
Abstract
Cancer stem cells (CSCs) are a subpopulation of tumour cells that possess the stem cell properties of self-renewal and differentiation. Stem cells might be the target cells responsible for malignant transformation, and tumour formation may be a disorder of stem cell self-renewal pathway. Epigenetic alterations and mutations of genes involved in signal transmissions may promote the formation of CSCs. These cells have been identified in many solid tumours including breast, brain, lung, prostate, testis, ovary, colon, skin, liver, and also in acute myeloid leukaemia. The CSC theory clarifies not only the issue of tumour initiation, development, metastasis and relapse, but also the ineffectiveness of conventional cancer therapies. Treatments directed against the bulk of the cancer cells may produce striking responses but they are unlikely to result in long-term remissions if the rare CSCs are not targeted. In this review, we consider the properties of CSCs and possible strategies for controlling the viability and tumourigenecity of these cells, including therapeutic models for selective elimination of CSCs and induction of their proper differentiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The traditional model of cancer development suggests that tumours arise from a series of sequential mutations resulting from genetic instability and/or environmental factors affecting normal cells. According to this model, many cancer cells have been considered to have tumourigenic potential. A major hurdle for the traditional model of cancer development is the prolonged period required to develop the first mutation that subsequently leads to malignant tumour formation. In many tissues in which tumours arise (e.g. gastrointestinal tract, epithelium, skin and blood), mature cells have a short lifespan and a limited opportunity to accumulate the multiple mutations required for tumour development [1, 2]. Therefore, the probability of an individual cell accumulating the necessary mutations is small.
An alternative explanation that has been proposed is the stem cell model of tumour formation. According to this model, tumours originate in either long-lived tissue stem cells or progenitor cells through misregulation of the normally tightly regulated process of self-renewal, leading to cancer stem cells (CSCs) [1, 3, 4]. Other terminologies for CSCs are tumour-initiating cells (TICs) and tumourigenic cancer cells. A hypothesis of CSCs stating that they have similar properties to stem cells was first explained by Rudolf Virchow and Julius Conheim in the nineteenth century. Virchow’s embryonal-rest hypothesis states that cancers arise from activation of dormant cells (which are remainders of embryonic cells) present in adult tissues. This hypothesis was based on the fact that there are some similarities between developing embryonic cells and some cancer cells including their ability to proliferate and differentiate [5, 6].
According to cancer stem cell model and based on experiments in which human cancer cells were xenotransplanted into non-obese diabetic/severe-combined immunodeficient (NOD/SCID) mice (that have a defect in T cells, B cells, natural killer cells, and complement), only small subpopulations of cancer cells retained the capacity to initiate, maintain and promote the development of the tumours [7–17]. Although xenotransplantation animal models have improved the possibility to study the cancer stem cell hypothesis, the critical role of tumour growth and its interactions with the local and extended microenvironment complicates the interpretation of such studies. The low frequency of tumour-producing cells from, for example, human acute myeloid leukaemia (AML) in NOD/SCID mice might be explained by the fact that rare tumour cells have been able to adapt to a foreign environment and continue to grow [18]. Modifications to the xenotransplantation assay have revealed that many more human cells have tumourigenic potential compared to the results obtained in NOD/SCID mice. For example, Quintana et al., showed that melanoma-initiating cells are rare in NOD/SCID mice, but more melanoma-initiating cells could be detected in NOD/SCID mice lacking the interleukin-2 gamma receptor (NSG, NOD scid gamma) owing in part to the lack of natural-killer cell activity in NSG mice. Therefore, more tumourigenic cells could be detected in NSG mice, which are more immunodeficient than NOD/SCID mice [19]. Additional modifications to xenotransplantation assays such as co-injection of tumour cells with Matrigel (gel medium containing growth factors and nutrients that could reinforce the tumour cells’ vitality) and increasing the length of observation for tumour formation show that frequency of TICs increases dramatically via more permissive xenotransplantation [13, 19]. These data indicate that some cancers, which appear to have rare tumourigenic cells in NOD/SCID mice, actually have more cells with tumourigenic capacity under other conditions, raising the possibility that the true frequency of CSCs has been greatly underestimated in most human tumours. Nevertheless, other cancers may still have infrequent tumourigenic cells, even when studied under optimised conditions. For example, Ishizawa et al. compared the growth of human pancreatic adenocarcinoma, lung adenocarcinoma, squamous cell lung carcinoma and head/neck squamous cell carcinoma cells in NOD/SCID and NSG mice. They showed that TICs frequency did not differ significantly for pancreatic carcinoma or head/neck squamous cell carcinoma, when measured in either strain of mice. Although CSCs frequency was up to tenfold higher in NSG mice for some tumours (lung adenocarcinoma and squamous cell lung carcinoma) but it remained relatively low in all cases [20]. In conclusion, whether cells with tumourigenic potential are common or rare within human cancers still remains a fundamental question in cancer biology.
