
Abstract
Lithium (Li+) is a first option treatment for adult acute episodes of Bipolar Disorder (BD) and for the prophylaxis of new depressed or manic episodes. It is also the preferred choice as maintenance treatment. Numerous studies have shown morphological abnormalities in the brains of BD patients, suggesting that this highly heritable disorder may exhibit progressive and deleterious changes in brain structure. Since treatment with Li+ ameliorates these abnormalities, it has been postulated that Li+ is a neuroprotective agent in the same way atypical antipsychotics are neuroprotective in patients diagnosed with schizophrenia spectrum disorders. Li+’s neuroprotective properties are related to its modulation of nerve growth factors, inflammation, mitochondrial function, oxidative stress, and programmed cell death mechanisms such as autophagy and apoptosis. Notwithstanding, it is not known whether Li+—induced neuroprotection is related to the inhibition of its putative molecular targets in a BD episode: the enzymes inositol-monophosphatase, (IMPase), glycogen-synthase-kinase 3β (GSK3), and Protein kinase C (PKC). Furthermore, it is uncertain whether these neuroprotective mechanisms are correlated with Li+’s clinical efficacy in maintaining mood stability. It is expected that in a nearby future, precision medicine approaches will improve diagnosis and expand treatment options. This will certainly contribute to ameliorating the medical and economic burden created by this devastating mood disorder.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bipolar Disorder (BD) (American Psychiatric Association. DSM-5 Task Force 2013) is a heritable mental illness (Stahl et al. 2019; Gordovez and McMahon 2020) characterized by cyclical disturbances in mood and behavior (Goodwin and Jamison 1990) and associated with well documented cortical brain abnormalities (Abe et al. 2020).
The efficacy of lithium (Li+) as a first option treatment for adult manic episodes and for maintenance of Bipolar Disorder (BD) is now well established (Curran and Ravindran 2014; Geddes et al. 2004; Severus et al. 2014; Licht 2012; Cousins et al. 2020). This is also the case for child and adolescent BD (Hafeman et al. 2019).
Li+ is also the preferred treatment for the prophylaxis of new episodes, either depressive or manic (Geddes and Miklowitz 2013; Licht 2012; Goodwin 2002). Although some clinical guidelines consider antiseizure and neuroleptic agents alongside Li+ as equally efficacious in BD (Graham et al. 2018), many psychiatrists adopt Li+ as the preferred first line treatment (NICE 2014; Goodwin 2002).
Being a Li+ responder runs in families (Grof et al. 1993) and this appears to have a specific genetic signature: response to Li+ is associated with genetic variations in glutamate decarboxylase-like protein 1 (GADL1) (Chen et al. 2014). These facts led some authors to postulate a distinct type of BD based on response to Li+ (Alda 2015).
It is important to point out that Li+ works best in patients exhibiting typical features of the disorder and that not every BD patient responds to Li+ (Tighe et al. 2011). In fact, only one third of BD patients are full responders (Kessing et al. 2016) and this response achieves better efficacy if treatment with Li+ is initiated early in life (Kessing et al. 2014).
Taking into consideration such a variability of responses to Li+, the Response to Lithium Network (R-LiNK) (Cousins et al. 2020) has been formed to identify which patients will respond to Li+ and also which ones will benefit from long-term treatment with this medication (see also (Pfennig et al. 2020).
This review focuses on neuroprotection, a term that is closely connected to the neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), or Huntington’s disease (HD) (Vajda 2002). These disorders are characterized by exhibiting a series of pathological changes in the brain that progress during the course of illness and ultimately lead to neuronal death, hence the term neuroprogression (Scearce-Levie et al. 2020).
Although research on neurodegenerative disorders has been traditionally reserved for pure neurological syndromes, there is now evidence that the concept of neuroprogression can also be applied to psychiatric entities such as the schizophrenia spectrum disorders (SCZ) or in BD (da Costa et al. 2016; Fries et al. 2012; Kapczinski et al. 2017). SCZ and BD are characterized by progressive structural brain changes, functional and cognitive decline, and a vulnerability to relapse (Dodd et al. 2013). Genome-Wide Association Studies (GWAS) have identified a panoply of genes showing that BD shares common genetic risk factors with schizophrenia and other mental disorders (Gordovez and McMahon 2020).
I will briefly detail possible mechanisms of action of Li+ in acute episodes of BD. Examples of neuroprogressive changes in BD brains will be given, and putative neuroprotective effects of Li+ in vitro as well as in vivo on these neuroprogressive processes will be discussed.
It is important to point out that the connection between Li+-induced neuroprotection in the human brain and the sustained efficacy of this agent in the long- term treatment of BD remains unresolved.
Methods
I undertook a PubMed search updated to March 2021 incorporating search terms such as bipolar, mania, hypomania, Li+, early intervention, prevention, neuroprogression and neuroprotection and using the Boolean operator AND.
An Example of Pharmacological-Induced Neuroprotection
Pharmacological neuroprotection is defined as the preventive effect of a medication on a neurodegenerative process that otherwise will inexorably progress in the absence of the medication.
Since their introduction in the 50’s, neuroleptic or antipsychotic medications have shown efficacy in the treatment of acute psychotic episodes and in the prevention of psychotic relapses (Leucht et al. 2009). However, long-term use of these medications raised concerns on damaging effects on the human brain (Gotzsche et al. 2015).
These effects include brain volume loss in monkeys (Dorph-Petersen et al. 2005) and humans (Haijma et al. 2013; Kubota et al. 2015) and upregulation of dopamine D2 receptors (Muller and Seeman 1977). The latter has been associated with loss of efficacy of neuroleptics over time (Chouinard and Jones 1980).
Nonetheless, there is no substantial evidence that long-term treatment with antipsychotics is deleterious to the brain (Goff et al. 2017). For a review on neuroprotective effects of second generation antipsychotic medications the reader is referred to a systematic review by Chen and Nasrallah (Chen and Nasrallah 2019).
We will apply these concepts of neuroprogression and neuroprotection when we discuss the effects of Li+ in the BD brain. But I will first discuss possible mechanisms of action of Li+ during a BD episode. This is because Li+’s well described molecular targets (see next section) are possibly the first link in a series of putative metabolic cascades regulated by this monovalent cation.
Possible Mechanisms of Action of Li+ in BD
The exact mechanism of action of Li+ in BD remains largely unknown. Li+ acts at molecular, cellular, and system levels, making a description of a single mechanism of action a daunting task (Alda 2015; Malhi et al. 2013; Haggarty et al. 2021).
The enzyme protein kinase C (PKC) is a molecular target for Li+ (Saxena et al. 2017; Zarate and Manji 2009). The term PKC designates in fact a family of enzymes expressed in mammalian brain structures involved in regulating mood (Wetsel et al. 1992; Alessenko et al. 1992). PKCs are involved in many metabolic cascades modifying protein function and organizing signal propagation within the cell (Rosse et al. 2010). PKC signaling modulates processes that could be operative in BD (Saxena et al. 2017).
PKC activity is inhibited by Li+ (Zarate and Manji 2009) both in vitro (Wang and Friedman 1989) and in vivo (Casebolt and Jope 1991) because it affects the translocation of the enzyme from the cytosol to the cell membrane, the region of the cell when it becomes active. Studies performed in patients with BD in the manic phase showed that Li+ inhibits PKC activity and its aforementioned translocation induced by serotonin (Friedman et al. 1993).
Support for the hypothesis of increased PKC signaling in BD was obtained from the use of the estrogen receptor modulator tamoxifen (TX). This drug crosses the blood–brain barrier (Carpenter et al. 2016) and inhibits PKC (Gunosewoyo et al. 2017). A proof-of-concept study showed the resolution of acute manic symptoms in BD patients treated with TX (Bebchuk et al. 2000). In the last 20 years many papers have been published regarding the antimanic properties of TX (Amrollahi et al. 2011; Zarate et al. 2007). The reader is referred to detailed reviews (Novick et al. 2020; Palacios et al. 2019; Talaei et al. 2016). To date, the FDA has not approved this promising drug for the treatment of manic episodes.
Li+ increases phosphorylation of glycogen synthase kinase 3ß (GSK3ß), an enzyme which is involved in a signaling pathway that modulates apoptosis and synaptic plasticity (Carter 2007). Increased phosphorylation of this enzyme leads to its inactivation and is associated with decreased excitotoxicity (Carter 2007; Haggarty et al. 2021). Furthermore, gene expression of GSK3ß is correlated with response to Li+ (Iwahashi et al. 2014).
Overexpression of GSK-3 correlates with neuronal degeneration (Lucas et al. 2001) and increases apoptosis (Bijur et al. 2000; King et al. 2001). The participation of GSK-3 in the therapeutic effects of Li+ is not well delineated. Instead, Li+ affects several cellular systems that use G proteins and second messenger systems (prominently the phosphatidylinositol (PI) cycle) that are involved in intracellular signaling cascades involved in cell functioning (Berridge 2016; Berridge et al. 1989).
The most widely accepted hypothesis regarding Li+ therapeutic efficacy in BD is the myo-inositol (henceforth inositol) depletion hypothesis. This posits that Li+ exerts its therapeutic effects by depleting inositol in specific areas of the brain. This stems from the pioneering work of Berridge who discovered signal transduction mechanisms involving the PI cycle (Berridge et al. 1989; Berridge 2016).
Of all the postulated targets for the effects of Li+ in the BD brain, I emphasize the PI cycle because it is a well-researched effect in vitro, and the postulated mechanism of action can be extended to human studies (Jope et al. 1996; Kato et al. 1991; Moore et al. 1999; Pacheco and Jope 1996). See also (Sharpley et al. 2020; Singh et al. 2013).
The Inositol Depletion Hypothesis
I will briefly review here the PI cycle and its special relevance to the mechanism of action of Li+ in BD. For further details, the reader is referred to comprehensive reviews (Berridge 2016; Berridge et al. 1989; Epand 2017; Harwood 2005). The PI cycle is the major pathway for the synthesis of this phospholipid (and its phosphorylated forms), located at the cytosolic side of eukaryotic cell membranes (Dickson and Hille 2019).
