
Abstract
Type A and B monoamine oxidases (MAO-A, -B) mediate and modulate intracellular signal pathways for survival or death of neuronal cells. MAO-A is associated with development of neuronal architecture, synaptic activity, and onset of psychiatric disorders, including depression, and antisocial aggressive impulsive behaviors. MAO-B produces hydrogen peroxide and plays a vital role in neuronal loss of neurodegenerative disorders, such as Parkinson’s and Alzheimer’s diseases. This review presents a novel role of MAO-A and B, their substrates and inhibitors, and hydrogen peroxide in brain function and neuronal survival and death. MAO-A activity is regulated not only by genetic factor, but also by environmental factors, including stress, hormonal deregulation, and food factors. MAO-A activity fluctuates by genetic–environmental factors, modulates the neuronal response to the stimuli, and affects behavior and emotional activities. MAO-B inhibitors selegiline and rasagiline protect neurons via increase expression of anti-apoptotic Bcl-2 and pro-survival neurotrophic factors in human neuroblastoma SH-SY5Y and glioblastoma U118MG cell lines. MAO-A knockdown suppressed the rasagiline-induced gene expression in SH-SY5Y cells, whereas MAO-B silencing enhanced the basal- and selegiline-induced gene expression in U118MG cells. MAO-A and B were shown to function as a mediator or repressor of gene expression, respectively. Further study on cellular mechanism underlying regulation of signal pathways by MAO-A and B may bring us a new insight on the role of MAOs in decision of neuronal fate and the development of novel therapeutic strategy may be expected for neuropsychiatric disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Monoamine oxidase [monoamine: oxygen oxidoreductase (deaminating), EC 1.4.3.4, MAO] catalyzes the oxidative deamination of monoamine neurotransmitters, dietary amines, and xenobiotics, and regulates their levels and functions in the brain. Oxidative deamination by MAO produces the corresponding aldehyde and hydrogen peroxide (H2O2), a potent reactive oxygen species (ROS). Oxidative stress and mitochondrial dysfunction are major risk factors common for neuronal loss in aging and age-related neurodegenerative disorders, such as Parkinson’s and Alzheimer’s diseases (PD and AD). MAO is classified into two classes, types A and B (MAO-A and MAO-B), according to the sensitivity to inhibitors and the affinity to substrates (Shih et al. 1999; Youdim and Bakhle 2006). MAO-A is selectively inhibited by clorgyline [3-(2,4-dichlorophenoxy)-N-methyl-N-prop-2-ylyl-propan-1-amine] and MAO-B by selegiline [(−)deprenyl, (2R)-N-methyl-1-phenyl-N-pro-2-ynyl-propan-2-amine] and rasagiline [(1R)-N-prop-2-ynyl-2,3-dihydro-1H-amine]. Serotonin (5-hydroxytryptamine, 5-HT) and norepinephrine (NE) are oxidized by MAO-A, whereas phenylethylamine, benzylamine, and octopamine are by MAO-B. Dopamine (DA) and tyramine are the substrates for both MAO-A and MAO-B.
MAO-A and B are expressed in distinct population of neuronal cells. MAO-A occurs predominantly in catecholaminergic neurons and MAO-B in serotonergic and histaminergic neurons and astrocytes (Riederer et al. 1989; Saura et al. 1996). MAO-A level in the brain is determined before the birth, and MAO-A regulates development of neuronal architecture coordinately with its major substrate 5-HT (Buckholtz and Mayer-Lindenberg 2008; Naoi et al. 2016, 2017a). MAO-B appears only in the postnatal stage and increases with age, suggesting its association with neuronal loss in aging and neurodegenerative disorders (Fowler et al. 1997). These isoenzymes are involved differentially in the brain function at the specified life stage, even though they share 70% common amino acid sequences and the same FAD coenzyme covalently bound to cysteine in the common pentapeptide sequence.
MAO-A has been proposed as a mediator or modifier of intracellular signal pathway directly and indirectly by regulation of the substrate monoamine levels and H2O2 production. This review will discuss mainly the recent research advances on the role of MAO-A and B in regulation of survival and death of neurons and in neuroprotection by MAO-B inhibitors and other bioactive compounds (Naoi et al. 2012; Finberg and Rabey 2016). MAO-A and MAO-B were confirmed to regulate expression of neuroprotective Bcl-2, neurotrophic factors, and the opposite MAO isoenzyme either in a promoting or suppressive way in human neuroblastoma SH-SY5Y and glioblastoma U118MG cells, respectively (Inaba-Hasegawa et al. 2012, 2017a). MAO-A activity fluctuates in respond to genetic and environmental stimuli, and the association with psychiatric disorders, such as depression and antisocial behavior, is discussed.
MAO-A and B are involved in neuronal death by different mechanisms
MAO-A in apoptosis
In the embryonic mouse brain, MAO-A is essentially required for apoptosis for development of neuronal architecture, as demonstrated by mao-A knockout (KO) (Wang et al. 2011). MAO-A is directly associated with death signaling in neuronal cells. A dopaminergic neurotoxin N-methyl(R)salsolinol was shown to bind to MAO-A at the substrate-binding site and induce apoptosis in SH-SY5Y cells, which mao-A knockdown (KD) with short interfering (siRNA) inhibited (Yi et al. 2006a). In apoptosis induced by NGF withdrawn in PC12 cells, MAO-A expression increased by activation of p38 mitogen-activated protein kinase (MAPK) pathway (De Zutter and Davis 2001). Increased MAO-A oxidized DA, enhanced H2O2 production and caused apoptosis, which was prevented by clorgyline. Decrease of an MAO-A repressor transcription factor R1 (RAM2/CDCA7L/JPO2) was reported to account for increased MAO-A expression (Ou et al. 2006a). Posttranslational increase of MAO-A mRNA, protein, and activity was detected in apoptosis induced by staurosporine, serum withdrawal, and inhibitors of complexes I, III, and IV in SH-SY5Y cells. MAO inhibitors, anti-oxidants, and mao-A KD with micro-RNA (miRNA) suppressed the cell death, suggesting that MAO-A-dependent ROS production caused cell death (Fitzgerald et al. 2007, 2014).
MAO-B in neurodegeneration
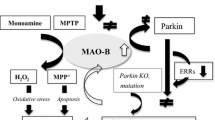
On the other hand, mainly, MAO-B oxidizes DA in the human brain (Glover et al. 1977) and it produces toxic 1-methyl-4-phenylpyridinium ion (MPP+) from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Heikkila et al. 1984). Therefore, MAO-B is proposed as a principal player in “oxidative stress hypothesis” for the pathogenesis of PD, AD, and other neurodegenerative disorders. MAO-B mRNA and enzymatic activity increased in the platelets from patients with PD, AD, and Huntington disease (HD) (Götz et al. 1998; Zhou et al. 2001). These results suggest the contribution of MAO-B to neurodegeneration via oxidative stress in the brain.
The toxic molecule hydrogen peroxide functions as a signaling molecule
Cytotoxicity of H2O2 in neurodegeneration
MAO localized at the outer mitochondrial membrane produces H2O2 and increases ROS levels in the mitochondrial matrix and cytosol (Fig. 1). Enzymatic oxidation of tyramine by MAO increased intra-mitochondrial H2O2 level to 48-folds of the basal level of H2O2 produced in complex II of the electron transfer chain (ETC) in the presence of antimycin A (Hauptman et al. 1996). Declined activity of complex I in the ETC in parkinsonian brain (Mizuno et al. 1989) and complexes I, III, and IV in Alzheimer’s disease (Valla et al. 2006) indicates that MAO mainly contributes to oxidative stress in mitochondria and subsequent neurodegeneration in these disorders. H2O2 is cleaved by transition metals bond to mitochondrial DNA into hydroxyl radical (OH.−), which causes single-strand breaks in mitochondrial DNA and impairs mitochondrial function (Giorgio et al. 2007). H2O2 induced mitochondrial permeability transition, an initial step of apoptosis (Marcocci et al. 2002) by oxidation of vital thiol residues in adenine–nucleotide translocator (ANT), a component of the mitochondrial permeability transition pore (Costantini et al. 1996). H2O2 activates ataxia–telangiectasia mutated (ATM) kinase and the tumor suppressor protein p53, and induces transcriptionally growth arrest and cell death.

H2O2 is produced in mitochondria mainly by the ETC and MAO, and activates various signal pathways. H2O2 activates receptors and enzymes (shown in boxes), and transcription factors (ovals) to increase gene expression and determine the fate of neurons. Figure presents protective signaling pathways detected in the brain, and abbreviations used here are as follows. ELK-1 Ets-like protein-1, ERK extracellular signal-regulated protein kinase, IAP inhibitor of apoptosis, JAK Janus protein kinase, JNK c-Jun N-terminal kinase, KLF Krüppel-like factor, MEK mitogen-activated protein/ERK kinase, Pi phosphate, PLCγ1 phospholipase C-γ1, STAT signal transducers and activators of transcription
H2O2 as a modulator in signal pathway
H2O2 functions as a signaling molecule for physiological processes to control cellular growth and death. H2O2 is membrane-permeable and diffusible, longer-lived than superoxide (O .−2 ) or OH.−, and serves as a redox signal and regulator of transcription factors (Marinho et al. 2014). Beneficial and harmful functions of H2O2 depend on the intracellular concentrations, the physiological range of which spans between 10 and 100 nM (Sies 2017). At lower concentrations about 10 nM, cells respond to H2O2 towards proliferation and adaptation to stress by activating signal pathways, such as the nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant response element (ARE) (Gan and Johnson Gan and Johnson 2014). H2O2 activates transcription factors, such as activator protein-1 (AP-1), cAMP-response-element-binding protein (CREB), heat shock factor 1 (HSF1), hypoxia-inducible factor 1 (HIF-1), NF-κB, NOTCH, and specificity protein 1 (Sp1) (Sies 2014). The activation is mediated by multiple diverse mechanisms, leading to cell death or survival (Martindale and Holbrook 2002). Figure 1 presents signal pathways activated by H2O2 to protect neuronal cells.