The existence of cancerous stem cells was first demonstrated in AML, that can be viewed as an aberrant hematopoietic tissue initiated by malignant leukaemic stem cells [21]; however, these cells were also observed in solid tumours such as breast [8], brain [9, 22], lung [23], prostate [24, 25], ovary [26], colon [27, 28], skin [29, 30], liver [14] and pancreas cancers [11]. The hypothesis that stem cells are origins of cancer is also observed in teratocarcinomas, which are a subtype of germ cell tumours. Teratocarcinomas are malignant tumours, which contain derivatives of all three germ layers and embryonal carcinoma (EC) cells. EC cells are the ‘pluripotent’ stem cells in these cancers with the ability of self-renewal as well as differentiation into different cell types. Therefore, EC cells can reconstruct the whole cancer including its differentiated cells [31]. Based on these properties, EC cells are the first experimental demonstration of a CSC, predating the current intense interest in CSCs by several decades.
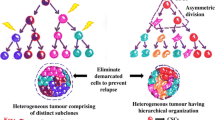
It has been hypothesised that CSCs persist in tumours as a distinct population and may cause relapse and metastasis, giving rise to new tumours. These cells may also explain why standard oncology treatments sometimes fail. Thus, development of therapeutic methods that can target CSCs holds promise for the improvement of survival and life quality of cancer patients, especially those with clinical metastasis. Different topics have been considered in this review as follows. “Characteristics of CSCs” section explains essential properties of CSCs and similarities between CSCs and normal stem cells. Hypotheses about origin of CSCs are described in “Origin of CSCs” section. “Genetic mutations and altered signalling pathways in CSCs” section describes mutations and altered pathways that convert a stem cell to a CSC. “Markers of CSCs” section covers markers of CSCs in hematopoietic system and solid tumours and finally “CSCs and perspectives in cancer therapy” section is devoted to cancer therapy by targeting CSCs.
Characteristics of CSCs
CSCs and normal stem cells (adult stem cells) have many similar properties, which may operate through similar molecular pathways, albeit aberrantly in CSCs. CSCs undergo self-renewal; this is an asymmetric division in which one copy has developmental potentials similar to that of the CSC [32] and another copy (progenitor cell) is a transit-amplifying cell, which terminally differentiates, similar to what happens in normal tissue renewal. However, the major difference between cancer growth and normal tissue renewal is that whereas normal transit amplifying cells usually differentiate and die, at various levels of differentiation, the cancer transit-amplifying cells fail to differentiate normally and instead accumulate (i.e. they undergo maturation arrest), resulting in cancer growth [21, 33]. Therefore, although pathways that regulate self-renewal are tightly controlled in normal stem cells, they might be constitutively activated or incorrectly regulated through genetic and/or epigenetic changes in CSCs, leading to uncontrolled growth [34, 35]. Furthermore, EC cells, which can be considered as an archetype of CSCs, in contrast to embryonic stem cells (ESCs), often have a limited capacity for differentiation. Therefore, even a small increase in the probability of self-renewal compared with differentiation could have a significant selective advantage during tumour progression [31].
In addition to the ability of self-renewal and differentiation, there are other similarities between normal and cancer stem cells which will be discussed. Current opinion is that CSCs are either dormant or in a proliferative phase. Dormant CSCs, like normal stem cells, exist in a quiescent state; therefore, they might be more resistant to the cytostatics that target dividing cells [36], whereas CSC proliferation would give rise to tumour mass/bulk [37]. For example, in AML at least a part of the leukaemia stem cells (LSCs)/leukaemia initiating cells (LICs) population is in the G0 phase of the cell cycle and these cells, like normal stem cells, are resistant to in vitro treatment with 5-fluorouracil, which destroys actively dividing cells [38]. Moreover, both types of stem cells highly express telomerase and have long lifespan (infinite replication potential) [36]. Generally, telomere shortening and activation of telomerase appear to have a dual role in tumour formation. On one hand, telomere shortening is associated with the development of chromosomal instability and initiation of cancer. On the other hand, telomerase activation is necessary for telomere stabilisation and prevention of chromosomal instability that finally leads to tumuor progression [39, 40].
Expression of OCT4 is another similarity between normal and cancer stem cells. OCT4, a member of the family of POU-domain transcription factors, is expressed in pluripotent embryonic stem and germ cells [41, 42]. Oct4 mRNA is normally found in totipotent and pluripotent stem cells of pregastrulation embryos [43]. Indeed, OCT4 expression is required to maintain the undifferentiated state of human EC and ES cells and we have shown that differentiation to trophectoderm occurs in its absence [44]. The expression of OCT4 has further been shown in human breast cancer stem-like cells, suggesting that its expression may be implicated in self-renewal and tumourigenesis via activating its downstream target genes [45]. One study on lung cancer showed that OCT4 expression plays a crucial role in maintaining self-renewal and cancer stem-like properties in CD133+ cells (CSCs in lung tumour cells), and also up-regulated expression of OCT4 in CSCs may contribute to the development of chemoradioresistance in patients with lung cancer [46]. In another study, Schoenhals et al. [47] showed that at least one of the genes coding for pluripotency factors OCT4, SOX2, KIF4, and c-MYC is overexpressed in many cancer types and expression of these genes is associated with tumour progression or bad prognosis. In fact, activation of an ES cell-like transcriptional programme in differentiated adult cells may induce pathological self-renewal characteristics of CSCs.