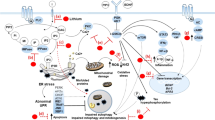
Specific first messengers (postulated neurotransmitters operative in a BD episode, especially GABA (Brady et al. 2013), dopamine (Berk et al. 2007) and glutamate (Beneyto et al. 2007; McCullumsmith et al. 2007) have been suggested as targeted by Li+. Upon binding to their specific membrane bound receptors, these messengers are linked via the Gq protein to phospholipase C (PLC) a membrane bound enzyme that catalyzes the breakdown of phosphatidylinositol-4,5-biphosphate (PIP2, a cell membrane phospholipid) into inositol-1-4-5-triphosphate (IP3) and diacylglycerol (DAG) (Figs. 1 and 2). The cycle proceeds clockwise for thermodynamical reasons and has as a distinctive characteristic: lipids are transferred between two membranes: the plasma membrane and the endoplasmic reticulum (ER) (Epand 2017) (see Fig. 1).

An extremely simplified diagram showing the phosphatidylinositol (PI) cycle as occurring in two separate membranes: the cell plasma membrane (rectangle) and the endoplasmic reticulum (solid oval). For a more comprehensive description of the cycle see recent reviews (Epand 2017; Harwood 2005). PI, a plasma membrane phospholipid located in the inner leaflet of the lipid bilayer, undergoes a series of phosphorylations to become Phosphatidylinositol-4,5-biphosphate (PIP2). The enzyme phospholipase C (PLC) becomes active when membrane bound and gives two products from its substrate PIP2: the lipid second messengers diacylglycerol (DAG) which remains membrane bound, and inositol-1-4-5 triphosphate (IP3) which is soluble and becomes associated with ER membranes. IP3 is sequentially dephosphorylated (not shown). The last enzyme in this sequence of dephosphorylations is inositol monophosphatase (IMPase), which has as substrate inositol monophosphate (IP), thus regenerating inositol. PI synthase (an ER enzyme, not shown) forms PI of inositol

Two important modulators of the PI cycle: (1) neurotransmitters (N) which bind to G protein coupled receptors (oblong shape) and activate phospholipase C (PLC) and (2) Li+ which inhibits (crossed circle) IMPase, thus depleting the cell from inositol (see text). The two boxed arrows and the two question marks represent still unknown downstream effects of IP3 and DAG that may relate to cellular mechanisms operative in a BD episode. Alternatively, these metabolic cascades may also be related to neuroprotective mechanisms. For more details see text
Both DAG and IP3 are lipid second messengers: DAG remains membrane bound and activates protein kinase C (PKC) which as described above, is involved in initiating signal transduction cascades. IP3 penetrates the cytosol and binds to a Ca2+ channel in the endoplasmic reticulum, promoting an efflux of Ca2+ from this organelle. This efflux of Ca2+ is also involved in initiating signal transduction cascades. It is hypothesized that the stimulus (neurotransmitter N in Fig. 2) magnifies the abovementioned metabolic cascades which are implied in the genesis and maintenance of a manic episode and that are in mood regulating areas of the brain.
A series of phosphatases dephosphorylate IP3. The enzyme inositol monophosphatase (IMPase) has inositol monophosphate (IP) as its substrate, regenerating inositol and ultimately PIP2 which allows the cycle to be perpetuated (Figs. 1 and 2).
Since the blood–brain barrier limits the availability of plasma inositol, inositol recycling as described above is critical for proper neuronal metabolism.
Among many other actions, Li+ inhibits IMPase, restricting recycling of inositol and depleting neurons of this molecule (Berridge et al. 1989). After prolonged administration of Li+, neurons have a reduced ability to re-synthesize PIP2 after its hydrolysis has been initiated by neurotransmitter receptor activation (Fig. 2). This is the postulated beneficial effect of Li+ in BD through its effects on IMPase.
In a very interesting paper by Williams et al. (Williams et al. 2002) the hypothesis of inositol depletion as a common mechanism of action for mood stabilizers was tested using cultured explants of sensory neurons from rat dorsal ganglia. The sensory neuron growth cone shows a phenomenon of collapse and expansion, two processes that appear to depend on the presence of inositol.
Drugs clinically used as mood stabilizers such as Li+, valproic acid, and carbamazepine were able to reduce the frequency of the collapse of neuron growth cones. This effect was reversed by inositol, indicating that inositol depletion is the common mechanism for the three drugs and that inositol phosphate was implicated in the response to the mood stabilizers (Williams et al. 2002).
Further evidence of inhibition of IMPase as an important mechanism of action of Li+ in BD came with the discovery of ebselen, a selenium-containing small molecule that has anti-inflammatory properties (Singh et al. 2013). Ebselen decreases inositol recycling in mice (Singh et al. 2013) and lowers the concentration of inositol in the anterior cingulate cortex (ACC) of humans as measured by magnetic resonance spectroscopy (Masaki et al. 2016; Singh et al. 2016). This suggests that the drug interacts with IMPase in the human brain.
Recently, a phase 2a randomized double-blind placebo-controlled trial where ebselen was added to anti-manic therapy (antipsychotics but not valproate, since the latter is known to inhibit inositol metabolism (Rosenberg 2007)) showed promising results on manic and hypomanic patients. Young Mania Rating Scale (YMRS) total scores were lower than when placebo was the add-on intervention (Sharpley et al. 2020).
As mentioned before, to date the involvement of the PI cycle in BD is well documented in vivo: this cycle is impaired in the brain of BD patients (Jope et al. 1996). The inositol transporter is overexpressed in BD and is downregulated by Li+ (Lubrich and van Calker 1999; van Calker and Belmaker 2000). Furthermore, there is an acute reduction of inositol induced by Li+ in the right frontal lobe of BD patients (Moore et al. 1999) albeit this acute reduction is not associated with a clinical response (Moore et al. 1999). This may explain why Li+ takes more that 2 or 3 weeks to exhibit efficacy, leaving open the hypothesis of a long-term effect of Li+ that does not depend on inositol depletion.
Although the emphasis of this review is on the inositol depletion hypothesis, there are also effects of Li+ in murine tissues that merit consideration since they open new avenues for human research. For example: dysregulation of mesolimbic dopamine neurotransmission has been implicated in BD, suggesting a hyper-dopaminergic state to explain the pathophysiology of the disorder (Ashok et al. 2017). In this respect, Ferrie et al. (Ferrie et al. 2008) showed that rats chronically treated with Li+ had decreased dopamine release in the nucleus accumbens. This effect was not due to increased auto receptor sensitivity or to decreased firing rate of dopaminergic neurons (Ferrie et al. 2008).
Norepinephrine is another catecholamine implied in the mechanism of action of Li+ (Kovács and Hernádi 2002). Li+ was iontophoretically applied to prefrontal cortical rat neurons located in the prelimbic cortical projection region. These neurons are targets for an ascending noradrenergic pathway. Single unit activity recorded from these neurons showed that Li+ suppressed discharge activity from this pathway, suggesting that it modulates the activity of noradrenergic neurons that target the prefrontal cortex (Kovács and Hernádi 2002).
Finally, an interesting effect of Li+ on Na+ and K+ channels that modulate cortical excitability was shown using brain slices of the mouse olfactory bulb. Treatment in vitro with lithium depolarized mitral cells and blocked action potential hyperpolarization (Butler-Munro et al. 2010). This opens a window into the interesting relationship between BD and epilepsy (Knott et al. 2015).
Neuroprogression in Bipolar Disorder
Accumulated evidence shows that mood episodes in BD have a deleterious effect on the brain, albeit to date no clear-cut neurodegenerative processes have been identified in BD brains (Frey et al. 2008; Sanches et al. 2008).
Compelling evidence from the Bipolar Disorder Working Group (Hibar et al. 2018) shows a cumulative degenerative effect of BD in the cortex of BD patients when compared to unaffected individuals, reinforcing the idea that neuroprogression is operative in the natural course of BD (Kapczinski et al. 2017).
The course of BD varies among patients (Passos et al. 2016). Along the years, especially in cases of treatment non-adherence, there is a worsening in cognitive capacity, refractoriness to pharmacotherapy, and shorter inter-episode intervals (Bauer et al. 2017; da Costa et al. 2016; Kapczinski et al. 2017).
This neuroprogression in the BD brain can be staged as much as staging is used in other branches of medicine (Fries et al. 2012; Grande et al. 2014; Kapczinski et al. 2014; van der Markt et al. 2020; Vieta et al. 2011). These stages are generally labeled as prodromal, early, middle and late and have been associated with specific biomarkers (Teixeira et al. 2019).
Genetic factors, a family history of BD, and sub-threshold mood and anxiety symptoms are characteristic of the prodromal phase. The other stages are associated with conspicuous biochemical changes: (1) activation of inflammatory pathways (Leboyer et al. 2012) (tumor necrosis factor alpha (TNFa) and interleukin 10 (IL10), (2) oxidative stress (glutathione reductase, glutathione N-transferase, (Andreazza et al. 2009); (3) changes in serum neurotrophic factors (prominently decrease of brain derived neurotrophic factor (BDNF) (Scola and Andreazza 2015), and (4) apoptotic mechanisms leading to neuronal death (Uribe and Wix 2012). This is correlated with reduced cortical thickness in the brain, as shown in the study by Hibar et al. (Hibar et al. 2018). The following section deals with this type of evidence at the neuroanatomical level.
Evidence for Neuroanatomical Changes in BD Brains
Imaging studies using magnetic resonance imaging (MRI) and neuropathological examination of BD brains show decreased volumes in hippocampus, amygdala, thalamus, and reduction of cortical thickness (Harrison et al. 2020; Hibar et al. 2018; Hibar et al. 2016; Kempton et al. 2011). Lyoo et al. (2006) reported decreased thickness in the dorsolateral prefrontal cortex (DLPFC) in BD patients. The structural changes in the amygdala may be related to progression of BD (Blumberg et al. 2005).
Diffusion tensor analysis shows changes in white matter tracts connecting the anterior cingulate cortex (ACC) with the amygdala and the hippocampus, and the frontal lobe with the amygdala, the hippocampus, the thalamus, and the cingulate gyrus. This suggests that BD patients have compromised connections between frontal-subcortical and prefrontal lobe-limbic brain regions (Nortje et al. 2013; Wise et al. 2016). A recently published systematic review confirmed that white matter abnormalities seen in BD patients could be prevented by treatment with Li+ (Espanhol and Vieira-Coelho 2021).
An MRI analysis of cortical volume, thickness, and surface area suggested a difference between patients diagnosed with either type of BD as specified in DSM5 (BD I or BD II) (Abe et al. 2016). This type of study is particularly relevant since both diagnoses present differently in clinical practice: while the manic episodes of BD I can be severe, persons with BD II do not show frank mania. Instead, they are depressed for longer periods of time and do not report their hypomanic episodes as often.
