
Abstract
Schizophrenia is a chronic and disabling mental disorder characterized by positive, negative and mood symptoms, disturbed coping abilities with elevated distress and a significant decline in cognition, quality of life and psychosocial functioning. About one-third of all patients with schizophrenia do not respond adequately to drug treatment. Today neuroscience and clinical research have sufficiently advanced to introduce a novel generation of compounds with neuroprotective properties. The use of neuroprotective agents in schizophrenia is not yet significantly established. An in-depth review of new compounds such as neurosteroids, estrogen, omega-3 fatty acids, S-adenosylmethionine, cannabinoids, piracetam, modafinil, L-theanine, bexarotene with neuroprotective properties is discussed. The mechanisms underlying the neuroprotective effects of these compounds vary and differ from classically defined dopamine and serotonin receptors. This review highlights selective evidence supporting a neuroprotective approach in the search for novel compounds, and suggests future directions for this exciting area. Neuroprotection strategy may be a useful paradigm for treatment of prodromal and first-episode schizophrenia patients and might have a significant impact on the subsequent course and outcome of the illness. The clinical effects of neuroprotective agents clearly merit further investigation in schizophrenia spectrum disorders.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Schizophrenia
- Neurodevelopmental model
- Neurodegenerative model
- Apoptosis
- Oxidative stress
- Excitotoxicity
- Stress sensitization
- Neurotrophic factor expression
- Alteration of neurosteroids
- Vulnerability model
- Neurocognitive domains
- Quality of life deficit
- Neuroprotective agents
- Neurosteroids
- Pregnenolone
- Dehydroepiandrosterone
- Bexarotene
- Theanine
1 Introduction
Schizophrenia is a clinical syndrome that affects about 1% of the general adult population. Whether schizophrenia represents a single disorder of markedly variable expressions or a family of clinically related brain disorders is unclear. Treatment of schizophrenia patients typically includes a combination of pharmacotherapy with antipsychotic agents and psychosocial interventions. Antipsychotic agents ameliorate symptoms in the early phases of disease but become less effective over time, as the underlying disease progresses [1, 2]. The clinical benefits of second-generation antipsychotic agents are modest; their greatest advantage is fewer extrapyramidal side effects. Furthermore, the earlier hope that these agents would have a primary effect on negative and cognitive symptoms is harder to sustain [3, 4]. Despite the effectiveness of antipsychotic medications in the treatment of schizophrenia, in a large scale multi-center study [5] about 74% of the patients discontinued study medication within the first 18 months: 64% of those assigned to olanzapine, 75% of those assigned to perphenazine, 82% of those assigned to quetiapine, 74% of those assigned to risperidone, and 79% of those assigned to ziprasidone. The majority of patients in each group discontinued their assigned treatment owing to inefficacy or intolerable side effects or for other reasons. Furthemore, about one-third of all patients with schizophrenia do not respond adequately to drug treatment. Thus, although antipsychotic agents heralded a major breakthrough in the treatment of positive symptoms in schizophrenia, negative symptoms and cognitive dysfunction continue to account for poor quality of life, reduced functioning and inferior employment status of the patients.
The most critical question facing clinical psychiatrists today is how to treat schizophrenia patients. Is the concept of brain protective therapy relevant for schizophrenia? What are the challenges and opportunities?
This chapter begins with a brief overview of the basic models of schizophrenia and a neuroprotective approach to establish a context for subsequent detailed discussions on several potential neuroprotective compounds, addressing a wide range of mechanisms.
2 Models of Schizophrenia
2.1 Psychopathological Models
Schizophrenia is characterized by psychopathological symptoms, a significant decline in cognition and quality of life, a decrease of psychosocial functioning, elevated emotional distress and disturbed coping abilities that exact considerable human and economic costs. The clinical picture includes a range of positive and negative symptoms such as delusions, hallucinations, agitation, hostility, emotional and social withdrawal, lack of spontaneity, poverty of speech and a wide range of mood symptoms and neurocognitive effects (Fig. 12.1) [6].

Phenotypic domains in schizophrenia and related disorders [6]. The term positive symptoms refers to symptoms that most individuals do not normally experience. They include delusions, auditory hallucinations, and thought disorder, and are typically regarded as manifestations of psychosis. Negative symptoms are so-named because they are considered to be the loss or absence of normal traits or abilities, and include features such as flat or blunted affect and emotion, poverty of speech (alogia), inability to experience pleasure (anhedonia), lack of desire to form relationships (asociality), and lack of motivation (avolition). For a significant portion of the time since the onset of the disturbance, one or more major areas of functioning such as work, interpersonal relations, or self-care, are markedly below the level achieved prior to the onset. A third symptom grouping, the disorganization syndrome, is commonly described, and includes chaotic speech, thought, and behavior
Positive symptoms usually emerge in adolescence or early adulthood, but are often preceded by varying degrees of negative symptoms, cognitive and quality of life impairments. Deterioration occurs primarily in the early stages of the illness and is generally confined to the first 5–7 years after onset. Schizophrenia’s course over time considerably differs from person to person with varying degrees of functional impairments, reduced quality of life, social disability, frequent comorbid substance abuse, and decreased longevity. Overall, schizophrenia tends to be a chronic and relapsing disorder, with only partial remissions (Fig. 12.2) [6].

The three-hit vulnerability model and course of schizophrenia and related disorders (Reproduced from [6]). Schizotaxia – term for the predisposition to schizophrenia
Categorical Models. Most major psychiatric classification systems are based on a categorical system of assessment and assume that individuals fall into distinct categories of pathology, each with unique configurations of symptom and behavioral expressions. For exapmle, two diagnostic systems – e DSM-IV [7] and ICD-10 [8] are based on a categorical model of the schizophrenia syndrome and its core symptoms, in which differences between psychotic symptoms and their normal counterparts are considered to be qualitative, an approach not dissimilar to that proposed by Kraepelin over a century ago. However, this categorical approach does not take into account several central concerns [9]:
-
1.
Many mental disorders are in fact on a dimensional spectrum such as an affective spectrum, an obsessional spectrum and in our case a psychotic spectrum.
-
2.
Each of these disorders actually includes several discrete dimensions such as a cognitive dimension, an impulsivity dimension, dimensions of positive, negative, and mood symptoms and so on.
-
3.
As such, by utilizing a dimensional approach we would be treating the particular pathological dimensional symptoms and not an entire categorical disease entity.
-
4.
Moreover, the validity of the categorical approach is further questioned by the vast heterogeneity of the diagnosis – the “x symptoms out of y” approach employed by the DSM-IV or ICD-10 leads to numerous different clinical combinations with little in common apart from the diagnosis.
The problem of non-homogeneous categories, the difficulty in drawing boundaries and individual progression of the severity of psychopathologic phenomena necessitate a change of paradigm from categorical to dimensional diagnostics [10].
Continuous or Dimensional Models. An alternative, dimensional approach assumes that schizophrenia is not a distinct illness entity, but that psychotic symptoms are diversions from normal experiences and behaviors [11, 12]. Clinical psychiatrists have developed numerous psychopathological models based on items from rating scales: the Positive and Negative Syndrome Scale [13–16] (PANSS), the Scale for the Assessment of Positive Symptoms [17] and the Scale for the Assessment of Negative Symptoms [17] (SANS), the Calgary Scale for Depression in schizophrenia [18], the Overt Aggression Scale [19], and others. These dimensional measures are usually used as outcome variables in clinical trials with psychopharmacological and neuroprotective compounds.
Inclusion of dimensional elements in psychiatric diagnostic systems have been advocated for many years, however the concept was resisted due to concerns of clinical utility. Kraemer et al. [20] argue that categorical and dimensional approaches are fundamentally equivalent, but that one or the other approach is more appropriate depending on the clinical circumstances and research questions being addressed. Using an example from the Infant Health and Development Program, the authors illustrate the importance of using dimensional approaches for hypothesis testing, identify the problems with power and with interpretation that arise from employing a categorical approach, and underscore the importance of identifying the appropriate cutpoints when a categorical approach is necessary. A comparison between categorical and dimensional approaches to the diagnosis of psychosis revealed that the categorical approach is beneficial primarily in terms of reliability, whereas the dimensional approach would enhance validity [21].
Thus, to date, diagnosis of schizophrenia remains problematic for the following reasons:
-
1.
diagnosis is based on a “categorical model” with inclusion and exclusion criteria for symptoms according to the DSM-IV and ICD-10 classification methods;
-
2.
psychopathological symptoms and pathological forms of behavior are evaluated through observation and clinical interview with or without psychiatric rating scales;
-
3.
similar symptoms may be found among schizophrenia patients and individuals with other mental disorders;
-
4.
no laboratory diagnostic tests or biomarkers are currently available to determine a diagnosis of schizophrenia.
2.2 Neurocognitive Domains and Tools
Neuropsychological studies of patients with schizophrenia have demonstrated that neurocognitive impairment is a prominent feature of the illness that is present at illness onset and generally remains stable over time [22]. Neurocognitive impairment may be prognostically important, and the enhancement of cognitive functioning has become an important target for both psychosocial and pharmacological interventions. As a result, the assessment of neurocognitive deficits in schizophrenia presents a major challenge to the clinician and researcher, and efforts are being made to develop a reliable and valid cognitive battery especially for use in clinical trials.
Computerized testing has multiple advantages, including increased time-efficiency, a wider range of stimulus options and response forms, and increased psychometric reliability. Computerized cognitive testing has the potential to effectively address the limitations posed by traditional paper-based neuropsychological measures. Technical innovations for accurate measurement of reaction time and frequency of errors enhance overall sensitivity, and on-line adjustment of level of difficulty may minimize ceiling or floor effects. A computerized testing session is usually of shorter duration and is less expensive than traditional paper-based neurocognitive testing. With rapid advances in technology and an emphasis on efficiency of neurocognitive testing, it has become necessary to further investigate the value of computerized cognitive examinations. Although there is no gold standard for assessing cognitive function in schizophrenia patients, a number of computerized cognitive batteries have been developed, such as the Computerized Neuropsychological Test Battery [23], the Computer Administered Neuropsychological Screen for Mild Cognitive Impairment [24], the ECO computerized cognitive battery [25], MicroCog Cognitive Battery [26], the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) [27], the Mindstreams Battery [28], and the Cambridge Neuropsychological Test Automated Battery [29] (CANTAB). The CANTAB tests run on an IBM-compatible personal computer with a touch-sensitive screen. The nonverbal nature of the CANTAB tests makes them largely language-independent and culture-free. Overall, neuropsychological tests are grouped into five cognitive domains: visual and movement skills, attention and memory, learning, sustained attention, and executive function. The CANTAB battery has been shown to be effective in detecting cognitive deficits in schizophrenia [28, 30, 31].
2.3 Health-Related Quality of Life Deficit Model
Despite increasing relevance of health-related quality of life (HRQL) measures in mental health, the theoretical conceptualization of the construct remains poorly developed (reviewed in [32]). Since many valued aspects of life, such as income, freedom and quality of the environment are not usually considered “health related”, the term HRQL came to refer to the physical, psychological, and social domains of health. HRQL is multidimensional in the sense that the subjects may simultaneously evaluate several dimensions to arrive at an overall assessment. Two persons with the same mental health status may have different HRQL levels since components such as personality differences and illness related factors influence one’s perception of health and satisfaction with life. Perceptions of HRQL are based on a cognitive process that involves identifying the domains relevant to one’s life quality, and integrating the various domain evaluations into an overall HRQL assessment. Each health-related domain has many components that need to be measured.
HRQL is a heterogeneous concept, as reflected in the different perceptions of this construct by psychiatrists and their patients. Such differences are reflected in observer rated versus self-report HRQL evaluation instruments. HRQL differs somewhat from subjective well-being, in that the latter concerns itself primarily with affective states, both positive and negative. According to the HRQL impairment concept, the HRQL deficit syndrome refers to the vulnerability to illness, and, consequently, should be viewed as a definitive expression or a particular syndrome of severe mental disorders, such as psychopathology or cognitive impairment [33].
The Distress/Protection Vulnerability Model [33], suggests that:
-
1.
HRQL impairment is a particular syndrome observed in most psychiatric and somatic disorders.
-
2.
This syndrome is an outcome of the interaction of an array of distressing factors, on the one hand, and putative stress process protective factors, on the other. HRQL impairment increases if distressing factors overweigh protective factors, and vice versa.
-
3.
There are primary and secondary factors that influence HRQL impairment. Primary or vulnerability related factors are those usually considered inborn or personal characteristics, while secondary factors are related to illness and environment. Primary factors such as harm avoidance, high levels of neuroticism, poor coping skills, elevated emotional distress, emotion-oriented coping, and weak self-constructs might lower the vulnerability threshold, and, consequently, result in severe HRQL impairment. Secondary factors influence HRQL impairment via primary factors.
-
4.
HRQL impairment syndrome is characterized based on underlying neurobiology that may lead to improved understanding of severe mental disorders and more effective treatment decisions.
Factors influencing the HRQL impairment syndrome in schizophrenia according to this model are summarized in Fig. 12.3 [6]. Since the year 2000, this model has been extensively used to compare HRQL impairment among patients with severe mental disorders [34, 35], to examine the role of side effects [36], to test the mediating effects of coping styles [37], to search for longitudinal predictors of general and domain-specific quality of life [38–40], to explore the association of HRQL impairment with suicide behavior [41], temperament factors [42], and sleep quality [43], and to examine the impact of antipsychotic and neuroprotective agents [44–46].

The Distress/Protection Vulnerability model of HRQL impairment and the three-hit vulnerability model of schizophrenia and related disorders [Reproduced from [6]]. HRQL – health-related quality of life
Thus, in addition to symptom dimensions, neurocognitive and HRQL outcome measures have reshaped our understanding of schizophrenia and should be essential tools for designing neuroprotective interventions.
The neurodevelopmental and neurodegenerative models are two alternative, but not mutually exclusive, pathophysiological hypotheses relating to schizophrenia.
2.4 Neurodevelopmental Model
Understanding the etiology and pathogenesis of schizophrenia is a major challenge facing psychiatry. In the last two decades schizophrenia has been increasingly viewed as a neurodevelopmental disorder [47–49] due to several advances, principally related to developments in neuroimaging, electrophysiological and neuropathological approaches (reviewed in [50]).
While multiple theories have been put forth regarding the origin of schizophrenia, by far the vast majority of evidence points to the neurodevelopmental model in which developmental insults as early as late first or early second trimester of preganancy cause the activation of pathologic neural circuits during adolescence or young adulthood leading to the emergence of positive or negative symptoms [47–49]. There is evidence from brain pathology (enlargement of the cerebroventricular system, changes in gray and white matters, and abnormal laminar organization), genetics (changes in the normal expression of proteins that are involved in early migration of neurons and glia, cell proliferation, axonal outgrowth, synaptogenesis, and apoptosis), environmental factors (increased frequency of obstetric complications and increased rates of schizophrenic births due to prenatal viral or bacterial infections), minor physical anomalies, and gene-environmental interactions, which support of the neurodevelopmental model [50–53]. Furthermore, premorbid characteristics of schizophrenia patients combined with structural brain changes observed in first-episode neuroleptic-naïve patients are consistent with a neurodevelopmental pathophysiology for schizophrenia [54, 55]. In addition, findings from both cross-sectional studies of first-episode patients and longitudinal studies in childhood-onset and adolescent onset schizophrenia support the concept of early-onset schizophrenia as a progressive neurodevelopmental disorder with both early and late developmental abnormalities [56].
2.5 Neurodegenerative Model
Investigation of the long-term course of schizophrenia with progression to different residual syndromes has suggested that schizophrenia may be a neuroregressive illness. In particular, various lines of evidence indicate the presence of progressive pathophysiological processes that occur in the brains of patients with schizophrenia (see review [57]):
-
(1)
Progressive MRI changes in longitudinal studies were revealed in childhood-onset schizophrenia [58], before and after transition to psychosis [59], and in the course of early psychosis [60, 61].
-
(2)
Progressive MRI changes were seen in subgroups of patients with chronic schizophrenia [60–63].
-
(3)
Some, though not all studies revealed more pronounced progressive brain changes in patients that are associated with poor outcome, more negative symptoms, and a decline in neuropsychological performance [64–66];
-
(4)
Neuroimaging studies documented progressive increases in ventricular size, accelerated loss of brain tissue, progressive delays in treatment response, and neurochemical (magnetic resonance spectroscopy) and neurophysiological (P300) indices, all of which are consistent with ongoing cerebral degeneration in a significant subgroup of schizophrenia patients [67].
-
(5)
In addition, the most-affected brain regions were consistently found to be the frontal and temporal cortices, the hippocampus, the amygdala, and the thalamus. Compared to healthy controls, the amygdala appears to be decreased in size among schizophrenia patients [68, 69].
-
(6)
Cerebellar abnormalities have also been noted in schizophrenia [70].
Although brain alterations in schizophrenia patients have contributed to the neurodevelopmental model of pathogenesis, a progressive neurodegenerative process has also been suggested. Genetic and prenatal adversity may underlie the pathophysiological process that led to a shift from a neurodevelopmental to a neurodegenerative model (a combined model) of schizophrenia with multiple biochemical abnormalities involving the dopaminergic, serotonergic, glutamate, and gamma-aminobutyric acidergic systems [71], and brain alterations.
2.6 Vulnerability Models
Though recent studies have shown that several neurobiological alterations in domains of brain structure, physiology and neurochemistry may reflect diverse pathophysiological pathways from the “genome to the phenome” (reviewed in [50, 72, 73]), a conclusive identification of specific etiological factors or pathogenic processes in the illness has remained elusive. Attention has focused on disease models involving genetic, neurodevelopment, neurodegenerative, and environmental factors and mechanisms. The stress-vulnerability models of schizophrenia and other psychotic disorders have dominated etiology theories for over three decades.
The stress-vulnerability model has become established as a framework for explaining how environmental factors interact with preexisting vulnerability in the etiology and course of the disorder [74]. This model postulates that vulnerability to illness is stable, enduring, and largely attributable to genetic and environmental factors. Greater vulnerability is associated with higher risk for developing schizophrenia, but the actual expression of this predisposition depends on a host of personal and environmental factors, some of which are noxious, while others are protective. The interaction of vulnerability, stressors and protective factors influences both the onset and the course of the disorder. More recently, with advances in our understanding of the biological processes that mediate the effects of stress, these models have incorporated mechanisms to account for the adverse impact of stress on brain function [75].
The neural diathesis–stress model proposes that the constitutional diathesis for schizophrenia depends on neuroendocrine pathways through which stress exposure, specifically cortisol release mediated by the hypothalamic-pituitary-adrenal (HPA) axis, influences dopamine transmission [74, 76]. The neural diathesis–stress model of schizophrenia can be expanded to account for the heterogeneity of effects of psychological stressors.