Transporters of multidrug resistance (MDR), which are energy-requiring efflux pumps with the function of pumping toxic chemotherapeutic drugs out of the cancer cells, are responsible for the tumour resistance to many drugs currently used for cancer therapy [48]. One property of the normal tissue stem cells and CSCs is the self-protection ability through innate MDR transporters [36, 49]. CSCs also have some properties that cause resistance to apoptosis and radiotherapy [50–53]. Finally, CSCs are able to initiate a tumour if they were implanted into immunodeficient mice models [7, 21]. Properties of normal and cancer stem cells are summarised in Table 1.
Origin of CSCs
The origin of CSCs is not completely identified yet, but there are some hypotheses about it. Figure 1 demonstrates various possible scenarios in which CSCs can originate in the tissues. Most adult tissues are maintained over the lifetime of the host by normal stem cells that undergo expansion and differentiation to provide the functional elements of the organ [54]. The genetic constrains on self-renewal limit the expansion of stem cells in normal tissues. Breakdown in this regulation is likely a key event in the development of cancer as demonstrated by the fact that several pathways involved in carcinogenesis also play a role in normal stem cell self-renewal decisions [55]. There is also a striking association between misregulation of stem cells function and carcinogenesis; many genes that promote self-renewal are also oncogenes and many genes that inhibit self-renewal are also tumour-suppressor genes [56]. Therefore, the first probability is that cancers originate from adult stem cells. This idea is supported by some other observations in biology: (1) these cells have been shown to exist in many tissues from which cancers often spread such as blood, brain, lung and prostate. (2) Multiple mutations are required for a cell to become cancerous, and stem cells can live longer than other cells, so they are better candidates for obtaining multiple mutations and becoming cancerous [57, 58]. (3) Tumour characteristics of monoclonality, unlimited proliferative capacity and phenotypic heterogeneity that includes a variety of differentiation states with some non-dividing cells, could be elucidated by tumours originating from a self-renewing, multipotent and slow-cycling cell. (4) Normal stem cells and cancer cells share many properties including induction of angiogenesis, resistance to apoptosis, cell migration and acquired drug resistance, implying the need for presence of stem cell-like cancer cells in initiation, recurrence and metastasis. (5) Since in stem cells, the machinery for self-renewal is already activated, preserving this activation may be simpler than turning it on de novo in a more differentiated cell [34, 57].

Various possible origins for cancer stem cells. 1 CSCs may originate from adult stem cells, which exist in many tissues. 2 CSCs might be generated from a population of more differentiated transit amplifying/progenitor cells. 3 Another possible origin is represented by embryonic stem cell-like cells that are abnormally left in the tissues during ontogenesis. 4 Finally, tumour-initiating mutations in terminally differentiated cells may produce CSCs
The second possibility is that CSCs can originate from a population of more differentiated transit-amplifying/progenitor cells [35]. Progenitors multiply for a much shorter period of time before terminal differentiation. Thus, progenitors would first need to acquire the extensive self-renewal capacity of stem cells in order to have the opportunity to accumulate additional mutations that would lead to their transformation [55]. Several strong lines of evidences support the concept that a committed progenitor can be the cancer-initiating cell as a result of oncogenic transformation. For example, committed myeloid progenitors with co-expression of Bcl-2 and Bcr/Abl protein (the fusion protein found in 90% of patients with chronic myelogenous leukaemia) are enough to drive leukaemia development in mice [59]. Studies of brain tumour development also indicate that some of the more committed neural progenitor cells are likely to arise from tumourigenic mutations. These mutated progenitor cells acquire the features believed to be specific to stem cells and undergo unlimited growth like cancer cells [60]. Results of a recent research by Molyneux et al. indicate that majority of human BRCA1-associated and sporadic basal-like tumours are derived from luminal progenitors rather than from basal stem cells. As a result, normal mammary gland stem cells are not common targets for transformation in breast but luminal progenitors are probably the cell type most commonly associated with the initiation of breast cancer [61]. Similarly, Persson et al. investigated and compared neural stem cells (NSCs) and oligodendrocyte progenitor cells (OPCs) as potential cells of origin in murine and human oligodendroglioma brain tumours. Their results indicated that in malignant oligodendroglial brain tumours, oligodendroglioma cells show hallmarks of OPCs, and that a progenitor rather than a neural stem cell is responsible for tumour formation [62]. The identity of the normal cell from which leukaemia originates has been the source of much debate. The similarity of AML initiating cells and normal repopulating cells (i.e. CD34+CD38−) led some authors to hypothesise that AML is derived from hematopoietic stem cells (HSCs). This theory provides an attractive model for leukaemogenesis since the long lifespan of the HSCs allows for multiple genetic hits to happen. Additionally, based on their physiologic ability for self-renewal, HSCs would require fewer genetic hits to become LSCs compared to other hematopoietic cells, which must aberrantly obtain self-renewal capacity [21]. Nevertheless, others argue that the features of myeloid differentiation that define AML, point to a progenitor origin. In support of this, it has been demonstrated that transient repopulating progenitors can initiate myeloid leukaemias in response to a mixed-lineage leukaemia (MLL) oncogene [63]. Therefore, leukaemic mutations may confer the self-renewal capacity to a mutant early progenitor cell.
