
Abstract
Cancer stem cells (CSCs) are sub-populated cells in the tumor and responsible for tumor growth, heterogeneity, relapse, and progression of cancer. They play a dynamic role in developing resistance of chemotherapeutics and promoting epithelial mesenchymal transition (EMT) and metastasis in tumors, which are accountable for approximately 90% of mortality. Thus, agents targeting CSCs or chemosensitizing CSCs have now gained significant importance in the regulation and inhibition of various malignancies. Nowadays, numerous dietary polyphenolic compounds such as flavonoids are being explored as potential candidates to be utilized in chemoprevention and treatment of various cancers by targeting CSCs. In multiple studies, flavonoids have shown an inhibitory effect on the self-renewal potential, stemness characteristics, EMT process, and survival of CSCs in different tumors. Literature shows that few flavonoids like genistein, quercetin, silibinin, and apigenin have been explored substantially for their role in inhibition of CSCs. However, there is paucity of data for some of the flavonoids such as broussoflavonol B, icaritin, morusin, casticin, wogonin, baicalein, luteolin, ugonin J and K, naringine, and pomiferin though they have also shown inhibition of CSCs. This chapter illustrates a descriptive information about CSCs, their characteristics, biomarkers, and pathways involved in their maintenance (Notch, Hedgehog, Wnt/β-catenin, PI3K/Akt, and NF-κB). In addition, the literature around several flavonoids and their effect in reduction or eradication of CSCs via attenuation of different signaling pathways have been reviewed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cancer stem cells (CSCs)
- Epithelial Mesenchymal Transition (EMT)
- Flavonoids
- Hedgehog
- Notch
- Polyphenolic compounds
- Wnt/β-catenin
7.1 Introduction
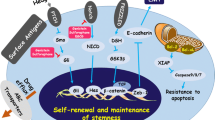
Cancer Stem Cells (CSCs), also known as cancer-initiating cells (CICs) or tumor-initiating cells (TICs), are sub-populated cells (0.1–10%) of the tumor and are mainly responsible for tumor heterogeneity, tumor growth, recurrence, self-renewal, and progression of various types of cancer depicted in Fig. 7.1 [1, 2]. Unlike normal stem cells, CSCs have indefinite potential of self-renewal that leads to tumorigenesis. The alteration in the metabolic and phenotypic characteristics of CSCs, mainly because of various genetic and epigenetic modifications, leads to the emergence of tumor heterogeneity which increases tumor survival and invasion into other tissues and further complicates the cancer treatment [1, 3]. CSCs were first identified in acute myeloid leukemia (AML) by Bonnet and Dick in 1994 [4]. In solid tumors, it was first derived from breast cancer cells in 2003 when a group of researchers injected the CD44+, CD24−/low populated cells in immune-deficient mice [5]. Thereafter, CSCs have also been found in brain, lung, prostate, colon, multiple myeloma, pancreatic, liver, head and neck, ovarian, cervical, gastric, and other cancers [1, 6,7,8,9].

Tumor initiation through cancer stem cells (CSCs)
The CSCs are accountable for the resistance development against chemoradiotherapies, epithelial mesenchymal transitions (EMT), and metastasis which are the main cause of approximately 90% of mortality [4, 10]. The resistance against treatments and disease progression may occur partly due to the lower proliferative rate of CSCs compared to non-CSCs [11]. They are nowadays targeted for cancer treatment due to their capability to initiate and propagate tumor growth and develop resistance [12].
In several in vitro and in vivo studies, dietary phytocompounds have been shown to inhibit tumor formation and progression in various malignancies [8, 13, 14]. The studies showing the potential role of phytocompounds against CSCs are limited. However, in recent years, studies have been conducted and demonstrated the anti-CSCs effect of some phytocompounds [8, 13,14,15,16,17]. Polyphenolic compounds, especially flavonoids have shown their role in inhibiting tumorigenesis due to their anti-CSCs effect indicating that they can be an attractive chemopreventive and chemotherapeutic candidate for cancer treatment [18,19,20]. In this chapter, the effects of different flavonoids and their derivatives on CSCs derived from various types of cancers have been illustrated.
7.2 Characteristics of Cancer Stem Cells (CSCs)
7.2.1 Salient Features
CSCs possess three unique characteristics that enable them for tumor initiation, propagation, and spread [2, 14, 18, 21,22,23,24]:
-
1.
The self-renewal capacity helps CSCs to preserve their pool.
-
2.
CSCs are multipotent and give rise to the heterogeneous population of cells through asymmetric division.
-
3.
The uncontrolled proliferative potential of CSCs supports the sustained development of tumors.
7.2.2 Promotion of Epithelial Mesenchymal Transition (EMT)
Epithelial mesenchymal transition is found as one of the significant characteristics of CSCs. During EMT, the features of epithelial cells converts to mesenchymal cell-like phenotypes (spindle-shaped appearance) which also enhances the invasive potential and motility of the cells [4, 25, 26]. During EMT progression, E-cadherin which is epithelial cell marker is downregulated whereas mesenchymal markers like N-cadherin and vimentin are upregulated [26]. EMT is a reversible change as it can induce intravasation to invade healthy group of cells followed by extravasation to form new tumors (Fig. 7.2). In intravasation process, the epithelial cancer cells change their phenotype to mesenchymal cells while entering into the bloodstream. Mesenchymal-epithelial transition (MET) is the reverse form of EMT occurring along with these changes after extravasation and can boost new tumors formation [26, 27].

Epithelial mesenchymal transition (EMT)
The molecular mechanism of EMT includes inducers, regulators, and effectors. When the tumors start to grow due to signal the transition order by the inducers like TGF- β, VEGF, IGF, Wnt, and Notch causes nutrient deficit and hypoxia in the cells of the tumor at the center [28]. The regulators are transcription factors or drivers that change the cell shape and make them more favorable to invade other healthy tissues by regulating the cytoskeleton [29]. Master regulators of the EMT are Twist and Snail1 which regulate repression of E-cadherin and enhances the tumor-initiating capacity of cells, respectively. Other regulators of EMT are SMAD, BMP, Slug, ZEB1, and ZEB2 which are found helpful in suppressing transcription by binding directly to E-cadherin at the promoter region [26, 30]. Vimentin and keratin act as a regulator to maintain overall cell shape towards mesenchymal cells which are more motile [3]. Enzymes such as collagenases and matrix metalloproteinases (MMPs) promote the escape of tumor cells from the primary tumor site and help them to enter the bloodstream [31]. Moreover, the transition of EMT to MET is an essential step in tumor formation to escape the CSCs from programmed cell death, an apoptosis that kills epithelial cells in the blood circulation [32].
7.2.3 Chemoradiotherapy Resistance
One of the significant characteristics of CSCs is the development of resistance against chemoradiotherapy. Resistance attained by CSCs is generally regulated by the high capacity for DNA repair mechanism and increased protection against reactive oxygen species (ROS). CSCs stay dormant during chemoradiotherapy and can regenerate cancer after therapy. Dormancy of CSCs is associated with multiple genes (e.g., TGF-β2) [29, 33] which are responsible for therapy resistance and contribute to cancer relapse [33, 34]. It has also been shown that conventional therapies target the high proliferative stem cells instead of dormant CSCs of tumor that can cause a recurrence of cancer [26]. Additionally, chemoradiotherapy resistance also develops due to the upregulation of anti-apoptotic pathways.
Recent studies suggested that abnormal chromatin package density is correlated with the survival rate of cancer cells which are resistant to chemotherapies [35]. Moreover, studies have indicated that the growing number of EMT cell states are regulated by epigenetic mechanisms [36].
Tumor heterogeneity is defined as a state when cancer cells possess distinct genotypes and phenotypes within a single tumor (intra-tumor heterogeneity) and are also responsible for complication or resistance to chemotherapy. An epigenetic mechanism is involved to cause the phenotypic differences between the CSCs and non-CSCs. In studies, CSCs have shown to cause tumor heterogeneity and promote new tumor development as compared to normal cancer cells [33, 37, 38]. In the present scenario, intra-tumor heterogeneity is a big challenge in the development of cancer therapy.
The overexpression of the telomerase enzyme, regulated by mutated hTERT gene, may cause unstable chromosome length in CSCs. Telomere repeats are akin to the self-renewal capacity of CSCs which is controlled by telomerase activity. Moreover, it has been shown that mitochondrial telomerase activity in CSCs protects nuclear DNA by reducing the level of ROS and causing therapy resistance [39, 40].
7.3 Biomarkers of CSCs
Biomarkers, proteins or glycoproteins, define the properties of CSCs and are upregulated, downregulated, or mutated in malignancies. The enhanced activity of these biomarkers is well correlated with CSCs biology and help in developing new strategies to treat various malignancies and resistance cases [29, 33, 41]. Few of the biomarkers of CSCs are cell surface antigens while others are located in the cytoplasm such as Aldehyde dehydrogenase 1 (ALDH1). ALDH1 oxidizes aldehyde into carboxylic acid to protect CSCs and increases their chemoresistance by detoxification of chemotherapeutics. It has three main isotypes such as ALDH1A1, ALDH1A2, and ALDH1A3 which are involved in self-renewal, differentiation, and self-protection of CSCs. CD44 is a hyaluronic acid receptor and is responsible for invasiveness, metastatic potential, and development of drug resistance in CD44 expressing CSCs. It has several isoforms with different significant roles in CSCs. CD44, CD24, and EpCAM have stemness properties and causing chemoresistance. CD117 is the stem cell growth factor receptor, encoded by the c-KIT gene and CD133 is a common marker of CSCs associated with a poor prognosis and chemoresistance [42]. Table 7.1 refers to the list of some common surface biomarkers of CSCs in various types of cancer [1, 32, 41, 43,44,45,46,47,48,49,50,51].
Specific biomarkers can be used to differentiate CSCs from normal cells and other tumor cells [52]. It is further difficult to identify CSCs by a single biomarker and understanding the role of specific biomarkers is pivotal to treat current issues of cancer. Moreover, to date, no universal CSCs biomarker has been discovered. The current methodologies to isolate and detect functional differences of CSCs from non-CSCs population are image-based, sphere formation, cytological sorting using flow cytometry, CRISPR-Cas9 3D sphere culture systems, magnetic-activated cell sorting (MACS), and xenotransplantation. Among all, xenotransplantation is the best method to confirm the existence of CSCs [53]. Thus, identifying and isolating specific CSCs biomarkers in conjunction with new technologies is imperative for the treatment of various malignancies [4]. This further promotes findings of the novel therapeutics in eradicating highly tumorigenic and therapy-resistant CSCs.
7.4 Possible Pathways Involved in Regulation of CSCs
The Wnt, Hedgehog, and Notch are the major evolutionarily conserved signaling pathways responsible for stemness and differentiation of CSCs. Other signaling pathways such as PI3K/AKT and NF-κB also play important role in the regulation of CSCs characteristics. The aberrant activation of these signaling pathways stimulates CSCs proliferation, restricts differentiation, and prevents apoptosis [54]. Therapeutic approaches targeting these aberrant signaling pathways are required to treat the various types of cancer [55]. Moreover, transcription factors such as SOX2, NANOG, OCT-4, KLF-4, and c-MYC are important for the self-renewal capacity of CSCs. These transcription factors also stands out as potential targets for cancer therapy [33, 56].