A tentative model of schizophrenia is presented, based on the evidence that certain personality characteristics may serve as vulnerability factors and that environmental stressors may precipitate psychotic periods in vulnerable individuals. Certain information-processing deficits, autonomic reactivity anomalies, and social competence and coping limitations are viewed as potential vulnerability factors. Stressors in the form of discrete life events as well as the prevailing level of social environmental stress are considered factors that interact with preexisting vulnerability characteristics to produce vicious cycles that lead, in turn, to psychotic episodes. A distinction among stable vulnerability indicators, mediating vulnerability factors, and episode indicators is suggested to differentiate types of abnormalities that characterize individuals prone to or manifesting schizophrenic disorder [75, 77].
“Multiple hit” models have been formulated including two and three hit models of schizophrenia, which suggest the importance of additive and interactive effects of environmental risk factors against a background of genetic predisposition [78–80]. Genetic factors, most likely multiple genes of modest effect, play a major role in its etiology, but an environmental “second hit” may be necessary for clinical expression. Stress has been postulated as a factor in so called “two hit” models of schizophrenia in which two independent insults (e.g., an aberrant genetic trait and stressful experience) are thought to be necessary for the occurrence of the disorder. In this model, genetic or environmental factors disrupt early central nervous system (CNS) development. The adaptive plasticity of chronic stress involves many mediators, including glucocorticoids, excitatory amino acids, brain neurotrophic factor (BDNF), polysialated neural cell adhesion molecule and tissue plasminogen activator [81]. Inappropriate neurotrophic support during brain development could lead to structural disorganization in which neuronal networks are not optimally established. Inadequate neurotrophic support in adult individuals could ultimately be an underlying mechanism that may lead to decreased capacity of the brain to adapt to changes and increased vulnerability to neurotoxic damage [82, 83]. Keshavan [78] proposed that these factors might interact cumulatively during successive critical “windows of vulnerability” during brain development and during the early course of the illness to lead to the clinical manifestations of the illness. Velakoulis et al. [84] suggest a three-hit model in which an early neurodevelopmental lesion renders the hippocampus vulnerable to further insult later in life during the transition phase to active illness. Figure 12.4 presents the integrative conceptualization of the four-hit model, which postulates that schizophrenia and other functional psychoses have caused by interaction between:
-
1.
genes with major and minor effects with the possibility of disorder specific and nonspecific effects, respectively, gene-gene interactions and a diversity of genetic causes in different families or populations (a genetic load first hit);
-
2.
a neuronal vulnerability to triggers during early neurodevelopment is a second hit (its underlying causes include both genes with major effects and environmental stress); and
-
3.
a stress sensitization that could act as a third hit, facilitating mechanisms of neurodegeneration such as apoptosis, excitotoxicity or oxygen radical formation due to environmental factors (a degeneration fourth hit).

The “multi-hit” vulnerability model of schizophrenia and other functional psychoses. Figure presents the four-hit model, which postulates that schizophrenia and other functional psychoses have caused by interaction between: (1) genes with major and minor effects with the possibility of disorder specific and nonspecific effects, respectively, gene-gene interactions and a diversity of genetic causes in different families or populations (a genetic load first hit); (2) a neuronal vulnerability to triggers during early neurodevelopment is a second hit (its underlying causes both genes with major effects and environmental stress); (3) a stress sensitization that could act as a third hit, facilitating (4) mechanisms of neurodegeneration such as apoptosis, excitotoxicity or oxygen radical formation due to environmental factors (a fourth hit)
Thus, currently schizophrenia is best conceptualized as a “multiple hit” illness or spectrum disorder similar to cancer.
3 Neuroprotective Approach
The neuroprotective approach is a treatment paradigm, that is theoretically based on both neurodevelopmental and neurodegenerative models of schizophrenia. This approach aims to protect against gray matter loss and slow functional decline following the onset of psychosis, and to maintain functional integrity of the brain in response to neurobiological stress. Neuroprotective therapy is the administration of an agent (medication, compound etc.) that can reverse some of the damage or prevent further damage. By definition, neuroprotection is an effect that may result in salvage, recovery or regeneration of the brain, its cells, structure and function [46, 85–88].
During the past few years research has focused on developing neuroprotective agents for the therapy of various degenerative diseases, including Alzheimer’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, and glaucoma [89]. Regarding schizophrenia and related disorders some neuroprotective agents (e.g., erythropoietin, glycine, D-serine, neurosteroids, memantine, celecoxib, and others) are currently being evaluated as add-on therapies. Ehrenreich et al. [90] reviewed the neuroprotective approach using erythropoietin that represents a novel frontier. The “Gottingen EPO-stroke trial” represents the first effective use in man of a neuroprotective therapy in an acute brain disease.The experimental erythropoietin therapy to combat cognitive decline in patients with schizophrenia was introduced as a neuroprotective strategy for a chronic brain disease. There is ample evidence that neurotrophins and endogenous cannabinoid systems have numerous neuroprotective effects (see below).
4 Targets for Neuroprotective Therapy
Although the molecular mechanisms of neurodegeneration and pathogenesis of schizophrenia remain largely unknown, a significant body of literature indicates that the main mechanisms implicated in the disease process may include apoptosis, excitotoxicity, oxidative stress, stress and others.
4.1 Apoptosis
Apoptosis (programmed cell death) is a normal physiologic process that occurs during embryonic development as well as in the maintenance of tissue homeostasis. Studies have shown that various neurochemical events at the synapse can induce apoptosis [91]. Triggering actions include excessive glutamate stimulation, calcium influx reactive oxygen species all of which may induce caspase activation in dendrites.
Apoptosis is a one of the most recent mechanisms implicated in the pathophysiology of schizophrenia [92, 93]. While schizophrenia is generally considered a neurodevelopmental disorder, evidence for progressive clinical deterioration and subtle neurostructural changes following the onset of psychosis has led to the hypothesis that apoptosis may contribute to the pathophysiology of schizophrenia [92]. A role for apoptosis in schizophrenia has long been hypothesized, but studies investigating this hypothesis have only recently begun. Several postmortem studies have demonstrated that apoptotic vulnerability may be increased in the brains of patients with chronic schizophrenia, even though active cell death does not occur [94]. Apoptosis appears to be downregulated in the cortex of patients with chronic schizophrenia that could reflect either a pathophysiological failure to mount an effective response to an apoptotic insult or an appropriate compensatory response to an earlier insult [95].
4.2 Oxidative Stress
Oxidative stress has been defined as “a disturbance in the pro-oxidant–antioxidant balance in favour of the former, leading to potential damage” [96]. Oxidative stress can cause cellular damage and subsequent cell death because the reactive oxygen species oxidize vital cellular components such as lipids, proteins, and DNA. Elaborate antioxidant defense systems exist to protect against oxidative stress (reviewed in [97]). Oxidative stress has been implicated in the pathophysiology of many neurodegenerative diseases, in particular, Parkinson, Huntington, and Alzheimer disorders, amyotrophic lateral sclerosis, and other disorders.
Accumulating evidence points to many interrelated mechanisms that increase production of reactive oxygen or decrease antioxidant protection in schizophrenia patients [53]. For example, there is evidence suggesting that peripheral activities of antioxidant enzymes and lipid peroxidation are abnormal in schizophrenic subjects [98]. Decreased activity of key antioxidant enzymes in schizophrenia was found [99]. Mahadik and Scheffer [100] found increased lipid peroxidation products and altered defence systems in both chronic and drug-naive first episode schizophrenic patients. Pavlović et al. [101] examined the erythrocyte levels of lipid peroxidation products and reduced glutathione and the activities of antioxidative defence enzymes – superoxide dismutase, glutathione peroxidase and catalase – as well as erythrocyte susceptibility to H2O2-induced oxidative stress in schizophrenia patients. The obtained results suggest a misbalance in pro/antioxidant status of chronic schizophrenics, which is more expressed in patients with positive symptoms of the disease. The accumulated results indicate that oxidative stress is integral to this disease and not the result of neuroleptic treatment [102]. Wood et al. [53] argue that a better understanding of the mechanisms and pathways underlying oxidative stress will assist in developing the therapeutic potential of this area.
4.3 Glutamate Excitotoxicity
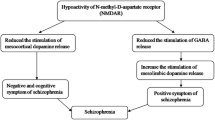
Excitotoxicity is the pathological process by which nerve cells are damaged and killed by glutamate and similar substances. In other words, too much glutamate release can be destructive and literally excite a neuron to death in a process called excitotoxicity. Glutamine synthetase constitutes an endogenous mechanism of protection against glutamate neurotoxicity in neural tissues by catalyzing the amidation of the neurotoxic amino acid glutamate to the non-toxic amino acid glutamine (for review see [103]). Deficits in N-methyl-d-aspartate (NMDA) receptor function play a critical role in the pathophysiology of schizophrenia. Patients who have excitotoxic damage would be expected to have poor outcomes characterized, perhaps, by anatomic evidence of progressive neurodegeneration, pronounced negative symptoms and cognitive deficits, and profound psychosocial deterioration [104]. Blocking the excitotoxicity process may be brain protective for schizophrenia patients.
4.4 Stress Sensitization
The brain is the key organ for responding to stress because it determines what is threatening and, therefore, potentially stressful. The brain also influences the physiological and behavioral responses which can be either adaptive or damaging [105]. The stress system orchestrates brain and body responses to the environment. Adverse conditions during early life are a risk factor for stress-related mental disorders. Psychosocial stress, such as life events, childhood trauma, or discriminatory experiences powerfully affect the brain and body and last throughout the entire life span, influencing brain function, behavior, and the risk for a number of systemic and mental disorders [81, 106, 107]. There is evidence that environmental factors, which interact with multiple genes, and epigenetic factors, psychological or physiological alterations, induce persistent sensitization to stress [107, 108]. Stress sensitization may be critical in the development or relapse of schizophrenia.
The neurobiological substrate of stress sensitization involves dysregulation of dopaminergic and noradrenergic systems. Glutamatergic regulation activates HPA axis in stress response [74, 109]. The HPA axis is one of the primary neural systems triggered by stress exposure, in the expression of vulnerability for schizophrenia. The results indicate that psychotic disorders are associated with elevated baseline and challenge-induced HPA activity; that antipsychotic medications reduce HPA activation, and that agents that augment stress hormone (cortisol) release exacerbate psychotic symptoms (reviewed in [75]).
Glucocorticoids modulate early life programming of stress reactivity and are a significant factor in brain plasticity underlying adaptation, the aging process and vulnerability to disease [110]. In rodents after a variety of experiences, even minor ones, during postnatal life, permanent changes in emotional and neuroendocrine reactivity have been observed. In particular, the results clearly demonstrate that early experiences trigger immediate changes in the stress system that may permanently alter the brain and behavior [111]. A fundamental question in the neuroendocrinology of stress-related psychopathology is why some individuals flourish and others perish under similar adverse conditions. We focus on the variants of mineralocorticorticoid and glucocorticoid receptors that operate in balance and coordinate behavioral, autonomic, and neuroendocrine response patterns involved in homeostasis and health. The data suggest that mineralocorticorticoid and glucocorticoid receptors contribute to individual differences in resilience and vulnerability to stressors [112]. Recent evidence shows that corticosteroid hormones exert rapid non-genomic effects on neurons in the hypothalamus and the hippocampal CA1 region [113]. Although many of the physiological effects of corticosteroid stress hormones on neuronal function are well recognised, the underlying genomic mechanisms are only beginning to be elucidated [114].
Brain regions such as the hippocampus, amygdala, and prefrontal cortex respond to acute and chronic stress by undergoing structural remodeling, which alters behavioral and physiological responses. Lyons et al. [115] suggest that small hippocampi reflect an inherited characteristic of the brain of monkeys. It has been reported that volume reductions in the amygdala, hippocampus, superior temporal gyrus, and anterior parietal cortex common to both patient groups may represent vulnerability to schizophrenia, while volume loss of the prefrontal cortex, posterior parietal cortex, cingulate, insula, and fusiform cortex preferentially observed in schizophrenia may be critical for overt manifestation of psychosis [108]. Genetically informed clinical studies should assess whether inherited variation in hippocampal morphology contributes to excessive stress levels of cortisol through diminished neuroendocrine regulation. In humans with mood and anxiety disorders, small hippocampal volumes have been taken as evidence that excessive stress levels of cortisol induce hippocampal volume loss. Translational studies in humans with structural and functional imaging reveal smaller hippocampal volume in stress-related conditions [116], and major depressive illness [117]. Laruelle [118] proposed that, in schizophrenia, neurodevelopmental abnormalities of prefrontal dopaminergic systems might result in a state of enhanced vulnerability to sensitization during late adolescence and early adulthood. It is also proposed that dopamine D2 receptor blockade, if sustained, might allow for an extinction of this sensitization process, with possible re-emergence upon treatment discontinuation.
Changes of protein expressions in the amygdala in the categories of synaptic, cytoskeletal, oxidative stress, apoptosis, and mitochondria related proteins could be associated with mechanisms underlying behavioral sensitization [119].
The burden of chronic stress and accompanying changes in personal behaviors (smoking, over eating, drinking, poor sleep quality; otherwise referred to as “lifestyle”) is called allostatic overload [81]. Behavioral sensitization to daily life (environmental) stress may therefore be a vulnerability marker for schizophrenia, reflecting dopaminergic hyper-responsivity in response to environmental stimuli [120]. There is evidence that emotional reactivity to daily life stress may be related to a familial liability to develop schizophrenia. In order to test a hypothesis that a persistent higher level of emotional distress in schizophrenia subjects is associated with a positive family history of schizophrenia, Ritsner and associates [121] recorded data for 69 multiplex family and 79 singleton patients at admission and about 16 months thereafter. Authors found that
-
patients with negative family history reported improvement in distress severity measured by the Talbieh Brief Distress Inventory and depression severity measured by the Montgomery-Asberg Depression Rating Scale 16 months after admission, while those with positive family history experienced persistent elevated emotional distress, mainly, on obsessiveness, and depression subscales;
-
both groups of patients are characterized by elevated emotional distress at follow-up examination compared to healthy subjects, and
-
familial schizophrenia is characterized by higher severity of dysphoric mood factors that also may represent impaired emotional reactivity [121].
Thus, it appears that there is a strong association between positive family history and persistent elevated emotional distress.
Stress sensitization is most often unspecific for schizophrenia and other brain disorders, since its can trigger high blood pressure, diabetes, ulcers, asthma and digestive and lung ailments among others.
4.5 Neurotrophic Factor Expression
Neurotrophic factors (or neurotrophins), known as the nerve growth factor, BDNF, neurotrophin-1, neurotrophin-3, and neurotrophin-4/5, are small proteins that exert survival-promoting, development and function of neuronal cells [82, 122, 123]. They belong to a class of growth factors, secreted proteins, which are capable of signaling particular cells to survive, differentiate, or grow [124].
Neurotrophins have established roles in neuronal development, synaptogenesis, response to stress stimuli, in the regulation of neuronal plasticity and neuron protection.These agents are neuromodulators of monoaminergic, gamma-aminobutyric acid (GABAA), and cholinergic systems [125]. This hypothesis is mainly based on new experimental evidence that psychiatric disorders are associated with neuronal atrophy and cell loss, impairments of structural plasticity and cellular resilience due to neurodevelopmental disturbances and morphological abnormalities of the brain.
The neurotrophin hypothesis proposes that repetitive neuronal activity enhances the expression, secretion and actions of neurotrophins to modify synaptic transmission and connectivity thereby providing a connection between neuronal activity and synaptic plasticity. Moreover, there is ample evidence that neurotrophins have numerous neuroprotective effects under pathological conditions, which might be important in particular for neurodegenerative diseases with a possible role in most psychiatric diseases including schizophrenia and mood disorders [126]. Indeed, since neurotrophic factors play a crucial role in neurodevelopment, they are plausible candidates for contributing to the pathophysiology of schizophrenia. In line with this hypothesis, accumulating preclinical and clinical data indicate that dysfunctions of neurotrophins may contribute to impaired brain development, neuroplasticity and synaptic “dysconnectivity” that lead to the schizophrenic syndrome, or at least some of its presentations [82, 127].
Moises et al. [128] proposed that functional deficiencies of glial growth factors and growth factors produced by glial cells are among the distal causes in the genotype-to-phenotype chain leading to the development of schizophrenia. The growth factor deficiency and synaptic destabilization hypothesis suggests that a functional deficiency of glial growth factors and of growth factors produced by glial cells such as neurotrophins and glutamate that lead to a weakening of synaptic strength may be implicated as one of the important causes of schizophrenia. This hypothesis suggests that glial cells are the locus of gene-environment interactions in schizophrenia, and that glial asthenia is an important factor for the genetic liability to the disorder. In addition, an increase of prolactin and/or insulin may be a putative working mechanism of traditional and atypical neuroleptic treatments.
There is evidence that the levels of growth factors in peripheral blood are disturbed in schizophrenia. The S100 protein family with pro- and antiapoptotic members, are mainly produced by astroglial cells, and essentially involved in the regulation of cell survival and death [129]. The most robust results are reported for S100B protein, which seems to be elevated in acute psychosis and in patients with predominant negative symptoms [130]. Large-scale longitudinal multivariate studies, that simultaneously investigate the levels of several growth factors might provide insight to etiological processes and may identify clinically useful subsets of patients within the heterogeneous schizophrenia sample. Since the mid-1960s, a wide variety of intracellular and extracellular activities of S100B has been elucidated, and it has also been implicated in an increasing number of CNS disorders. S100B is a calcium-binding peptide produced mainly by astrocytes that exert paracrine and autocrine effects on neurons and glia. It regulates the balance between proliferation and differentiation in neurons and glial cells by affecting protective and apoptotic mechanisms. Findings from in vitro and in vivo animal experiments relevant for human neurodegenerative diseases and brain damage are reviewed together with the results of studies on traumatic, ischemic, and inflammatory brain damage as well as neurodegenerative and psychiatric disorders. Information about the functional implication of S100B secretion by astrocytes into the extracellular space is scant but there is substantial evidence that secreted glial S100B exerts trophic or toxic effects depending on its concentration. Recent findings relating S100B to a diversity of CNS pathologies such as traumatic brain injury, Alzheimer’s disease, Down’s syndrome, schizophrenia, and Tourette’s syndrome were discussed [131].
