
Abstract
While high levels of glucose and saturated fatty acids are known to have detrimental effects on beta cell function and survival, the signalling pathways mediating these effects are not entirely known. In a previous study, we found that ADP regulates beta cell insulin secretion and beta cell apoptosis. Using MIN6c4 cells as a model system, we investigated if autocrine/paracrine mechanisms of ADP and purinergic receptors are involved in this process. High glucose (16.7 mmol/l) and palmitate (100 μmol/l) rapidly and potently elevated the extracellular ATP levels, while mannitol was without effect. Both tolbutamide and diazoxide were without effect, while the calcium channel blocker nifedipine, the volume-regulated anion channels (VRAC) inhibitor NPPB, and the pannexin inhibitor carbenoxolone could inhibit both effects. Similarly, silencing the MDR1 gene also blocked nutrient-generated ATP release. These results indicate that calcium channels and VRAC might be involved in the ATP release mechanism. Furthermore, high glucose and palmitate inhibited cAMP production, reduced cell proliferation in MIN6c4 and increased activated Caspase-3 cells in mouse islets and in MIN6c4 cells. The P2Y13-specific antagonist MRS2211 antagonized all these effects. Further studies showed that blocking the P2Y13 receptor resulted in enhanced CREB, Bad and IRS-1 phosphorylation, which are known to be involved in beta cell survival and insulin secretion. These findings provide further support for the concept that P2Y13 plays an important role in beta cell apoptosis and suggest that autocrine/paracrine mechanisms, related to ADP and P2Y13 receptors, contribute to glucolipotoxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Besides genetic influence, it is generally accepted that environmental factors are the main cause for the rapidly increasing worldwide prevalence of type 2 diabetes. Lifestyle and hypercaloric diets rich in large amounts of carbohydrate and saturated fats are known risk factors.
Elevated glucose and increased FFA have been shown to trigger pancreatic beta cell loss and dysfunction in type 2 diabetes [1], and increasing evidences demonstrate that long-term exposure to high glucose or FFA generates peripheral insulin resistance, both in vivo and in vitro resulting in pancreatic stress and increased susceptibility to beta cell apoptosis and, as a consequence, suppression of insulin secretion [2]. Although the overall picture is known, the detailed mechanisms underlying these effects are only partially understood.
During the last two decades, it has become generally accepted that the intracellular energy mediator ATP also mediates cell-to-cell signalling, modulating a wide range of cell and organ functions through activation of purinergic receptors on the plasma membrane [3, 4]. Many cells exhibit regulated release of ATP, which is rapidly degraded by ectonucleotidases to ADP, AMP and adenosine thus extending and modulating the signal to a wider spectrum of receptor subtypes. Once outside of the cell, ATP and its metabolites in nanomolar concentrations function as an autocrine/paracrine signal modulating a broad range of cell and organ functions.
In our previous studies, we demonstrated that extracellular ADP inhibits beta cell insulin secretion [5] and causes beta cell apoptosis by acting on P2Y13 receptors [6]. The aim of the present study was to put this mechanism into a functional perspective, investigating if autocrine/paracrine activation of purinergic receptors might be involved in the proapoptotic effects of high glucose and FFA. To do so, we first examined if high glucose and palmitate could induce ATP release in MIN6c4 cells. The nature of the release mechanism was evaluated, and we found that, in the case of both glucose and palmitate, it was dependent on the gene MDR1 encoding multidrug resistance P-glycoprotein and activation of volume-regulated anionic channels (VRAC). We then studied if the released ATP/ADP had effects on cAMP, Caspase-3 and on cell proliferation, and through which receptors this effect was mediated. We further studied if autocrine/paracrine ATP release had effects on CREB, Bad and IRS-1 activation. Mouse insulinoma cell line MIN6c4 was used as our primary model system and isolated murine pancreatic islets as our secondary model. Our results support the hypothesis that high glucose and free fatty acids induce beta cell apoptosis via autocrine effects of ADP acting on the P2Y13 receptor.
Materials and methods
Materials
MRS2211 were purchased from Tocris (UK). Lentiviral MDR1 siRNA (m) and scrambled control siRNA were from Santa Cruz Biotechnology, Inc. (USA). The cyclic AMP kit was obtained from Cayman Chemical (USA). Anti-phospho-CREB, anti-CREB, anti-phospho-Bad, anti-Bad antibodies, anti-phospho-IRS-1, anti-IRS-1 and anti-activated-Caspase-3 were all obtained from Cell Signalling Technology (Danvers, MA, USA). The targefect-PCL kit was purchased from BioSite (USA). The EnzChek® Caspase-3 Assay kit #2 was purchased from Molecular Probes (USA). All other chemicals, unless otherwise stated, were obtained from Sigma-Aldrich and were either of analytical or laboratory grade.
Cell culture
The mouse MIN6 pancreatic beta cell line subclone MIN6c4, a generous gift from Professor Jun-Ichi Miyazaki, Osaka, Japan, was used for the present study. MIN6c4 cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing Glutamax-1 and 25 mmol/l of glucose (Invitrogen, Paisley, UK) in the presence of 15 % heat-inactivated FBS (Invitrogen, Paisley, UK), 60 μmol/l β-mercaptothanol, 5 × 104 U/l penicillin, and 50 mg/l streptomycin under 5 % CO2 and 95 % air at 37 °C.
Animals
Eight female C57B6 mice from Charles River (Sulzfeld, Germany) were used in the experiments. Animals had free access to standard chow and water. Experimental designs were approved by the Animal Ethics Committee, Lund and Malmö, Sweden. Animals were used in accordance with the European Community Council Directive (86/609/EEC and 2010/63/EU) and the Swedish Animal Welfare Act (SFS 1988:534).
