
Abstract
Epidermal growth factor (EGF) module-containing mucin–like receptor 2 (EMR2) is a member of the seven span transmembrane adhesion G-protein coupled receptor subclass. This protein is expressed in a subset of glioblastoma (GBM) cells and associated with an invasive phenotype. The expression pattern and functional significance of EMR2 in low grade or anaplastic astrocytomas is unknown and our goal was to expand and further define EMR2’s role in gliomas with an aggressive invasive phenotype. Using the TCGA survival data we describe EMR2 expression patterns across histologic grades of gliomas and demonstrate an association between increased EMR2 expression and poor survival (p < 0.05). This data supports prior functional data depicting that EMR2-positive neoplasms possess a greater capacity for infiltrative and metastatic spread. Genomic analysis suggests that EMR2 overexpression is associated with the mesenchymal GBM subtype (p < 0.0001). We also demonstrate that immunohistorchemistry is a feasible method for screening GBM patients for EMR2 expression. Protein and mRNA analysis demonstrated variable expression of all isoforms of EMR2 in all glioma grades, however GBM displayed the most diverse isoforms expression pattern as well as the highest expression of the EGF1–5 isoform of EMR2. Finally, a correlation of an increased EMR2 expression after bevacizumab treatment in glioma cells lines is identified. This observation should serve as the impetus for future studies to determine if this up-regulation of EMR2 plays a role in the observation of the diffuse and increasingly invasive recurrence patterns witnessed in a subset of GBM patients after bevacizumab treatment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM) is the most frequent, aggressive, and invasive brain tumor and despite aggressive therapy, prognosis remains poor, with a median survival of <15 months [1]. GBM typically grows by invading surrounding neural structures, and it is arguably this feature that contributes to such a poor prognosis: invasion in gliomas restricts curative resection, limits effective delivery of chemotherapy and radiation, and can activate mechanisms to evade treatment effects [2]. Further understanding of the molecular mechanisms of glioma invasion could prove to have great potential in the treatment of GBM.
Epidermal growth factor (EGF) module-containing mucin-like receptor 2 (EMR2) is a member of the EGF seven span transmembrane (TM7) protein family, which is almost universally characterized by the presence of alternative splicing, producing isoforms with variable number and arrangement of EGF domains [3]. Initially identified due to its high molecular homology to CD97 and similar functioning [4], EMR2 has been demonstrated to mediate cellular adhesion, migration, and signaling [5–9]. In neoplastic processes like breast cancer, EMR2 appears to have a functional role in disease aggressiveness, and relapse-free survival as well as tumor grade [10]. In that manner, EMR2’s associated role with cellular migration, which can increase a tumor invasiveness, is our focus in understanding EMR2’s expression in gliomas [5–9] We therefore continued our prior research, which showed that EMR2 mediates tumor invasion and correlates with poor survival in GBM patients, by focusing on EMR2’s influence on invasion and survival in high-grade gliomas and their genomic subtypes as well as low-grade gliomas(LGG). We hypothesized that, given EMR2’s role in cellular adhesion, migration, and signaling [5–9], EMR2 would likely be up-regulated in tumors with more aggressive genomic subtypes in high-grade gliomas and those with more aggressive phenotypes in LGGs. We also investigated whether EMR2 plays a role in the highly invasive features of recurrence seen after development of resistance to bevacizumab [11–14].
Methods and reagents
Immunohistochemistry
Tumor sections were cut, deparaffinized in xylene, and rehydrated through graded alcohols to deionized water. Antigen retrieval and endogenous peroxidase block was performed. Samples were stained with EMR2 antibody(1:500;Abcam), phosphorlyated-S6 ribosomal protein (P-S6) (1:2,000 m, Cell Signaling), and phosphorhylated PRAS40 (1:2,000 m, Cell Signaling). For tissue array analysis, samples were obtained from the Brain Tissue Research Center(BTRC) at UCSF, under the appropriate institutional review board(IRB) approval. Each tissue had 2 specimens prepped, stained, and independently read by a pathologist. EMR2 expression was recorded and positive staining defined as >25 % and negative staining as ≤25 % cells with EMR2 presence. Normal brain and thyroid tissue served as negative and positive controls, respectively.
