
Graphical Abstract

Abstract
Alterations in the homeostasis of several adhesion GPCRs (aGPCRs) have been observed in cancer. The main cellular functions regulated by aGPCRs are cell adhesion, migration, polarity, and guidance, which are all highly relevant to tumor cell biology. Expression of aGPCRs can be induced, increased, decreased, or silenced in the tumor or in stromal cells of the tumor microenvironment, including fibroblasts and endothelial and/or immune cells. For example, ADGRE5 (CD97) and ADGRG1 (GPR56) show increased expression in many cancers, and initial functional studies suggest that both are relevant for tumor cell migration and invasion. aGPCRs can also impact the regulation of angiogenesis by releasing soluble fragments following the cleavage of their extracellular domain (ECD) at the conserved GPCR-proteolytic site (GPS) or other more distal cleavage sites as typical for the ADGRB (BAI) family. Interrogation of in silico cancer databases suggests alterations in other aGPCR members and provides the impetus for further exploration of their potential role in cancer. Integration of knowledge on the expression, regulation, and function of aGPCRs in tumorigenesis is currently spurring the first preclinical studies to examine the potential of aGPCR or the related pathways as therapeutic targets.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Cancer arises through the sequential accumulation of driver gene mutations which ultimately confer a growth advantage upon the cells in which they have occurred, leading to tumor formation through a clonal selection process. An important cancer hallmark is the ability of cancer cells to acquire invasive and metastatic properties which is associated with progression to systemic disease and reduced survival. Invasion and metastasis require tumor cell detachment and migration, extravasation into the circulation, and subsequent seeding at distant sites accompanied by altered extracellular matrix (ECM) turnover and initiation of tumor neo-angiogenesis. Each of these steps involves direct tumor cell-cell and tumor cell-microenvironment interactions, including stromal elements such as tumor-associated fibroblasts, endothelial cells, and tumor-infiltrating immune cells as well as the ECM. aGPCRs regulate cell adhesion, migration, polarity, and guidance; cellular functions that are also required for tumorigenesis. Characteristic for the aGPCRs is the large ECD with multiple adhesive folds, which is coupled to a seven-span transmembrane (7TM) domain and an intracellular domain (ICD) (see also [1–4]). Many aGPCRs are cleaved at the GPS in the juxtamembrane region which is part of and regulated by a GPCR autoproteolysis-inducing (GAIN) domain and results in an N- (NTF) and a C-terminal fragment (CTF), which remain associated at the cell surface (discussed in depth in [3, 5]). The various possible signaling scenarios resulting from this bipartite structure have been reviewed recently [6, 7] and in [8–10].
2 Software for the In Silico Interrogation of Cancer-Related Databases
A number of online resources provide user-friendly software for the rapid interrogation of large-scale cancer genomics data sets, and this in silico data mining provides useful starting information on aGPCR expression or mutation in a variety of cancer samples and cell lines.
-
1.
The Cancer Cell Line Encyclopedia (CCLE) (www.broadinstitute.org/ccle) contains detailed genetic characterization for more than 1000 human cancer cell lines [11].
-
2.
The cBioPortal for Cancer Genomics (www.cbioportal.org) provides analysis tools to query, analyze, and visualize large-scale cancer genomics data sets, including The Cancer Genome Atlas (TCGA) [12, 13].
-
3.
The Human Protein Atlas (HPA) database (www.proteinatlas.org) contains high-resolution images showing the spatial distribution of proteins in dozens of normal and malignant tissues [14]. Note: the specificity of the used paraffin-suitable antibodies is not verified in depth.
-
4.
The Catalogue of Somatic Mutations in Cancer (COSMIC) (cancer.sanger.ac.uk/cosmic) is designed to store and display somatic mutation information .
-
5.
The SurvExpress is a biomarker validation tool and database for cancer gene expression with survival analysis [15].
3 Identification of Adhesion GPCRs in Cancer
In the last two decades, it has become apparent that changes in aGPCRs occur in cancer, either at the gene or protein expression level. Studies addressing the relevance of these changes to the tumorigenesis process are currently limited. CD97 was the first aGPCR identified to be correlated to cancer [16]. While it is not expressed in normal thyrocytes and well-differentiated papillary and in part follicular thyroid cancers, the protein was found to be elevated in anaplastic carcinomas. In the following years other aGPCRs such as GPR56 were identified to be cancer related [17]. Non-biased screens such as mRNA microarrays, genome-wide association (GWA) studies, or proteome analyses confirmed previously identified cancer-related changes in aGPCRs [18], showed alterations in new tumor entities [19], or identified new aGPCRs involved in cancer [20]. Recently, a whole-genome sequencing (WGS) approach in 441 human breast, lung, ovarian, and prostate cancers identified the aGPCRs ADGRB3 (BAI3) and ADGRL3 (LPHN3) among 77 significantly mutated genes [21]. Further studies are warranted to determine whether these mutations are functionally significant to cancer development or progression or simply passenger mutations.
4 Alterations in Adhesion GPCR Expression in Tumors Compared to Normal Tissue
aGPCRs may be induced, increased, decreased, or silenced in tumor compared to the corresponding normal cells. A quick way to determine whether a particular aGPCR may be present in tumor cells is to interrogate the CCLE [11]. This database shows high levels of mRNA for ADGRA3 (GPR125), ADGRB2 (BAI2), ADGRC2 (CELSR2) and ADGRC3 (CELSR3), CD97, GPR56, ADGRG6 (GPR126), ADGRL1 (LPHN1), and ADGRL2 (LPHN2) genes in hundreds of cell lines derived from nearly all tumor entities (note: the normal corresponding cells are not included in the database). The mRNA of other aGPCRs is absent in these cell lines, including many members of the ADGRF and ADGRG families. Whether expression in cultured cell lines reflects expression in tumor cells in vivo needs to be addressed in each individual aGPCR and tumor type.
There is also evidence for changes in posttranslational modification of aGPCRs associated with cancer, which could have functional consequences. This was first shown for CD97. In normal smooth and skeletal muscle cells, the non-modified CD97 protein core is present, whereas in their malignant counterparts, i.e., in leiomyosarcoma and rhabdomyosarcoma cells, CD97 is found N-glycosylated [66, 67]. It is well known that glycosylation patterns are modified in cancer [88, 89]; therefore, it is important to determine whether such changes are relevant to the tumorigenic process for each individual aGPCR. N-glycosylation of CD97 is mapped to the adhesive EGF-like domains and is needed for the binding of the interaction partner CD55 [67], which is consistent with CD97 and CD55 often being co-expressed in cancer [53, 54, 90–93].
Regulation of aGPCR expression in stromal cells, such as tumor-associated fibroblasts and endothelial or tumor-infiltrating immune cells, as shown for ADGRA2 (GPR124) and all BAIs, may also support or suppress tumor progression. For example, BAI1 has been shown to play a role as an engulfment receptor on macrophages that can recognize apoptotic cells through binding of membrane-exposed phosphatidylserine groups to BAI1’s extracellular thrombospondin repeats (TSRs) [94]. Variation in BAI1 expression on macrophages or possibly tumor cells may change the dynamics of dying tumor cell clearance from tumors, related inflammation, and cancer metabolism.
5 Soluble Adhesion GPCR and Cleavage Fragments in Tumorigenesis
As a direct consequence of the autocatalytic cleavage of aGPCRs at the GPS, as well as through (additional) independent proteolysis events within their ECD, the NTF or smaller fragments may be released from the receptor and appear in body fluids. The existence of circulating soluble NTFs is reported for GPR124 [33], BAI1 [69, 85], BAI2 [95], CD97 [96], ADGRF5 (GPR116) [97], GPR126 [98], and LPHN1 [99].
Theoretically, soluble NTFs could engage with interaction partners over large distances far away from the site at which they were released and thereby modulate heterotypic tumor-stromal interactions that may be relevant to tumorigenesis. This has been shown experimentally for the NTFs of the various isoforms of CD97, which differ in the number of EGF-like domains in their ECDs. Soluble CD97 stimulates tumor angiogenesis in vitro and in vivo through binding of integrins and chondroitin sulfate, a constituent of the ECM, in an isoform-specific manner [100]. Soluble CD97 is found at sites of inflammation [96, 101, 102], most likely released from CD97-positive immune cells, but not in body fluids of patients suffering from CD97-positive tumors [48]. These data suggest either that unknown cell-specific mechanisms control the release of the NTF or that an additional independent proteolysis event, restricted to immune cells, is necessary for this release.
In contrast to the pro-tumorigenic activity of CD97 NTFs, the NTFs of BAI1 have anti-angiogenic and anti-tumorigenic activities [69, 70] as further discussed below.
6 Biological Functions of Adhesion GPCRs in Cancer
6.1 Proliferation , Apoptosis , and Cell Cycle Regulation
Little is known about the role of aGPCRs in tumor growth, either through alteration in tumor cell division or cell death mechanisms including apoptosis. A few aGPCRs have been shown to promote cell proliferation and/or prevent apoptosis, although their role in signaling is not clarified. Downregulation of ADGRL4 (ELTD1) by microRNA-139-5p leads to an inhibition of glioblastoma cell proliferation in vitro [103]. CD97 protects fibrosarcoma HT1080 and cervical HeLa cancer cell lines from serum starvation- and staurosporine-induced intrinsic apoptosis [104].