Islet isolation
Islets of Langerhans were isolated according to a previously described protocol [7]. In brief, animals were killed by cervical dislocation, and abdominal cavity was opened. Sternum and liver was removed to allow access to the biliary tract. The biliary tract was clamped using a micro-vascular clip. Pancreatic islets were isolated by retrograde injection of a cold collagenase solution via the bile–pancreatic duct. The pancreas was excised, and islets were isolated by digestion for 20 min at 37 °C. Tissues were dispersed by manual shaking, and islets were washed 3 × 5 min in cold HBSS with 10 mmol/l HEPES. The islets were then picked manually using a stereo microscope.
ATP release
ATP release was measured using an adaptation of the luciferase method previously described by Taylor et al. [8]. Briefly, 35,000 of MIN6c4 cells per well were plated in 35-mm Petri dish and were allowed to grow in 25 mmol/l glucose DMEM containing 15 % FBS for 24 h. The medium was then replaced with 1 ml serum-free DMEM containing 5.6 mmol/l glucose, and the cells were cultivated for another 24 h. One-millilitre 2 × ATP SL reagent (Biothema, Sweden), in serum-free DMEM containing 5.6 mmol/l glucose, was added, and the Petri dish was incubated for 30 min at RT. After recording the baseline luciferase activity in the Petri dish for 1 min in a GloMax 20/20 Luminometer (Promega Corporation, Madison, WI, USA), 400 μl of the medium was removed. The test substances were added to the removed medium, which subsequently was returned to the Petri dish where the luminescence was recorded for another 10 min. The applied routine for sample addition was chosen since it minimises disturbing the cells, which in turn, decreases the background signal.
Insulin secretion
MIN6c4 cells were cultured in a 35-mm Petri dish in 2 ml of 25 mmol/l glucose DMEM containing 15 % FBS for 72 h. Just prior to the experiment, the medium was replaced with 1 ml of Krebs-Ringer bicarbonate HEPES buffer (KRBH buffer; 135 mM NaCl, 3.6 mM KCl, 2 mM NaHCO3, 0.5 mM NaH2PO4, 0.5 mM MgCl2, 1.5 mM CaCl2, 10 mM HEPES and 3.0 mM glucose, pH 7.4), and the cells were incubated for 30 min. The buffer was then exchanged for KRBH containing 16.7 mM glucose or 100 μM palmitate. Samples (50 μl) were collected every 30 s and replaced with an equal amount of fresh buffer. The insulin content of the samples was determined using an ELISA kit (Mercodia, Uppsala, Sweden), according to the manufacturer's instructions.
Lentiviral transduction of MIN6c4 cells
MIN6c4 cells were seeded in 12-well dishes and grown to obtain 50–60 % confluency. The medium was replaced with 1 ml fresh medium, containing polybrene (5 mg/l), and shRNA lentiviral particles (Santa Cruz Biotechnology, Santa Cruz, CA, USA) to give a multiplicity of infection (MOI) of approximately 0.05. The cells were incubated for 24 h at 37 °C when the medium was replaced with selection medium containing 1.5 mg/l puromycin. The puromycin selection was continued until a stable puromycin-resistant cell population was obtained.
Relative MDR1 mRNA measurement by quantitative real-time PCR
Total RNA was extracted from MIN6c4 (control siRNA) and MIN6c4 (MDR1 siRNA) cells using TRIZOL reagent (Invitrogen, Grand Island, NY, USA) according to manufacturer's instructions. Total RNA was quantified using nanodrop ND-1000 spectrophotometer (Nanodrop Technologies Inc., Wilmington, DE, USA). cDNA was synthesized from 3 μg of total RNA using the RevertAid™ H Minus First Strand cDNA Synthesis Kit (Fermentas, Waltham, MA, USA) according to the manufacturer's instructions.
Real-time quantitative PCR was performed using an ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). A specific set of primers was chosen and described before to obtain a product close to 200 bp [9]. The forward primers were 5′-CATGACAGATAGCTTTGCAAGTGTAG-3′ (MDR1), 5′-CTACCTCATGAAGATCCTCACCGA-3′ (Beta-actin), and the reversed primers were 5′-GGCAAACATGGCTCTTTTATCG-3′ (MDR1), 5′-CGCTCATTGCCAATGGTGAT-3′ (Beta-actin). All quantitative real-time PCR (Q-RT-PCR) reactions were carried out in MicroAmp Fast 96-well Reaction Plate (Applied Biosystems, Foster City, CA, USA) in 20-μl reactions containing 1 μl cDNA template, 0.2 μmol/l of forward and reverse primers and 1 × Faster SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA). The PCR enzyme was activated by initial 20 s incubation at 95 °C, followed by 40 PCR cycles (3 s denaturation at 95 °C, 30 s annealing and elongation at 60 °C). Results were expressed using the comparative C t method as described in User Bulletin 2 (Applied Biosystems). Briefly, the ΔΔC t values were calculated as follows: (C t gene of interest − C t reference gene)treated − (C t gene of interest − C t reference gene)untreated, with Beta-actin as the reference gene, a well-known housekeeping gene. The fold change in the level of target mRNA between untreated and treated cells was then expressed as 2−ΔΔCt with ΔΔC t ± SE.
cAMP real-time luciferase analysis
Real time of cAMP production was analysed by using transient transfection of MIN6c4 cells with the experimental plasmid Glosensor L9, a kind gift from Dr. Neal Cosby, Promega. MIN6c4 cells, 5 × 105, were plated in a 35-mm Petri dish and cultivated to get a confluence of 30–70 %. The Glosensor A1 plasmid was transfected into the MIN6c4 cells using Targefect-PCL (Targeting Systems, El Cajon, CA, USA) according to the manufacturer's instructions. Forty-eight hours after transfection, the cell culture medium was replaced with 1-ml low glucose DMEM (containing 10 mmol/l HEPES, pH 7.5) prepared cAMP reagent medium (pH 7.5), which contains 10 mmol/l HEPES and 2 % (v/v) Glosensor cAMP reagent (Promega Corporation, Madison, WI, USA). After equilibration, for 2 h at RT, the baseline luciferase activity was recorded for 3 min in a GloMax 20/20 Luminometer (Promega Corporation, Madison, WI, USA). The test substances were then added, and the luminescence was recorded and measured for another 10 min.