Frozen tissue analysis
Flash-frozen glioma specimens were acquired from the UCSF BTRC, including pathologically confirmed GBMs(WHO grade IV), anaplastic astrocytomas(WHO grade III), and LGGs(WHO grade II). Tissue was homogenized to extract RNA and protein. For protein isolation, homogenate was resuspended in 1× RIPA buffer(Cell-Signaling) and placed on a rotating rack for 1 h at 4 °C.
Culturing of Human Glioma Lines
Human glioma cell lines U251 and U87MG were provided by Dr. Russell Pieper (UCSF) and grown in DMEM with 4.5 g/L glucose, 0.584 g/L l-glutamine, 0.11 g/L sodium pyruvate, 3.7 g/L NaHCO3, 10 % FBS, and 1 % penicillin/streptomycin.
Immunoblotting
Cell lysates were generated using 10x lysis buffer (Cell-Signaling) containing 20 mM Tris–HCl(pH7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1 % Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, and 1 μg/ml leupeptin. Protease and phosphatase inhibitor (Roche) and 1 mM phenylmethasnesulfonylfluoride were added. Lysates were cleared of insoluble materials by centrifugation. Protein concentrations were normalized by bicinchoninic acid assay (Thermo Scientific) to 1 μg/μl in 1XSDS loading buffer. Protein samples were heated to 90 °C for 5 min then electrophoresed through a 4-20 % Tris–Glycine(Invitrogen). Resolved proteins were then transferred to an Immun-Blot PVDF membrane, blocked in 5 % milk in Tris-buffered saline and incubated in primary antibody overnight at 4 °C. EMR2 antibody was applied at 1:500(Abcam) and anti-GAPDH antibody at 1:10,000 (cell-vignaling). After incubation a secondary antibody was conjugated to horseradish peroxidase (cell-signaling) diluted to 1:2,000 in 5 % milk. Membranes were developed using the Amersham ECL Western blotting detection system (GE Healthcare).
RNA analysis
RNA was extracted using the RNeasy Mini Kit (Qiagen) and reverse transcribed using Superscript III kit (Invitrogen). Gene expression was detected by PCR using Platinum Taq DNA Polymerase with the following primers: EMR2 (5′-TGGAT-GGCCT-AGAGG-ATGTC/CTTCT-CAGGT-TCCAG-GACCA-3′), EMR2 EGF isoforms (12,345(1–5), 1,235, 125, 12) 5′-TTTCT-GGGGC-AAAAA-CATTC/GCCTG-GCTTG-TAGTC-TCTGC-3′). Primers were selected to identify the EGF-binding domains of EMR2. PCR products were run on a 2 % agarose gel in 1xTAE buffer stained with ethidium bromide and visualized under UV light. EMR2 and isoform expression were confirmed with genome sequencing. For qPCR, RNA was extracted then reverse transcribed using SuperScript® III reverse transcriptase (Life Technologies) to generate a cDNA library. EMR2 was quantified using the primer: 5′-TGGAT-GGCCT-AGAGG-ATGTC/CTTCT-CAGGT-TCCAG-GACCA-3′.
Treatment with bevacizumab
U251 and U87 EMR2-expressing cell lines were grown in culture and left untreated (control) or treated with 0.25 or 0.5 μg/mL of Bevacizumab for 24 h. Cells were harvested and analysis as described above. To model acquired anti-angiogenic therapy resistance, U87MG xenografts were treated with bevacizumab or IgG during serial in vivo passaging of tumors in nude athymic mice. These U87-IgG and U87-BevR xenografts represent a model of paired isogenic bevacizumab-responsive vs. resistant GBM, as described previously [11, 12]. Cells were treated with either IgG or 10 mg/kg of bevacizumab 2x/week. Endpoint was subcutaneous tumor growth to a diameter of 2.1 cm in any dimension, at which point the tumor was collected. RNA was collected utilizing the TRIzol® Plus RNA Purification Kit, after which cDNA was generated with the iScript™ cDNA Synthesis Kit. Protein was extracted as noted above.