Preliminary studies examining the effect of GPR56 on in vitro tumor cell proliferation and apoptosis have yielded contradictory results and may reflect the different cancer types and studies performed. One study reported that the knockdown of GPR56 expression induces a transient activation of apoptosis in several cancer cell lines, suggesting that GPR56 may be important for cell survival and have pro-tumorigenic functions [32]. This pro-apoptotic response could be overcome, as this research group was able to generate stable cell lines with GPR56 knockdown. These clones show a reduction in anchorage-independent growth in vitro, and some also have decreased tumor growth in vivo [32]. Similar observations were reported in the EVI-1high leukemia cell line, in which GPR56 knockdown induced apoptosis although its effects on leukemia progression in vivo were not explored [22]. This pro-growth function of GPR56 contrasts with observations by another group in metastatic melanoma (A375P and MC-1) cell lines. This study suggests that GPR56 is necessary for the subcutaneous growth of these cells in mice, but does not affect their in vitro proliferation [60, 105]. The mechanisms linking GPR56 to in vivo tumor growth are found to depend on regulation of proper ECM deposition by the melanoma cells [60]. A larger panel of cell lines as well as a combination of in vitro and in vivo studies will be needed to fully understand the function of GPR56 during cancer progression.
6.2 Cell Adhesion and Interaction with ECM
Some aGPCRs have been implicated in regulating tumor cell adhesion, but potential mechanisms of these effects have not been investigated in depth. Knocking down Gpr125 in murine myeloid sarcoma lines that harbor KRASG12C and MLL/AF10(OM-LZ) oncogenes leads to reduced myeloid sarcoma burden in mice, and a decrease in cell adhesion in culture, possibly contributing to their reduced tumorigenicity [106].
ECM proteins are large peptides that assemble into highly ordered structures modulating various aspects of cell behavior, including cell adhesion, migration, survival, and proliferation, which are key factors during tumorigenesis [107]. GPR56 NTF was discovered to bind tissue transglutaminase (TG2) [105] and collagen III [108]. It was reported that GPR56 internalizes TG2 from the surface of melanoma cells, resulting in defects in ECM deposition in melanomas [60]. This defect may contribute to the inhibitory function of GPR56 on melanoma growth and metastasis. Knocking down GPR56 inhibits the adhesion of EVI-1high leukemia cells to ECM [22], and granule cells from the rostral cerebellum of Gpr56 −/− mice are defective in adhering to fibronectin and laminin [109]. The adhesion effect of GPR56 could be blocked by its NTF: purified recombinant GPR56 NTF inhibits adhesion of glioma cells to fibronectin [82]. Taken together, the above studies point to a promoting role of GPR56 in cell-ECM adhesion, but whether this is a shared mechanism among different tumor cells and whether it contributes to the function of GPR56 in cancer progression remain to be investigated.
Several other aGPCRs such as ADGRE2 (EMR2) and GPR126 have also been shown to interact with the ECM [110–112], but the impact of these interactions on cancer has not been explored.
6.3 Cell Migration and Invasion
Among all aGPCRs, CD97 has been the most studied in the context of cell migration and invasion (see also [113] on this topic). Its association with tumor invasion is reported in several studies. It is upregulated at the invasive tumor front [42, 48, 91], and its high expression correlates with lymph node invasion [48, 54], advanced tumor stages [48, 53, 54, 91, 93], and patient survival [53, 77, 93]. The positive effects of CD97 on tumor invasion are verified in experimental models both in vitro and in vivo [49, 58]. Consistent with its function in promoting migration and invasion, CD97 is reported to enhance trans-well cell migration toward fetal calf serum in 15 different colorectal cancer cell lines [48] and toward fetal calf serum and lysophosphatidic acid (LPA) in AML cells (MV4-1 cell line) [29]. Consistently, directed cell migration and invasion stimulated by fetal calf serum are decreased following CD97 knockdown in a prostate cancer cell line (DU145) [58]. Conflicting results have been published on the effect of CD97 in a fibrosarcoma cell line (HT1080) on in vitro migration and in vivo formation of tumors and metastases: in one study CD97 overexpression increases migration in vitro and induces earlier tumor growth [49], whereas another study found the opposite [114].
Other aGPCRs have also been reported to regulate cancer cell migration. GPR56 inhibits cell migration toward stromal cell-derived factor 1 in leukemia cell lines [22], and GPR116 promotes cell migration and invasion toward serum in a breast cancer cell line (MDA-MB-231) through the Gαq-p63RhoGEF-RhoA/Rac1 pathway [65].
6.4 Angiogenesis
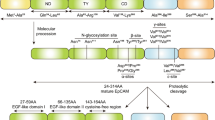
Several aGPCRs are found to regulate tumor angiogenesis, the process through which tumors elicit vessel formation that is critical for their sustained growth (see also [115] on this topic). The most studied one is BAI1, which inhibits angiogenesis in glioblastomas, the most malignant primary intracranial brain tumor which is incurable [69, 70, 116, 117] as well as other cancer types [36, 37, 118, 119]. The ECD of BAIs contains TSRs, and some TSRs are known to negatively regulate angiogenesis [120, 121]. Cleavage of the BAI1 ECD leads to the release of two fragments, dubbed vasculostatin-40 (Vstat40) and vasculostatin-120 (Vstat120) due to their molecular size (Fig. 1), and both were shown to suppress blood vessel formation in a variety of in vitro and in vivo assays [69, 70, 116]. When overexpressed, GPR56 can also inhibit tumor angiogenesis in melanoma, probably by blocking the secretion of vascular endothelial growth factor (VEGF) [61].

Structure and functions of BAIs (ADGRBs). (a) Schematic of the three BAI proteins, representing the major known structural and functional features as well as known proteolysis events that generate either Vstat120 [69] or Vstat40 [71], and cancer-associated somatic mutations [21, 122]. Abbreviations: TSR thrombospondin type 1 repeat, GAIN GPCR autoproteolysis-inducing domain [123], GPS GPCR-proteolytic site, 7TM seven-transmembrane region, RGD Arg-Gly-Asp integrin-binding motif, PRR proline-rich region. (b) Multiple functions of BAI1: BAI1 inhibits tumor cell growth in part by inhibiting angiogenesis. Moreover, its TSRs interact with exposed phosphatidylserine at the outer leaflet on apoptotic cells and elicit the engulfment in macrophages (Mϕ). BAI1 also promotes myogenesis. Deficiency of BAI1 promotes PSD-95 degradation at the synapses and induces enhanced long-term potentiation. Figure adapted from [124]
A number of aGPCRs also stimulate neovascularization in tumors. GPR124 was described first as a tumor endothelial marker (TEM5) [125]. ELTD1 is identified as part of a core angiogenic signature composed of genes whose expression jointly correlates with that of several well-recognized angiogenesis and/or endothelial cells “seed” genes, in more than 1000 human primary tumors, and is subsequently found to be essential for angiogenesis and tumor growth [34]. The NTF of CD97 promotes angiogenesis via binding to integrin α5β1 and αvβ3 and elicits a chemotactic response in endothelial cells leading to their recruitment to the tumor [100].
7 Families of Adhesion GPCR in Tumorigenesis
Table 1 gives an overview on published studies of aGPCRs in human tumors.
7.1 ADGRA
GPR124 was originally identified as an endothelial marker that is upregulated during tumor angiogenesis [126]. A soluble GPR124 fragment, probably the NTF, is shed from GPR124-transfected endothelial cells. Further proteolytic processing creates a protein subunit that mediates endothelial survival and subsequent tumor angiogenesis via interactions with glycosaminoglycans and the integrin αvβ3 [33]. Only cell lines derived from Ewing sarcoma, chondrosarcoma, and osteosarcoma strongly express GPR124 [11]. microRNA miR-138-5p, found to be decreased in gefitinib-resistant non-small cell lung cancer (NSCLC) cell lines (PC9GR, H1975), directly targets GPR124. Thus, downregulation of GPR124 is discussed as a therapeutic approach to overcome NSCLC gefitinib resistance [127]. Knockdown of Gpr125 reduces tumor cell aggregation and diminishes myeloid sarcoma formation induced by transplantation of immortalized acute myeloid leukemia [106].
7.2 ADGRB
The BAI1–3 proteins are predominantly expressed in the brain [116, 128]. The human ADGRB1 (BAI1) gene is located on chromosome 8q24, and BAI1 is the most studied member in this aGPCR subfamily. BAI1 contains several well-defined protein modules in the N-terminus such as an integrin-binding Arg-Gly-Asp (RGD) motif followed by five TSRs , a hormone binding domain and a GAIN domain with a GPS (Fig. 1) [85]. BAI1 can be cleaved at the GPS releasing a soluble, 120 kDa anti-angiogenic NTF called vasculostatin (Vstat120), which was reported to suppress the proliferation of endothelial cells by blocking αvβ5 integrin [129], reduce the migration of cultured microvascular endothelial cells in a CD36-dependent manner [70], and inhibit angiogenesis and glioma growth in vivo [69, 71, 72, 116, 130]. Subsequently, a novel more distal proteolytic processing event was identified in BAI1, and it generates a more abundant cleaved NTF containing only the RGD and the first TSR (Vstat40). This fragment was also able to inhibit angiogenesis in vitro and in vivo [71]. At the other end of the protein, the C-terminus of BAI1 contains an intracellular proline-rich region (PRR) and a terminal PDZ-binding domain (Fig. 1), which associates with a number of intracellular signaling and scaffolding proteins [131, 132], but the role of these signaling events in cancer has not been studied to date. BAI1 also plays an important role in myogenesis, synaptic plasticity, and phagocytosis [124], but it is unclear whether any of these functions can also intersect with cancer.