Measurement of cAMP concentrations
MIN6c4 cells were plated in 24-well plates at a density of 4 × 105 cells/well and were then allowed to grow for 3 days. The medium was replaced with 250 μl OptiMEM containing 25 μmol/l Rolipram. The cells were first pre-incubated for 5 min at 37 °C, the test substances were then added and the incubation was extended to 25 min. The medium was removed and the monolayer extracted using 200 μl 0.1 mol/l hydrochloric acid per well. The cell lysates were centrifuged at 1,000 rpm for 10 min at 4 °C, and the cyclic AMP content of the supernatant was measured using a Cyclic AMP EIA kit (Cayman Chemical, USA) according to manufacturer's instructions.
Measurement of Caspase-3 activity in MIN6c4 cells
Caspase-3 activity was determined by using the EnzChek Caspase-3 Assay Kit #2 (Molecular Probes) according to the manufacturer's instructions. Briefly, after incubation with test substances for 36 h, cells were washed with PBS and lysed. The Caspase-3 activity in the extracts was then measured using a fluorometric assay. The fluorescent product of the substrate Z-DEVD-rhodamine 110 generated by Caspase-3 in the cell extract was detected in a microplate fluorometer (VICTOR, PerkinElmer, Finland) using an excitation of 496 nm and emission of 520 nm. Background fluorescence was determined by including a specific Caspase-3 inhibitor (Ac-DEVD-CHO).
Islet treatment
Isolated mouse islets were equilibrated over night at 37 °C in a humidified incubator holding 5 % CO2 in 5.6 mmol/l glucose baseline medium [RPMI 1640 glutamax medium (GIBCO), 10 % FBS (GIBCO), 5 × 104 U/l penicillin and 50 mg/l streptomycin (A2213, BioChrom AG, Berlin, Germany)]; islets from each animal were not pooled. The islets were then divided into groups and moved to wells containing different treatment medium. Islets were incubated in treatment wells for 24 h in a humidified incubator holding 5 % CO2. The incubation was terminated by fixation in Stefanini's fixative (0.2 % picric acid, 2 % formaldehyde in 0.1 mol/l phosphate buffer, pH 7.2) for 30 min, followed by washing 2 × 10 min in Tyrode's solution containing 10 % sucrose prior to embedding in cryomount (Histolab, Gothenburg, Sweden) and stored at −80 °C.
Immunocytochemistry on isolated islets
The embedded islets were cut into 10-μm cryosections and mounted onto slides. Slides were washed in PBS containing 0.25 % Triton X-100 and stained using antibodies against activated Caspase-3 (Cell Signalling pre-diluted #8120) overnight at 4 °C. Slides were washed once in PBS containing 0.25 % Triton X-100. Antigens were visualized using 1:1,000 diluted Dylight 594 donkey anti-rabbit (Jackson laboratories 711-515-152) in PBS containing 0.25 % Triton X-100 and 0.25 % BSA, incubated at RT for 1 h. Hoechst staining (Life Technologies, Stockholm, Sweden) was performed as a counter stain to visualize the nucleus of cells. Islets were visualized using fluorescent microscope (Olympus BX42), and pictures were captured using a camera (Olympus XC30). Pictures were analysed using Image J cell counter application (rsbweb.nih.gov/ij), and total numbers of cells as well as Caspase-3-activated cells were counted. The frequency of Caspase-3-activated cells were calculated and normalized to the control value of the same animal.
Western blot analysis
MIN6c4 cells, growing in 6-well plates, were incubated for 30 min in the presence of different test agents. Cells were lysed in ice-cold SDS buffer containing a cocktail of protease inhibitors. Cellular lysates were prepared by sonication (15 s), heating for 5 min at 99 °C and then centrifugation for 5 min. The protein content of the lysate was determined by DC protein assay (BIO-RAD), and samples, representing 20 μg of total protein, were separated on a Precase 8–6 % SDS-polyacrylamide gel (Lonza, USA) and transferred to Immobilon-P membranes (Millipore Corporation, Billerica, MA, USA). The intensities of bands were quantitated by scanning densitometry (Fluor-S Multi-Imager).
Cellular viability
The effects of a P2-receptor agonist or antagonist on cell viability of MIN6c4 cell line were studied using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H tetrazolium bromide (MTT) colorimetric assay. The MIN6c4 cells were seeded in 96-well plates at a concentration of 1 × 104 cells per well, in a volume of 200 μl of cell culture medium per well. After 24 h, test substances were added, and the plates were kept in the 5 % CO2 incubator for 3 days, then 20 μl of the MTT reagent (5 g/l in PBS buffer) was added to each well, and the plates were further incubated in the 5 % CO2 incubator at 37 °C for an additional 4 h. The medium was aspirated and replaced with 200 μl of DMSO (dimethyl sulfoxide solubilisation solution) to dissolve the insoluble purple formazan product. The absorbance was quantified by measuring at 490 nm in a microplate reader (VICTOR, PerkinElmer, Finland).