Several studies have reported elevated S100B serum levels in schizophrenia patients. Rothermundt et al. [132] examined S100B serum levels and psychopathology (PANSS) among 98 chronic schizophrenic patients with negative symptoms upon study admission and after 12 and 24 weeks. They showed significantly increased S100B concentrations upon admission and after 12 and 24 weeks of treatment. High PANSS negative scores were correlated with high S100B levels. Regression analysis comparing psychopathology subscales and S100B identified negative symptomatology as the predicting factor for S100B. S100B is not just elevated during acute stages of disease since it remains elevated for at least 6 months following acute exacerbation. This might indicate that S100B in schizophrenia patients either promotes apoptotic mechanisms by itself or is released from astrocytes as part of an attempt to repair a degenerative or destructive process. In the next study of this group the relationship between astrocyte activation and cognitive performance, S100B serum concentration, memory performance, and psychopathology were assessed in 40 first-episode and 35 chronic schizophrenia patients upon admission and after 4 weeks of treatment [133]. Chronic schizophrenia patients with high S100B were impaired concerning verbal memory performance (Auditory Verbal Learning Test) compared to chronic and first-episode patients with low S100B levels. These findings support the hypothesis that astrocyte activation might contribute to the development of cognitive dysfunction in schizophrenia.
Thus, various aspects of the potential role of neurotrophins in psychiatric disorders have been studied [82, 126, 127, 130].
-
1.
Animal studies indicate the involvement of neurotrophins in psychopharmacological therapies and show that gene expression of cerebral neurotrophins is altered in animal models of several psychiatric disorders.
-
2.
A reduction in BDNF production and availability in the dorsolateral prefrontal cortex of schizophrenics, suggests that intrinsic cortical neurons, afferent neurons, and target neurons may receive less trophic support in this disorder [134].
-
3.
Neurotrophin serum changes have been observed in most psychiatric disorders. Whether or not these alterations represent primary-causal or secondary-reactive changes remains to be determined.
Although many issues related to the role of neurotrophic factors in schizophrenia are still unclear, these factors are attractive candidates for therapeutic agents in many clinical conditions and chronic neurodegenerative diseases including schizophrenia.
4.6 Neurosteroids: Modes of Action and Alterations
Pregnenolone (PREG) and its metabolites such as pregnenolone sulfate (PREGS) [together abbreviated PREG(S)], dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS) [together abbreviated DHEA(S)] are neurosteroids. Neurosteroids are synthesized in the central and peripheral nervous system, particularly but not exclusively in myelinating glial cells, from cholesterol or steroidal precursors imported from peripheral sources [135]. DHEA is formed from PREG by the microsomal cytochrome P450c17 enzyme (17a-hydroxylase/17,20-desmolase) in the brain and the adrenals. DHEA(S) concentrations in the human brain were found to be much higher than in peripheral circulation but also exceeded their very low cerebrospinal fluid (CSF) levels, ranging from about 1 to 5% of the corresponding plasma concentrations. DHEA(S) levels decrease markedly with age in humans, and levels in elderly populations are reduced to 20–30% of peak levels in young adulthood (reviewed in [136]).
Biological actions of PREG(S) and DHEA(S) include neuroprotection, neurite growth, and antagonistic effects on oxidants and glucocorticoids, a modulatory effect on neuronal excitability and synaptic plasticity, they have many functions associated with response to stress, mood regulation and cognitive performance [136–138] (reviewed in [136–138]).
Neuroprotective effects. The main effect of PREG and DHEA and their sulfates is neuroprotective [139–142]. Indeed, many lines of investigation demonstrate that neurosteroids have neuroprotective properties.
DHEA(S) inhibits apoptosis in human peripheral blood lymphocytes through a mechanism independent of either androgen receptors or estrogen receptors [143]. These neurosteroids also protect sympathoadrenal medulla cells against apoptosis via antiapoptotic Bcl-2 proteins [144]. DHEA(S) exhibit reduction of neurodegeneration [145, 146]. Yapanoglu et al. [147] evaluated the effects of DHEA on apoptosis of testicular germ cells after repair of testicular torsion in rats. The results suggest that DHEA may be a protective agent for preventing apoptosis caused by testicular torsion. Animal and in-vitro studies have shown that DHEA and DHEAS stimulate neuronal outgrowth and development [148, 149] and improve glial survival, learning and memory [141, 148]. Recent studies described a protective effect of DHEA on neuronal survival after oxidative, ischemic or traumatic damage [144, 150]. This specifically extends to a protective effect of DHEA on the hippocampus [151], which supports the proposed anti-glucocorticoid effect of DHEA, as glucocorticoids are known to affect hippocampal structure and function. DHEA(S) protect chromaffin cells and the sympathoadrenal PC12 cells (an established model for the study of neuronal cell apoptosis and survival) against serum deprivation-induced apoptosis [152].
PREG(S) and DHEA(S) modulate HPA axis activity and cerebral BDNF protein levels in adult male rats. In detail, they induced corticotropin-releasing hormone and/or arginine vasopressin synthesis and release at the hypothalamic level, thus enhancing plasma adrenocorticotropin hormone and corticosterone concentrations. This stimulation of the HPA axis occurred concomitantly with BDNF modifications at the hippocampus, amygdala and hypothalamus levels [142]. These results highly suggest that part of the HPA axis and antidepressant effects of neuroactive steroids could be mediated by BDNF, particularly at the amygdala level. They also suggest that neurosteroid effects on central BDNF could partially explain the trophic properties of these molecules.
PREG has neuroprotective effects against both glutamate and amyloid beta protein neuropathology and glutamate neurotoxicity [140]. Likewise, DHEA(S) demonstrated neuroprotective effects on NMDA-induced neurotoxicity in primary cultured rat hippocampal neurons [153]. In addition, DHEA(S) block the neurotoxic effects of cortisol on hippocampal cells [154], protect neurons against glutamate and amyloid ß-protein toxicity [155], and glucocorticoid toxicity [156]. The participation of aromatase in the neuroprotective effect of these neurosteroids was assessed in a study that suggested that estradiol formation by aromatase mediates neuroprotective effects of PREG and DHEA against excitotoxic-induced neuronal death in the hippocampus [157]. Recently, Akan et al. [158] investigated the effects of PREG and PREGS on cell viability and amyloid beta peptide toxicity in a concentration and exposure time-dependent manner in rat PC-12 cells. PREG showed a dose-dependent protective effect against amyloid beta peptide in PC-12 cells. But its sulfate ester did not have the same effect on amyloid beta peptide toxicity. Leskiewicz et al. [159] examined the effect of PREG, DHEA(S), and allopregnanolone on staurosporine-, glutamate-, and NMDA-induced damage in primary cortical neuronal culture. It was shown that glutamate-induced cell damage was attenuated by PREG, DHEA, and DHEAS, but not by allopregnanolone. The results of the present in vitro studies suggest that excitatory neurosteroids PREG and DHEA(S) at physiological concentrations participate in the inhibition of cortical neuronal degeneration elicited by staurosporine and glutamate. DHEA(S) supplement greatly increases neuronal survival and differentiation and reduces astroglial proliferation rates in mouse brain cells in cultures [160]. Treating adult male rats with subcutaneous pellets of DHEA increased the number of newly formed cells in the dentate gyrus of the hippocampus, and also antagonized the suppression of corticosterone. In other words, DHEA regulates neurogenesis in the hippocampus and modulates the inhibitory effect of increased corticoids on both the formation of new neurons and their survival [161].
DHEA has been shown to display antioxidant properties. DHEA protects hippocampal cells from oxidative stress-induced damage [162].
DHEA and DHEAS exhibit anti-stress properties [163, 164], in particular, as a mediator of the HPA axis adaptation to stress and other psychiatric symptoms [165].
CNS receptors. Neurosteroids directly affect major CNS receptors, especially the gamma-aminobutyric acid (GABAA), NMDA and sigma receptors [145, 166]. More specifically, certain naturally occurring PREG can enhance GABAA receptor function in a direct manner, and consequently have anxiolytic, analgesic, anticonvulsant, sedative, hypnotic and anaesthetic properties [167]. In particular, they may act as potential signaling molecules for neocortical organization during brain development, regulate the neuronal function by affecting the neuronal excitability through prominent modulatory effects on the GABAA, sigma-1, and NMDA [145, 168, 169], cholinergic [170], and dopaminergic [171] systems. These modulations may lead to important changes for neuronal excitability.
There is evidence supporting a receptor-dependent basis for the direct physiological effects of DHEA(S). The data supporting an intracellular receptor for DHEA(S) are relatively weak and do not allow us to determine whether DHEA(S) directly, or a metabolite of DHEA(S), acts as a direct receptor ligand [172].
Neurosteroids demonstrate cognitive-enhancing effects and have been reported to improve memory [173]. In preclinical studies, memory-enhancing effects of PREGS and DHEAS have been attributed to their NMDA-agonistic properties [174].
While several neurotransmitter systems have been linked to neurocognitive abnormalities in schizophrenia, including prefrontal glutamatergic, cortical dopaminergic and cholinergic neurotransmission function, the association between neurosteroids and neurocognitive function is not yet fully understood and has been sparsely investigated. Early DHEA studies that investigated neurocognitive functions in schizophrenia patients noted that neurocognitive impairment was associated with low DHEAS levels [175, 176], high DHEAS levels [177] or high DHEA levels [175], or no association was found altogether [178].
Alterations. There is accumulating evidence that alterations in PREG(S) and DHEA(S) may be involved in the pathophysiology of schizophrenia, mood and cognitive disorders [73, 136, 178–180]. Previous clinical studies demonstrated low circulating levels of PREG in the elderly, including those with dementia [181], in individuals with schizophrenia [182], in male patients with generalized anxiety disorder [183], and in non-medicated male patients with generalized social phobia [184].
Comparison of the values of blood DHEA and DHEAS levels of schizophrenia patients with healthy controls were found to differ between studies, ranging from normal to low, and to high levels. Overall, serum DHEA and DHEAS concentrations range from 15.7 to 90.9 nmol/L, and from 4,928 to 12,777 nmol/L, respectively, among schizophrenia patients, as well as, from 24.0 to 68.8 nmol/L, and from 5,375 to 13,477 nmol/L among healthy subjects, respectively. Meta-analysis of differences in mean concentrations of serum DHEA(S) between schizophrenia patients and control subjects show significant non-zero effect (p <0.001), and significant heterogeneity of data (p <0.001; see more details and references in [136]).
Contradictory and confusing reports on serum DHEA levels in schizophrenia led us to compare the serum concentration of PREG, and DHEA between 15 medicated schizophrenia patients and 12 healthy subjects at four time points: at the start of the study, after 2, 4 and 8 weeks [182]. Controlling for age, serum concentrations of PREG were lower, while the DHEA level and the molar ratio values of DHEA to PREG were higher in schizophrenia patients compared to healthy controls. PREG and DHEA levels and their molar ratio did not change significantly during the study period either among schizophrenia patients or healthy controls. The blood levels of PREG appear to be associated with trait-anxiety scores in schizophrenia patients, while associations of clinical symptoms with two neurosteroids did not reach a significant level when the confounding effect of emotional distress, and anxiety scores was controlled. Thus, low serum PREG concentrations in schizophrenia appear to be associated with trait-anxiety scores independent of symptoms. PREG and DHEA to PREG molar ratio may represent a trait-like marker of impaired hormonal response to stress in schizophrenia. Although, the role of PREG and DHEA in non-specific response to distress and anxiety or in the pharmacotherapy of schizophrenia is as yet unclear, this report adds evidence to the assumption that, for example PREG and DHEA concentrations, are not related to specific diagnoses, but to more general psychological states such as anxiety, that can occur in various disorders. A further longitudinal large-scale case-control comparison of PREG and DHEA levels in treated and an untreated, as well as in washed-out schizophrenia patients is warranted.
Thus, some neurosteroids may act as endogenous neuroprotective factors. The decline of neurosteroid levels during aging and schizophrenia may leave the brain unprotected against neurotoxic challenges. Therefore, PREG and DHEA may be suitable candidates for the treatment of schizophrenia and schizoaffective disorder patients.
5 Brain Protective Compounds
There are some compounds with neuroprotective properties that may be able to protect brain maturational processes disturbed in schizophrenia, mood and cognitive disorders, such as neurosteroids (PREG, DHEA), estrogen, omega-3 fatty acids, memantine, erythropoietin, S-adenosylmethionine, cannabinoids, piracetam, modafinil, L-theanine, bexarotene. The main targets for neuroprotective therapy may be divided on: (1) neurodegenerative processes in schizophrenia (e.g., apoptosis, excitotoxicity, oxidative stress, stress sensitization, neurotrophic factor expression, and alteration of neurosteroids); and (2) psychopathological symptoms, and behavioral characteristics of schizophrenia patients, which used as “outcome measures” in order to test efficacy and safety of a new candidate for treatment agent (Fig. 12.5).

Neurodegenerative, clinical and behavioral targets for neuroprotective therapy in schizophrenia and related disorders (Reproduced from [6])
5.1 Pregnenolone and DHEA
Several clinical trials have been conducted with these neurosteroids for treatment of schizophrenia patients. DHEA augmentations (50–200 mg/day) for a period of 1–12 weeks were examined in cross-sectional [185–187] and crossover [188–190] designs. Comparative analyses of the obtained findings are presented in the review [191]. Briefly, the first compared patients receiving DHEA (n=15) and placebo (n=12) and indicated a significant efficacy of DHEA augmentation (100 mg/day) after 6 weeks in the management of negative, depressive, and anxiety symptoms of schizophrenia [185]. Subjects receiving DHEA demonstrated a significant increase in DHEA(S) plasma levels that were correlated with improvement in negative symptoms, but not with improvement in depressive and anxiety symptoms. Limitations of the small sample size and lack of cognitive and quality of life assessments are noted.
A second study investigated the effect of DHEA administration during 7 days on medication-induced extrapyramidal symptoms (EPS) among inpatients with schizophrenia or schizoaffective disorder that were randomized in a double-blind fashion to receive either 100 mg DHEA or placebo in addition to a constant dosage of antipsychotic medication [186]. Parkinsonism showed a favorable effect of DHEA with a significant time effect, as well as a significant group by time interaction and with no change noted on akathisia. Change of DHEA blood levels was negatively associated with change of Parkinsonism (p < 0.05) as well as with change of total EPS ratings (p < 0.05). Authors concluded that DHEA appears to demonstrate a significant effect on EPS, with improvement observed particularly in Parkinsonian symptoms.
A third study performed by the same research group included 40 patients with chronic schizophrenia stabilized on olanzapine. The subjects were randomized in a double-blind fashion to receive either DHEA (150 mg/day) or placebo augmentation for a period of 12-weeks [187]. 16 patients who received DHEA and 15 patients who received placebo completed the study. DHEA augmentation was not superior to placebo in improving the scores on rating scales (SANS, PANSS), measures of side effects, in cognitive performance, and aggressive behavior. Thus, these cross-sectional DHEA trials [185–187] did not replicate one another in terms of the depressive and anxiety symptoms, and in the medication-induced adverse side effects. They did not show a consistent and unequivocal significant favorable effect of DHEA administration on negative symptoms compared to placebo.
In order to resolve some of the concerns that have risen in the cross-sectional trials, a randomized, double-blind, placebo-controlled crossover study was conducted in two mental health centers [188]. During this trial 55 patients received either DHEA (200 mg/day) or placebo in identical capsules for 6 weeks following which they were switched to either placebo or DHEA for a further 6 weeks. Patients continued to receive their regular treatment with daily doses of antipsychotic medication kept constant for at least 2 weeks prior to entering the study and throughout the study period. The crossover analysis revealed no statistically significant treatment effect of DHEA on severity of illness symptoms (PANSS), side effects, or on quality of life measures compared with placebo treatment. However, this investigation, while preliminary, supports prior findings of some improvement noted in visual sustained attention, visual and motor skills due to DHEA administration. DHEA treatment was well tolerated without any serious adverse effects.
Recently, Ritsner and Strous [190] explored changes in circulating neurosteroids and neurocognitive deficits in schizophrenia. In order to study the association, they conducted multiple regression analysis for predicting sustained attention, memory, and executive function scores across three examinations from circulating levels of DHEA, DHEAS, androstenedione, and cortisol through DHEA administration in schizophrenia. Data were collected among 55 schizophrenia patients for a double-blind, randomized, placebo-controlled, crossover trial with DHEA at three intervals: upon study entry, after 6 weeks of DHEA administration (200 mg/day), and after 6 weeks of placebo [188, 189]. DHEA augmentation was associated with elevations of both DHEA and DHEAS serum concentrations. Six weeks of DHEA treatment was associated with significant improvement in cognitive functions of visual sustained attention, and motor skills compared to placebo conditions, while DHEA administration did not produce significant improvement in clinical symptoms, side effects and quality of life scores. Obtained findings indicated that circulating DHEAS and androstenedione levels were positive predictors of cognitive functioning, and DHEA levels was a negative predictor. Overall, blood neurosteroid levels and their molar ratios accounted for 16.5% of the total variance in sustained attention, 8–13% in visual memory tasks, and about 12% in executive functions. In addition, effects of symptoms, illness duration, daily doses of antipsychotic agents, side effects, education, and age of onset accounted for variability in cognitive functioning in schizophrenia. Thus, this study suggests that alterations in circulating levels of neurosteroids and their molar ratios may reflect pathophysiological processes, which, at least in part, underlie cognitive dysfunction in schizophrenia.
Early human trials with PREG conducted on healthy volunteers under stressful conditions, demonstrated significant improvements in mood, general well-being, psychomotor performance and learning [192–194]. Low-doses of oral PREG (30 mg/day) were generally well tolerated in 17 healthy volunteers who received pregnenolone for 4 weeks [195].
Given the neuroprotective potential roles of PREG, it was hypothesized that PREG augmentation to ongoing and unchanged antipsychotic therapy may be able to improve psychotic symptoms and cognitive performance in chronic schizophrenia and schizoaffective disorder patients compared to DHEA and placebo administration. An 8-week, controlled, double-blind, randomized, parallel-group trial with “low dose” and “high dose” of PREG (30 and 200 mg/day, respectively), and 400 mg/day DHEA augmentation to on-going antipsychotics in the treatment of chronic schizophrenia and schizoaffective disorders patients was conducted for the first time in two large state referral institutions: Sha’ar Menashe Mental Health Center and Be’er-Sheva Mental Health Center [196]. Data were collected from February 2005 until June 2007 (70 patients). A total of 58 patients were randomized, 44 patients (12/13 women and 32 /45 men) completed the trial. Ten patients met criteria for schizoaffective disorders; all other subjects met criteria for schizophrenia. After an 8-week period, compared with placebo, PREG-30 administration was associated with significant reduction in positive symptom scores, EPS, and improvement in attention, and working memory performance, whereas subjects treated with PREG-200 did not differ on outcome variable scores for the study period. The condition of patients receiving placebo and PREG-30 improved more than those subjects treated with DHEA- in general psychopathology severity, and general functioning, while DHEA was superior to placebo in improving EPS. No significant main effect of the type of antipsychotics or type of antipsychotics x time interaction on the Clinical Global Impression – Severity scale (CGI-S), PANSS subscale, the Global Assessment of Functioning Scale, Extrapyramidal Symptom Rating Scale (ESRS) and Barnes Akathisia Rating Scale ratings was observed for patients receiving PREG-30, PREG-200, DHEA and placebo (all p values >0.05). Interestingly, a significant efficacy of DHEA augmentation was observed with 50–150 mg/day [185, 187], but not with 200 mg/day [188] or 400 mg/day [196]. Moreover, the augmentation of 400 mg/day of DHEA resulted in significantly less improvement of CGI-S, and PANSS general psychopathology scores compared to placebo. Therefore, we suggest an inverted-U clinical response on a daily dose of PREG and DHEA augmentations (although there could be other reasons for the inconsistent results: methodological issues, different sample characteristics, different baseline severity of illness, and varying durations of combination treatment). Negative symptoms and akathisia did not significantly benefit from any treatment. The administration of PREG and DHEA was well tolerated.