BAI1 mRNA levels are consistently downregulated in primary glioma specimens and cell lines [72, 73], and in brain metastases from lung adenocarcinoma [37]. Pulmonary adenocarcinomas, gastric, and colorectal cancers also show reduced BAI1 expression compared with normal tissue [36, 37, 44, 46]. A recent study also suggests that BAI1 expression is significantly reduced in breast cancer and correlates with poorer patient survival [62].
The human BAI2 and BAI3 genes were discovered as a result of their sequence identity with BAI1 [128] and are localized on chromosomes 1p35 and 6q12, respectively. The amino acid sequence is highly conserved in all three members; however, BAI1 contains five TSRs, while BAI2 or BAI3 each contains only four. In addition, the extracellular RGD motif and the intracellular proline-rich region (PRR), a domain known to interact with Src homology 3 (SH3) and WW domain-containing proteins [133], are unique to BAI1. Unlike BAI1, the expression of BAI2 or BAI3 is not silenced in glioma cells [72]. Moreover, recent studies determined higher BAI2 expression in tumor metastasis and advanced stages [134, 135].
Importantly, recent discoveries have shown that the BAI genes are silenced and/or undergo somatic mutations in several cancers (Fig. 1), including the lung, breast, ovarian, and brain [21, 72, 122], suggesting that eliminating their function might be required for tumor formation. Whether the identified mutations alter BAI1 function and may contribute to tumor formation has not been investigated to date. The function of BAI2 and BAI3 in tumorigenesis needs to be further investigated.
7.3 ADGRC
Although the role of ADGRCs (CELSRs) as key components in planar cell polarity (PCP) was clarified elegantly in knockout mice , our knowledge on their expression and function in human malignancies is rather limited. Unbiased screening approaches for cancer-related proteins often scored CELSRs as interesting hits, but most of them await further validation as to their role in cancer. CELSR1 is upregulated in B cells of patients with chronic lymphocytic leukemia (CLL) [25]; CELSR1 and CELSR3 are found to be present in gastrointestinal and brain tumors, respectively [47]; and Celsr1 is overexpressed in Graffi murine leukemia virus (MuLV)-induced hematologic malignancies with a lymphoid subtype [24]. Gene expression profiling of mantle cell lymphoma shows that CELSR1 is downregulated in the non-nodal form compared to other lymphomas [27], and CELSR1 expression is increased in pure ductal breast carcinomas in situ (DCIS) compared to such tumors with invasive components [63]. Moreover, the gene locus of CELSR1 is hypermethylated in hepatocellular carcinoma (HCC) compared to control tissue [51].
7.4 ADGRE
ADGRE1–3 (EMR1–3) expression is restricted to leukocytes. Thus, almost only cell lines derived from hematologic malignancies express them [11]. EMR2 is not or rarely expressed in gastric, pancreatic, esophageal, and colorectal carcinomas [42, 136]. Here, only a subpopulation of tumor-infiltrating macrophages is strongly EMR2 positive . Aberrant expression of EMR2 protein is reported in breast carcinomas. It is associated with poor patient survival [64]. EMR2 is found upregulated in glioblastoma, in particular in the mesenchymal subtype [74], and is associated with poor overall survival and an invasive phenotype [75].
EMR3 is also found to be increased in glioblastoma and associated with poor survival [76]. In colon cancer EMR3 is restricted to tumor-infiltrating immune cells [137].
CD97 is the only ADGRE family member whose expression is not restricted to immune cells. In human, its expression varies in cells of epithelial and mesenchymal origin from negative, as in keratinocytes and thyrocytes, to low in enterocytes, and high in pneumocytes and leukocytes . Cancer cell lines are almost all moderately or strongly CD97 positive, suggesting that, compared to normal tissues, CD97 is frequently induced and/or increased in the corresponding malignancies. Low CD97 levels are found only in cell lines derived from neuroblastomas. Small cell lung cancer (SCLC) cell lines are nearly all CD97 negative, while non-small cell lung cancer (NSCLC) cell lines are strongly CD97 positive, a fact that could be helpful in discriminant analysis on lung cancers.
In thyroid cancer, CD97 is induced and expression levels correlate with malignant progression [16, 57], i.e., only few papillary and follicular thyroid carcinomas are CD97 positive, whereas most anaplastic cancers strongly express CD97. In normal human intestinal epithelial cells, CD97 resides at low levels in lateral cell contacts, whereas in colorectal cancer, the molecule is increased. In part, this upregulation parallels its new cellular location within the cytoplasm [48, 49]. Overexpression of CD97 in scattered tumor cells is observed at the tumor invasion front [48]. The presence of these CD97-positive scattered tumor cells correlates with higher tumor stage and higher lymphatic vessel infiltration, both prognostic factors in colorectal cancer. CD97 is also upregulated in gastric cancer [42]. Further supporting an oncogenic role, in mouse models of colorectal and gastric cancer, CD97 supports local tumor growth and promoted metastatic spread [49, 138].
In gall bladder carcinoma, CD97 expression is an independent risk factor for overall survival [53]. CD97 is highly expressed in tumor cells of poorly differentiated pancreatic ductal adenocarcinoma and in tumor-infiltrating leukocytes in chronic pancreatitis samples, but not on normal pancreatic epithelial cells [139]. In a study of 37 pancreatic cancer patients, CD97 correlates with aggressiveness and was associated with prognosis [54]. In esophageal cancer CD97 is among the 19 genes with promoter hypomethylation and upregulation [140].
CD97 mediates invasion in prostate cancer cells, at least in part, by associating with lysophosphatidic acid receptor 1 (LPAR1), leading to enhanced LPA-dependent RHO and extracellular signal-regulated kinase (ERK) activation. Consistent with its role in invasion, depletion of CD97 in prostate cancer cells (PC3 cell line) results in decreased bone metastasis without affecting subcutaneous tumor growth. CD97 and LPAR1 are significantly co-expressed in clinical prostate cancer specimens.
CD97 is highly expressed in glioblastoma (WHO grade IV) and has prognostic significance in two independent cohorts of 187 and 539 glioblastoma patients, respectively [18, 77]. CD97 is also identified as a potential biomarker for glioma-initiating cells through an in vivo phage display screen [78], suggesting it represents a potential new therapeutic target [77]. In contrast, CD97 expression is minimal in WHO grade II and III astrocytomas [79]. Experimental data suggest that CD97 regulates tumor cell invasion in glioblastomas . Decreasing CD97 by siRNA reduces migration and invasion, but not proliferation in two glioblastoma cell lines [77]. Mass spectroscopy-based proteomic analysis showed that CD97 is enriched in membrane fractions of invadopodia, actin-rich protrusions, of invasive glioblastoma cells [18]. The mechanism underlying CD97-mediated promotion of invasion of glioblastoma cells is under investigation. Suppression of Wilms’ tumor gene product WT1 by siRNA led to a decrease in the invasiveness of glioblastoma cell lines paralleled by a suppression of CD97 RNA [80].
Further evidence for a role of CD97 in tumorigenesis comes from screening studies demonstrating that it is a direct target of the tumor suppressor microRNA-126 in the breast cancer cell line MDA-MB-231 [141].
Additionally, evaluation of CD97 expression in leukemia is highly informative. Gene expression studies and characterization of the leukemia cell surface proteome identified CD97 as a marker for minimal residual disease in acute lymphoblastic leukemia (ALL) [19]. CD97 expression also accounts for the most informative differences between normal and malignant cells in ALL [26]. CD97 has further been identified as a leukemic stem cell marker in acute myeloid leukemia (AML) [28]. In AML, expression levels of CD97 are associated with internal tandem duplications within the juxtamembrane region of the FMS-like tyrosine kinase receptor FLT3 (LFT3-ITD) [29].
Whether the function of CD97 in tumorigenesis could be modulated by other interaction partners such as LPAR1 is unknown. CD55 [142], chondroitin sulfate B [110], α5β1 and αvβ3 integrins [100], and CD90 [143] bind distinct sites within the NTF of CD97 at immune cells; nothing is known whether these interaction partners also bind CD97 in tumors. A few articles have been published on co-expression of CD55 and CD97 in solid tumors but not on the resulting functional consequence [53, 54, 91–93].
7.5 ADGRF
GPR116, highly enriched in fetal and adult lung [144–146], is part of a gene expression signature that differentiates adenocarcinoma of lung and breast origin in effusions [20]. Knockdown of GPR116 suppresses migration and invasion, whereas ectopic expression enhances invasion of the breast cancer cell line MDA-MB-231 [65]. GPR116 promotes constitutively breast cancer metastasis via the Gαq-p63RhoGEF-Rho GTPase pathway [65]. Its expression is significantly correlated with breast tumor progression, recurrence, and poor prognosis.