Statistical analysis
All statistical analysis was performed using JMP-11.1.1. Kaplan–Meier plots were used to generate survival estimates, and differences were evaluated by log-rank test. qPCR analysis and association of GBM subtypes with EMR2 was performed using ANOVA. Oncoprint gene analysis was performed using z-scores to determine up/down-regulated expression and likelihood ratio analysis was performed comparing gene expression. Statistical significance was defined as p < 0.05.
Results
EMR2 expression inversely correlates with overall and disease-free survival in GBM and low grade glioma
Following Kaplan–Meier analysis of survival data for GBM using the Cancer Genome Atlas (TCGA, Nature 2008) [13] we found that any EMR2 overexpression is found in 49 % of the samples and using a z-score threshold of 1.5 there is an overexpression of EMR2 in 12 % which was significantly associated with decreased disease-free survival (p < 0.01) as well as decreased time until mortality (p < 0.04) (Fig. 1a–b) [13]. The median disease-free survival and overall survival decreased. In low-grade gliomas (LGG) using the TCGA [13] and a z-score threshold of 1.5 we found that there is an overexpression of EMR2 in 6 % of patients and was also significantly associated with decreased time until mortality (p < 0.01) but not disease-free survival (p < 0.36)(data not shown). However at this threshold there were a very small number of tumors with followup data in the TCGA dataset. Therefore we reviewed tumors that show any EMR2 overexpression(9.5 % of cases), and a significant association was noted with decreased disease-free survival (p < 0.025) as well as decreased time until mortality (p < 0.0005) (Fig. 1c, d) [13]. The average z-score of the tumors with EMR2 overexpression was 1.31 and without EMR2 overexpression was −0.41. With increased EMR2 expression, median disease-free survival and overall survival also decreased for LGG’s. Finally RNA expression of EMR2 in various histological grades of gliomas was studied. Six samples of pathologically confirmed WHO grade 2 and grade three tissue samples were prepared and analyzed for EMR2 expression. In Fig. 1e, qPCR analysis revealed significantly higher EMR2 expression in GBM when compared to grade 2 and 3 astrocytomas (p = 0.037). Later, we also show increased protein expression of EMR2 in the GBM samples (Fig. 4).

Kaplan–Meier analysis of TCGA z-score data in April 2014 depicting increased EMR2 expression associated with decreased disease-free survival (a) and overall survival (b) in GBM. Kaplan–Meier analysis depicting increased EMR2 expression associated with decreased disease-free survival (c) and overall survival (d) in LGG. e Quantitative PCR of EMR2 expression in grade 2 astrocytomas, grade 3 astrocytomas, and glioblastoma. Standard deviation noted with error bars
EMR2 overexpressing GBM tumors have similar genomic profiles to the mesenchymal GBM subtype
Recently, Verhaak et al. [14], using data obtained from TCGA [13], re-classified GBMs into four genetic subtypes characterized by aberrations in genes. Using the TCGA, the z-score expression of EMR2 was found for 54 Classical, 56 Mesenchymal, 29 Neural, and 56 Proneural tumors [13]. This search revealed that EMR2 overexpression is most associated with the mesenchymal subtype (p < 0.0001) (Fig. 2a). More specifically, using the TCGA cBioportal gene which are up-regulated or down-regulated in association with EMR2 were identified (Fig. 3b). For instance up-regulated EMR2 associated with a decrease in NF1 (p = 0.0004) and EGFR genes (p = 0.04), which further supports the association with the mesenchymal subtype. In addition, there were alterations with the PTEN, PIP, and AKT1 gene that were also associated with EMR2 (p = 0.002). Because of the association with EGFR and PTEN we included additional genes associated with the PI3K/AKT/PTEN pathway with glioma survival [14–17].