Circulatory pregnenolone was found significantly higher among the patients treated by both neurosteroids compared to the placebo group, however, it was significantly higher among those receiving 200 mg/day PREG compared to PREG-30 and the DHEA groups. This study demonstrates no effects of PREG administration on the other hormones measured in this trial, while treatment with DHEA significantly elevated blood levels of pregnenolone (but to a lesser extent than PREG 200 mg/day), as well as DHEA, DHEAS, androstenedione, 3a-androstane-3a-17b-diol-glucoronide, testosterone, and estradiol compared to PREG-30, PREG-200 and placebo. No between group differences in the levels of progesterone, 17-OH-progesterone, and cortisol were demonstrated. The patients receiving PREG do not have a risk for elevation of androgenic metabolites, like DHEA, which may in turn potentially predispose them to various problems such as prostatic hypertrophy in men and hirsutism in women. PREG’s treatment effects cannot be explained by an impact of their neuroactive metabolites like DHEA, DHEAS, androstenedione, 3a-androstane-3a-17b-diol-glucoronide, testosterone, and estradiol. Considering the lack of any significant effect of PREG on the measured hormonal profile in this study, it may be suggested that PREGs therapeutic effects as noted, are mediated by other mechanisms, including further potential hormonal influences not investigated in this study. In addition, direct neuromodulatory effects on the GABAA, NMDA, sigma-1, dopaminergic, cholinergic or neurotrophic systems may mediate PREG’s effect.
Thus, although based on a relatively small sample size, this study suggests that low-dose PREG treatment for 8 weeks, used as an adjunct to antipsychotics, has a valuable ameliorating effect on positive symptoms, attention and memory impairments and antipychotic-induced extrapyramidal side effects, in chronic schizophrenia and schizoaffective patients. Although the results of this study are notable, it is crucial to replicate the trial with a larger sample of chronic and non-chronic schizophrenia or schizoaffective patients, and for a longer duration of treatment. Further double-blind controlled studies are needed in order to investigate the clinically significant benefits of pregnenolone augmentation.
5.2 Estrogen
In recent years, we have become increasingly aware that estrogen is a gonadal hormone that exerts diverse non-reproductive actions on multiple organs and in multiple physiological systems. Three major forms of estrogen exist in humans and rodents: the biologically most prevalent and potent estrogen 17b-estradiol and, in order of decreasing potency, estrone and estriol. These estrogens are known to exert their actions through members of the nuclear hormone receptor superfamily, estrogen receptor-a, and the more recently identified estrogen receptor-b [197, 198]. Estrogen is now recognized to have centrally mediated neuromodulatory actions in both in males and females [199–201]. Estrogen receptors are expressed in brain regions that are involved in sex differentiation and maturation. Wise et al. [202, 203] found that estrogen receptors play a pivotal functional role in neuroprotection. However, the cellular mechanisms by which estrogens exert neuroprotective effects are not clearly understood. In vitro models of neuroprotection, 17bestradiol treatment exerts neuroprotective effects on diverse neuronal cell types under b-amyloid–induced toxicity, excitotoxicity, and oxidative stress [204]. Signaling mechanisms underlying estrogen-induced neuroprotection and synaptic plasticity, including the important concepts of genomic versus nongenomic mechanisms, types of estrogen receptor involved and their subcellular targeting, and implicated downstream signaling pathways and mediators were reviewed by Brann et al. [205]. This review demonstrates the remarkable body of work that has been conducted on the neuroprotective and neurotrophic actions of estrogen in the brain with particular emphasis on estrogen actions in the hippocampus, cerebral cortex and striatum.
In addition estradiol exerts rapid membrane effects on neural cells, modulating ion channels, neurotransmitter transporters, levels of intracellular calcium and other second messengers and phosphorylation of different kinases [206].
Several clinical observations support the estrogen protection hypothesis, which proposes:
-
fluctuation of psychotic symptoms in women with schizophrenia, during their menstruation cycle;
-
indications of a higher efficacy of antipsychotic treatment in women with schizophrenia than in men [207];
-
reduction of levels of plasma estrogen in both male [208] and female [209] schizophrenia patients.
Estrogen has now been used as an adjunct to standard antipsychotic medication in quite a few studies of female schizophrenia patients [210]. However, most of these are not double-blind, randomized, controlled trials. Three randomized double-blind placebo-controlled trials and an open-label study showed that adding estradiol to women’s usual antipsychotic medications was associated with significant abatement of schizophrenia symptoms [211]. Several trials indicate a protective effect of estrogen against onset of schizophrenia and the severity of negative symptoms [212] (reviewed in [213]). Although estrogen appears to be a useful treatment for schizophrenia, further research is required to determine the appropriate dose and duration of use of estradiol augmentation.
5.3 S-Adenosylmethionine
S-Adenosyl-Methionine (SAMe) is a naturally occurring molecule distributed in virtually all body tissues and fluids [214]. It is naturally synthesized in the body during the metabolism of methionine to cysteine, taurine, glutathione and other polyamine compounds in the presence of methionine-adenosyl-transferase and adenosine-5’-triphosphate [215]. SAMe’s predominant function is as a primary methyl group donor for a wide range of compounds including catecholamines, membrane phospholipids, fatty acids, nucleic acids, porphyrins, choline carnitine and creatinine. Following release of its methyl group, SAMe is converted to S-adenosyl-homocysteine which, in turn, acts as a competitive inhibitor of SAMe-mediated methylation reactions. An important function of SAMe involves methylation of certain phospholipids, particularly phosphatidylethanolamine, and proteins which aid in the maintenance/control of the fluidity and microviscosity of cell membranes. Intact SAM-e metabolism is also considered vital for myelin maintenance [216].
SAM-e is able to cross the blood-brain barrier. CSF levels of homovanillic acid and 5-hydroxyindolacetic acid increase in the brain following its administration [217]. While the bioavailability of oral administered SAM-e, as opposed to intravenous or intramuscular administration, is incomplete, with a significant first-pass effect and subsequent rapid liver metabolism [218], oral SAM-e does increase levels in blood [219] and CSF [220]. This is especially so when administered in an enteric-coated form, which is now the simplest, preferred method of administration.
SAMe provides neuroprotection against various aspects of neurotoxicity in normal and apolipoprotein E-deficient mice and in cultured neuronal cells deprived of dietary folate and vitamin E and subjected to iron overload. For instance, SAMe (AdoMet; 1 mM) protects the stationary phase cells of saccharomyces cerevisiae against the killing effect of acid (10 mM HCl) by increased the cell survival of the acid stressed cells [221, 222].
It has been suggested that since SAMe plays an important function in several metabolic processes, its administration could influence the course of a variety of disorders. James et al. [223] evaluated plasma concentrations of metabolites in the methionine transmethylation and transsulfuration pathways in children diagnosed with autism. Relative to the control children, those with autism had significantly lower baseline plasma concentrations of methionine, SAMe, homocysteine, cystathionine, cysteine, and total glutathione and significantly higher concentrations of S-adenosylhomocysteine, adenosine, and oxidized glutathione. This metabolic profile is consistent with impaired capacity for methylation (significantly lower ratio of SAMe to S-adenosylhomocysteine) and increased oxidative stress (significantly lower redox ratio of reduced glutathione to oxidized glutathione) in children with autism.
The intervention trial was effective in normalizing the metabolic imbalance in autistic children. An increased vulnerability to oxidative stress and a decreased capacity for methylation may contribute to the development and clinical manifestation of autism. SAMe was proposed as a treatment for aging [224], gastric illness [225], liver disease [226], migraine [227], Alzheimer’s disease [228], epilepsy, multiple sclerosis, HIV-associated neurological implications and spinal cord disease [229]. Parenteral SAM-e, a methyl group donor, was shown to be an effective antidepressant (e.g. [230]).
Strous et al. [231] investigated the efficacy of SAMe in managing schizophrenia symptomatology in patients with the low activity catechol-O-methyltransferase enzyme activity polymorphism. Eighteen patients with chronic schizophrenia were randomly assigned to receive either SAMe (800 mg) or placebo for 8 weeks in a double-blind design. Results indicated some reduction in aggressive behavior and improved quality of life following SAMe administration. Female patients showed improvement of depressive symptoms. Clinical improvement did not correlate with serum SAMe levels. This preliminary pilot short-term study cautiously supports SAMe as an adjunct in management of aggressive behavior and quality of life impairment in schizophrenia. However, like all medications, SAMe may result in unwanted side-effects mostly mild to moderate in nature and of brief duration. Most side effects appear to be gastrointestinal in nature (nausea, vomiting, diarrhea, heartburn), but may include hypomanic switch (in depressed individuals with underlying bipolar disorder), anaphylaxis, dizziness, insomnia and headache [215].
5.4 Cannabinoids
Cannabidiol is a major constituent of the Cannabis sativa plant. The endocannabinoid system consists of cannabinoid receptors, their endogenous ligands and enzymes for synthesis and degradation of endocannabinoids and represents a local messenger system within and between the nervous and immune system (e.g. [232, 233]). Two subtypes of G-protein coupled cannabinoid receptors (CB1 and CB2) have been cloned and several putative endogenous ligands (endocannabinoids) have been detected during the past 15 years [234].
Since the discovery of an endogenous cannabinoid system, research into the pharmacology and therapeutic potential of cannabinoids has steadily increased. Although it is now apparent that cannabinoids have neuroprotective properties, many of the mechanisms involved in the process have yet to be characterized. Cannabinoids have antioxidant, anti-inflammatory, anti-excitotoxic, and anxiolytic-like properties that allow them to afford neuroprotection in different neurodegenerative disorders [234–237]. Cannabinoids have been shown to protect against neurotoxicity in a number of different cellular, animal, and human experimental paradigms [238–241]. Cannabinoids produce neuroprotection by reducing intracellular calcium release from ryanodine-sensitive stores [242].
Delta(9)-tetrahydrocannabinol (Delta9-THC), the main Cannabis sativa component, increased serum BDNF levels in healthy controls but not light users of cannabis, which had lower basal BDNF levels [243]. The modulation of mediotemporal and ventrostriatal function by Delta9-THC may underlie the effects of Cannabis sativa on verbal learning and psychotic symptoms, respectively [244]. Delta9-THC induces anxiety and psychotic-like symptoms in healthy volunteers; these effects of delta9-tetrahydrocannabinol are significantly reduced by cannabidiol, which is devoid of the typical effects of the plant. Studies in animal models and in healthy volunteers clearly suggest an anxiolytic-like effect of cannabidiol [236].
Recent advances in knowledge about cannabinoid receptor function have renewed interest in the association between cannabis and psychosis (reviewed in [245–249]):
-
Case series, autobiographical accounts, and surveys of cannabis users in the general population suggest an association between cannabis and psychosis.
-
Cross-sectional studies document an association between cannabis use and psychotic symptoms, and longitudinal studies suggest that early exposure to cannabis confers a close to two-fold increase in the risk of developing schizophrenia.
-
Pharmacological studies show that cannabinoids can induce a full range of transient positive, negative, and cognitive symptoms in healthy individuals that are similar to those seen in schizophrenia.
-
There is considerable evidence that in individuals with an established psychotic disorder such as schizophrenia, exposure to cannabis can exacerbate symptoms, trigger relapse, and worsen the course of the illness. For instance, Delta9-THC in a 3-day, double-blind, randomized, placebo-controlled study (0, 2.5, and 5 mg intravenous) transiently increased learning and recall deficits; positive, negative, and general schizophrenia symptoms; perceptual alterations; akathisia, rigidity, and dyskinesia; deficits in vigilance; and plasma prolactin and cortisol [250]. However, Schwarcz et al. [251] reported improvement of symptoms of schizophrenia in a small group of patients who received the cannabinoid agonist dronabinol (synthetic Delta9-THC). They found that 4 of 6 treatment-refractory patients with severe chronic schizophrenia but who had a self-reported history of improving with marijuana abuse improved with dronabinol. This improvement seems to have been a reduction of core psychotic symptoms in 3 of the 4 responders and not just nonspecific calming. These results complement the recent finding that the cannabinoid blocker rimonabant does not improve schizophrenic symptoms and suggest that the role of cannabinoids in psychosis may be more complex than previously thought.
-
Only a very small proportion of the general population exposed to cannabis develops a psychotic illness. It is likely that cannabis exposure is a “component cause” that interacts with other factors to “cause” schizophrenia or other psychotic disorder, but is neither necessary nor sufficient to do so alone.
-
Although there is evidence that cannabis use increases the risk of developing psychotic symptoms, the causal nature of this association remains unclear [252].
-
It is unclear if research findings support the clinical opinion that cannabis use leads to worse outcomes in people with psychosis, or whether this impression is confounded by other factors [253]. Rathbone et al. [248] evaluated the effects of cannabis use on people with schizophrenia and schizophrenia-like illnesses. At present, there is insufficient evidence to support or refute the use of cannabis/cannabinoid compounds for people suffering with schizophrenia.
Thus, highlighting the association between schizophrenia and Cannabis sativa and the endogenous cannabinoid receptor system, respectively, there are two opposing relevant findings. On the one hand, there is substantial evidence that cannabis is an independent risk factor for psychosis that may lead to a worse outcome of the disease. This risk seems to be increased in genetically predisposed people and may depend on the amount of cannabis used. On the other hand, during the last few years, an endogenous cannabinoid receptor system has been discovered [254].
Medications that activate cannabinoid receptors are already available: these are Cesamet (nabilone), Marinol (dronabinol; Delta9-THC) and Sativex (Delta9-THC with cannabidiol). The first two of these agents can be prescribed to reduce chemotherapy-induced nausea and vomiting. Marinol can also be prescribed to stimulate appetite, while Sativex is prescribed for the symptomatic relief of neuropathic pain in adults with multiple sclerosis and as an adjunctive analgesic treatment for adult patients with advanced cancer [255]. One challenge now is to identify therapeutic targets for cannabinoid receptor agonists in psychiatry, next is to identify the factors that underlie individual vulnerability to cannabinoid-related psychosis and to elucidate the biological mechanisms underlying this risk.
5.5 Omega-3 Fatty Acids
The two main omega-3 fatty acids in fish oil, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) have important biological functions in the CNS [256]:
-
DHA is a major structural component of neuronal membranes, and changing the fatty acid composition of neuronal membranes leads to functional changes in the activity of receptors and other proteins embedded in the membrane phospholipid.
-
EPA has important physiological functions that can affect neuronal activity.
-
Omega-3 polyunsaturated fatty acids are important for cell protection after ischemia and also seem to play an important role in the activation of antiapoptotic signaling pathways [257];
-
DHA pretreatment effectively reduces cell-associated methylmercury (MeHg)-induced neurotoxicity and prooxidant response from MeHg in both cerebellar astrocytes and neurons [258], improves functional outcome and reduces volume loss after hypoxia-ischemia in neonatal rats [259].
For schizophrenia or schizoaffective disorder patients treated with 3 g/day of ethyl EPA, improvement in residual symptoms and cognitive impairment was no greater than for those patients treated with placebo [260]. There are contradictory findings regarding therapeutic benefits of omega-3 fatty acids, particularly when EPA is added to existing antipsychotic therapy of schizophrenia patients [261–263].
5.6 Piracetam
Piracetam is a nootropic agent with potent neuroprotective properties that improves cognitive and mental abilities [264, 265]. The mechanism of action of piracetam is not well understood. It facilitates cholinergic and excitatory amine neurotransmission without specific binding to receptors [266]. Glutamate alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors mediate most of the excitatory neurotransmission in the CNS and also participate in forms of synaptic plasticity thought to underlie memory and learning, and the formation of neural networks during development [267]. Interestingly, the neurogenic activity of piracetam is mediated through the AMPA receptor [268]. Piracetam molecules surround the phospholipid polar heads in the cell membrane, forming mobile drug-phospholipid complexes. Consequently, they restore membrane integrity and fluidity, resulting in better cell membrane viscosity and improved cell function [269]. This mechanism works at different sites: neuronal, red cell, and platelet [270, 271].
Piracetam has been used experimentally or clinically to treat a wide range of diseases and conditions, mainly in treatment of organic brain syndrome, myoclonus, memory impairment, post-concussion syndrome, vertigo, alcohol withdrawal, cerebrovascular insufficiency, hypoxia, intoxications of different origins or mechanic brain injuries [272]. Piracetam has been used successfully in treatment of alcoholism, recovery of aphasia after stroke, improvement or reduction of deterioration in dementia and Alzheimer’s disease (http://www.piracetam.com/index.htm). Piracetam has increased reading comprehension, accuracy, memory and verbal learning in dyslexic children [265], it has improved mental performance in “aging, nondeteriorated individuals” suffering only from “middle-aged forgetfulness” [264].
Libov et al. [273] examined the efficacy of piracetam (4800 mg/day) in the treatment of tardive dyskinesia using an oral preparation in a 9-week, double-blind, crossover (4 weeks), placebo-controlled trial assessing 40 schizophrenia and schizoaffective patients. Authors concluded that piracetam appears to be effective in reducing symptoms of tardive dyskinesia. The specific mechanism by which piracetam may attenuate symptoms of tardive dyskinesia needs to be further evaluated.
5.7 Modafinil
Modafinil (2-[(Diphenylmethyl) sulfinyl] acetamide, Provigil) is a USA Food and Drug Administration-approved medication that promotes wakefulness. It is a novel cognitive enhancer, has neuroprotective effects [274], and selectively improves neuropsychological task performance in healthy volunteers and adult patients with attention deficit hyperactivity disorder. Modafinil exhibits robust effects on catecholamines, serotonin, glutamate, gamma amino-butyric acid, orexin, and histamine systems in the brain. It is associated with a number of neurochemical actions in the brain that may be related to cognitive processes [275].
Turner et al. [276] examined the potential of modafinil (200 mg) as a cognitive enhancer in schizophrenia among 20 chronic schizophrenia patients using a double-blind, randomized, placebo-controlled crossover design. Modafinil had some cognitive enhancing properties in schizophrenia similar to those observed in healthy adults and adult patients with attention deficit hyperactivity disorder.