7.6 ADGRG
Among the ADGRG members, GPR56 is the most studied in the context of cancer progression. It was reported in 1999 to be downregulated in highly metastatic melanoma cell lines compared with poorly metastatic lines [17]. Its inverse correlation with metastatic potential was evaluated in more depth later in an experimental metastasis model which shows the downregulation of GPR56 in several highly metastatic melanoma derivatives compared with their poorly metastatic parental line (A375P). Re-expression of GPR56 inhibits the growth and metastasis of melanoma cells [105], supporting a tumor suppressor role for GPR56 in melanoma. Mechanistic studies in one of the metastatic derivatives (MC-1) revealed that GPR56 overexpression inhibits the activation of PKCα, resulting in the suppression of VEGF secretion from melanoma cells, leading to angiogenesis inhibition [61]. In contrast to the full-length receptor, expression of the CTF of GPR56 or deletion of the serine threonine proline (STP)-rich segment (ΔSTP-GPR56) leads to enhanced activation of PKCα, VEGF secretion, and angiogenesis. These opposing functions of GPR56 and its CTF indicate that GPR56 might exist in different activation states, although the signaling mechanisms of GPR56 in cancer have not been elucidated. The NTF of GPR56 binds to tissue transglutaminase (TG2) [105] and collagen III [108]. The interaction between GPR56 and collagen III in cancer progression is not reported. TG2 is a cross-linking enzyme in the ECM and thought to play pleiotropic roles in cancer progression [147]. The interaction of GPR56 with TG2 in melanoma growth was evaluated recently in a xenograft model using the immunodeficient Tg2 −/− mice [60]. TG2-knockdown melanomas growing in these mice are depleted of TG2 in both cancer cells and stroma and found to be much smaller than the control tumors growing in Tg2 +/+ mice, arguing that TG2 promotes melanoma growth. This tumor-promoting function is abolished by GPR56 overexpression, probably via receptor-mediated TG2 internalization from the cell surface [60]. Whether GPR56 functions similarly in metastasis and in other cancer types remains to be determined.
In contrast to the situation in melanoma, GPR56 is often found upregulated in other cancer types relative to the corresponding normal tissues [32, 43, 148, 149], so perhaps its function in cancer is cell of origin and/or stage specific. Knocking down of GPR56 induces transient apoptosis and reduces the in vitro growth of several cancer cell lines derived from colon (HCT116), melanoma (M14), and cervix (HeLa). Notwithstanding this pro-apoptotic response, the authors were able to derive stable GPR56 knockdown in all the cancer cell lines they tested, including those from the cervix (HeLa), colon (HCT116), melanoma (A2058 and M14), ovary (OVCAR3 and OVCAR8), prostate (PC3), pancreas (AsPC1), and lung (NCI-H460). A reduction in anchorage-independent growth in vitro was observed in all the knockdown lines, and decreased tumor growth in vivo was observed in the knockdown of A2058, PC3, and HCT116 cell lines [32]. This study suggests that GPR56 plays a tumor-promoting role in certain cancer cells. Similar pro-proliferation effects of GPR56 are observed in EVI1high leukemia cell lines AML1 and HNT34, in which knocking down of GPR56 enriches cells at the subG1 phase of the cell cycle and induces apoptosis [22].
The discrepancies among the above studies need to be followed up in future investigations. It is possible that GPR56 has both pro- and anti-growth functions during cancer progression, for example, via distinct binding partners. In fact, in a recent study [60], knocking down of both GPR56 and TG2 leads to a much more severe reduction in subcutaneous growth of MC-1 cells than TG2-knockdown alone, suggesting that GPR56 may have tumor-promoting functions in MC-1 cells in the absence of TG2. This effect of GPR56 was not observed in the WM115 melanoma cell line, pointing again to the complexity of GPR56 function in cancer.
Very recently, GPR56 was identified as a novel and stable marker for leukemic subpopulations with high repopulating capacity, a key feature attributed to leukemia stem cells, for the majority of acute myeloid leukemia (AML) samples [30]. High GPR56 expression was significantly associated with high-risk genetic subgroups and poor outcome.
The second member of the ADGRG group, GPR64, was found upregulated in Ewing sarcomas (ES) compared with normal tissues and other sarcomas [68]. Gpr64 knockdown in Ewing sarcoma lines led to a reduction in tumor growth and metastasis. This tumor-promoting function of Gpr64 was mediated by the induction of placental growth factor (PGF) and matrix metalloproteinase 1 (MMP1) [68]. The mRNA of GPR64 was detected at high levels in cell lines from prostate cancer, non-small cell lung cancer (NSCLC), and melanomas and at moderate to low levels in cell lines from brain, ovary, breast, and colon cancers [68]. It was also identified as a marker for a subgroup of medulloblastomas characterized by overactive WNT signaling [87]. GPR64 silencing in the highly motile cancer cell lines Hs578T and MDA-MB-231 resulted in a reduction of cell adhesion and migration [150].
ADGRG4 (GPR112) was identified as a marker for neuroendocrine carcinoma cells [151] and was predicted to be one of the candidate genes for targeted therapy of ileal carcinoids [50].
None of the other ADGRG members (ADGRG3, GPR97; ADGRG5, GPR114; GPR126; ADGRG7, GPR128) have been reported to affect tumorigenesis or be dysregulated in cancer samples.
7.7 ADGRL
The CCLE predicts high expression of LPHN1 and LPHN2 in many tumors [11]. A gene identification study in breast tumors led to Latrophilin- 1 [59]. Indeed, a number of breast tumor cell lines apparently overexpress the gene [152]. An invasion-associated four-gene signature including Latrophilin-1 obtained from the NCI-60 cell line panel has significant prediction in non-small cell lung cancer (NSCLC) [40]. A genome-wide screen between a recurrent muscle-invasive cisplatin-resistant urothelial bladder carcinoma and its adjacent non-tumor tissue found cancer-associated alternative splicing with differential exon usage for LPHN2 [153]. Linkage studies have revealed numerous Latrophilin- 3 loss-of-function mutations in breast, lung, ovarian, and prostate cancers [21].
ELTD1 is a regulator of physiological and tumor angiogenesis in vitro and in vivo [34]. ELTD1 showed higher expression on endothelium in peritumoral vessels compared to vessels of matched normal tissues [34]. Silencing of ELTD1 in human ovarian and colorectal cancer xenografts implanted in mice inhibited tumor growth [34]. The transcriptome of the microvasculature associated with glioblastoma identified ETLD1 as 1 of 95 upregulated genes [35]. Indeed, ELTD1 can be used as a vascular biomarker in glioblastoma [84]. It displays higher expression in high-grade compared with low-grade gliomas. miR-139-5p suppresses glioma cell proliferation by targeting ELTD1 and regulating the cell cycle [103].
7.8 ADGRV
To identify molecular biomarkers associated with low-grade glioma-associated epileptic seizures, one of the initial symptoms of this tumor, RNA sequence data were collected [154]. A lower expression level of ADGRV1 (VLGR1) was found in patients with epileptic seizures compared to seizure-free patients and was confirmed by quantitative RT-PCR.
8 Tumor Therapy and Adhesion GPCRs
Accumulating evidence of the direct involvement of aGPCRs in tumor pathogenesis provides a solid foundation for their further study as potential therapeutic targets. Some aGPCRs are silenced with tumor formation and appear to have tumor suppressor activity (BAI1), while others are overexpressed in cancer and may act as oncogenic factors (CD97, GPR116, ELTD1).
The observation that BAI1 is epigenetically silenced in malignant glioma [72, 155], along with the recent findings of multiple somatic point mutations in the BAI1–3s in several cancers [21, 122], suggests that tumorigenesis may select for BAI1 silencing or inactivation. Importantly, exogenous restoration of BAI1 expression reduces growth and vascularization of tumors derived from gliomas and pancreatic and renal cell carcinomas [36, 119, 156, 157]. Taken together, these studies suggest that BAI1 fulfills the criteria for a bona fide tumor suppressor, and there is significant potential for BAI1 and its extracellular fragments as therapies for the treatment of human cancers. BAI1 is silenced in glioblastoma multiforme due to methylation of a CpG island in the gene regulatory region, which leads to binding of methyl-CpG-binding domain protein 2 (MBD2) and transition to a suppressive chromatin conformation [72]. Treatment of glioma cells with 5-aza-2′-deoxycytidine (5-Aza-dC) or knockdown of MBD2 by shRNA resulted in reactivation of BAI1 expression and restoration of BAI1 functional activity in that it conferred potent anti-angiogenic activity to glioma cell conditioned media in vitro and in vivo [72]. These findings have therapeutic implications since inhibiting MBD2 could offer a strategy to reactivate BAI1 expression and suppress tumor growth. Sequence-specific antisense inhibitors of MBD2 have been shown to inhibit both anchorage-independent growth of human cancer cell lines in vitro and the growth of human tumor xenografts in vivo [158, 159]. At present, epigenetic approaches in cancer therapy have focused primarily on inhibitors of the DNA methyltransferases and histone modifiers (e.g., HDACs). Thus, targeting MBD2 to reactivate BAI1 may represent a novel promising cancer therapeutic intervention.
In contrast to BAI1, some aGPCRs are overexpressed in tumors. Expression of CD97 was found in human thyroid carcinomas, but not in the corresponding normal thyrocytes [16, 57]. In human thyroid cancer cell lines, CD97 depletion reduced Rho-GTP and decreased lysophosphatidic acid (LPA)-stimulated invasion [57]. Similarly, GPR116 protein expression correlates with clinical progression stages in breast cancer and peaks in tumors with distant metastases [20, 65]. Knockdown of GPR116 in highly metastatic breast cancer cells (MDA-MB-231) suppressed cell migration, invasion, and metastasis in vivo [65]. These studies suggest that modulation of aGPCR expression in tumors may be effective therapeutics. A recent study identified ELTD1 as a novel angiogenesis regulator [34, 35, 84]. ELTD1 is upregulated in tumor endothelial cells and is a good prognostic marker. Moreover, targeting Eltd1 blocks tumor angiogenesis and substantially inhibits tumor growth in vivo [34]. The function of GPR56 in different cancer types was found to differ, with both pro- and anti-tumorigenic properties, so the therapeutic implication of targeting GPR56 in cancer in general awaits careful evaluation. ADGRG4 (GPR112) was reported to serve as a marker for neuroendocrine carcinoma cells [151] and proposed a therapeutic target for treating ileal carcinoids [50].
