
Abstract
The distribution of nucleotide P2Y receptors across different tissues suggests that they fulfil key roles in a number of physiological and pathological conditions. P2Y13 is one of the latest P2Y receptors identified, a novel member of the Gi-coupled P2Y receptor subfamily that responds to ADP, together with P2Y12 and P2Y14. Pharmacological studies drew attention to this new ADP receptor, with a pharmacology that overlaps that of P2Y12 receptors but with unique features and roles. The P2RY12–14 genes all reside on human chromosome 3 at 3q25.1 and their strong sequence homology supports their evolutionary origin through gene duplication. Polymorphisms of P2Y13 receptors have been reported in different human populations, yet their consequences remain unknown. The P2Y13 receptor is versatile in its signalling, extending beyond the canonical signalling of a Gi-coupled receptor. Not only can it couple to different G proteins (Gs/Gq) but the P2Y13 receptor can also trigger several intracellular pathways related to the activation of MAPKs (mitogen-activated protein kinases) and the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3 axis. Moreover, the availability of P2Y13 receptor knockout mice has highlighted the specific functions in which it is involved, mainly in the regulation of cholesterol and glucose metabolism, bone homeostasis and aspects of central nervous system function like pain transmission and neuroprotection. This review summarizes our current understanding of this elusive receptor, not only at the pharmacological and molecular level but also, in terms of its signalling properties and specific functions, helping to clarify the involvement of P2Y13 receptors in pathological situations.
Raquel Pérez-Sen and Rosa Gómez-Villafuertes contributed equally to this work.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
P2Y13 is a G-protein-coupled receptor (GPCR) that is included along with P2Y12 and P2Y14 in the P2Y subgroup of receptors, of which the main physiological agonist is ADP. GPCRs are a very large family of membrane proteins that account for approximately 2% of all the genes in the human genome. These receptors control a wide range of key physiological functions and they are also the pharmacological target to treat a large number of prevalent human diseases. P2Y receptors belong to the rhodopsin family, also known as Class A GPCRs, and based on sequence homology they have been included in the δ subfamily of Class A GPCRs, which also contains glycoprotein receptors, protease-activated receptors and olfactory receptors. Based on their pharmacology, signal transduction and structure, P2Y receptors are classified into two main subfamilies. The first subgroup are coupled to phospholipase C (PLC) via Gq proteins and it includes the P2Y1, P2Y2, P2Y4, P2Y6 and P2Y11 receptors. Conversely, the “P2Y12-like” subfamily that contains the P2Y12, P2Y13 and P2Y14 receptors preferentially signal through Gi proteins, and they mainly inhibit adenylate cyclase activity or they regulate ion channel activity (von Kugelgen and Hoffmann 2016). Currently, the only crystallographic structure available for these “P2Y12-like” receptors is that of P2Y12 (Zhang et al. 2014a; b), confirming to a large extent its inclusion in the δ subfamily receptors, although some of their specific features do not exactly match those of the δ or α subfamilies of Class A GPCRs. Whether the structural characteristics of P2Y12 are shared by all P2Y family members, or if they are specific to the P2Y12-like ADP-Gi coupled receptors, will require studies of the structure of these other receptors. Notably, the P2Y13 receptor is closely related to P2Y12.
Historically the discovery of ADP receptors is closely associated with platelet function. There is an abundance of adenine nucleotides inside the dense granules of platelets, mainly ADP together with serotonin, and the release of the content of these granules induces platelet aggregation and clot formation. In fact, ADP was the first known aggregating agent and hence, a search began for specific ADP receptors in platelets. P2Y1 was the first P2Y receptor family described in a platelet model and it was shown to be activated by ADP, changing the commonly accepted idea that ATP was its main agonist. This receptor was isolated by hybridization screening of a cDNA library generated from the embryonic chick brain (Webb et al. 1993). P2Y1 is coupled to the Gq protein, activating the PLC pathway and mobilizing internal calcium stores in most cellular models, including platelets (Hechler et al. 1998; Savi et al. 1998). Platelets challenged with ADP also produce Gi-mediated inhibition of adenylyl cyclase, which lowers the cAMP available (Gachet et al. 1997). However, the specific ADP-receptor subtype coupled to this inhibition of adenylyl cyclase remained elusive until it was associated with clinical bleeding disorders, and to the absence of a response to the ADP-selective anti-aggregating drugs ticlopidine and clopidogrel (Cattaneo and Gachet 1999). Thus, the association of familial bleeding disorders with the anti-aggregant pharmacology was the key to hunt this ADP receptor.
The identification of a platelet ADP receptor targeted by antithrombotic drugs was preceded by the isolation of a functional P2 receptor that responded equally to ATP and UTP, leading to it being named P2U, currently P2Y2 (Alexander et al. 2015; Lustig et al. 1993). P2Y12 was the first nucleotide receptor to be associated with a genetic defect, a familial bleeding disorder (Hollopeter et al. 2001), and its identification facilitated the development of powerful anti-platelet aggregating agents that are among the most effective drugs in preventing cardiovascular diseases and ictus. Subsequently, the orphan GPCR SP1999 was shown to be the cognate receptor for ADP (Zhang et al. 2001), also corresponding in sequence to that described by Hollopeter and co-workers (2001). This receptor was linked to Gαi and it is expressed strongly in neural tissues and blood platelets. Since then the search for other homologous P2Y receptors became more intense.
Following a similar strategy as for P2Y12, the orphan GPCRs GPR86 and SP174 were identified as ADP receptors and named P2Y13 (Communi et al. 2001; Zhang et al. 2002). This new receptor couples to Gi proteins and it shares a high degree of sequence homology to P2Y12, as well as a similar rank order of potency for ADP and analogues. The comparison of human and mouse P2Y13 receptors demonstrated approximately 75% sequence homology and a similar pharmacological profile, although ADP and nucleotide analogues appear to act more potently on the murine receptor (Zhang et al. 2002). More extensive pharmacological characterization of the human P2Y13 receptor has also been carried out, allowing further functional studies to discriminate the distinct P2Y-Gi-coupled receptors (Marteau et al. 2003).
The clinical and pharmacological relevance of P2Y12 has somehow cast a shadow on the important physiological role of P2Y13. Thus, in this review, we will try to provide an overview of this less well known family member, bringing together the data available regarding different aspects of this multi-faceted receptor. The review is organized in sections to make its content more comprehensible and accessible. The pharmacology of P2Y13 with respect to other P2Y receptors represents a good starting point, which is followed by a comparative study of the sequence of P2Y13 and its known single nucleotide polymorphisms (SNPs). The use of genetically modified animals and a better understanding of the mechanisms that control its expression will be particularly useful to assign specific cellular responses to P2Y13. This is particularly relevant when attempting to understand the signals generated by this receptor given the number of complex pathways it can activate. This signalling is clearly related to the wide range of essential functions that P2Y13 performs in the control of relevant physiological functions, such as protection from oxidative and genotoxic stress, lipoprotein mobilization in cholesterol metabolism or pain, to mention just a few. All these examples indicate that P2Y13 is a key receptor in the purinergic field, and that better understanding its physiology will provide us with useful tools to cope with pathophysiological situations that could be relevant to human disease.
2 Pharmacology of the P2Y13 Receptor
Regarding the pharmacological profile of P2Y13 receptors, they share a characteristic preference for ADP. ADP is the most potent agonist of the naturally occurring nucleotides that act on the P2Y13 receptor, stimulating the receptor at an EC50 in the nanomolar range (Communi et al. 2001; Zhang et al. 2002; Marteau et al. 2003) a pharmacological feature shared with the P2Y12 receptor (Hollopeter et al. 2001; Zhang et al. 2001). Another P2Y receptor that prefers ADP and that is potently activated by this nucleoside diphosphate is P2Y1 (Leon et al. 1997). The P2Y13 receptor also responds to adenine diphosphate analogues adenosine such as 2-methylthio-ADPβS (2MeSADP) or 5′-O-(2-thiodiphosphate) (Fig. 1) (Communi et al. 2001; Zhang et al. 2002; Marteau et al. 2003). In some cellular systems, 2MeSADP proved to be more potent than ADP, whereas under other experimental conditions both compounds were equipotent, possibly reflecting the distinct affinity for 2MeSADP or ADP of multiple active conformations of the P2Y13 receptor, as well as differences in their preference for G proteins (Marteau et al. 2003). ATP and 2-methylthio-ATP appear to be partial and weak agonists of the P2Y13 receptor (Marteau et al. 2003). Although the interaction of the P2Y12 and P2Y13 receptors with nucleotide analogues follows a similar pharmacological profile, inosine diphosphate is about fivefold more potent for human P2Y13 than for P2Y12 receptors. Moreover, inosine diphosphate acts more potently on murine P2Y13 than human P2Y13 and P2Y12 receptors, with an EC50 of 9.2, 552 and 3180 nM, respectively (Zhang et al. 2002).

Structures of agonists and antagonists of ADP receptors
In addition to conventional mononucleotide agonists, dinucleotides have also been tested for their ability to stimulate the P2Y13 receptor. Diadenosine triphosphate (Ap3A) potently activates the P2Y13 receptor, whereas higher diadenosine polyphosphate homologues (Ap4A, Ap5A and Ap6A) are inactive (Zhang et al. 2002; Marteau et al. 2003). A similar profile has been observed with the P2Y1 receptor (Patel et al. 2001), suggesting that selective sensitivity to Ap3A is a common feature of ADP receptors. It has been hypothesized that dinucleoside triphosphates can structurally mimic nucleoside diphosphates (Shaver et al. 2005). Indeed, Ap3A can be stored in secretory vesicles in neural and neuroendocrine tissues through the activity of a broad specificity vesicular nucleotide transporter capable of internalizing a wide variety of nucleotides, as well as the diadenosine polyphosphates (Gualix et al. 1997). Moreover, Ap3A has been identified in microdialysis samples from the cerebellum of conscious, freely moving rats under basal conditions (i.e.: in the absence of any exogenously added stimuli). The extracellular concentration of Ap3A in cerebellar interstitial fluid (10.5 nM) is double that of the other diadenosine polyphosphates detected (Ap4A and Ap5A) and it is in a range that allows this dinucleotide to interact with the P2Y13 receptor (Gualix et al. 2014).