Most of the available studies on BD were performed on BD I and measured cortical volume. Abe et al. used MRI to analyze abnormalities in BD I and BD II patients and healthy controls and simultaneously measured cortical volume, thickness, and surface area to describe the neurobiology underlying these two types of BD (Abe et al. 2016). Decreased cortical volume, thickness, and surface area were decreased in frontal, temporal, and medial occipital brain regions in both BD I and BD II patients as compared to controls. Interestingly, only BD I patients showed low cortical volume and thickness in temporal and medial prefrontal areas as compared to BD II patients (Abe et al. 2016). This study provides evidence that both types of BD have real neurobiological differences and could serve as a template to develop biomarkers for this disorder.
A recent systematic review and meta-analysis on the neuropathology of BD (Harrison et al. 2020) shows that there is not a signature pathological lesion that distinguishes BD from other brain disorders. Instead, there is decreased cortical thickness in the subgenual anterior cingulate cortex (ACC), along with reduced neuronal density in some amygdala nuclei and decreased density of calbindin-positive neurons in the prefrontal cortex (Harrison et al. 2020).
Postmortem human brain studies have yielded interesting results. There is dendritic spine loss in the dorsolateral prefrontal cortex of BD brains (Konopaske et al. 2014) and reduction in the expression profiles of cortical fast-spiking parvalbumin interneurons (Toker et al. 2018) and parvalbumin- and somatostatin-positive interneurons in the parahippocampal region (Wang et al. 2011).
As mentioned before, the course of illness in BD is variable, and progression is not always demonstrable in BD. From a practical point of view, the number of manic episodes is to date the best predictor of neuroprogression in BD (Passos et al. 2016).
Biomarkers can be Used to Track Neuroprotection Effected by Li+
Neuroprotective effects of Li+ have been demonstrated in vitro and possibly in vivo (Alda 2015; Manji et al. 2000; Abe et al. 2020; Hibar et al. 2018). These can be evidenced using specific biomarkers such as: (1) imaging techniques that show morphological changes in BD brains, (2) proton magnetic resonance spectroscopy measuring metabolites such as N-acetylaspartate (NAA) and glutamine plus glutamate (GLx) that provide evidence for cytotoxic processes; and (3) measuring serum levels of neurotrophins (especially brain-derived neurotrophic factor (BDNF)).
Morphological Changes
Human grey matter is increased in patients treated with Li+ (Moore et al. 2000). In this respect, the reader is referred to a meta-analysis by Sun et al. (Sun et al. 2018).
BD patients on Li+ show greater grey matter density in the cingulate and paralimbic cortices as compared to healthy controls (Bearden et al. 2007) and an increase in subgenual ACC, hippocampus, insula, and amygdala (Germana et al. 2010).
A study performed on patients that responded to Li+ showed an increase in prefrontal and subgenual grey matter volume (Moore et al. 2009). A longitudinal study (Lyoo et al. 2010) showed that this Li+-induced increase in human brain grey matter can be identified as an anatomical substrate of treatment response in BD (Lyoo et al. 2010).
The hippocampus of BD patients treated with Li+ for 2 years showed volumes that did not differ from those of normal persons. In contrast, BD patients with limited exposure to Li+ had lower hippocampal volumes as compared to healthy controls (Hajek et al. 2014, 2012b). Nevertheless, this effect of Li+ on hippocampal volume is not correlated with the ability of Li+ to prevent bipolar episodes (Hajek et al. 2014).
Interestingly, patients with BD which were naïve to medication, and were acutely treated with Li+ (1–8 weeks) showed a volumetric increase of the hippocampus, especially in its head, when compared to healthy controls (Yucel et al. 2008). The same effects of Li+ on the hippocampus were demonstrated using long-term (2–4 years) treatment with Li+ (Yucel et al. 2007).
Compelling evidence regarding protective effects of Li+ in BD brains was obtained by the ENIGMA working group study performed on 6503 BD patients (Hibar et al. 2018). Patients on Li+ (and not on antiseizure or neuroleptic medications Hafeman et al. 2012; Hibar et al. 2018)) had increased cortical thickness when compared with patients not treated with Li+.
Measuring Brain Metabolites
Proton magnetic resonance spectroscopy (MRS) allows for the quantitative noninvasive assessment of regional brain biochemistry (Novotny et al. 1998).
N-acetylaspartate (NAA) is a unique metabolite found in the vertebrate brain and second only in concentration to glutamate. It has been used in proton magnetic resonance spectroscopy (Baslow 2003), and it was identified in neurons (Simmons et al. 1991) as a putative marker of neuronal integrity (Baslow 2003).
Hajek et al. (Hajek et al. 2012a) showed that prefrontal NAA levels in Li+-treated patients are comparable to those of healthy controls. This supports the notion for neuroprotective effects of Li+ on prefrontal cortex in patients with BD (Hajek et al. 2012a).
Abnormal glutamatergic transmission in the frontal lobe has also been implicated in the BD brain. Proton magnetic resonance spectroscopy ((1)H-MRS) studies have reported increased levels of combined glutamate and glutamine ("Glx"), which have been linked to impairments in N-methyl-D-aspartate (NMDA) receptor function (Chitty et al. 2013, 2015). Finally, there is a probable direct effect of Li+ on NMDA receptors (Amiri et al. 2020) that merits further inquiry.
Measuring Neurotrophic Factors
There are many in vitro pieces of evidence indicating that Li+ regulates brain growth factors (Hashimoto et al. 2004) and programmed cell death such as apoptosis (Dodd et al. 2013) and autophagy (Motoi et al. 2014). Li+ decreases apoptosis through inhibition of glycogen synthase kinase 3β activity (Klein and Melton 1996).
The BNDF gene (Maisonpierre et al. 1991) is a risk locus for the development of BD (Neves-Pereira et al. 2002; Sklar et al. 2002). Additionally, chronic administration of Li+ increases BDNF expression in rat brain (Fukumoto et al. 2001).
Neurotrophins have been identified as participating in various stages of BD: BDNF, insulin-like growth factor (IGF-1) and vascular endothelial growth factor (VEGF) are present at different stages of the disorder, suggesting the existence of BD subtypes (Scola and Andreazza 2015). Interestingly, BDNF levels are reduced in the serum of bipolar patients while experiencing a manic episode (Machado-Vieira et al. 2007).
Li+ differentially accumulates in brain regions known to be neurogenic (for example, the hippocampus). When Li+ is administered for 28 days to juvenile mice, cell proliferation (but not neurogenesis) increases in their hippocampus. This was determined by the novel imaging procedure Time-of-Flight Secondary Ion Mass Spectrometry (Zanni et al. 2017). Importantly, steady state serum concentrations of Li+ were analogous to those clinically relevant for treating BD (around 1.2 mM).
A discussion of the mechanisms targeted by Li+ that could be implicated in neuroprotection are detailed in a review by Niciu et al. (Niciu et al. 2013). What is still unknown is how these effects are correlated with mood stabilization and with the maintenance of Li+ ‘s efficacy in the long-term treatment of BD.
Prophylactic Effects of Li+: When to Treat
Early detection of BD and identifying its proper treatment mirrors the dilemmas emerging in treating SCZ patients. The most accepted model for SCZ is the neurodevelopmental hypothesis. This posits that interactions between multiple genes initiate a series of neuropathological events during gestation which progress into adolescence and adulthood, and that are influenced by environmental factors (Fatemi and Folsom 2009; Murray and Lewis 1987; Rapoport et al. 2012).
The impetus for early detection and early intervention in psychosis (Fusar-Poli et al. 2020; Leopold et al. 2020) offers hope for treating this serious mental condition and serves as a template for other mental disorders such as BD.
Some authors suggest the existence of neurodevelopmental changes in BD. These may be expressed as premorbid neuro-behavioral changes demonstrated by neuroimaging differences comparing the children of BD parents and age matched healthy controls (Sanches et al. 2008). An abnormal maturation of the brain structures involved in the regulation of affect has been postulated to explain the pathophysiology of BD (Sanches et al. 2008). However, the case for neurodevelopmental factors leading to neuroprogression in BD is not as clear as it is the case for SCZ (Valli et al. 2019).
Having discussed the neuroprotective effects of Li+ in BD patients, the question of a truly preventive or prophylactic use of Li+ surges spontaneously. It is important to identify in the child and adolescent population and as accurately as possible, who will develop BD and who will respond to pharmacological treatment (Cousins et al. 2020). As mentioned before, starting Li+ early in life vs. starting late shows that early treatment improves BD outcomes (Kessing et al. 2016).
It remains a crucial clinical question how early psychiatrists should treat BD with Li+ and the associated ethical concerns need to be examined, as pointed in a paper by Ratheesh et al. (2017).
From a clinical standpoint, there are some indicators that suggest a diagnosis of BD: earlier age of onset (Lish et al. 1994), family history of bipolar disorder (Bowden 2005; Hirschfeld et al. 2003; Manning et al. 1998) or family history of lithium responsiveness in a first-degree relative (Manning et al. 1998). Multiple failed antidepressant trials, rapidly occurring episodes of recurrent depression, and history of prompt antidepressant response followed by sudden decline in response, have also been reported to suggest bipolarity (Perlis 2005).
Despite these clinical indicators, several factors conspire against an accurate diagnosis: (1) a young person may spend most of his/her life in depression (Kupka et al. 2007) and experience late onset of mania (Bolton et al. 2020; Dols and Beekman 2018); (2) differentiating between classical unipolar depression and bipolar depression still remains a dauting task (Cuellar et al. 2005) and because of this, (3) there are delays in diagnosis (Fritz et al. 2017).
Finally, comorbidities such as medical non-psychiatric conditions (Crump et al. 2013), personality disorders (Fan and Hassell 2008) and/or illicit drug use (Levin and Hennessy 2004; Sherwood Brown et al. 2001) influence diagnostic and treatment decisions.
It is expected that in a nearby future, precision medicine approaches will improve diagnosis and expand treatment options (Cousins et al. 2020; Pfennig et al. 2020). This will certainly contribute to ameliorating the medical and economic burden created by this devastating mood disorder.