Cellular proliferation
MIN6c4 cells were seeded in 24-well plates at a concentration of 1 × 104 cells per well in cell culture medium. The cells were counted every 2 days by using a Bürkert chamber.
Statistical analysis
Results were presented as mean and standard error of the mean (SEM) unless otherwise stated. The level of significance for the difference between sets of data was assessed by Student's unpaired t test using GraphPad InStat, Version 5.0 (GraphPad Prism Software, San Diego, CA, USA). For immunocytochemical Caspase-3 activity in mouse islets, statistical analysis was performed using one-way ANOVA followed by Dunnett's post hoc test. Statistically significant differences were considered at p < 0.05 (*), p < 0.01 (**) or p < 0.001 (***) (two-tailed test).
Results
High glucose and palmitate stimulates ATP release from MIN6c4 cells
The release of extracellular ATP upon treatment with nutrients was investigated by stimulating MIN6c4 cells with glucose or palmitate. Both the glucose (16.7 mmol/l)- and the palmitate (100 μmol/l)-triggered ATP release were fast, reaching about 80 % of maximal release within seconds after stimulation preceding the insulin release by half a minute (Fig. 1a–d). Furthermore, the shape of the ATP release time courses, both for glucose and for palmitate, was different from the corresponding insulin curves. However, no effect was found when stimulating the cells with 16.7 mmol/l mannitol (Fig. 1e). The ATP release, induced by glucose, was significantly inhibited (Fig. 1e) by the calcium channel blocker nifedipine (20 μmol/l), and the palmitate-induced release was similarly inhibited (Fig. 1f). The KATP channel inhibitor tolbutamide (100 μmol/l) failed to stimulate ATP release, while the KATP channel activator diazoxide (10 μmol/l) was unable to block glucose-stimulated ATP release (Fig 1e). 2-Bromo palmitate (100 μmol/l), previously reported to inhibit fatty acid-stimulated insulin release [10, 11], was unable to block palmitate (100 μmol/l)-stimulated ATP release (Fig. 1f). On the other hand, exposure of the cells to the pannexin-1 inhibitor carbenoxolone (20 μmol/l) or to the VRAC inhibitor NPPB (100 μmol/l) both resulted in a substantial attenuation of high glucose/palmitate-induced ATP release (Fig. 1e, f).

ATP release and insulin secretion from MIN6c4 cells. ATP release analysis was performed as described under “Materials and methods” section. ATP release stimulated by 16.7 mmol/l glucose (a) or 100 μmol/l palmitate (c) was faster than the 16.7-mmol/l glucose- or 100-μmol/l palmitate-induced insulin secretion (b, d) under same conditions. e Addition of 16.7 mmol/l glucose, but not 16.7 mmol/l mannitol, resulted in a significant increase (p < 0.001) in ATP release that was decreased by the addition of the Ca2+ channel blocker nifedipine (20 μmol/l), the pannexin-1 blocker carbenoxolone (20 μmol/l) or the VRAC inhibitor NPPB (100 μmol/l). Diazoxide did not affect the glucose-induced ATP release, and tolbutamide itself did not cause ATP release. f The addition of 100 μmol/l palmitate resulted in a significant increase (p < 0.001) in ATP release that was attenuated by addition of the Ca2+ channel blocker nifedipine (20 μmol/l), the pannexin-1 blocker carbenoxolone (20 μmol/l) or VRAC inhibitor NPPB. 2-Bromopalmitate had no influence on palmitate-mediated ATP release. ATP release levels were normalized to baseline and expressed as means ± SEM was shown (n = 3). Asterisks denote probability level of random difference, by Student's t test. *p < 0.05, **p < 0.01, ***p < 0.001
Both glucose- and palmitate-stimulated ATP release are dependent on MDR1 expression
While the molecular identity of VRACs is still unknown, activation was reported being regulated by the multidrug resistance protein, MDR1 protein [12]. To investigate this connection, we generated MIN6c4 cells where the expression of the MDR1 gene was suppressed. Silencing of MDR1 was confirmed using real-time RT-PCR (Fig. 2a). Knockdown of the MDR1 gene substantially reduced both glucose- and palmitate-stimulated ATP release compared to control cells (Fig. 2b, c).

ShRNA-mediated knockdown of MDR1. a mRNA quantification of MDR1 gene expression in MIN6c4 and MIN6c4 MDR1 silenced cells. Changes in MDR1 mRNA levels normalized with respect to Beta-actin mRNA levels. The mRNA expression levels were expressed as fold expression relative to control (MIN6c4 cell) and are reported as the means ± SEM (n = 3); *p < 0.05 versus control. Silencing of MDR1 gene substantially reduced 16.7 mmol/l glucose- (b) and 100 μmol/l palmitate- (c) induced ATP release in MIN6c4 cells. ATP release analysis of high glucose or palmitate was performed as described under “Materials and methods” section. ATP release levels were normalized to baseline and expressed as means ± SEM (n = 3) was shown. *p < 0.05, **p < 0.01, ***p < 0.001
ATP release triggered by high glucose or palmitate inhibits cAMP production
We next investigated the effect of autocrine release on the cAMP levels. This was done in real time by transient expression of a cAMP-sensitive luciferase (Glosensor, Promega) in MIN6c4 cells. The results show that addition of glucose (16.7 mmol/l) or palmitate (100 μmol/l) resulted in an inhibited cAMP production, which was reversed by stimulation of the endogenous GLP-1 (1 μmol/l) receptors (Fig. 3a, b). To further confirm that P2Y13 is involved in the autocrine inhibition of adenylate cyclase, cAMP content was also measured in MIN6c4 cells, which were treated with either high glucose or palmitate in the presence and in the absence of the P2Y13-specific antagonist MRS2211. We found that in a high glucose (25 mmol/l) medium containing the P2Y13 antagonist, MRS2211 (10 μmol/l), the cAMP accumulation increased by 33 % (p < 0.01, Fig. 4a). The effect of palmitate (100 μmol/l) was even more profound reducing cAMP levels by about 70 % (p < 0.001, Fig. 4b), which was partially reversed by MRS2211, increasing the cAMP levels (p < 0.05, Fig. 4b).