The third possible origin is that some cancers such as teratocarcinomas or some of the paediatric sarcomas (e.g. nephroblastoma) may develop from very early ES cell-like cells that are abnormally left in the tissues during ontogenesis [64].
Finally, another probable origin of CSCs is that tumour-initiating mutations may occur in mature progenitor or even terminally differentiated cells. These cells subsequently become unstable with respect to their phenotype and may dedifferentiate and produce CSCs in certain conditions [65]. Whatever the origin of CSCs, transformation of original cells to CSCs needs genetic modifications and epigenetic alterations. In the following section, some genetic changes and altered signalling pathways that can convert a normal stem cell to CSC are discussed.
Genetic mutations and altered signalling pathways in CSCs
Understanding the genetic alterations that convert a stem cell to a CSC is crucial for developing effective anti-cancer therapies. In fact, many types of genetic variations that might lead to malignant cell proliferation have been identified in various types of tumours [66]. Two main classes of genes are involved in the process of carcinogenesis: activated proto-oncogenes and inactivated tumour suppressor genes [67]. Both types of genes are required for normal cell division and differentiation, and their aberrant expression results in abnormal cell proliferation [66]. The transformation of a normal stem cell into a CSC is also due to the accumulation of genetic modifications (mutations in oncogenes, tumour suppressor genes and mismatch repair genes) and epigenetic alterations (abnormal methylation, histone modifications). For example, loss of the Pten tumour suppressor (which is a phosphatase that negatively regulates signalling through the PI-3 kinase pathway, attenuating proliferation and survival signals) and subsequent up regulation of β-catenin activity are thought to be critical steps in transformation of stem cells to CSCs in some carcinomas [68, 69]. Bmi-1 protein also plays a critical role in regulating the self-renewal process of stem cells and CSCs [70]. In normal conditions, Bmi-1 inhibits the transcription of the Ink4a locus that encodes two cyclin-dependent kinase inhibitors: p16Ink4a and p14Ink4a [71]. A lack of p16 inhibitor, accompanied by abnormal Bmi-1 function, stimulates cell proliferation by increasing its self-renewal potential, whereas a lack of the p14 inhibitor prevents proapoptotic gene expression [72]. Advances in stem cell research have identified some key signalling pathways, which might be abnormal in CSCs and thus would be candidate targets for future cancer therapies (Fig. 2) [32]. Some of these pathways including Wnt, Hedgehog and Notch are explained in this section.

Signalling pathways that regulate self-renewal during development of normal stem cells and cancer transformation. Activation of the hedgehog (Hh) signalling pathway is started by binding of an Hh ligand to protein patched homologue (PTCH). This leads to suppression of Smoothened homologue (Smo), activating a cascade that leads to the translocation of glioma-associated oncogene homologue (Gli) into the nucleus and the activation of target genes. The Wnt signalling pathway is activated by the binding of Wnt ligands to their receptors Frizzled (Fzd) and low-density lipoprotein receptor-related protein 5 (LRP5) and LRP6, leading to the release of β-catenin (β-cat) from the ‘multifactorial cytoplasmic complex’, which is composed of adenomatous polyposis coli protein (APC), axis inhibition protein (axin), glycogen synthase kinase 3β (GSK3β) and casein kinase 1α (CK1α). Then β-catenin (β-cat) migrates inside the nucleus and, interaction of β-cat with transcription factor TCF–LEF (lymphoid enhancer binding factor) activates transcription of the indicated target genes. The core Notch pathway is activated by interaction between the Notch receptor on one cell with the Notch ligand (delta-like or jagged) on another cell, resulting in two proteolytic cleavages of the receptor. This mediates the release of the Notch intracellular domain, which goes into the nucleus and interacts with the CBF1 transcription factor
Wnt pathway
Wnt proteins are a large family of secreted glycoproteins that bind to Frizzled receptors and LRP5/6 coreceptors. By stabilising the mediator β-catenin, they start a complex signalling cascade that plays a significant role in regulating cell proliferation and differentiation. Wnt cascade has appeared as a critical regulator of stem cells self-renewal [73]. Misregulation of Wnt/β-catenin signalling pathway mainly by inactivating mutations of APC tumour suppressor or oncogenic mutations of β-catenin, in cancer cells or most likely CSCs leads to malignant proliferation [74, 75]. For example, β-catenin which is largely dispensable for normal function of HSCs, is frequently activated in MLL. Therefore, activation of the Wnt/β-catenin pathway plays critical roles for establishment and drug-resistant properties of MLL stem cells [76].