H2O2 signaling is cellular-specific, and H2O2 activated the pro-survival zinc finger transcription factor Sp1 in neurons, but did not in glia (Ryu et al. 2003). H2O2 increased Sp1 level and its binding to DNA in nuclei of cortical neurons, and enhanced gene expression for neuroprotection. Dexamethasone activated H2O2–Sp1 pathway and increased MAO-A expression transcriptionally and translationally, but did not affect MAO-B (Manoli et al. 2005). H2O2 increased glial cell line derived neurotrophic factor (GDNF) mRNA and protein in the substantia nigra neuron-glia cell cultures by activation of phosphatidylinisitol-3 kinase (PI3K) and MAPK pathway (Saavedra et al. 2006; Fonseca et al. 2014). H2O2 increased Bcl-2/Bax ratio and neuronal apoptosis inhibitory protein (NAIP) in PC12 cells by activation of extracellular signal-regulated protein kinase (ERK) 5—Krüppel-like factor (KLF) 4 signaling (Su et al. 2014). H2O2 activated p38 MAPK, c-JUN amino-terminal kinase (JNK), and ERK, which further activated Ets-like protein-1 (ELK-1), leading to transcriptional activation of c-FOS. On the other hand, a transitional increase in intracellular H2O2 level activated receptor tyrosine kinases (RTKs) of epidermal growth factor (EGF) receptor and platelet-derived growth factor (PDGF) receptor and activated downstream MAPK and PI3K/Akt pathway to promote proliferation, differentiation, and chemotaxis in cancer and atherosclerosis (Catarzi et al. 2005; Truong and Carroll 2012).
MAO-B inhibitors, monoamines, and MAO-B regulate MAO-A expression
R1 a transcription repressor binds to Sp1/KLF-binding sites in mao-A core promoter, and inhibits MAO-A promoter and enzymatic activity (Fig. 2). Increased MAO-A in depression was mediated by R1–Sp1 pathway (Johnson et al. 2011; Harris et al. 2015). KLF11 [also called transforming growth factor β-inducible early gene 2 (TIEG2)] is an mao-A transcriptional activator. Sp1/KLF pathway takes part in cell proliferation, apoptosis, differentiation, and neoplastic transformation. KLF11 and related transcription factors interact with histone acetyl transferase (HAT) and upregulate mao-A expression in chronic social defeat stress (Grunewald et al. 2012).

In SH-SY5Y cells, various factors affect MAO-A expression. a Rasagiline and H2O2 increase MAO-A expression transcriptionally by reduction of R1 suppressor and activation of KLF11 transcription factor. MAO-A substrates, 5-HT, NE, and DA increase MAO-A by the receptors and activate signals, such as diacetyl glycol (DAG), cAMP, and PI3K. Stress, glucocorticoid, and estrogen increase MAO-A by mean of the receptors in nucleus. Modification and degradation of MAO protein by the ubiquitin–proteasome system (UPS) decrease MAO-A activity. b MAO-B inhibitors increased MAO-A protein. Rasagiline upregulated MAO-A mRNA, protein, and activity by R1-Sp1/KLF11 transcription pathway, but the increase by selegiline did not depend on this pathway (Inaba-Hasegawa et al. 2013)
Rasagiline and selegiline increased MAO-A distinctly depending on MAO-A and B in neuronal and glial cells
In human neuroblastoma SH-SY5Y cells, rasagiline and selegiline (10−6 to 10−12 M) increased MAO-A mRNA, protein, and activity (Fig. 2) (Inaba-Hasegawa et al. 2013). R1–Sp1/KLF11 pathway mediated rasagiline-induced MAO-A expression, but this pathway did not mediate selegiline-increased MAO-A expression.
In SH-SY5Y cells, siRNA against MAO-A (siMao-A) treatment downregulated MAO-A expression, but did not affect MAO-B (Inaba-Hasegawa et al. 2013). On the other hand, in human glioblastoma U118MG cells, treatment with siRNA against MAO-B (siMao-B) significantly upregulated mao-A expression, and selegiline (10−6 to 10−10 M) synergistically increased mao-A expression, whereas siMao-A did not affect mao-B (Inaba-Hasegawa et al. 2017a).
MAO-A substrates increased MAO-A activity
The substrate availability affects MAO-A expression. DA and NE dynamically enhanced MAO-A activity by D2-like receptor in rat mesangial cells (Pizzinat et al. 2003). 5-HT reduction by tryptophan depletion decreased MAO-A binding in the prefrontal cortex, whereas DA increase by carbidopa–levodopa administration enhanced MAO-A in the striatum of healthy volunteers measured with [11C]-harmine positron emission tomography (PET) (Sacher et al. 2012). In primary cultured astrocytes, MAO oxidized DA, produces H2O2, activates Ca2+ signaling, and increased MAO-A activity (Vaarmann et al. 2010). 5-HT, NE, and DA have been presented to activate signal pathways, increase MAO-A expression, and affect brain architecture in developmental period and adulthood neurogenesis in the hippocampus, and impact affective and aggressive behaviors (McCarthy et al. 2007; Yu et al. 2014).
However, in mao-A and mao-B KO mice, no compensatory increase in MAO-B or MAO-A was observed (Holschneider et al. 2001). Induction of mao-A mRNA expression by rasagiline was transit (Inaba-Hasegawa et al. 2013), suggesting that MAO-A expression and activity may fluctuate transitionally and reversely in response to changes in monoamine and H2O2 levels in the brain.
Genetic, biological, and environmental factors regulate MAO-A activity
MAO-A activity is regulated by gene–environment interaction
Altered expression of MAO-A is recognized in psychiatric disorders (Shih et al. 2011; Mousseau and Baker 2012; Godar et al. 2016), and even modest increase in MAO-A activity was associated with depression (Meyer et al. 2006). Association between functional polymorphism of MAO and environmental factors has been confirmed. A VNTR polymorphism of mao-A promoter with low transcription activity was detected in impulsive, aggressive behavior, and alcoholism (Ducci et al. 2008; Sjöberg et al. 2008). Environmental factors, such as abuse exposure in childhood, sexual abuse, and maternal stress, have been reported to decrease MAO-A activity and cause aggressive, impulsive, and antisocial behaviors (Huang et al. 2004; Fergusson et al. 2011; Byrd and Manuck 2014). Fluctuation of MAO-A activity at the distinct period of life may affect behavioral and emotional function during later life stages.
Stress and hormone affect MAO-A activity
MAO-A expression is regulated by hormonal system. Acute stress significantly decreases MAO-A activity in the human brain, and acute dexamethasone exposure decreased MAO-A protein and activity by 30–39% in SH-SY5Y and 1242-MG cells (Soliman et al. 2012). Chronic stress deregulates the hypothalamic–pituitary–adrenal (PHA) axis, activates Sp1/KLF11 signal pathway, and upregulates MAO-A and MAO-B mRNA and enzymatic activity (Chen et al. 2011; Harris et al. 2015). Glucocorticoid (GC) and androgen increased MAO-A activity by direct interaction of glucocorticoid/androgen receptors with the third glucocorticoid/androgen response element (GRE) in the promoter (Ou et al. 2006b). MAO-A expression increased in depression of postpartum or perimenopausal period by age-dependent reduction of estrogen and progesterone (Sacher et al. 2010, 2015; Rekkas et al. 2014).
Genes related to AD and PD are involved in MAO-A expression
Genes related to the familiar forms of PD and AD affect MAO-A expression. Parkin suppressed MAO-A and MAO-B activities in SH-SY5Y cells (Jiang et al. 2006). Parkin-induced degradation of estrogen-related receptors (ERRs) and inhibited MAO expression, whereas the PD-linked mutants did not affect MAO activity (Ren et al. 2011). Wild and AD-related presenilin-1 (PS-1) variants physically interacted with MAO-A and affected the activity in mouse hippocampal HT-22 cells and PS-1 knock-in mice, and the ΔEx9, A431E, and A235V variants increased MAO-A activity (Pennington et al. 2011; Wei et al. 2012a). Increased MAO-A activity was proposed to cause depressive state in AD.
Modification of MAO protein
Modification of MAO protein also affects the enzymatic activity. Ca2+ increased MAO-A activity in monkey brain, mouse, and rat (Egashira et al. 2003; Samantaray et al. 2003), which might increase ROS and promote aging process (Cao et al. 2007). Ca2+ bound to serine 209 residue and increased MAO-A, which was inhibited by the phosphorylation with activated p38(MAPK) (Cao et al. 2009). Rines/RNF180, the RING finger-type E3 ubiquitin ligase, interacted with MAO-A, and promoted its ubiquitination and degradation, whereas Rines KO increased MAO-A activity in the locus coeruleus of mice (Kabayama et al. 2013).
MAO-B expression is elevated in PD, AD, alcoholism, and other psychiatric disorders
MAO-B in PD and AD
As discussed above, MAO-B has been proposed as a pathogenic factor of PD, but the increased activity is mainly due to massive gliosis in the substantia nigra, especially in the recessive forms of PD caused by mutation in PINK-1, parkin, and DJ-1 (Haneka et al. 2010). MAO-B activity increased in reactive astrocytes of senile plaques (Nakamura et al. 1990) and oxidative stress and loss of nigra-striatal were induced in dopaminergic neurons of PD mouse model (Liu et al. 2013). Occurrence of the intron 13 single-nucleotide polymorphism (SNPs) (rs1799836) of mao-B was reported in the female parkinsonian patients (Kang et al. 2006). Allele G of intron 13 has significantly higher transcriptional activity than allele A (Costa-Mallen et al. 2005), and A/G dimorphism in intron 13 sequence increased MAO-B mRNA and protein in PD and AD (Balciuniene et al. 2002; Jakubauskiene et al. 2012).
MAO-B expression increased in the brain and platelet of patients with AD (Gulyas et al. 2011; Zellner et al. 2012). MAO-B was associated with γ-secretase in the human brain, and increased with Aβ42 level in pyramidal neurons of the AD brain. Silencing MAO-B with siRNA reduced intraneuronal Aβ42 in mouse primary cultured cortical neurons, and MAO-B overexpression increased it in HerpG2 cells (Schedin-Weiss et al. 2017).