a Plot of EMR2 expression in each glioblastoma subtype using z-score values from TGCA database queried in April 2014. Mesenchymal subtype shows the most consistent up-regulation of EMR2. b OncoPrint plot generate by TCGA database showing tumors with up-regulation and down regulation of EMR2 and other significant genes of glioblastoma subtypes and the AKT/PTEN/PI3K pathway. Likelihood ratio of each genes association with the up/down expression of EMR2

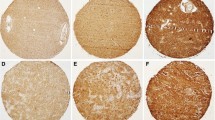
a IHC staining of a low grade glioma and GBM human samples showing negative (<25 %) and positive (≥25 %) expression of EMR2 throughout tumor cells. b Kaplan–Meier analysis comparing post-diagnosis overall survival of the 46 gliobastoma patients where IHC staining was performed. IHC positive staining of EMR2 (blue) and negative staining of EMR2 (red)
Detection of EMR2 in GBM samples can be performed with IHC
Given the association between EMR2 expression and deceased survival, rapid quantification of EMR2 has the potential to provide prognostic data for patients and physicians. To investigate IHC as a possible screening assay for EMR2, we stained 2 representative sections from 46 GBM specimens on a tissue array generated by the UCSF BTRC. Of 92 samples, 26 had positive staining for EMR2 and 66 samples had negative staining for EMR2 (Fig. 3a). Survival data from the time of tissue acquisition was obtained from our tissue bank and used to plot a Kaplan–Meier analysis. Patients with >25 % staining of EMR2 did have a lower mean survival (Fig. 3b), however a log-rank analysis did not find this difference to be statistically significant (p = 0.378). Four LGG specimens were also prepared for IHC, and 1 stained positive for EMR2. The LGG patient who stained positive for EMR2 survived for 9.6 months after tissue acquisition and the patients who stained negative had an average survival of 50.9 months. The sample size was too small to perform statistics.
Variable EMR2 isoform expression occurs in gliomas
Protein and RT-PCR analysis of 18 glioma samples were performed and a variation in the bands expressed was noted. Through sequencing analysis, these PCR products were found to represent the 4 isoforms of EMR2 (Fig. 4a) and are labeled on the agarose gels in Fig. 4b–d. Many of these isoforms were also seen in the protein form in the matched protein samples suggesting that translation of the associated transcript occurred. These were identified based on size. EMR2 (1–5) the only isoform to contain the 4th EGF domain, is expressed in a greatest number of GBM samples.

a EMR2 EGF binding domain variations. b Protein and mRNA EMR2 isoform expression in human grade 2 astrocytomas shown with western blot (top) and RT-PCR blot (bottom). c Protein and mRNA EMR2 isoform expression in human grade three astrocytomas shown with western blot (top) and RT-PCR blot (bottom). d Protein and mRNA EMR2 isoform expression in human glioblastomas shown with western blot (top) and RT-PCR blot (bottom)
EMR2 expression increases in bevacizumab exposure
Finally, it was of interest to see how EMR2 expression changed based on treatment of GBM. Recently in the literature there have been several retrospective studies have described atypical patterns of GBM progression after treatment with bevacizumab, a humanized monoclonal antibody against VEGF-A, including multifocal and widely disseminated disease [18–24]. In addition after analysis of our OncoPrint, a possible association between EMR2 and the PI3K pathway was hypothesized. To investigate the correlation with EMR2 and the PI3K pathway we performed additional IHC staining of 2 key proteins, phosphorylated PRAS40 and phosphorlyated-S6 ribosomal protein (P-S6), involved in the AKT/PI3K pathway on our GBM tissue array of 54 patient samples. We found that 88 % of tumors with upregulated EMR2 also had upregulation of PRAS40-phosphorylation and 75 % had an upregulated P-S6 protein. Therefore, 88 % (p = 0.02) of the GBM’s with upregulated EMR2 also expressed an upregulation of the PI3K/AKT pathway. Prior data shows a decreased survival of patients treated with bevacizumab who had an increased activation of the PI3K pathway [25]. On the basis that both bevacizumab and EMR2 can be linked to increased invasiveness and increased activation of the PI3K pathway, we decided to investigate if EMR2 expression was upregulated after bevacizumab resistance. Primary cell line U251 was treated with bevacizumab and Fig. 5a shows that the EMR2 (1–5) isoform upregulated. This result was confirmed using U87 cells (not shown). Finally, in vivo GBM U87 cells were grown in 4 xenograft mice and treated with bevacizumab or IgG. Protein and RNA expression was significantly increased in the bevacizumab treated group compared to the control group (Fig. 5b–c) (p < 0.041). Taken together, these findings suggest that bevacizumab increased EMR2 expression by GBM cells, with the occurrence of this effect in cell culture suggesting that it reflected direct effects of bevacizumab on tumor cells occurring independent of the established effects of bevacizumab on endothelial cells in the tumor microenvironment in vivo.