Sevy et al. [277] examined 24 patients with a DSM-IV diagnosis of schizophrenia or schizoaffective disorder who were randomly assigned to modafinil up to 200 mg a day or placebo as an adjunct therapy in an 8-week, double-blind, placebo-controlled study. At the end of the trial, fatigue improved in both groups, and there were no between group differences on changes in fatigue, symptoms, attention, working memory, or executive functioning. Lack of between group differences may be due to small sample size.
Saavedra-Velez et al. [278] reviewed available data on trials of modafinil published in English up to January 2008 (6 trials were identified). Authors concluded that while the available data suggest that modafinil is generally well tolerated and may have some efficacy in the treatment of antipsychotic-induced sedation and cognitive domains, the small sample sizes, contradictory results, and methodological differences between trials, especially with respect to cognitive testing, make it difficult to draw firm conclusions about the overall effectiveness of adjunctive modafinil in the treatment of schizophrenia. Therefore, further research is required to address the potential benefits and risks of co-administration of modafinil to patients with schizophrenia.
5.8 L-Theanine
L-Theanine (gamma-glutamylethylamide) is a unique amino acid present almost exclusively in the tea plant (Camellia sinensis), where it typically occurs in amounts estimated from 1 to 2% by dry weight [46, 279]. L-theanine is absorbed through the intestinal tract and is hydrolyzed to glutamic acid and ethylamine in the kidney.Peak plasma concentration was found 30 minutes after oral dosing [280]. When 200 mg of L-theanine was orally administered to rats, the plasma concentrations of L-theanine and ethylamine reached their highest levels about 0.5 and 2 h after administration, respectively. When theanine becomes catabolized in the liver [281, 282] or kidney [280] it decomposes into two components: glutamic acid and ethylamine. The main effects of theanine may be grouped as follows: neuroprotective, mood-enhancing, supporting the immune system, possessing anti-obesity and anti-tumor activity, hypotensive, and reducing liver injury effects resulting from alcohol.
The neuroprotective effects of L-theanine are the focus of considerable attention. For instance, animal studies indicate possible neuroprotective effects of L-theanine in the hippocampus through blockade of multiple glutamate receptor subtypes, NMDA, and AMPA receptors [283, 284]. L-theanine was found to inhibit ischemic delayed neuronal death in gerbils. Furthermore, L-theanine is effective in protecting nerve cells from injury caused by low levels of oxygen, a condition known as ischemia, which is also characterized by excessive glutamate release [283]. L-Theanine was also reported to stimulate the release of nerve growth factor, a protein needed by cholinergic brain cells for survival [285].
The antioxidant activity of L-theanine has been studied with regard to its effect on the oxidation of low-density lipoprotein. In vitro testing, using malondialdehyde as a marker of lipid peroxidation, demonstrated inhibition of low-density lipoprotein oxidation with L-theanine, although the effect was weaker than the potent antioxidant effect of green tea polyphenols [286]. L-Theanine attenuated the doxorubicin -induced adverse reactions involved in oxidative damage, due to increased glutamate and the recovery of glutathione levels in normal tissues [287].
L-Theanine bears structural similarity to glutamic acid and hence competes with it in binding to glutamate receptors, offering protection against glutamate neurotoxicity [288]. Nagasawa et al. [289] investigated the molecular mechanism underlying the neuroprotective effect of L-theanine using primary cultured rat cortical neurons. Their findings strongly support the notion that neuroprotection by L-theanine can be observed when it is administered orally in vivo.
Theanine may have mood-modulating activity and play a role in reducing emotional distress (anti-stress activity). This amino acid actually acts antagonistically against the stimulatory effects of caffeine on the nervous system [283, 288]. Research on human volunteers has demonstrated that L-theanine induces a sense of relaxation approximately 30–40 minutes after ingestion via at least two different mechanisms. First, this amino acid directly stimulates the brain alpha wave activity [290], creating a state of deep relaxation and mental alertness similar to what is achieved through meditation. Second, the relaxing effects of L-theanine partly depend on its ability to interact with the brain’s glutamatergic system [291].
Whereas historically, L-theanine has been shown to have relaxing properties [284, 292], it also has a reputation for counteracting the anxious jitters associated with caffeine without interfering with its ability to fight fatigue or sharpen mental focus [283, 293], although, the anxiolytic effects of theanine have not been established scientifically in animal or human studies. The pharmacological effects of L-theanine reported in animals suggest that it may have some anxiolytic properties, given that both serotonin and GABA play a fundamental role in the neurobiology of anxiety and are molecular targets in the treatment of various anxiety disorders [294]. Supporting the preclinical pharmacological effects of L-theanine, one electrophysiological study using healthy human subjects reported possible relaxing effects of L-theanine (200 mg), as indicated by increased alpha activity in the occipital and parietal cortex [295].
L-Theanine has become a promising candidate for management of schizophrenia because L-theanine may influence neurotransmitters in the brain such as GABA, dopamine, and serotonin [296, 297], and ameliorate attention and learning [292], and emotional distress [298].
Ritsner et al. [299] conducted the first trial aimed to evaluate the efficacy and tolerability of L-theanine augmentation of antipsychotic treatment in patients with chronic schizophrenia or schizoaffective disorders. L-Theanine was added-on to ongoing antipsychotic treatment (400 mg/day) of 60 patients that participated in an 8-week, double-blind, randomized, placebo-controlled study (40 patients completed the study protocol). Compared with placebo, L-theanine augmentation is associated with reduction of anxiety, positive, and general psychopathology scores measured by Hamilton Scale for Anxiety (HAM-A) scale and by the PANSS three-dimensional model, respectively. According to the five-dimension model of psychopathology, L-theanine produced significant reductions on PANSS positive, and activation factor scores compared to placebo. The effect sizes for these differences ranged from modest to moderate (0.09–0.39). PANSS negative and CANTAB task scores, general functioning, side effect and quality of life measures were not affected by L-theanine augmentation. L-theanine was found to be a safe and well-tolerated medication. Thus, L-theanine augmentation to antipsychotic therapy can ameliorate positive, activation, and anxiety symptoms in schizophrenia and schizoaffective disorder patients. Further long-term studies of L-theanine are needed to substantiate the clinically significant benefits of L-theanine augmentation.
5.9 Retinoids (Bexarotene)
Vitamin A derivatives, retinoids, play a critical role in normal development and physiology by modulating cell growth, division, reproduction, differentiation, and immune function [300], they are involved in a complex signaling pathway that regulates gene expression and, in the CNS, controls neuronal differentiation and neural tube patterning [301]. Retinoids are also capable of inhibiting cell growth, inducing differentiation, and triggering apoptosis in a variety of tumor cell lines [302].
There are two types of retinoid nuclear hormone receptors: retinoic acid receptors (RARs) and retinoid X receptors (RXRs). Both belong to the corticosteroid receptor superfamily and co-exist in most cells. The alpha, beta, and gamma subtypes of the RARs and RXRs have distinct and conserved amino and carboxy terminal domains. Each receptor subtype has a specific pattern of expression during embryonal development and a different distribution in adult tissues. This differential expression of receptor subtypes is thought to regulate the expression of distinct sets of genes. Heterodimers of the RARs and RXRs bind and regulate a specific DNA sequence known as the retinoic acid response element, which is located in the promoter region of genes such as the RAR-b2 gene (reviewed in [303]).
Rexinoid receptor (RAR, RXR) families have been shown to mediate genes associated with both growth and differentiation. Gorgun and Foss [304] explored the immunomodulatory effects of RAR and RXR rexinoids in human T- and B-cell leukemia cells and demonstrated that RXR rexinoids are capable of up-regulating high-affinity interleukin-2 receptor expression.
Several studies reported that retinoids are involved in neurodevelopment [305, 306] and regulation of genes thought to be important in the pathogenesis of schizophrenia [307–309]. Although a major functional implication of retinoic signaling has been repeatedly suggested in synaptic plasticity, learning and memory, sleep, schizophrenia, depression, Parkinson disease, and Alzheimer disease, the targets and the underlying mechanisms in the adult brain remain elusive (see reviews [46, 310]).
The retinoid hypothesis in schizophrenia, developed by Ann Goodman [311, 312] is supported by three independent lines of evidence:
-
congenital anomalies similar to those caused by retinoid dysfunction, are found in schizophrenic patients and their relatives;
-
the loci which have been suggestively linked to schizophrenia are the same as the genes of the retinoid cascade (convergent loci); and
-
the transcriptional activation of dopamine D2 receptors and numerous other schizophrenia candidate genes are regulated by retinoic acid [311].
Several recent reports at the molecular level now suggest that altered transport and lowered synthesis of retinoic acid may be fundamental mechanisms in schizophrenia [312]. Vitamin A (retinoid) deficiency induces selective memory impairment further supporting the hypothesis in that the fine regulation of retinoid-mediated gene expression is important for optimal brain and higher cognitive functions [313]. Animal experiments, which disrupt retinoid-signalling pathways, compromise the regulation of synaptic plasticity and related learning and memory behaviours [314–319]. These pathways have also been connected with the pathophysiology of Alzheimer’s disease, schizophrenia and depression. Retinoid analogs have therefore been proposed as potential treatment for schizophrenia [311, 320].
Bexarotene, LGD1069 (Targretin) belongs to the group of synthetic medicines derived from vitamin A (retinoid). Its chemical name is 4-[1-(5,6,7,8-tetrahydro-3,5,5,8,8-pentamethyl-2-naphthalenyl)ethenyl] benzoic acid [321]. To date this medication has been exclusively used as treatment of neoplastic or dermatological diseases using a high daily dose (more than 300 mg/day) [322–324]. Adverse events potentially related to bexarotene include lipid abnormalities, hypothyroidism, headache, asthenia, rash, leucopenia, anemia, nausea, and increased risk of infection, peripheral edema, abdominal pain, dry skin, dizziness, hyperesthesia, hypoesthesia, and neuropathy [325].
Based on the retinoid hypothesis in schizophrenia, our group conducted a 6-week open label trial in two mental health centers [326]. It was assumed that the combined effect of antipsychotic agents and bexarotene would have a beneficial effect in treatment of psychopathological symptoms in chronic schizophrenia patients. Since high daily doses of bexarotene can produce numerous adverse effects, the first trial was aimed to examine safety and preliminary efficacy of a low daily dose (75 mg/day) of bexarotene in an open label pilot study. Twenty-five patients with chronic schizophrenia received a low dose of bexarotene (75 mg/day) augmentation. Significant improvement from baseline to endpoint was observed on the total PANSS score, general psychopathology, and on the positive and the dysphoric mood factor scores. Furthermore, a trend to a diminishing ESRS score was found. Low doses of bexarotene were well tolerated. Bexarotene was found to be a safe medication as measured by all laboratory parameters with the exception of increased total cholesterol serum levels. This short-term pilot study supports bexarotene as a potential valuable adjunct in management of schizophrenia. A double-blind controlled study is currently underway to replicate these preliminary results (ClinicalTrials.gov Identifier: NCT00141947).
6 Conclusions and Future Directions
Despite increases in our understanding of the pathophysiology and environmental and genetic influences, etiology oriented treatment of schizophrenia remains an elusive goal. The evidence of the “stressed brain” together with neurodegeneration (apoptosis, oxidative stress, excitotoxicity, stress sensitization, neurotrophic factor expression, and alteration of neurosteroids) in the pathophysiology of schizophrenia suggests that there may be treatment opportunities using neural protection. Beyond the use of antipsychotic agents, it will be of interest to examine whether agents that demonstrate neuroprotective properties in preclinical studies may prove to have neuroprotective effects in schizophrenia. In recent years several potential neuroprotective compounds for treatment of schizophrenia patients were investigated. There are at least three steps for searching and testing neuroprotective agents in clinical practice:
-
1.
searching for candidates with neuroprotective properties using findings from basic research studies;
-
2.
assessment of safety and efficacy of suitable candidates for augmentation to antipsychotic therapy in “add-on” randomized, double-blind, placebo-controlled studies; and
-
3.
assessment of safety and efficacy of promising compounds in randomized, double-blind, placebo-controlled multicenter studies.
Clinical trials for the evaluation of neuroprotective agents pose unique challenges in terms of experimental design and data interpretation [89]. There are many methodological issues that have limited the clinical application of therapies that have shown effectiveness in various animal models of acute neurodegeneration. Inadequate preclinical pharmacological evaluation is most likely a major drawback [327]. In order to generate meaningful results, clinical trials on neuroprotective agents should ideally be designed to minimize the potential for bias and optimize the ability to detect the neuroprotective effect of a therapeutic intervention in as short a time as possible. Key issues for the design of clinical trials of neuroprotective therapies include identifying appropriate endpoints and determining the ideal timing of the intervention.
Pharmacotherapy needs to be thoroughly integrated with neuroprotection strategy, which may be a useful paradigm for the treatment of schizophrenia patients. Neuroprotection therapy may putatively have a significant impact on the subsequent course and outcome of schizophrenia. Several potential neuroprotective compounds, representing a wide range of mechanisms, are available and merit further investigation in schizophrenia.
Abbreviations
- AMPA:
-
alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- BDNF:
-
Brain-derived neurotrophic factor
- CANTAB:
-
Cambridge Automated Neuropsychological Test Battery
- CGI-S:
-
Clinical Global Impression – Severity scale
- CNS:
-
Central Nervous System
- CSF:
-
Cerebrospinal fluid
- Delta9-THC:
-
Delta(9)-tetrahydrocannabinol
- DHA:
-
docosahexaenoic acid
- DHEA:
-
dehydroepiandrosterone
- DHEAS:
-
dehydroepiandrosterone sulfate
- DHEA(S):
-
DHEA and DHEAS together
- DSM-IV:
-
Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition
- EPA:
-
eicosapentaenoic acid
- EPS:
-
Extrapyramidal symptoms
- ESRS:
-
Extrapyramidal Symptom Rating Scale
- GABAA :
-
gamma-aminobutyric acid
- HPA:
-
hypothalamic-pituitary-adrenal axis
- HRQL:
-
the health-related quality of life
- ICD-10:
-
International Classification of Mental and Behavioural Disorders
- NMDA:
-
N-methyl-D-aspartate
- PREG:
-
pregnenolone
- PREGS:
-
pregnenolone sulphate
- PREG(S):
-
PREG and PREGS together
- PANSS:
-
Positive and Negative Syndrome Scale
- RARs:
-
retinoic acid receptors
- RXRs:
-
retinoid X receptors
- SANS:
-
Scale for the Assessment of Negative Symptoms
References
Scolnick EM. Mechanisms of action of medicines for schizophrenia and bipolar illness: status and limitations. Biol Psychiatry. 2006; 59:1039–1045
Buckley PF. Update on the treatment and management of schizophrenia and bipolar disorder. CNS Spectr 2008; 13(2 Suppl 1):1–10; quiz 11–12
Agid O, Kapur S, Remington G. Emerging drugs for schizophrenia. Expert Opin Emerg Drugs 2008; 13:479–495
Gründer G, Hippius H, Carlsson A. The ‘atypicality’ of antipsychotics: a concept re-examined and re-defined. Nat Rev Drug Discov 2009; 8:197–202