Regarding antagonists, the human P2Y13 receptor is blocked by suramin, reactive blue-2 and high concentrations of the selective purinergic P2X antagonist, PPADS (Marteau et al. 2003). In recent years, PPADS analogues have been designed in an effort to identify more potent and/or selective P2Y13 receptor antagonists. Among them, the 2-chloro-5-nitro analogue MRS2211 (2-[(2-Chloro-5-nitrophenyl)azo]-5-hydroxy-6-methyl-3-[(phosphonooxy)methyl]-4-pyridinecarboxaldehyde) proved to be 45-fold more potent than PPADS as a competitive antagonist of the human P2Y13 receptor, with a pA2-value of 6.3 (Kim et al. 2005). Moreover, MRS2211 is >20-fold more selective as an antagonist at the P2Y13 receptor than of the P2Y1 and P2Y12 receptors, cangrelor (Kim et al. 2005). The P2Y12 antagonists and AR-C67085 also block the human P2Y13 receptors, and cangrelor apparently acts 100 times more potently than AR-C67085. By contrast to its competitive interaction with the P2Y12 receptor, cangrelor depressed the maxima of the agonist dose-response curves in studies on the recombinant human P2Y13 receptor, compatible with a non-competitive interaction (Marteau et al. 2003). The rat P2Y13 receptor is also blocked by nanomolar concentrations of cangrelor but not by the selective P2Y1 antagonist MRS2179 (2′-Deoxy-N6-methyl adenosine 3′,5′-diphosphate), even when used at a concentration as high as 100 μM (Fumagalli et al. 2004). However, cangrelor is a partial agonist of the mouse P2Y13 receptor, enhancing P2Y13-mediated high density lipoprotein (HDL) endocytosis by hepatocytes more potently than its endogenous agonist, ADP (Jacquet et al. 2005). Ticagrelor is an antagonist of the P2Y12 receptor approved for the prevention of thromboembolic events in patients with acute coronary syndrome. Ticagrelor and its active metabolite, TAM, act as P2Y13 antagonists in a transfected cell system in vitro, although they had no impact on P2Y13-regulated pro-platelet formation by human megakaryocytes in culture (Bjorquist et al. 2016). Ap4A has also been described as a complete antagonist of the human P2Y13 receptor, with an IC50 of 216 nM (Marteau et al. 2003).
Given the important roles that P2Y13 and P2Y12 receptors play, it will be crucial to obtain selective ligands that can discriminate between them, capable of distinguishing the influence of P2Y13 receptors on lipid transport and metabolism, and on bone formation, as deduced from studies on knockout animals (see Sect. 4). Due to the similarities between the P2Y12 and P2Y13 receptors in terms of their activation by nucleotide analogues, possibly reflecting a similarity in their agonist binding sites, allosteric effectors would be one possible approach to develop selective modulators of the P2Y13 receptor, as has already been achieved for other GPCRs (May et al. 2007; Melancon et al. 2012).
3 Sequence Analysis of the P2Y13 Receptor
Considering that the P2Y12 and P2Y13 receptors exhibit similar pharmacological features, we looked into the molecular structure of this receptor subfamily. Alignments of amino acid sequences of human P2Y13 with either P2Y12 or P2Y14 receptors reveal approximately 40–45% identity (Fig. 2, Table 1). Moreover, the P2RY12, P2RY13 and P2RY14 genes reside on human chromosome 3 at 3q25.1, which would be consistent with gene duplication having led to their evolutionary origin. Conversely, Gq-coupled P2Y receptors share less than 20% identity with P2Y13 receptors, even the P2Y1 receptor that has a similar pharmacological profile and chromosomal location (the P2RY1 gene is situated at human chromosome 3q25.2, in close proximity to the P2RY12, P2RY13 and P2RY14 genes). Notably, the P2Y12, P2Y13 and P2Y14 receptors cluster with GPR87 in the same region of human chromosome 3, an orphan GPCR that shares 38%, 36% and 44% amino acid identity with the P2Y12, P2Y13 and P2Y14 receptors, respectively (Fig. 3). Although the protein encoded by the GPR87 gene is still to be identified, it would not be a surprise if it were a new P2Y-like receptor, as occurred with GPR17. From a phylogenetic point of view, GPR17 lies in an apparently intermediate position between P2Y and cysteinyl leukotriene receptors, as GPR17 can bind both uracil-nucleotide sugars (UDP, UDP-galactose and UDP-glucose) and cysteinyl leukotrienes (LTD4, LTC4 and LTE4) (Marucci et al. 2016). Interestingly, inhibition of GPR17 by montelukast, a well-known anti-asthmatic drug that antagonizes CysLT1R, reduces neuroinflammation, it elevates hippocampal neurogenesis and it improves learning and memory in aged rats (Marschallinger et al. 2015).

Comparative alignment of the human P2Y receptor amino acid sequences

Chromosomal location of the P2RY genes on human chromosome 3. P2RY12, P2RY13 and P2RY14 genes cluster on the long arm (band 25.1) of chromosome 3, together with the orphan GPCR GPR87. The P2RY1 gene is also located nearby (band 25.2) (Modified from (Milewicz and Seidman 2000))
Regarding interspecific variation, the alignment of P2Y13 receptors from 13 different species reveals that both their nucleotide and amino acid sequences are highly conserved, especially in mammals (Fig. 4). Humans, great apes (chimpanzee Pan troglodytes, orangutan Pongo pygmaeus, and gorilla Gorilla gorilla), and old world monkeys (rhesus macaque Macaca mulatta) show more than 95% identity at both the nucleotide and amino acid level. Human and new world monkeys (Ma’s night monkey Aotus nancymaae), rodents (rat Rattus norvegicus and mouse Mus musculus) and other mammals with diverse diets (carnivorous dog Canis familiaris, herbivorous cow Bos taurus and omnivorous pig Sus scrofa) show approximately 75–85% sequence identity. Finally, humans and birds (chicken Gallus gallus) or fish (zebrafish Danio rerio) share less than 60% and 45% identity, respectively (Table 2).

Alignment of the P2Y13 receptors from 13 different species, including mammals, birds and fish
As mentioned previously, the first crystallographic assessment of a “P2Y12-like” subfamily member of purinergic receptors was that of human P2Y12 receptor, both the agonist- and antagonist-bound structures (Zhang et al. 2014a; b). Key residues involved in both 2MeSADP and AZD1283 binding were Y105, F106, L155, N159, N191, R256, Y259 and K280. Other residues specifically participate in 2MeSADP binding, including R19, R93, C97, S156, T163, C175, K179, H187 and Q263, whereas V102, Y109, V190, Q195, F252 and L276 contribute to the interaction with AZD1283. In addition to the AZD1283 binding site (pocket 1), an analysis of the extracellular interface revealed an adjacent ligand-binding region (pocket 2). Both pockets seem to be required for Ap4A recognition and binding to the receptor. One nucleotide may bind in pocket 1 while the second half of the dinucleotide molecule was predicted to reach pocket 2, the polyphosphate moiety occupying a highly cationic region of the binding site (Zhang et al. 2014a, b). Crystal structure data can be very helpful in optimizing receptor models for structure-guided drug design (Jacobson et al. 2015). Thus, a model of ticagrelor binding to the human P2Y12 receptor and homology models of the human P2Y14 receptor for ligand docking have recently been described (Kiselev et al. 2014; Paoletta et al. 2015). A similar strategy could be used to study the P2Y13 receptor, since neither crystal structure nor homology models of this receptor are as yet available. Remarkably, alignment of the human P2Y12 and P2Y13 amino acid sequences reveals that most of the key residue positions implicated in agonist/antagonist binding to the P2Y12 receptor are conserved in the P2Y13 receptor, with the exception of F106E, L155I and T163S. It is noteworthy that the relative distances between all the residues are also conserved (Fig. 5, Table 3). It is not inconceivable that the change of these 3 amino acids is involved in the pharmacological differences observed between P2Y12 and P2Y13 receptors.

Alignment of the amino acid sequences of human P2Y12 and P2Y13 receptors. Amino acids with similar chemical characteristics are colour-coded: R, H, K – positively charged side chains (blue); D, E – negatively charged chains (red); S, C, T, N, Q, Y – polar uncharged side chains (green); G, A, I, L, M, P, F, W, V – hydrophobic side chains (black). Purple asterisks indicate the location of key residues involved in agonist/antagonist binding to P2Y12 receptor (Zhang et al. 2014a, b)
4 Single Nucleotide Polymorphisms (SNPs) of the Human P2RY13 Gene
Many human diseases have a causative association with genetic components and recent advances have significantly improved our understanding of the causal effects of genetic changes associated with a growing number of diseases. There is currently a great deal of information regarding the SNPs in genes associated with disease, including those affecting the genes encoding human purinergic receptors. SNPs are widespread in genes encoding ionotropic nucleotide receptors, particularly the P2RX7 gene (Caseley et al. 2014), variants of which are associated with altered chronic pain sensitivity, cardiovascular risk, susceptibility to affective mood disorders, multiple sclerosis, childhood febrile seizure, osteoporosis, tuberculosis, toxoplasmosis and sepsis. Furthermore, SNPs in the P2RX2 gene are associated with susceptibility to hearing loss, and a P2XR4 gene variant is related to high pulse pressure and age-related macular degeneration.
Concerning P2Y receptors, non-synonymous SNPs in the P2RY1 and P2RY12 genes are mainly associated to alterations in platelet aggregation. Thus, the A1622G mutation of the P2RY1 gene could contribute to an inadequate platelet response to anti-coagulants, which would be associated with a higher risk of cardio- and cerebrovascular diseases (Lordkipanidze et al. 2011; Timur et al. 2012). Haplotype variants of the P2RY12 gene have also been related to platelet aggregation, peripheral arterial disease, venous thromboembolism, myocardial infarction and cerebrovascular accidents (Lordkipanidze et al. 2011; Cavallari et al. 2007; Kim et al. 2013; Li et al. 2015; Oestreich et al. 2014; Ou et al. 2016; Siasos et al. 2016; Zee et al. 2008). Moreover, several SNPs and haplotypes of the P2RY12 gene are associated with lung function in patients with asthma (Bunyavanich et al. 2012). Polymorphisms in P2RY2 gene have been related to cerebral and myocardial infarction, essential hypertension (Wang et al. 2009a, b, 2010) and decreased risk of osteoporosis (Wesselius et al. 2013). Recently, a gene-based analysis of regulatory variants identified P2RY13 and P2RY14 as putative asthma risk genes, and functional studies in mice demonstrated that selective agonists of both receptors can promote airway inflammation (Ferreira et al. 2016). However, the pathophysiological consequences of P2RY13 polymorphisms in human populations remain unclear.