Autocrine effects of high glucose and palmitate on real-time luciferase cAMP production. MIN6c4 growing in 35-mm Petri dishes were transfected with Glosensor vector. After equilibrating the cells with reagent, real-time cAMP-stimulated luciferase luminescence was followed for 15 min: (a) high glucose (16.7 mmol/l) decreased cAMP levels, which was reversed by GLP-1 (1 μmol/l); (b) palmitate (100 μmol/l) inhibited cAMP production, which was reversed by GLP-1 (1 μmol/l). The shown data represent two independent experiments

Autocrine effects of high glucose or palmitate on cAMP production in MIN6c4 cells. MIN6c4 cells were treated with (a) high glucose (25 mmol/l), in the presence or absence of MRS2211 (10 μmol/l), and (b) palmitate (100 μmol/l), in the presence or absence of MRS2211 (10 μmol/l). Cells growing in 6-well plates were treated with the compounds for 30 min, in the presence of 25 μmol/l Rolipram, and were then extracted with 200 μl of 0.1-mol/l hydrochloric acid and analysed for cAMP. Data are given as the means ± SEM (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001
High glucose and palmitate stimulation of MIN6c4 cells causes autocrine activation of Caspase-3 through P2Y13
We have previously reported that activation of the purinergic receptor P2Y13, on MIN6c4 cells, results in activation of proapoptotic pathways [6]. We next investigated if high glucose or palmitate could influence Caspase-3 level in MIN6c4 insulinoma cells cultured for 36 h and to what extent this effect could be inhibited by MRS2211. The Caspase-3 activity induced by high glucose (25 mmol/l) was suppressed by 10 μmol/l of MRS2211 by 15 % (p < 0.01, Fig. 5a), while diazoxide was without effect (Fig. 5a). Similarly, 100 μmol/l of palmitate induced a significant increase in Caspase-3 activity in MIN6c4 cells, and this effect was markedly inhibited by 10 μmol/l of MRS2211 by 26 % (p < 0.001, Fig. 5b). Both the glucose- and the palmitate-induced Caspase-3 activity were significantly inhibited by treating the cells with the membrane-permeable cAMP analogue 8-bromo cAMP (p < 0.0012, Fig. 5a and p < 0.0395, Fig. 5b).

Effects of the P2Y13 antagonist MRS2211 on high glucose- and palmitate-induced Caspase-3 activity in MIN6c4 cells. MIN6c4 cells growing in 6-well dishes were treated for 36 h with (a) high glucose (25 mmol/l) in the presence or absence of MRS2211 (10 μmol/l), 8-Bromo cAMP decreased high glucose-induced Caspase-3 activity while diazoxide (10 μmol/l) was without effect; and (b) palmitate (100 μmol/l) in the presence or absence of MRS2211 (10 μmol/l), 8-Bromo-cAMP could also rescue palmitate-induced Caspase-3 activity. After completing the incubation, the cells were washed and lysed on ice and then centrifuged at 500×g for 3 min. Supernatants were transferred to 96-well plates and analysed for Caspase-3 activity using a commercial kit. Result of Caspase-3 was normalized to protein concentration and expressed as the percentage change in specific activity. Data are given as the means ± SEM (n = 4). **p < 0.01, ***p < 0.001
High glucose and palmitate stimulation of mouse islets causes an autocrine activation of Caspase-3 that is partly mediated by P2Y13
To confirm that the P2Y13-mediated Caspase-3 activation, which is observed in the MIN6c4 cells, also takes place in natural cells, we cultured isolated mouse islets with either 16.7 mmol/l glucose or with 150 μmol/l of palmitate in the presence or absence of 10 μmol/l of MRS2211. Staining the islets for activated Caspase-3 clearly confirmed our previous results, as both glucose- and palmitate-mediated activation of Caspase-3 (p < 0.01 and p < 0.05, respectively, Fig. 6a–c) was suppressed in the presence of MRS2211 (p > 0.05 and p > 0.05, Fig. 6e, f)

Effect of the P2Y13 antagonist MRS2211 on activated Caspase-3 frequency in isolated mouse islets. Images shown are representative for their treatments; inserts show Hoechst staining for the represented islet. a Control treatment, islet treated with low glucose (5.6 mmol/l). b Glucose treatment, islet treated with high glucose (16.7 mmol/l). c Palmitate treatment, islet treated with palmitate (150 μmol/l). d Control + MRS2211 treatment, islets treated as a with MRS2211 (10 μmol/l). e Glucose + MRS2211 treatment, islets treated as b with MRS2211 (10 μmol/l). f Palmitate + MRS2211 treatment, islets treated as c with MRS2211 (10 μmol/l). g Frequency analysis of Caspase-3 activated islets cells to total number of islet cells. Bars are normalized to control treatment. Statistical analysis, one-way ANOVA (p = 0.004) followed by Dunnett's post hoc test with control (n = 3–8). *p < 0.05, **p < 0.01. Bar equals 20 μm
CREB, Bad and IRS-1 are activated upon blocking the P2Y13 receptor
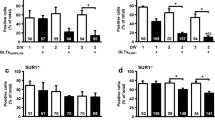
To determine if pathways, important for cellular survival, are activated by autocrine/paracrine activation of the P2Y13 receptor, we carried out western blot analysis using antibodies against Ser-133 phospho-CREB, Ser-612 phospho-IRS-1 and Ser-112 phospho-Bad. MIN6c4 cells incubated in high glucose (25 mmol/l) medium in the presence of 10 μmol/l of the P2Y13 receptor antagonist, MRS2211, displayed an enhanced CREB activation (Fig. 7a, b). Blocking P2Y13 receptor by MRS2211, in the presence of palmitate (100 μmol/l), also elevated the CREB activation (Fig. 7c, d). These results were paralleled by the results of the analysis of Bad, since P2Y13 inhibition also produced a strong phosphorylation of Bad (Fig. 7e, f). The effect on the IRS-1 phosphorylation state was significant although less pronounced (Fig. 7g, h).