Abbreviations
- AD:
-
Alzheimer’s disease
- BD:
-
Bipolar disorder
- BD I:
-
Bipolar disorder I
- BD II:
-
Bipolar disorder II
- BDNF:
-
Brain-derived neurotrophic factor
- DAG:
-
Diacylglycerol
- HD:
-
Huntington’s disease
- IMPase:
-
Inositol monophosphatase
- IP:
-
Inositol monophosphate
- IP3 :
-
Inositol-1-4-5 triphosphate
- Li+ :
-
Lithium
- MRI:
-
Magnetic resonance imaging
- PD:
-
Parkinson’s disease
- PI:
-
Phosphatidylinositol
- PIP2 :
-
Phosphatidylinositol-4,5-biphosphate
- PKC:
-
Protein kinase C
- SCZ:
-
Schizophrenia/SCZ spectrum disorders
- GSK3:
-
Glycogen-synthase-kinase 3β
References
Abe C, Ekman CJ, Sellgren C, Petrovic P, Ingvar M, Landen M (2016) Cortical thickness, volume and surface area in patients with bipolar disorder types I and II. J Psychiatry Neurosci 41(4):240–250. https://doi.org/10.1503/jpn.150093
Abe C, Liberg B, Song J, Bergen SE, Petrovic P, Ekman CJ, Sellgren CM, Ingvar M, Landen M (2020) Longitudinal cortical thickness changes in bipolar disorder and the relationship to genetic risk, mania, and lithium use. Biol Psychiatry 87(3):271–281. https://doi.org/10.1016/j.biopsych.2019.08.015
Alda M (2015) Lithium in the treatment of bipolar disorder: pharmacology and pharmacogenetics. Mol Psychiatry 20(6):661–670. https://doi.org/10.1038/mp.2015.4
Alessenko A, Khan WA, Wetsel WC, Hannun YA (1992) Selective changes in protein kinase C isoenzymes in rat liver nuclei during liver regeneration. Biochem Biophys Res Commun 182(3):1333–1339. https://doi.org/10.1016/0006-291x(92)91879-u
Amiri S, Jafari-Sabet M, Keyhanfar F, Falak R, Shabani M, Rezayof A (2020) Hippocampal and prefrontal cortical NMDA receptors mediate the interactive effects of olanzapine and lithium in memory retention in rats: the involvement of CAMKII-CREB signaling pathways. Psychopharmacology 237(5):1383–1396. https://doi.org/10.1007/s00213-020-05465-4
Amrollahi Z, Rezaei F, Salehi B, Modabbernia AH, Maroufi A, Esfandiari GR, Naderi M, Ghebleh F, Ahmadi-Abhari SA, Sadeghi M, Tabrizi M, Akhondzadeh S (2011) Double-blind, randomized, placebo-controlled 6-week study on the efficacy and safety of the tamoxifen adjunctive to lithium in acute bipolar mania. J Affect Disord 129(1–3):327–331. https://doi.org/10.1016/j.jad.2010.08.015
Andreazza AC, Kapczinski F, Kauer-Sant’Anna M, Walz JC, Bond DJ, Gonçalves CA, Young LT, Yatham LN (2009) 3-Nitrotyrosine and glutathione antioxidant system in patients in the early and late stages of bipolar disorder. J Psychiatry Neurosci 34(4):263–271
Ashok AH, Marques TR, Jauhar S, Nour MM, Goodwin GM, Young AH, Howes OD (2017) The dopamine hypothesis of bipolar affective disorder: the state of the art and implications for treatment. Mol Psychiatry 22(5):666–679. https://doi.org/10.1038/mp.2017.16
American Psychiatric Association. DSM-5 Task Force (2013) Diagnostic and statistical manual of mental disorders: DSM-5, 5th edn. American Psychiatric Association, Washington, DC
Baslow MH (2003) N-acetylaspartate in the vertebrate brain: metabolism and function. Neurochem Res 28(6):941–953. https://doi.org/10.1023/a:1023250721185
Bauer IE, Soares JC, Selek S, Meyer TD (2017) The link between refractoriness and neuroprogression in treatment-resistant bipolar disorder. Mod Trends Pharmacopsychiatry 31:10–26. https://doi.org/10.1159/000470803
Bearden CE, Thompson PM, Dalwani M, Hayashi KM, Lee AD, Nicoletti M, Trakhtenbroit M, Glahn DC, Brambilla P, Sassi RB, Mallinger AG, Frank E, Kupfer DJ, Soares JC (2007) Greater cortical gray matter density in lithium-treated patients with bipolar disorder. Biol Psychiatry 62(1):7–16. https://doi.org/10.1016/j.biopsych.2006.10.027
Bebchuk JM, Arfken CL, Dolan-Manji S, Murphy J, Hasanat K, Manji HK (2000) A preliminary investigation of a protein kinase C inhibitor in the treatment of acute mania. Arch Gen Psychiatry 57(1):95–97. https://doi.org/10.1001/archpsyc.57.1.95
Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH (2007) Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 32(9):1888–1902. https://doi.org/10.1038/sj.npp.1301312
Berk M, Dodd S, Kauer-Sant’anna M, Malhi GS, Bourin M, Kapczinski F, Norman T (2007) Dopamine dysregulation syndrome: implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr Scand Suppl 434:41–49. https://doi.org/10.1111/j.1600-0447.2007.01058.x
Berridge MJ (2016) The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol Rev 96(4):1261–1296. https://doi.org/10.1152/physrev.00006.2016
Berridge MJ, Downes CP, Hanley MR (1989) Neural and developmental actions of lithium—a unifying hypothesis. Cell 59(3):411–419. https://doi.org/10.1016/0092-8674(89)90026-3
Bijur GN, De Sarno P, Jope RS (2000) Glycogen synthase kinase-3beta facilitates staurosporine- and heat shock-induced apoptosis. Protection by lithium. J Biol Chem 275(11):7583–7590. https://doi.org/10.1074/jbc.275.11.7583
Blumberg HP, Fredericks C, Wang F, Kalmar JH, Spencer L, Papademetris X, Pittman B, Martin A, Peterson BS, Fulbright RK, Krystal JH (2005) Preliminary evidence for persistent abnormalities in amygdala volumes in adolescents and young adults with bipolar disorder. Bipolar Disord 7(6):570–576. https://doi.org/10.1111/j.1399-5618.2005.00264.x
Bolton S, Warner J, Harriss E, Geddes J, Saunders KEA (2020) Bipolar disorder: trimodal age-at-onset distribution. Bipolar Disord. https://doi.org/10.1111/bdi.13016
Bowden CL (2005) A different depression: clinical distinctions between bipolar and unipolar depression. J Affect Disord 84(2–3):117–125. https://doi.org/10.1016/S0165-0327(03)00194-0
Brady RO Jr, McCarthy JM, Prescot AP, Jensen JE, Cooper AJ, Cohen BM, Renshaw PF, Ongur D (2013) Brain gamma-aminobutyric acid (GABA) abnormalities in bipolar disorder. Bipolar Disord 15(4):434–439. https://doi.org/10.1111/bdi.12074
Butler-Munro C, Coddington EJ, Shirley CH, Heyward PM (2010) Lithium modulates cortical excitability in vitro. Brain Res 1352:50–60. https://doi.org/10.1016/j.brainres.2010.07.021
Carpenter C, Sorenson RJ, Jin Y, Klossowski S, Cierpicki T, Gnegy M, Showalter HD (2016) Design and synthesis of triarylacrylonitrile analogues of tamoxifen with improved binding selectivity to protein kinase C. Bioorg Med Chem 24(21):5495–5504. https://doi.org/10.1016/j.bmc.2016.09.002
Carter CJ (2007) Multiple genes and factors associated with bipolar disorder converge on growth factor and stress activated kinase pathways controlling translation initiation: implications for oligodendrocyte viability. Neurochem Int 50(3):461–490. https://doi.org/10.1016/j.neuint.2006.11.009
Casebolt TL, Jope RS (1991) Effects of chronic lithium treatment on protein kinase C and cyclic AMP-dependent protein phosphorylation. Biol Psychiatry 29(3):233–243. https://doi.org/10.1016/0006-3223(91)91285-y
Chen AT, Nasrallah HA (2019) Neuroprotective effects of the second generation antipsychotics. Schizophr Res 208:1–7. https://doi.org/10.1016/j.schres.2019.04.009
Chen CH, Lee CS, Lee MT, Ouyang WC, Chen CC, Chong MY, Wu JY, Tan HK, Lee YC, Chuo LJ, Chiu NY, Tsang HY, Chang TJ, Lung FW, Chiu CH, Chang CH, Chen YS, Hou YM, Chen CC, Lai TJ, Tung CL, Chen CY, Lane HY, Su TP, Feng J, Lin JJ, Chang CJ, Teng PR, Liu CY, Chen CK, Liu IC, Chen JJ, Lu T, Fan CC, Wu CK, Li CF, Wang KH, Wu LS, Peng HL, Chang CP, Lu LS, Chen YT, Cheng AT, Taiwan Bipolar C (2014) Variant GADL1 and response to lithium therapy in bipolar I disorder. N Engl J Med 370(2):119–128. https://doi.org/10.1056/NEJMoa1212444
Chitty KM, Lagopoulos J, Lee RS, Hickie IB, Hermens DF (2013) A systematic review and meta-analysis of proton magnetic resonance spectroscopy and mismatch negativity in bipolar disorder. Eur Neuropsychopharmacol 23(11):1348–1363. https://doi.org/10.1016/j.euroneuro.2013.07.007
Chitty KM, Lagopoulos J, Hickie IB, Hermens DF (2015) Hippocampal glutamatergic/NMDA receptor functioning in bipolar disorder: a study combining mismatch negativity and proton magnetic resonance spectroscopy. Psychiatry Res 233(2):88–94. https://doi.org/10.1016/j.pscychresns.2015.05.002
Chouinard G, Jones BD (1980) Neuroleptic-induced supersensitivity psychosis: clinical and pharmacologic characteristics. Am J Psychiatry 137(1):16–21. https://doi.org/10.1176/ajp.137.1.16
Cousins DA, Squarcina L, Boumezbeur F, Young AH, Bellivier F (2020) Lithium: past, present, and future. Lancet Psychiatry 7(3):222–224. https://doi.org/10.1016/S2215-0366(19)30365-7
Crump C, Sundquist K, Winkleby MA, Sundquist J (2013) Comorbidities and mortality in bipolar disorder: a Swedish national cohort study. JAMA Psychiatry 70(9):931–939. https://doi.org/10.1001/jamapsychiatry.2013.1394
Cuellar AK, Johnson SL, Winters R (2005) Distinctions between bipolar and unipolar depression. Clin Psychol Rev 25(3):307–339. https://doi.org/10.1016/j.cpr.2004.12.002