In summary, alterations in the expression pattern of different aGPCRs have been documented in several cancers, suggesting that they may potentially serve as biomarkers of disease or therapeutic targets. Undoubtedly, the interest in the therapeutic targeting of the aGPCR family will continue to grow in the upcoming years, but first requires a more comprehensive characterization of their fundamental biological function.
Abbreviations
- CCLE:
-
Cancer Cell Line Encyclopedia
- CTF:
-
C-terminal fragment
- ECD:
-
Extracellular domain
- ECM:
-
Extracellular matrix
- GAIN:
-
GPCR autoproteolysis inducing
- GPS:
-
GPCR-proteolytic site
- NTF:
-
N-terminal fragment
- TG2:
-
Tissue transglutaminase
- TSR:
-
Thrombospondin repeat
References
Krishnan A, Nijmeijer S, de Graaf C, Schiöth HB (2016) Classification, nomenclature and structural aspects of adhesion GPCRs. In: Langenhan T, Schöneberg T (eds) Adhesion G protein-coupled receptors: molecular, physiological and pharmacological principles in health and disease. Springer, Heidelberg
Kovacs P, Schöneberg T (2016) The relevance of genomic signatures at adhesion GPCR loci in humans. In: Langenhan T, Schöneberg T (eds) Adhesion G protein-coupled receptors: molecular, physiological and pharmacological principles in health and disease. Springer, Heidelberg
Araç D, Sträter N, Seiradake E (2016) Understanding the structural basis of adhesion GPCR functions. In: Langenhan T, Schöneberg T (eds) Adhesion G protein-coupled receptors: molecular, physiological and pharmacological principles in health and disease. Springer, Heidelberg
Nijmeijer S, Wolf S, Ernst OP, de Graaf C (2016) 7TM domain structure of adhesion GPCRs. In: Langenhan T, Schöneberg T (eds) Adhesion G protein-coupled receptors: molecular, physiological and pharmacological principles in health and disease. Springer, Heidelberg
Nieberler M, Kittel RJ, Petrenko AG, Lin H-H, Langenhan T (2016) Control of adhesion GPCR function through proteolytic processing. In: Langenhan T, Schöneberg T (eds) Adhesion G protein-coupled receptors: molecular, physiological and pharmacological principles in health and disease. Springer, Heidelberg
Langenhan T, Aust G, Hamann J (2013) Sticky signaling – Adhesion class G protein-coupled receptors take the stage. Sci Signal 6(276):re3. doi:10.1126/scisignal.2003825
Liebscher I, Schon J, Petersen SC, Fischer L, Auerbach N, Demberg LM et al (2014) A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep 9:2018–2026
Liebscher I, Schöneberg T (2016) Tethered agonism: a common activation mechanism of adhesion GPCRs. In: Langenhan T, Schöneberg T (eds) Adhesion G protein-coupled receptors: molecular, physiological and pharmacological principles in health and disease. Springer, Heidelberg
Kishore A, Hall RA (2016) Versatile signaling activity of adhesion GPCRs. In: Langenhan T, Schöneberg T (eds) Adhesion G protein-coupled receptors: molecular, physiological and pharmacological principles in health and disease. Springer, Heidelberg
Scholz N, Monk KR, Kittel RJ, Langenhan T (2016) Adhesion GPCRs as a putative class of metabotropic mechanosensors. In: Langenhan T, Schöneberg T (eds) Adhesion G protein-coupled receptors: molecular, physiological and pharmacological principles in health and disease. Springer, Heidelberg
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S et al (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483:603–607
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6(269):pl1. doi:10.1126/scisignal.2004088
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A et al (2015) Proteomics. Tissue-based map of the human proteome. Science 347(6220):1260419. doi:10.1126/science.1260419
Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E, Martinez-Torteya A, Chacolla-Huaringa R, Rodriguez-Barrientos A et al (2013) SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One 8(9), e74250. doi:10.1371/journal.pone.0074250
Aust G, Eichler W, Laue S, Lehmann I, Heldin N-E, Lotz O et al (1997) CD97: a dedifferentiation marker in human thyroid carcinomas. Cancer Res 57:1798–1806
Zendman AJ, Cornelissen IM, Weidle UH, Ruiter DJ, van Muijen GN (1999) TM7XN1, a novel human EGF-TM7-like cDNA, detected with mRNA differential display using human melanoma cell lines with different metastatic potential. FEBS Lett 446:292–298
Mallawaaratchy DM, Buckland ME, McDonald KL, Li CC, Ly L, Sykes EK et al (2015) Membrane proteome analysis of glioblastoma cell invasion. J Neuropathol Exp Neurol 74:425–441
Coustan-Smith E, Song G, Clark C, Key L, Liu P, Mehrpooya M et al (2011) New markers for minimal residual disease detection in acute lymphoblastic leukemia. Blood 117:6267–6276
Davidson B, Stavnes HT, Risberg B, Nesland JM, Wohlschlaeger J, Yang Y et al (2012) Gene expression signatures differentiate adenocarcinoma of lung and breast origin in effusions. Hum Pathol 43:684–694
Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM et al (2010) Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466:869–873
Saito Y, Kaneda K, Suekane A, Ichihara E, Nakahata S, Yamakawa N et al (2013) Maintenance of the hematopoietic stem cell pool in bone marrow niches by EVI1-regulated GPR56. Leukemia 27:1637–1649
Silveira VS, Scrideli CA, Moreno DA, Yunes JA, Queiroz RG, Toledo SC et al (2013) Gene expression pattern contributing to prognostic factors in childhood acute lymphoblastic leukemia. Leuk Lymphoma 54:310–314
Charfi C, Edouard E, Rassart E (2014) Identification of GPM6A and GPM6B as potential new human lymphoid leukemia-associated oncogenes. Cell Oncol 37:179–191
Kaucka M, Plevova K, Pavlova S, Janovska P, Mishra A, Verner J et al (2013) The planar cell polarity pathway drives pathogenesis of chronic lymphocytic leukemia by the regulation of B-lymphocyte migration. Cancer Res 73:1491–1501
Mirkowska P, Hofmann A, Sedek L, Slamova L, Mejstrikova E, Szczepanski T et al (2013) Leukemia surfaceome analysis reveals new disease-associated features. Blood 121:e149–e159
Del Giudice I, Messina M, Chiaretti S, Santangelo S, Tavolaro S, De Propris MS et al (2012) Behind the scenes of non-nodal MCL: downmodulation of genes involved in actin cytoskeleton organization, cell projection, cell adhesion, tumour invasion, TP53 pathway and mutated status of immunoglobulin heavy chain genes. Br J Haematol 156:601–611
Bonardi F, Fusetti F, Deelen P, van Gosliga D, Vellenga E, Schuringa JJ (2013) A proteomics and transcriptomics approach to identify leukemic stem cell (LSC) markers. Mol Cell Proteomics 12:626–637
Wobus M, Bornhauser M, Jacobi A, Krater M, Otto O, Ortlepp C et al (2015) Association of the EGF-TM7 receptor CD97 expression with FLT3-ITD in acute myeloid leukemia. Oncotarget 6:38804–38815
Pabst C, Bergeron A, Lavallee VP, Yeh J, Gendron P, Norddahl GL et al (2016) GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood 27:2018–27
Hricik T, Federici G, Zeuner A, Alimena G, Tafuri A, Tirelli V et al (2013) Transcriptomic and phospho-proteomic analyzes of erythroblasts expanded in vitro from normal donors and from patients with polycythemia vera. Am J Hematol 88:723–729
Ke N, Sundaram R, Liu G, Chionis J, Fan W, Rogers C et al (2007) Orphan G protein-coupled receptor GPR56 plays a role in cell transformation and tumorigenesis involving the cell adhesion pathway. Mol Cancer Ther 6:1840–1850
Vallon M, Essler M (2006) Proteolytically processed soluble tumor endothelial marker (TEM) 5 mediates endothelial cell survival during angiogenesis by linking integrin alpha(v)beta3 to glycosaminoglycans. J Biol Chem 281:34179–34188
Masiero M, Simoes FC, Han HD, Snell C, Peterkin T, Bridges E et al (2013) A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 24:229–241
Dieterich LC, Mellberg S, Langenkamp E, Zhang L, Zieba A, Salomaki H et al (2012) Transcriptional profiling of human glioblastoma vessels indicates a key role of VEGF-A and TGFbeta2 in vascular abnormalization. J Pathol 228:378–390
Fukushima Y, Oshika Y, Tsuchida T, Tokunaga T, Hatanaka H, Kijima H et al (1998) Brain-specific angiogenesis inhibitor 1 expression is inversely correlated with vascularity and distant metastasis of colorectal cancer. Int J Oncol 13:967–970