Over recent years, the availability of high-throughput DNA sequencing technologies has allowed whole genomes or exomes (protein-coding regions) of a large numbers of humans to be analysed. The recent generation of an extensive catalogue of human protein-coding genetic variation, the Exome Aggregation Consortium (ExAC), provides a powerful source of information about the patterns of low-frequency exome variants from nearly 70,000 individuals of diverse geographic ancestries, representing a public resource for the clinical interpretation of genetic variants observed in disease patients (Lek et al. 2016). Using the ExAC database (http://exac.broadinstitute.org; version 0.3, January 2015), we identified 184 candidate polymorphisms in the human P2RY13 gene, 166 of which lie in exons. When only high-quality filtered variants were analysed, most (98.4%) had an allele frequency of less than 0.01% (very low-frequency variants) and 57% of them were singletons (only observed once in the data set). In terms of the mutational properties of exon variants, there are 48 synonymous (non-protein-altering) SNPs and 118 non-synonymous variants, the latter including 111 missense SNPs, 1 in-frame deletion, 1 stop lost and 3 protein-truncating variants (2 frame-shift and 1 stop gained mutations). In addition, 43 of the missense variants were predicted to be probable/possible damaging variants (Table 4).
Although the consequences of loss-of-function mutations in the human P2RY13 receptor remain unknown, the availability of a P2Y13 knockout mice has proved to be an invaluable resource to assess both the physiological role of this receptor and its potential involvement in specific pathologies (Fabre et al. 2010). Mice lacking the P2Y13 receptor display altered bone remodelling, with less complex bone structures. In addition, these mice suffer a significant reduction in the osteoclast and osteoblast populations on the surfaces of the cortical bone, leading to a reduced capacity for mineralization. These osteoblasts proliferate normally in vitro, although their mineralization capacity is dampened. Moreover, their gene expression is altered, with an upregulation of osteoprotegerin and a downregulation of the RhoA genes. Conversely, osteoclasts maintain their function, although the number of resorbing osteoclast is decreased. However, the bone phenotype of the P2Y13 null mice protects against the loss of bone linked to oestrogen-deficiency (Orriss et al. 2011; Wang et al. 2012).
Another major feature of the P2Y13 knockout mice is related to lipoprotein metabolism. Even though HDL levels in plasma are normal (Fabre et al. 2010) or slightly decreased (Blom et al. 2010), hepatic uptake of HDL holo-particles is impaired in the absence of the receptor. Furthermore, mice exhibit deficient biliary cholesterol excretion and a reduced hepatic cholesterol content. Remarkably, the reverse cholesterol transport (RCT) that is crucial for the atheroprotective role of HDL proteins is also affected, with a striking reduction in macrophage-to-faeces RCT, making them more sensitive to a high cholesterol diet (Fabre et al. 2010; Lichtenstein et al. 2013). Despite the strong reduction in RCT, P2Y13 null mice do not exhibit enhanced atherosclerosis, possibly to the notable differences between human and mouse lipoprotein metabolism. However, dual P2Y13/apoE knockout mice develop enhanced aortic sinus lesions with more infiltrated macrophages (Lichtenstein et al. 2015). Importantly, the atherosclerotic pathology is independent of the P2Y13 blood cell receptors. Moreover, a high fat diet is also responsible of neuropathies that affect the enteric nervous system, crucial for the maintenance and regulation of gastrointestinal activity. Experiments performed on P2Y13 null mice fed with a high fat diet reveal that the purinergic receptor is responsible for the myenteric neuronal loss induced by the high fat diet or palmitic acid (Voss et al. 2014).
In summary, there is strong evidence that the P2Y13 receptor constitutes a promising therapeutic target for pathologies like osteoporosis, atherosclerosis and intestinal neuropathies. However, the role of the receptor in distinct tissues in the organism implies that its involvement in other pathologies should not be ruled out, encouraging further research into this receptor.
5 P2Y13 Receptor Signaling
The range of signals transmitted by P2Y13 receptors extend beyond their versatility in G protein coupling, and several intracellular pathways can be activated independently of the canonical inhibition of adenylate cyclase.
5.1 G Protein Signalling
Like the P2Y12 receptor, the P2Y13 receptor is an ADP receptor mainly coupled to Gi proteins. Activation of the P2Y13 receptor with ADP or the analogue 2MeSADP leads to the inhibition of adenylate cyclase and a decrease in cAMP production triggered by forskolin, a direct adenylate cyclase activator or following Gs-coupled GPCR stimulation. The EC50 values for these events are in the nanomolar range (Communi et al. 2001; Zhang et al. 2002; Marteau et al. 2003; Carrasquero et al. 2005), yet such inhibition was reversed at higher agonist concentrations. From the very first studies, biphasic dose-response curves for the effect of ADP on forskolin-cAMP accumulation were described, with inhibition at nanomolar agonist concentrations and potentiation at micromolar levels, clearly demonstrating the versatility of P2Y13 receptor coupling to different G proteins (Gi/Gs) (Communi et al. 2001; Marteau et al. 2003).
Studies with Gα16/Gαq in heterologous expression systems (Communi et al. 2001; Zhang et al. 2002; Marteau et al. 2003) and native tissues, such as immature human dendritic cells and rat cerebellar astrocytes (Carrasquero et al. 2005; Marteau et al. 2004), revealed that P2Y13 can also couple to PLC (Fig. 6). Functional P2Y13 receptors are present in cerebellar astrocytes as ADP elicits calcium transients that are not sensitive to the P2Y1 specific antagonist, MRS2179. The presence of Gq-coupled P2Y13 receptors is restricted to astrocytic populations other than those expressing P2Y1 receptors, indicating some degree of specialization (Carrasquero et al. 2005; Jimenez et al. 1999, 2000). A link to the canonical pathway of adenylate cyclase inhibition further demonstrated the identity of the P2Y13 receptor in this glial model. Moreover, when P2Y13 and P2X7 purinergic receptors are functionally expressed in rat cerebellar astrocytes they mediate the increase in intracellular calcium elicited by BzATP in these cells (Carrasquero et al. 2009). Moreover, some populations of rat cerebellar granule neurons exhibit 2MeSADP-mediated calcium mobilization with the pharmacological profile of a P2Y13 receptor (Hervas et al. 2003).

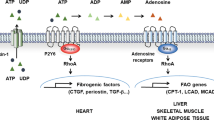
Schematic representation of the intracellular signalling cascades activated by P2Y13 receptor stimulation. P2Y13 couples to adenylate cyclase inhibition through the canonical mechanism described for Gi-coupled receptors. P2Y13 receptor stimulation via the Gi protein also triggers the PI3K/Akt/GSK3 axis. Phosphorylation of GSK3 causes enzyme inactivation and releases two key proteins, β-catenin and Nrf2, which act as transcription factors. The activation of the antioxidant Nfr2/HO-1 axis promotes cell survival in the face of oxidative stress. The P2Y13 receptor also couples to Gq proteins in some cell types, promoting phospholipase C activation that in turn stimulates intracellular calcium mobilization and DAG production. These second messengers activate different PKC isoforms and induce ERK activation, which is required for Dusp2 phosphatase gene transcription. DUSP2 could be responsible for the neuroprotection displayed by the P2Y13 receptor to counteract the neurotoxic actions of cisplatin. In some cell models, ERK activation can also be achieved via PI3K (broken lines). The P2Y13 receptor also couples to RhoA activation and cytoskeleton reorganization, and it may inhibit Ca2+ channel activity via the βγ subunits of the activated Gi protein and modulate neurotransmitter release
5.2 MAP Kinase Activation
Downstream of the first membrane effectors, P2Y13 receptor stimulation induces Gi-dependent Mitogen Activated Protein Kinase (MAPK) activation (Communi et al. 2001). The P2Y13 receptor was first seen to couple to the ERK1/2 MAPK in native human dendritic cells (Marteau et al. 2004). ERK activation triggered by P2Y13 receptor agonists was sensitive to ARC-699931MX and intracellular calcium chelation by BAPTA-AM. Again, rat cerebellar astrocytes and granule neurons provided examples of Gi-dependent 2MeSADP-mediated ERK activation, which was completely inhibited by Pertussis toxin (Carrasquero et al. 2005; Ortega et al. 2011; Perez-Sen et al. 2015). In addition, the sensitivity to the specific antagonist MRS2211, together with lack of effect of MRS2179, a P2Y1 antagonist, confirmed the participation of P2Y13 receptors in this signalling.
ERK activation by P2Y13 receptors in cerebellar astrocytes and granule neurons resembles that of tyrosine kinase receptors, with maximal activation reached after 10–15 min. In astrocytes, the EC50 value of ERK activation correlated with that obtained in experiments where cAMP production was inhibited (around 40 nM). Interestingly, the activation of ERKs was dependent on nProtein Kinase C and src-like kinase activation (Carrasquero et al. 2005). Alternatively, PI3 kinase (PI3K) seemed to lie upstream of ERK activation in granule neurons, as its activity was completely abolished by the PI3K inhibitors wortmannin and LY294002. In this neuronal model the CREB (cAMP response element-binding) transcription factor was activated in an ERK-dependent manner, conferring neuroprotection against apoptotic stimuli (Fig. 6) (Ortega et al. 2011; Perez-Sen et al. 2015).
In recent studies, new targets of ERK signalling activated by P2Y13 receptors have been identified (Morente et al. 2014). The stimulation of granule neurons by 2MeSADP induced the P2Y13 dependent expression of an early gene, Dusp2, which was sensitive to the P2Y13 antagonist, MRS2211. DUSP2 is a dual specificity protein phosphatase that is involved in regulating MAPK activity. As described below, DUSP2 may be responsible for some neuroprotection associated to P2Y13 receptor stimulation. Opposite effects were found in pancreatic β cells, where the P2Y13 receptor inhibition activated ERK/Akt/CREB signalling (Tan et al. 2010).