7.4.1 The Notch Pathway
The Notch signaling pathway is complex and multifaceted, reflecting its roles in diverse functional activities. The loss of Notch activity favors the EMT process [1]. For the maintenance of stemness of CSCs, upregulation of Notch pathway is responsible along with overexpression of Notch signaling genes (Notch1, Notch3, Jag1, and Jag2) and Notch target gene (Hes1) [57]. Notch signaling via transmembrane ligands and receptors is primarily involved in the communication between adjoining cells. Interaction between ligand on one cell and a transmembrane receptor on a neighboring cell triggers a two-step proteolytic cleavage of the receptor [58]. The first cleavage is mediated by a disintegrin and metalloproteinase enzymes (ADAM 10 or 17) also known as tumor necrosis factor-α converting enzyme (TACE) and the second cleavage is mediated by γ-secretase. This cleavage releases an intracellular fragment which interacts with nuclear factors to regulate target gene expression. The Notch pathway comprises of five canonical Notch ligands (Delta-like ligand 1 [DLL1], DLL3, DLL4, Jagged1, and Jagged2) and four Notch receptor paralogues (Notch1–4) [59]. Different tumors and tumor subtypes can express different Notch receptors and ligands. Furthermore, posttranslational modifications of Notch receptors can change their affinity for ligands and their intracellular half-lives. The non-canonical Notch signaling pathway also has relevance in cancer. Thus, targeting Notch signaling has the potential to simultaneously affect multiple cell types within a tumor, from CSCs to immune cells, vascular endothelial cells, and tumor cells. Additionally, the mechanistic understanding of the role of Notch signaling in specific cancers is required for the successful development of agents targeting the Notch pathway.
The Notch pathway is associated with CSCs in various cancers such as breast cancer, medulloblastoma, and other gliomas. CSCs can be eliminated by Gamma-secretase inhibitors (GSIs) which decrease the subpopulation and tumor sphere formation frequency of CSCs. However, GSIs are relatively nonselective drugs and sometimes also produce toxicity like secretory diarrhea. Highly specialized monoclonal antibodies (mAbs) that specifically antagonize Notch ligands and receptors provide single-target specificity. Knockdown of Hes1 of the CSCs decreases tumor sphere formation, suggesting that Notch signaling activity is required for stemness and promoting cell survival of CSCs.
Hence, it can be speculated that inhibition of Notch signaling pathway in CSCs can play a great role in the treatment of various types of tumors via reducing the population of CSCs. With anti-Delta-like 4 ligand antibodies, either alone or in combination with the chemotherapeutic agents, we can reduce the frequency of CSCs (EpCAM+/CD44+/CD166+). Si-RNA targeted to Notch4 is also found active in suppressing breast cancer recurrence [60].
Flavonoids also target the Notch signaling pathway for eradication and reduction of CSCs. This activity of flavonoids might be due to the regulation of ϒ-secretase, Notch ligands and receptors, knockdown of Hes1, si-RNA targeted to Notch4, or inhibition of DLL-4 ligand.
7.4.2 The Hedgehog Pathway
The Hedgehog pathway is considered to modulate tumorigenesis through tissue patterning, propagation, differentiation, and EMT [61, 62]. Atypical activation of this pathway is responsible for maintenance and tumorigenesis of CSCs as seen in various cancers like myeloid leukemia, myeloma, glioma, colorectal, and gastric cancer [63, 64].
The major troupes in the Hedgehog pathway are the three secreted ligands including Sonic, Desert, and Indian. Smoothened (transmembrane protein) and 3 Gli transcription factors (Gli1-3) along with ligands regulate the suppression or activation of Hedgehog pathway. Islam and team have demonstrated the indispensable role of Sonic hedgehog pathway in the promotion of the EMT, tumorigenicity, and stemness in both in vitro and in vivo studies [65].
When Patched receptor is unoccupied, it acts as a constitutive inhibitor of Smoothened. At this state, Gli3 and Gli2-R repress the target gene transcription. However, when the ligand binds to Patched receptor, the suppression on Smoothened is released allowing transcription of target genes [66]. Overexpression of Smoothened, Gli1, Sonic hedgehog, and Patched1 gene with decreased expression of the stemness genes (SOX-2, NANOG, and OCT-4) are found to be responsible for survival, stemness, proliferation, self-renewal, and clonogenicity of CSCs both in vivo and in vitro [67].
Cyclopamine and IPI269609, which are antagonist of Smoothened, have been shown to reduce the populations or eradicate CSCs and induce tumor suppression in pancreatic and brain cancer [68,69,70]. The combined chemotherapeutics targeting Hedgehog pathway to eradicate CSCs have attracted general attention [71, 72]. In studies, flavonoids alone or in combination with chemotherapeutic agents have shown to target CSCs or sensitize CSCs possibly via hedgehog signaling pathway by regulating their receptors, ligands, smoothened, or transcriptional factors. This has been further described in section 5.
7.4.3 The Wnt/β-catenin Pathway
It is an enormously evolutionarily conserved signaling pathway which plays a dynamic role in modulating cell propagation and differentiation. In carcinogenesis, the aberrant signaling of this pathway facilitates the clonal expansion or tumor heterogeneity which ultimately causes self-renewal, metastasis, multidrug resistance, and invasiveness of CSCs [54, 73, 74].
This is a highly complex pathway comprising of 19 different Wnt ligands and more than 15 receptors. Conventionally, this pathway comprises of 2 signaling pathways: canonical (mediated through β-catenin, a transcriptional regulator) and non-canonical (independent to β-catenin) [74]. The canonical pathway gets triggered when one cell secreted Wnt ligands binds to Frizzled receptors or LRP 5 (low-density lipoprotein-related protein) and LRP 6 co-receptors of the adjacent cell [75]. Signaling through these two Wnt pathways is necessary for embryonic development and homeostasis of various tissues [74, 76]. In general, the canonical Wnt pathway is involved in regulation of proliferation, survival, and cell fate decisions while the non-canonical pathway is involved in regulation of asymmetrical divisions in cells, cell polarity, and migration. It is observed that stem cells of various postnatal tissues are controlled through the canonical signaling pathway [75].
Along with tumorigenesis, Wnt signaling has been associated with CSCs-mediated metastasis and maintenance of its stemness. A significantly higher level of Wnt signaling proteins such as LEF-1, cyclin D1, β-catenin, and TCF-4 along with Wnt-responsive gene transcription are found in breast CSCs compared with normal cancer cells. Moreover, the knockdown of canonical Wnt pathway in CSCs diminishes the expression of genes involved in stemness (CD44, ALDH1, and Sca-1), CSCs subpopulation, and inhibits tumor sphere formation. This indicates that Wnt signaling is essential for CSCs stemness maintenance [77]. Furthermore, a higher expression of Wnt genes (TCF-4 and Disheveled) is present in metastatic CSCs [78].
Non-canonical pathway may also be responsible for tumor instigation through Wnt5a actions, a non-canonical Wnt ligand. An in vivo study (ErbB2-driven mammary tumorigenesis on mouse model) showed that Wnt5a ligand limited the expansion of basally located CSCs in tumor [79].
The canonical Wnt signaling cascade is involved in self-renewal of stem cells and production or differentiation of ancestor cells [80,81,82] whereas non-canonical Wnt signaling pathway is involved in the conservation of stem cells, guidance of cell movement, or inhibition of the canonical signaling cascade [9, 83,84,85]. Both Wnt signaling cascades play crucial roles in the growth and progression of CSCs [86].
Deviant activation of this pathway in CSCs was severely linked with tumorigenesis in various tissues. Chemotherapeutic agents that can be specific to a Wnt receptor frizzled7, essential co-receptor binder for LRP6, and Wnt signaling antagonist are responsible for depletion of clonal expansion and tumorigenicity of CSCs in various kinds of tumors [87]. Knocking down miR-142, which is a potent effector for activating this signaling is also helpful in diminishing tumor-initiating ability and sphere formation of CSCs [88]. Moreover, suppressors of Wnt/β-catenin pathway significantly lessen the population, stemness, and self-renewal capacity of CSCs [89]. Additionally, inhibiting Wnt/β-catenin makes CSCs more chemosensitive to conventional drugs along with reduction of self-renewal and tumorigenic ability [90]. Thus, targeting Wnt/β-catenin signaling would be a promising approach to conquer CSCs.
7.4.4 Role of PI3K/Akt and NF-kB Pathways in CSCs
The aberrant PI3K/Akt signaling pathway boosts up the cellular proliferation and survival of the CSCs [91, 92]. PI3K is a heterodimer consisting of a regulatory subunit—p85 and a catalytic subunit—p110 and Akt, a protein kinase. Both can regulate the EMT process by modulating a series of relevant transcription factors such as Twist, Snail, and Slug; inducing integrin-linked kinase activities and stimulating MMPs. Moreover, PI3K/Akt might induce the EMT in CSCs in cooperation with TGF-β, NF-κB, RAS, and Wnt/β-catenin [93].
Studies reported that microRNAs (miR-126, miR-10b) are helpful in the maintenance of CSCs state via PI3K signaling through inhibition of PTEN. They promote maintenance of CSCs by increasing tumor sphere formation along with overexpression of stemness genes OCT-4 and Snail1 [94, 95]. These findings show that PTEN signaling plays a suppressive role in the maintenance of CSCs stemness [95].
Aberrant activation and overexpression of the proinflammatory transcription factor (NF-κB) protect CSCs from the programmed cell death (apoptosis) by direct upregulation of anti-apoptotic genes or antagonistic effect on p53 pathway and promote self-renewal characteristics of CSCs [58]. Transcription factors consist of five different proteins that function as dimers which are normally inactivated in the cytoplasm through binding to IκB proteins. Activation of this pathway occurs due to binding of tumor necrosis factor alpha (TNF-α), IL-1β, and bacterial cell wall components to their respective receptors (TNF receptor, IL-1 receptor, and toll-like receptors also known as TLRs), respectively [96]. In case of canonical NF-κB pathway, adapter proteins are recruited, facilitating the phosphorylation and activation of IκB kinase (IKKβ) proteins which subsequently initiate the phosphorylation of IκB proteins, marking them for ubiquitination and degradation [96]. Degradation of IκB releases NF-κB which translocates to the nucleus and activates transcription of target genes [58]. In case of non-canonical NF-κB pathway, activation occurs through different receptors, such as receptor activator of NF-κB (RANK) and CD40, signaling through NF-κB-inducing kinase and IKKα. Then p100/RelB dimers are processed into p52/RelB dimers which translocate to the nucleus and activates transcription. The NF-kB pathway is a highly complex and critical signaling pathway and has role in cellular proliferation, survival, and differentiation of CSCs [96]. Hence, we can conclude that NF-κB signaling constitutes an important pathway controlling the self-renewal and tumorigenesis of CSCs [97, 98]. NF-κB signaling has also been implicated in enabling CSCs to facilitate metastasis by downregulation of IKKβ. Genetic silencing or chemical inhibition of IKKβ reduced the expression of the stemness proteins LIN-28, OCT-4, SOX-2, and NANOG. The NF-κB signaling pathway may support CSCs stemness and promote tumor metastasis in cancers [99].