Effects of high glucose and palmitate on the phosphorylation status of CREB, IRS-1 and Bad in MIN6c4 cells. MIN6c4 cells were incubated for 30 min in medium containing control, high glucose (25 mmol/l) or palmitate (100 μmol/l) with or without MRS2211 (10 μmol/l). Cell lysates were prepared and then subjected to western blot analysis (20 μg protein/lane), using antibodies against phosphorylated ser-133-CREB, ser-112-Bad and ser-612-IRS-1, respectively. a, b ser-133-CREB. c, d ser-112-Bad. e, f ser-612-IRS-1 . Membranes were re-probed with the anti-CREB, anti-Bad and anti-IRS-1 antibody. Each bar represents the fold increase of phospho-CREB, phospho-Bad or phospho-IRS-1 relative to control after normalizing against total non-phosphorylated CREB, Bad or IRS-1. Data for western blots (b, d, f) are given as the means ± SEM in each group (n = 3). *p < 0.05, **p < 0.01
Autocrine mechanisms involving the P2Y13 receptor influence the viability and proliferation of MIN6c4 cells
To investigate the functional consequences of autocrine P2Y13 activation, created by palmitate or by high glucose, the changes in the cell viability and proliferation were determined by means of MTT assay and by cell growth. MIN6c4 cells were seeded at a low cell density and cultured in cell culture medium, in the presence or absence of different stimulants. As measured by the MTT assay 3 days after treatment, the growth of MIN6c4 cells was inhibited by treatment with 100 μmol/l palmitate (Fig. 8b). However, when the cells were incubated with 100 μmol/l palmitate in the presence of MRS2211, at a concentration of 10 μmol/l, the effect was inverted, and cells proliferated more efficiently (Fig. 8b). A similar effect was obtained when MIN6c4 cells were incubated in 25 mmol/l glucose (Fig. 8a). These results were confirmed by the cell growth that showed an increased proliferation in the presence of MRS2211 (Fig. 9a, b). The results of the MIN6c4 cell MTT assay and the cell proliferation study show that blocking the P2Y13 receptor promotes both the viability and proliferation.

Effects of high glucose and palmitate on the viability of MIN6c4 cells. Cell viability was determined by MTT assay. MIN6c4 cells (1 × 104 cells/well) growing in 96-well plate were incubated with high glucose (25 mmol/l) (a) or palmitate (100 μmol/l) (b) with or without MRS2211 (10 μmol/l). After 3 days in culture, the cellular viability was determined using a colorimetric MTT assay. Data are given as the means ± SEM (n = 4). **p < 0.01, ***p < 0.001

Effects of high glucose and palmitate on the proliferation of MIN6c4 cells. Cell proliferation was estimated by cell growth assay. MIN6c4 cells (1 × 104 cells/well) seeded in 24-well plates were allowed to grow in the presence of high glucose (25 mmol/l) (a) or palmitate (100 μmol/l) (b) with or without MRS2211 (10 μmol/l). The cell proliferation curves were obtained by counting the cells every 2 days. Data are given as the means ± SEM (n = 4). Asterisks, **p < 0.01, ***p < 0.001, indicate cells receiving treatment versus cells receiving treatment + MRS2211
Discussion
While chronic exposure to high levels of glucose or free fatty acids is well known to have detrimental effects on beta cell function and survival [1, 2, 13], the mechanisms leading to initiation of the cell death program are complex, and the details remain to be clarified. In a previous study, we showed that stimulation of purinergic P2Y13 receptors on beta cells activates apoptotic pathways [6]. The aim of the present study was to determine if high glucose and free fatty acids contribute to beta cell apoptosis via autocrine/paracrine activation of the P2Y13 receptor.
Studies conducted in multiple cell models and tissues have demonstrated that ATP can be released via several mechanisms, including exocytosis from neurotransmitter-containing vesicles, diffusion through nucleotide transporters and large transmembrane pores (e.g. connexin hemi-channels and anion channels) [3, 4]. Purinergic signalling in the endocrine pancreas is well described and known to involve insulin release, coordination of beta cell calcium signals and synchronization of calcium-dependent events [14].