Curran G, Ravindran A (2014) Lithium for bipolar disorder: a review of the recent literature. Expert Rev Neurother 14(9):1079–1098. https://doi.org/10.1586/14737175.2014.947965
da Costa SC, Passos IC, Lowri C, Soares JC, Kapczinski F (2016) Refractory bipolar disorder and neuroprogression. Prog Neuropsychopharmacol Biol Psychiatry 70:103–110. https://doi.org/10.1016/j.pnpbp.2015.09.005
Dickson EJ, Hille B (2019) Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem J 476(1):1–23. https://doi.org/10.1042/bcj20180022
Dodd S, Maes M, Anderson G, Dean OM, Moylan S, Berk M (2013) Putative neuroprotective agents in neuropsychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry 42:135–145. https://doi.org/10.1016/j.pnpbp.2012.11.007
Dols A, Beekman A (2018) Older age bipolar disorder. Psychiatr Clin N Am 41(1):95–110. https://doi.org/10.1016/j.psc.2017.10.008
Dorph-Petersen KA, Pierri JN, Perel JM, Sun Z, Sampson AR, Lewis DA (2005) The influence of chronic exposure to antipsychotic medications on brain size before and after tissue fixation: a comparison of haloperidol and olanzapine in macaque monkeys. Neuropsychopharmacology 30(9):1649–1661. https://doi.org/10.1038/sj.npp.1300710
Epand RM (2017) Features of the phosphatidylinositol cycle and its role in signal transduction. J Membr Biol 250(4):353–366. https://doi.org/10.1007/s00232-016-9909-y
Espanhol JCL, Vieira-Coelho MA (2021) Effects of lithium use on the white matter of patients with bipolar disorder—a systematic review. Nord J Psychiatry. https://doi.org/10.1080/08039488.2021.1921264
Fan AH, Hassell J (2008) Bipolar disorder and comorbid personality psychopathology: a review of the literature. J Clin Psychiatry 69(11):1794–1803. https://doi.org/10.4088/jcp.v69n1115
Fatemi SH, Folsom TD (2009) The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull 35(3):528–548. https://doi.org/10.1093/schbul/sbn187
Ferrie LJ, Gartside SE, Martin KM, Young AH, McQuade R (2008) Effect of chronic lithium treatment on D2/3 autoreceptor regulation of dopaminergic function in the rat. Pharmacol Biochem Behav 90(2):218–225. https://doi.org/10.1016/j.pbb.2007.10.013
Frey BN, Zunta-Soares GB, Caetano SC, Nicoletti MA, Hatch JP, Brambilla P, Mallinger AG, Soares JC (2008) Illness duration and total brain gray matter in bipolar disorder: evidence for neurodegeneration? Eur Neuropsychopharmacol 18(10):717–722. https://doi.org/10.1016/j.euroneuro.2008.04.015
Friedman E, Hoau Yan W, Levinson D, Connell TA, Singh H (1993) Altered platelet protein kinase C activity in bipolar affective disorder, manic episode. Biol Psychiatry 33(7):520–525. https://doi.org/10.1016/0006-3223(93)90006-y
Fries GR, Pfaffenseller B, Stertz L, Paz AV, Dargél AA, Kunz M, Kapczinski F (2012) Staging and neuroprogression in bipolar disorder. Curr Psychiatry Rep 14(6):667–675. https://doi.org/10.1007/s11920-012-0319-2
Fritz K, Russell AMT, Allwang C, Kuiper S, Lampe L, Malhi GS (2017) Is a delay in the diagnosis of bipolar disorder inevitable? Bipolar Disord 19(5):396–400. https://doi.org/10.1111/bdi.12499
Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S (2001) Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology 158(1):100–106. https://doi.org/10.1007/s002130100871
Fusar-Poli P, Salazar de Pablo G, Correll CU, Meyer-Lindenberg A, Millan MJ, Borgwardt S, Galderisi S, Bechdolf A, Pfennig A, Kessing LV, van Amelsvoort T, Nieman DH, Domschke K, Krebs MO, Koutsouleris N, McGuire P, Do KQ, Arango C (2020) Prevention of psychosis: advances in detection, prognosis, and intervention. JAMA Psychiatry 77(7):755–765. https://doi.org/10.1001/jamapsychiatry.2019.4779
Geddes JR, Miklowitz DJ (2013) Treatment of bipolar disorder. Lancet 381(9878):1672–1682. https://doi.org/10.1016/S0140-6736(13)60857-0
Geddes JR, Burgess S, Hawton K, Jamison K, Goodwin GM (2004) Long-term lithium therapy for bipolar disorder: systematic review and meta-analysis of randomized controlled trials. Am J Psychiatry 161(2):217–222. https://doi.org/10.1176/appi.ajp.161.2.217
Germana C, Kempton MJ, Sarnicola A, Christodoulou T, Haldane M, Hadjulis M, Girardi P, Tatarelli R, Frangou S (2010) The effects of lithium and anticonvulsants on brain structure in bipolar disorder. Acta Psychiatr Scand 122(6):481–487. https://doi.org/10.1111/j.1600-0447.2010.01582.x
Goff DC, Falkai P, Fleischhacker WW, Girgis RR, Kahn RM, Uchida H, Zhao J, Lieberman JA (2017) The long-term effects of antipsychotic medication on clinical course in Schizophrenia. Am J Psychiatry 174(9):840–849. https://doi.org/10.1176/appi.ajp.2017.16091016
Goodwin FK (2002) Rationale for long-term treatment of bipolar disorder and evidence for long-term lithium treatment. J Clin Psychiatry 63(Suppl 10):5–12
Goodwin FK, Jamison KR (1990) Manic-depressive illness. Oxford University Press, New York
Gordovez FJA, McMahon FJ (2020) The genetics of bipolar disorder. Mol Psychiatry 25(3):544–559. https://doi.org/10.1038/s41380-019-0634-7
Gotzsche PC, Young AH, Crace J (2015) Does long term use of psychiatric drugs cause more harm than good? BMJ 350:h2435. https://doi.org/10.1136/bmj.h2435
Graham RK, Tavella G, Parker GB (2018) Is there consensus across international evidence-based guidelines for the psychotropic drug management of bipolar disorder during the perinatal period? J Affect Disord 228:216–221. https://doi.org/10.1016/j.jad.2017.12.022
Grande I, Magalhães PV, Chendo I, Stertz L, Panizutti B, Colpo GD, Rosa AR, Gama CS, Kapczinski F, Vieta E (2014) Staging bipolar disorder: clinical, biochemical, and functional correlates. Acta Psychiatr Scand 129(6):437–444. https://doi.org/10.1111/acps.12268
Grof P, Alda M, Grof E, Fox D, Cameron P (1993) The challenge of predicting response to stabilising lithium treatment. The importance of patient selection. Br J Psychiatry Suppl 21:16–19
Gunosewoyo H, Yu L, Munoz L, Kassiou M (2017) Kinase targets in CNS drug discovery. Future Med Chem 9(3):303–314. https://doi.org/10.4155/fmc-2016-0214
Hafeman DM, Chang KD, Garrett AS, Sanders EM, Phillips ML (2012) Effects of medication on neuroimaging findings in bipolar disorder: an updated review. Bipolar Disord 14(4):375–410. https://doi.org/10.1111/j.1399-5618.2012.01023.x
Hafeman DM, Rooks B, Merranko J, Liao F, Gill MK, Goldstein TR, Diler R, Ryan N, Goldstein BI, Axelson DA, Strober M, Keller M, Hunt J, Hower H, Weinstock LM, Yen S, Birmaher B (2019) Lithium versus other mood-stabilizing medications in a longitudinal study of youth diagnosed with Bipolar. J Am Acad Child Adolesc Psychiatry 59(10):1146–1555. https://doi.org/10.1016/j.jaac.2019.06.013
Haggarty SJ, Karmacharya R, Perlis RH (2021) Advances toward precision medicine for bipolar disorder: mechanisms & molecules. Mol Psychiatry 26(1):168–185. https://doi.org/10.1038/s41380-020-0831-4
Haijma SV, Van Haren N, Cahn W, Koolschijn PC, Hulshoff Pol HE, Kahn RS (2013) Brain volumes in schizophrenia: a meta-analysis in over 18 000 subjects. Schizophr Bull 39(5):1129–1138. https://doi.org/10.1093/schbul/sbs118
Hajek T, Bauer M, Pfennig A, Cullis J, Ploch J, O’Donovan C, Bohner G, Klingebiel R, Young LT, Macqueen GM, Alda M (2012a) Large positive effect of lithium on prefrontal cortex N-acetylaspartate in patients with bipolar disorder: 2-centre study. J Psychiatry Neurosci 37(3):185–192. https://doi.org/10.1503/jpn.110097
Hajek T, Cullis J, Novak T, Kopecek M, Hoschl C, Blagdon R, O’Donovan C, Bauer M, Young LT, Macqueen G, Alda M (2012b) Hippocampal volumes in bipolar disorders: opposing effects of illness burden and lithium treatment. Bipolar Disord 14(3):261–270. https://doi.org/10.1111/j.1399-5618.2012.01013.x
Hajek T, Bauer M, Simhandl C, Rybakowski J, O’Donovan C, Pfennig A, Konig B, Suwalska A, Yucel K, Uher R, Young LT, MacQueen G, Alda M (2014) Neuroprotective effect of lithium on hippocampal volumes in bipolar disorder independent of long-term treatment response. Psychol Med 44(3):507–517. https://doi.org/10.1017/S0033291713001165
Harrison PJ, Colbourne L, Harrison CH (2020) The neuropathology of bipolar disorder: systematic review and meta-analysis. Mol Psychiatry 25(8):1787–1808. https://doi.org/10.1038/s41380-018-0213-3
Harwood AJ (2005) Lithium and bipolar mood disorder: the inositol-depletion hypothesis revisited. Mol Psychiatry 10(1):117–126. https://doi.org/10.1038/sj.mp.4001618
Hashimoto K, Shimizu E, Iyo M (2004) Critical role of brain-derived neurotrophic factor in mood disorders. Brain Res Brain Res Rev 45(2):104–114. https://doi.org/10.1016/j.brainresrev.2004.02.003