Hedgehog pathway
Hedgehog (Hh) genes encode secreted proteins, which signal through autocrine and paracrine mechanisms to regulate cell proliferation, differentiation, and morphology [77]. The Hh proteins exert their function by binding to a 12-pass transmembrane protein called patched (Ptch) [78]. Hh proteins relieve the inhibitory effect of Ptch on a serpentine protein called Smoothened (Smo), leading to hyper-phosphorylation of Smo [79, 80]. The signal pathway induces activation and repression of target genes through the Gli family of transcription factors, Gli-1, 2 and 3, which regulate the transcription of target genes. There are three hedgehog family members namely: Desert, Indian, and Sonic [77]. Hh pathway plays a central role in the proliferative control and differentiation of both ESCs and adult stem cells. Therefore, an alteration in the Hh pathway, either by misexpression of components of that pathway or by changes in the expression of other cellular components that interfere with the Hh signalling system, may trigger the development of several types of cancers [81, 82].
Notch pathway
Notch signalling is important for cell–cell communications and regulates a broad spectrum of cell fate decisions during embryonic development and in the adult organism via stem cell proliferation, differentiation, and cell death [83, 84]. Notch proteins (Notch1–4) are members of the conserved transmembrane receptor family. The Notch genes encode transmembrane receptors, which include a large extracellular domain, composed of a variable number of epidermal growth factor-like repeats and an intracellular signalling domain, which consists of six ankyrin/cdc10 motifs and nuclear localization signals [83]. Notch receptors interact through their extracellular domain with other membrane-associated ligands, Delta and Serrate/Jagged families, including, Jagged 1 and 2, and Delta-like 1, 3, and 4 [83]. Notch signalling is activated by ligand–receptor interactions and gives rise to proteolytic cleavage by the gamma-secretase complex, which releases the Notch intracellular domain into the nucleus. The Notch intracellular domain binds to the CBF1 DNA binding protein of the transcriptional activator complex, the activation of which can cause the expression of target genes, such as Hes family. These are involved in cell growth and differentiation [37, 85]. NOTCH pathway provides a cell–cell proliferation signal in human ES cells and their malignant counterparts, EC cells and also plays a crucial role in the maintenance of undifferentiated human EC and ES cells [86]. A number of studies with gene modifications in animal models have proved the role of Notch signalling pathway in stem cells and early progenitor cells [87]. Notch signalling has also been shown to be involved in tumours of various origins such as glioblastoma multiforme and B-cell lymphomas [88–91].
Markers of CSCs
The most critical issue in the field of CSCs is to develop phenotypic assays that can be used to reliably identify CSCs [66]. The most widely used method to identify these cells is through their expression of special cell surface markers. Some of these markers are shared by CSCs from malignant tissues and corresponding normal tissue stem cells [66]. There are two possible explanations for this overlap. The first explanation is that many CSCs originate directly from normal stem cells thus, the surface markers expressed by CSCs could also be found on normal stem cells [2, 32]. The second explanation is that some CSCs are transformed from committed cells or precursors that are similar to normal stem cells in their expression of cell surface markers [10]. With the development of advanced assays to identify gene expression profile of CSCs, it will be possible to determine surface markers that are more unique to CSCs and therefore allow the development of more target-specific cancer therapies [66].
CSCs in hematopoietic system
Immunodeficient mice are increasingly used to assay human hematopoietic stem and progenitor cells as well as LSCs. One method commonly used to isolate these rare cells is to sort cells stained with fluorochrome-conjugated antibodies into fractions via fluorescent-activated cell sorting (FACS), then transplant different fractions into immunodeficient mice to test their repopulating ability. Distinct cell surface markers profiles that would allow for prospective isolation of normal mouse HSCs by FACS became known in the late 1980s and early 1990s [92]. Experiments in humans also indicated that normal human HSCs are enriched in the CD34+CD38− fraction [93]. Using NOD/SCID mice strains with enhanced immunosuppression as recipients, it was shown that the CD34+/CD38+ cell fraction also possesses some repopulating activity [94]. However, CD34+/CD38+ cells possess only a short-term SCID-repopulating activity, while the long-term repopulating activity is limited to the CD34+/CD38− cell population [94]. It is important to mention that several studies have characterised a rare SCID-repopulating fraction observed at the level of CD34−Lin− cells. These cells, similar to CD34+/CD38− cells, have a long-term repopulating capacity [95, 96].