7.5 Flavonoids Targeting CSCs
In recent years, several dietary compounds derived from natural sources have been found effective in chemoprevention and treatment of various types of cancers. Flavonoids are a class of polyphenolic secondary metabolites consisting of a C6-C3-C6 skeleton (15-carbon structure that consists of two phenyl rings and a heterocyclic ring) that are found abundantly in dietary plants and some medicinal herbs. On the basis of their chemical structures, they are categorized as flavones, flavanones, flavonols, and isoflavones which are commonly present in the human diet [1, 100,101,102]. They possess anticancer activity both in preclinical and cellular model systems (Fig. 7.3). They have also shown an inhibitory effect on the self-renewal potential and survival of CSCs in various tumors [1, 54]. Moreover, several recent studies have suggested that flavonoids can also play important role in targeting the CSCs and may sensitize them towards conventional anticancer therapies [58, 103,104,105]. We have reviewed the available literature of flavonoids targeting CSCs responsible for progression of disease along with their attenuating signaling pathways.

Flavonoids targeting CSCs
Mostly flavonoids regulate or eradicate CSCs of tumors by targeting various pathways which might be associated with maintenance of CSCs such as Wnt/β-catenin, Hedgehog, Notch, PI3K/Akt, and NF-κB signaling pathways. Few of the flavonoids such as genistein, quercetin, silibinin, and apigenin have been explored substantially in the literature for their role in inhibition of CSCs. However, there is paucity of data for some of the flavonoids such as broussoflavonol B, icaritin, morusin, casticin, wogonin, baicalein, luteolin, ugonin J and K, naringine, and pomiferin though they have also shown inhibition of CSCs. These flavonoids have been shown to directly or indirectly modulate these signaling pathways and contribute to the reduction of CSCs growth and maintenance.
7.5.1 Genistein
Genistein or Prunetol (4’,5,7-Trihydroxyisoflavone) is an isoflavone type of flavonoid. Perkin and Newbury were the first to isolate genistein in 1899 from Genista tinctoria (Leguminosae) [106,107,108,109]. Genistein is a highly active anticancer phytocompound used in the treatment of various types of malignancies [110,111,112].
Genistein is known to act via attenuating some signaling pathways like Notch, Hedgehog, and Wnt/β-catenin of CSCs. Moreover, there are other cellular targets of genistein through which it can inhibit stemness of CSCs. In recent years, the inhibitory action of genistein against CSCs was established in colon, breast, prostate, and pancreatic cancer [16, 113,114,115]. Downregulation of expression of cyclin B1, Bcl-2, and Bcl-xL via Notch pathway in breast cancer was also exemplified [114]. In one in vitro study, Chen and colleagues demonstrated that genistein can cause overexpression of ARHI tumor suppressor gene thereby inhibiting cell proliferation and inducing apoptosis in CSCs in prostate cancer [15]. It has also exhibited an antagonistic role against prostate CSCs through inhibition of Hedgehog-Gli1 pathway [16]. Sekar and coworkers conducted in vivo experiment in 1, 2-dimethyl hydrazine (DMH) induced colon cancer model in mice. They observed that genistein had reduced Argyrophilic nuclear organizer region (AgNOR) and proliferating cell nucleolar antigen (PCNA) along with suppression of colonic stem cell markers [113]. Xia and colleagues showed that genistein was capable to upregulate miR-34a along with downregulation of Notch-1 in pancreatic cancer [115].
The inhibitory role of genistein on Wnt/β-catenin signaling pathway in CSCs has also been well-established in various studies. In an ex vivo study, genistein was found to have an inhibitory effect on Wnt/β-catenin pathway by regulating miR-1260b expression in renal cancer cells [116]. In a study, genistein prevented self-renewal of breast CSCs via attenuation of Wnt/β-catenin pathway [117].
Furthermore, genistein has the potential to inhibit ovarian CSCs via suppression of FOXO3a and FOXM1 along with downregulation of expression of stem cell markers (CD133, CD44, and ALDH1) responsible for self-renewal [14]. This finding was further supported by another study in which ovarian tumor suppression was due to decreased expression of CD163 and p-STAT3 [118]. Genistein also inhibited the self-renewal capacity and reduced the resistance of therapy in gastric cancer by suppression of CSCs markers [119]. It has the ability to reverse EMT process in colon cancer by inhibiting cell migration via downregulation of EMT markers (ZEB1, ZEB2, FOXC1, FOXC2, Snail2/slug, and TWIST1) along with suppression of Notch-1, p-NF-κB, and NF-κB signaling in in vitro study [120]. The inhibitory role of genistein in CSCs was tested in renal and nasopharyngeal cancer and was found to suppress of Hedgehog signaling pathway [121, 122].
Recently, genistein has also shown an inhibitory effect in lung cancer by decreasing cell viability, migration, and invasion of lung CSCs through suppression of protein expression levels of CD133, CD44, Bmi1, and Nanog [123]. The role of genistein in head and neck cancer was too studied and was found to downregulate EMT. It also synergized the effect of doxorubicin, cisplatin, and 5-flourouracil to cause cell death in CSCs [124] (Table 7.2). Genistein has also produced a synergistic effect with other chemotherapeutic drugs and is helpful in chemosensitizing the CSCs to treat resistance cases. Sanaei et al. studied the combined effect of genistein and Tamoxifen in hepatocellular cancer cell line (HepG2). It showed that the combination synergistically inhibited proliferation and induced apoptosis [125]. Genistein has a synergistic effect when used in combination with oxaliplatin since the combination exhibited suppression of the expression of CSCs marker (CD44) and inhibited cell proliferation in oral squamous cell carcinoma [126]. Genistein has also shown a synergistic effect when given in combination with doxorubicin and 5-FU. They targeted CSCs and chemosensitize them [127, 128]. The role of genistein to retard CSCs has been explored in various studies which are presented in Table 7.2 with their cellular pathways.
7.5.2 Quercetin
Quercetin (C15H10O7), a flavonol from the class of flavonoids, is dietary polyphenolic compound found in many dietary plants and also found in medicinal botanicals [Ginkgo biloba (Ginkgoaceae) and Hypericum perforatum (Hypericaceae)] displays excellent antitumor activity [129]. It induces apoptosis and downregulates protein expression of EMT, angiogenesis, and stemness of CSCs population in many cancer [130, 131]. In studies, quercetin has been shown to inhibit breast cancer via targeting CSCs. Recently, upregulation of small heat shock proteins 27 (Hsp27) was found to be beneficial in maintaining CSCs along with their stemness [131,132,133]. Quercetin could act as an inhibitor of Hsp27 which causes a decrease in self-renewal capacity of CSCs which eventually reduces the population of ALDH+ breast CSCs. Quercetin further displayed the synergistic effect with geldanamycin (Hsp90 inhibitor) and reduced the migration, tumorigenesis, and population of ALDH+ breast CSCs via the suppression of Hsp90 and Hsp27 [134]. In another study, quercetin suppressed vascularization of tumors by targeting epidermal growth factor (EGF)/Hsp27 signaling [135]. In addition to target Hsp27, quercetin has shown an inhibitory effect on PI3K/Akt/mTOR signaling pathway which is responsible for self-renewal and stemness of CSCs in breast cancer [136]. Quercetin has also demonstrated an improvement in chemosensitivity of resistance cases and inhibited population of breast CSCs by blocking nuclear translocation of Y-box binding protein 1 and hence downregulating P-glycoprotein. This is one of the reasons for its effect in reducing the multidrug resistance and stemness of CSCs [136, 137]. The use of anticancer agents in combination with quercetin has resulted in reduced target toxicity, induction of apoptosis, lowering the cancer recurrence, and inhibition of EMT in CSCs.
Quercetin has also shown promising result in head and neck cancer by showing inhibitory effect on stemness signature, self-renewal capacity, migration ability, and EMT along with reduction in CSCs number which were derived from SAS and OECM1 cell lines (Table 7.3) [138]. It has also shown anticancer effect against teratocarcinoma via antagonizing the Wnt/β-catenin signaling pathway in CSCs of NT2/D1 human cell line [139]. When quercetin was used with other flavonoid, such as luteolin, the combination reversed the EMT process by downregulating the EMT markers in epidermal carcinoma [140]. These combinations are also shown to inhibit the JNK signaling pathway, which further explain their effects on stemness, vasculogenic mimicry properties, and metastatic potential in Du145-III cells (CSCs) derived from Du144-Parental cell line of prostate cancer [141].
Quercetin and EGCG (Epigallocatechin gallate) found in tea, act synergistically and inhibited self-renewal potential along with migration and invasion properties of CSCs of prostate carcinoma by inhibiting TCF/LEF and Gli activities. Quercetin with EGCG lowers the viability of prostate tumor spheroids and lessens the migratory, invasiveness, and colony-forming potential of CD44+/CD133+ prostate CSCs [18]. The anti-CSCs activity of sulforaphane in combination with quercetin has been found more effective in treatment of pancreatic cancer in the MIA-aCa2 CSCs via inhibiting tumor growth [130]. Quercetin has further drawn attention as a potential CSCs targeting therapeutic agent in colon cancer by inhibiting the proliferation of CD133+ colon CSCs and also increasing the chemosensitivity to doxorubicin in in vitro study [142].
Furthermore, the combined effect of cisplatin and quercetin in head and neck cancer was found promising in drug-resistant cases of cisplatin therapy. SCC25 oral squamous cisplatin-resistant CSCs were implanted into nude mice. They significantly inhibited the tumor growth compared with cisplatin or control alone and chemosensitized the CSCs [143] (Table 7.3).
7.5.3 Silibinin
Silibinin is a flavonolignan obtained from the seeds and fruits of milk thistle plant Silybum marianum (Asteraceae) and has been used for the treatment of various types of liver ailment [144]. Previous investigations have shown its strong chemopreventive abilities in various types of cancers [6, 145,146,147,148,149] (Table 7.4). Silibinin has exemplified its action to inhibit colon CSCs in in vitro and ex vivo models and prevent the self-renewal and sphere formation of CSCs by suppressing the PP2Ac/AKT Ser473/mTOR pathway [150]. This is further supported by another study in which silibinin decreased the number and colon sphere formation of CSCs in colorectal cancer by interfering with kinetics and shifted the cell division process towards asymmetric type (generating one CSCs and one first-generation progenitor cell) [151]. Silibinin was found to be effective in colon cancer cell line via blockage of β-catenin Wnt signaling pathway. It downregulates β-catenin gene and protein expression in CSCs. Silibinin also significantly suppressed the proliferation of CSCs by inducing apoptosis by increasing the Bax/Bcl-2 ratio. It has further shown downregulation of stemness markers of CSCs like CD133, CD44, BMI1, ALDH1, and doublecortin-like kinase 1. Additionally, it has the ability to inhibit migration by attenuation of EMT through decreased expression of N-cadherin and vimentin along with increased expression of E-cadherin [152].
The nanoformulation of silibinin has inhibited proliferation and migration of CSCs by induction of apoptosis using MIA-PaCa pancreatic cell line through suppression of some onco-miRs (miR-155, miR-222, and miR-21) and upregulation of some tumor suppressive miRs (miR-34a, miR-126, and miR-let7b) [153]. Moreover, silibinin also has a synergistic effect with other therapeutics. Silibinin in combination with sorafenib has shown a synergistic effect through inhibition of phosphorylation of STAT3/ERK/AKT pathway. This leads to inhibited sphere formation and self-renewal of CSCs in hepatic carcinoma [144] (Table 7.4). The combination of silibinin and 5-FU has demonstrated inhibition of CD44v6 (isoform of CD44) which resulted in weakened stemness characteristic of colon CSCs. CD44v6 is a functional biomarker responsible for cancer progression, initiation of metastatic process, resistance to conventional therapeutics, relapse, and associated with poor survival in patients with colon cancer [154].