5.3 PI3K/Akt/GSK3 Activation
Another interesting feature of the P2Y13 receptor is its specific coupling to the PI3K/Akt/GSK3 axis in granule neurons. Accordingly, 2MeSADP induces Thr308 phosphorylation and activation of Akt in these cells, which is sensitive to Pertussis toxin and PI3K inhibition. This mechanism connects P2Y13 receptors to important intracellular pathways in granule neurons. As such, 2MeSADP induces the rapid and transient phosphorylation of one of the main Akt targets, GSK3 (Ser21 and Ser9 residues of the α and β isoforms, respectively), which inhibits its catalytic activity. The EC50 value for 2MeSADP is close to 20 nM, and both the P2Y13 antagonist MRS2211 and the PI3K inhibitors wortmaninn and LY294002, prevent this effect (Ortega et al. 2008).
The signalling triggered by P2Y13 receptors that inhibits GSK3 deserves some attention, particularly as some key GSK3 substrates normally retained in the cytosol escape from GSK3-mediated phosphorylation and their subsequent proteasomal degradation (Fig. 6). The nuclear translocation of these GSK3 substrates occurs very rapidly in granule neurons, after a 10–30 min stimulation with 1 μM 2MeSADP, such as for the transcriptional regulator β-catenin (Ortega et al. 2008). Likewise, the nuclear factor (erythroid-derived 2)-like 2 (Nrf2), which regulates the expression of antioxidant genes, is also regulated by GSK3, and a new mechanism by which GSK3 can phosphorylate Nrf2 to promote its further degradation has been demonstrated (Cuadrado 2015; Rada et al. 2012; Rojo et al. 2008). The inhibition of GSK3 induced by P2Y13 receptors lies upstream of the increase in Nrf2, and it accumulates in the nucleus of granule neurons after a 6 h stimulation with 2MeSADP, which also turned out to provide neuroprotection (see below). Furthermore, GSK3 has a key role in the formation of amyloid plaques and neurofibrillary tangles, being one of the most relevant therapeutic targets for Alzheimer’s disease (Diaz-Hernandez et al. 2012; Maqbool et al. 2016).
The activation of the PI3K/Akt signalling pathway is not exclusive to P2Y13 receptors, as it is also induced by the Gi-coupled P2Y12 receptor. In C6 glioma cells, the intracellular cascade triggered by 2MeSADP to induce proliferation involves Rap1 activation by Gβγ subunits, which leads to PI3K-dependent Akt activation. In this pathway, Ca2+ mobilization and assembly of the PyK2/Src/PLD2 complex is required to achieve the final effect, and this signalling contributes to the proliferation elicited by P2Y12 receptors in glioma cells (Van Kolen et al. 2006; Van Kolen and Slegers 2004).
As described for other GPCRs, including the P2Y12 receptor, P2Y13 stimulation also couples to Rho signalling and subsequent cytoskeleton reorganization. The P2Y13 receptor activates RhoA and ROCK I in hepatocytes and osteoblasts, inducing relevant physiological effects (as discussed below). The mechanism of action for RhoA activation is still not completely understood, although it could involve Gi subunits binding to Rho specific guanine exchange factors (Rho-GEF) (Wang et al. 2012; Malaval et al. 2009).
5.4 Molecular Mechanisms Regulating P2Y13 Receptor Expression
It is well known that basal ubiquitination and deubiquitination are important to control the cell surface expression of several GPCRs (Dores and Trejo 2012). In terms of the P2Y13 receptor, constitutive ubiquitination of its C- terminus tail is directly modulated in the endoplasmic reticulum. Consequently, the P2Y13 receptor is degraded through the proteasome pathway, thereby controlling the density of functional receptors at the cell surface and cellular responsiveness (Pons et al. 2014). In this context, the discovery of deubiquitinating enzymes that specifically regulate P2Y13 receptor ubiquitination might provide the basis for a novel therapeutic approach to improve hepatic HDL uptake and bile acid secretion, for instance preventing or impairing the development of atherosclerosis (Serhan et al. 2013).
6 Physiological Relevance of P2Y13 Receptors
Given the widespread distribution of the purinergic P2Y13 receptor, its signalling could be involved in regulating multiple activities in different tissues and organs. The P2Y13 receptor is mainly expressed in the spleen, bone marrow cells, peripheral leukocytes, brain, liver, pancreas and heart. Here, we will summarize the direct evidence for the physiological roles of the P2Y13 receptor.
6.1 Metabolic Disorders: Atherosclerosis and Diabetes
One of the most promising activities of P2Y13 receptor is related to its influence on atherosclerosis. The P2Y13 receptor plays a pivotal role in HDL metabolism, which transports cholesterol from peripheral tissues to the liver for elimination. The identification of a new pathway in the liver involving the F1-ATPase and the P2Y13 receptor, which regulates the removal of HDL-cholesterol (HDL-c), has enhanced our understanding of HDL metabolism (Jacquet et al. 2005). HDL endocytosis triggered by the P2Y13 receptor is dependent on activation of the small GTPase RhoA and ROCK1, producing the cytoskeletal arrangements that drive endocytosis. This step is followed by an increase in biliary lipid secretion (Malaval et al. 2009). The participation of the P2Y13 receptor is clearly evident when the effects of its agonists are studied, such as cangrelor and the new available drug, CT1007900 (6-[1-(2-Dimethylaminopyrimidin-5-ylmethyl)-piperidin-4-yl]-2-morpholin-4-yl-pyrimidin-4-ol monohydrate). Acute administration of these compounds increases RCT in the same way as ADP, and in the case of cangrelor, its effect was greater than that found with the physiological agonist. This was unexpected for a compound initially designed as a P2Y12 and P2Y13 antagonist, and that turned out to act as a partial agonist for P2Y13 receptors (Jacquet et al. 2005; Serhan et al. 2013; Goffinet et al. 2014). The fact that impaired HDL clearance occurred in P2Y13 null mouse as well as in-loss-of function experiments (using P2Y13-shRNA) favours a central role of the P2Y13 receptor in RCT (Fabre et al. 2010; Lichtenstein et al. 2013).
The therapeutic potential of P2Y13 agonists was also tested in long-term studies carried out through their continuous delivery, which efficiently induced the clearance of circulating cholesterol in the form of HDL particles and its elimination in the form of bile acid secreted by the liver. Moreover, after 1 month of oral treatment with the new agonist CT1007900, HDL particle size was reduced and fewer atherosclerotic plaques were deposited in a mouse model of atherosclerotic pathology (Lichtenstein et al. 2015; Goffinet et al. 2014). These studies provided clues that the P2Y13 receptor is a promising therapeutic target for the treatment of atherosclerosis, in mice at least.
Therefore, in clinical trials to assess cangrelor as an anti-aggregating therapy, it should be borne in mind that it may act on hepatic P2Y13 receptors and reduce the size of HDL particles. Indeed, it seems that the same effects as those observed in mice will take place in humans. However, while treatment with cangrelor and the P2Y13 agonist CT1007900 exerts a significant effect on circulating HDL, it does not substantially affect plasma HDL-C levels (Martinez et al. 2015). In addition, the P2Y13 receptor may also be involved in insulin secretion as P2Y1 and P2Y13 receptors modulate insulin release from pancreatic β cells. The activation of P2Y1 Gq-coupled receptors increases intracellular calcium levels and induces insulin release from isolated pancreatic β cells, whereas P2Y13 receptor activation in these cells had the opposite effect. Administration of MRS2211, a P2Y13 antagonist during glucose injection in mice results in both increased insulin secretion and reduced glucose levels (Amisten et al. 2010), providing a therapeutic opportunity for P2Y13 receptor antagonists in the treatment of diabetes. Similarly, blocking P2Y13 receptors protects pancreatic beta cells from apoptosis (Tan et al. 2013).
6.2 Bone Homeostasis
The P2Y13 receptor also contributes to bone formation and remodelling, a new and important function for this receptor. The first clue regarding this activity of P2Y13 came from the striking bone phenotype and altered bone turnover in P2Y13 null mice (Wang et al. 2012). The lack of the P2Y13 receptor mainly affected the osteogenic response, since activation of this receptor was crucial to obtain adequate differentiation of bone marrow cells into osteoblasts (Biver et al. 2013) due to the activation of RhoA/ROCK (see below) (Wang et al. 2012). These changes in bone phenotype were age-dependent and while P2Y13 receptors modulate bone remodelling in mature animals, at younger ages this receptor affects hormonal regulators of phosphate homeostasis. Indeed, in young mice, the increase in trabecular bone formation is correlated to higher serum phosphate levels and increased FGF23 production (Wang et al. 2014).
Another important aspect of P2Y13 receptor activity in bone homeostasis is associated to its coordinated activity with other nucleotide receptors. In red blood cells, the P2Y13 receptor provides negative feedback modulation of ATP release and thus, increased ATP production in P2Y13 null mice may facilitate an osteogenic response of osteoblasts upon mechanical stimulation. The enhanced osteogenic response together with the protective role of the P2Y13 receptor in conditions of oestrogen deprivation, suggest that specific inactivation of this receptor could have therapeutic applications in preventing bone loss in diseases like osteoporosis (Wang et al. 2013).
6.3 The Nervous System and Neurological Implications
Prior to the cloning of Gi coupled ADP receptors, several studies reported the inhibitory effect of P2Y-like receptors in modulating neurotransmitter release in both the peripheral sympathetic and central nervous system (Koch et al. 1997; von Kugelgen et al. 1994). Later on, the identification of the P2Y12 and P2Y13 receptors, and specific antagonists, helped to identify P2Y13 as the receptor responsible for inhibiting noradrenaline release in the rat vas deferens and in rat brain hippocampal slices (Csolle et al. 2008; Queiroz et al. 2003). Moreover, P2Y13 receptors were also involved in fine-tuning cholinergic transmission at mammalian neuromuscular junctions, where a specific P2Y13 antagonist abolishes the effect of 2MeSADP, inhibiting spontaneous and evoked presynaptic acetylcholine release (Guarracino et al. 2016). Indeed, direct negative modulation of N-type Ca2+ channels by Gi protein subunits may underlie these effects (Wirkner et al. 2004).