MAO-B expression in alcoholism and other psychiatric disorders
Increased platelet MAO-B activity was detected in subjects with alcohol dependence, with cognitive deficiency and loss of neurons and glia (Erjavec et al. 2014). MAO-B protein and KLF11 were upregulated in the prefrontal cortex of human alcohol dependence, leading to neuronal loss (Udemgba et al. 2014). Human mao-B core promoter fragment contains two clusters of overlapping Sp/KLF-binding sites separated by a CSCCC element and a TATA box, whereas mao-A core promoter consists of three Sp1 binding sites in reversed orientation without a TATA box. Sp1 sites contribute positively to the transcriptional activity, whereas the CACCC element negatively. Sp1 and Sp4 activate MAO-B promoter activity, and Sp3 represses (Wong et al. 2001). Decrease in methylation of the CpG sites and Sp3 upregulated MAO-B expression (Wong et al. 2003). Selegiline and rasagiline prevented the increase in KLF11–MAO-B activity by ethanol and protected SH-SY5Y cells and brain injury of rats exposed to binge ethanol (Lu et al. 2008; Duncan et al. 2016).
A sex-specific association between mao-B rs1799836 with increased frequency of G allele was detected in Spanish female patients with schizophrenia (Gasso et al. 2008). Platelet MAO-B activity increased in patients with post-traumatic stress disorder (PTSD) (Strac et al. 2016). Two mao-B SNPs, rs10521432 and rs6651806, out of 12 SNPs, were reported in negative emotionality (Dlugos et al. 2009). Platelet MAO-B activity was higher in subjects with severe agitation than non-agitated subjects, but no association was found between severe agitation and mao-b rs1799836 polymorphism in Caucasian male subjects (Perkovic et al. 2016).
Phorbol-12-myristale-13-acetate (PMA) is an extracellular stress inducer and increased MAO-B expression via activation of protein kinase C (PKC) and MAPK involving Sp1, Sp3, c-Jun, and early growth response 1 (Egr-1) (Wong et al. 2002). The fourth estrogen response element in mao-B promoter overlaps with a consensus retinoic acid receptor element (RARE), and retinoic acid activated mao-B promoter through activation of retinoic acid receptor α (RARα) and retinoid X receptor α (RXRα) in BE(2)C cells (Wu et al. 2009a).
Are MAO-A and B the principal player or bystander in neuroprotection by MAO-B inhibitors?
Neuroprotective activity of selegiline and rasagiline has been proved in animal and cellular models of neurodegenerative disorders. Clinical trials of selegiline and rasagiline in parkinsonian patients have been reported to prevent disease progression and ameliorate symptoms (Riederer and Laux 2011). The neuroprotective activity is mainly attributed to the direct suppression of apoptosis signaling triggered by pore formation at the mitochondrial membrane (Wu et al. 2015) and the activation of endogenous biosynthesis of anti-apoptotic Bcl-2 protein family and NTFs (Naoi et al. 2013; Bar-Am et al. 2016).
MAO-B inhibitors bind to MAO-B and also to MAO-A
However, it remains to be elusive whether MAO-B itself is involved in neuroprotection of MAO-B inhibitors. In mao-B KO mice, selegiline could not prevent brain damage by ischemia and age-related deficient spatial learning, suggesting the essential role of MAO-B in neuroprotection (Holschneider et al. 1999a, b). In mao-B KO mice, binding of [3H]-l-deprenyl in the cortex, striatum and corpus callossum decreased markedly to 3.5, 4.0, and 2.7% of control, which was further downregulated by clorgyline (Ekblom et al. 1998). After daily administration of selegiline and rasagiline, MAO-A activity reduced by 70% in the plasma of patients treated with MAO-B inhibitors (Bartl et al. 2014). Systematic administration of Zydas and transdermal selegiline downregulated MAO-A to one-third of control in healthy men (Fowler et al. 2015). These results present that rasagiline and selegiline bind also to MAO-A, not only MAO-B, which may be relevant with the neuroprotection of MAO-B inhibitors in MAO-A-expressed cells.
Enzymatically “dead” MAOs may be involved in neuroprotection by MAO-B inhibitors
Inhibition of MAO-B enzymatic activity is not essentially required for the neuroprotective function of MAO-B inhibitors (Klegeris and McGeer 2000). Selegiline and rasagiline (10−4–10−6 M) irreversibly inhibit the enzymatic activity and protected cells at these concentrations, suggesting that catalytically inactive MAO protein may be associated with neuroprotection. Substitution of aspartic acid 328 residue of MAO-A completely inhibited the enzymatic activity, but catalytic “dead” MAO-A still affected cell viability and proliferation (Wei et al. 2012b), suggesting the different effects of genetic mao KO and MAO inactivation with the inhibitors on regulation of neuronal viability and function.
MAO-B inhibiters bind to MAO apart from the active site and also to other protein
MAO inhibitors bind to MAO at site different from the active site and trigger downstream pro-survival signaling. TVP1022, the S-enantiomer of rasagiline, a very week MAO-B inhibitor, bound to imidazolines 1 and 2 (I1 and I2) binding sites in MAO-A and protected PC12 cells and neonatal rat ventricular myocytes, through activation of p42/44 MAPK (Barac et al. 2012). Other MAO inhibitors, clorgyline, moclobemide, transcypromine, and phenelzine, also show affinity for I2 site (Alemany et al. 1995; MacInnes and Handley 2002). MAO inhibitors bind to other amine oxidases [semicarbazide-sensitive amine oxidase (SSAO), diamine oxidase (DAO), plasma amine oxidase (PAO)], alcohol, and aldehyde dehydrogenases (Holt et al. 2004). Clorgyline, Ro41-1049 (a reversible MAO-A inhibitor), and phenelzine have very high affinity to D2 receptors (Levant et al. 2010). However, there is no direct evidence to support that binding to other protein can contribute neuroprotection by MAO-B inhibitors.
MAO-A mediates Bcl-2 and NTF induction by MAO-B inhibitors in SH-SY5Y cells
Neuroprotective activity of NTFs, especially brain-derived neurotrophic factor (BDNF) and GDNF, has been demonstrated in clinical studies and also in cellular and animal models of neurodegenerative disorders. BDNF, a member of the neurotrophin family (BDNF, NGF, and 3-NT), activates tropomycin-related kinase (Trk) receptor B (TrkB), and promotes neurogenesis, synaptic plasticity, and cell survival. Reduced BDNF levels and BDNF functional polymorphism in major depressive disorder are proposed to account for impaired neurogenesis in the hippocampus (Michel et al. 2008). GDNF family (GDNF, neurturin, artemin, and persephin) functions in cellular growth, differentiation, and survival, and the activity is mediated by a multicomponent receptor complex composed of GDNF family receptor α1 (GFRα1), RET (rearranged during transfection) receptor tyrosine kinase (TK), and phosphatidyl inositol-linked protein. GDNF is expressed in the striatum (caudate putamen) and thalamus and protects selectively dopaminergic neurons.
Rasagiline, selegiline, and related compounds increased Bcl-2 and NTFs
Selegiline, rasagiline, N-propargylamine, aminoindan (a rasagiline metabolite), and befloxantine (a reversible MAO-A inhibitor) increased Bcl-2 expression and suppressed apoptosis (Akao et al. 2002; Yi et al. 2006b; Weinreb et al. 2004, 2010). In cultured cells, selegiline and rasagiline enhanced the levels of GDNF, BDNF, and other NTFs (Tatton et al. 2002; Maruyama et al. 2004; Nakaso et al. 2006). Rasagiline and selegiline (10−7 to 10−10 M) increased GDNF mRNA and protein more markedly than BDNF in SH-SY5Y cells (Maruyama et al. 2004; Maruyama and Naoi 2013). Oral administration of selegiline (5 mg/day for 7–8 weeks) to parkinsonian patients and subcutaneous injection of rasagiline (0.25 mg/day for 4 weeks) in non-human primates increased BDNF and GDNF in the cerebrospinal fluid (CSF) (Maruyama and Naoi 2013). Rasagiline increased BDNF and GDNF also in the rodent brain (Gyarfas et al. 2010; Ledreux et al. 2016). MAO inhibitors permeable though the blood–brain barrier (BBB) may be applicable for the NTF supplement therapy in neurodegenerative disorders.
Rasagiline, aminoindan, and MT-031 (an MAO-A and acetylcholine esterase inhibitor) induced TrkB receptor, activated downstream cell signal mediators, and increased PI3K protein in animal models of PD, inflammation, and aging (Mandel et al. 2007; Badinter et al. 2015; Liu et al. 2017). However, binding of rasagiline or selegiline to Trk receptors and GFRα has been not reported.
MAO-A mediates gene induction by rasagiline and selegiline
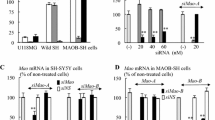
MAO-A mediates Bcl-2 and NTF induction by rasagiline in SH-SY5Y cells. MAO-A KD with siRNA inhibited rasagiline-dependent Bcl-2 protein and BDNF and NGF mRNA expression, whereas selegiline (10−6–10−10 M) increases BDNF expression more markedly in mao-A KD cells than cells treated with non-specific (NS) siRNA (Fig. 4). In mao-B-overexpressed SH-SY5Y cells, MAO-B was found to mediate Bcl-2 induction by selegiline, but not by rasagiline (Inaba-Hasegawa et al. 2012). Rasagiline and selegiline increased Bcl-2 protein and NTF mRNA expression either in MAO-A dependent or independent way (Inaba-Hasegawa et al. 2017a, b).
MAO-B represses the constitutional and selegiline-enhanced expression of genes in U119MG cells
Glial cells induce cell death in neurons by production of pro-inflammatory cytokines and chemokines, and phagocytosis. However, protoplasmic astrocytes contain also protective NTFs and glutathione and inhibit disease progression (Halliday and Stevens 2011). Various neuroprotective compounds, such as selegiline (Mizuta et al. 2000), dopamine agonists (Ohta et al. 2010), memantine (Wu et al. 2009b), valproate (Chen et al. 2006), amantadine (Ossola et al. 2011), and antidepressant (Hisaoka et al. 2008), induced NTF expression in astrocytes.
In U118MG cells, MAO-B was involved in constitutional expression and induction by selegiline and rasagiline of Bcl-2 and NTFs (Inaba-Hasegawa et al. 2017a, b). Figures 3, 4 show that mao-B KD with siMao-B increased the basal expression of Bcl-2 mRNA and protein, BDNF, NGF, and GDNF mRNA, whereas mao-A KD decreased them. In control U118MG cells, selegiline (10−6–10−10 M) and rasagiline (10−7–10−10 M) enhanced BDNF and GDNF. In siMao-B treated cells, selegiline (10−6 to 10−10 M) further increased Bcl-2, BDNF, and GDNF expressions, but rasagiline did not, suggesting that selegiline and rasagiline-activated distinct signal pathways to increase gene expression.