7.5.4 Apigenin
Apigenin, a common polyphenolic dietary flavone, is abundantly present in many fruits, vegetables, and Chinese medicinal herbs. Evidence from in vitro and in vivo studies has shown its anticancer potential in multiple types of malignancies such as brain tumor, ovarian cancer, lung carcinoma, prostate cancer, breast cancer, and other tumors [104, 105, 155,156,157]. Recently, the anticancer effect of apigenin has been widely investigated via targeting sub-populated CSCs. Also, it reduced the toxicity of chemotherapeutic agents. Apigenin has been reported to suppress various human cancers in in vitro and in vivo models by targeting multiple biological processes such as triggering cell apoptosis and autophagy, inducing cell cycle arrest, and suppressing cell migration and invasion. This chapter also includes the most recent advancement of apigenin and its synergistic effect with other chemotherapeutic agents by targeting CSCs along with attenuation of involved signaling pathways (Table 7.5). The use of apigenin with chemotherapeutics has overcome the cancer drug resistance or may reduce the toxicities [158]. The glycosidal form of apigenin, Isovitexin (apigenin-6-C-glucoside), has also exhibited its anticancer potential against CSCs in hepatic carcinoma. It decreases the progression of carcinogenicity and stemness by downregulating FoxM1 via inhibition of manganese superoxide dismutase [159]. Isovitexin also suppressed sphere, colony formation, and decreased CD44+ cell population along with suppressed the level of ABCG2, ALDH1, and NANOG mRNA in SK-Hep-1 spheroids of hepatocellular carcinoma by upregulating miR-34a expression [160]. It has the ability to inhibit osteosarcoma by decreasing CSCs population in in vivo model. It has shown to repressed sphere formation, induced apoptotic cell death, and reduced mRNA levels in CSCs derived from U2OS-SC and MG63-SC cells [161]. Studies of apigenin in CSCs are presented in Table 7.5.
7.5.5 Miscellaneous Flavonoids Targeting CSCs
There is limited evidence exists on other flavonoids which have shown their preventive effect against CSCs via modulating signaling pathways involved in the maintenance of CSCs. These flavanoids are broussoflavonol B, icaritin, casticin, pomiferin, morusin, baicalein, ugonin, wogonin, luteolin, and kaempferol.
Broussoflavonol B (5,7,3′,4′- Tetrahydroxy-3-methoxy-6,8-diprenylflavone) is chemically prenylflavone isolated from Broussonetia papyrifera (Moraceae) commonly known as Paper mulberry. It inhibits the growth of ER-positive (estrogen positive) breast cancer in MCF7 cells probably through downregulation of ER-α36 expression. [64, 103, 162]. The knockdown of expression of ER-α36 by broussoflavonol B inhibits tumor sphere formation and reduced the count of HER2-CSCs which help in treating the therapy-resistant cases [103]. Jeong and Ryu reported its anticancer potential in pancreatic cancer via suppression of the FoxM1 and its target genes to induce G0/G1 phase arrest in p53 mutant PANC-1 cells. It also inhibited cell migration and invasion by reducing ERK activity and MMP-2 expression [163] Table 7.6.
Icaritin is a mono-prenylflavonoid derivative (flavonoid skeleton with a lipophilic prenyl side chain) obtained from Chinese herb Epimedium Genus having estrogen receptor modulator effect and hence called phytoestrogen. Icaritin and its analogs regulate cell growth of various types of cancers such as breast cancer, esophageal cancer, chronic myeloid leukemia (CML), and lung carcinoma [103, 164,165,166,167] Table 7.6.
Morusin, a prenylated flavonoids obtained from root bark of Morus australis (Moraceae) possess anticancer effect on various type of malignancies [168,169,170]. It showed inhibition of the growth and migration of human cervical CSCs from HeLa cell line through attenuation of NF-kBp65 activity mediated apoptotic induction [168]. Further, it showed promising anticancer potential in aggressive type of brain cancer, i.e., glioblastoma. Morusin inhibits glioblastoma CSCs by induction of apoptosis by upregulating the protein expressions of PPARϒ, Bax, and caspase-3. Additionally, it downregulates the expressions of Bcl-2 and stemness markers such as CD133, nestin, Oct4, and Sox2 and attenuates adipocyte trans-differentiation [171]. Recently, morusin was found to be a potential anticancer agent in laryngeal cancer by inhibiting the stemness and proliferation of CSCs [172] (Table 7.6).
Casticin (3′,5-dihydroxy-3,4′,6,7-tetramethoxyflavone) is a natural poly-methoxy-flavone also called as vitexicarpin, isolated from the fruits of Vitex trifolia (Lamiaceae) [173]. Casticin has exemplified its anticancer potential via targeting CSCs and modulating their stemness related proteins, AMPK/FoxO3 signaling pathway activation, blocking Wnt/catenin signaling pathways and inhibiting EMT process by regulating expressions of E-cadherin, MMPs and N-cadherin in various types of cancers like liver cancer, lung cancer, and nasopharyngeal cancer [173,174,175] Table 7.6.
Other flavonoids having anti-CSCs effect are pomiferin which is isolated from the fruit of the Maclura pomifera (Moraceae) effective in glioblastoma [176]. Ugonin J and K (two cyclohexylmethyl flavonoids) isolated from the rhizomes of Helminthostachys zeylanica (Ophioglossaceae) are effective in breast cancer [177]. Naringenin which is obtained from tomato and citrus fruits acts as a phytoestrogen and is effective in inhibition of ER+ breast cancer CSCs [178]. Its seminatural derivative, named 6-C-(E-phenylethenyl) naringenin was found effective in the treatment of hepatocellular carcinoma by suppressing Wnt/β-catenin signaling [179]. Baicalein is 5,6,7-trihydroxyflavone, originally isolated from the roots of Scutellaria baicalensis (Lamiaceae) possessing anticancer effect by targeting CSCs of pancreatic, liver, multiple myeloma, and breast cancer [180,181,182,183] Table 7.6. Luteolin is 3,4,5,7-tetrahydroxyflavone obtained from many dietary plants such as chamomile tea, celery, perilla leaf, and green peppers. The in vitro and in vivo studies showed that it inhibits cancer initiation and progression by interfering with transcription factors and kinases, regulating cell cycle, apoptosis, and inhibiting cell transformation, migration, invasion, and angiogenesis [184,185,186,187,188,189]. It also has the potential to target CSCs via attenuation of different pathways [167] and also produce a synergistic effect to enhance the anticancer potential of the other chemotherapeutic drugs [190]. Moreover, it is sensitizing the CSCs and treating therapy resistance cases of cancer [141, 191] (Tables 7.3 and 7.6).
Wogonin is an O-methylated flavone found in the roots of Scutellaria baicalensis (Lamiaceae). Wogonin has been also used to target CSCs in various malignancies such as osteosarcoma, multiple myeloma, and breast cancer [183, 192,193,194] by attenuation of EMT markers (MMP-9), regulation of ROS signaling. Wogonoside which is a glycoside of wogonin has shown anticancer potential against cutaneous squamous carcinoma via suppression of PI3K/AKT and Wnt/β-catenin pathway of CSCs [195].
Kaempferol (3,5,7-trihydroxy-2-(4-hydroxyphenyl)-4H-1-benzopyran-4-one) is a phytoestrogen, obtained abundantly from tea, broccoli, apples, strawberries, and beans. It showed an anti-CSCs effect by decreasing breast CSCs derived from MCF-7 cell line and downregulated the markers such as Oct4, Nanog, ABCB1, and ALDH1A1 [196].
The details of studies regarding the inhibitory action of miscellaneous flavonoids in CSCs have been described in Table 7.6.
7.6 Future Prospects of Flavonoids Targeting CSCs in Malignancies
In recent years, there has been much attention towards the inhibition of CSCs to reduce the severeness and resistant cases of cancer. Hence, polyphenolic flavonoids are used to prevent cancer progression via targeting CSCs. Flavonoids are regarded as multifacet phytocompounds possessing plethora of therapeutic effects [181, 197, 198]. There is substantial data available which have shown their potential to eradicate CSCs. However, no evaluation has been conducted in the clinical setting targeted CSCs. Furthermore, the major issue to target the CSCs is the identification of specific markers for a particular type of tumor. The specific markers would provide novel strategies to target the CSCs and inhibit the progression of cancer. Flavonoids also act as epigenetic modifiers by inhibiting early epigenetic alterations and inhibit cancer cell proliferation in in vitro models using cell lines. In various in vitro studies, flavonoids activate the expression of different tumor suppressor genes by epigenetic modifications [199, 200]. However, there is a lack of studies of flavonoids as epigenetic modifiers targeting CSCs maintenance. Hence, there is a need for studies on flavonoids as natural epigenetic modulators for the treatment of cancers targeting CSCs which could represent a promising and valid strategy to inhibit chemoresistance and carcinogenesis. Flavonoids are able to eradicate and chemosensitize the CSCs of various tumors via attenuating many pathways but they suffer from certain limitations such as poor solubility, poor permeability, bitter taste, extensive intestinal metabolism, and instability which diminish their bioavailability. Due to these issues, relatively high dose of flavonoids is required to produce a significant biological response. Strategies are needed to overcome these issues of solubility and thereby improving its oral bioavailability. Chemical modification in the structure of flavonoids may enhance the stability of flavonoids [109, 111, 201,202,203]. This will help to conduct their clinical trials and enhance their clinical usage. This chapter also described that the combination of flavonoids with conventional therapies could enhance the therapeutic effects and chemoradiosensitize the CSCs in various malignancies [182]. Hence, they may enhance the anticancer potential along with a reduction in resistance.
References
Sak K. Cytotoxicity of dietary flavonoids on different human cancer types. Pharmacogn Rev. 2014;8(16):122–46.
Chen D, Pamu S, Cui Q, Chan TH, Dou QP. Novel epigallocatechin gallate (EGCG) analogs activate AMP-activated protein kinase pathway and target cancer stem cells. Bioorg Med Chem. 2012;20(9):3031–7.
O'Brien-Ball C, Biddle A. Reprogramming to developmental plasticity in cancer stem cells. Dev Biol. 2017;430(2):266–74.
Kuşoğlu A, Biray AÇ. Cancer stem cells: a brief review of the current status. Gene. 2019;681:80–5.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–8.
Ting H, Deep G, Agarwal R. Molecular mechanisms of silibinin-mediated cancer chemoprevention with major emphasis on prostate cancer. AAPS J. 2013;15(3):707–16.
Hu Y, Fu L. Targeting cancer stem cells: a new therapy to cure cancer patients. Am J Cancer Res. 2012;2(3):340–56.
Li Y, Wicha MS, Schwartz SJ, Sun D. Implications of cancer stem cell theory for cancer chemoprevention by natural dietary compounds. J Nutr Biochem. 2011;22(9):799–806.
Wang MT, Holderfield M, Galeas J, Delrosario R, To MD, Balmain A, et al. K-ras promotes tumorigenicity through suppression of non-canonical Wnt signaling. Cell. 2015;163(5):1237–51.