In addition to HSC markers, investigators have recognised cell surface markers in LSCs. Dick et al. provided the first evidence for existence of LSCs by using FACS to separate cells from human AML that were able to initiate leukaemia in transplanted NOD/SCID mice. They indicated that the CD34+/CD38− fraction was highly enriched for leukaemia-initiating activity in transplanted recipients, while both the CD34+/CD38+ and CD34− fractions did not initiate leukaemia [7, 21]. Moreover, an engrafted leukaemia could be serially transplanted into secondary recipients providing functional evidence for self-renewal [7, 21]. However, some recent studies have shown that LSCs are present also in the CD34+/CD38+ fraction. In fact, it was shown that, in a significant proportion of AMLs, cells contained in the CD34+/CD38+ fraction are capable of initiating and maintaining the leukaemic process when grafted in to NOD/SCID mice [97]. The discrepancy between these observations and previous studies relies in the observation that the anti-CD38 monoclonal antibody used for cell fractionation studies has a marked negative effect on the engraftment of AML repopulating cells in NOD/SCID mice [97]. Very recently, it was shown that, LICs are observed within the CD34- fraction in a significant proportion of AMLs [98]. These studies were based on the analysis of LICs in a group of patients bearing nucleophosmin mutations using the most immunodeficient SCID mice available. These AMLs are classified as a separate entity and are characterised by a low CD34 expression. In half of these AMLs, the CD34− fraction contained LICs, while the CD34+ fraction gave rise to normal multilineage hematopoiesis. In the remaining half of the patients, LICs are observed among both CD34+ and CD34− AML cells [98]. These observations further reinforce the concept that the membrane phenotype of LICs is heterogeneous in various AMLs.
Solid tumours CSCs
A number of markers for different types of solid tumours have been recognised so far. CD44 and CD133 have been reported as cell surface CSC markers in several solid tumour types [99]. For example, CD133 has been considered as an important marker representing the subpopulation of CSCs in prostate carcinoma, hepatocellular carcinoma and lung cancer [100, 101]. It was also shown that CD133 is a temporary marker of CSCs in small cell lung cancer, but not in non-small cell lung cancer [102]. CD44+ cells in head/neck squamous cell carcinoma and colon cancer were enriched for tumourigenic CSCs able to proliferate and produce tumours in mice, whereas CD44− cells were not able to do so [12, 103]. In pancreatic cancer, CD44+/CD24+/ESA+ cells (named pancreatic CSCs) were also forming tumour, whereas CD44− CD24− ESA− cells did not have this ability [11].
Some CSCs have been identified in different and sometimes non-overlapping subpopulations such as human breast and ovarian CSCs. Research showed that CD44/CD24 and also CD133 can be regarded as CSCs markers in ovarian carcinoma [104, 105]. In human breast carcinoma, Al-Hajj et al. separated tumourigenic cells using FACS [8]. These cells were fractionated based on their expression of CD44 and CD24, the two markers were shown to be heterogeneously expressed among tumour cells. Fractionated samples were implanted into the mammary pads of SCID mice and it was shown that only the cells expressing a CD44+/CD24− profile in human breast cancer could form tumours, whereas 100-fold more cells from the CD44+/CD24+ or CD24− fractions did not produce tumours [8, 106]. Engrafted tumours exhibited similar morphology and immunophenotypic heterogeneity to the original sample containing CD44+/CD24− cells as well as CD44+/CD24+ and CD24− cells. Finally, engrafted tumours could be serially transplanted, providing rigorous proof for self-renewal [8, 106]. The activity of aldehyde dehydrogenase isoform 1 (ALDH1) that can be assessed by the ALDEFLUOR assay has also been identified as a common functional marker of normal and malignant human breast and colonic stem cells [107, 108]. In breast carcinomas, high ALDH activity identifies the tumourigenic cell fraction, capable of self-renewal and generating tumours that recapitulate the heterogeneity of the parental tumour [107]. Indeed, CD24−/CD44+/ALDH+ cells are regarded as having the most prominent tumour-initiating activity in breast cancers. Furthermore, immunohistochemical analysis of ALDH1 using human breast cancer samples revealed that high expression of ALDH1 is significantly correlated with the shorter survival of breast cancer patients [107]. ALDH1+ cell population is also enriched in bladder TICs and associated with progression of bladder cancer [109]. Conversely, in ovarian tumour, Penumatsa et al. indicated ALDH1 expression was significantly reduced in malignant tumours compared to normal ovaries and benign tumours and ALDH1 did not appear to be co-expressed with the CSC markers CD44 and CD133. Thus, ALDH1 expression in the ovary does not appear to be similar to breast or colon cancer suggesting possible functional differences in these cancers [110].