In U118MG cells, MAO-B functions as a repressor of gene expression coding Bcl-2, BDNF, and other NTFs. a MAO-B suppressed the basal expression of Bcl-2, BDNF, NGF, and GDNF, and MAO-B silencing with siMao-B enhanced it. Selegiline increased these genes synergistically. Selegiline triggers MAO-B-mediated signal pathways, and monoamines activate the receptors and downstream transcription factors, including NF-κB, CREB, CREB-binding protein (CBP), FoxO, forkhead in rhabdomyosarcoma (FKHR), and NGF-inducible factor A (NGFI-A). Finally, increased Bcl-2 and NTFs increase cell survival, neurogenesis, and synaptic plasticity. Ethanol activates KLF11 and upregulates mao-B expression. MAO-B expression is regulated also by glucocorticoid, estrogen, and retinoic acid via their receptor element, GRE/estrogen response element (ERE), and retinoic acid receptor element (RARE). b siMao-B treatment enhanced the constitutional expression of BDNF, NGF, and GDNF in U118MG cells, whereas siMao-A suppressed it (Inaba-Hasegawa et al. 2017a, b)

Contrasting effects of mao-A and mao-B KD on the gene induction by rasagiline and selegiline in SH-SY5Y and U118MG cells. a, b In SH-SY5Y cells, MAO-A mediates the induction of Bcl-2, BDNF, and GDNF. Rasagiline and selegiline increased BDNF mRNA, and siMao-A decreased BDNF expression by rasagiline, but enhanced that by selegiline. c, d In U118MG cells, MAO-B represses signal pathways to gene expression. Rasagiline and selegiline increased BDNF mRNA and siMao-B treatment synergistically enhanced selegiline-dependent induction, whereas siMao-A reduced selegiline and rasagiline-induced BDNF level (Inaba-Hasegawa et al. 2017a, b)
As summarized in Fig. 4, rasagiline induced pro-survival genes by activation of signal pathways mediated by MAO-A in neuronal cells. In glial cells, MAO-B functioned as a repressor of mao-A, bcl-2, and NTFs and the gene induction by selegiline, whereas mao-A KO suppresses it.
MAO-A substrates, 5-HT, NE, and DA, induce BDNF and GDNF expressions
MAO-A substrates, 5-HT and NE, stimulate BDNF synthesis and affect neuronal plasticity in aging and neurodegenerative disorders (Mattson et al. 2004). β-Adrenergic receptors mediated NE-dependent BDNF induction by exercise and antidepressants, and 5-HT1A and 5-HT2A/C were associated with antidepressant-induced BDNF expression (Ivy et al. 2003). NE and nitric oxide (NO) promoted BDNF level and survival of cultured hippocampal neurons through activation of cAMP-response element binding (CREB) and Akt-MAP signal pathways (Patel et al. 2010). NE induced BDNF in embryonic rat hippocampal neurons by PI3K and MAPK cascades (Chen et al. 2007).
In astrocytes, monoamine receptors are also expressed (Pav et al. 2008), and DA and NE stimulated biosynthesis of endogenous BDNF (Juric et al. 2006). In cultured rat cortical astrocytes, DA upregulated BDNF protein level (Miklic et al. 2004). NE increased BDNF through binding to α1- and β1/β2-adrenergic receptors, and activation of ligand-G-protein-coupled receptor-PI3K–ERK–CREB cascades or Ca2+-dependent protein kinase (Juric et al. 2008). NE, epinephrine, and DA increased 3-NT expression in primary cultured cerebellar astrocytes by cAMP/PKA and PKC pathways and Ca2+ mobilization (Mele et al. 2010). However, little is known about the expression of BDNF transcripts by 5-HT in astrocytes.
DA increased also GDNF via activation of D1 receptors in human fetal astrocytes (Kinor et al. 2001). 5-HT increased GDNF in C6 glioblastoma cells by binding to 5-HT2A and activation of MEK–MAPK pathway (Hisaoka et al. 2004, 2008), and also via fibroblast growth factor (FGF) receptor 2 (FGFR2) (Tsuchioka et al. 2008). In contrast to BDNF, GDNF induction by NE has been not reported in astrocytes.
Epigenetic regulation of MAO-A expression in gene–environmental interaction
Recently, genotype-dependent environmental influence has been proposed as the pathogenic factor for effective disorders (Ludwig and Dwivedi 2016). Epigenetic regulation of MAO activity influences the vulnerability to environmental stress, and affects social cognition, learning and memory, and stress-related behaviors (Roth and Sweatt 2011). In depression, high levels of MAO-A expression are proposed to impair neurogenesis in the hippocampus and cause molecular changes. However, MAO-A genotype did not fully correspond to MAO-A activity in the brain (Fowler et al. 2007), suggesting involvement of epigenetic modification of MAO-A activity. By epigenetic modification, specific gene is manipulated in the interaction through DNA methylation, hypomethylation, histone modifications, and non-coding RNAs.
DNA methylation in epigenetic regulation of MAO-A
The CpG site-specific methylation state of mao-A promoter predicts MAO-A activity in the brain of healthy men (Shumay et al. 2012). Alteration of DNA methylation in mao-A promoter was reported in female patients with depression and panic disorder (Domschke et al. 2012; Melas and Forsell 2015; Ziegler et al. 2016) and antisocial personality disorder (Checknita et al. 2015), and with nicotine and alcohol dependence (Philibert et al. 2008). Increased level of methylation at the CpG residues in mao-A promoter was reported in male patients with paranoid schizophrenia (Chen et al. 2012).
Chromatin modification in MAO-A expression
Posttranslational reversible modification of histone, such as acetylation, phosphorylation, ubiquitination, and sumoylation, rearranges chromatin and affects the transcription. SIRT1 an NAD+-dependent deacetylase regulates gene expression through histone acetylation, and enhances memory and learning, cognitive function, and synaptic plasticity (Michan et al. 2010). It showed neuroprotective activity in animal models of AD and amyotrophic lateral sclerosis (ALS) (Chen et al. 2005; Kim et al. 2007). SIRT1 activated MAO-A in the brain and induced anxiety and exploratory drive, whereas SIRT1 KO mice showed less susceptibility to depression (Libert et al. 2011). SIRT1 deacetylated nescient helix–loop–helix 2 (NHLH2), a brain-specific transcription factor, and increased the transcriptional activity on mao-A promoter. Micro-RNA-142 (miR-142) was shown to suppress SIRT1–NHLH2 pathways and decrease MAO-A mRNA, protein, and the activity in BE(2)M17 cells, and might be associated with the pathogenesis of several neurodegenerative disorders and HIV-associated cognitive deficient (Chaudhuri et al. 2013). SIRT1 regulates MAO-A activity, but not MAO-B, and serves as a stress sensor signaling for MAO-A to respond to environmental stimuli.
Diet and food-derived phytochemicals regulate MAO activity and affect behavior and emotion
Dietary habits and food factors regulate lifespan, age-dependent decline of cognition and incidence of neuropsychiatric disorders (Mattson et al. 2002). Food-derived polyphenolic compounds, such as (−)-epigallocatechin-3-gallate (EGCG), and genistein (4′,5,7-rihydroxyisoflavone), inhibited DNA methyltransferase and reactivated genes (Yang et al. 2008). Bioactive phytochemicals inhibit MAO activity and show NTF-mimic activity (Vina et al. 2012; Naoi et al. 2017b). Flavonoids with catechol structure, such as quercetin (3′,4′,5,7-tetrahydroflavonol), ginkgolide B, and EGCG, inhibit MAO-A and induces BDNF expression, whereas non-flavonoid phytochemicals, resveratrol (trans-3,4′,5-trihydroxstilbene) and curcumin [(1E, 6E)-1,7-bis(4-hydroxy-3-methoxypheny-hepta-1,6-diene-3,5-dione), inhibit MAO-B and increase GDNF. Phytochemicals affect MAO expression and vice versa MAO-A and MAO-B regulate pro-survival gene induction by phytochemicals. Ginkgolide B and curcumin increase mao-A expression, and tetrahydrocurcumin and sesamin mao-B expression in U118MG cells (Inaba-Hasegawa et al. 2017b). Ginkgolide B, EGCG, and curcumin increase the expression of neuroprotective Bcl-2, GDNF, NGF, and NT-3 mRNA (Naoi et al. 2017b), which was synergistically enhanced by mao-B KD, but inhibited by mao-A KD, as in the case with selegiline. Phytochemicals capable of inhibition of MAO and selective induction of GDNF or BDNF may be expected as neuroprotective and antidepressant compounds for the therapy in neurodegenerative disorders, cognitive decline, and depressive disorders.
Discussion
This review presents the fluctuation of MAO-A activity by genetic and environmental factors and the association with neurodevelopment and brain functions, including mood, motor, cognition, substance abuse, and aggressive and asocial behaviors. Transient and reversible changes in MAO-A activity in combination with the substrates and H2O2 modulate intracellular signaling systems and expression of genes related to neuronal survival and death. For this study, the in vivo assay for MAO-A enzyme activity is essentially required. PET imaging can demonstrate in situ MAO activity using a [11C]-labeled irreversible propargylamine MAO inhibitors, clorgyline, and reversible MAO-A inhibitors, harmine and befloxatone, and [18F]fluoroethyl-harmol, and MAO-B activity with [11C]-selegiline, and [18F]fluororasagiline (Dolle et al. 2003; Fowler et al. 2005; Nag et al. 2012; Kersemans et al. 2013; Maschauer et al. 2015). Human PET studies demonstrated in vivo the effects of MAO-A substrates on MAO-A activity (Sacher et al. 2012), inhibition of MAO-A and -B by smoking (Leroy et al. 2009), MAO-B elevation in aging, and AD (Gulyas et al. 2011), the distribution in the brain, and binding of rasagiline to MAO-B (Freedman et al. 2005). PET imaging of MAO activity and genotype analysis in peripheral samples are expected to present the fluctuation of MAO activity and clarify its role in gene–environment interaction and neuropsychiatric disorders.