Gooding AJ, Schiemann WP. Epithelial-mesenchymal transition programs and cancer stem cell phenotypes: mediators of breast cancer therapy resistance. Mol Cancer Res. 2020;18(9):1257–70.
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–68.
Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea – a paradigm shift. Cancer Res. 2006;66(4):1883–90. discussion 95-6
Dai Z, Nair V, Khan M, Ciolino HP. Pomegranate extract inhibits the proliferation and viability of MMTV-Wnt-1 mouse mammary cancer stem cells in vitro. Oncol Rep. 2010;24(4):1087–91.
Ning Y, Luo C, Ren K, Quan M, Cao J. FOXO3a-mediated suppression of the self-renewal capacity of sphere-forming cells derived from the ovarian cancer SKOV3 cell line by 7-difluoromethoxyl-5,4'-di-n-octyl genistein. Mol Med Rep. 2014;9(5):1982–8.
Chen Y, Zaman MS, Deng G, Majid S, Saini S, Liu J, et al. MicroRNAs 221/222 and genistein-mediated regulation of ARHI tumor suppressor gene in prostate cancer. Cancer Prev Res (Philadelphia, PA). 2011;4(1):76–86.
Zhang L, Li L, Jiao M, Wu D, Wu K, Li X, et al. Genistein inhibits the stemness properties of prostate cancer cells through targeting Hedgehog-Gli1 pathway. Cancer Lett. 2012;323(1):48–57.
Nishimura N, Hartomo TB, Pham TV, Lee MJ, Yamamoto T, Morikawa S, et al. Epigallocatechin gallate inhibits sphere formation of neuroblastoma BE(2)-C cells. Environ Health Prev Med. 2012;17(3):246–51.
Tang SN, Singh C, Nall D, Meeker D, Shankar S, Srivastava RK. The dietary bioflavonoid quercetin synergizes with epigallocatechin gallate (EGCG) to inhibit prostate cancer stem cell characteristics, invasion, migration and epithelial-mesenchymal transition. J Mol Signal. 2010;5:14.
Tang SN, Fu J, Nall D, Rodova M, Shankar S, Srivastava RK. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int J Cancer. 2012;131(1):30–40.
Sharif T, Auger C, Bronner C, Alhosin M, Klein T, Etienne-Selloum N, et al. Selective proapoptotic activity of polyphenols from red wine on teratocarcinoma cell, a model of cancer stem-like cell. Invest New Drugs. 2011;29(2):239–47.
Lin CH, Shen YA, Hung PH, Yu YB, Chen YJ. Epigallocatechin gallate, polyphenol present in green tea, inhibits stem-like characteristics and epithelial-mesenchymal transition in nasopharyngeal cancer cell lines. BMC Complement Altern Med. 2012;12:201.
Quan MF, Xiao LH, Liu ZH, Guo H, Ren KQ, Liu F, et al. 8-bromo-7-methoxychrysin inhibits properties of liver cancer stem cells via downregulation of β-catenin. World J Gastroenterol. 2013;19(43):7680–95.
Alvero AB, Montagna MK, Holmberg JC, Craveiro V, Brown D, Mor G. Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells. Mol Cancer Ther. 2011;10(8):1385–93.
Hsu HS, Lin JH, Huang WC, Hsu TW, Su K, Chiou SH, et al. Chemoresistance of lung cancer stemlike cells depends on activation of Hsp27. Cancer. 2011;117(7):1516–28.
Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45.
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–29.
Chiang SP, Cabrera RM, Segall JE. Tumor cell intravasation. Am J Physiol Cell Physiol. 2016;311(1):C1–c14.
Jiang J, Tang YL, Liang XH. EMT: a new vision of hypoxia promoting cancer progression. Cancer Biol Ther. 2011;11(8):714–23.
Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6(13):10697–711.
De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110.
Leopold PL, Vincent J, Wang H. A comparison of epithelial-to-mesenchymal transition and re-epithelialization. Semin Cancer Biol. 2012;22(5-6):471–83.
Reid PA, Wilson P, Li Y, Marcu LG, Bezak E. Current understanding of cancer stem cells: Review of their radiobiology and role in head and neck cancers. Head Neck. 2017;39(9):1920–32.
Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23(10):1124–34.
Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, et al. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat Cell Biol. 2013;15(11):1351–61.
You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22(1):9–20.
Avgustinova A, Benitah SA. The epigenetics of tumour initiation: cancer stem cells and their chromatin. Curr Opin Genet Dev. 2016;36:8–15.
Sun XX, Yu Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin. 2015;36(10):1219–27.
Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–37.
Lipinska N, Romaniuk A, Paszel-Jaworska A, Toton E, Kopczynski P, Rubis B. Telomerase and drug resistance in cancer. Cell Mol Life Sci. 2017;74(22):4121–32.
Yan J, Zhou Y, Chen D, Li L, Yang X, You Y, et al. Effects of mitochondrial translocation of telomerase on drug resistance in hepatocellular carcinoma cells. J Cancer. 2015;6(2):151–9.
Gopalan V, Islam F, Lam AK. Surface markers for the identification of cancer stem cells. Methods Mol Biol (Clifton, NJ). 2018;1692:17–29.
Nunes T, Hamdan D, Leboeuf C, El Bouchtaoui M, Gapihan G, Nguyen TT, et al. Targeting cancer stem cells to overcome chemoresistance. Int J Mol Sci. 2018;19(2):4036.
Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18(5):460–6.
Carvalho MJ, Laranjo M, Abrantes AM, Torgal I, Botelho MF, Oliveira CF. Clinical translation for endometrial cancer stem cells hypothesis. Cancer Metastasis Rev. 2015;34(3):401–16.
Leccia F, Del Vecchio L, Mariotti E, Di Noto R, Morel AP, Puisieux A, et al. ABCG2, a novel antigen to sort luminal progenitors of BRCA1- breast cancer cells. Mol Cancer. 2014;13:213.
Soltanian S, Matin MM. Cancer stem cells and cancer therapy. Tumour Biol. 2011;32(3):425–40.
Vilchez V, Turcios L, Zaytseva Y, Stewart R, Lee EY, Maynard E, et al. Cancer stem cell marker expression alone and in combination with microvascular invasion predicts poor prognosis in patients undergoing transplantation for hepatocellular carcinoma. Am J Surg. 2016;212(2):238–45.
Kure S, Matsuda Y, Hagio M, Ueda J, Naito Z, Ishiwata T. Expression of cancer stem cell markers in pancreatic intraepithelial neoplasias and pancreatic ductal adenocarcinomas. Int J Oncol. 2012;41(4):1314–24.
Salnikov AV, Gladkich J, Moldenhauer G, Volm M, Mattern J, Herr I. CD133 is indicative for a resistance phenotype but does not represent a prognostic marker for survival of non-small cell lung cancer patients. Int J Cancer. 2010;126(4):950–8.
Svachova H, Pour L, Sana J, Kovarova L, Raja KR, Hajek R. Stem cell marker nestin is expressed in plasma cells of multiple myeloma patients. Leuk Res. 2011;35(8):1008–13.
Isfoss BL, Busch C, Hermelin H, Vermedal AT, Kile M, Braathen GJ, et al. Stem cell marker-positive stellate cells and mast cells are reduced in benign-appearing bladder tissue in patients with urothelial carcinoma. Virchows Arch. 2014;464(4):473–88.
Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013;108(3):479–85.
Fulawka L, Donizy P, Halon A. Cancer stem cells – the current status of an old concept: literature review and clinical approaches. Biol Res. 2014;47(1):66.
Kandhari K, Agraval H, Sharma A, Yadav UC, Singh RP. Flavonoids and cancer stem cells maintenance and growth. Functional food and human health. New York: Springer; 2018. p. 587–622.
Koury J, Zhong L, Hao J. Targeting signaling pathways in cancer stem cells for cancer treatment. Stem Cells Int. 2017;2017:2925869.
Chi HC, Tsai CY, Tsai MM, Yeh CT, Lin KH. Roles of long noncoding RNAs in recurrence and metastasis of radiotherapy-resistant cancer stem cells. Int J Mol Sci. 2017;18(9) https://doi.org/10.3390/ijms18091903.
Paryan M, Mohammadi-Yeganeh S, Samiee SM, Soleimani M, Arefian E, Azadmanesh K, et al. Investigation of deregulated genes of Notch signaling pathway in human T cell acute lymphoblastic leukemia cell lines and clinical samples. Mol Biol Rep. 2013;40(10):5531–40.
Matsui WH. Cancer stem cell signaling pathways. Medicine. 2016;95(1 Suppl 1):S8–s19.
Venkatesh V, Nataraj R, Thangaraj GS, Karthikeyan M, Gnanasekaran A, Kaginelli SB, et al. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018;5:5.
Pan Y, Ma S, Cao K, Zhou S, Zhao A, Li M, et al. Therapeutic approaches targeting cancer stem cells. J Cancer Res Ther. 2018;14(7):1469–75.
Katoh Y, Katoh M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review). Int J Mol Med. 2008;22(3):271–5.
He HC, Chen JH, Chen XB, Qin GQ, Cai C, Liang YX, et al. Expression of hedgehog pathway components is associated with bladder cancer progression and clinical outcome. Pathol Oncol Res. 2012;18(2):349–55.
Takezaki T, Hide T, Takanaga H, Nakamura H, Kuratsu J, Kondo T. Essential role of the Hedgehog signaling pathway in human glioma-initiating cells. Cancer Sci. 2011;102(7):1306–12.
Zhang C, Li C, He F, Cai Y, Yang H. Identification of CD44+CD24+ gastric cancer stem cells. J Cancer Res Clin Oncol. 2011;137(11):1679–86.
Islam SS, Mokhtari RB, Noman AS, Uddin M, Rahman MZ, Azadi MA, et al. Sonic hedgehog (Shh) signaling promotes tumorigenicity and stemness via activation of epithelial-to-mesenchymal transition (EMT) in bladder cancer. Mol Carcinog. 2016;55(5):537–51.
Merchant AA, Matsui W. Targeting Hedgehog – a cancer stem cell pathway. Clin Cancer Res. 2010;16(12):3130–40.
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17(2):165–72.
Zhang B, Jiang T, Shen S, She X, Tuo Y, Hu Y, et al. Cyclopamine disrupts tumor extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials. 2016;103:12–21.
Iovine V, Mori M, Calcaterra A, Berardozzi S, Botta B. One hundred faces of cyclopamine. Curr Pharm Des. 2016;22(12):1658–81.
Fu Q, Liu P, Sun X, Huang S, Han F, Zhang L, et al. Ribonucleic acid interference knockdown of IL-6 enhances the efficacy of cisplatin in laryngeal cancer stem cells by down-regulating the IL-6/STAT3/HIF1 pathway. Cancer Cell Int. 2017;17:79.
Ulasov IV, Nandi S, Dey M, Sonabend AM, Lesniak MS. Inhibition of Sonic hedgehog and Notch pathways enhances sensitivity of CD133(+) glioma stem cells to temozolomide therapy. Mol Med (Cambridge, MA). 2011;17(1-2):103–12.
Bahra M, Kamphues C, Boas-Knoop S, Lippert S, Esendik U, Schüller U, et al. Combination of hedgehog signaling blockage and chemotherapy leads to tumor reduction in pancreatic adenocarcinomas. Pancreas. 2012;41(2):222–9.
Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26.
Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13(7):513–32.
Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science (New York, NY). 2014;346(6205):1248012.
Holland JD, Klaus A, Garratt AN. Birchmeier WJCoicb. Wnt signal stem cancer stem cells. 2013;25(2):254–64.
Jang GB, Kim JY, Cho SD, Park KS, Jung JY, Lee HY, et al. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep. 2015;5:12465.
Pandit TS, Kennette W, Mackenzie L, Zhang G, Al-Katib W, Andrews J, et al. Lymphatic metastasis of breast cancer cells is associated with differential gene expression profiles that predict cancer stem cell-like properties and the ability to survive, establish and grow in a foreign environment. Int J Oncol. 2009;35(2):297–308.
Borcherding N, Kusner D, Kolb R, Xie Q, Li W, Yuan F, et al. Paracrine WNT5A signaling inhibits expansion of tumor-initiating cells. Cancer Res. 2015;75(10):1972–82.
Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol. 2014;15(1):19–33.
Van Camp JK, Beckers S, Zegers D, Van Hul W. Wnt signaling and the control of human stem cell fate. Stem Cell Rev Rep. 2014;10(2):207–29.
Yang K, Wang X, Zhang H, Wang Z, Nan G, Li Y, et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: implications in targeted cancer therapies. Lab Invest. 2016;96(2):116–36.
Qin L, Yin YT, Zheng FJ, Peng LX, Yang CF, Bao YN, et al. WNT5A promotes stemness characteristics in nasopharyngeal carcinoma cells leading to metastasis and tumorigenesis. Oncotarget. 2015;6(12):10239–52.
Webster MR, Kugel CH 3rd, Weeraratna AT. The Wnts of change: How Wnts regulate phenotype switching in melanoma. Biochim Biophys Acta. 2015;1856(2):244–51.
Kumawat K, Gosens R. WNT-5A: signaling and functions in health and disease. Cell Mol Life Sci. 2016;73(3):567–87.
Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol. 2017;51(5):1357–69.
Ribeiro AS, Paredes J. P-cadherin linking breast cancer stem cells and invasion: a promising marker to identify an “Intermediate/Metastable” EMT State. Front Oncol. 2014;4:371.
Isobe T, Hisamori S, Hogan DJ, Zabala M, Hendrickson DG, Dalerba P, et al. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. eLife. 2014;3:e01977.
Xu L, Zhang L, Hu C, Liang S, Fei X, Yan N, et al. WNT pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int J Oncol. 2016;48(3):1175–86.
Sun X, Xu C, Xiao G, Meng J, Wang J, Tang SC, et al. Breast cancer stem-like cells are sensitized to tamoxifen induction of self-renewal inhibition with enforced Let-7c dependent on Wnt blocking. Int J Mol Med. 2018;41(4):1967–75.
Lunardi A, Webster KA, Papa A, Padmani B, Clohessy JG, Bronson RT, et al. Role of aberrant PI3K pathway activation in gallbladder tumorigenesis. Oncotarget. 2014;5(4):894–900.
Ye B, Jiang LL, Xu HT, Zhou DW, Li ZS. Expression of PI3K/AKT pathway in gastric cancer and its blockade suppresses tumor growth and metastasis. Int J Immunopathol Pharmacol. 2012;25(3):627–36.
Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. 2015;9(4):317–24.
Lechman ER, Gentner B, Ng SWK, Schoof EM, van Galen P, Kennedy JA, et al. miR-126 regulates distinct self-renewal outcomes in normal and malignant hematopoietic stem cells. Cancer Cell. 2016;29(4):602–6.
Bahena-Ocampo I, Espinosa M, Ceballos-Cancino G, Lizarraga F, Campos-Arroyo D, Schwarz A, et al. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep. 2016;17(5):648–58.
Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86.
Pellegrini P, Cordero A, Gallego MI, Dougall WC, Muñoz P, Pujana MA, et al. Constitutive activation of RANK disrupts mammary cell fate leading to tumorigenesis. Stem cells (Dayton, Ohio). 2013;31(9):1954–65.
Zhang W, Tan W, Wu X, Poustovoitov M, Strasner A, Li W, et al. A NIK-IKKα module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell. 2013;23(5):647–59.
Chen C, Cao F, Bai L, Liu Y, Xie J, Wang W, et al. IKKβ enforces a LIN28B/TCF7L2 positive feedback loop that promotes cancer cell stemness and metastasis. Cancer Res. 2015;75(8):1725–35.
Tran TA, Ahn KS, Song YW, Moon JY, Cho M, Lim Y, et al. Mechanism of 2',3'-dimethoxyflavanone-induced apoptosis in breast cancer stem cells: role of ubiquitination of caspase-8 and LC3. Arch Biochem Biophys. 2014;562:92–102.
Xu SL, Zhu KY, Bi CW, Yan L, Men SW, Dong TT, et al. Flavonoids, derived from traditional Chinese medicines, show roles in the differentiation of neurons: possible targets in developing health food products. Birth Defects Res C Embryo Today. 2013;99(4):292–9.
Haar CP, Hebbar P, Wallace GC, Das A, Vandergrift WA 3rd, Smith JA, et al. Drug resistance in glioblastoma: a mini review. Neurochem Res. 2012;37(6):1192–200.
Yin L, Pan X, Zhang XT, Guo YM, Wang ZY, Gong Y, et al. Downregulation of ER-α36 expression sensitizes HER2 overexpressing breast cancer cells to tamoxifen. Am J Cancer Res. 2015;5(2):530–44.
Li Y, Chen X, He W, Xia S, Jiang X, Li X, et al. Apigenin enhanced antitumor effect of cisplatin in lung cancer via inhibition of cancer stem cells. Nutr Cancer. 2020:1–9. https://doi.org/10.1080/01635581.2020.1802494.
Erdogan S, Turkekul K, Serttas R, Erdogan Z. The natural flavonoid apigenin sensitizes human CD44(+) prostate cancer stem cells to cisplatin therapy. Biomed Pharmacother. 2017;88:210–7.
Bansal M, Singh N, Pal S, Dev I, Ansari KM. Chemopreventive role of dietary phytochemicals in colorectal cancer, Advances in molecular toxicology, vol. 12. Amsterdam: Elsevier; 2018. p. 69–11.
Evans WCED. Phenols and phenolic glycosides. Trease and Evans’ Pharmacognosy. Philadelphia, PA: WB Saunders; 2009.
Walter ED. EJJotAcS. Genistin (an isoflavone glucoside) and its aglucone, genistein, from soybeans. J Am Chem Soc. 1941;63(12):3273–6.
Spagnuolo C, Russo GL, Orhan IE, Habtemariam S, Daglia M, Sureda A, et al. Genistein and cancer: current status, challenges, and future directions. Adv Nutr (Bethesda, MD). 2015;6(4):408–19.
Lee JY, Kim HS, Song YS. Genistein as a potential anticancer agent against ovarian cancer. J Tradit Complement Med. 2012;2(2):96–104.
Tuli HS, Tuorkey MJ, Thakral F, Sak K, Kumar M, Sharma AK, et al. Molecular mechanisms of action of genistein in cancer: recent advances. Front Pharmacol. 2019;10:1336.
Aarushi G, Sahoo P, Tejpal AJ. Genistein - a potential boon for cancer therapy. Pharm Innov. 2016;5(6, Part B):81.
Sekar V, Anandasadagopan SK, GanapasamSJB. Genistein regulates tumor microenvironment and exhibits anticancer effect in dimethyl hydrazine-induced experimental colon carcinogenesis. Biofactors. 2016;42(6):623–37.
Pan H, Zhou W, He W, Liu X, Ding Q, Ling L, et al. Genistein inhibits MDA-MB-231 triple-negative breast cancer cell growth by inhibiting NF-κB activity via the Notch-1 pathway. Int J Mol Med. 2012;30(2):337–43.
Xia J, Duan Q, Ahmad A, Bao B, Banerjee S, Shi Y, et al. Genistein inhibits cell growth and induces apoptosis through up-regulation of miR-34a in pancreatic cancer cells. Curr Drug Targets. 2012;13(14):1750–6.
Hirata H, Ueno K, Nakajima K, Tabatabai ZL, Hinoda Y, Ishii N, et al. Genistein downregulates onco-miR-1260b and inhibits Wnt-signalling in renal cancer cells. Br J Cancer. 2013;108(10):2070–8.
Montales MT, Rahal OM, Nakatani H, Matsuda T, Simmen RC. Repression of mammary adipogenesis by genistein limits mammosphere formation of human MCF-7 cells. J Endocrinol. 2013;218(1):135–49.
Ning Y, Feng W, Cao X, Ren K, Quan M, Chen A, et al. Genistein inhibits stemness of SKOV3 cells induced by macrophages co-cultured with ovarian cancer stem-like cells through IL-8/STAT3 axis. J Exp Clin Cancer Res. 2019;38(1):19.
Huang W, Wan C, Luo Q, Huang Z, Luo Q. Genistein-inhibited cancer stem cell-like properties and reduced chemoresistance of gastric cancer. Int J Mol Sci. 2014;15(3):3432–43.
Zhou P, Wang C, Hu Z, Chen W, Qi W, Li A. Genistein induces apoptosis of colon cancer cells by reversal of epithelial-to-mesenchymal via a Notch1/NF-κB/slug/E-cadherin pathway. BMC Cancer. 2017;17(1):813.
Zhang Q, Cao WS, Wang XQ, Zhang M, Lu XM, Chen JQ, et al. Genistein inhibits nasopharyngeal cancer stem cells through sonic hedgehog signaling. Phytother Res. 2019;33(10):2783–91.
Li E, Zhang T, Sun X, Li Y, Geng H, Yu D, et al. Sonic hedgehog pathway mediates genistein inhibition of renal cancer stem cells. Oncol Lett. 2019;18(3):3081–91.
Fu Z, Cao X, Liu L, Cao X, Cui Y, Li X, et al. Genistein inhibits lung cancer cell stem-like characteristics by modulating MnSOD and FoxM1 expression. Oncol Lett. 2020;20(3):2506–15.
Hsieh PL, Liao YW, Hsieh CW, Chen PN, Yu CC. Soy isoflavone genistein impedes cancer stemness and mesenchymal transition in head and neck cancer through activating miR-34a/RTCB axis. Nutrients. 2020;12(7):1924.
Sanaei M, Kavoosi F, Atashpour S, Haghighat S. Effects of genistein and synergistic action in combination with tamoxifen on the HepG2 human hepatocellular carcinoma cell line. Asian Pac J Cancer Prev. 2017;18(9):2381–5.
Hussein AM, El-Sheikh SM, Darwish ZE, Hussein KA, Gaafar AI. Effect of genistein and oxaliplatin on cancer stem cells in oral squamous cell carcinoma: an experimental study. Alexandria Dent J. 2018;43(1):117–23.
Montales MT, Simmen RC, Ferreira ES, Neves VA, Simmen FA. Metformin and soybean-derived bioactive molecules attenuate the expansion of stem cell-like epithelial subpopulation and confer apoptotic sensitivity in human colon cancer cells. Genes Nutr. 2015;10(6):49.
Xue JP, Wang G, Zhao ZB, Wang Q, Shi Y. Synergistic cytotoxic effect of genistein and doxorubicin on drug-resistant human breast cancer MCF-7/Adr cells. Oncol Rep. 2014;32(4):1647–53.