Credibility of many determined CSC markers has been questioned and rejected by recent studies. Some studies have shown that CD133, previously thought to be a robust brain tumour stem cell marker [9, 22, 51], does not consistently distinguish tumourigenic from nontumourigenic cells and CD133- population can also be tumourigenic contrary to what was originally detected [111, 112]. The CD133 has also been used as a phenotypic marker of colon CSCs [27, 28], but also in this cancer type, both CD133-negative and -positive populations have been reported to induce tumour growth in vivo [113]. Moreover, both CD133+ and CD133− fractions from melanomas exhibited very high frequencies of tumourigenic cells and no phenotypic differences that can distinguish tumourigenic from non-tumourigenic melanoma cells have been identified [19].
In all of these studies, it must be remembered that specific cell surface markers can enrich for cells having CSC properties of self-renewal and differentiation. There might be markers in each respective system yet to be recognised that could help define each CSC population more precisely [114]. While the recognition of surface markers expressed by CSCs will be useful, this is clearly not sufficient to specify these cells in the absence of a test for self-renewal. As in normal stem cells, functional assays for CSCs need to be evaluated for their ability of both self-renewal and tumour propagation [66]. The assay that best suits these criteria is serial transplantation in animals, which although imperfect, is now regarded as the gold standard [66, 115].
CSCs and perspectives in cancer therapy
Conventional anti-cancer treatments (e.g. chemotherapy and radiation) can often transiently shrink tumours by targeting the tumour bulk, but these therapies fail to target and kill CSCs, leading to treatment failure, relapse and ultimately death. Some properties of CSCs make them difficult cells to kill: CSCs maintain the property of self-protection through the activity of multiple drug resistance transporters such as ABCB1 (P-glycoprotein) and/or ABCG2 (breast cancer resistance protein-1) [48, 116, 117]. Activation of these transporters that pump substrate drugs out of the cells, decreases the effective drug concentration within the cells and is responsible for their relative resistance to chemotherapy [118]. Furthermore, CSCs divide much more slowly, which allows them to escape from traditional radio- and chemotherapies that hit fast-multiplying cells [72]. CSCs also have some properties that lead to their resistance to apoptosis, radiotherapy and anticancer drugs: they show over expression of antiapoptotic proteins such as BCL-2 and survivin [50] and also a great ability for DNA repair [51, 52]. Many anticancer drugs, such as platinum compounds, alkylating agents and nitrosoureas cause direct damage to the structure of DNA, and resistance to these compounds results from activation of DNA repair systems [53]. CD133+ cancer-initiating neural stem cell population in glioblastoma multiforme (an aggressive brain tumour) is also resistant to gamma radiation through preferential activation of DNA double-strand break response machinery [119]. Moreover, recently it has been demonstrated that polycomb group protein BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery [120]. Bao et al. also reported that checkpoint proteins play an essential role in resistance of CSCs to radiotherapy and DNA damage. In response to DNA damage, the checkpoint proteins are activated and their expression is increased [51]. The study of CD133+ glioma stem cells that are known as CSCs indicated that the radioresistance of CD133+ CSCs can be reversed with a specific inhibitor of the Chk1 and Chk2 checkpoint kinases [51]. It is known that the Chk1 and Chk2 proteins play roles in the execution of checkpoint response to delay or arrest the cell cycle, which elicits the repair of the DNA damage [121, 122]. Furthermore, cell cycle restriction through the expression of cyclin-dependent kinase inhibitor 1A (CDKN1A; also known as p21) limits DNA damage and maintains the self-renewal of LICs [123]. These preliminary experiments highlight the potential of inhibiting DNA damage responses to overcome the resistance of TICs to therapy [124]. Moreover, expression of high levels of oxidative stress-responsive genes in CSCs could confer part of their ability to resist anticancer therapy [125, 126].
For efficient cancer treatment, one must be able to target exclusively the CSCs and not normal stem cells [66, 127]. Researches to distinguish CSCs from normal stem cells are being done at the level of identifying markers at the cell surface or discovering functional differences in signalling or structural proteins [128]. Therefore, by comparing gene expression profiles of CSCs, the bulk tumour cell population, normal stem cells and normal tissues, it may be possible to recognise therapeutic approaches that preferentially attack CSCs [129]. Therapies could be designed to target CSCs in order to induce the differentiation of these cells [130, 131] or eliminate CSCs by inhibiting the maintenance of stem cell state [66]. These two approaches are discussed in the following parts.