a Protein and mRNA EMR2 expression of U251 glioblastoma cells treated with increasing bevizcumab shown with western blot (top) and RT-PCR blot (bottom). b Protein expression of EMR2 in U87 cell lines grown in vivo with and without treatment of bevacizumab. c Quantitiative PCR data of EMR2 expression in U87 cells treated in vivo with and without bevacizumab
Discussion
Expression of EMR2 and clinical outcomes
Among patients with EMR2-positive cancers, the clinicopathologic significance of this protein varies by tumor type. In colorectal cancer, EMR2 did not affect tumor stage or systemic invasion [26], however in breast cancer, EMR2 appears to have a functional role in tumor grade, disease aggressiveness and invasion [10]. By analyzing the TCGA dataset for both GBM and low grade glioma, we demonstrate that EMR2 overexpression is associated with both decreased disease-free survival as well as decreased overall survival, suggesting that EMR2 expression may represent a clinically relevant biomarker in this disease especially since we have now shown that this can be feasibly accomplished by the use of IHC [27, 28]. Our IHC results indicate that GBM and LGG samples contain variable amounts of EMR2 expression relative to one another which can be easily detected. A large sample size of tumors need to be screened with IHC to ensure the sensitivity to assay for EMR2 is as strong for predicting outcomes as with the TCGA data. IHC testing for EMR2 can prove in the future to be an important method for providing prognostic data for patients with gliomas if this correlation is further investigated. Overall, we show, however, that the expression of EMR2 is increased in high grade gliomas when compared to low grade gliomas.
Genomic profile of tumors with up-regulated EMR2
Recent analysis by Verlak et al. [14] described four genetic subtypes of GBM. The mesenchymal subtype is characterized by high rates of mutations or other alterations in NF1, and less expression of EGFR than other types [14]. In addition, the mesenchymal subtype has been identified as the most aggressive subtype, that it has with the poorest prognosis among affected patients and can be resistant to radiation [29]. This subtype, however, also shows increased response to chemotherapy [14]. We show a potential correlation between overexpressing EMR2 tumors and the mesenchymal GBM subtype which could help guide future treatment decisions. The survival discrepancy among patients with high versus low EMR2 expressing tumors could be related to the invasive potential of their tumors [27]. The mechanisms underlying EMR2 expression in GBM are not well understood, however we find a strong correlation with EMR2 and the PI3K gene.
The PI3K–Akt pathway is believed to play a critical role in cell survival, and its activation is linked to tumorigenesis [30]. Up-regulation of Akt and its upstream regulator PI3K has been observed in various human malignancies, and the negative regulator of this pathway PTEN is a tumor suppressor [31, 32]. One of the four isoforms of PI3K is type-1B PI3K p110γ(PIK3CG) which is activated G-protein-coupled receptors [33]. Prior studies have shown that increased expression of PI3KCG gene can induce invasion and migration of breast cancer [34]. In our TCGA analysis of tumors with increased expression of EMR2 there is a strong correlation with up-regulation of PIK3CG. Up-regulation of EMR2 may represent a unique genetic GBM subtype characterized by a highly invasive phenotype, with an up-regulated PI3K–Akt pathway that provides a selective advantage due to its invasive potential. Two proteins indicative of activation of the PI3K–Akt pathway are phosphorylated PRAS40 and phosphorlyated-S6 ribosomal protein (P-S6) [35]. Activation of this pathway has previously been shown to be associated with poor survival in low grade gliomas [35]. As evidence by IHC staining of our GBM array, we demonstrate a possible correlation between the PI3K–Akt pathway and EMR2.