Hatanaka H, Oshika Y, Abe Y, Yoshida Y, Hashimoto T, Handa A et al (2000) Vascularization is decreased in pulmonary adenocarcinoma expressing brain-specific angiogenesis inhibitor 1 (BAI1). Int J Mol Med 5:181–183
Hao C, Wang L, Peng S, Cao M, Li H, Hu J et al (2015) Gene mutations in primary tumors and corresponding patient-derived xenografts derived from non-small cell lung cancer. Cancer Lett 357:179–185
Guo R, Wu G, Li H, Qian P, Han J, Pan F et al (2013) Promoter methylation profiles between human lung adenocarcinoma multidrug resistant A549/cisplatin (A549/DDP) cells and its progenitor A549 cells. Biol Pharm Bull 36:1310–1316
Hsu YC, Yuan S, Chen HY, Yu SL, Liu CH, Hsu PY et al (2009) A four-gene signature from NCI-60 cell line for survival prediction in non-small cell lung cancer. Clin Cancer Res 15:7309–7315
Bari MF, Brown H, Nicholson AG, Kerr KM, Gosney JR, Wallace WA et al (2014) BAI3, CDX2 and VIL1: a panel of three antibodies to distinguish small cell from large cell neuroendocrine lung carcinomas. Histopathology 64:547–556
Steinert M, Wobus M, Schütz A, Aust G (2002) CD97, but not its closely related EGF-TM7 family member EMR2, is expressed on gastric pancreatic and esophageal carcinomas. Am J Clin Pathol 118(5):699–707
Kausar T, Sharma R, Hasan MR, Tripathi SC, Saraya A, Chattopadhyay TK et al (2011) Clinical significance of GPR56, transglutaminase 2, and NF-kappaB in esophageal squamous cell carcinoma. Cancer Invest 29:42–48
Miyamoto N, Yamamoto H, Taniguchi H, Miyamoto C, Oki M, Adachi Y et al (2007) Differential expression of angiogenesis-related genes in human gastric cancers with and those without high-frequency microsatellite instability. Cancer Lett 254:42–53
Miao R, Guo X, Zhi Q, Shi Y, Li L, Mao X et al (2013) VEZT, a novel putative tumor suppressor, suppresses the growth and tumorigenicity of gastric cancer. PLoS One 8:e74409
Yoshida Y, Oshika Y, Fukushima Y, Tokunaga T, Hatanaka H, Kijima H et al (1999) Expression of angiostatic factors in colorectal cancer. Int J Oncol 15:1221–1225
Katoh M, Katoh M (2007) Comparative integromics on non-canonical WNT or planar cell polarity signaling molecules: transcriptional mechanism of PTK7 in colorectal cancer and that of SEMA6A in undifferentiated ES cells. Int J Mol Med 20:405–409
Steinert M, Wobus M, Boltze C, Schütz A, Wahlbuhl M, Hamann J et al (2002) Expression and regulation of CD97 in colorectal carcinoma cell lines and tumor tissues. Am J Pathol 161:1657–1667
Galle J, Sittig D, Hanisch I, Wobus M, Wandel E, Loeffler M et al (2006) Individual cell-based models of tumor–environment interactions. Multiple effects of CD97 on tumor invasion. Am J Pathol 169:1802–1811
Nilsson O (2013) Profiling of ileal carcinoids. Neuroendocrinology 97:7–18
Ammerpohl O, Pratschke J, Schafmayer C, Haake A, Faber W, von Kampen O et al (2012) Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma. Int J Cancer 130:1319–1328
Tomimaru Y, Koga H, Yano H, de la Monte S, Wands JR, Kim M (2013) Upregulation of T-cell factor-4 isoform-responsive target genes in hepatocellular carcinoma. Liver Int 33:1100–1112
Wu J, Lei L, Wang S, Gu D, Zhang J (2012) Immunohistochemical expression and prognostic value of CD97 and its ligand CD55 in primary gallbladder carcinoma. J Biomed Biotechnol 2012:587672. doi:10.1155/2012/587672
He Z, Wu H, Jiao Y, Zheng J (2015) Expression and prognostic value of CD97 and its ligand CD55 in pancreatic cancer. Oncol Lett 9:793–797
Huang Y, Fan J, Yang J, Zhu GZ (2008) Characterization of GPR56 protein and its suppressed expression in human pancreatic cancer cells. Mol Cell Biochem 308:133–139
Maina EN, Morris MR, Zatyka M, Raval RR, Banks RE, Richards FM et al (2005) Identification of novel VHL target genes and relationship to hypoxic response pathways. Oncogene 24:4549–4558
Ward Y, Lake R, Martin PL, Killian K, Salerno P, Wang T et al (2013) CD97 amplifies LPA receptor signaling and promotes thyroid cancer progression in a mouse model. Oncogene 32:2726–2738
Ward Y, Lake R, Yin JJ, Heger CD, Raffeld M, Goldsmith PK et al (2011) LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res 71:7301–7311
White GR, Varley JM, Heighway J (1998) Isolation and characterization of a human homologue of the latrophilin gene from a region of 1p31.1 implicated in breast cancer. Oncogene 17:3513–3519
Yang L, Friedland S, Corson N, Xu L (2014) GPR56 inhibits melanoma growth by internalizing and degrading its ligand TG2. Cancer Res 74:1–10
Yang L, Chen G, Mohanty S, Scott G, Fazal F, Rahman A et al (2011) GPR56 regulates VEGF production and angiogenesis during melanoma progression. Cancer Res 71:5558–5568
Meisen WH, Dubin S, Sizemore ST, Mathsyaraja H, Thies K, Lehman NL et al (2015) Changes in BAI1 and nestin expression are prognostic indicators for survival and metastases in breast cancer and provide opportunities for dual targeted therapies. Mol Cancer Ther 14:307–314
Liao S, Desouki MM, Gaile DP, Shepherd L, Nowak NJ, Conroy J et al (2012) Differential copy number aberrations in novel candidate genes associated with progression from in situ to invasive ductal carcinoma of the breast. Genes Chromosomes Cancer 51:1067–1078
Davies JQ, Lin HH, Stacey M, Yona S, Chang GW, Gordon S et al (2011) Leukocyte adhesion-GPCR EMR2 is aberrantly expressed in human breast carcinomas and is associated with patient survival. Oncol Rep 25:619–627
Tang X, Jin R, Qu G, Wang X, Li Z, Yuan Z et al (2013) GPR116, an adhesion G-protein-coupled receptor, promotes breast cancer metastasis via the Galphaq-p63RhoGEF-Rho GTPase pathway. Cancer Res 73:6206–6218
Zyryanova T, Schneider R, Adams V, Sittig D, Kerner C, Gebhardt C et al (2014) Skeletal muscle expression of the adhesion-GPCR CD97: CD97 deletion induces an abnormal structure of the sarcoplasmatic reticulum but does not impair skeletal muscle function. PLoS One 9, e100513
Aust G, Wandel E, Boltze C, Sittig D, Schütz A, Horn LC et al (2006) Diversity of CD97 in smooth muscle cells (SMCs). Cell Tissue Res 323:1–9
Richter GH, Fasan A, Hauer K, Grunewald TG, Berns C, Rossler S et al (2013) G-Protein coupled receptor 64 promotes invasiveness and metastasis in Ewing sarcomas through PGF and MMP1. J Pathol 230:70–81
Kaur B, Brat DJ, Devi NS, Van Meir EG (2005) Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene 24:3632–3642
Kaur B, Cork SM, Sandberg EM, Devi NS, Zhang Z, Klenotic PA et al (2009) Vasculostatin inhibits intracranial glioma growth and negatively regulates in vivo angiogenesis through a CD36-dependent mechanism. Cancer Res 69:1212–1220
Cork SM, Kaur B, Devi NS, Cooper L, Saltz JH, Sandberg EM et al (2012) A proprotein convertase/MMP-14 proteolytic cascade releases a novel 40 kDa vasculostatin from tumor suppressor BAI1. Oncogene 31:5144–5152
Zhu D, Hunter SB, Vertino PM, Van Meir EG (2011) Overexpression of MBD2 in glioblastoma maintains epigenetic silencing and inhibits the antiangiogenic function of the tumor suppressor gene BAI1. Cancer Res 71:5859–5870
Kaur B, Brat DJ, Calkins CC, Van Meir EG (2003) Brain angiogenesis inhibitor 1 is differentially expressed in normal brain and glioblastoma independently of p53 expression. Am J Pathol 162:19–27
Ivan ME, Safaee M, Oh T, Clark AJ, Sun MZ, Kim J et al (2015) Epidermal growth factor-like module containing mucin-like hormone receptor 2 expression in gliomas. J Neurooncol 121:53–61
Rutkowski MJ, Sughrue ME, Kane AJ, Kim JM, Bloch O, Parsa AT (2011) Epidermal growth factor module-containing mucin-like receptor 2 is a newly identified adhesion G protein-coupled receptor associated with poor overall survival and an invasive phenotype in glioblastoma. J Neurooncol 105:165–171
Kane AJ, Sughrue ME, Rutkowski MJ, Phillips JJ, Parsa AT (2010) EMR-3: a potential mediator of invasive phenotypic variation in glioblastoma and novel therapeutic target. Neuroreport 21:1018–1022
Safaee M, Clark AJ, Oh MC, Ivan ME, Bloch O, Kaur G et al (2013) Overexpression of CD97 confers an invasive phenotype in glioblastoma cells and is associated with decreased survival of glioblastoma patients. PLoS One 8(4), e62765. doi:10.1371/journal.pone.0062765
Liu JK, Lubelski D, Schonberg DL, Wu Q, Hale JS, Flavahan WA et al (2014) Phage display discovery of novel molecular targets in glioblastoma-initiating cells. Cell Death Differ 21:1325–1339