We started by examining if glucose and palmitate can induce ATP release in MIN6c4 cells and to study which release mechanisms that might be involved. The results showed that high glucose (16.7 mmol/l) and palmitate (100 μmol/l) within seconds of the exposure potently elevated the extracellular ATP levels and that the calcium channel blocker nifedipine, VRAC inhibitor NPPB and the pannexin-1 blocker carbenoxolone all inhibited this process. Even though the general view is that islet ATP is co-released with insulin, we were not able to detect any increase in insulin secretion during the peak of the ATP release. Furthermore, as tolbutamide was unable to trigger ATP release and diazoxide had no effect on glucose-stimulated release of ATP, we conclude that this early part of ATP release is not dependent on modulation of KATP channels, which is an essential step in insulin secretion. The inability of 2-bromopalmitate to block palmitate-stimulated ATP release rules out the involvement of the islet FFA1 receptor [11]. The effect of carbenoxolone was surprising, as Scemes et al. [15] previously reported that MIN6 cells, although expressing Cx36 and pannexin1, are lacking hemi-channel activity. However, the unexpected effect of carbenoxolone can be explained by that this compound has been described as a potent inhibitor of volume-regulated anionic channels also known as VRACs [16]. There is an accumulating body of evidence that the VRAC is involved in the nutrient-sensing mechanism of the pancreatic beta cell, as both glucose [17, 18] and fatty acids [18] have been shown to cause VRAC activation. While the molecular identity of the VRAC is still unknown, it has been suggested as a critical component in an alternate route for depolarization of the pancreatic beta cell [19], a route that is independent of metabolic coupling and subsequent activation of KATP channels. Several investigators have also suggested VRACs to be instrumental in the mechanism responsible for triggering cellular ATP release [3, 20]. Although the exact mechanism remains to be described, Valverde et al. [12] has reported that the ABCB1 transporter, also known as the multidrug resistance protein MDR1 or P-glycoprotein, is a potent VRAC regulator. MDR1 is expressed in beta cells [21], and although mainly implicated in extruding xenochemicals, it has been reported to be important in the insulin release mechanism [22–24]. In order to confirm the MDR1-VRAC connection, we silenced the MDR1 expression, which resulted in a drastically decreased release of ATP upon stimulation with high glucose or with palmitate. The conclusion of the first part of this study is that glucose as well as palmitate can stimulate ATP release from MIN6c4 cells through a mechanism involving VRACs and the MDR1 transporter. Having shown that common nutrients like glucose and fatty acids are capable of stimulating beta cells to release ATP, we next tried to answer if the released ATP also is sufficient to exercise an autocrine or paracrine effect.
Cellular release of ATP is extracellular degraded to adenosine and ADP that activate membrane-bound adenosine and P2Y receptors thus modulating the cellular function through an autocrine–paracrine mechanism [3]. To investigate if the P2Y13 receptor can be similarly stimulated, we next examined the effects of glucose and palmitate on the production of cAMP. We found that the cAMP production was inhibited in the presence of either high glucose or palmitate and that the P2Y13 receptor antagonist MRS2211 could inhibit this effect. Although this observation may seem to be in contrast with the current view that glucose induces cAMP oscillations, it is important to understand that while this view is based on the use of compartment-specific biosensor techniques [25, 26], reflecting cAMP fluctuations in specific cellular compartments, our results are generated with methods that show more general changes.
As we previously shown that ADP induced Caspase-3 activation, which could be inhibited by MRS2211 in MIN6c4 cells, we decided to find out if the ATP release, generated by either high glucose or palmitate, is sufficient to stimulate a P2Y13-mediated Caspase-3 activation. We found that this is indeed the case, as MRS2211 partially inhibited both palmitate- and glucose-stimulated Caspase-3 activation. Furthermore, 8-Bromo cAMP, a membrane permeable cAMP analogue, also suppressed caspase-3 activation thus supporting the results obtained with MRS2211. The indifference of glucose/palmitate-mediated Caspase-3 activation to diazoxide is well in line with the results of the release experiments.
The fact that we were able to repeat these results, using isolated mouse islets instead of cells, further strengthens the idea that purinergic mechanisms involving P2Y13 modulate beta cell apoptosis. Furthermore, the inhibition of Caspase-3 activation was reflected by the phosphorylation status of the apoptotic marker proteins CREB and Bad. Phosphorylation of CREB, by PKA, is a major route of survival in pancreatic beta cells [27]. Bad is a proapoptotic member of the Bcl-2 that is inactivated upon phosphorylation at Ser112 [28]. We found that blocking P2Y13 receptors with MRS2211, in the presence of glucose or palmitate, resulted in a significant increase in both the Ser-133-CREB and the Ser-112-Bad phosphorylation.
In conclusion, the present study suggests that nutrients, like glucose and free fatty acids, induce a release of ATP into the extracellular space, through a mechanism dependent on VRAC and MDR1. Ectonucleotidases present on the plasma membrane subsequently convert ATP to ADP that, through stimulation of P2Y13, activates proapoptotic pathways. Thus, activation of P2Y13 receptors by nutrient-stimulated ATP release is likely to constitute a contributing component of the glucolipotoxicity effect.
