padbury J, Ross MG (1992) Fetal catecholamine, cardiovascular, and neurobehavioral responses to cocaine. Am J Obstet Gynecol 167(6):1616–1623
Bzoskie L, Blount L, Kashiwai K, Humme J, Padbury JF (1997) The contribution of transporter-dependent uptake to fetal catecholamine clearance. Biol Neonate 71(2):102–110
Jensen A, Hohmann M, Kunzel W (1987) Redistribution of fetal circulation during repeated asphyxia in sheep: effects on skin blood flow, transcutaneous PO2, and plasma catecholamines. J Dev Physiol 9(1):41–55
Jones S, Kornblum JL, Kauer JA (2000) Amphetamine blocks long-term synaptic depression in the ventral tegmental area. J Neurosci 20(15):5575–5580. pii: 20/15/5575
Taylor MJ, Freemantle N, Geddes JR, Bhagwagar Z (2006) Early onset of selective serotonin reuptake inhibitor antidepressant action: systematic review and meta-analysis. Arch Gen Psychiatry 63(11):1217–1223. doi:10.1001/archpsyc.63.11.1217
Hendrick V, Stowe ZN, Altshuler LL, Hwang S, Lee E, Haynes D (2003) Placental passage of antidepressant medications. Am J Psychiatry 160(5):993–996
Weissman AM, Levy BT, Hartz AJ, Bentler S, Donohue M, Ellingrod VL, Wisner KL (2004) Pooled analysis of antidepressant levels in lactating mothers, breast milk, and nursing infants. Am J Psychiatry 161(6):1066–1078
Chambers CD, Hernandez-Diaz S, Van Marter LJ, Werler MM, Louik C, Jones KL, Mitchell AA (2006) Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med 354(6):579–587. doi:10.1056/NEJMoa052744
Wogelius P, Norgaard M, Gislum M, Pedersen L, Munk E, Mortensen PB, Lipworth L, Sorensen HT (2006) Maternal use of selective serotonin reuptake inhibitors and risk of congenital malformations. Epidemiology 17(6):701–704. doi:10.1097/01.ede.0000239581.76793.ae
Noorlander CW, Ververs FF, Nikkels PG, van Echteld CJ, Visser GH, Smidt MP (2008) Modulation of serotonin transporter function during fetal development causes dilated heart cardiomyopathy and lifelong behavioral abnormalities. PLoS One 3(7):e2782. doi:10.1371/journal.pone.0002782
Hemels ME, Einarson A, Koren G, Lanctot KL, Einarson TR (2005) Antidepressant use during pregnancy and the rates of spontaneous abortions: a meta-analysis. Ann Pharmacother 39(5):803–809. doi:10.1345/aph.1E547
Wen SW, Yang Q, Garner P, Fraser W, Olatunbosun O, Nimrod C, Walker M (2006) Selective serotonin reuptake inhibitors and adverse pregnancy outcomes. Am J Obstet Gynecol 194(4):961–966. doi:10.1016/j.ajog.2006.02.019
Williams KE, Marsh WK, Rasgon NL (2007) Mood disorders and fertility in women: a critical review of the literature and implications for future research. Hum Reprod Update 13(6):607–616. doi:10.1093/humupd/dmm019
Glanzman DL (1994) Postsynaptic regulation of the development and long-term plasticity of Aplysia sensorimotor synapses in cell culture. J Neurobiol 25(6):666–693. doi:10.1002/neu.480250608
Homberg JR, Schubert D, Gaspar P (2010) New perspectives on the neurodevelopmental effects of SSRIs. Trends Pharmacol Sci 31(2):60–65. doi:10.1016/j.tips.2009.11.003
Lira A, Zhou M, Castanon N, Ansorge MS, Gordon JA, Francis JH, Bradley-Moore M, Lira J, Underwood MD, Arango V, Kung HF, Hofer MA, Hen R, Gingrich JA (2003) Altered depression-related behaviors and functional changes in the dorsal raphe nucleus of serotonin transporter-deficient mice. Biol Psychiatry 54(10):960–971. pii: S0006322303006966
Holmes A, Murphy DL, Crawley JN (2003) Abnormal behavioral phenotypes of serotonin transporter knockout mice: parallels with human anxiety and depression. Biol Psychiatry 54(10):953–959. pii: S0006322303009521