Kashyap D, Mittal S, Sak K, Singhal P, Tuli HS. Molecular mechanisms of action of quercetin in cancer: recent advances. Tumour Biol. 2016;37(10):12927–39.
Zhou W, Kallifatidis G, Baumann B, Rausch V, Mattern J, Gladkich J, et al. Dietary polyphenol quercetin targets pancreatic cancer stem cells. Int J Oncol. 2010;37(3):551–61.
Lu KT, Wang BY, Chi WY, Chang-Chien J, Yang JJ, Lee HT, et al. Ovatodiolide inhibits breast cancer stem/progenitor cells through SMURF2-mediated downregulation of Hsp27. Toxins. 2016;8(5):127.
Yasuda K, Hirohashi Y, Mariya T, Murai A, Tabuchi Y, Kuroda T, et al. Phosphorylation of HSF1 at serine 326 residue is related to the maintenance of gynecologic cancer stem cells through expression of HSP27. Oncotarget. 2017;8(19):31540–53.
Wei L, Liu TT, Wang HH, Hong HM, Yu AL, Feng HP, et al. Hsp27 participates in the maintenance of breast cancer stem cells through regulation of epithelial-mesenchymal transition and nuclear factor-κB. Breast Cancer Res. 2011;13(5):R101.
Lee CH, Hong HM, Chang YY, Chang WW. Inhibition of heat shock protein (Hsp) 27 potentiates the suppressive effect of Hsp90 inhibitors in targeting breast cancer stem-like cells. Biochimie. 2012;94(6):1382–9.
Lee CH, Wu YT, Hsieh HC, Yu Y, Yu AL, Chang WW. Epidermal growth factor/heat shock protein 27 pathway regulates vasculogenic mimicry activity of breast cancer stem/progenitor cells. Biochimie. 2014;104:117–26.
Li S, Zhao Q, Wang B, Yuan S, Wang X, Li K. Quercetin reversed MDR in breast cancer cells through down-regulating P-gp expression and eliminating cancer stem cells mediated by YB-1 nuclear translocation. Phytother Res. 2018;32(8):1530–6.
To K, Fotovati A, Reipas KM, Law JH, Hu K, Wang J, et al. Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res. 2010;70(7):2840–51.
Chang WW, Hu FW, Yu CC, Wang HH, Feng HP, Lan C, et al. Quercetin in elimination of tumor initiating stem-like and mesenchymal transformation property in head and neck cancer. Head Neck. 2013;35(3):413–9.
Mojsin M, Vicentic JM, Schwirtlich M, Topalovic V, Stevanovic M. Quercetin reduces pluripotency, migration and adhesion of human teratocarcinoma cell line NT2/D1 by inhibiting Wnt/β-catenin signaling. Food Funct. 2014;5(10):2564–73.
Lin YS, Tsai PH, Kandaswami CC, Cheng CH, Ke FC, Lee PP, et al. Effects of dietary flavonoids, luteolin, and quercetin on the reversal of epithelial-mesenchymal transition in A431 epidermal cancer cells. Cancer Sci. 2011;102(10):1829–39.
Tsai PH, Cheng CH, Lin CY, Huang YT, Lee LT, Kandaswami CC, et al. Dietary flavonoids luteolin and quercetin suppressed cancer stem cell properties and metastatic potential of isolated prostate cancer cells. Anticancer Res. 2016;36(12):6367–80.
Atashpour S, Fouladdel S, Movahhed TK, Barzegar E, Ghahremani MH, Ostad SN, et al. Quercetin induces cell cycle arrest and apoptosis in CD133(+) cancer stem cells of human colorectal HT29 cancer cell line and enhances anticancer effects of doxorubicin. Iran J Basic Med Sci. 2015;18(7):635–43.
Chen SF, Nieh S, Jao SW, Liu CL, Wu CH, Chang YC, et al. Quercetin suppresses drug-resistant spheres via the p38 MAPK-Hsp27 apoptotic pathway in oral cancer cells. PLoS One. 2012;7(11):e49275.
Mao J, Yang H, Cui T, Pan P, Kabir N, Chen D, et al. Combined treatment with sorafenib and silibinin synergistically targets both HCC cells and cancer stem cells by enhanced inhibition of the phosphorylation of STAT3/ERK/AKT. Eur J Pharmacol. 2018;832:39–49.
Vue B, Chen QH. The potential of flavonolignans in prostate cancer management. Curr Med Chem. 2016;23(34):3925–50.
Bosch-Barrera J, Queralt B, Menendez JA. Targeting STAT3 with silibinin to improve cancer therapeutics. Cancer Treat Rev. 2017;58:61–9.
Jahanafrooz Z, Motamed N, Rinner B, Mokhtarzadeh A, Baradaran B. Silibinin to improve cancer therapeutic, as an apoptotic inducer, autophagy modulator, cell cycle inhibitor, and microRNAs regulator. Life Sci. 2018;213:236–47.
Mateen S, Raina K, Agarwal R. Chemopreventive and anti-cancer efficacy of silibinin against growth and progression of lung cancer. Nutr cancer. 2013;65 Suppl 1(01):3–11.
Liakopoulou C, Kazazis C, Vallianou NG. Silimarin and cancer. Anticancer Agents Med Chem. 2018;18(14):1970–4.
Wang JY, Chang CC, Chiang CC, Chen WM, Hung SC. Silibinin suppresses the maintenance of colorectal cancer stem-like cells by inhibiting PP2A/AKT/mTOR pathways. J Cell Biochem. 2012;113(5):1733–43.
Kumar S, Raina K, Agarwal C, Agarwal R. Silibinin strongly inhibits the growth kinetics of colon cancer stem cell-enriched spheroids by modulating interleukin 4/6-mediated survival signals. Oncotarget. 2014;5(13):4972–89.
Sameri S, Saidijam M, Bahreini F, Najafi R. Cancer chemopreventive activities of silibinin on colorectal cancer through regulation of E-cadherin/β-catenin pathway. Nutr Cancer. 2020:1–11. https://doi.org/10.1080/01635581.2020.1800764.
Khakinezhad Tehrani F, Ranji N, Kouhkan F, Hosseinzadeh S. Apoptosis induction and proliferation inhibition by silibinin encapsulated in nanoparticles in MIA PaCa-2 cancer cells and deregulation of some miRNAs. Iran J Basic Med Sci. 2020;23(4):469–82.
Patel S, Waghela B, Shah K, Vaidya F, Mirza S, Patel S, et al. Silibinin, a natural blend in polytherapy formulation for targeting Cd44v6 expressing colon cancer stem cells. Sci Rep. 2018;8(1):16985.
Erdogan S, Doganlar O, Doganlar ZB, Serttas R, Turkekul K, Dibirdik I, et al. The flavonoid apigenin reduces prostate cancer CD44(+) stem cell survival and migration through PI3K/Akt/NF-κB signaling. Life Sci. 2016;162:77–86.
Ketkaew Y, Osathanon T, Pavasant P, Sooampon S. Apigenin inhibited hypoxia induced stem cell marker expression in a head and neck squamous cell carcinoma cell line. Arch Oral Biol. 2017;74:69–74.
Tang AQ, Cao XC, Tian L, He L, Liu F. Apigenin inhibits the self-renewal capacity of human ovarian cancer SKOV3-derived sphere-forming cells. Mol Med Rep. 2015;11(3):2221–6.
Yan X, Qi M, Li P, Zhan Y, Shao H. Apigenin in cancer therapy: anti-cancer effects and mechanisms of action. Cell Biosci. 2017;7:50.
Cao X, Liu L, Yuan Q, Li X, Cui Y, Ren K, et al. Isovitexin reduces carcinogenicity and stemness in hepatic carcinoma stem-like cells by modulating MnSOD and FoxM1. J Exp Clin Cancer Res. 2019;38(1):264.
Xu C, Cao X, Cao X, Liu L, Qiu Y, Li X, et al. Isovitexin inhibits stemness and induces apoptosis in hepatocellular carcinoma SK-Hep-1 spheroids by upregulating miR-34a expression. Anticancer Agents Med Chem. 2020;20(14):1654–63.
Liang X, Xu C, Cao X, Wang W. Isovitexin suppresses cancer stemness property and induces apoptosis of osteosarcoma cells by disruption of The DNMT1/miR-34a/Bcl-2 axis. Cancer Manag Res. 2019;11:8923–36.
Guo M, Wang M, Zhang X, Deng H, Wang ZY. Broussoflavonol B restricts growth of ER-negative breast cancer stem-like cells. Anticancer Res. 2013;33(5):1873–9.
Jeong JH, Ryu JH. Broussoflavonol B from broussonetia kazinoki siebold exerts anti-pancreatic cancer activity through downregulating FoxM1. Molecules (Basel, Switzerland). 2328;25(10):2020.
Guo Y, Zhang X, Meng J, Wang ZY. An anticancer agent icaritin induces sustained activation of the extracellular signal-regulated kinase (ERK) pathway and inhibits growth of breast cancer cells. Eur J Pharmacol. 2011;658(2-3):114–22.
Chen M, Turhan AG, Ding H, Lin Q, Meng K, Jiang X. Targeting BCR-ABL+ stem/progenitor cells and BCR-ABL-T315I mutant cells by effective inhibition of the BCR-ABL-Tyr177-GRB2 complex. Oncotarget. 2017;8(27):43662–77.
Liu S, Guo Y, Wang J, Zhu H, Han Y, Jin M, et al. A novel anticancer agent SNG1153 inhibits growth of lung cancer stem/progenitor cells. Oncotarget. 2016;7(29):45158–70.
Han K, Lang T, Zhang Z, Zhang Y, Sun Y, Shen Z, et al. Luteolin attenuates Wnt signaling via upregulation of FZD6 to suppress prostate cancer stemness revealed by comparative proteomics. Sci Rep. 2018;8(1):8537.
Wang L, Guo H, Yang L, Dong L, Lin C, Zhang J, et al. Morusin inhibits human cervical cancer stem cell growth and migration through attenuation of NF-κB activity and apoptosis induction. Mol Cell Biochem. 2013;379(1-2):7–18.
Lee HJ, da Lyu H, Koo U, Nam KW, Hong SS, Kim KO, et al. Protection of prenylated flavonoids from Mori Cortex Radicis (Moraceae) against nitric oxide-induced cell death in neuroblastoma SH-SY5Y cells. Arch Pharm Res. 2012;35(1):163–70.
Dat NT, Binh PT, Quynh LTP, Van Minh C, Huong HT, Lee JJ. Cytotoxic prenylated flavonoids from Morus alba. Fitoterapia. 2010;81(8):1224–7.
Guo H, Liu C, Yang L, Dong L, Wang L, Wang Q, et al. Morusin inhibits glioblastoma stem cell growth in vitro and in vivo through stemness attenuation, adipocyte transdifferentiation, and apoptosis induction. Mol Carcinog. 2016;55(1):77–89.
Chen C, Xu ZH, Wang L. [The effect of morusin on stemness phenotype of laryngeal cancer stem cell]. Sichuan Da Xue Xue Bao Yi Xue Ban. 2020;51(5):650–7.
Gong Q, Cao X, Cao J, Yang X, Zeng W. Casticin suppresses the carcinogenesis of small cell lung cancer H446 cells through activation of AMPK/FoxO3a signaling. Oncol Rep. 2018;40(3):1401–10.