Safaee M, Fakurnejad S, Bloch O, Clark AJ, Ivan ME, Sun MZ et al (2015) Proportional upregulation of CD97 isoforms in glioblastoma and glioblastoma-derived brain tumor initiating cells. PLoS One 10, e0111532
Chidambaram A, Fillmore HL, Van Meter TE, Dumur CI, Broaddus WC (2012) Novel report of expression and function of CD97 in malignant gliomas: correlation with Wilms tumor 1 expression and glioma cell invasiveness. J Neurosurg 116:843–853
Somasundaram A, Ardanowski N, Opalak CF, Fillmore HL, Chidambaram A, Broaddus WC (2014) Wilms tumor 1 gene, CD97, and the emerging biogenetic profile of glioblastoma. Neurosurg Focus 37:E14
Shashidhar S, Lorente G, Nagavarapu U, Nelson A, Kuo J, Cummins J et al (2005) GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene 24:1673–1682
Taatjes DJ (2010) The human Mediator complex: a versatile, genome-wide regulator of transcription. Trends Biochem Sci 35:315–322
Towner RA, Jensen RL, Colman H, Vaillant B, Smith N, Casteel R et al (2013) ELTD1, a potential new biomarker for gliomas. Neurosurgery 72:77–90
Cork SM, Van Meir EG (2011) Emerging roles for the BAI1 protein family in the regulation of phagocytosis, synaptogenesis, neurovasculature, and tumor development. J Mol Med (Berl) 89:743–752
Fujii K, Kurozumi K, Ichikawa T, Onishi M, Shimazu Y, Ishida J et al (2013) The integrin inhibitor cilengitide enhances the anti-glioma efficacy of vasculostatin-expressing oncolytic virus. Cancer Gene Ther 20:437–444
Whittier KL, Boese EA, Gibson-Corley KN, Kirby PA, Darbro BW, Qian Q et al (2013) G-protein coupled receptor expression patterns delineate medulloblastoma subgroups. Acta Neuropathol Commun 1:66
Boligan KF, Mesa C, Fernandez LE, von Gunten S (2015) Cancer intelligence acquired (CIA): tumor glycosylation and sialylation codes dismantling antitumor defense. Cell Mol Life Sci 72:1231–1248
Pinho SS, Reis CA (2015) Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 15:540–555
Durrant LG, Chapman MA, Buckley DJ, Spendlove I, Robins RA, Armitage NC (2003) Enhanced expression of the complement regulatory protein CD55 predicts a poor prognosis in colorectal cancer patients. Cancer Immunol Immunother 52:638–642
Liu Y, Chen L, Peng S, Chen Z, Gimm O, Finke R et al (2005) The expression of CD97EGF and its ligand CD55 on marginal epithelium is related to higher stage and depth of tumor invasion of gastric carcinomas. Oncol Rep 14:1413–1420
Loberg RD, Wojno KJ, Day LL, Pienta KJ (2005) Analysis of membrane-bound complement regulatory proteins in prostate cancer. Urology 66:1321–1326
Han SL, Xu C, Wu XL, Li JL, Liu Z, Zeng QQ (2010) The impact of expressions of CD97 and its ligand CD55 at the invasion front on prognosis of rectal adenocarcinoma. Int J Colorectal Dis 25:695–702
Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z et al (2007) BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450:430–434
Okajima D, Kudo G, Yokota H (2010) Brain-specific angiogenesis inhibitor 2 (BAI2) may be activated by proteolytic processing. J Recept Signal Transduct Res 30:143–153
Gray JX, Haino M, Roth MJ, Maguire JE, Jensen PN, Yarme A et al (1996) CD97 is a processed, seven-transmembrane, heterodimeric receptor associated with inflammation. J Immunol 157:5438–5447
Abe J, Fukuzawa T, Hirose S (2002) Cleavage of Ig-Hepta at a “SEA” module and at a conserved G protein-coupled receptor proteolytic site. J Biol Chem 277:23391–23398
Moriguchi T, Haraguchi K, Ueda N, Okada M, Furuya T, Akiyama T (2004) DREG, a developmentally regulated G protein-coupled receptor containing two conserved proteolytic cleavage sites. Genes Cells 9:549–560
Krasnoperov V, Deyev IE, Serova OV, Xu C, Lu Y, Buryanovsky L et al (2009) Dissociation of the subunits of the calcium-independent receptor of alpha-latrotoxin as a result of two-step proteolysis. Biochemistry 48:3230–3238
Wang T, Ward Y, Tian L, Lake R, Guedez L, Stetler-Stevenson WG et al (2004) CD97, an adhesion receptor on inflammatory cells, stimulates angiogenesis through binding integrin counter receptors on endothelial cells. Blood 105:2836–2844
Hamann J, Wishaupt JO, van Lier RA, Smeets TJ, Breedveld FC, Tak PP (1999) Expression of the activation antigen CD97 and its ligand CD55 in rheumatoid synovial tissue. Arthritis Rheum 42:650–658
de Groot DM, Vogel G, Dulos J, Teeuwen L, Stebbins K, Hamann J et al (2009) Therapeutic antibody targeting of CD97 in experimental arthritis: the role of antigen expression, shedding, and internalization on the pharmacokinetics of anti-CD97 monoclonal antibody 1B2. J Immunol 183:4127–4134
Dai S, Wang X, Li X, Cao Y (2015) MicroRNA-139-5p acts as a tumor suppressor by targeting ELTD1 and regulating cell cycle in glioblastoma multiforme. Biochem Biophys Res Commun 467:204–210
Hsiao CC, Keysselt K, Chen HY, Sittig D, Hamann J, Lin HH et al (2015) The Adhesion GPCR CD97 inhibits apoptosis. Int J Biochem Cell Biol 65:197–208
Xu L, Begum S, Hearn JD, Hynes RO (2006) GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci U S A 103:9023–9028
Fu JF, Yen TH, Chen Y, Huang YJ, Hsu CL, Liang DC et al (2013) Involvement of Gpr125 in the myeloid sarcoma formation induced by cooperating MLL/AF10(OM-LZ) and oncogenic KRAS in a mouse bone marrow transplantation model. Int J Cancer 133:1792–1802
Mouw JK, Ou G, Weaver VM (2014) Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol 15:771–785
Luo R, Jin Z, Deng Y, Strokes N, Piao X (2012) Disease-associated mutations prevent GPR56-collagen III interaction. PLoS One 1, e29818. doi:10.1371/journal.pone.0029818
Koirala S, Jin Z, Piao X, Corfas G (2009) GPR56-regulated granule cell adhesion is essential for rostral cerebellar development. J Neurosci 29:7439–7449
Stacey M, Chang GW, Davies JQ, Kwakkenbos MJ, Sanderson RD, Hamann J et al (2003) The epidermal growth factor-like domains of the human EMR2 receptor mediate cell attachment through chondroitin sulphate glycosaminoglycans. Blood 102:2916–2924
Paavola KJ, Sidik H, Zuchero JB, Eckart M, Talbot WS (2014) Type IV collagen is an activating ligand for the adhesion G protein-coupled receptor GPR126. Sci Signal 7(338):ra76. doi:10.1126/scisignal.2005347
Petersen SC, Luo R, Liebscher I, Giera S, Jeong SJ, Mogha A et al (2015) The adhesion GPCR GPR126 has distinct, domain-dependent functions in Schwann cell development mediated by interaction with laminin-211. Neuron 85:755–769
Strutt D, Schnabel R, Fiedler F, Prömel S (2016) Adhesion GPCRs govern polarity of epithelia and cell migration. In: Langenhan T, Schöneberg T (eds) Adhesion G protein-coupled receptors: molecular, physiological and pharmacological principles in health and disease. Springer, Heidelberg
Hsiao CC, Wang WC, Kuo WL, Chen HY, Chen TC, Hamann J et al (2014) CD97 inhibits cell migration in human fibrosarcoma cell by modulating TIMP-2/MT1-MMP/MMP-2 activity. FEBS J 281:4878–4891
Musa G, Engel FB, Niaudet C (2016) Heart development, angiogenesis and blood-brain barrier function is modulated by adhesion GPCRs. In: Langenhan T, Schöneberg T (eds) Adhesion G protein-coupled receptors: molecular, physiological and pharmacological principles in health and disease. Springer, Heidelberg
Nishimori H, Shiratsuchi T, Urano T, Kimura Y, Kiyono K, Tatsumi K et al (1997) A novel brain-specific p53-target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene 15:2145–2150
Kang X, Xiao X, Harata M, Bai Y, Nakazaki Y, Soda Y et al (2006) Antiangiogenic activity of BAI1 in vivo: implications for gene therapy of human glioblastomas. Cancer Gene Ther 13:385–392
Lee JH, Koh JT, Shin BA, Ahn KY, Roh JH, Kim YJ et al (2001) Comparative study of angiostatic and anti-invasive gene expressions as prognostic factors in gastric cancer. Int J Oncol 18:355–361
Kudo S, Konda R, Obara W, Kudo D, Tani K, Nakamura Y et al (2007) Inhibition of tumor growth through suppression of angiogenesis by brain-specific angiogenesis inhibitor 1 gene transfer in murine renal cell carcinoma. Oncol Rep 18:785–791
de Fraipont F, Nicholson AC, Feige JJ, Van Meir EG (2001) Thrombospondins and tumor angiogenesis. Trends Mol Med 7:401–407