6.3.1 Pain Transmission
P2Y13 receptors have also been implicated in pain transmission, and both pro-nociceptive and anti-nociceptive actions have been described in different experimental pain models. In peripheral sensory neurons, the expression of several types of P2Y receptors responding to ADP following peripheral nerve injury and they modulate nociceptive signalling in different ways. While P2Y1 is pro-nociceptive and facilitates pain transmission, the stimulation of a Gi-coupled ADP receptor with the pharmacological profile of P2Y13 produces analgesic effects. Indeed, ADP and other P2Y13 agonists reduce the magnitude of depolarization-evoked Ca2+ transients in dorsal root ganglia (DRG) sensory neurons, and this effect was also reproduced in P2Y1 knockout mice (Malin and Molliver 2010). Notably, P2Y1 was required for the full expression of inflammatory hyperalgesia after peripheral nerve injury, and when antagonized it occluded the action of Gi-coupled ADP receptors. Only after P2Y1 blockade was the anti-nociceptive action of P2Y13 fully revealed. The integration of these opposing signals adjusts nociceptor sensitivity. However, P2Y1 receptors may exert anti-nociceptive effects in other pain models (Ando et al. 2010; Selden et al. 2007) and the selective P2Y1 receptor agonist MRS2365 has potent analgesic activity against neuropathic and acute pain.
In line with the anti-nociceptive effects of ADP, new roles for P2Y1 and P2Y13 receptors have been described in the regulation of inhibitory glycinergic neurotransmission in the spinal cord. Interestingly, P2Y13 acts in conjunction with P2Y1 receptors to regulate glycine transporters in primary neuronal cultures of the spinal cord and in brainstem preparations (Jimenez et al. 2011). They reduced the activity of the neuronal glycine transporter, GLYT2, to increase the levels of inhibitory glycine neurotransmitter in the synaptic cleft. Conversely, both receptors, along with the P2Y12 receptor, activate the glial GLYT1 transporter, reducing glycine levels at glycinergic and glutamatergic synapses, and thereby decreasing glycine concentrations in the NMDA receptor milieu. This produced a net increase in the inhibitory over the excitatory pathways that may contribute to anti-nociception.
The intracellular mechanisms triggered by P2Y1/P2Y13 receptor stimulation involved PLC/PKC activation, nitric oxide (NO) release and paracrine PKG-I activation. Regulation of GLYT activity by this pathway was corroborated in heterologous COS cell systems expressing recombinant glycine transporters and the P2Y1 receptor. In fact, the regulatory activity of P2Y1 receptors was lost after siRNA knockdown of NO synthase activity. This common mechanism of action reflects how PKG mediates the phosphorylation of key residues in either glycine transporters or adaptor proteins, which explains the contrasting changes in transport activity reported. These studies reveal a paracrine regulation of GLYT1 and GLYT2 by ADP-P2Y receptors in the spinal cord that contributes to the processing of nociceptive information. This paracrine regulatory mechanism extends the role of ADP receptors to the regulation of nociceptive signalling, an activity involving the modulation of the glycine levels that influences both excitatory and inhibitory neurotransmission at spinal cord synapses. Indeed, GLYT2 pharmacological blockade in the spinal cord produces pain relief in models of acute pain (Dohi et al. 2009).
In comparison to the anti-nociceptive action of P2Y13 receptors in sensory neurons, P2Y12 receptors expressed in spinal cord microglia also participate in the establishment of neuropathic pain and mechanical allodynia following peripheral nerve injury. Activation of p38 signalling by P2Y12 receptors contributes to the generation and maintenance of hyperalgesia (Kobayashi et al. 2008; Tatsumi et al. 2015; Tozaki-Saitoh et al. 2008). Although P2Y13 receptors may be expressed sporadically in microglial cells, several P2Y receptors are up-regulated in response to nerve injury, including P2Y13, which can then contribute to the development of neuropathic pain (Kobayashi et al. 2012). In this respect, the increases in calcium promoted by Gq-coupling of P2Y13 receptors in dorsal spinal cord microglia contribute to the early phase of pain hyper-sensitization and to the changes in size of microglia (Kobayashi et al. 2013; Zheng et al. 2014).
All these findings indicate that purinergic receptors fine tune different populations of sensory neurons and glial cells in the spinal cord and dorsal horn to modulate nociceptive sensitivity. Before ADP formation and activation of P2Y receptors, ATP can exert a direct and acute stimulation of nociceptive signalling through the activation of ionotropic P2X receptors, mainly P2X2/3 receptors present in DRG neurons (Lewis et al. 1995). In addition, P2X4 receptors participate in the maintenance of neuropathic pain by acting on microglial cells (Inoue and Tsuda 2012). A similar interaction between P2Y1 and P2Y13 receptors occurs at the axonal growth cone, where both receptors modulate intracellular signalling triggered by P2X7 receptors to control axonal elongation and sprouting (del Puerto et al. 2012). Moreover, the P2Y13 receptor antagonist MRS2211 accelerates neurite outgrowth in PC12, Neuro2a and MEB5 cells (Yano et al. 2012).
6.3.2 Cell Survival and Neuroprotection
In contrast to the pro-apoptotic role of P2Y13 receptors in enteric neurons and pancreatic β cells, these receptors play a predominant survival role in the central nervous system. P2Y13 receptors promoted the survival of cerebellar astrocytes and granule neurons, both neural models in which P2Y13 is co-expressed with P2Y1 receptors, and where specialized functions are elicited by their coupling to different signalling targets. In granule neurons, P2Y13 receptors provide neuroprotection against different types of apoptotic stimuli and their activation protects granule neurons from oxidative stress induced by hydrogen peroxide. Indeed, both the production of reactive oxygen species and cell death induced by H2O2 treatment diminish after a 2 h pre-treatment with the P2Y13 agonist, 2MeSADP. The intracellular mechanism responsible for this neuroprotective effect involves activation of the antioxidant axis Nrf2/heme oxygenase-1 (HO-1). Nrf2 transcriptional activity induces the expression of HO-1, whose levels increase 6 h after P2Y13 receptor activation. Both HO-1 expression and survival in response to oxidative stress are prevented in cultured neurons obtained from Nrf2 knockout mice, demonstrating that the antioxidant Nrf2/HO-1 axis is functional in granule neurons and that it is regulated by P2Y13 receptors (Perez-Sen et al. 2015; Espada et al. 2010). Similar neuroprotection against oxidative stress is elicited by ADP-P2Y receptors in astrocytes (unpublished results).
Along similar lines, P2Y13 receptors also prevent cell death induced by toxic extracellular concentrations of glutamate. The neuroprotection elicited by 2MeSADP is not as potent as that exerted by well-known trophic factors in granule neurons, such as the neurotrophin BDNF (brain derived neurotrophic factor), although it shares a similar mechanism of action. Indeed, a 2 h pre-treatment with both 2MeSADP and BDNF prevents caspase-3 activation in a manner dependent on the activation of the ERK/CREB pathway. The increase in neuron survival promoted by 2MeSADP is also abolished by the antagonist MRS2211 and the PI3K inhibitor wortmannin, again validating the contribution of a Gi-coupled PI3K activity triggered by the P2Y13 receptor (Ortega et al. 2011).
P2Y13 receptors also protect against other kinds of stress. Genotoxic stress induced by both UV exposure and cisplatin, the cytotoxic drug employed in chemotherapy, also produces apoptotic cell death of granule neurons. Again, P2Y13 receptor activation enhances cell survival in these conditions and this protection was abolished by MRS2211 (Morente et al. 2014). In addition, to the PI3K and ERK signalling activated by P2Y13 receptors, other signalling mechanisms appear to contribute to the increase in cell survival. Interestingly, these stress conditions promote MAPK p38 over-activation and long-term accumulation of phosphorylated p38 in the nucleus. 2MeSADP pre-treatment before exposure to the cytotoxic stimuli decreases nuclear p38 phosphorylation towards basal levels, indicative of an activation of protein phosphatase activity. In particular, the induction of the expression of an immediate early gene, Dusp2, is promoted by 2MeSADP-mediated ERK1,2 signalling in granule neurons (Morente et al. 2014). DUSP2 is a dual specificity phosphatase with a nuclear localization that is selective for p38 and JNK MAPK. Therefore, P2Y13 receptors participate in homeostatic mechanisms in granule neurons contributing to bidirectional regulation of MAPK signalling cascades (Perez-Sen et al. 2015; Marin-Garcia et al. 2009).
7 Concluding Remarks
The data available regarding the P2Y13 receptor reflects the great efforts and advances made by scientists working in this area. It is currently accepted that the protein sequence of the P2Y13 receptor is very similar to that of the P2Y12 receptor, for which a crystallographic structure is available. Thus, plausible explanations for the similarities in their pharmacological properties can be drawn, which in turn make their clear and unequivocal characterization more difficult. However, the presence of two pockets in the receptor structure could explain the agonistic effect of the Ap3A diadenosine triphosphate, and the possible existence of an allosteric modulatory site near to the orthosteric site where the endogenous agonistic ligand, ADP, binds. Hence, the development of so-called biotopic orthosteric/allosteric ligands could define a functionally selective ligand able to specifically discriminate the members of this receptor subfamily, mainly P2Y12, although this field remains unexplored. Nevertheless, such biotopic GPCR agonists and antagonists have recently been reported for the muscarinic receptor M2 and A1 adenosine receptor (Keov et al. 2011). Beyond the similarities between the P2Y13 and P2Y12 receptor, their expression does not seem to overlap in different tissues, which favours their specialized functions. In addition, while they share some signalling properties, P2Y13 is distinct in its ability to switch to Gq proteins and calcium mobilization pathways, as well as to activate signalling cascades that seem to be exclusive to them.
On the other hand, there are no data available concerning the formation of homo- or heterodimers of P2Y13, even though this receptor coexists in most cells with other P2Y receptors and GPCRs. Indeed, the complexity of the signalling cascades highlights the possibility of a broad crosstalk among these receptors. The co-expression of P2Y13 together with P2Y1 receptors in many cellular models seems to be a general rule of particular interest. The best-known example of interactions between two P2Y ADP-responding receptors is that previously described in platelets for P2Y1 and P2Y12 receptors. Both these receptors can act in a coordinated way to regulate complementary functions, or even to behave in an antagonistic fashion. Thus, P2Y13 appears to interact with the P2Y1 receptor in the axonal growth cone, modulating the signalling of the ionotropic P2X7 receptor and resulting in axonal elongation, just one such example among many (del Puerto et al. 2012). These interactions testify to the complexity and broad possibilities of the signalling cascades that regulate metabolism. It is noteworthy that beyond the classical pathways regulated by protein kinases, P2Y13 can also induce the expression of different genes involved in protection against oxidative stress. Likewise, it can drive the expression of protein phosphatases like the dual phosphatases family member, DUSP, which can restore the steady cell state and allow the cell to respond to new agonist challenges.