Nulman I, Rovet J, Stewart DE, Wolpin J, Gardner HA, Theis JG, Kulin N, Koren G (1997) Neurodevelopment of children exposed in utero to antidepressant drugs. N Engl J Med 336(4):258–262. doi:10.1056/NEJM199701233360404
McElhatton PR, Garbis HM, Elefant E, Vial T, Bellemin B, Mastroiacovo P, Arnon J, Rodriguez-Pinilla E, Schaefer C, Pexieder T, Merlob P, Dal Verme S (1996) The outcome of pregnancy in 689 women exposed to therapeutic doses of antidepressants. A collaborative study of the European Network of Teratology Information Services (ENTIS). Reprod Toxicol 10(4):285–294. pii: 0890623896000573
Paykel ES (1995) Clinical efficacy of reversible and selective inhibitors of monoamine oxidase A in major depression. Acta Psychiatr Scand Suppl 386:22–27
Fulton B, Benfield P (1996) Moclobemide. An update of its pharmacological properties and therapeutic use. Drugs 52(3):450–474
Boyer EW, Shannon M (2005) The serotonin syndrome. N Engl J Med 352(11):1112–1120. doi:10.1056/NEJMra041867
Mihalik J, Kravcukova P, Spakovska T, Marekova M, Schmidtova K (2008) Study of high deprenyl dose on the preimplantation embryo development and lymphocyte DNA in rat. Gen Physiol Biophys 27(2):121–126
Stimmel GL, Dopheide JA, Stahl SM (1997) Mirtazapine: an antidepressant with noradrenergic and specific serotonergic effects. Pharmacotherapy 17(1):10–21
Nezvalova-Henriksen K, Spigset O, Nordeng H (2010) Triptan exposure during pregnancy and the risk of major congenital malformations and adverse pregnancy outcomes: results from the Norwegian Mother and Child Cohort Study. Headache 50(4):563–575. doi:10.1111/j.1526-4610.2010.01619.x
Bzoskie L, Yen J, Tseng YT, Blount L, Kashiwai K, Padbury JF (1997) Human placental norepinephrine transporter mRNA: expression and correlation with fetal condition at birth. Placenta 18(2–3):205–210
Pratt NC, Lisk RD (1989) Effects of social stress during early pregnancy on litter size and sex ratio in the golden hamster (Mesocricetus auratus). J Reprod Fertil 87(2):763–769
Nepomnaschy PA, Welch KB, McConnell DS, Low BS, Strassmann BI, England BG (2006) Cortisol levels and very early pregnancy loss in humans. Proc Natl Acad Sci USA 103(10):3938–3942. doi:10.1073/pnas.0511183103
Peyronnet J, Roux JC, Geloen A, Tang LQ, Pequignot JM, Lagercrantz H, Dalmaz Y (2000) Prenatal hypoxia impairs the postnatal development of neural and functional chemoafferent pathway in rat. J Physiol 524 Pt 2:525–537. pii: PHY_0151
Weinstock M (1997) Does prenatal stress impair coping and regulation of hypothalamic-pituitary-adrenal axis? Neurosci Biobehav Rev 21(1):1–10. pii: S0149-7634(96)00014-0
Savelieva KV, Zhao S, Pogorelov VM, Rajan I, Yang Q, Cullinan E, Lanthorn TH (2008) Genetic disruption of both tryptophan hydroxylase genes dramatically reduces serotonin and affects behavior in models sensitive to antidepressants. PLoS One 3(10):e3301. doi:10.1371/journal.pone.0003301
Rosini E, Pollegioni L, Ghisla S, Orru R, Molla G (2009) Optimization of d-amino acid oxidase for low substrate concentrations—towards a cancer enzyme therapy. FEBS J 276(17):4921–4932. doi:10.1111/j.1742-4658.2009.07191.x
Ludwig P, Holzhutter HG, Colosimo A, Silvestrini MC, Schewe T, Rapoport SM (1987) A kinetic model for lipoxygenases based on experimental data with the lipoxygenase of reticulocytes. Eur J Biochem 168(2):325–337