Genes coding mao-A and mao-B exhibit the identical exon–intron organization and are derived from duplication of a common ancestral gene (Grimsby et al. 1991). The two isoenzymes share the common protein structure and function in many aspects, even though they are expressed in different types of cells. Mainly MAO-A and B protein and activity are expressed in SH-SY5Y and U118MG cells, respectively, but mRNA of both MAO isoenzymes is detected in either cells. Crosstalk between the MAO isoenzymes has been shown by the substrates, inhibitors, and H2O2 between neuronal and glial cells. In glial cells, MAO-A expression was found to be suppressed by MAO-B and this issue should be further clarified to find role of MAO-A and B in the brain.
MAO-A gene and environmental factors determine the MAO expression and enzymatic activity, which may be associated with development of neural architecture and brain function throughout the life stages. MAO-B was found to repress the constitutional expression and selegiline- and phytochemical-sensitive increase of Bcl-2 and NTFs, and mao-A itself in glial cells. Further studies on these novel functions of MAOs should bring us new strategy for elucidation of the pathogenesis and development of new therapy for neuropsychiatric disorders.
Abbreviations
- ERK:
-
Extracellular signal-regulated protein kinase
- ETC:
-
Electron transfer chain
- KLF:
-
Krüppel-like factor
- MAO-A and MAO-B:
-
Type A and B monoamine oxidase
- mao-A and mao-B KD, KO:
-
MAO-A and MAO-B knockdown, knockout
- NHLH2:
-
Nescient helix-loop-helix 2
- NTF:
-
Neurotrophic factor
- PI3K:
-
Phosphatidylinisitol-3 kinase
- siMao-A, siMao-B, siNS :
-
siRNA against mao-A and mao-B, and non-specific
- Sp1:
-
Specificity protein 1
References
Akao Y, Maruyama W, Yi H, Shamoto-Nagai M, Youdim MB, Naoi M (2002) An anti-Parkinson’s disease drug, N-propargyl-1(R)-aminoindan (rasagiline) enhances expression of antiapoptotic bcl-2 in human dopaminergic SH-SY5Y cells. Neurosci Lett 326(2):105–108
Alemany R, Olmos G, Garcia-Sevilla JA (1995) The effects of phenelzine and other monoamine oxidase inhibitor anti-depressants on brain and liver I2 imidazoline-preferring receptors. Br J Pharmacol 114(4):837–845
Badinter F, Amit T, Bar-Am O, Youdim BH, Weinreb O (2015) Beneficial behavioral, neurochemical and molecular effects of 1-(R)-aminoindan in aged mice. Neuropharmacology 99:264–272
Balciuniene J, Emiilsson L, Oreland L, Pettersson U, Jazin E (2002) Investigation of the functional effect of monoamine oxidase polymorphisms in human brain. Hum Genet 110(1):1–7
Barac YD, Bar-Am O, Liani E, Amit T, Frolov L, Ovcharenko W, Angel O, Youdim MBH, Binah O (2012) I1 imidazoline receptor: novel potential cytoprotective target of TV1022, the S-enantiomer of rasagiline. PLoS ONE 7(11):e47890
Bar-Am O, Amit T, Youdim MB, Weinreb O (2016) Neuroprotective and neurorestorative potential of propargylamine derivatives in ageing: focus on mitochondrial targets. J Neural Transm 123(2):125–135
Bartl J, Müler T, Grünblatt E, Gerlach M, Riederer P (2014) Chronic monoamine oxidase-B inhibitor treatment blocks monoamine oxidase-A enzyme activity. J Neural Transm 121(4):379–383
Buckholtz JW, Mayer-Lindenberg A (2008) MAOA and the neurogenic architecture of human aggression. Trends Neurosci 31(3):120–129
Byrd AL, Manuck SB (2014) MAOA, childhood maltreatment, and antisocial behavior: meta-analysis of a gene–environmental interaction. Biol Psychiatry 75(1):9–17
Cao X, Wei Z, Gabriel GG, Li XM, Mousseau DD (2007) Calcium-sensitive regulation of monoamine oxidase-A contributes to the production of peroxyradicals in hippocampal cultures: implications for Alzheimer disease-related pathology. BMC Neurosci 8:73
Cao X, Rui L, Pennington PR et al (2009) Serine 209 resides with a putative p38(MAPK) consensus motif and regulates monoamine oxidase-A activity. J Neurochem 111(1):101–110
Catarzi S, Biagioni C, Favilli F, Marcucci T, Iantomasi T, Vincenzini MT (2005) Redox regulation of platelet-derived-growth-factor-receptor: role of NADPH-oxidase and c-Src tyrosine kinase. Biochim Biophys Acta 1745:166–175
Chaudhuri AD, Yelamanchili SV, Fox HS (2013) MicroRNA-142 reduces monoamine oxidase A expression and activity in neuronal cells by downregulating SIRT1. PLoS ONE 8(1):e79579
Checknita D, Maussion G, Labonte B et al (2015) Monoamine oxidase A gene promoter methylation and transcriptional downregulation in an offender population with antisocial personality disorder. Br J Psychiatry 206(3):216–222
Chen J, Zhou Y, Müler-Steiner S et al (2005) SIRT1 protects against microglia- dependent amyloid-β toxicity through inhibiting NF-κB signaling. J Biol Chem 280(48):40364–40374
Chen PS, Peng GS, Li G et al (2006) Valproate protects dopaminergic neurons in midbrain neuron/glia culture by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry 11(12):1116–1125
Chen MJ, Nguyen TV, Pike CJ, Rosso-Neustadt AA (2007) Norepinephrine induces BDNF and activates the PI-3K and MAPK cascades in embryonic hippocampal neurons. Cell Signal 19(1):114–128
Chen K, Ou XM, Wu JB, Shih JC (2011) Transcription factor E2F-associated phosphoprotein (EAPP), RAM2/CDCA7L/JPO2 (R1), and Simian virus 40 promoter factor 1 (Sp1) cooperatively regulate glucocorticoid activation of monoamine oxidase B. Mol Pharmacol 79(2):308–317
Chen Y, Zhang J, Zhang L, Shen Y, Xu Q (2012) Effects of MAOA promoter methylation on susceptibility to paranoid schizophrenia. Hum Genet 131(7):1081–1087
Costa-Mallen P, Kelada SN, Costa LG, Checkoway H (2005) Characterization of the in vitro transcriptional activity of polymorphic allele of the human monoamine oxidase-B gene. Neurosci Lett 283(1–2):171–175
Costantini P, Chernyak BV, Petronilli V, Bernardi P (1996) Modulation of the mitochondrial permeability transition pore by pyrimidine nucleotides and dithiol oxidation at two separate sites. J Biol Chem 271(12):6746–6751
De Zutter GS, Davis RJ (2001) Pro-apoptotic gene expression mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Proc Natl Acad Sci USA 98(11):6168–6173
Dlugos AM, Palmer AA, de Wit H (2009) Negative emotionality: monoamine oxidase B gene variants modulate personality traits in healthy humans. J Neural Transm 116(10):1323–1334
Dolle F, Valette H, Bramoulle Y et al (2003) Synthesis and in vivo imaging properties of [11C]befloxatone: a novel highly potent position emission tomography ligand for mono-amine oxidase-A. Bioorg Med Chem 13(10):1771–1775
Domschke K, Tidow N, Kuithan H et al (2012) Monoamine oxidase A gene DNA hypomethylation—a risk factor for panic disorder? Int J Neuropychopharmacol 15(9):1217–1228
Ducci F, Enoch MA, Hodgkinson C, Xu K, Catena M, Robin RW, Goldman D (2008) Interaction between a functional MAOA locus and childhood sexual abuse predicts alcoholism and antisocial personal disorder in adult women. Mol Psychiatry 13(3):334–347
Duncan JW, Zhang X, Wang N et al (2016) Binge ethanol exposure the Krüppel-like factor 11-monoamine oxidase (MAO) pathway in rats: examining the use of MAO inhibitors to prevent ethanol-induced brain injury. Neuropharmacology 105:329–340
Egashira T, Sakai K, Sakurai M, Takayama F (2003) Calcium disodium edetate enhances type A monoamine oxidase activity in monkey brain. Biol Trace Elem Res 94(3):203–211
Ekblom J, Oreland L, Chen K, Shih JC (1998) Is there a “non-MAO” macromolecular target for l-deprenyl?: Studies on MAOB mutant mice. Life Sci 63(12):PL161–PL186
Erjavec GN, Sviglin KN, Perkovic MN, Muck-Seler D, Jovanovic T, Pivac N (2014) Association of gene polymorphisms encoding dopaminergic system components and platelet MAO-B activity with alcohol dependence and alcohol dependence-related phenotypes. Prog Neuropsychopharmacol Biol Psychiatry 54:321–327
Fergusson DM, Boden JM, Honwood LJ, Miller AL, Kennedy MA (2011) MAOA, abuse exposure and antisocial behaviour: 30-year longitudinal study. Br J Psychiatry 198(6):457–463
Finberg JPM, Rabey JM (2016) Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front Pharmacol 7:340
Fitzgerald JC, Ufer C, De Girolamo LA, Kuhn H, Billett EE (2007) Monoamine oxidase-A modulates apoptosis cell death induced by staurosporine in human neuroblastoma cells. J Neurochem 103(6):2189–2199
Fitzgerald KC, Ugun-Klusek A, Allen G, De Girolamo LA, Hargreaves I, Ufer C, Abramov AY, Billett WW (2014) Monoamine oxidase-A knockdown in human neuroblastoma cells reveals protection against mitochondrial toxins. FASEB J 28(1):218–229
Fonseca CP, Gama S, Saavedra A, Baltazar G (2014) H2O2- or l-DOPA-injured dopaminergic neurons trigger the release of double mediators that up-regulate striatal GDNF through different signaling pathways. Biochim Biophys Acta 1842(7):927–934
Fowler JS, Volkow ND, Wang GJ, Logan J, Pappas N, Shea C, MacGregor R (1997) Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol Aging 18(4):431–435
Fowler JS, Logan J, Volkow ND, Wang GJ (2005) Translational neuroimaging: positron emission tomography studies of monoamine oxidase. Mol Imaging Biol 7:377–387