In the near future, more precise and specific pharmacological agonists and antagonists may become available, as well as molecular tools. Thus, the pharmacological potential of P2Y13 receptors as a target in pathophysiological situations will be revealed in full, advancing the biomedical horizons for this purinergic receptor.
Abbreviations
- ADP:
-
Adenosine 5′-diphosphate.
- Ap3A:
-
P1,P3-Di(adenosine-5′) triphosphate.
- Ap4A:
-
P1,P4-Di(adenosine-5′) triphosphate.
- ATP:
-
Adenosine 5′-triphosphate
- cAMP:
-
Adenosine 3′,5′-cyclic monophosphate.
- CT1007900:
-
(6-[1-(2-Dimethylaminopyrimidin-5-ylmethyl)-piperidin-4-yl]-2-morpholin-4-yl-pyrimidin-4-ol monohydrate).
- GPCRs:
-
G protein–coupled receptors.
- GSK3:
-
Glycogen synthase kinase 3.
- HDL:
-
High density lipoprotein.
- 2MeSADP:
-
2-methylthio-adenosine 5′-diphosphate.
- MAP kinases:
-
Mitogen-activated protein kinases.
- MRS2211:
-
2-[(2-Chloro-5-nitrophenyl)azo]-5-hydroxy-6-methyl-3-[(phosphonooxy)methyl]-4-pyridinecarboxaldehyde.
- MRS2179:
-
2′-Deoxy-N6-methyl adenosine 3′,5′-diphosphate.
- PLC:
-
Phospholipase C.
- PI3K:
-
Phosphatidylinositol 3-kinase.
- PPADS:
-
Pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid).
- RCT:
-
Reverse cholesterol transport
- SNP:
-
Single-nucleotide polymorphism.
References
Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Southan C, Davies JA, Collaborators C (2015) The concise guide to PHARMACOLOGY 2015/16: G protein-coupled receptors. Br J Pharmacol 172(24):5744–5869
Amisten S, Meidute-Abaraviciene S, Tan C, Olde B, Lundquist I, Salehi A, Erlinge D (2010) ADP mediates inhibition of insulin secretion by activation of P2Y13 receptors in mice. Diabetologia 53(9):1927–1934
Ando RD, Mehesz B, Gyires K, Illes P, Sperlagh B (2010) A comparative analysis of the activity of ligands acting at P2X and P2Y receptor subtypes in models of neuropathic, acute and inflammatory pain. Br J Pharmacol 159(5):1106–1117
Biver G, Wang N, Gartland A, Orriss I, Arnett TR, Boeynaems JM, Robaye B (2013) Role of the P2Y13 receptor in the differentiation of bone marrow stromal cells into osteoblasts and adipocytes. Stem Cells 31(12):2747–2758
Bjorquist A, Di Buduo CA, Femia EA, Storey RF, Becker RC, Balduini A, Nylander S, Cattaneo M (2016) Studies of the interaction of ticagrelor with the P2Y13 receptor and with P2Y13-dependent pro-platelet formation by human megakaryocytes. Thromb Haemost 116(6):1079–1088
Blom D, Yamin TT, Champy MF, Selloum M, Bedu E, Carballo-Jane E, Gerckens L, Luell S, Meurer R, Chin J, Mudgett J, Puig O (2010) Altered lipoprotein metabolism in P2Y(13) knockout mice. Biochim Biophys Acta 1801(12):1349–1360
Bunyavanich S, Boyce JA, Raby BA, Weiss ST (2012) Gene-by-environment effect of house dust mite on purinergic receptor P2Y12 (P2RY12) and lung function in children with asthma. Clin Exp Allergy 42(2):229–237
Carrasquero LM, Delicado EG, Jimenez AI, Perez-Sen R, Miras-Portugal MT (2005) Cerebellar astrocytes co-express several ADP receptors. Presence of functional P2Y(13)-like receptors. Purinergic Signal 1(2):153–159
Carrasquero LM, Delicado EG, Bustillo D, Gutierrez-Martin Y, Artalejo AR, Miras-Portugal MT (2009) P2X7 and P2Y13 purinergic receptors mediate intracellular calcium responses to BzATP in rat cerebellar astrocytes. J Neurochem 110(3):879–889
Caseley EA, Muench SP, Roger S, Mao HJ, Baldwin SA, Jiang LH (2014) Non-synonymous single nucleotide polymorphisms in the P2X receptor genes: association with diseases, impact on receptor functions and potential use as diagnosis biomarkers. Int J Mol Sci 15(8):13344–13371
Cattaneo M, Gachet C (1999) ADP receptors and clinical bleeding disorders. Arterioscler Thromb Vasc Biol 19(10):2281–2285
Cavallari U, Trabetti E, Malerba G, Biscuola M, Girelli D, Olivieri O, Martinelli N, Angiolillo DJ, Corrocher R, Pignatti PF (2007) Gene sequence variations of the platelet P2Y12 receptor are associated with coronary artery disease. BMC Med Genet 8:59
Communi D, Gonzalez NS, Detheux M, Brezillon S, Lannoy V, Parmentier M, Boeynaems JM (2001) Identification of a novel human ADP receptor coupled to G(i). J Biol Chem 276(44):41479–41485
Csolle C, Heinrich A, Kittel A, Sperlagh B (2008) P2Y receptor mediated inhibitory modulation of noradrenaline release in response to electrical field stimulation and ischemic conditions in superfused rat hippocampus slices. J Neurochem 106(1):347–360
Cuadrado A (2015) Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radic Biol Med 88(Pt B):147–157
del Puerto A, Diaz-Hernandez JI, Tapia M, Gomez-Villafuertes R, Benitez MJ, Zhang J, Miras-Portugal MT, Wandosell F, Diaz-Hernandez M, Garrido JJ (2012) Adenylate cyclase 5 coordinates the action of ADP, P2Y1, P2Y13 and ATP-gated P2X7 receptors on axonal elongation. J Cell Sci 125(Pt 1):176–188
Diaz-Hernandez JI, Gomez-Villafuertes R, Leon-Otegui M, Hontecillas-Prieto L, Del Puerto A, Trejo JL, Lucas JJ, Garrido JJ, Gualix J, Miras-Portugal MT, Diaz-Hernandez M (2012) In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer’s disease through GSK3beta and secretases. Neurobiol Aging 33(8):1816–1828
Dohi T, Morita K, Kitayama T, Motoyama N, Morioka N (2009) Glycine transporter inhibitors as a novel drug discovery strategy for neuropathic pain. Pharmacol Ther 123(1):54–79
Dores MR, Trejo J (2012) Ubiquitination of G protein-coupled receptors: functional implications and drug discovery. Mol Pharmacol 82(4):563–570
Espada S, Ortega F, Molina-Jijon E, Rojo AI, Perez-Sen R, Pedraza-Chaverri J, Miras-Portugal MT, Cuadrado A (2010) The purinergic P2Y(13) receptor activates the Nrf2/HO-1 axis and protects against oxidative stress-induced neuronal death. Free Radic Biol Med 49(3):416–426
Fabre AC, Malaval C, Ben Addi A, Verdier C, Pons V, Serhan N, Lichtenstein L, Combes G, Huby T, Briand F, Collet X, Nijstad N, Tietge UJ, Robaye B, Perret B, Boeynaems JM, Martinez LO (2010) P2Y13 receptor is critical for reverse cholesterol transport. Hepatology 52(4):1477–1483
Ferreira MA, Jansen R, Willemsen G, Penninx B, Bain LM, Vicente CT, Revez JA, Matheson MC, Hui J, Tung JY, Baltic S, Le Souef P, Montgomery GW, Martin NG, Robertson CF, James A, Thompson PJ, Boomsma DI, Hopper JL, Hinds DA, Werder RB, Phipps S, Australian Asthma Genetics Consortium C (2016) Gene-based analysis of regulatory variants identifies 4 putative novel asthma risk genes related to nucleotide synthesis and signaling. J Allergy Clin Immunol 139(4):1148–1157
Fumagalli M, Trincavelli L, Lecca D, Martini C, Ciana P, Abbracchio MP (2004) Cloning, pharmacological characterisation and distribution of the rat G-protein-coupled P2Y(13) receptor. Biochem Pharmacol 68(1):113–124
Gachet C, Hechler B, Leon C, Vial C, Leray C, Ohlmann P, Cazenave JP (1997) Activation of ADP receptors and platelet function. Thromb Haemost 78(1):271–275
Goffinet M, Tardy C, Boubekeur N, Cholez G, Bluteau A, Oniciu DC, Lalwani ND, Dasseux JL, Barbaras R, Baron R (2014) P2Y13 receptor regulates HDL metabolism and atherosclerosis in vivo. PLoS One 9(4):e95807
Gualix J, Fideu MD, Pintor J, Rotllan P, Garcia-Carmona F, Miras-Portugal MT (1997) Characterization of diadenosine polyphosphate transport into chromaffin granules from adrenal medulla. FASEB J 11(12):981–990
Gualix J, Gomez-Villafuertes R, Pintor J, Llansola M, Felipo V, Miras-Portugal MT (2014) Presence of diadenosine polyphosphates in microdialysis samples from rat cerebellum in vivo: effect of mild hyperammonemia on their receptors. Purinergic Signal 10(2):349–356
Guarracino JF, Cinalli AR, Fernandez V, Roquel LI, Losavio AS (2016) P2Y13 receptors mediate presynaptic inhibition of acetylcholine release induced by adenine nucleotides at the mouse neuromuscular junction. Neuroscience 326:31–44
Hechler B, Leon C, Vial C, Vigne P, Frelin C, Cazenave JP, Gachet C (1998) The P2Y1 receptor is necessary for adenosine 5′-diphosphate-induced platelet aggregation. Blood 92(1):152–159
Hervas C, Perez-Sen R, Miras-Portugal MT (2003) Coexpression of functional P2X and P2Y nucleotide receptors in single cerebellar granule cells. J Neurosci Res 73(3):384–399
Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB (2001) Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 409(6817):202–207
Inoue K, Tsuda M (2012) P2X4 receptors of microglia in neuropathic pain. CNS Neurol Disord Drug Targets 11(6):699–704
Jacobson KA, Gao ZG, Paoletta S, Kiselev E, Chakraborty S, Jayasekara PS, Balasubramanian R, Tosh DK (2015) John Daly lecture: structure-guided drug design for adenosine and P2Y receptors. Comput Struct Biotechnol J 13:286–298