Lieberman JA, Stroup TS, McEvoy JP, et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005; 353:1209–1223
Ritsner MS. Health-related Quality of Life Impairment in Schizophrenia and Related Disorders as a Target for Neuroprotective Therapy. Int J Neuroprotection Neuroregeneration 2010 (in press)
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed. American Psychiatric Press, Washington, DC; 1994
The ICD-10 Classification of Mental and Behavioural Disorders. Clinical descriptions and diagnostic guidelines. Geneva, World Health Organization; 1992
Ritsner MS, Susser E. Molecular genetics of schizophrenia: focus on symptom dimensions. In: Ritsner MS (ed) The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes. Springer, Vol IV, 2009; pp. 95–124
Musalek M, Scheibenbogen O. From categorical to dimensional diagnostics: deficiency-oriented versus person-centred diagnostics. Eur Arch Psychiatry Clin Neurosci 2008; 258(Suppl 5):18–21
van Os J. Is there a continuum of psychotic experiences in the general population? Epidemiol Psichiatr Soc 2003; 12:242–252
Lincoln TM. Relevant dimensions of delusions: continuing the continuum versus category debate. Schizophr Res 2007; 93:211–220
Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull 1987; 13:261–276
Kay SR, Sevy S. Pyramidical model of schizophrenia. Schizophr Bull 1990; 16:537–545
White L, Harvey PD, Opler L, Lindenmayer JP. Empirical assessment of the factorial structure of clinical symptoms in schizophrenia. A multisite, multimodel evaluation of the factorial structure of the Positive and Negative Syndrome Scale. The PANSS Study Group. Psychopathology 1997; 30:263–274
Shafer A. Meta-analysis of the brief psychiatric rating scale factor structure. Psychol Assess 2005; 17:324–35
Andreasen NC. The Scale for the Assessment of Negative Symptoms (SANS): conceptual and theoretical foundations. Br J Psychiatry 1989; Suppl:49–58
Addington D, Addington J, Matincka-Tyndale E. Reliability and validity of a depression rating scale for schizophrenics, Schizophr Res 1992; 6:201–208
Yudofsky SC, Silver JM, Jackson W et al. The Overt Aggression Scale for the objective rating of verbal and physical aggression. Am J Psychiatry 1986; 143:35–39
Kraemer HC, Noda A, O’Hara, R. Categorical versus dimensional approaches to diagnosis: methodological challenges. J Psychiatric Res 2004; 38:17–25
Esterberg ML, Compton MT. The psychosis continuum and categorical versus dimensional diagnostic approaches. Curr Psychiatry Rep 2009; 11:179–184
Heinrichs RW. The primacy of cognition in schizophrenia. Am Psychol 2005; 60:229–242
Sachs G, Steger-Wuchse D, Kryspin-Exner I, et al. Facial recognition deficits and cognition in schizophrenia. Schizophr Res 2004; 68:27–35
Tornatore JB, Hill E, Laboff JA, McGann ME. Self-administered screening for mild cognitive impairment: initial validation of a computerized test battery. J Neuropsychiatry Clin Neurosci 2005; 17:98–105
Merrick PI, Secker DI, Fright R, Melding P. The ECO computerized cognitive battery: collection of normative data using elderly New Zealanders. Int Psychogeriatr 2004; 16:93–105
Extermann M, Chen H, Booth-Jones M, et al. Pilot testing of the computerized cognitive test Microcog in chemotherapy-treated older cancer patients. Crit Rev Oncol Hematol 2005; 54:137–143
Green MF, Nuechterlein KH, Gold JM, et al. Approaching a consensus cognitive battery for clinical trials in schizophrenia: the NIMH-MATRICS conference to select cognitive domains and test criteria. Biol Psychiatry 2004; 56:301–307
Ritsner MS, Blumenkrantz H, Dubinsky T, Dwolatzky T. The detection of neurocognitive decline in schizophrenia using the Mindstreams Computerized Cognitive Test Battery. Schizophr Res 2006; 82:39–49
Robbins TW, James M, Owen AM, Sahakian BJ, McInnes L, Rabbitt P. Cambridge Neuropsychological Test Automated Battery (CANTAB): a factor analytic study of a large sample of normal elderly volunteers. Dementia 1994; 5:266–281
Ritsner MS. Predicting quality of life impairment in chronic schizophrenia from cognitive variables. Qual Life Res 2007; 16:929–937
Levaux MN, Potvin S, Sepehry AA, Sablier J, Mendrek A, Stip E. Computerized assessment of cognition in schizophrenia: promises and pitfalls of CANTAB. Eur Psychiatry 2007; 22:104–115
Ritsner MS, Awad AG (eds) Quality of Life Impairment in Schizophrenia, Mood and Anxiety Disorders. New Perspectives on Research and Treatment. Springer, 2007; 388 pp
Ritsner MS. The Distress/Protection Vulnerability Model of the quality of life impairment syndrome: current evidence and new directions for research. In: Ritsner MS, Awad AG (eds) Quality of Life Impairment in Schizophrenia, Mood and Anxiety Disorders. New Perspectives on Research and Treatment. Springer, 2007; pp. 3–19
Ritsner M, Modai I, Endicott J, et al. Differences in quality of life domains and psychopathologic and psychosocial factors in psychiatric patients. J Clin Psychiatry 2000; 61:880–889
Ritsner M, Kurs R, Gibel A, et al. Predictors of quality of life in major psychoses: a naturalistic follow-up study. J Clin Psychiatry 2003; 64:308–315
Ritsner M, Ponizovsky A, Endicott J, et al. The impact of side-effects of antipsychotic agents on life satisfaction of schizophrenia patients: a naturalistic study. Eur Neuropsychopharmacol 2002; 12:31–38
Ritsner M, Ben-Avi I, Ponizovsky A, et al. Quality of life and coping with schizophrenia symptoms. Qual Life Res 2003; 12:1–9
Ritsner M. Predicting changes in domain-specific quality of life of schizophrenia patients. J Nerv Ment Dis 2003; 191:287–294
Ritsner M, Gibel A, Ratner Y. Determinants of changes in perceived quality of life in the course of schizophrenia. Qual Life Res 2006; 15:515–526
Ritsner M, Modai I, Kurs R, et al. Subjective quality of life measurements in severe mental health patients: measuring quality of life of psychiatric patients: comparison two questionnaires. Quality Life Res 2002; 11:553–561
Ponizovsky AM, Grinshpoon A, Levav I, Ritsner MS. Life satisfaction and suicidal attempts among persons with schizophrenia. Comprehensive Psychiatry 2003; 44:442–447
Kurs R, Farkas H, Ritsner M. Quality of life and temperament factors in schizophrenia: comparative study of patients, their siblings and controls. Quality Life Res 2005; 14:433–440
Ritsner M, Kurs R, Ponizovsky A, Hadjez J. Perceived quality of life in schizophrenia: relationships to sleep quality. Quality Life Res 2004; 13:783–791
Ritsner M, Perelroyzen G, Kurs R, Ratner Y, Jabarin M, Gibel A. Quality of life outcomes in schizophrenia patients treated with atypical and typical antipsychotic agents: A naturalistic comparative study. Int Clin Psychopharmacol 2004; 24:582–591
Ritsner M, Kurs R. Quality-of-life impairment in severe mental illness: focus on schizoaffective disorders. In: Murray WH (ed) Schizoaffective Disorder: New Research. NOVA Publishers, NY, 2009; Chapter 3, pp. 69–107
Ritsner MS. Novel neuroprotective agents for schizophrenia: neurosteroids, memantine, bexarotene and L-theanine. In: Lerner V, Miodownik C (eds) New Hope for Mental Disturbances. NOVA Publisher, New-York, 2009, pp. 119–151
Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci 2002; 25:409–432
Rapoport JL, Addington AM, Frangou S, Psych MR. The neurodevelopmental model of schizophrenia: update 2005. Mol Psychiatry 2005; 10:434–449
Lakhan SE, Vieira KF. Schizophrenia pathophysiology: are we any closer to a complete model? Ann Gen Psychiatry 2009; 8:12
Ritsner MS (ed) The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes. Springer, Vol. I–IV; 2009
Fatemi SH, Folsom TD. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull 2009; 35:528–548
Compton MT, Walker EF. Physical manifestations of neurodevelopmental disruption: are minor physical anomalies part of the syndrome of schizophrenia? Schizophr Bull 2009; 35:425–436
Wood SJ, Pantelis C, Yung AR, et al. Brain changes during the onset of schizophrenia: implications for neurodevelopmental theories. Med J Aust 2009; 190(4 Suppl):S10–S13
Gur RE, Maany V, Mozley PD, et al. Subcortical MRI volumes in neuroleptic-naive and treated patients with schizophrenia. Am J Psychiatry 1998; 155:1711–1717
Zipursky R, Lambe EK, Kapur S, Mikulis DJ. Cerebral gray matter volume deficits in first episode psychosis. Arch Gen Psychiatry 1998; 55:540–546
Arango C, Moreno C, Martínez S, et al. Longitudinal brain changes in early-onset psychosis. Schizophr Bull 2008; 34:341–353
Berger GE, Wood S, McGorry PD. Incipient neurovulnerability and neuroprotection in early psychosis. Psychopharmacol Bull 2003; 37:79–101
Rapoport JL, Giedd J, Kumra S, et al. Childhood-onset schizophrenia. Progressive ventricular change during adolescence. Arch Gen Psychiatry 1997; 54:897–903
Pantelis C, Velakoulis D, McGorry PD, et al. Neuroanatomical abnormalities before and after onset of psychosis: a cross-sectional and longitudinal MRI comparison. Lancet 2003; 361:281–288
Gur RE, Cowell P, Turetsky BI, et al. A follow-up magnetic resonance imaging study of schizophrenia. Relationship of neuroanatomical changes to clinical and neurobehavioral measures. Arch Gen Psychiatry 1998; 55:145–152
Lieberman JA, Perkins D, Belger A, et al. The early stages of schizophrenia: speculations on pathogenesis, pathophysiology, and therapeutic approaches. Biol Psychiatry 2001; 50:884–897
Velakoulis D, Stuart GW, Wood SJ, et al. Selective bilateral hippocampal volume loss in chronic schizophrenia. Biol Psychiatry 2001; 50:531–539
Mathalon DH, Sullivan EV, Lim KO, Pfefferbaum A. Progressive brain volume changes and the clinical course of schizophrenia in men: a longitudinal magnetic resonance imaging study. Arch Gen Psychiatry 2001; 58:148–157
Hulshoff Pol HE, Kahn RS. What happens after the first episode? A review of progressive brain changes in chronically ill patients with schizophrenia. Schizophr Bull 2008; 34:354–366
Takahashi T, Wood SJ, Yung AR, et al. Progressive gray matter reduction of the superior temporal gyrus during transition to psychosis. Arch Gen Psychiatry 2009; 66:366–376
Thompson PM, Bartzokis G, Hayashi KM, et al. Time-lapse mapping of cortical changes in schizophrenia with different treatments. Cereb Cortex 2009; 19:1107–1123
Knoll JLt, Garver DL, Ramberg JE, et al. Heterogeneity of the psychoses: is there a neurodegenerative psychosis? Schizophr Bull 1998; 24:365–379
Wright IC, Rabe-Hesketh S, Woodruff PWR, et al. Meta-analysis of regional brain volumes in schizophrenia. Am J Psychiatry 2000; 157:16–25
Clinton SM, Meador-Woodruff JH. Thalamic dysfunction in schizophrenia: neurochemical, neuropathological, and in vivo imaging abnormalities. Schizophr Res 2004; 69:237–253
Ende G, Hubrich P, Walter S, et al. Further evidence for altered cerebellar neuronal integrity in schizophrenia. Am J Psychiatry 2005; 162:790–792
Miyamoto S, LaMantia AS, Duncan GE, et al. Recent advances in the neurobiology of schizophrenia. Mol Interv 2003; 3:27–39
Keshavan MS, Tandon R, Boutros NN, Nasrallah HA. Schizophrenia, “just the facts”: what we know in 2008 Part 3: neurobiology. Schizophr Res 2008; 106:89–107
Ritsner MS, Weizman A (eds) Neuroactive Steroids in Brain Functions, and Mental Health. New Perspectives for Research and Treatment. Springer Springer-Verlag, New York, LLC, 2008; 564 pp
Walker EF, Diforio D. Schizophrenia: a neural diathesis-stress model. Psychol Rev 1997; 104:667–685
Walker E, Mittal V, Tessner K. Stress and the hypothalamic pituitary adrenal axis in the developmental course of schizophrenia. Annu Rev Clin Psychol 2008; 4:189–216
Jones SR, Fernyhough C. A new look at the neural diathesis–stress model of schizophrenia: the primacy of social-evaluative and uncontrollable situations. Schizophr Bull 2007; 33:1171–1177
Nuechterlein KH, Dawson ME. A heuristic vulnerability/stress model of schizophrenic episodes. Schizophr Bull 1984; 10:300–312
Keshavan MS. Development, disease and degeneration in schizophrenia: a unitary pathophysiological model. J Psychiatr Res 1999; 33:513–521
Bayer TA, Falkai P, Maier W. Genetic and nongenetic vulnerability factors in schizophrenia: The basis of the “two hit hypothesis.” J Psychiatric Res 1999; 33:543–548
Maynard TM, Sikich L, Lieberman JA, LaMantia AS. Neural development, cell-cell signaling, and the “two-hit” hypothesis of schizophrenia. Schizophr Bull 2001; 27:457–476
McEwen BS. Central effects of stress hormones in health and disease: Understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol 2008; 583:174–185
Angelucci F, Brenè S, Mathé AA. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry 2005; 10:345–352
Durany N, Thome J. Neurotrophic factors and the pathophysiology of schizophrenic psychoses. Eur Psychiatry 2004; 19:326–337
Velakoulis D, Wood SJ, McGorry PD, Pantelis C. Evidence for progression of brain structural abnormalities in schizophrenia: beyond the neurodevelopmental model. Austr NZ J Psychiatry 2000; 34(Suppl):113–126
Ehrenreich H, Siren AL. Neuroprotection – what does it mean? – What means do we have? Eur Arch Psychiatry Clin Neurosci 2001; 251:149–151
Krebs M, Leopold K, Hinzpeter A, Schaefer M. Neuroprotective agents in schizophrenia and affective disorders. Expert Opin Pharmacother 2006; 7:837–848
Jarskog LF, Lieberman JA. Neuroprotection in schizophrenia. J Clin Psychiatry 2006; 67(9):e09
Berger G, Dell’Olio M, Amminger P, et al. Neuroprotection in emerging psychotic disorders. Early Intervention Psychiatry 2007; 1:114–127
Whitcup SM. Clinical trials in neuroprotection. Prog Brain Res 2008; 173:323–335
Ehrenreich H, Aust C, Krampe H, et al. Erythropoietin: novel approaches to neuroprotection in human brain disease. Metab Brain Dis 2004; 19:195–206
Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem 2009; 109 Suppl 1:133–138
Jarskog LF, Glantz LA, Gilmore JH, Lieberman JA. Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2005; 29:846–858
Csernansky JG. Neurodegeneration in schizophrenia: evidence from in vivo neuroimaging studies. Scientific World J 2007; 7:135–143
Jarskog LF, Selinger ES, Lieberman JA, Gilmore JH. Apoptotic proteins in the temporal cortex in schizophrenia: high Bax/Bcl-2 ratio without caspase-3 activation. Am J Psychiatry 2004; 161:109–115
Jarskog LF. Apoptosis in schizophrenia: pathophysiologic and therapeutic considerations. Curr Opin Psychiatry 2006; 19:307–312
Sies H. Oxidative Stress II: Oxidants and Antioxidants. Academic Press, London; 1991
David S, Warner DS, Sheng H, et al. Oxidants, antioxidants and the ischemic brain. J Exp Biol 2004; 207:3221–3231
Cadet JL, Kahler LA. Free radical mechanisms in schizophrenia and tardive diskynesia. NeurosciBiobehav Rev 1994; 18:457–467
Fendri C, Mechri A, Khiari G, Othman A, Kerkeni A, Gaha L. Oxidative stress involvement in schizophrenia pathophysiology: a review. Encephale 2006; 32:244–252. [Article in French]
Mahadik SP, Scheffer RE. Oxidative injury and potential use of antioxidants in schizophrenia. Prostaglandins Leukot Essent Fatty Acids 1996; 55:45–54
Pavlović D, Tamburić V, Stojanović I, et al. Oxidative stress as marker of positive symptoms in schizophrenia. Medicine Biol 2002; 9:157–161
Yao JK, Reddy R, McElhinny LG, van Kammen DP. Reduced status of plasma total antioxidant capacity in schizophrenia. Schizohr Res 1998; 32:1–8
Vardimon L. Neuroprotection by glutamine synthetase. Isr Med Assoc J 2000; 2(Suppl):46–51
Deutsch SI, Rosse RB, Schwartz BL, Mastropaolo J. A revised excitotoxic hypothesis of schizophrenia: therapeutic implications. Clin Neuropharmacol. 2001; 24:43–49
McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev 2007; 87:873–904
van Winkel R, Stefanis NC, Myin-Germeys I. Psychosocial stress and psychosis. A review of the neurobiological mechanisms and the evidence for gene-stress interaction. Schizophr Bull 2008; 34:1095–1105
Collip D, Myin-Germeys I, Van Os J. Does the concept of “sensitization” provide a plausible mechanism for the putative link between the environment and schizophrenia? Schizophr Bull 2008; 34:220–225
Yuii K, Suzuki M, Kurachi M. Stress sensitization in schizophrenia. Ann NY Acad Sci 2007; 1113:276–290
Phillips LJ, McGorry PD, Garner B, et al. Stress, the hippocampus and the hypothalamic-pituitary-adrenal axis: implications for the development of psychotic disorders. Aust NZ J Psychiatry 2006; 40:725–741
De Kloet ER. Hormones and the stressed brain. Ann NY Acad Sci 2004; 1018:1–15
de Kloet ER, Karst H, Joëls M. Corticosteroid hormones in the central stress response: quick-and-slow. Front Neuroendocrinol 2008; 29:268–272
DeRijk R, de Kloet ER. Corticosteroid receptor genetic polymorphisms and stress responsivity. Endocrine 2005; 28:263–270
de Kloet ER, Sibug RM, Helmerhorst FM, Schmidt MV. Stress, genes and the mechanism of programming the brain for later life. Neurosci Biobehav Rev 2005; 29:271–281
Datson NA, Morsink MC, Meijer OC, de Kloet ER. Central corticosteroid actions: Search for gene targets. Eur J Pharmacol 2008; 583:272–289
Lyons DM, Chou Yang, Sawyer-Glover AM, et al. Early Life Stress and Inherited Variation in Monkey Hippocampal Volumes. Arch Gen Psychiatry 2001; 58:1145–1151