Fowler JS, Alia-Klein N, Kriplani A et al (2007) Evidence that brain MAO A activity does not correspond to MAO genotype in healthy male subjects. Biol Psychiatry 62(4):355–358
Fowler JS, Logan J, Volkow ND et al (2015) Evidence that formulation of the elective MAO-B inhibitor, selegiline, which bypass first-pass metabolism, also inhibit MAO-A in the human brain. Neuropharmacology 40(3):650–657
Freedman NMT, Mishani E, Krausz Y, Weininger J, Lester H, Blaugrund E, Ehrlich D, Chisin R (2005) In vivo measurement of rain monoamine oxidase B occupancy by rasagiline, using 11C-l-deprenyl and PET. J Nucl Med 46(10):1618–1624
Gan L, Johnson JA (2014) Oxidative damage and the Nrf2–ARE pathway in neurodegenerative disorders. Biochim Biophys Acta 1842(8):1208–1218
Gasso P, Bernardo M, Mas S, Crescenti A, Garcia C, Parellada E, Lafuente A (2008) Association of A/G polymorphism in intron 13 of the monoamine oxidase B gene with schizophrenia in a Spanish population. Neuropsychobiology 58(2):65–70
Giorgio M, Trinei M, Migliaccio E, Pelicci PG (2007) Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 8(9):722–728
Glover V, Sandler M, Owen F, Riley GJ (1977) Dopamine is a monoamine oxidase B substrate in man. Nature 265(5589):80–81
Godar SC, Fite PJ, McFarlin KM, Bortolato M (2016) The role of monoamine oxidase A in aggression: current translational developments and future challenges. Prog Neuropsychopharmacol Biol Psychiatry 69:90–100
Götz ME, Fischer P, Gsell W, Riederer P, Streifler M, Simanyi M, Müller F, Danielczyk W (1998) Platelet monoamine oxidase B activity in dementia. A 4-year follow-up. Dement Geriatr Cogn Disord 9(2):74–77
Grimsby J, Chen K, Wang LJ, Lan NC, Shih JC (1991) Human monoamine oxidase A and B genes exhibit identical exon-intron organization. Proc Natl Acad Sci USA 88(9):3637–3641
Grunewald M, Johnson S, Lu D et al (2012) Mechanistic role of a novel gluocorticoid-KLF11 (TIEG2) protein pathway in stress-induced monoamine oxidase A expression. J Biol Chem 287(29):24195–24206
Gulyas B, Pavlova E, Kasa P et al (2011) Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-l-deprenyl using whole hemisphere autoradiography. Neurochem Int 58(1):60–68
Gyarfas T, Knuuttila J, Lindholm P, Rantamäki T, Castren E (2010) Regulation of brain-derived neurotrophic factor (BDNF) and cerebral dopamine neurotrophic factor (CDNF) by anti-parkinsonian drug therapy in vivo. Cell Mol Neurobiol 30(3):361–369
Halliday GM, Stevens CH (2011) Glia: initiators and progressors of pathology in Parkinson’s disease. Mov Disord 26(1):6–17
Haneka MT, Rodriguez JJ, Verkharatsky A (2010) Neuroglia in neurodegeneration. Br Res Rev 63(1–2):189–211
Harris S, Johnson S, Duncan JW et al (2015) Evidence revealing deregulation of the KLF11–MAO A pathway in association with chronic stress and depressive disorders. Neuropsychopharmacology 40:1373–1382
Hauptman N, Grmsby J, Shi JC, Cadenas E (1996) The metabolism of tyramine by monoamine oxidase A/B causes oxidative damage to mitochondrial DNA. Arch Biochem Biophys 335(2):295–304
Heikkila RE, Manzino L, Cabbat FS, Duvoisin RC (1984) Protection against the dopaminergic neurotoxicity of 1-methyl-1,2,3,6-tetrahydropyridine (MPTP) by monoamine inhibitors. Nature 311(5985):467–469
Hisaoka K, Nishida A, Takebayashi M, Koda T, Yamawaki S, Nakata Y (2004) Serotonin increases glial cell line-derived neurotrophic factor release in rat C6 glioblastoma cells. Br Res 1002:167–170
Hisaoka K, Maeda N, Tsuchioka M, Takebayashi M (2008) Antidepressants induce acute CREB phosphorylation and CRE-mediated gene expression in glial cells: a possible contribution to GDNF production. Br Res 1196:53–58
Holschneider DP, Scremin QU, Huynh L, Chen K, Shih JC (1999a) Lack of protection from ischemic injury of monoamine oxidase B-deficient mice following middle cerebral artery occlusion. Neurosci Lett 259(3):161–164
Holschneider DP, Scremin QU, Chen K, Shih JC (1999b) Lack of protection of monoamine oxidase B-deficient mice from age-related spatial learning deficits in the Morris water maze. Life Sci 65(17):1757–1763
Holschneider DP, Chen K, Shi JC (2001) Biochemical, behavioral, physiologic, and neurodevelopmental changes in mice deficient in monoamine oxidase A or B. Brain Res Bull 56(5):453–462
Holt A, Berry MD, Boulton AA (2004) On the binding of monoamine oxidase inhibitors to some sites distinct from the MAO active site, and effects thereby elicited. Neurotoxicology 25(1–2):251–266
Huang Y, Cate SP, Battistuzz C, Oquendo MA, Brent D, Man JJ (2004) An association between a functional polymorphism in the monoamine oxidase A gene promoter, impulsive traits and early abuse experiences. Neuropsychopharmacology 29(8):1498–1505
Inaba-Hasegawa K, Akao Y, Maruyama W, Naoi M (2012) Type A monoamine oxidase is associated with induction of neuroprotective Bcl-2 by rasagiline, an inhibitor of type B monoamine oxidase. J Neural Transm 119(4):405–414
Inaba-Hasegawa K, Akao Y, Maruyama W, Naoi M (2013) Rasagiline and selegiline, inhibitors of type B monoamine oxidase, induce type A monoamine oxidase in human SH-SY5Y cells. J Neural Transm 120(3):435–444
Inaba-Hasegawa K, Shamoto-Nagai M, Maruyama W, Naoi M (2017a) Type B and A monoamine oxidase and their inhibitors regulate the gene expression of Bcl-2 and neurotrophic factors in human glioblastoma U118MG cells: different signal pathways for neuroprotection by selegiline and rasagiline. J Neural Transm 124(9):1055–1066
Inaba-Hasegawa K, Shamoto-Nagai M, Maruyama W, Naoi M (2017b) Phytochemicals induce genes coding Bcl-2 and neurotrophic factors in human glioblastoma U118MG cells: suppression by type B monoamine oxidase (in preparation)
Ivy AS, Rodriguez FG, Garcia C, Chen MJ, Russo-Neustadt AA (2003) Noradrenergic and serotonergic blockage inhibits BDNF mRNA activation following exercise and antidepressant. Pharmacol Biochem Behav 75(1):81–88
Jakubauskiene E, Janaviciute V, Peciuliene I, Söderkvist P, Kanopka A (2012) G/A polymorphism in intronic sequence affects the processing of MAO-B in patients with Parkinson disease. FEBS Lett 586(20):3698–3704
Jiang H, Jiang Q, Liu W, Feng J (2006) Parkin suppresses the expression of monoamine oxidases. J Biol Chem 281(13):8591–8599
Johnson S, Stockmeyer CA, Meyer JH et al (2011) The reduction of R1, a novel repressor protein for monoamine oxidase A, in major depressive disorder. Neuropsychopharmacology 36(10):2139–2148
Juric DM, Miklic S, Carman-Krzan M (2006) Monoaminergic neuronal activity up-regulates BDNF synthesis in cultured neonatal rat astrocytes. Brain Res 1108:54–62
Juric DM, Loncar D, Carman-Lrzan M (2008) Noradrenergic stimulation of BDNF synthesis in astrocytes: mediation via α1- and β1/β2-adrenergic receptors. Neurochem Int 52(1–2):297–306
Kabayama M, Swkoori K, Yamada K, Ornthanalai VG, Ota M, Morimura N, Katayama K, Murphy NP, Aruga J (2013) Rines E3 ubiquitin ligase regulates MAO-A levels and emotional responses. J Neurosci 33(32):12940–12953
Kang SJ, Scott WK, Li YJ et al (2006) Family-based case-control study of MAOA and MAOB polymorphisms in Parkinson disease. Mov Dis 21(12):2175–2180
Kersemans K, Laeken NV, de Vos F (2013) Padiochemistry devoted to the production monoamine oxidase (MAO-A and MAO-B) ligands for brain imaging with positron emission tomography. Label Compd Radiopharm 56(3–4):78–88
Kim D, Nguyen MD, Dobbin MM et al (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J 26(13):3169–3179
Kinor N, Geffen R, Golomb E, Zinman T, Yadid G (2001) Dopamine increases glial cell line-derived neurotrophic factor in human fetal astrocytes. Glia 33(2):143–150
Klegeris A, McGeer PL (2000) R-(−)-Deprenyl inhibits monocytic THP-1 cell neurotoxicity independently of monoamine oxidase inhibition. Exp Neurol 166(2):658–664
Ledreux A, Boger HA, Hinson VK, Cantwell K, Granholm AC (2016) BDNF levels are increased by aminoindan and rasagiline in a double lesion model of Parkinson’s disease. Br Res 1631:34–45
Leroy C, Bragulat V, Berlin I et al (2009) Cerebral monoamine oxidase A inhibition in tobacco smokers confirmed with PET and [11C]befloxatone. J Clin Psychopharmacol 29(1):86–88
Levant B, Morgan KA, Ahlgren-Beckendorf JA, Grandy DK, Chen K, Shih JC, Self I (2010) Modulation of [3H]quinopirole binding at striatal D2 dopamine receptor by a monoamine oxidaseA-like site: evidence from radioligand studies and D2-receptor- and MAO(A)-deficient mice. Life Sci 70(2):229–241
Libert S, Pointer K, Bell EL et al (2011) SIRT1 activates MAO-A in the brain to mediate anxiety and exploratory drive. Cell 147(7):1459–1472
Liu CA, Chinta SJ, Rane A, Andersen JK (2013) Age-related behavioral phenotype of astrocytic monoamine oxidase-B transgenic mouse model of Parkinson’s disease. PLoS ONE 8(1):e54200
Liu W, Rabinpvich A, Nash Y, Frenkel D, Wang Y, Youdim MBH, Weinreb O (2017) Anti-inflammatory and protective effects of MT-031, a novel multitarget MAO-A and AChE/BuChE inhibitor in scopolamine mouse model and inflammatory cells. Neuropharmacology 113(Pt A):445–456