Jacquet S, Malaval C, Martinez LO, Sak K, Rolland C, Perez C, Nauze M, Champagne E, Terce F, Gachet C, Perret B, Collet X, Boeynaems JM, Barbaras R (2005) The nucleotide receptor P2Y13 is a key regulator of hepatic high-density lipoprotein (HDL) endocytosis. Cell Mol Life Sci 62(21):2508–2515
Jimenez AI, Castro E, Mirabet M, Franco R, Delicado EG, Miras-Portugal MT (1999) Potentiation of ATP calcium responses by A2B receptor stimulation and other signals coupled to Gs proteins in type-1 cerebellar astrocytes. Glia 26(2):119–128
Jimenez AI, Castro E, Communi D, Boeynaems JM, Delicado EG, Miras-Portugal MT (2000) Coexpression of several types of metabotropic nucleotide receptors in single cerebellar astrocytes. J Neurochem 75(5):2071–2079
Jimenez E, Zafra F, Perez-Sen R, Delicado EG, Miras-Portugal MT, Aragon C, Lopez-Corcuera B (2011) P2Y purinergic regulation of the glycine neurotransmitter transporters. J Biol Chem 286(12):10712–10724
Keov P, Sexton PM, Christopoulos A (2011) Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology 60(1):24–35
Kim YC, Lee JS, Sak K, Marteau F, Mamedova L, Boeynaems JM, Jacobson KA (2005) Synthesis of pyridoxal phosphate derivatives with antagonist activity at the P2Y13 receptor. Biochem Pharmacol 70(2):266–274
Kim KA, Song WG, Lee HM, Joo HJ, Park JY (2013) Effect of P2Y1 and P2Y12 genetic polymorphisms on the ADP-induced platelet aggregation in a Korean population. Thromb Res 132(2):221–226
Kiselev E, Barrett MO, Katritch V, Paoletta S, Weitzer CD, Brown KA, Hammes E, Yin AL, Zhao Q, Stevens RC, Harden TK, Jacobson KA (2014) Exploring a 2-naphthoic acid template for the structure-based design of P2Y14 receptor antagonist molecular probes. ACS Chem Biol 9(12):2833–2842
Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K (2008) P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci 28(11):2892–2902
Kobayashi K, Yamanaka H, Yanamoto F, Okubo M, Noguchi K (2012) Multiple P2Y subtypes in spinal microglia are involved in neuropathic pain after peripheral nerve injury. Glia 60(10):1529–1539
Kobayashi K, Yamanaka H, Noguchi K (2013) Expression of ATP receptors in the rat dorsal root ganglion and spinal cord. Anat Sci Int 88(1):10–16
Koch H, von Kugelgen I, Starke K (1997) P2-receptor-mediated inhibition of noradrenaline release in the rat hippocampus. Naunyn Schmiedeberg’s Arch Pharmacol 355(6):707–715
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation C (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616):285–291
Leon C, Hechler B, Vial C, Leray C, Cazenave JP, Gachet C (1997) The P2Y1 receptor is an ADP receptor antagonized by ATP and expressed in platelets and megakaryoblastic cells. FEBS Lett 403(1):26–30
Lewis C, Neidhart S, Holy C, North RA, Buell G, Surprenant A (1995) Coexpression of P2X2 and P2X3 receptor subunits can account for ATP-gated currents in sensory neurons. Nature 377(6548):432–435
Li MP, Tang J, Wen ZP, Zhang YJ, Zhang W, Zhou HH, Zhang ZL, Chen XP (2015) Influence of P2Y12 polymorphisms on platelet activity but not ex-vivo antiplatelet effect of ticagrelor in healthy Chinese male subjects. Blood Coagul Fibrinolysis 26(8):874–881
Lichtenstein L, Serhan N, Annema W, Combes G, Robaye B, Boeynaems JM, Perret B, Tietge UJ, Laffargue M, Martinez LO (2013) Lack of P2Y13 in mice fed a high cholesterol diet results in decreased hepatic cholesterol content, biliary lipid secretion and reverse cholesterol transport. Nutr Metab (Lond) 10(1):67
Lichtenstein L, Serhan N, Espinosa-Delgado S, Fabre A, Annema W, Tietge UJ, Robaye B, Boeynaems JM, Laffargue M, Perret B, Martinez LO (2015) Increased atherosclerosis in P2Y13/apolipoprotein E double-knockout mice: contribution of P2Y13 to reverse cholesterol transport. Cardiovasc Res 106(2):314–323
Lordkipanidze M, Pharand C, Schampaert E, Palisaitis DA, Diodati JG (2011) Heterogeneity in platelet cyclooxygenase inhibition by aspirin in coronary artery disease. Int J Cardiol 150(1):39–44
Lustig KD, Shiau AK, Brake AJ, Julius D (1993) Expression cloning of an ATP receptor from mouse neuroblastoma cells. Proc Natl Acad Sci U S A 90(11):5113–5117
Malaval C, Laffargue M, Barbaras R, Rolland C, Peres C, Champagne E, Perret B, Terce F, Collet X, Martinez LO (2009) RhoA/ROCK I signalling downstream of the P2Y13 ADP-receptor controls HDL endocytosis in human hepatocytes. Cell Signal 21(1):120–127
Malin SA, Molliver DC (2010) Gi- and Gq-coupled ADP (P2Y) receptors act in opposition to modulate nociceptive signaling and inflammatory pain behavior. Mol Pain 6:21
Maqbool M, Mobashir M, Hoda N (2016) Pivotal role of glycogen synthase kinase-3: a therapeutic target for Alzheimer’s disease. Eur J Med Chem 107:63–81
Marin-Garcia P, Sanchez-Nogueiro J, Diez A, Leon-Otegui M, Linares M, Garcia-Palencia P, Bautista JM, Miras-Portugal MT (2009) Altered nucleotide receptor expression in a murine model of cerebral malaria. J Infect Dis 200(8):1279–1288
Marschallinger J, Schaffner I, Klein B, Gelfert R, Rivera FJ, Illes S, Grassner L, Janssen M, Rotheneichner P, Schmuckermair C, Coras R, Boccazzi M, Chishty M, Lagler FB, Renic M, Bauer HC, Singewald N, Blumcke I, Bogdahn U, Couillard-Despres S, Lie DC, Abbracchio MP, Aigner L (2015) Structural and functional rejuvenation of the aged brain by an approved anti-asthmatic drug. Nat Commun 6:8466
Marteau F, Le Poul E, Communi D, Communi D, Labouret C, Savi P, Boeynaems JM, Gonzalez NS (2003) Pharmacological characterization of the human P2Y13 receptor. Mol Pharmacol 64(1):104–112
Marteau F, Communi D, Boeynaems JM, Suarez Gonzalez N (2004) Involvement of multiple P2Y receptors and signaling pathways in the action of adenine nucleotides diphosphates on human monocyte-derived dendritic cells. J Leukoc Biol 76(4):796–803
Martinez LO, Najib S, Perret B, Cabou C, Lichtenstein L (2015) Ecto-F1-ATPase/P2Y pathways in metabolic and vascular functions of high density lipoproteins. Atherosclerosis 238(1):89–100
Marucci G, Dal Ben D, Lambertucci C, Santinelli C, Spinaci A, Thomas A, Volpini R, Buccioni M (2016) The G protein-coupled receptor GPR17: overview and update. ChemMedChem 11(23):2567–2574
May LT, Leach K, Sexton PM, Christopoulos A (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51
Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW (2012) Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J Med Chem 55(4):1445–1464
Milewicz DM, Seidman CE (2000) Genetics of cardiovascular disease. Circulation 102(20 Suppl 4):IV103–IV111
Morente V, Perez-Sen R, Ortega F, Huerta-Cepas J, Delicado EG, Miras-Portugal MT (2014) Neuroprotection elicited by P2Y13 receptors against genotoxic stress by inducing DUSP2 expression and MAPK signaling recovery. Biochim Biophys Acta 1843(9):1886–1898
Oestreich JH, Steinhubl SR, Ferraris SP, Loftin CD, Akers WS (2014) Effect of genetic variation in P2Y12 on TRAP-stimulated platelet response in healthy subjects. J Thromb Thrombolysis 38(3):372–379
Orriss I, Syberg S, Wang N, Robaye B, Gartland A, Jorgensen N, Arnett T, Boeynaems JM (2011) Bone phenotypes of P2 receptor knockout mice. Front Biosci (Schol Ed) 3:1038–1046
Ortega F, Perez-Sen R, Miras-Portugal MT (2008) Gi-coupled P2Y-ADP receptor mediates GSK-3 phosphorylation and beta-catenin nuclear translocation in granule neurons. J Neurochem 104(1):62–73
Ortega F, Perez-Sen R, Delicado EG, Teresa Miras-Portugal M (2011) ERK1/2 activation is involved in the neuroprotective action of P2Y13 and P2X7 receptors against glutamate excitotoxicity in cerebellar granule neurons. Neuropharmacology 61(8):1210–1221
Ou W, He Y, Li A, Liu B, Jin L (2016) Genotype frequencies of CYP2C19, P2Y12 and GPIIIa polymorphisms in coronary heart disease patients of Han ethnicity, and their impact on Clopidogrel responsiveness. Int Heart J 57(5):586–592
Paoletta S, Sabbadin D, von Kugelgen I, Hinz S, Katritch V, Hoffmann K, Abdelrahman A, Strassburger J, Baqi Y, Zhao Q, Stevens RC, Moro S, Muller CE, Jacobson KA (2015) Modeling ligand recognition at the P2Y12 receptor in light of X-ray structural information. J Comput Aided Mol Des 29(8):737–756
Patel K, Barnes A, Camacho J, Paterson C, Boughtflower R, Cousens D, Marshall F (2001) Activity of diadenosine polyphosphates at P2Y receptors stably expressed in 1321N1 cells. Eur J Pharmacol 430(2–3):203–210
Perez-Sen R, Queipo MJ, Morente V, Ortega F, Delicado EG, Miras-Portugal MT (2015) Neuroprotection mediated by P2Y13 nucleotide receptors in neurons. Comput Struct Biotechnol J 13:160–168
Pons V, Serhan N, Gayral S, Malaval C, Nauze M, Malet N, Laffargue M, Gales C, Martinez LO (2014) Role of the ubiquitin-proteasome system in the regulation of P2Y13 receptor expression: impact on hepatic HDL uptake. Cell Mol Life Sci 71(9):1775–1788
Queiroz G, Talaia C, Goncalves J (2003) ATP modulates noradrenaline release by activation of inhibitory P2Y receptors and facilitatory P2X receptors in the rat vas deferens. J Pharmacol Exp Ther 307(2):809–815
Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, Tobon-Velasco JC, Devijver H, Garcia-Mayoral MF, Van Leuven F, Hayes JD, Bertho G, Cuadrado A (2012) Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol 32(17):3486–3499
Rojo AI, Rada P, Egea J, Rosa AO, Lopez MG, Cuadrado A (2008) Functional interference between glycogen synthase kinase-3 beta and the transcription factor Nrf2 in protection against kainate-induced hippocampal cell death. Mol Cell Neurosci 39(1):125–132
Savi P, Beauverger P, Labouret C, Delfaud M, Salel V, Kaghad M, Herbert JM (1998) Role of P2Y1 purinoceptor in ADP-induced platelet activation. FEBS Lett 422(3):291–295
Selden NR, Carlson JD, Cetas J, Close LN, Heinricher MM (2007) Purinergic actions on neurons that modulate nociception in the rostral ventromedial medulla. Neuroscience 146(4):1808–1816
Serhan N, Cabou C, Verdier C, Lichtenstein L, Malet N, Perret B, Laffargue M, Martinez LO (2013) Chronic pharmacological activation of P2Y13 receptor in mice decreases HDL-cholesterol level by increasing hepatic HDL uptake and bile acid secretion. Biochim Biophys Acta 1831(4):719–725
Shaver SR, Rideout JL, Pendergast W, Douglass JG, Brown EG, Boyer JL, Patel RI, Redick CC, Jones AC, Picher M, Yerxa BR (2005) Structure-activity relationships of dinucleotides: potent and selective agonists of P2Y receptors. Purinergic Signal 1(2):183–191
Siasos G, Kioufis S, Oikonomou E, Zaromitidou M, Maniatis K, Vavuranakis M, Kokkou E, Tousoulis D (2016) Impact of C34T P2Y12 ADP receptor polymorphism and smoking status on cardiovascular outcome in coronary artery disease patients receiving clopidogrel. Int J Cardiol 210:161–163
Tan C, Salehi A, Svensson S, Olde B, Erlinge D (2010) ADP receptor P2Y(13) induce apoptosis in pancreatic beta-cells. Cell Mol Life Sci 67(3):445–453
Tan C, Voss U, Svensson S, Erlinge D, Olde B (2013) High glucose and free fatty acids induce beta cell apoptosis via autocrine effects of ADP acting on the P2Y(13) receptor. Purinergic Signal 9(1):67–79
Tatsumi E, Yamanaka H, Kobayashi K, Yagi H, Sakagami M, Noguchi K (2015) RhoA/ROCK pathway mediates p38 MAPK activation and morphological changes downstream of P2Y12/13 receptors in spinal microglia in neuropathic pain. Glia 63(2):216–228
Timur AA, Murugesan G, Zhang L, Aung PP, Barnard J, Wang QK, Gaussem P, Silverstein RL, Bhatt DL, Kottke-Marchant K (2012) P2RY1 and P2RY12 polymorphisms and on-aspirin platelet reactivity in patients with coronary artery disease. Int J Lab Hematol 34(5):473–483
Tozaki-Saitoh H, Tsuda M, Miyata H, Ueda K, Kohsaka S, Inoue K (2008) P2Y12 receptors in spinal microglia are required for neuropathic pain after peripheral nerve injury. J Neurosci 28(19):4949–4956
Van Kolen K, Slegers H (2004) P2Y12 receptor stimulation inhibits beta-adrenergic receptor-induced differentiation by reversing the cyclic AMP-dependent inhibition of protein kinase B. J Neurochem 89(2):442–453
Van Kolen K, Gilany K, Moens L, Esmans EL, Slegers H (2006) P2Y12 receptor signalling towards PKB proceeds through IGF-I receptor cross-talk and requires activation of Src, Pyk2 and Rap1. Cell Signal 18(8):1169–1181
von Kugelgen I, Hoffmann K (2016) Pharmacology and structure of P2Y receptors. Neuropharmacology 104:50–61
von Kugelgen I, Spath L, Starke K (1994) Evidence for P2-purinoceptor-mediated inhibition of noradrenaline release in rat brain cortex. Br J Pharmacol 113(3):815–822
Voss U, Turesson MF, Robaye B, Boeynaems JM, Olde B, Erlinge D, Ekblad E (2014) The enteric nervous system of P2Y13 receptor null mice is resistant against high-fat-diet- and palmitic-acid-induced neuronal loss. Purinergic Signal 10(3):455–464
Wang Z, Nakayama T, Sato N, Yamaguchi M, Izumi Y, Kasamaki Y, Ohta M, Soma M, Aoi N, Ozawa Y, Ma Y, Doba N, Hinohara S (2009a) Purinergic receptor P2Y, G-protein coupled, 2 (P2RY2) gene is associated with cerebral infarction in Japanese subjects. Hypertens Res 32(11):989–996
Wang ZX, Nakayama T, Sato N, Izumi Y, Kasamaki Y, Ohta M, Soma M, Aoi N, Matsumoto K, Ozawa Y, Ma YT, Doba N, Hinohara S (2009b) Association of the purinergic receptor P2Y, G-protein coupled, 2 (P2RY2) gene with myocardial infarction in Japanese men. Circ J 73(12):2322–2329
Wang Z, Nakayama T, Sato N, Izumi Y, Kasamaki Y, Ohta M, Soma M, Aoi N, Ozawa Y, Ma Y (2010) The purinergic receptor P2Y, G-protein coupled, 2 (P2RY2) gene associated with essential hypertension in Japanese men. J Hum Hypertens 24(5):327–335
Wang N, Robaye B, Agrawal A, Skerry TM, Boeynaems JM, Gartland A (2012) Reduced bone turnover in mice lacking the P2Y13 receptor of ADP. Mol Endocrinol 26(1):142–152
Wang N, Rumney RM, Yang L, Robaye B, Boeynaems JM, Skerry TM, Gartland A (2013) The P2Y13 receptor regulates extracellular ATP metabolism and the osteogenic response to mechanical loading. J Bone Miner Res 28(6):1446–1456
Wang N, Robaye B, Gossiel F, Boeynaems JM, Gartland A (2014) The P2Y13 receptor regulates phosphate metabolism and FGF-23 secretion with effects on skeletal development. FASEB J 28(5):2249–2259
Webb TE, Simon J, Krishek BJ, Bateson AN, Smart TG, King BF, Burnstock G, Barnard EA (1993) Cloning and functional expression of a brain G-protein-coupled ATP receptor. FEBS Lett 324(2):219–225
Wesselius A, Bours MJ, Henriksen Z, Syberg S, Petersen S, Schwarz P, Jorgensen NR, van Helden S, Dagnelie PC (2013) Association of P2Y(2) receptor SNPs with bone mineral density and osteoporosis risk in a cohort of Dutch fracture patients. Purinergic Signal 9(1):41–49
Wirkner K, Schweigel J, Gerevich Z, Franke H, Allgaier C, Barsoumian EL, Draheim H, Illes P (2004) Adenine nucleotides inhibit recombinant N-type calcium channels via G protein-coupled mechanisms in HEK 293 cells; involvement of the P2Y13 receptor-type. Br J Pharmacol 141(1):141–151
Yano S, Tsukimoto M, Harada H, Kojima S (2012) Involvement of P2Y13 receptor in suppression of neuronal differentiation. Neurosci Lett 518(1):5–9
Zee RY, Michaud SE, Diehl KA, Chasman DI, Emmerich J, Gaussem P, Aiach M, Ridker PM (2008) Purinergic receptor P2Y, G-protein coupled, 12 gene variants and risk of incident ischemic stroke, myocardial infarction, and venous thromboembolism. Atherosclerosis 197(2):694–699
Zhang FL, Luo L, Gustafson E, Lachowicz J, Smith M, Qiao XD, Liu YH, Chen GD, Pramanik B, Laz TM, Palmer K, Bayne M, Monsma FJ (2001) ADP is the cognate ligand for the orphan G protein-coupled receptor SP1999. J Biol Chem 276(11):8608–8615
Zhang FL, Luo L, Gustafson E, Palmer K, Qiao X, Fan X, Yang S, Laz TM, Bayne M, Monsma F Jr (2002) P2Y(13): identification and characterization of a novel Galphai-coupled ADP receptor from human and mouse. J Pharmacol Exp Ther 301(2):705–713
Zhang J, Zhang K, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Muller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q (2014a) Agonist-bound structure of the human P2Y12 receptor. Nature 509(7498):119–122
Zhang K, Zhang J, Gao ZG, Zhang D, Zhu L, Han GW, Moss SM, Paoletta S, Kiselev E, Lu W, Fenalti G, Zhang W, Muller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q (2014b) Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 509(7498):115–118
Zheng X, Liang Y, Kang A, Ma SJ, Xing L, Zhou YY, Dai C, Xie H, Xie L, Wang GJ, Hao HP (2014) Peripheral immunomodulation with ginsenoside Rg1 ameliorates neuroinflammation-induced behavioral deficits in rats. Neuroscience 256:210–222
Acknowledgments
We are grateful to Dr. Mark Sefton for the english copy-editing of the manuscript. The work in the authors’ laboratory is funded by the Spanish Ministerio de Economia y Competitividad (MINECO, BFU 2014-53654-P) and Red de excelencia Consolider-Ingenio Spanish Ion Channel Initiative” (BFU2015-70067REDC); by the Comunidad de Madrid (BRADE-CM S2013/ICE-2958), UCM-Santander (PR26/16-18B-3) and by the Fundación Ramón Areces Grant (PR2018/16-02). F. Ortega is a recipient of a Ramón y Cajal contract (RYC-2013-13290).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Ethical Approvals
This article does not contain any studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Pérez-Sen, R., Gómez-Villafuertes, R., Ortega, F., Gualix, J., Delicado, E.G., Miras-Portugal, M.T. (2017). An Update on P2Y13 Receptor Signalling and Function. In: Atassi, M. (eds) Protein Reviews. Advances in Experimental Medicine and Biology(), vol 1051. Springer, Singapore. https://doi.org/10.1007/5584_2017_91
Download citation
DOI: https://doi.org/10.1007/5584_2017_91
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-7610-7
Online ISBN: 978-981-10-7611-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)