Winter H, Irle E. Hippocampal volume in adult burn patients with and without posttraumatic stress disorder. Am J Psychiatry 2004; 161:2194–2200
Vythilingam et al. Childhood trauma associated with smaller hippocampal volume in women with major depression. Am J Psychiatry 2002; 159:2072–2080
Laruelle M. The role of endogenous sensitization in the pathophysiology of schizophrenia: implications from recent brain imaging studies. Brain Res Brain Res Rev 2000; 31:371–384
Iwazaki T, McGregor IS, Matsumoto I. Protein expression profile in the amygdala of rats with methamphetamine-induced behavioral sensitization. Neurosci Lett 2008; 435:113–119
Myin-Germeys I, Delespaul P, van Os J. Behavioural sensitization to daily life stress in psychosis. Psychol Med 2005; 35:733–741
Ritsner MS, Ratner Y, Gibel A, Weizman R. Positive family history is associated with persistent elevated emotional distress in schizophrenia: evidence from a 16-month follow-up study. Psychiatry Res 2007; 153:217–223
Arévalo JC, Wu SH. Neurotrophin signaling: many exciting surprises! Cell Mol Life Sci 2006; 63:1523–1537
Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond, B Biol Sci 2006; 361:1545–1564
Allen SJ, Dawbarn D. Clinical relevance of the neurotrophins and their receptors. Clin. Sci 2006; 110:175–191
Buckley PF, Mahadik S, Pillai A, Terry A Jr. Neurotrophins and schizophrenia. Schizophr Res 2007; 94:1–11
Lang UE, Jockers-Scherubl MC, Hellweg R. State of the art of the neurotrophin hypothesis in psychiatric disorders: implications and limitations. J Neural Transm 2004; 111:387–411
Shoval G, Weizman A. The possible role of neurotrophins in the pathogenesis and therapy of schizophrenia. Eur Neuropsychopharmacol 2005; 15:319–329
Moises HW, Zoega T, Gottesman, II. The glial growth factors deficiency and synaptic destabilization hypothesis of schizophrenia. BMC Psychiatry 2002; 2:8
Zimmer DB, Cornwall EH, Landar A, Song W. The S100 protein family: history, function, and expression. Brain Res Bull 1995; 37:417–429
van Beveren NJ, van der Spelt JJ, de Haan L, Fekkes D. Schizophrenia-associated neural growth factors in peripheral blood. A review. Eur Neuropsychopharmacol 2006; 16:469–480
Sen J, Belli A. S100B in neuropathologic states: the CRP of the brain? J Neurosci Res 2007; 85:1373–1380
Rothermundt M, Ponath G, Glaser T, et al. S100B serum levels and long-term improvement of negative symptoms in patients with schizophrenia. Neuropsychopharmacology 2004; 29:1004–1011
Pedersen A, Diedrich M, Kaestner F, et al. Memory impairment correlates with increased S100B serum concentrations in patients with chronic schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32:1789–1792
Weickert CS, Hyde TM, Lipska BK, et al. Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry 2003; 8:592–610
Baulieu EE. Neurosteroids: a novel function of the brain. Psychoneuroendocrinology 1998; 23:963–987
Ritsner MS, Gibel A, Ratner Y, Weizman A. Dehydroepiandrosterone and pregnenolone alterations in schizophrenia. In: Ritsner MS, Weizman A (eds) Neuroactive Steroids in Brain Functions, and Mental Health. New Perspectives for Research and Treatment. Springer Springer-Verlag, New York, LLC, 2008; pp. 251–298
Mellon SH. Neurosteroid regulation of central nervous system development. Pharmacol Ther 2007; 116:107–124
Maninger N, Wolkowitz OM, Reus VI, Epel ES, Mellon SH. Neurobiological and neuropsychiatric effects of dehydroepiandrosterone (DHEA) and DHEA sulfate (DHEAS). Front Neuroendocrinol 2009; 30:65–91
Wolf OT, Kirschbaum C. Actions of dehydroepiandrosterone and its sulfate in the central nervous system: effects on cognition and emotion in animals and humans. Brain Res Brain Res Rev 1999; 30:264–288
Gursoy E, Cardounel A, Kalimi M. Pregnenolone protects mouse hippocampal (HT-22) cells against glutamate and amyloid beta protein toxicity. Neurochem Res 2001; 26:15–21
Lhullier FL, Nicolaidis R, Riera NG, et al. Dehydroepiandrosterone increases synaptosomal glutamate release and improves the performance in inhibitory avoidance task. Pharmacol Biochem Behav 2004; 77:601–606
Naert G, Maurice T, Tapia-Arancibia L, Givalois L. Neuroactive steroids modulate HPA axis activity and cerebral brain-derived neurotrophic factor (BDNF) protein levels in adult male rats. Psychoneuroendocrinology 2007; 32:1062–1078
Takahashi H, Nakajima A, Sekihara H. Dehydroepiandrosterone (DHEA) and its sulfate (DHEAS) inhibit the apoptosis in human peripheral blood lymphocytes. J Steroid Biochem Mol Biol 2004; 88:261–264
Charalampopoulos I, Tsatsanis C, Dermitzaki E, et al. Dehydroepiandrosterone and allopregnanolone protect sympathoadrenal medulla cells against apoptosis via antiapoptotic Bcl-2 proteins. Proc Natl Acad Sci USA 2004; 101:8209–8214
Majewska MD. Neurosteroids: endogenous bimodal modulators of the GABAA receptor. Mechanism of action and physiological significance. Prog Neurobiol 1992; 38:379–95
Majewska MD. Neuronal actions of dehydroepiandrosterone. Possible roles in brain development, aging, memory, and affect. Ann NY Acad Sci 1995; 774:111–120
Yapanoglu T, Aksoy Y, Gursan N, Ozbey I, Ziypak T, Calik M. Antiapoptotic effects of dehydroepiandrosterone on testicular torsion/detorsion in rats. Andrologia 2008; 40:38–43
Compagnone NA, Mellon SH. Dehydroepiandrosterone: a potential signaling molecule for neocortical organization during development. Proc Natl Acad Sci USA 1998; 95:4678–4683
Suzuki M, Wright LS, Marwah P, et al. Mitotic and neurogenic effects of dehydroepiandrosterone (DHEA) on human neural stem cell cultures derived from the fetal cortex. Proc Natl Acad Sci USA 2004; 101:3202–3207
Aragno M, Parola S, Brignardello E, et al. Dehydroepiandrosterone prevents oxidative injury induced by transient ischemia/reperfusion in the brain of diabetic rats. Diabetes 2000; 49:1924–1931
Lapchak PA, Araujo DM. Preclinical development of neurosteroids as neuroprotective agents for the treatment of neurodegenerative diseases. Int Rev Neurobiol 2001; 46:379–397
Charalampopoulos I, Alexaki VI, Tsatsanis C, et al. Neurosteroids as endogenous inhibitors of neuronal cell apoptosis in aging. Ann NY Acad Sci 2006; 1088:139–152
Kurata K, Takebayashi M, Morinobu S, Yamawaki S. Beta-estradiol, dehydroepiandrosterone, and dehydroepiandrosterone sulfate protect against N-methyl-D-aspartate-induced neurotoxicity in rat hippocampal neurons by different mechanisms. J Pharmacol Exp Ther 2004; 311:237–245
Kimonides VG, Khatibi NH, Svendsen CN, et al. Dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEAS) protect hippocampal neurons against excitatory amino acid-induced neurotoxicity. Proc Natl Acad Sci USA 1998; 95:1852–1857
Cardounel A, Regelson W, Kalimi M. Dehydroepiandrosterone protects hippocampal neurons against neurotoxin-induced cell death: mechanism of action. Proc Soc Exp Biol Med 1999; 222:145–149
Kimonides VG, Spillantini MG, Sofroniew MV, et al. Dehydroepiandrosterone antagonizes the neurotoxic effects of corticosterone and translocation of stress-activated protein kinase 3 in hippocampal primary cultures. Neuroscience 1999; 89:429–436
Veiga S, Garcia-Segura LM, Azcoitia I. Neuroprotection by the steroids pregnenolone and dehydroepiandrosterone is mediated by the enzyme aromatase. J Neurobiol 2003; 56:398–406
Akan P, Kizildag S, Ormen M, Genc S, Oktem MA, Fadiloglu M. Pregnenolone protects the PC-12 cell line against amyloid beta peptide toxicity but its sulfate ester does not. Chem Biol Interact 2009; 177:65–70
Leskiewicz M, Jantas D, Budziszewska B, Lason W. Excitatory neurosteroids attenuate apoptotic and excitotoxic cell death in primary cortical neurons. J Physiol Pharmacol 2008; 59:457–475
Bologa L, Sharma J, Roberts E. Dehydroepiandrosterone and its sulfated derivative reduce neuronal death and enhance astrocytic differentiation in brain cell cultures. J Neurosci Res 1987; 17:225–234
Karishma KK, Herbert J. Dehydroepiandrosterone (DHEA) stimulates neurogenesis in the hippocampus of the rat, promotes survival of newly formed neurons and prevents corticosterone-induced suppression. Eur J Neurosci 2002; 16:445–453
Bastianetto S, Ramassamy C, Poirier J, Quirion R. Dehydroepiandrosterone (DHEA) protects hippocampal cells from oxidative stress-induced damage. Brain Res Mol Brain Res 1999; 66:35–41
Hu Y, Cardounel A, Gursoy E, Anderson P, Kalimi M. Anti-stress effects of dehydroepiandrosterone: protection of rats against repeated immobilization stress-induced weight loss, glucocorticoid receptor production, and lipid peroxidation. Biochem Pharmacol 2000; 59:753–762
Boudarene M, Legros JJ, Timsit-Berthier M. Study of the stress response: role of anxiety, cortisol and DHEAs. Encephale 2002; 28:139–146
Rasmusson AM, Vythilingam M, Morgan CA 3rd. The neuroendocrinology of posttraumatic stress disorder: new directions. CNS Spectr 2003; 8:651–656, 665–667
Debonnel G, Bergeron R, de Montigny C. Potentiation by dehydroepiandrosterone of the neuronal response to N-methyl-D-aspartate in the CA3 region of the rat dorsal hippocampus: an effect mediated via sigma receptors. J Endocrinol 1996; 150(Suppl):S33–S42
Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABA(A) receptor. Nat Rev Neurosci 2005; 6:565–575
Holsboer F, Grasser A, Friess E, Wiedemann K. Steroid effects on central neurons and implications for psychiatric and neurological disorders. Ann NY Acad Sci 1994; 746:345–359
Rupprecht R. The neuropsychopharmacological potential of neuroactive steroids. J Psychiatr Res 1997; 31:297–314
George O, Vallée M, Le Moal M, Mayo W. Neurosteroids and cholinergic systems: implications for sleep and cognitive processes and potential role of age-related changes. Psychopharmacology 2006; 186:402–413
Pérez-Neri I, Montes S, Ojeda-López C, Ramírez-Bermúdez J, Ríos C. Modulation of neurotransmitter systems by dehydroepiandrosterone and dehydroepiandrosterone sulfate: Mechanism of action and relevance to psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32:1118–1130
Widstrom RL, Dillon JS. Is there a receptor for dehydroepiandrosterone or dehydroepiandrosterone sulfate? Semin Reprod Med 2004; 22:289–298
Flood JF, Morley JE, Roberts E. Memory-enhancing effects in male mice of pregnenolone and steroids metabolically derived from it. Proc Natl Acad Sci USA 1992; 89:1567–1571
Mathis C, Vogel E, Cagniard B, Criscuolo F, Ungerer A. The neurosteroid pregnenolone sulfate blocks deficits induced by a competitive NMDA antagonist in active avoidance and lever-press learning tasks in mice. Neuropharmacology 1996; 35:1057–1064
Yanase T, Fukahori M, Taniguchi S. Serum dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEA-S) in Alzheimer’s disease and in cerebrovascular dementia. Endocr J 1996; 43:119–123
Silver H, Knoll G, Isakov V, et al. Blood DHEAS concentrations correlate with cognitive function in chronic schizophrenia patients: a pilot study. J Psychiatr Res 2005; 39: 569–575
Morrison MF, Redei E, TenHave T. Dehydroepiandrosterone sulfate and psychiatric measures in a frail, elderly residential care population. Biol Psychiatry 2000; 47:144–150
Vallee M, Mayo W, Le Moal M. Role of pregnenolone, dehydroepiandrosterone and their sulfate esters on learning and memory in cognitive aging. Brain Res Brain Res Rev 2001; 37:301–312
Ritsner M, Maayan R, Gibel A, et al. Elevation of the cortisol/dehydroepiandrosterone ratio in schizophrenia patients. Eur Neuropsychopharmacol 2004; 14:267–273
Gallagher P., Ritsner MS. Can the cortisol to DHEA molar ratio be used as a peripheral biomarker for schizophrenia and mood disorders? In: Ritsner MS (ed) The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes. Springer, Vol. III, 2009; pp. 27–45
Roberts E, Fitten LJ. Serum steroid levels in two old men with Alzheimer’s disease (AD) before and after oral administration of dehydroepiandrosterone (DHEA). Pregnenolone synthesis may be ratelimiting in aging. In: Kalimi M, Regelson W (eds) The Biological Role of Dehydroepiandrosterone (DHEA). de Gruyter, Berlin, 1990; pp. 43–63
Ritsner M, Maayan R, Gibel A, Weizman A. Differences in blood pregnenolone and dehydroepiandrosterone levels between schizophrenia patients and healthy subjects. Eur Neuropsychopharmacol 2007; 17:358–365
Semeniuk T, Jhangri GS, Le Melledo JM. Neuroactive steroid levels in patients with generalized anxiety disorder. J Neuropsychiatry Clin Neurosci 2001; 13:396–398
Heydari B, Le Melledo JM. Low pregnenolone sulphate plasma concentrations in patients with generalized social phobia. Psychol Med 2002; 32:929–933
Strous RD, Maayan R, Lapidus R, et al. Dehydroepiandrosterone augmentation in the management of negative, depressive, and anxiety symptoms in schizophrenia. Arch Gen Psychiatry 2003; 60:133–141
Nachshoni T, Ebert T, Abramovitch Y, et al. Improvement of extrapyramidal symptoms following dehydroepiandrosterone (DHEA) administration in antipsychotic treated schizophrenia patients: a randomized, double-blind placebo controlled trial. Schizophr Res 2005; 79:251–256
Strous RD, Stryjer R, Maayan R, et al. Analysis of clinical symptomatology, extrapyramidal symptoms and neurocognitive dysfunction following dehydroepiandrosterone (DHEA) administration in olanzapine treated schizophrenia patients: a randomized, double-blind placebo controlled trial. Psychoneuroendocrinology 2007; 32:96–105
Ritsner MS, Gibel A, Ratner Y, et al. Improvement of sustained attention and visual and movement skills, but not clinical symptoms, after dehydroepiandrosterone augmentation in schizophrenia: a randomized, double-blind, placebo-controlled, crossover trial. J Clin Psychopharmacol 2006; 26:495–499
Strous RD, Gibel A, Maayan R, Weizman A, Ritsner MS. Hormonal response to dehydroepiandrosterone administration in schizophrenia: findings from a randomized, double-blind, placebo-controlled, crossover study. J Clin Psychopharmacol 2008; 28:456–459
Ritsner MS, Strous RD. Neurocognitive deficits in schizophrenia are associated with alterations in blood levels of neurosteroids: A multiple regression analysis of findings from a double-blind, randomized, placebo-controlled, crossover trial with DHEA. Journal of Psychiatric Research (2009), doi:10.1016/j.jpsychires.2009.07.002
Ritsner MS. Dehydroepiandrosterone administration in treating medical and neuropsychiatric disorders: high hopes, disappointing results, and topics for future research. In: Ritsner MS, Weizman A (eds) Neuroactive Steroids in Brain Functions, and Mental Health. New Perspectives for Research and Treatment. Springer Springer-Verlag, New York, LLC, 2008; pp. 337–368
Pincus G, Hoagland H. Effects of administered pregnenolone on fatiguing psychomotor performance. J Aviation Med 1944; 15:98–111
Pincus G, Hoagland H. Effects on industrial production of the administration of pregnenolone to factory workers. J Psychosom Med 1945; 7:342–346
McGavack T, Chevalley J, Weissberg J. The use of delta 5-pregnenolone in various clinical disorders. J Clin Endocrinol Metab 1951; 11:559–577
Meieran SE, Reus VI, Webster R, Shafton R, Wolkowitz OM. Chronic pregnenolone effects in normal humans: attenuation of benzodiazepine-induced sedation. Psychoneuroendocrinology 2004; 29:486–500
Ritsner MS, Gibel A, Shleifer T, et al. Pregnenolone and dehydroepiandrosterone as an adjunctive treatment in schizophrenia: an 8-week, double-blind, randomized, controlled, two-center, parallel-group trial. J Clin Psychiatry 2010 (in press)
Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA 1996; 93: 5925–5930
Ogawa S, Inoue S, Watanabe T, Hiroi H, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. The complete primary structure of human estrogen receptor beta (hER beta) and its heterodimerization with ER alpha in vivo and in vitro. Biochem Biophys Res Commun 1998; 243:122–126
Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci 2000; 18:347–358
Lee SJ, McEwen BS. Neurotrophic and neuroprotective actions of estrogens and their therapeutic implications. Annu Rev Pharmacol Toxicol 2001; 41:569–591
Mendez P, Azcoitia I, Garcia-Segura LM. Interdependence of oestrogen and insulin-like growth factor-I in the brain: potential for analysing neuroprotective mechanisms J Endocrinol 2005; 185:11–17
Wise PM, Dubal DB, Wilson ME, et al. Estradiol is a protective factor in the adult and aging brain: understanding of mechanisms derived from in vivoand in vitrostudies. Brain Res Rev 2001; 37:313–319
Wise PM, Dubal DB, Rau SW, Brown CM, Suzuki S. Are Estrogens protective or risk factors in brain injury and neurodegeneration? Reevaluation after the women’s health initiative. Endocrine Rev 2005; 26:308–312
Simpkins JW, Green PS, Gridley JS, Monck EK. Neuroprotective effects of estrogens. In: Bellino FL (ed) Biology of Menopause. Springer-Verlag, New York, 2000; pp.103–111
Brann DW, Dhandapani K, Wakade C, Mahesh VB, Khan MM. Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids 2007; 72:381–405
Singh M, Dykens JA, Simpkins JW. Novel mechanisms for estrogen-induced neuroprotection. Exp Biol Med (Maywood) 2006; 231:514–521
Salokangas RK. Gender and the use of neuroleptics in schizophrenia. Further testing of the oestrogen hypothesis. Schizophr Res 1995; 16:7–16
Huber TJ, Tettenborn C, Leifke E, Emrich HM: Sex hormones in psychotic men. Psychoneuroendocrinology 2005, 30:111–114.