Nicholson AC, Malik SB, Logsdon JM Jr, Van Meir EG (2005) Functional evolution of ADAMTS genes: evidence from analyses of phylogeny and gene organization. BMC Evol Biol 5:11
Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD et al (2010) A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463:191–196
Arac D, Boucard AA, Bolliger MF, Nguyen J, Soltis SM, Sudhof TC et al (2012) A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J 31:1364–1378
Zhu D, Van Meir EG (2016) BAI1: from cancer to neurological disease. Oncotarget
St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E et al (2000) Genes expressed in human tumor endothelium. Science 289:1197–1202
Carson-Walter EB, Watkins DN, Nanda A, Vogelstein B, Kinzler KW, St CB (2001) Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res 61:6649–6655
Gao Y, Fan X, Li W, Ping W, Deng Y, Fu X (2014) miR-138-5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating G protein-coupled receptor 124. Biochem Biophys Res Commun 446:179–186
Shiratsuchi T, Nishimori H, Ichise H, Nakamura Y, Tokino T (1997) Cloning and characterization of BAI2 and BAI3, novel genes homologous to brain-specific angiogenesis inhibitor 1 (BAI1). Cytogenet Cell Genet 79:103–108
Koh JT, Kook H, Kee HJ, Seo YW, Jeong BC, Lee JH et al (2004) Extracellular fragment of brain-specific angiogenesis inhibitor 1 suppresses endothelial cell proliferation by blocking alphavbeta5 integrin. Exp Cell Res 294:172–184
Klenotic PA, Huang P, Palomo J, Kaur B, Van Meir EG, Vogelbaum MA et al (2010) Histidine-rich glycoprotein modulates the anti-angiogenic effects of vasculostatin. Am J Pathol 176:2039–2050
Shiratsuchi T, Futamura M, Oda K, Nishimori H, Nakamura Y, Tokino T (1998) Cloning and characterization of BAI-associated protein 1: a PDZ domain-containing protein that interacts with BAI1. Biochem Biophys Res Commun 247:597–604
Stephenson JR, Paavola KJ, Schaefer SA, Kaur B, Van Meir EG, Hall RA (2013) Brain-specific angiogenesis inhibitor-1 signaling, regulation, and enrichment in the postsynaptic density. J Biol Chem 288:22248–22256
Oda K, Shiratsuchi T, Nishimori H, Inazawa J, Yoshikawa H, Taketani Y et al (1999) Identification of BAIAP2 (BAI-associated protein 2), a novel human homologue of hamster IRSp53, whose SH3 domain interacts with the cytoplasmic domain of BAI1. Cytogenet Cell Genet 84:75–82
Kim JC, Kim SY, Roh SA, Cho DH, Kim DD, Kim JH et al (2008) Gene expression profiling: canonical molecular changes and clinicopathological features in sporadic colorectal cancers. World J Gastroenterol 14:6662–6672
Wang Q (2015) Identification of biomarkers for metastatic osteosarcoma based on DNA microarray data. Neoplasma 62:365–371
Aust G, Hamann J, Schilling N, Wobus M (2003) Detection of alternatively spliced EMR2 mRNAs in colorectal tumor cell lines but rare expression of the molecule in the corresponding adenocarcinomas. Virchows Arch 443:32–37
Grone J, Lenze D, Jurinovic V, Hummel M, Seidel H, Leder G et al (2011) Molecular profiles and clinical outcome of stage UICC II colon cancer patients. Int J Colorectal Dis 26:847–858
Liu D, Trojanowicz B, Ye L, Li C, Zhang L, Li X et al (2012) The invasion and metastasis promotion role of CD97 small isoform in gastric carcinoma. PLoS One 7, e39989
Boltze C, Schneider-Stock R, Aust G, Mawrin C, Dralle H, Roessner A et al (2002) CD97, CD95 and Fas-L clearly discriminate between chronic pancreatitis and pancreatic ductal adenocarcinoma in perioperative evaluation of cryocut sections. Pathol Int 52:83–88
Singh V, Singh LC, Vasudevan M, Chattopadhyay I, Borthakar BB, Rai AK et al (2015) Esophageal cancer epigenomics and integrome analysis of genome-wide methylation and expression in high risk northeast Indian population. OMICS 19:688–699
Lu YY, Sweredoski MJ, Huss D, Lansford R, Hess S, Tirrell DA (2013) Prometastatic GPCR CD97 is a direct target of tumor suppressor microRNA-126. ACS Chem Biol 9:334–338
Hamann J, Vogel B, van Schijndel GM, van Lier RA (1996) The seven-span transmembrane receptor CD97 has a cellular ligand (CD55, DAF). J Exp Med 184:1185–1189
Wandel E, Saalbach A, Sittig D, Gebhardt C, Aust G (2012) Thy-1 (CD90) is an interaction partner for CD97 on activated endothelial cells. J Immunol 188:1442–1450
Fukuzawa T, Ishida J, Kato A, Ichinose T, Ariestanti DM, Takahashi T et al (2013) Lung surfactant levels are regulated by Ig-Hepta/GPR116 by monitoring surfactant protein D. PLoS One 8, e69451
Yang MY, Hilton MB, Seaman S, Haines DC, Nagashima K, Burks CM et al (2013) Essential regulation of lung surfactant homeostasis by the orphan G protein-coupled receptor GPR116. Cell Rep 3:1457–64
Bridges JP, Ludwig MG, Mueller M, Kinzel B, Sato A, Xu Y et al (2013) Orphan G protein-coupled receptor GPR116 regulates pulmonary surfactant pool size. Am J Respir Cell Mol Biol 49:348–57
Yang L, Xu L (2012) GPR56 in cancer progression: current status and future perspective. Future Oncol 8:431–440
Mori A, Arii S, Furutani M, Hanaki K, Takeda Y, Moriga T et al (1999) Vascular endothelial growth factor-induced tumor angiogenesis and tumorigenicity in relation to metastasis in a HT1080 human fibrosarcoma cell model. Int J Cancer 80:738–743
Duda DG, Sunamura M, Lozonschi L, Yokoyama T, Yatsuoka T, Motoi F et al (2002) Overexpression of the p53-inducible brain-specific angiogenesis inhibitor 1 suppresses efficiently tumour angiogenesis. Br J Cancer 86:490–496
Peeters MC, Fokkelman M, Boogaard B, Egerod KL, van de Water B, Ijzerman AP et al (2015) The adhesion G protein-coupled receptor G2 (ADGRG2/GPR64) constitutively activates SRE and NFkappaB and is involved in cell adhesion and migration. Cell Signal 27:2579–2588
Leja J, Essaghir A, Essand M, Wester K, Oberg K, Totterman TH et al (2009) Novel markers for enterochromaffin cells and gastrointestinal neuroendocrine carcinomas. Mod Pathol 22:261–272
White GR, Varley JM, Heighway J (2000) Genomic structure and expression profile of LPHH1, a 7TM gene variably expressed in breast cancer cell lines. Biochim Biophys Acta 1491:75–92
Zhang S, Liu Y, Liu Z, Zhang C, Cao H, Ye Y et al (2014) Transcriptome profiling of a multiple recurrent muscle-invasive urothelial carcinoma of the bladder by deep sequencing. PLoS One 9, e91466
Wang Y, Fan X, Zhang W, Zhang C, Wang J, Jiang T et al (2015) Deficiency of very large G-protein-coupled receptor-1 is a risk factor of tumor-related epilepsy: a whole transcriptome sequencing analysis. J Neurooncol 121:609–616
Mosmann TR, Li L, Sad S (1997) Functions of CD8 T-cell subsets secreting different cytokine patterns. Semin Immunol 9:87–92
Nam DH, Park K, Suh YL, Kim JH (2004) Expression of VEGF and brain specific angiogenesis inhibitor-1 in glioblastoma: prognostic significance. Oncol Rep 11:863–869
Yoon KC, Ahn KY, Lee JH, Chun BJ, Park SW, Seo MS et al (2005) Lipid-mediated delivery of brain-specific angiogenesis inhibitor 1 gene reduces corneal neovascularization in an in vivo rabbit model. Gene Ther 12:617–624
Slack A, Bovenzi V, Bigey P, Ivanov MA, Ramchandani S, Bhattacharya S et al (2002) Antisense MBD2 gene therapy inhibits tumorigenesis. J Gene Med 4:381–389
Campbell PM, Bovenzi V, Szyf M (2004) Methylated DNA-binding protein 2 antisense inhibitors suppress tumourigenesis of human cancer cell lines in vitro and in vivo. Carcinogenesis 25:499–507
Acknowledgments
G.A. was supported by grants of the German Research Foundation (AU 132/7-3; FOR2149 Project 8 AU 132/8-1), L.X. by the National Institute of General Medicine Sciences (NIGMS; grant R01GM098591), D.Z. and E.G.V.M. in part by the US National Cancer Institute (grants CA086335 and NS096236), and the Southeastern Brain Tumor, Cure Childhood Cancer, and St. Baldrick’s Foundations.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Aust, G., Zhu, D., Van Meir, E.G., Xu, L. (2016). Adhesion GPCRs in Tumorigenesis. In: Langenhan, T., Schöneberg, T. (eds) Adhesion G Protein-coupled Receptors. Handbook of Experimental Pharmacology, vol 234. Springer, Cham. https://doi.org/10.1007/978-3-319-41523-9_17
Download citation
DOI: https://doi.org/10.1007/978-3-319-41523-9_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41521-5
Online ISBN: 978-3-319-41523-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)