Lu D, Johnson C, Johnson S, Tazil S, Ou XM (2008) The neuroprotective effect of antidepressant drug via inhibition of TIEG2–MAO B mediated cell death. Dug Disc Ther 2(5):289–295
Ludwig B, Dwivedi Y (2016) Dissecting bipolar disorder complexity through epigenomic approach. Mol Psychiatry 21(11):1490–1498
MacInnes N, Handley SL (2002) Characterization of the discriminable stimulus produced by 2-BFI: effects of imidazoline I2-site ligands, MAOIs, β-carbolines, agmatine and ibogaine. Br J Pharm 135(5):1227–1234
Mandel S, Sagi Y, Amit T (2007) Rasagiline promotes regeneration of substantia nigra dopaminergic neurons in post-MPTP-induced parkinsonism via activation of tyrosine kinase receptor signaling pathway. Neurochem Res 32(10):1694–1699
Manoli I, Le H, Alesci S, McFann KK, Su YA, Kino T, Chrousos GP, Blackman MR (2005) Monoamine oxidase-A is a major target for glucocorticoids in human skeletal muscle cells. FASEB J 19(10):1359–1361
Marcocci L, De March U, Salvi M, Nocera S, Agostinelli E, Mondovi B, Toninelllo A (2002) Tyramine and monoamine oxidase inhibitors as modulators of the mitochondrial membrane permeability transition. J Membr Biol 188(1):23–32
Marinho HS, Reak C, Cyrne L, Soares H, Antunes F (2014) Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol 2:535–562
Martindale JL, Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192(1):1–15
Maruyama W, Naoi M (2013) “70th Birthday Professor Riederer” Induction of glial cell line-derived and brain-derived neurotrophic factors by rasagiline and (−)deprenyl: a way to a disease-modifying therapy? J Neural Transm 120(1):83–89
Maruyama W, Nitta A, Shamoto-Nagai M, Hirata Y, Akao Y, Youdim M, Furukawa S, Nabeshima T, Naoi M (2004) N-Propargyl-1-(R)-aminoindan, rasagiline, increases glial cell line-derived neurotrophic factor (GDNF) in neuroblastoma SH-SY5Y cells through activation of NF-κB transcription factor. Neurochem Int 44(6):293–400
Maschauer S, Haller A, Riss PJ, Kuwert T, Prante O, Cumming P (2015) Specific binding of [18F]fluoroethyl-harmol to monoamine oxidase A in rat brain cytostat sections, and compartmental analysis of binding in living brain. J Neurochem 135(5):908–917
Mattson MP, Chan SL, Duan W (2002) Modification of brain aging and neurodegenerative disorders by genes, diet, and behavior. Physiol Rev 82:637–672
Mattson MP, Maudsley S, Martin B (2004) BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci 27(10):588–594
McCarthy D, Lueras P, Bhide P (2007) Elevated dopamine levels during gestation reduced region-specific decreases in neurogenesis and subtle deficits in neuronal numbers. Br Res 1182:11–25
Melas PA, Forsell Y (2015) Hypomethylation of MAOA’s first exon region in depression: a replication study. Psychiatry Res 226(1):389–391
Mele T, Carman-Krzan M, Juric DM (2010) Regulatory role of monoamine transmitters in astrocytic NT-3 synthesis. Int J Dev Neurosci 28(1):13–19
Meyer JH, Ginovart N, Boovariwala A et al (2006) Elevated monoamine oxidase A levels in the brain. An explanation for the monoamine imbalance of major depression. Arch Gen Psychiatry 63(11):1209–1216
Michan S, Li Y, Chou MMH et al (2010) SIRT1 is essential for normal cognitive function and synaptic plasticity. J Neurosci 30(29):9695–9707
Michel TM, Frangou S, Camara S, Thiemeyer D, Jecel J, Tatschner T, Zoechling R, Gruunblatt E (2008) Altered glial cell line-derived neurotrophic factor (GDNF) concentrations in the brain of patients with depressive disorder: a comprehensive post-mortem study. Eur Psychiatry 23(6):413–420
Miklic S, Juric DM, Caman-Krzan M (2004) Differences in the regulation of BDNF and NGF synthesis in cultured neonatal rat astrocytes. Int J Dev Neurosci 22(3):119–130
Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y (1989) Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun 163(3):1450–1455
Mizuta I, Ohta M, Ohta K, Nishimura M, Mizuta E, Hayashi K, Kuno S (2000) Selegiline and desmethylselegiline stimulate NGF, BDNF, and GDNF synthesis in cultured mouse astrocytes. Biochem Biophys Res Commun 279(3):751–755
Mousseau DD, Baker GB (2012) Recent developments in the regulation of monoamine oxidase form and function: is the current model restricting our understanding of the breath of contribution of monoamine oxidase to brain dysfunction? Curr Topics Med Chem 12(20):2163–2176
Nag S, Lehmann L, Kettschau G, Heinrich T, Thiele A, Varrone A, Gulyas B, Halldin C (2012) Synthesis and evaluation of [18F]fluororasagiline, a novel position emission tomography (PET) radioligand for monoamine oxidase B (MAO-B). Bioorg Med Chem 20(9):3065–3071
Nakamura S, Kawamata T, Akiguchi I, Kameyama M, Nakamura N, Kimura H (1990) Expression of monoamine oxidase B activity in astrocytes of senile plaques. Acta Neuropathol 80(4):419–425
Nakaso K, Nakamura C, Sato H, Imamura K, Takeshima T, Nakashima K (2006) Novel cytoprotective mechanism of anti-parkinsonian drug deprenyl: pIK3 and Nrf2-derived induction of antioxidant proteins. Biochim Biophys Res Acta 339(3):915–922
Naoi M, Maruyama W, Inaba-Hasegawa K (2012) Type A and B monoamine oxidase in age-related neurodegenerative disorders: their distinct roles in neuronal death and survival. Curr Top Med Chem 12(20):2177–2188
Naoi M, Maruyama W, Inaba-Hasegawa K (2013) Revelation in the neuroprotective functions of rasagiline and selegiline: the induction of distinct genes by different mechanisms. Expert Rev Neurother 13(6):1233–1250
Naoi M, Riederer P, Maruyama W (2016) Modulation of monoamine oxidase (MAO) expression in neuropsychiatric disorders: genetic and environmental factors involved in type A MAO expression. J Neural Transm 123(2):91–106
Naoi M, Maruyama W, Shamoto-Nagai M (2017a) Type A monoamine oxidase and serotonin are coordinately involved in depressive disorders: from neurotransmitter imbalance to impaired neurogenesis. J Neural Transm. https://doi.org/10.1007/s00702-017-1709-8
Naoi M, Inaba-Hasegawa K, Shamoto-Nagai M, Maruyama W (2017b) Neurotrophic function of phytochemicals for neuroprotection in aging and neurodegenerative disorders: modulation of intracellular signaling and gene expression. J Neural Transm. https://doi.org/10.1007/s00702-1797-5
Ohta K, Kuno S, Inoue S, Ikeda E, Fujinami A, Ohta M (2010) The effect of dopamine agonists: the expression of GDNF, NGF, and BDNF in cultured mouse astrocytes. J Neurol Sci 291(1–2):12–16
Ossola B, Schendzielorz N, Chen SH, Bird GS, Tuominen RK, Manniströ PT, Hong JS (2011) Amantadine protects dopamine neurons from a dual action: reducing activation of microglia and inducing expression of GDNF in astroglia. Neuropharmacology 61(4):574–582
Ou XM, Chen K, Shih JC (2006a) Monoamine oxidase A and repressor R1 are involved in apoptotic signaling pathway. Proc Natl Acad Sci USA 103(29):10923–10928
Ou XM, Chen K, Shih JC (2006b) Glucocorticoid and androgen activation of monoamine oxidase A is regulated by R1 and Sp1. J Biol Chem 281(30):21512–21525
Patel NJ, Chen MJ, Russo-Neustadt AA (2010) Norepinephrine and nitric oxide promote cell survival signaling in hippocampal neurons. Eur J Pharmacol 633(1–3):1–9
Pav M, Kovaru H, Fiserova A, Havrdova E, Lisa V (2008) Neurobiological aspects of depressive disorder and antidepressant treatment: role of glia. Physiol Res 57(2):151–164
Pennington PR, Wei Z, Rui L, Doing JA, Graham B, Kuski K, Gabriel GG, Mousseau DD (2011) Alzheimer disease-related presenilin-1 variants exert distinct effects on monoamine oxidase-A activity in vitro. J Neural Transm 118(7):987–995
Perkovic MN, Strac DS, Erjavec GN, Uzun S, Podobnik J, Kozumplik O, Vlatkovic S, Pivac N (2016) Monoamine oxidase and agitation in psychiatric patients. Prog Neuropsychopharmacol Biol Psychiatry 69:131–146
Philibert RA, Gunter TD, Beach SRH, Brody GH, Madan A (2008) MAOA methylation is associated with nicotine and alcohol dependence in women. Am J Med Genet B Neuropsychiatr Genet 147B(5):565–570
Pizzinat N, Marchal-Victorion S, Maurel A, Ordener C, Bompart G, Parini A (2003) Substrate-dependent regulation of MAO-A in rat mesangial cells: involvement of dopamine D2-like receptors. Am J Physiol Renal Physiol 284(1):F167–F174
Rekkas PV, Wilson AA, Lee VWH et al (2014) Greater monoamine oxidase A binding in perimenopausal age as measured with carbon 11-labelled harmine position emission tomography. JAMA Psychiatry 71(8):873–879
Ren Y, Jiang H, Ma D, Nakaso K, Feng J (2011) Parkin degrades estrogen-related receptors to limit the expression of monoamine oxidases. Hum Mol Genet 20(6):1074–1083
Riederer P, Laux G (2011) MAO-inhibitors in Parkinson’s disease. Exp Neurobiol 20(1):1–17
Riederer P, Konradi C, Habenstreit G, Youdim MBH (1989) Neurochemical perspectives to the function of monoamine oxidase. Acta Neurol Scand 126(1):41–45
Roth TL, Sweatt JD (2011) Annual research review: Epigenetic mechanisms and environmental shaping of the brain during sensitive periods of development. J Child Psychol Psychiatry 52(4):398–408