Hibar DP, Westlye LT, van Erp TG, Rasmussen J, Leonardo CD, Faskowitz J, Haukvik UK, Hartberg CB, Doan NT, Agartz I, Dale AM, Gruber O, Kramer B, Trost S, Liberg B, Abe C, Ekman CJ, Ingvar M, Landen M, Fears SC, Freimer NB, Bearden CE, Costa Rica/Colombia Consortium for Genetic Investigation of Bipolar E, Sprooten E, Glahn DC, Pearlson GD, Emsell L, Kenney J, Scanlon C, McDonald C, Cannon DM, Almeida J, Versace A, Caseras X, Lawrence NS, Phillips ML, Dima D, Delvecchio G, Frangou S, Satterthwaite TD, Wolf D, Houenou J, Henry C, Malt UF, Boen E, Elvsashagen T, Young AH, Lloyd AJ, Goodwin GM, Mackay CE, Bourne C, Bilderbeck A, Abramovic L, Boks MP, van Haren NE, Ophoff RA, Kahn RS, Bauer M, Pfennig A, Alda M, Hajek T, Mwangi B, Soares JC, Nickson T, Dimitrova R, Sussmann JE, Hagenaars S, Whalley HC, McIntosh AM, Thompson PM, Andreassen OA (2016) Subcortical volumetric abnormalities in bipolar disorder. Mol Psychiatry 21(12):1710–1716. https://doi.org/10.1038/mp.2015.227
Hibar DP, Westlye LT, Doan NT, Jahanshad N, Cheung JW, Ching CRK, Versace A, Bilderbeck AC, Uhlmann A, Mwangi B, Kramer B, Overs B, Hartberg CB, Abe C, Dima D, Grotegerd D, Sprooten E, Boen E, Jimenez E, Howells FM, Delvecchio G, Temmingh H, Starke J, Almeida JRC, Goikolea JM, Houenou J, Beard LM, Rauer L, Abramovic L, Bonnin M, Ponteduro MF, Keil M, Rive MM, Yao N, Yalin N, Najt P, Rosa PG, Redlich R, Trost S, Hagenaars S, Fears SC, Alonso-Lana S, van Erp TGM, Nickson T, Chaim-Avancini TM, Meier TB, Elvsashagen T, Haukvik UK, Lee WH, Schene AH, Lloyd AJ, Young AH, Nugent A, Dale AM, Pfennig A, McIntosh AM, Lafer B, Baune BT, Ekman CJ, Zarate CA, Bearden CE, Henry C, Simhandl C, McDonald C, Bourne C, Stein DJ, Wolf DH, Cannon DM, Glahn DC, Veltman DJ, Pomarol-Clotet E, Vieta E, Canales-Rodriguez EJ, Nery FG, Duran FLS, Busatto GF, Roberts G, Pearlson GD, Goodwin GM, Kugel H, Whalley HC, Ruhe HG, Soares JC, Fullerton JM, Rybakowski JK, Savitz J, Chaim KT, Fatjo-Vilas M, Soeiro-de-Souza MG, Boks MP, Zanetti MV, Otaduy MCG, Schaufelberger MS, Alda M, Ingvar M, Phillips ML, Kempton MJ, Bauer M, Landen M, Lawrence NS, van Haren NEM, Horn NR, Freimer NB, Gruber O, Schofield PR, Mitchell PB, Kahn RS, Lenroot R, Machado-Vieira R, Ophoff RA, Sarro S, Frangou S, Satterthwaite TD, Hajek T, Dannlowski U, Malt UF, Arolt V, Gattaz WF, Drevets WC, Caseras X, Agartz I, Thompson PM, Andreassen OA (2018) Cortical abnormalities in bipolar disorder: an MRI analysis of 6503 individuals from the ENIGMA Bipolar Disorder Working Group. Mol Psychiatry 23(4):932–942. https://doi.org/10.1038/mp.2017.73
Hirschfeld RM, Calabrese JR, Weissman MM, Reed M, Davies MA, Frye MA, Keck PE Jr, Lewis L, McElroy SL, McNulty JP, Wagner KD (2003) Screening for bipolar disorder in the community. J Clin Psychiatry 64(1):53–59. https://doi.org/10.4088/jcp.v64n0111
Iwahashi K, Nishizawa D, Narita S, Numajiri M, Murayama O, Yoshihara E, Onozawa Y, Nagahori K, Fukamauchi F, Ikeda K, Ishigooka J (2014) Haplotype analysis of GSK-3beta gene polymorphisms in bipolar disorder lithium responders and nonresponders. Clin Neuropharmacol 37(4):108–110. https://doi.org/10.1097/WNF.0000000000000039
Jope RS, Song L, Li PP, Young LT, Kish SJ, Pacheco MA, Warsh JJ (1996) The phosphoinositide signal transduction system is impaired in bipolar affective disorder brain. J Neurochem 66(6):2402–2409. https://doi.org/10.1046/j.1471-4159.1996.66062402.x
Kapczinski F, Magalhães PV, Balanzá-Martinez V, Dias VV, Frangou S, Gama CS, Gonzalez-Pinto A, Grande I, Ha K, Kauer-Sant’Anna M, Kunz M, Kupka R, Leboyer M, Lopez-Jaramillo C, Post RM, Rybakowski JK, Scott J, Strejilevitch S, Tohen M, Vazquez G, Yatham L, Vieta E, Berk M (2014) Staging systems in bipolar disorder: an International Society for Bipolar Disorders Task Force Report. Acta Psychiatr Scand 130(5):354–363. https://doi.org/10.1111/acps.12305
Kapczinski NS, Mwangi B, Cassidy RM, Librenza-Garcia D, Bermudez MB, Kauer-Sant’anna M, Kapczinski F, Passos IC (2017) Neuroprogression and illness trajectories in bipolar disorder. Expert Rev Neurother 17(3):277–285. https://doi.org/10.1080/14737175.2017.1240615
Kato T, Shioiri T, Takahashi S, Inubushi T (1991) Measurement of brain phosphoinositide metabolism in bipolar patients using in vivo 31P-MRS. J Affect Disord 22(4):185–190. https://doi.org/10.1016/0165-0327(91)90064-y
Kempton MJ, Salvador Z, Munafo MR, Geddes JR, Simmons A, Frangou S, Williams SC (2011) Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Arch Gen Psychiatry 68(7):675–690. https://doi.org/10.1001/archgenpsychiatry.2011.60
Kessing LV, Vradi E, Andersen PK (2014) Starting lithium prophylaxis early v. late in bipolar disorder. Br J Psychiatry 205(3):214–220. https://doi.org/10.1192/bjp.bp.113.142802
Kessing LV, Vradi E, Andersen PK (2016) Nationwide and population-based prescription patterns in bipolar disorder. Bipolar Disord 18(2):174–182. https://doi.org/10.1111/bdi.12371
King TD, Bijur GN, Jope RS (2001) Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3beta and attenuated by lithium. Brain Res 919(1):106–114. https://doi.org/10.1016/s0006-8993(01)03005-0
Klein PS, Melton DA (1996) A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA 93(16):8455–8459. https://doi.org/10.1073/pnas.93.16.8455
Knott S, Forty L, Craddock N, Thomas RH (2015) Epilepsy and bipolar disorder. Epilepsy Behav 52(Pt A):267–274. https://doi.org/10.1016/j.yebeh.2015.07.003
Konopaske GT, Lange N, Coyle JT, Benes FM (2014) Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 71(12):1323–1331. https://doi.org/10.1001/jamapsychiatry.2014.1582
Kovács P, Hernádi I (2002) Iontophoresis of lithium antagonizes noradrenergic action on prefrontal neurons of the rat. Brain Res 947(1):150–156. https://doi.org/10.1016/s0006-8993(02)03150-5
Kubota M, van Haren NE, Haijma SV, Schnack HG, Cahn W, Hulshoff Pol HE, Kahn RS (2015) Association of IQ changes and progressive brain changes in patients with Schizophrenia. JAMA Psychiatry 72(8):803–812. https://doi.org/10.1001/jamapsychiatry.2015.0712
Kupka RW, Altshuler LL, Nolen WA, Suppes T, Luckenbaugh DA, Leverich GS, Frye MA, Keck PE Jr, McElroy SL, Grunze H, Post RM (2007) Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord 9(5):531–535. https://doi.org/10.1111/j.1399-5618.2007.00467.x
Leboyer M, Soreca I, Scott J, Frye M, Henry C, Tamouza R, Kupfer DJ (2012) Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord 141(1):1–10. https://doi.org/10.1016/j.jad.2011.12.049
Leopold K, Becker T, Förstl J, Kiefer F, de Millas W, Janetzky W, Lambert M, Pfennig A, Bechdolf A (2020) Early intervention in Schizophrenia—an update. Fortschr Neurol Psychiatr 88(6):387–397. https://doi.org/10.1055/a-0918-6071
Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM (2009) Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet 373(9657):31–41. https://doi.org/10.1016/s0140-6736(08)61764-x
Levin FR, Hennessy G (2004) Bipolar disorder and substance abuse. Biol Psychiatry 56(10):738–748. https://doi.org/10.1016/j.biopsych.2004.05.008
Licht RW (2012) Lithium: still a major option in the management of bipolar disorder. CNS Neurosci Ther 18(3):219–226. https://doi.org/10.1111/j.1755-5949.2011.00260.x
Lish JD, Dime-Meenan S, Whybrow PC, Price RA, Hirschfeld RM (1994) The National Depressive and Manic-depressive Association (DMDA) survey of bipolar members. J Affect Disord 31(4):281–294. https://doi.org/10.1016/0165-0327(94)90104-x
Lubrich B, van Calker D (1999) Inhibition of the high affinity myo-inositol transport system: a common mechanism of action of antibipolar drugs? Neuropsychopharmacology 21(4):519–529. https://doi.org/10.1016/S0893-133X(99)00037-8
Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20(1–2):27–39. https://doi.org/10.1093/emboj/20.1.27
Lyoo IK, Sung YH, Dager SR, Friedman SD, Lee JY, Kim SJ, Kim N, Dunner DL, Renshaw PF (2006) Regional cerebral cortical thinning in bipolar disorder. Bipolar Disord 8(1):65–74. https://doi.org/10.1111/j.1399-5618.2006.00284.x
Lyoo IK, Dager SR, Kim JE, Yoon SJ, Friedman SD, Dunner DL, Renshaw PF (2010) Lithium-induced gray matter volume increase as a neural correlate of treatment response in bipolar disorder: a longitudinal brain imaging study. Neuropsychopharmacology 35(8):1743–1750. https://doi.org/10.1038/npp.2010.41
Machado-Vieira R, Dietrich MO, Leke R, Cereser VH, Zanatto V, Kapczinski F, Souza DO, Portela LV, Gentil V (2007) Decreased plasma brain derived neurotrophic factor levels in unmedicated bipolar patients during manic episode. Biol Psychiatry 61(2):142–144. https://doi.org/10.1016/j.biopsych.2006.03.070
Maisonpierre PC, Le Beau MM, Espinosa R 3rd, Ip NY, Belluscio L, de la Monte SM, Squinto S, Furth ME, Yancopoulos GD (1991) Human and rat brain-derived neurotrophic factor and neurotrophin-3: gene structures, distributions, and chromosomal localizations. Genomics 10(3):558–568. https://doi.org/10.1016/0888-7543(91)90436-i