Van Os CP, Rijke-Schilder GP, Van Halbeek H, Verhagen J, Vliegenthart JF (1981) Double dioxygenation of arachidonic acid by soybean lipoxygenase-1. Kinetics and regio-stereo specificities of the reaction steps. Biochim Biophys Acta 663(1):177–193
Wu G, Lu JM, van der Donk WA, Kulmacz RJ, Tsai AL (2011) Cyclooxygenase reaction mechanism of prostaglandin H synthase from deuterium kinetic isotope effects. J Inorg Biochem 105(3):382–390. doi:10.1016/j.jinorgbio.2010.11.015
Zhao X, Ma W, Das SK, Dey SK, Paria BC (2000) Blastocyst H(2) receptor is the target for uterine histamine in implantation in the mouse. Development 127(12):2643–2651
Shuey DL, Sadler TW, Tamir H, Lauder JM (1993) Serotonin and morphogenesis. Transient expression of serotonin uptake and binding protein during craniofacial morphogenesis in the mouse. Anat Embryol (Berl) 187(1):75–85
Cote F, Thevenot E, Fligny C, Fromes Y, Darmon M, Ripoche MA, Bayard E, Hanoun N, Saurini F, Lechat P, Dandolo L, Hamon M, Mallet J, Vodjdani G (2003) Disruption of the nonneuronal tph1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc Natl Acad Sci USA 100(23):13525–13530. doi:10.1073/pnas.2233056100
Cote F, Fligny C, Fromes Y, Mallet J, Vodjdani G (2004) Recent advances in understanding serotonin regulation of cardiovascular function. Trends Mol Med 10(5):232–238. doi:10.1016/j.molmed.2004.03.007
Audero E, Coppi E, Mlinar B, Rossetti T, Caprioli A, Banchaabouchi MA, Corradetti R, Gross C (2008) Sporadic autonomic dysregulation and death associated with excessive serotonin autoinhibition. Science 321(5885):130–133. doi:10.1126/science.1157871
Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, Miller GW, Wightman RM, Caron MG (1997) Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron 19(6):1285–1296. pii: S0896-6273(00)80419-5
Nebigil CG, Choi DS, Dierich A, Hickel P, Le Meur M, Messaddeq N, Launay JM, Maroteaux L (2000) Serotonin 2B receptor is required for heart development. Proc Natl Acad Sci USA 97(17):9508–9513. pii: 97/17/9508
Herve P, Launay JM, Scrobohaci ML, Brenot F, Simonneau G, Petitpretz P, Poubeau P, Cerrina J, Duroux P, Drouet L (1995) Increased plasma serotonin in primary pulmonary hypertension. Am J Med 99(3):249–254. pii: S0002934399801569
Szczypka MS, Rainey MA, Kim DS, Alaynick WA, Marck BT, Matsumoto AM, Palmiter RD (1999) Feeding behavior in dopamine-deficient mice. Proc Natl Acad Sci USA 96(21):12138–12143
Zhou QY, Palmiter RD (1995) Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 83(7):1197–1209. pii: 0092-8674(95)90145-0
Thomas SA, Matsumoto AM, Palmiter RD (1995) Noradrenaline is essential for mouse fetal development. Nature 374(6523):643–646. doi:10.1038/374643a0
Markova LN, Sadykova KA, Sakharova N (1990) The effect of biogenic monoamine antagonists on the development of preimplantation mouse embryos cultured in vitro. Zh Evol Biokhim Fiziol 26(5):726–732
Ufer C, Wang CC, Fahling M, Schiebel H, Thiele BJ, Billett EE, Kuhn H, Borchert A (2008) Translational regulation of glutathione peroxidase 4 expression through guanine-rich sequence-binding factor 1 is essential for embryonic brain development. Genes Dev 22(13):1838–1850. doi:10.1101/gad.466308
Acknowledgments
The authors would like to thank Thomas Horn at Charité-Universitätsmedizin Berlin for producing the structural figures used in this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, C.C., Billett, E., Borchert, A. et al. Monoamine oxidases in development. Cell. Mol. Life Sci. 70, 599–630 (2013). https://doi.org/10.1007/s00018-012-1065-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1065-7