References
Talchai C, Lin HV, Kitamura T, Accili D (2009) Genetic and biochemical pathways of beta-cell failure in type 2 diabetes. Diabetes Obes Metab 11(Suppl 4):38–45
Newsholme P, Gaudel C, McClenaghan NH (2010) Nutrient regulation of insulin secretion and beta-cell functional integrity. Adv Exp Med Biol 654:91–114
Corriden R, Insel PA (2010) Basal release of ATP: an autocrine-paracrine mechanism for cell regulation. Sci Signal 3:re1
Fitz JG (2007) Regulation of cellular ATP release. Trans Am Clin Climatol Assoc 118:199–208
Amisten S, Meidute-Abaraviciene S, Tan C, Olde B, Lundquist I, Salehi A, Erlinge D (2010) ADP mediates inhibition of insulin secretion by activation of P2Y13 receptors in mice. Diabetologia 53:1927–1934
Tan C, Salehi A, Svensson S, Olde B, Erlinge D (2010) ADP receptor P2Y(13) induce apoptosis in pancreatic beta-cells. Cell Mol Life Sci 67:445–453
Winzell MS, Ahren B (2007) Role of VIP and PACAP in islet function. Peptides 28:1805–1813
Taylor AL, Kudlow BA, Marrs KL, Gruenert DC, Guggino WB, Schwiebert EM (1998) Bioluminescence detection of ATP release mechanisms in epithelia. Am J Physiol 275:C1391–C1406
Menez C, Mselli-Lakhal L, Foucaud-Vignault M, Balaguer P, Alvinerie M, Lespine A (2012) Ivermectin induces P-glycoprotein expression and function through mRNA stabilization in murine hepatocyte cell line. Biochem Pharmacol 83:269–278
Warnotte C, Gilon P, Nenquin M, Henquin JC (1994) Mechanisms of the stimulation of insulin release by saturated fatty acids. A study of palmitate effects in mouse beta-cells. Diabetes 43:703–711
Salehi A, Flodgren E, Nilsson NE, Jimenez-Feltstrom J, Miyazaki J, Owman C, Olde B (2005) Free fatty acid receptor 1 (FFA(1)R/GPR40) and its involvement in fatty-acid-stimulated insulin secretion. Cell Tissue Res 322:207–215
Valverde MA, Bond TD, Hardy SP, Taylor JC, Higgins CF, Altamirano J, Alvarez-Leefmans FJ (1996) The multidrug resistance P-glycoprotein modulates cell regulatory volume decrease. EMBO J 15:4460–4468
El-Assaad W, Buteau J, Peyot ML, Nolan C, Roduit R, Hardy S, Joly E, Dbaibo G, Rosenberg L, Prentki M (2003) Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 144:4154–4163
Petit P, Lajoix AD, Gross R (2009) P2 purinergic signalling in the pancreatic beta-cell: control of insulin secretion and pharmacology. Eur J Pharm Sci 37:67–75
Scemes E, Bavamian S, Charollais A, Spray DC, Meda P (2008) Lack of “hemichannel” activity in insulin-producing cells. Cell Commun Adhes 15:143–154
Benfenati V, Caprini M, Nicchia GP, Rossi A, Dovizio M, Cervetto C, Nobile M, Ferroni S (2009) Carbenoxolone inhibits volume-regulated anion conductance in cultured rat cortical astroglia. Channels (Austin) 3:323–336
Jakab M, Grundbichler M, Benicky J, Ravasio A, Chwatal S, Schmidt S, Strbak V, Furst J, Paulmichl M, Ritter M (2006) Glucose induces anion conductance and cytosol-to-membrane transposition of ICln in INS-1E rat insulinoma cells. Cell Physiol Biochem 18:21–34
Best L (1997) Glucose and alpha-ketoisocaproate induce transient inward currents in rat pancreatic beta cells. Diabetologia 40:1–6
Best L, Brown PD, Sener A, Malaisse WJ (2010) Electrical activity in pancreatic islet cells: the VRAC hypothesis. Islets 2:59–64
Roman RM, Lomri N, Braunstein G, Feranchak AP, Simeoni LA, Davison AK, Mechetner E, Schwiebert EM, Fitz JG (2001) Evidence for multidrug resistance-1 P-glycoprotein-dependent regulation of cellular ATP permeability. J Membr Biol 183:165–173
Bani D, Brandi ML, Axiotis CA, Bani-Sacchi T (1992) Detection of P-glycoprotein on endothelial and endocrine cells of the human pancreatic islets by C 494 monoclonal antibody. Histochemistry 98:207–209
Li CJ, Zhou HL, Li J, Yao HT, Su R, Li WP (2011) Roles of sulfonylurea receptor 1 and multidrug resistance protein 1 in modulating insulin secretion in human insulinoma. Hepatobiliary Pancreat Dis Int 10:88–94
Barg S, Renstrom E, Berggren PO, Bertorello A, Bokvist K, Braun M, Eliasson L, Holmes WE, Kohler M, Rorsman P, Thevenod F (1999) The stimulatory action of tolbutamide on Ca2+-dependent exocytosis in pancreatic beta cells is mediated by a 65-kDa mdr-like P-glycoprotein. Proc Natl Acad Sci U S A 96:5539–5544
Tang YZ, Li DQ, Sun FJ, Li L, Yu DM (2009) P-glycoprotein regulating biphasic insulin secretion in rat pancreatic beta cells. Chin Med J (Engl) 122:2587–2592
Tian G, Sandler S, Gylfe E, Tengholm A (2011) Glucose- and hormone-induced cAMP oscillations in alpha- and beta-cells within intact pancreatic islets. Diabetes 60:1535–1543
Landa LR Jr, Harbeck M, Kaihara K, Chepurny O, Kitiphongspattana K, Graf O, Nikolaev VO, Lohse MJ, Holz GG, Roe MW (2005) Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 beta-cell line. J Biol Chem 280:31294–31302
Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M, Montminy M (2003) cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev 17:1575–1580
Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ (1995) Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80:285–291
Acknowledgments
This study was supported by the Swedish Heart and Lung Foundation, Swedish Scientific Research Council, Diabetes Wellness Network, Sweden, The Vascular Wall Program and Lund University.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tan, C., Voss, U., Svensson, S. et al. High glucose and free fatty acids induce beta cell apoptosis via autocrine effects of ADP acting on the P2Y13 receptor. Purinergic Signalling 9, 67–79 (2013). https://doi.org/10.1007/s11302-012-9331-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-012-9331-6