Ryu H, Lee J, Zaman K, Kubilis J, Ross BD, Neve R, Ratan RR (2003) Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci 23(9):3597–3606
Saavedra A, Baltazar G, Santos P, Carvalhp CM, Duarte EP (2006) Selective injury to dopaminergic neurons up-regulates GDNF in substantia nigra postnatal cell cultures: role of neuron-glia crosstalk. Neurobiol Dis 23(3):533–542
Sacher J, Wilson AA, Houle S, Rusjan P, Hassan S, Bloomfield PM, Stewart DE, Maeyer JH (2010) Elevated brain monoamine oxidase A binding in the early postpartum period. Arch Gen Psychiatry 67(5):468–474
Sacher J, Rabiner EA, Clark M et al (2012) Dynamic, adaptive changes in MAO-A binding after alterations in substrate availability: an in vivo [11C]-harmine position emission tomography study. J Cereb Blood Flow Metab 32(3):443–446
Sacher J, Rekkas PV, Wilson AA et al (2015) Relationship of monoamine oxidase-A distribution volume to postpartum depression and postpartum crying. Neuropsychopharmacology 40(2):427–435
Samantaray S, Chandra G, Mohanakumar KP (2003) Calcium channel agonist, (±)-Bay K8644, causes a transient increase in striatal monoamine oxidase activity in Balb/c mice. Neurosci Lett 342(1–2):73–76
Saura J, Bleuel Z, Ulrich J et al (1996) Molecular neuroanatomy of human monoamine oxidases A and B revealed by quantitative enzyme radioautography and in situ hybridization histochemistry. Neuroscience 70(3):755–774
Schedin-Weiss S, Inoue M, Hromadkova L et al (2017) Monoamine oxidase B in elevated in Alzheimer disease neurons, is associated with γ-secretase and regulates neuronal amyloid β-peptide levels. Alzheimers Res Ther 9(1):57
Shih JC, Chen K, Ridd MJ (1999) Monoamine oxidase: from genes to behavior. Annu Rev Neurosci 22:197–217
Shih JC, Boyang J, Chen K (2011) Transcriptional regulation and multiple functions of MAO genes. J Neural Transm 118(7):979–986
Shumay E, Logan J, Volkow ND, Fowler JS (2012) Evidence that the methylation state of the monoamine oxidase A (MAOA) gene predicts brain activity of MAOA enzyme in healthy men. Epigenetics 7(10):1151–1160
Sies H (2014) Role of metabolic H2O2 generation, redox signaling and oxidative stress. J Biol Chem 289(13):8735–8741
Sies H (2017) Hydrogen peroxide as central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol 11:613–619
Sjöberg RL, Ducci F, Barr CS, Newman T, Dell’Osso L, Virkkunen M, Foldman D (2008) A non-additive interaction of a functional MAO-A VNTR and testosterone predicts antisocial behavior. Neuropsychopharmacology 33(2):425–430
Soliman A, Udemgba C, Fan I et al (2012) Convergent effects of acute stress and glucocorticoid exposure upon MAO-A in humans. J Neurosci 32(48):17120–17127
Strac DS, Petrovic ZK, Perkovic MN, Molac D, Erjavec GN, Pivac N (2016) Platelet monoamine oxidase type B, MAOB intron 13 and MAOA-uVNTR polymorphism and symptoms of post-traumatic stress disorders. Stress 29(4):362–373
Su C, Sun F, Cunningham RL, Rybalchenko N, Sigh M (2014) ERK5/KLF4 signaling as a common mediator of the neuroprotective effects of both nerve growth factor and hydrogen peroxide preconditioning. Age (Dordr) 36(4):9685
Tatton WG, Chalmers-Redman RME, Ju WJ, Mammen M, Carlile GW, Pong AW, Tatton NA (2002) Propargylamines induce antiapoptotic new protein synthesis in serum- and nerve growth factor (NGF)-withdrawn, NGF-differentiated PC-12 cells. J Pharmacol Exp Ther 301(12):753–764
Truong TH, Carroll KS (2012) Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry 51(50):9954–9965
Tsuchioka M, Takebayashi M, Hisaoka K, Maeda N, Nakata Y (2008) Serotonin (5-HT) induces glial cell line-derived neurotrophic factor (GDNF) mRNA expression via the transactivation of fibroblast growth factor 2 (FGR2) in rat C6 glioma cells. J Neurochem 106(1):244–257
Udemgba C, Johnson S, Stockmeier CA et al (2014) The expression of KLF11(TIEG2), a monoamine oxidase B transcription activator in the prefrontal cortex of human alcohol dependence. Alcohol Clin Exp Res 38(1):144–151
Vaarmann A, Gandhi S, Abramov AY (2010) Dopamine induces Ca2+ signaling in astrocytes through reactive oxygen species generated by monoamine oxidase. J Biol Chem 85(32):25018–25023
Valla J, Schneider L, Niedzielko T et al (2006) Impaired platelet mitochondrial activity in Alzheimer’s disease and mild cognitive impairment. Mitochondrion 6(6):323–330
Vina D, Serra S, Lamela M, Delogu G (2012) Herbal natural products as a source of monoamine oxidase inhibitors: a review. Curr Topics Ned Chem 12(20):2131–2144
Wang CC, Borchert A, Ugun-Klusek A et al (2011) Monoamine oxidase A expression is vital for embryonic brain development by modulating developmental apoptosis. J Biol Chem 286(32):28322–28330
Wei Z, Gabriel GG, Rui L, Cao X, Pennington PR, Chlan-Fourney J, Nazaralli A, Baker GB, Mousseau DD (2012a) Monoamine oxidase-A physically interacts with presenilin-1 (M146V) in the mouse cortex. J Alzheimer’s Dis 28(2):403–422
Wei Z, Satram-Maharaj T, Chaharyn B, Kuski K, Pennington PR, Cao X, Chlan J, Mousseau DD (2012b) Aspartic acid substitution in monoamine oxidase-A reveal both catalytic-dependent and -independent influences on cell viability and proliferation. J Neural Transm 119(11):1285–1294
Weinreb O, Bar-Am O, Amit T, Chillag-Talmor O, Youdim MBH (2004) Neuroprotection via pro-survival protein kinase C isoforms associated with Bcl-2 family members. FASEB J 18(12):1471–1473
Weinreb O, Amit T, Bar-Am O, Youdim MB (2010) Rasagiline: a novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog Neurobiol 92(3):330–344
Wong WK, Chen K, Shih JC (2001) Regulation of human monoamine oxidase B gene by Sp1 and Sp3. Mol Psychiatry 59(4):852–859
Wong WK, Chen K, Shih JC (2002) Activation of human monoamine oxidase B gene expression by a protein kinase C MAP signal transduction pathway involves c-Jun and Egr-1. J Biol Chem 277(25):22222–22230
Wong WK, Ou XM, Chen K, Shih JC (2003) Decreased methylation and transcription repressor Sp3 up-regulated human monoamine oxidase (MAO) B expression during Caco-2 differentiation. J Biol Chem 278(38):36227–36235
Wu JB, Chen K, Ou XM, Shi JC (2009a) Retinoic acid activates monoamine oxidase-B promoter in human neuronal cells. J Biol Chem 284(25):16723–16735
Wu HM, Tzeng NS, Qian L et al (2009b) Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglia activation. Neuropsychopharmacology 34(10):2344–2357
Wu Y, Kazumura K, Maruyama W, Osawa T, Naoi M (2015) Rasagiline and selegiline suppress calcium efflux from mitochondria by PK11195-induced opening of mitochondrial permeability transition pore: a novel antiapoptotic function for neuroprotection. J Neural Transm 122(10):1399–1407
Yang CS, Fang M, Lambert JD, Yan P, Huang HM (2008) Reversal of hypomethylation and reactivation of genes by dietary polyphenolic compounds. Nutr Res 66(Suppl 1):S18–S20
Yi H, Akao Y, Maruyama W, Chen K, Shih Naoi M (2006a) Type A monoamine oxidase is the target of an endogenous dopaminergic neurotoxin, N-methyl(R)salsolinol, leading to apoptosis in SH-SY5Y cells. J Neurochem 96(2):541–549
Yi H, Maruyama W, Akao Y, Takahashi T, Iwasa K, Youdim MB, Naoi M (2006b) N-Propargylamine protects SH-SY5Y cells from apoptosis induced by an endogenous neurotoxin, N-methyl(R)salsolinol, through stabilization of mitochondrial membrane and induction of anti-apoptotic Bcl-2. J Neural Transm 113(1):21–32
Youdim MBH, Bakhle YS (2006) Monoamine oxidase: isoforms and inhibitors in Parkinson’s disease and depressive illness. Br J Pharmacol 147(Suppl 1):S287–S296
Yu Q, Teixeira CM, Mahadevia D, Huang Y, Balsam D, Mann JJ, Gingrich JA, Ansorge MS (2014) Dopamine and serotonin signaling during two sensitive developmental periods differentially impact adult aggressive and affective behaviors in mice. Mol Psychiatry 19(6):688–698
Zellner M, Baureder M, Rappold E et al (2012) Comparative platelet proteome analysis reveals an increase of monoamine oxidase-B protein expression in Alzheimer’s disease but not in non-demented Parkinson’s disease patients. J Proteom 75(7):2080–2092
Zhou G, Miura Y, Shoji H, Yamada S, Matsuishi T (2001) Platelet monoamine oxidase B and plasma β-phenylethanolamine in Parkinson’s disease. J Neurol Neurosurg Psychiatry 70(2):229–231
Ziegler C, Richter J, Mahr M et al (2016) MAOA gene hypomethylation in panic disorder-reversibility of an epigenetic risk pattern by psychotherapy. Transl Psychiatry 6:e773
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no competing financial interests in relation to the work described.
Rights and permissions
About this article

Cite this article
Naoi, M., Maruyama, W. & Shamoto-Nagai, M. Type A and B monoamine oxidases distinctly modulate signal transduction pathway and gene expression to regulate brain function and survival of neurons. J Neural Transm 125, 1635–1650 (2018). https://doi.org/10.1007/s00702-017-1832-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-017-1832-6