Liu J, Yang J, Hou Y, Zhu Z, He J, Zhao H, et al. Casticin inhibits nasopharyngeal carcinoma growth by targeting phosphoinositide 3-kinase. Cancer Cell Int. 2019;19:348.
He G, Cao X, He M, Sheng X, Wu Y, Ai X. Casticin inhibits self-renewal of liver cancer stem cells from the MHCC97 cell line. Oncol Lett. 2014;7(6):2023–8.
Zhao D, Yao C, Chen X, Xia H, Zhang L, Liu H, et al. The fruits of Maclura pomifera extracts inhibits glioma stem-like cell growth and invasion. Neurochem Res. 2013;38(10):2105–13.
Liao WY, Liaw CC, Huang YC, Han HY, Hsu HW, Hwang SM, et al. Cyclohexylmethyl flavonoids suppress propagation of breast cancer stem cells via downregulation of NANOG. Evidence-based Complem Altern Med. 2013;2013:170261.
Peiffer DS, Ma E, Wyatt D, Albain KS, Osipo C. DAXX-inducing phytoestrogens inhibit ER+ tumor initiating cells and delay tumor development. NPJ Breast Cancer. 2020;6:37.
Kang Q, Gong J, Wang M, Wang Q, Chen F, Cheng KW. 6-C-(E-Phenylethenyl)naringenin attenuates the stemness of hepatocellular carcinoma cells by suppressing Wnt/β-catenin signaling. J Agric Food Chem. 2019;67(50):13939–47.
Song L, Chen X, Wang P, Gao S, Qu C, Liu L. Effects of baicalein on pancreatic cancer stem cells via modulation of sonic Hedgehog pathway. Acta Biochim Biophys Sin. 2018;50(6):586–96.
Wu R, Murali R, Kabe Y, French SW, Chiang YM, Liu S, et al. Baicalein targets GTPase-mediated autophagy to eliminate liver tumor-initiating stem cell-like cells resistant to mTORC1 inhibition. Hepatology (Baltimore, MD). 2018;68(5):1726–40.
Koh SY, Moon JY, Unno T, Cho SK. Baicalein suppresses stem cell-like characteristics in radio- and chemoresistant MDA-MB-231 human breast cancer cells through up-regulation of IFIT2. Nutrients. 2019;11(3):624.
Lin MG, Liu LP, Li CY, Zhang M, Chen Y, Qin J, et al. Scutellaria extract decreases the proportion of side population cells in a myeloma cell line by down-regulating the expression of ABCG2 protein. Asian Pac J Cancer Prev. 2013;14(12):7179–86.
Cao D, Zhu GY, Lu Y, Yang A, Chen D, Huang HJ, et al. Luteolin suppresses epithelial-mesenchymal transition and migration of triple-negative breast cancer cells by inhibiting YAP/TAZ activity. Biomed Pharmacother. 2020;129:110462.
Cai X, Ye T, Liu C, Lu W, Lu M, Zhang J, et al. Luteolin induced G2 phase cell cycle arrest and apoptosis on non-small cell lung cancer cells. Toxicol In Vitro. 2011;25(7):1385–91.
Dia VP, Pangloli P. Epithelial-to-mesenchymal transition in paclitaxel-resistant ovarian cancer cells is downregulated by luteolin. J Cell Physiol. 2017;232(2):391–401.
Choi EM, Lee YS. Luteolin suppresses IL-1beta-induced cytokines and MMPs production via p38 MAPK, JNK, NF-kappaB and AP-1 activation in human synovial sarcoma cell line, SW982. Food Chem Toxicol. 2010;48(10):2607–11.
Chian S, Thapa R, Chi Z, Wang XJ, Tang X. Luteolin inhibits the Nrf2 signaling pathway and tumor growth in vivo. Biochem Biophys Res Commun. 2014;447(4):602–8.
Rao PS, Satelli A, Moridani M, Jenkins M, Rao US. Luteolin induces apoptosis in multidrug resistant cancer cells without affecting the drug transporter function: involvement of cell line-specific apoptotic mechanisms. Int J Cancer. 2012;130(11):2703–14.
Davies AH, Reipas K, Hu K, Berns R, Firmino N, Stratford AL, et al. Inhibition of RSK with the novel small-molecule inhibitor LJI308 overcomes chemoresistance by eliminating cancer stem cells. Oncotarget. 2015;6(24):20570–7.
Tu DG, Lin WT, Yu CC, Lee SS, Peng CY, Lin T, et al. Chemotherapeutic effects of luteolin on radio-sensitivity enhancement and interleukin-6/signal transducer and activator of transcription 3 signaling repression of oral cancer stem cells. J Formos Med Assoc. 2016;115(12):1032–8.
Huynh DL, Kwon T, Zhang JJ, Sharma N, Gera M, Ghosh M, et al. Wogonin suppresses stem cell-like traits of CD133 positive osteosarcoma cell via inhibiting matrix metallopeptidase-9 expression. BMC Complement Altern Med. 2017;17(1):304.
Huynh DL, Sharma N, Kumar Singh A, Singh Sodhi S, Zhang JJ, Mongre RK, et al. Anti-tumor activity of wogonin, an extract from Scutellaria baicalensis, through regulating different signaling pathways. Chin J Nat Med. 2017;15(1):15–40.
Koh H, Sun HN, Xing Z, Liu R, Chandimali N, Kwon T, et al. Wogonin influences osteosarcoma stem cell stemness through ROS-dependent signaling. In Vivo (Athens, Greece). 2020;34(3):1077–84.
Wang X, Chang Y, Gao M, Zhang F. Wogonoside attenuates cutaneous squamous cell carcinoma by reducing epithelial-mesenchymal transition/invasion and cancer stem-like cell property. OncoTargets Ther. 2020;13:10097–109.
Soltanian S, Riahirad H, Pabarja A, Karimzadeh MR, Saeidi KJB. Kaempferol and docetaxel diminish side population and down-regulate some cancer stem cell markers in breast cancer cell line MCF-7. Biocell. 2017;41(2&3):33.
Khaw AK, Yong JW, Kalthur G, Hande MP. Genistein induces growth arrest and suppresses telomerase activity in brain tumor cells. Genes Chromosomes Cancer. 2012;51(10):961–74.
Ozturk SA, Alp E, Yar Saglam AS, Konac E, Menevse ES. The effects of thymoquinone and genistein treatment on telomerase activity, apoptosis, angiogenesis, and survival in thyroid cancer cell lines. J Cancer Res Ther. 2018;14(2):328–34.
Selvakumar P, Badgeley A, Murphy P, Anwar H, Sharma U, Lawrence K, et al. Flavonoids and other polyphenols act as epigenetic modifiers in breast cancer. Nutrients. 2020;12(3):761.
Izzo S, Naponelli V, Bettuzzi S. Flavonoids as epigenetic modulators for prostate cancer prevention. Nutrients. 2020;12(4):1010.
Zhang Z, Zhou L, Xie N, Nice EC, Zhang T, Cui Y, et al. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct Targeted Ther. 2020;5(1):113.
Jefferson WN, Williams CJ. Circulating levels of genistein in the neonate, apart from dose and route, predict future adverse female reproductive outcomes. Reprod Toxicol (Elmsford, NY). 2011;31(3):272–9.
Motlekar N, Khan MA, Youan BBC. Preparation and characterization of genistein containing poly (ethylene glycol) microparticles. J Appl Plym Sci. 2006;101(3):2070–8.
Nwaeburu CC, Abukiwan A, Zhao Z, Herr I. Quercetin-induced miR-200b-3p regulates the mode of self-renewing divisions in pancreatic cancer. Mol Cancer. 2017;16(1):23.
Cao L, Yang Y, Ye Z, Lin B, Zeng J, Li C, et al. Quercetin-3-methyl ether suppresses human breast cancer stem cell formation by inhibiting the Notch1 and PI3K/Akt signaling pathways. Int J Mol Med. 2018;42(3):1625–36.
Shen X, Si Y, Wang Z, Wang J, Guo Y, Zhang X. Quercetin inhibits the growth of human gastric cancer stem cells by inducing mitochondrial-dependent apoptosis through the inhibition of PI3K/Akt signaling. Int J Mol Med. 2016;38(2):619–26.
Hoca M, Becer E, Kabadayı H, Yücecan S, Vatansever HS. The effect of resveratrol and quercetin on epithelial-mesenchymal transition in pancreatic cancer stem cell. Nutr Cancer. 2020;72(7):1231–42.
Wang R, Yang L, Li S, Ye D, Yang L, Liu Q, et al. Quercetin inhibits breast cancer stem cells via downregulation of aldehyde dehydrogenase 1A1 (ALDH1A1), chemokine receptor type 4 (CXCR4), Mucin 1 (MUC1), and epithelial cell adhesion molecule (EpCAM). Med Sci Monit. 2018;24:412–20.
Erdogan S, Turkekul K, Dibirdik I, Doganlar O, Doganlar ZB, Bilir A, et al. Midkine downregulation increases the efficacy of quercetin on prostate cancer stem cell survival and migration through PI3K/AKT and MAPK/ERK pathway. Biomed Pharmacother. 2018;107:793–805.
Appari M, Babu KR, Kaczorowski A, Gross W, Herr I. Sulforaphane, quercetin and catechins complement each other in elimination of advanced pancreatic cancer by miR-let-7 induction and K-ras inhibition. Int J Oncol. 2014;45(4):1391–400.
Li Y, Wang Z, Jin J, Zhu SX, He GQ, Li SH, et al. Quercetin pretreatment enhances the radiosensitivity of colon cancer cells by targeting Notch-1 pathway. Biochem Biophys Res Commun. 2020;523(4):947–53.
Srivastava RK, Tang SN, Zhu W, Meeker D, Shankar S. Sulforaphane synergizes with quercetin to inhibit self-renewal capacity of pancreatic cancer stem cells. Front Biosci (Elite Edition). 2011;3:515–28.
Kim B, Jung N, Lee S, Sohng JK, Jung HJ. Apigenin inhibits cancer stem cell-like phenotypes in human glioblastoma cells via suppression of c-Met signaling. Phytother Res. 2016;30(11):1833–40.
Li YW, Xu J, Zhu GY, Huang ZJ, Lu Y, Li XQ, et al. Apigenin suppresses the stem cell-like properties of triple-negative breast cancer cells by inhibiting YAP/TAZ activity. Cell Death Dis. 2018;4:105.
Liu F, Cao X, Liu Z, Guo H, Ren K, Quan M, et al. Casticin suppresses self-renewal and invasion of lung cancer stem-like cells from A549 cells through down-regulation of pAkt. Acta Biochim Biophys Sin. 2014;46(1):15–21.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive licence to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Siddiqui, N., Abidin, L., Nisar, N., Ahmad, I., Siddiqui, A.N. (2021). Flavonoids Targeting Cancer Stem Cells: A Paradigm to Anticancer Efficacy. In: Tabrez, S., Imran Khan, M. (eds) Polyphenols-based Nanotherapeutics for Cancer Management. Springer, Singapore. https://doi.org/10.1007/978-981-16-4935-6_7
Download citation
DOI: https://doi.org/10.1007/978-981-16-4935-6_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-4934-9
Online ISBN: 978-981-16-4935-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)