Variable EMR2 isoform expression
The EGF-TM7 family is known to have alternative splicing that selectively combines different exons, producing isoforms with variable EGF domains [3]. Although EGF domains mediate the interactions between these receptors and their ligands in a calcium-dependent fashion, the intricacies of ligand binding are not fully understood but seem ideally suited for adhesion or migration [36]. One example of this is shown when one EMR2 isoform localizes to the leading edge of neutrophils undergoing chemotaxis and is upregulated in inflammatory states [5]. Cells with different isoforms have also shown to form different morphology based on activation of their intracellular actin filaments [8]. Each of the isoforms increase the functional repertoire of EMR2, but one study shows that the major ligand-binding site is located at the EGF-like domain 4 [8, 36]. Although we have only evaluated a small sample size, the EMR2(1–5) isoform, the only isoform containing the EGF-like domain 4, had the most protein and RNA expression in GBM than LGG, and therefore could have a more pro-invasive associated phenotype. Therefore, the total amount of expression of EMR2 may be as important as the amount of EMR2 with EGF-like domain 4 expression in gliomas. If there is an isoform such as EMR2(1-5) that proves to truly be pro-invasive, this can ultimately act as a therapeutic target for monoclonal antibodies similar to what has been developed for CD97 [37].
EMR2 expression in bevacizumab treated tumors
Regulation of VEGF activity by the PI3K pathway has been reported in breast cancer patients and endothelial cells [25, 38]. A subset of patients that were treated with bevacizumab experienced an activation of the PI3K pathway which resulted in a decrease in overall survival. In this study, we demonstrate a correlation between EMR2 and the PI3K–Akt pathway and an up-regulation of EMR2 expression after treatment of bevacizumab in GBM cell lines. GBMs which overexpress EMR2 may exhibit increased activation of the PI3K pathway which could play a role in the evolution of resistance to bevacizumab. Recently, there have been multiple clinical cancer trials, including one involving GBM, which have focused on using PI3K inhibitors in combination with bevacizumab for a combined benefit [39, 40]. Breast cancer models have also shown that combining bevacizumab with PI3K inhibitors had an additive anti-invasive effect [41]. Identifying the subset of patients that will experience the most benefit from this combined therapy is critical, and understanding the role of EMR2, PI3K pathway and regulation by bevacizumab can assist in this goal.
Our functional and descriptive data suggest that EMR2 may serve as a biomarker of increased malignancy or invasive potential in gliomas. Inhibition of EMR2 may represent one strategy for limiting local and systemic invasion of neoplastic cells. Given that EMR2 expression has been tied to patient survival and cellular invasion, such therapies could conceivably improve clinical outcomes for patients with EMR2-positive tumors, or aid in selecting those patient with the most benefit from a particular treatment method.
Conclusion
EMR2 is a critically important member of the EGF-TM7 family that may help stratify patients by tumor phenotype, allow for more accurate prognoses, and serve as a target for novel therapeutic agents. In this study, we describe a pattern of increasing EMR2 expression in higher grade gliomas and demonstrate an association of overexpressing EMR2 tumors with poor survival in all grades of glioma. This data supports prior functional data depicting that EMR2-positive neoplasms possess a greater capacity for infiltrative spread. We also describe the variations of the unique EGF-like domains of EMR2 in gliomas, and discuss that one particular isoform may be responsible for a more invasive phenotype. Finally, a correlation of an increased EMR2 expression after bevacizumab treatment in glioma cells lines is identified. Further understanding of EMR2’s role in the PI3K pathway, regulation by bevacizumab, and in tumor invasion could have predictive survival properties for glioma patients and deserves future investigation.