Bergemann N, Mundt C, Parzer P, et al. Estrogen as an adjuvant therapy to antipsychotics does not prevent relapse in women suffering from schizophrenia: results of a placebo-controlled double-blind study. Schizophr Res 2005; 74:125–134
Mortimer AM. Relationship between estrogen and schizophrenia. Expert Rev Neurother 2007; 7:45–55
Kulkarni J. Oestrogen – a new treatment approach for schizophrenia? Med J Aust 2009; 190(4 Suppl):S37–S38
Seeman MV, Lang M. The role of estrogens in schizophrenia gender differences. Schizophr Bull 1990; 16:185–194
Riecher-Rössler A. Estrogens and schizophrenia. In: Bergemann N, Riecher-Rössler A (eds) Oestrogen Effects in Psychiatric Disorders. Springer, Wien, 2005; pp. 31–52
Cantoni GL. S-adenosylmethionine: a new intermediate formed enzymatically from L-methionine and adenosine-triphosphate. J Biol Chem 1953; 204:403–416
Fetrow CW, Avila JR. Efficacy of the dietary supplement S-adenosyl-L-methionine. Ann Pharmacother 2001; 35:1414–1425
Surtees R, Leonard J, Austin S. Association of demyelination with deficiency of cerebrospinal-fluid S-adenosylmethionine in inborn errors of methyl-transfer pathway. Lancet 1991; 338:1550–1554
Bell KM, Potkin SG, Carreon D, Plon L. S-adenosylmethionine blood levels in major depression: changes with drug treatment. Acta Neurol Scand Suppl 1994; 154:15–18
Stramentinoli G, Gualano M, Galli-Kienle M, Intestinal absorption of S-adenosyl-L-methionine. J Pharmacol Exp Ther 1979; 209:323–326
Stramentinoli G. Pharmacologic aspects of S-adenosylmethionine. Pharmacokinetics and pharmacodynamics. Am J Med 1987; 83:35–42
Bottiglieri T, Godfrey P, Flynn T, et al. Cerebrospinal fluid S-adenosylmethionine in depression and dementia: effects of treatment with parenteral and oral S-adenosylmethionine. J Neurol Neurosurg Psychiatry 1990; 53:1096–1098
Malakar D, Dey A, Ghosh AK. Protective role of S-adenosyl-L-methionine against hydrochloric acid stress in Saccharomyces cerevisiae. Biochim Biophys Acta 2006; 1760:1298–1303
Malakar D, Dey A, Basu A, Ghosh AK. Antiapoptotic role of S-adenosyl-l-methionine against hydrochloric acid induced cell death in Saccharomyces cerevisiae. Biochim Biophys Acta 2008; 1780:937–947
James SJ, Cutler P, Melnyk S, et al. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr 2004; 80: 1611–1617
Stramentinoli G, Gualano M, Catto E, Algeri S.Tissue levels of S-adenosylmethionine in aging rats. J Gerontol 1977; 32:392–394
Laudanno OM. Cytoprotective effect of S-adenosylmethionine compared with that of misoprostol against ethanol-, aspirin-, and stress-induced gastric damage. Am J Med 1987; 83:43–47
Mato JM, Camara J, Fernandez de Paz J, et al. S-adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo-controlled, double-blind, multicenter clinical trial. J Hepatol 1999; 30:1081–1089
Gatto G, Caleri D, Michelacci S, Sicuteri F. Analgesizing effect of a methyl donor (S-adenosylmethionine) in migraine: an open clinical trial. Int J Clin Pharmacol Res 1986; 6:15–17
Morrison LD, Smith DD, Kish SJ. Brain S-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J Neurochem 1996; 67:1328–1331
Friedel HA, Goa KL, Benfield P. S-adenosyl-L-methionine. A review of its pharmacological properties and therapeutic potential in liver dysfunction and affective disorders in relation to its physiological role in cell metabolism. Drugs 1089; 38:389–416
Papakostas GI, Alpert JE, Fava M. S-adenosyl-methionine in depression: a comprehensive review of the literature. Curr Psychiatry Rep 2003; 5:460–466
Strous RD, Ritsner MS, Adler S, et al. Improvement of aggressive behavior and quality of life impairment following S-adenosyl-methionine (SAM-e) augmentation in schizophrenia. Eur Neuropsychopharmacol 2009; 19:14–22
Ullrich O, Merker K, Timm J, Tauber S. Immune control by endocannabinoids – new mechanisms of neuroprotection? J Neuroimmunol 2007; 184:127–135
Roser P, Vollenweider FX, Kawohl W. Potential antipsychotic properties of central cannabinoid (CB(1)) receptor antagonists. World J Biol Psychiatry 2008; 7:1–12
Grotenhermen F. Cannabinoids. Curr Drug Targets CNS Neurol Disord 2005; 4:507–530
Moreira FA, Aguiar DC, Guimarães FS. Anxiolytic-like effect of cannabidiol in the rat Vogel conflict test. Prog Neuropsychopharmacol Biol Psychiatry 2006; 30:1466–1471
Zuardi AW, Crippa JA, Hallak JE, et al. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz J Med Biol Res 2006; 39:421–429
de Lago E, Fernández-Ruiz J. Cannabinoids and neuroprotection in motor-related disorders. CNS Neurol Disord Drug Targets 2007; 6:377–387
Davies SN, Pertwee RG, Riedel G. Functions of cannabinoid receptors in the hippocampus. Neuropharmacology 2002; 42:993–1007
Fride E, Shohami E. The endocannabinoid system: function in survival of the embryo, the newborn and the neuron. Neuroreport 2002; 13:1833–1841
Pryce G, Ahmed Z, Hankey DJ, et al. Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain 2003; 126:2191–2202
van der Stelt M, Veldhuis WB, Maccarrone M, et al. Acute neuronal injury, excitotoxicity, and the endocannabinoid system. Mol Neurobiol 2002; 26:317–346
Zhuang SY, Bridges D, Grigorenko E, et al. Cannabinoids produce neuroprotection by reducing intracellular calcium release from ryanodine-sensitive stores. Neuropharmacology 2005; 48:1086–1096
D’Souza DC, Pittman B, Perry E, Simen A. Preliminary evidence of cannabinoid effects on brain-derived neurotrophic factor (BDNF) levels in humans. Psychopharmacology (Berl) 2009; 202:569–578
Bhattacharyya S, Fusar-Poli P, Borgwardt S, et al. Modulation of mediotemporal and ventrostriatal function in humans by Delta9-tetrahydrocannabinol: a neural basis for the effects of Cannabis sativa on learning and psychosis. Arch Gen Psychiatry 2009; 66:442–451
Semple DM, McIntosh AM, Lawrie SM. Cannabis as a risk factor for psychosis: systematic review. J Psychopharmacol 2005; 19:187–194
Ben Amar M, Potvin S. Cannabis and psychosis: what is the link? J Psychoactive Drugs 2007; 39:131–142
Cohen M, Solowij N, Carr V. Cannabis, cannabinoids and schizophrenia: integration of the evidence. Aust NZ J Psychiatry 2008; 42:357–368
Rathbone J, Variend H, Mehta H. Cannabis and schizophrenia. Cochrane Database Syst Rev 2008; (3):CD004837
Sewell RA, Ranganathan M, D’Souza DC. Cannabinoids and psychosis. Int Rev Psychiatry 2009; 21:152–162
D’Souza DC, Abi-Saab WM, Madonick S, et al. Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis, and addiction. Biol Psychiatry 2005; 57:594–608
Schwarcz G, Karajgi B, McCarthy R. Synthetic delta-9-tetrahydrocannabinol (dronabinol) can improve the symptoms of schizophrenia. J Clin Psychopharmacol 2009; 29:255–258
Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 2007; 370:319–328
Zammit S, Moore TH, Lingford-Hughes A, et al. Effects of cannabis use on outcomes of psychotic disorders: systematic review. Br J Psychiatry 2008; 193:357–363
Müller-Vahl KR, Emrich HM. Cannabis and schizophrenia: towards a cannabinoid hypothesis of schizophrenia. Expert Rev Neurother 2008; 8:1037–1048
Pertwee RG. Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br J Pharmacol 2009 Feb; 156(3):397–411
Peet M, Stokes C. Omega-3 fatty acids in the treatment of psychiatric disorders. Drugs 2005; 65:1051–1059
Moreira JD, Knorr L, Thomazi AP, et al. Dietary omega-3 fatty acids attenuate cellular damage after a hippocampal ischemic insult in adult rats. J Nutr Biochem 2009 May 1. [Epub ahead of print]
Kaur P, Heggland I, Aschner M, Syversen T. Docosahexaenoic acid may act as a neuroprotector for methylmercury-induced neurotoxicity in primary neural cell cultures. Neurotoxicology 2008; 29:978–987
Berman DR, Mozurkewich E, Liu Y, Barks J. Docosahexaenoic acid pretreatment confers neuroprotection in a rat model of perinatal cerebral hypoxia-ischemia. Am J Obstet Gynecol 2009; 200:305.e1–6
Fenton WS, Dickerson F, Boronow J, Hibbeln JR, Knable M. A placebo-controlled trial of omega-3 fatty acid (ethyl eicosapentaenoic acid) supplementation for residual symptoms and cognitive impairment in schizophrenia. Am J Psychiatry 2001; 158:2071–2074
Freeman MP, Hibbeln JR, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry 2006; 67:1954–1967
Peet M. Omega-3 polyunsaturated fatty acids in the treatment of schizophrenia. Isr J Psychiatry Relat Sci 2008; 45:19–25
Emsley R, Niehaus DJ, Oosthuizen PP, et al. Safety of the omega-3 fatty acid, eicosapentaenoic acid (EPA) in psychiatric patients: results from a randomized, placebo-controlled trial. Psychiatry Res 2008; 161:284–291
Mindus P, Cronholm B, Levander SE, Schalling D. Piracetam-induced improvement of mental performance. A controlled study on normally aging individuals. Acta Psychiatr Scand 1976; 54:150–160
Deberdt W. Interaction between psychological and pharmacological treatment in cognitive impairment. Life Sci 1994; 55:2057–2066
Giurgea C. Piracetam: nootropic pharmacology of neurointegrative activity. Curr Dev Psychopharmacol 1976; 3:223–723
O’Neill MJ, Bleakman D, Zimmerman DM, Nisenbaum ES. AMPA receptor potentiators for the treatment of CNS disorders. Curr Drug Targets CNS Neurol Disord 2004; 3:181–194
Taupin P. Nootropic agents stimulate neurogenesis. Expert Opin Ther Pat 2009; 19: 727–730
Muller WE, Eckert GP, Eckert A. Piracetam: novelty in a unique mode of action. Pharmacopsychiatry 1999; 32 Suppl 1:2–9
Cohen S, Mueller W. Interaction of piracetam with several neurotransmitter receptors in central nervous system – relative specificity for 3H-glutamate sites. Arzneimittelforschung 1985; 35:1350–1352
Muller WE, Koch S, Scheuer K, Rostock A, Bartsch R. Effects of piracetam on membrane fluidity in the aged mouse, rat, and human brain. Biochem Pharmacol 1997; 53:135–140
Gouliaev AH, Senning A. Piracetam and other structurally related nootropics. Brain Res Brain Res Rev 1994; 19:180–222
Libov I, Miodownik C, Bersudsky Y, Dwolatzky T, Lerner V. Efficacy of piracetam in the treatment of tardive dyskinesia in schizophrenic patients: a randomized, double-blind, placebo-controlled crossover study. J Clin Psychiatry 2007; 68:1031–1037
van Vliet SA, Blezer EL, Jongsma MJ, et al. Exploring the neuroprotective effects of modafinil in a marmoset Parkinson model with immunohistochemistry, magnetic resonance imaging and spectroscopy. Brain Res 2008; 1189:219–228
Minzenberg MJ, Carter CS. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology 2008; 33:1477–1502
Turner DC, Clark L, Pomarol-Clotet E, et al. Modafinil improves cognition and attentional set shifting in patients with chronic schizophrenia. Neuropsychopharmacology 2004; 29:1363–1373
Sevy S, Rosenthal MH, Alvir J, et al. Double-blind, placebo-controlled study of modafinil for fatigue and cognition in schizophrenia patients treated with psychotropic medications. J Clin Psychiatry 2005; 66:839–843
Saavedra-Velez C, Yusim A, Anbarasan D, Lindenmayer JP. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J Clin Psychiatry 2009; 70:104–112
Ekborg-Ott KH, Taylor A, Armstrong DW. Varietal differences in the total and enantiomeric composition of theanine in tea. J Agric Food Chem 1997; 45:353–363
Unno T, Suzuki Y, Kakuda T, et al. Metabolism of theanine, gamma-glutamylethylamide, in rats. J Agric Food Chem 1999; 47:1593–1596
Bukowski JF, Morita CT, Brenner MB. Human gamma delta T cells recognize alkylamines derived from microbes, edible plants, and tea: implications for innate immunity. Immunity 1999; 11:57–65
Terashima T, Takido J, Yokogoshi H. Time-dependent changes of amino acids in the serum, liver, brain and urine of rats administered with theanine. Biosci Biotechnol Biochem 1999; 63:615–618
Kakuda T, Nozawa A, Unno T, Okamura N, Okai O. Inhibiting effects of theanine on caffeine stimulation evaluated by EEG in the rat. Biosci Biotechnol Biochem 2000; 64:287–293
Lu K, Gray MA, Oliver C, et al. The acute effects of L-theanine in comparison with alprazolam on anticipatory anxiety in humans. Hum Psychopharmacol 2004; 19:457–465
Sadzuka Y, Sugiyama T, Miyagishima A, et al. The effects of theanine, as a novel biochemical modulator, on the antitumor activity of adriamycin. Cancer Lett 1996; 105:203–209
Yokozawa T, Dong E. Influence of green tea and its three major components upon low-density lipoprotein oxidation. Exp Toxicol Pathol 1997; 49:329–335
Sugiyama T, Sadzuka Y. Theanine, a specific glutamate derivative in green tea, reduces the adverse reactions of doxorubicin by changing the glutathione level. Cancer Lett 2004; 212:177–184
Kakuda T, Nozawa A, Sugimoto A, Niino H. Inhibition by theanine of binding of [3H]AMPA, [3H]kainate, and [3H]MDL 105,519 to glutamate receptors. Biosci Biotechnol Biochem 2002; 66:2683–2686
Nagasawa K, Aoki H, Yasuda E, Nagai K, Shimohama S, Fujimoto S. Possible involvement of group I mGluRs in neuroprotective effect of theanine. Biochem Biophys Res Commun 2004; 320:116–122
Mason R. 200 mg of Zen. L-theanine boosts alpha waves, promotes alert relaxation. Alternative & Complementary Therapies 2001; 7:91–95
Kimura R, Murata T. Influence of alkylamides of glutamic acid and related compounds on the central nervous system. II. Syntheses of amides of gutamic acid and related compounds, and their effects on the central nervous system. Chem Pharm Bull (Tokyo) 1971; 19:1301–7
Juneja LR, Chu DC, Okubo T, et al. L-theanine-a unique amino acid of green tea and its relaxation effect in humans. Trends Food Sci Technol 1999; 10:199–204
Kakuda T, Matsuura T, Sagesaka Y, Kawasaki T. Product and method for inhibiting caffeine stimulation with theanine. vol 5,501,866. USA, 1996
Kent JM, Mathew SJ, Gorman JM. Molecular targets in the treatment of anxiety. Biol Psychiatry 2002; 52:1008–1030
Ito K, Nagato Y, Aoi N, et al. Effects of L-theanine on the release of alpha-brain waves in human volunteers. Nippon Nogeikagaku Kaishi 1998; 72:153–157
Yokogoshi H, Kobayashi M, Mochizuki M, Terashima T. Effect of theanine, r-glutamylethylamide, on brain monoamines and striatal dopamine release in conscious rats. Neurochem Res 1998; 23:667–673
Yokogoshi H, Mochizuki M, Saitoh K. Theanine-induced reduction of brain serotonin concentration in rats. Biosci Biotechnol Biochem 1998; 62:816–817
Kobayashi K, Nagato Y, Aoi N, Juneja LR, Kim M, et al. Effects of L-theanine on the release of α-brain waves in human volunteers. Nippon Nogeikagaku Kaishi 1998; 72:153–157
Ritsner MS, Miodownik C, Ratner Y, et al. L-theanine relieves positive, activation, and anxiety symptoms in patients with schizophrenia and schizoaffective disorders: an 8-week, randomized, double-blind, placebo-controlled, two-center study. J Clin Psychiatry 2010 (in press)
Sporn MB, Roberts AB. Role of retinoids in differentiation and carcinogenesis. J Natl Cancer Inst 1984; 73:1381–1387
Tafti M, Ghyselinck NB. Functional implication of the vitamin A signaling pathway in the brain. Arch Neurol 2007; 64:1706–1711
Oridate N, Lotan D, Xu XC, Hong WK, Lotan R. Differential induction of apoptosis by all-trans-retinoic acid and N-(4-hydroxyphenyl)retinamide in human head and neck squamous cell carcinoma cell lines. Clin Cancer Res 1996; 2:855–863
Sarah J. Bailey SJ, McCaffery PJ. Retinoic acid signalling in neuropsychiatric disease: possible markers and treatment agents. In: Ritsner MS (ed) The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes. Springer, Vol III, 2009; pp. 171–189
Gorgun G, Foss F. Immunomodulatory effects of RXR rexinoids: modulation of high-affinity IL-2R expression enhances susceptibility to denileukin diftitox. Blood 2002; 100:1399–1403
McCaffery P, Drager UC. High levels of a retinoic acid-generating dehydrogenase in the meso-telencephalic dopamine system. Proc Natl Acad Sci USA 1994; 91:7772–7776
Wagner E, Luo T, Drager UC. Retinoic acid synthesis in the postnatal mouse brain marks distinct developmental stages and functional systems. Cereb Cortex 2002; 12:1244–1253
Arinami T, Gao M, Hamaguchi H, Toru M. A functional polymorphism in the promoter region of the dopamine D2 receptor gene is associated with schizophrenia. Hum Mol Genet 1997; 6:577–582
Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res 2002; 43:1773–1808
Krezel W, Ghyselinck N, Samad TA, Dupe V, Kastner P, Borrelli E, Chambon P. Impaired locomotion and dopamine signaling in retinoid receptor mutant mice. Science 1998; 279:863–867
Palha JA, Goodman AB. Thyroid hormones and retinoids: a possible link between genes and environment in schizophrenia. Brain Res Rev 2006; 51:61–71
Goodman AB. Three independent lines of evidence suggest retinoids as causal to schizophrenia. Proc Natl Acad Sci USA 1998; 95:7240–7244
Goodman AB. Chromosomal locations and modes of action of genes of the retinoid (vitamin A) system support their involvement in the etiology of schizophrenia. Am J Med Genet 1995; 60:335–48
Etchamendy N, Enderlin V, Marighetto A, et al. Vitamin A deficiency and relational memory deficit in adult mice: relationships with changes in brain retinoid signalling. Behl Brain Res 2003; 145:37–49
Misner DL, Jacobs S, Shimizu Y, et al. Vitamin deprivation results in reversible loss of hippocampal long-term synaptic plasticity. Proc Natl Acad Sci USA 2001; 98:11714–11719
Alfos S, Boucheron C, Pallet V, et al. A retinoic acid receptor antagonist suppresses brain retinoic acid receptor overexpression and reverses a working memory deficit induced by chronic ethanol consumption in mice. Alcohol Clin Exp Res 2001; 25:1506–1514
Corcoran JP, So PL, Maden M. Disruption of the retinoid signalling pathway causes a deposition of amyloid beta in the adult rat brain. Eur J Neurosci 2004; 20:896–902
Goodman AB, Pardee AB. Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc Natl Acad Sci USA 2003; 100:2901–2905
Lane MA, Bailey SJ. Role of retinoid signalling in the adult brain. Prog Neurobiol 2005; 75:275–293
Mey J, McCaffery P. Retinoic acid signaling in the nervous system of adult vertebrates. Neuroscientist 2004; 10:409–421
Sharma, R.P.Schizophrenia, epigenetics and ligand-activated nuclear receptors: a framework for chromatin therapeutics. Schizophr Res 2005; 72:79–90
Boehm MF, Zhang L, Badea BA, et al. Synthesis and structure-activity relationships of novel retinoid X receptor-selective retinoids. J Med Chem 1994; 37:2930–2941
Bischoff ED, Heyman RA, Lamph WW. Effect of the retinoid X receptor-selective ligand LGD1069 on mammary carcinoma after tamoxifen failure. J Natl Cancer Inst 1999; 91(24):2118
Hurst RE. Bexarotene ligand pharmaceuticals. Curr Opin Investig Drugs 2000; 1:514–523
Prince HM, McCormack C, Ryan G, et al. Bexarotene capsules and gel for previously treated patients with cutaneous T-cell lymphoma: results of the Australian patients treated on phase II trials. Australas J Dermatol 2001; 42:91–97
Rigas JR, Maurer LH, Meyer LP, Hammond SM, Crisp MR, Parker BA, Truglia JA. Targretin, a selective retinoid X receptor ligand (LGD1069), vinorelbine and cisplatin for the treatment of non small cell lung cancer (NSCLC): a phase I/II trial. Paper presented at: Proc Annu Meet Am Soc Clin Oncol, 1997
Lerner V, Miodownik C, Gibel A, Kovalyonok E, Shleifer T, Goodman AB, Ritsner MS. Bexarotene as add-on to antipsychotic treatment in schizophrenia patients: a pilot open-label trial. Clin Neuropharmacol 2008; 31:25–33
Faden AI, Bogdan Stoica B. Neuroprotection. Challenges and Opportunities. Arch Neurol 2007; 64:794–800
Acknowledgments
The author would like to express gratitude to my colleagues Drs. Anatoly Gibel, Yael Ratner, Professor Vladimir Lerner, and Professor Abraham Weizman for fruitful cooperation. Mrs. Rena Kurs provided outstanding editorial assistance. The author gratefully acknowledges the support of the team of clinical departments of Shaar Menashe Mental Health Center. Clinical trials with neuroprotective compounds were supported by generous grants from the Stanley Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Ritsner, M.S. (2010). Is a Neuroprotective Therapy Suitable for Schizophrenia Patients?. In: Ritsner, M. (eds) Brain Protection in Schizophrenia, Mood and Cognitive Disorders. Springer, Dordrecht. https://doi.org/10.1007/978-90-481-8553-5_12
Download citation
DOI: https://doi.org/10.1007/978-90-481-8553-5_12
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-90-481-8552-8
Online ISBN: 978-90-481-8553-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)