Malhi GS, Tanious M, Das P, Coulston CM, Berk M (2013) Potential mechanisms of action of lithium in bipolar disorder. Current understanding. CNS Drugs 27(2):135–153. https://doi.org/10.1007/s40263-013-0039-0
Manji HK, Moore GJ, Chen G (2000) Lithium up-regulates the cytoprotective protein Bcl-2 in the CNS in vivo: a role for neurotrophic and neuroprotective effects in manic depressive illness. J Clin Psychiatry 61(Suppl 9):82–96
Manning JS, Connor PD, Sahai A (1998) The bipolar spectrum: a review of current concepts and implications for the management of depression in primary care. Arch Fam Med 7(1):63–71. https://doi.org/10.1001/archfami.7.1.63
Masaki C, Sharpley AL, Godlewska BR, Berrington A, Hashimoto T, Singh N, Vasudevan SR, Emir UE, Churchill GC, Cowen PJ (2016) Effects of the potential lithium-mimetic, ebselen, on brain neurochemistry: a magnetic resonance spectroscopy study at 7 tesla. Psychopharmacology 233(6):1097–1104. https://doi.org/10.1007/s00213-015-4189-2
McCullumsmith RE, Kristiansen LV, Beneyto M, Scarr E, Dean B, Meador-Woodruff JH (2007) Decreased NR1, NR2A, and SAP102 transcript expression in the hippocampus in bipolar disorder. Brain Res 1127(1):108–118. https://doi.org/10.1016/j.brainres.2006.09.011
Moore GJ, Bebchuk JM, Parrish JK, Faulk MW, Arfken CL, Strahl-Bevacqua J, Manji HK (1999) Temporal dissociation between lithium-induced changes in frontal lobe myo-inositol and clinical response in manic-depressive illness. Am J Psychiatry 156(12):1902–1908. https://doi.org/10.1176/ajp.156.12.1902
Moore GJ, Bebchuk JM, Wilds IB, Chen G, Manji HK (2000) Lithium-induced increase in human brain grey matter. Lancet 356(9237):1241–1242. https://doi.org/10.1016/s0140-6736(00)02793-8
Moore GJ, Cortese BM, Glitz DA, Zajac-Benitez C, Quiroz JA, Uhde TW, Drevets WC, Manji HK (2009) A longitudinal study of the effects of lithium treatment on prefrontal and subgenual prefrontal gray matter volume in treatment-responsive bipolar disorder patients. J Clin Psychiatry 70(5):699–705. https://doi.org/10.4088/JCP.07m03745
Motoi Y, Shimada K, Ishiguro K, Hattori N (2014) Lithium and autophagy. ACS Chem Neurosci 5(6):434–442. https://doi.org/10.1021/cn500056q
Muller P, Seeman P (1977) Brain neurotransmitter receptors after long-term haloperidol: dopamine, acetylcholine, serotonin, alpha-noradrenergic and naloxone receptors. Life Sci 21(12):1751–1758. https://doi.org/10.1016/0024-3205(77)90155-2
Murray RM, Lewis SW (1987) Is schizophrenia a neurodevelopmental disorder? Br Med J 295(6600):681–682. https://doi.org/10.1136/bmj.295.6600.681
Neves-Pereira M, Mundo E, Muglia P, King N, Macciardi F, Kennedy JL (2002) The brain-derived neurotrophic factor gene confers susceptibility to bipolar disorder: evidence from a family-based association study. Am J Hum Genet 71(3):651–655. https://doi.org/10.1086/342288
NICE (2014) Bipoiar disorder: assessment and management. https://www.niceorguk/guidance/cg185. Accessed Sept 2020
Niciu MJ, Ionescu DF, Mathews DC, Richards EM, Zarate CA Jr (2013) Second messenger/signal transduction pathways in major mood disorders: moving from membrane to mechanism of action, part I: major depressive disorder. CNS Spectr 18(5):231–241. https://doi.org/10.1017/s1092852913000059
Nortje G, Stein DJ, Radua J, Mataix-Cols D, Horn N (2013) Systematic review and voxel-based meta-analysis of diffusion tensor imaging studies in bipolar disorder. J Affect Disord 150(2):192–200. https://doi.org/10.1016/j.jad.2013.05.034
Novick AM, Scott AT, Neill Epperson C, Schneck CD (2020) Neuropsychiatric effects of tamoxifen: Challenges and opportunities. Front Neuroendocrinol 59:100869. https://doi.org/10.1016/j.yfrne.2020.100869
Novotny E, Ashwal S, Shevell M (1998) Proton magnetic resonance spectroscopy: an emerging technology in pediatric neurology research. Pediatr Res 44(1):1–10. https://doi.org/10.1203/00006450-199807000-00001
Pacheco MA, Jope RS (1996) Phosphoinositide signaling in human brain. Prog Neurobiol 50(2–3):255–273. https://doi.org/10.1016/s0301-0082(96)00035-4
Palacios J, Yildiz A, Young AH, Taylor MJ (2019) Tamoxifen for bipolar disorder: systematic review and meta-analysis. J Psychopharmacol 33(2):177–184. https://doi.org/10.1177/0269881118822167
Passos IC, Mwangi B, Vieta E, Berk M, Kapczinski F (2016) Areas of controversy in neuroprogression in bipolar disorder. Acta Psychiatr Scand 134(2):91–103. https://doi.org/10.1111/acps.12581
Perlis RH (2005) Misdiagnosis of bipolar disorder. Am J Manag Care 11(9 Suppl):S271-274
Pfennig A, Leopold K, Martini J, Boehme A, Lambert M, Stamm T, Bermpohl F, Reif A, Kittel-Schneider S, Juckel G, Fallgatter AJ, Kircher T, Jansen A, Pfeiffer S, Berndt C, Rottmann-Wolf M, Sauer C, Ritter P, Correll CU, Bechdolf A, Falkenberg I, Bauer M (2020) Improving early recognition and intervention in people at increased risk for the development of bipolar disorder: study protocol of a prospective-longitudinal, naturalistic cohort study (Early-BipoLife). Int J Bipolar Disord 8(1):22. https://doi.org/10.1186/s40345-020-00183-4
Rapoport JL, Giedd JN, Gogtay N (2012) Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry 17(12):1228–1238. https://doi.org/10.1038/mp.2012.23
Ratheesh A, Cotton SM, Davey CG, Adams S, Bechdolf A, Macneil C, Berk M, McGorry PD (2017) Ethical considerations in preventive interventions for bipolar disorder. Early Interv Psychiatry 11(2):104–112. https://doi.org/10.1111/eip.12340
Rosenberg G (2007) The mechanisms of action of valproate in neuropsychiatric disorders: can we see the forest for the trees? Cell Mol Life Sci 64(16):2090–2103. https://doi.org/10.1007/s00018-007-7079-x
Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ (2010) PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol 11(2):103–112. https://doi.org/10.1038/nrm2847
Sanches M, Keshavan MS, Brambilla P, Soares JC (2008) Neurodevelopmental basis of bipolar disorder: a critical appraisal. Prog Neuropsychopharmacol Biol Psychiatry 32(7):1617–1627. https://doi.org/10.1016/j.pnpbp.2008.04.017
Saxena A, Scaini G, Bavaresco DV, Leite C, Valvassori SS, Carvalho AF, Quevedo J (2017) Role of protein kinase C in bipolar disorder: a review of the current literature. Mol Neuropsychiatry 3(2):108–124. https://doi.org/10.1159/000480349
Scearce-Levie K, Sanchez PE, Lewcock JW (2020) Leveraging preclinical models for the development of Alzheimer disease therapeutics. Nat Rev Drug Discov 19(7):447–462. https://doi.org/10.1038/s41573-020-0065-9
Scola G, Andreazza AC (2015) The role of neurotrophins in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 56:122–128. https://doi.org/10.1016/j.pnpbp.2014.08.013
Severus E, Taylor MJ, Sauer C, Pfennig A, Ritter P, Bauer M, Geddes JR (2014) Lithium for prevention of mood episodes in bipolar disorders: systematic review and meta-analysis. Int J Bipolar Disord 2:15. https://doi.org/10.1186/s40345-014-0015-8
Sharpley AL, Williams C, Holder AA, Godlewska BR, Singh N, Shanyinde M, MacDonald O, Cowen PJ (2020) A phase 2a randomised, double-blind, placebo-controlled, parallel-group, add-on clinical trial of ebselen (SPI-1005) as a novel treatment for mania or hypomania. Psychopharmacology 237(12):3773–3782. https://doi.org/10.1007/s00213-020-05654-1
Sherwood Brown E, Suppes T, Adinoff B, Rajan Thomas N (2001) Drug abuse and bipolar disorder: comorbidity or misdiagnosis? J Affect Disord 65(2):105–115. https://doi.org/10.1016/s0165-0327(00)00169-5
Simmons ML, Frondoza CG, Coyle JT (1991) Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies. Neuroscience 45(1):37–45. https://doi.org/10.1016/0306-4522(91)90101-s
Singh N, Halliday AC, Thomas JM, Kuznetsova OV, Baldwin R, Woon EC, Aley PK, Antoniadou I, Sharp T, Vasudevan SR, Churchill GC (2013) A safe lithium mimetic for bipolar disorder. Nat Commun 4:1332. https://doi.org/10.1038/ncomms2320
Singh N, Sharpley AL, Emir UE, Masaki C, Herzallah MM, Gluck MA, Sharp T, Harmer CJ, Vasudevan SR, Cowen PJ, Churchill GC (2016) Effect of the putative lithium mimetic ebselen on brain myo-inositol, sleep, and emotional processing in humans. Neuropsychopharmacology 41(7):1768–1778. https://doi.org/10.1038/npp.2015.343
Sklar P, Gabriel SB, McInnis MG, Bennett P, Lim Y, Tsan G, Schaffner S, Kirov G, Jones I, Owen M, Craddock N, DePaulo JR, Lander ES (2002) Family-based association study of 76 candidate genes in bipolar disorder: BDNF is a potential risk locus. Brain-derived neutrophic factor. Mol Psychiatry 7(6):579–593. https://doi.org/10.1038/sj.mp.4001058
Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, Mattheisen M, Wang Y, Coleman JRI, Gaspar HA, de Leeuw CA, Steinberg S, Pavlides JMW, Trzaskowski M, Byrne EM, Pers TH, Holmans PA, Richards AL, Abbott L, Agerbo E, Akil H, Albani D, Alliey-Rodriguez N, Als TD, Anjorin A, Antilla V, Awasthi S, Badner JA, Baekvad-Hansen M, Barchas JD, Bass N, Bauer M, Belliveau R, Bergen SE, Pedersen CB, Boen E, Boks MP, Boocock J, Budde M, Bunney W, Burmeister M, Bybjerg-Grauholm J, Byerley W, Casas M, Cerrato F, Cervantes P, Chambert K, Charney AW, Chen D, Churchhouse C, Clarke TK, Coryell W, Craig DW, Cruceanu C, Curtis D, Czerski PM, Dale AM, de Jong S, Degenhardt F, Del-Favero J, DePaulo JR, Djurovic S, Dobbyn AL, Dumont A, Elvsashagen T, Escott-Price V, Fan CC, Fischer SB, Flickinger M, Foroud TM, Forty L, Frank J, Fraser C, Freimer NB, Frisen L, Gade K, Gage D, Garnham J, Giambartolomei C, Pedersen MG, Goldstein J, Gordon SD, Gordon-Smith K, Green EK, Green MJ, Greenwood TA, Grove J, Guan W, Guzman-Parra J, Hamshere ML, Hautzinger M, Heilbronner U, Herms S, Hipolito M, Hoffmann P, Holland D, Huckins L, Jamain S, Johnson JS, Jureus A, Kandaswamy R, Karlsson R, Kennedy JL, Kittel-Schneider S, Knowles JA, Kogevinas M, Koller AC, Kupka R, Lavebratt C, Lawrence J, Lawson WB, Leber M, Lee PH, Levy SE, Li JZ, Liu C, Lucae S, Maaser A, MacIntyre DJ, Mahon PB, Maier W, Martinsson L, McCarroll S, McGuffin P, McInnis MG, McKay JD, Medeiros H, Medland SE, Meng F, Milani L, Montgomery GW, Morris DW, Muhleisen TW, Mullins N, Nguyen H, Nievergelt CM, Adolfsson AN, Nwulia EA, O’Donovan C, Loohuis LMO, Ori APS, Oruc L, Osby U, Perlis RH, Perry A, Pfennig A, Potash JB, Purcell SM, Regeer EJ, Reif A, Reinbold CS, Rice JP, Rivas F, Rivera M, Roussos P, Ruderfer DM, Ryu E, Sanchez-Mora C, Schatzberg AF, Scheftner WA, Schork NJ, Shannon Weickert C, Shehktman T, Shilling PD, Sigurdsson E, Slaney C, Smeland OB, Sobell JL, Soholm Hansen C, Spijker AT, St Clair D, Steffens M, Strauss JS, Streit F, Strohmaier J, Szelinger S, Thompson RC, Thorgeirsson TE, Treutlein J, Vedder H, Wang W, Watson SJ, Weickert TW, Witt SH, Xi S, Xu W, Young AH, Zandi P, Zhang P, Zollner S, e QC, Consortium B, Adolfsson R, Agartz I, Alda M, Backlund L, Baune BT, Bellivier F, Berrettini WH, Biernacka JM, Blackwood DHR, Boehnke M, Borglum AD, Corvin A, Craddock N, Daly MJ, Dannlowski U, Esko T, Etain B, Frye M, Fullerton JM, Gershon ES, Gill M, Goes F, Grigoroiu-Serbanescu M, Hauser J, Hougaard DM, Hultman CM, Jones I, Jones LA, Kahn RS, Kirov G, Landen M, Leboyer M, Lewis CM, Li QS, Lissowska J, Martin NG, Mayoral F, McElroy SL, McIntosh AM, McMahon FJ, Melle I, Metspalu A, Mitchell PB, Morken G, Mors O, Mortensen PB, Muller-Myhsok B, Myers RM, Neale BM, Nimgaonkar V, Nordentoft M, Nothen MM, O’Donovan MC, Oedegaard KJ, Owen MJ, Paciga SA, Pato C, Pato MT, Posthuma D, Ramos-Quiroga JA, Ribases M, Rietschel M, Rouleau GA, Schalling M, Schofield PR, Schulze TG, Serretti A, Smoller JW, Stefansson H, Stefansson K, Stordal E, Sullivan PF, Turecki G, Vaaler AE, Vieta E, Vincent JB, Werge T, Nurnberger JI, Wray NR, Di Florio A, Edenberg HJ, Cichon S, Ophoff RA, Scott LJ, Andreassen OA, Kelsoe J, Sklar P, Bipolar Disorder Working Group of the Psychiatric Genomics C (2019) Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet 51(5):793–803. https://doi.org/10.1038/s41588-019-0397-8
Sun YR, Herrmann N, Scott CJM, Black SE, Khan MM, Lanctot KL (2018) Global grey matter volume in adult bipolar patients with and without lithium treatment: a meta-analysis. J Affect Disord 225:599–606. https://doi.org/10.1016/j.jad.2017.08.078
Talaei A, Pourgholami M, Khatibi-Moghadam H, Faridhosseini F, Farhoudi F, Askari-Noghani A, Sadeghi R (2016) Tamoxifen: a protein kinase C inhibitor to treat mania: a systematic review and meta-analysis of randomized, placebo-controlled trials. J Clin Psychopharmacol 36(3):272–275. https://doi.org/10.1097/JCP.0000000000000492
Teixeira AL, Colpo GD, Fries GR, Bauer IE, Selvaraj S (2019) Biomarkers for bipolar disorder: current status and challenges ahead. Expert Rev Neurother 19(1):67–81. https://doi.org/10.1080/14737175.2019.1550361
Tighe SK, Mahon PB, Potash JB (2011) Predictors of lithium response in bipolar disorder. Ther Adv Chronic Dis 2(3):209–226. https://doi.org/10.1177/2040622311399173
Toker L, Mancarci BO, Tripathy S, Pavlidis P (2018) Transcriptomic evidence for alterations in astrocytes and parvalbumin interneurons in subjects with Bipolar Disorder and Schizophrenia. Biol Psychiatry 84(11):787–796. https://doi.org/10.1016/j.biopsych.2018.07.010
Uribe E, Wix R (2012) Neuronal migration, apoptosis and bipolar disorder. Rev Psiquiatr Salud Ment 5(2):127–133. https://doi.org/10.1016/j.rpsm.2011.11.005
Vajda FJ (2002) Neuroprotection and neurodegenerative disease. J Clin Neurosci 9(1):4–8. https://doi.org/10.1054/jocn.2001.1027
Valli I, Fabbri C, Young AH (2019) Uncovering neurodevelopmental features in bipolar affective disorder. Br J Psychiatry 215(1):383–385. https://doi.org/10.1192/bjp.2019.117
van Calker D, Belmaker RH (2000) The high affinity inositol transport system–implications for the pathophysiology and treatment of bipolar disorder. Bipolar Disord 2(2):102–107. https://doi.org/10.1034/j.1399-5618.2000.020203.x
van der Markt A, Klumpers UMH, Dols A, Draisma S, Boks MP, van Bergen A, Ophoff RA, Beekman ATF, Kupka RW (2020) Exploring the clinical utility of two staging models for bipolar disorder. Bipolar Disord 22(1):38–45. https://doi.org/10.1111/bdi.12825
Vieta E, Reinares M, Rosa AR (2011) Staging bipolar disorder. Neurotox Res 19(2):279–285. https://doi.org/10.1007/s12640-010-9197-8
Wang HY, Friedman E (1989) Lithium inhibition of protein kinase C activation-induced serotonin release. Psychopharmacology 99(2):213–218. https://doi.org/10.1007/BF00442810
Wang AY, Lohmann KM, Yang CK, Zimmerman EI, Pantazopoulos H, Herring N, Berretta S, Heckers S, Konradi C (2011) Bipolar disorder type 1 and schizophrenia are accompanied by decreased density of parvalbumin- and somatostatin-positive interneurons in the parahippocampal region. Acta Neuropathol 122(5):615–626. https://doi.org/10.1007/s00401-011-0881-4
Wetsel WC, Khan WA, Merchenthaler I, Rivera H, Halpern AE, Phung HM, Negro-Vilar A, Hannun YA (1992) Tissue and cellular distribution of the extended family of protein kinase C isoenzymes. J Cell Biol 117(1):121–133. https://doi.org/10.1083/jcb.117.1.121
Williams RSB, Cheng LL, Mudge AW, Harwood AJ (2002) A common mechanism of action for three mood-stabilizing drugs. Nature 417(6886):292–295. https://doi.org/10.1038/417292a
Wise T, Radua J, Nortje G, Cleare AJ, Young AH, Arnone D (2016) Voxel-based meta-analytical evidence of structural disconnectivity in Major Depression and Bipolar Disorder. Biol Psychiatry 79(4):293–302. https://doi.org/10.1016/j.biopsych.2015.03.004
Yucel K, McKinnon MC, Taylor VH, Macdonald K, Alda M, Young LT, MacQueen GM (2007) Bilateral hippocampal volume increases after long-term lithium treatment in patients with bipolar disorder: a longitudinal MRI study. Psychopharmacology 195(3):357–367. https://doi.org/10.1007/s00213-007-0906-9
Yucel K, Taylor VH, McKinnon MC, Macdonald K, Alda M, Young LT, MacQueen GM (2008) Bilateral hippocampal volume increase in patients with bipolar disorder and short-term lithium treatment. Neuropsychopharmacology 33(2):361–367. https://doi.org/10.1038/sj.npp.1301405
Zanni G, Michno W, Di Martino E, Tjarnlund-Wolf A, Pettersson J, Mason CE, Hellspong G, Blomgren K, Hanrieder J (2017) Lithium accumulates in neurogenic brain regions as revealed by high resolution ion imaging. Sci Rep 7:40726. https://doi.org/10.1038/srep40726
Zarate CA, Manji HK (2009) Protein kinase C inhibitors: rationale for use and potential in the treatment of bipolar disorder. CNS Drugs 23(7):569–582. https://doi.org/10.2165/00023210-200923070-00003
Zarate CA Jr, Singh JB, Carlson PJ, Quiroz J, Jolkovsky L, Luckenbaugh DA, Manji HK (2007) Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord 9(6):561–570. https://doi.org/10.1111/j.1399-5618.2007.00530.x
Acknowledgements
I wish to thank Drs. Jose S. Aguilar-Marquez, Francisco Barrantes and Robert Schneider for critically reviewing the manuscript.
Author information
Authors and Affiliations
Contributions
E.L.M.O conceived the idea and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The author declares not to have competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ochoa, E.L.M. Lithium as a Neuroprotective Agent for Bipolar Disorder: An Overview. Cell Mol Neurobiol 42, 85–97 (2022). https://doi.org/10.1007/s10571-021-01129-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-021-01129-9