References
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
Mikheeva SA, Mikheev AM, Petit A, Beyer R, Oxford RG et al (2010) TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol Cancer 9:194
Bjarnadottir TK, Geirardsdottir K, Ingemansson M, Mirza MA, Fredriksson R et al (2007) Identification of novel splice variants of Adhesion G protein-coupled receptors. Gene 387:38–48
Lin HH, Stacey M, Hamann J, Gordon S, McKnight AJ (2000) Human EMR2, a novel EGF-TM7 molecule on chromosome 19p13.1, is closely related to CD97. Genomics 67:188–200
Yona S, Lin HH, Dri P, Davies JQ, Hayhoe RP et al (2008) Ligation of the adhesion-GPCR EMR2 regulates human neutrophil function. FASEB J 22:741–751
Chang GW, Davies JQ, Stacey M, Yona S, Bowdish DM et al (2007) CD312, the human adhesion-GPCR EMR2, is differentially expressed during differentiation, maturation, and activation of myeloid cells. Biochem Biophys Res Commun 353:133–138
Kwakkenbos MJ, Pouwels W, Matmati M, Stacey M, Lin HH et al (2005) Expression of the largest CD97 and EMR2 isoforms on leukocytes facilitates a specific interaction with chondroitin sulfate on B cells. J Leukoc Biol 77:112–119
Stacey M, Chang GW, Davies JQ, Kwakkenbos MJ, Sanderson RD et al (2003) The epidermal growth factor-like domains of the human EMR2 receptor mediate cell attachment through chondroitin sulfate glycosaminoglycans. Blood 102:2916–2924
Kwakkenbos MJ, Chang GW, Lin HH, Pouwels W, de Jong EC et al (2002) The human EGF-TM7 family member EMR2 is a heterodimeric receptor expressed on myeloid cells. J Leukoc Biol 71:854–862
Davies JQ, Lin HH, Stacey M, Yona S, Chang GW et al (2011) Leukocyte adhesion-GPCR EMR2 is aberrantly expressed in human breast carcinomas and is associated with patient survival. Oncol Rep 25:619–627
Carbonell WS, DeLay M, Jahangiri A, Park CC, Aghi MK (2013) beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res 73:3145–3154
Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL et al (2013) Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin Cancer Res 19:1773–1783
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110
Shen R, Mo Q, Schultz N, Seshan VE, Olshen AB et al (2012) Integrative subtype discovery in glioblastoma using iCluster. PLoS ONE 7:e35236
Lino MM, Merlo A (2011) PI3 Kinase signaling in glioblastoma. J Neurooncol 103:417–427
Tso CL, Shintaku P, Chen J, Liu Q, Liu J et al (2006) Primary glioblastomas express mesenchymal stem-like properties. Mol Cancer Res 4:607–619
Iwamoto FM, Abrey LE, Beal K, Gutin PH, Rosenblum MK et al (2009) Patterns of relapse and prognosis after bevacizumab failure in recurrent glioblastoma. Neurology 73:1200–1206
Norden AD, Young GS, Setayesh K, Muzikansky A, Klufas R et al (2008) Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology 70:779–787
de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K et al (2010) Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol 12:233–242
DeLay M, Jahangiri A, Carbonell WS, Hu YL, Tsao S et al (2012) Microarray analysis verifies two distinct phenotypes of glioblastomas resistant to antiangiogenic therapy. Clin Cancer Res 18:2930–2942
Narayana A, Golfinos JG, Fischer I, Raza S, Kelly P et al (2008) Feasibility of using bevacizumab with radiation therapy and temozolomide in newly diagnosed high-grade glioma. Int J Radiat Oncol Biol Phys 72:383–389
Narayana A, Kunnakkat SD, Medabalmi P, Golfinos J, Parker E et al (2012) Change in pattern of relapse after antiangiogenic therapy in high-grade glioma. Int J Radiat Oncol Biol Phys 82:77–82
Pope WB, Xia Q, Paton VE, Das A, Hambleton J et al (2011) Patterns of progression in patients with recurrent glioblastoma treated with bevacizumab. Neurology 76:432–437
Vidal M, Di Cosimo S, Torrejon D et al (2011) Survival outcome with bevacizumab: activation of the phosphatidylinositol-3 kinase (PI3K) pathway due to PIK3CA Mutations or PTEN loss makes a difference. Cancer Res 71: Abstract nr P5-13-01
Aust G, Hamann J, Schilling N, Wobus M (2003) Detection of alternatively spliced EMR2 mRNAs in colorectal tumor cell lines but rare expression of the molecule in colorectal adenocarcinomas. Virchows Arch 443:32–37
Rutkowski MJ, Sughrue ME, Kane AJ, Kim JM, Bloch O et al (2011) Epidermal growth factor module-containing mucin-like receptor 2 is a newly identified adhesion G protein-coupled receptor associated with poor overall survival and an invasive phenotype in glioblastoma. J Neurooncol 105:165–171
Kane AJ, Sughrue ME, Rutkowski MJ, Phillips JJ, Parsa AT (2010) EMR-3: a potential mediator of invasive phenotypic variation in glioblastoma and novel therapeutic target. NeuroReport 21:1018–1022
Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K et al (2013) Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 24:331–346
Toker A, Cantley LC (1997) Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 387:673–676
Cantley LC, Neel BG (1999) New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA 96:4240–4245
Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296:1655–1657
Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M et al (1995) Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269:690–693
Xie Y, Abel PW, Kirui JK, Deng C, Sharma P et al (2013) Identification of upregulated phosphoinositide 3-kinase gamma as a target to suppress breast cancer cell migration and invasion. Biochem Pharmacol 85:1454–1462
McBride SM, Perez DA, Polley MY, Vandenberg SR, Smith JS et al (2010) Activation of PI3K/mTOR pathway occurs in most adult low-grade gliomas and predicts patient survival. J Neurooncol 97:33–40
Chiu PL, Ng BH, Chang GW, Gordon S, Lin HH (2008) Putative alternative trans-splicing of leukocyte adhesion-GPCR pre-mRNAs generates functional chimeric receptors. FEBS Lett 582:792–798
de Groot DM, Vogel G, Dulos J, Teeuwen L, Stebbins K et al (2009) Therapeutic antibody targeting of CD97 in experimental arthritis: the role of antigen expression, shedding, and internalization on the pharmacokinetics of anti-CD97 monoclonal antibody 1B2. J Immunol 183:4127–4134
Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA et al (1998) Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 273:30336–30343
Hainsworth J (2011) Combination of BKM120 and bevacizumab in refractory solid tumors and relapsed/refractory glioblastoma multiforme. U.S. National Institutes of Health, Bethesda
Health USNIo (ed) (2011) GDC-0980 in combination with a fluoropyrimidine, oxaliplatin, and bevacizumab in patients with advanced solid tumors
Lindholm EM, Krohn M, Iadevaia S, Kristian A, Mills GB et al (2014) Proteomic characterization of breast cancer xenografts identifies early and late bevacizumab-induced responses and predicts effective drug combinations. Clin Cancer Res 20:404–412
Acknowledgments
Dr. Ivan was supported by the National Research and Education Foundation from the American Association of Neurological Surgeons. Mr. Safaee was supported by a grant from the Doris Duke Charitable Foundation. Mr. Sun were supported by grants from the Howard Hughes Medical Institute. Mr. Jahangiri is a Howard Hughes Medical Institute Advanced Research Fellow. Dr. Parsa is partially supported by the Michael J. Marchese Endowed Chair in Neurological Surgery.
Conflict of interest
The authors declare no potential conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ivan, M.E., Safaee, M., Oh, T. et al. Epidermal growth factor-like module containing mucin-like hormone receptor 2 expression in gliomas. J Neurooncol 121, 53–61 (2015). https://doi.org/10.1007/s11060-014-1606-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-014-1606-z