Differentiation therapy
One way to control the tumour progression is to treat cancer by inducing differentiation of CSCs. Differentiation therapy causes CSCs to differentiate and lose their self-renewal property [6, 127]. Some anticancer drugs/agents that can affect cancer cell differentiation will be discussed in this section [127]. Vitamin A and its analogue (retinoid) can reverse the malignant progression process through signal modulations mediated by nuclear retinoid receptors. All-trans retinoic acid leads to frequent remission of acute promyelocytic leukaemia by inducing promyelocyte differentiation [132]. In solid tumours, RA also increases differentiation and apoptosis, and reduces proliferation, invasiveness and metastasis [133]. For example, in glioma, efficacy of retinoic acid-induced differentiation to target the stem-like tumour cells has been demonstrated recently [131]. Some types of cancer cells are prevented from entering the differentiation pathway partly because of abnormal chromatin modification enzymes, which keep cancer cells in the cycling state. In this case, effective differentiation can be induced by agents that act directly or indirectly to convert abnormal chromatin modifying enzymes into normal ones, enabling cancer cells to undergo terminal differentiation [66]. For example, histone deacetylase regulates histone acetylation by catalysing the removal of acetyl groups on the N-terminal lysine residues of core nucleosomal histones. Regulation of the acetylation status of core histones is involved in the regulation of transcription activity of certain genes. Abnormal recruitment of histone deacetylase activity has been associated with the development of definite human cancers [134]. The histone deacetylase inhibitor, suberoylanilide hydroxamic acid, which was initially recognised as a differentiation inducer in cultured murine erythroleukaemia cells, has been used experimentally in cancer differentiation therapy [135]. Piccirillo et al. noticed a reduction of the number of CSCs which are initiating glioma development in culture after exposing them to bone morphogenetic proteins (BMPs). BMPs under normal situations induce differentiation of glioma CSCs to non-malignant cells [136].
Elimination therapy
Another way to intensify the efficacy of cancer therapy is to eliminate CSCs. This can be achieved using different approaches, which are explained in this section. Exposure of CSCs to sufficiently high levels of conventional cytotoxic agents, and the development of novel therapeutic agents that are targeted to CSCs can be used for this purpose [137]. For example, findings indicated that CSCs within breast cancer cell populations are resistant to paclitaxel, a commonly used breast cancer chemotherapeutic drug, but salinomycin selectively kills breast CSCs [138].
One of the main restrictions to overcome in cancer treatment is the resistance to chemotherapy through the activity of multiple drug resistance transporters. Understanding the anticancer drug transport properties of these transporters, as well as their physiological functions will lead to more effective therapeutics for oncology. Increased exposure can be obtained at the cellular level if transport-related resistance mechanisms can be overcome. Since the finding of P-glycoprotein in the early 1980s, most agents tested for the reversal of MDR in the clinic, have been aimed for inhibiting P-glycoprotein function. Some of the P-glycoprotein inhibitors include verapamil, quinine and cyclosporine [48]. Salinomycin, a polyether antibiotic acting as a highly selective potassium ionophore and widely used as an anticoccidial drug, was recently shown to act as a specific inhibitor of P-glycoprotein, and treatment of the MDR cell lines with salinomycin, restored a normal drug sensitivity of these cells [139]. Natural products also show great potential as anti-MDR agents. For example, polyphenol compounds are active agents in green tea. In Chinese hamster ovary cells, green tea-derived polyphenols, inhibited activities of P-glycoprotein transport [140]. Curcumin is the most potent polyphenol in turmeric, a spice widely used in Southeast Asian countries. Combinatorial treatment of curcumin with a variety of chemotherapeutic drugs increases the cellular accumulation of these agents, therefore effectively sensitising drug-resistant cells [141, 142]. Sesquiterpene coumarins have also been shown to enhance the cytotoxicity of chemotherapeutic agents. For example, mogoltacin, conferone and feselol, sesquiterpene coumarins from Ferula badrakema, can significantly enhance the cytotoxicity of vincristine in transitional cell carcinoma cells [143–145]. Other alternative approaches to target P-glycoprotein-mediated drug resistance could involve the development of agents to interfere with any one of the regulatory steps in expression of P-glycoprotein: transcription, mRNA turnover, translation, protein processing, and turnover [53, 146]. There are other inhibitors, which act on a larger range of ABC transporters. These include biricodar and GF-120918, which inhibit not only P-glycoprotein but also MRP1 and ABCG2, respectively [53, 147].
In addition to drug resistance, CSCs are expected to express, at high levels, genes involved in anti-apoptotic mechanisms. For example, NF-κB plays a critical role in anti-apoptotic responses, and carcinogenesis in LSCs. In particular, the constitutive activation of NF-κB was observed in AML cell populations enriched in leukaemic stem cells, but not in normal HSCs [148]. According to these observations, it seemed clear that NF-κB could be a potential therapeutic target for LSCs eradication. Indeed the pharmacological inhibition of NF-κB was effective in killing LSCs [149]. It was shown that use of NF-κB inhibitors in combination with classical chemotherapeutic agents is useful for the treatment of AMLs resistant to standard therapy [150].
In addition to protecting the host from invading pathogens, the immune system is accepted to protect the host from developing tumours. Patients with cancer usually produce circulating antibodies or cytotoxic T cells against tumour antigens, which in some instances may lead to regression of tumours [128]. Therefore, another way for elimination therapy is antigen-based therapy that targets different aspects of CSCs progress and growth. Antigen-based immune therapy, established on the immune system’s spontaneous response to cancer, can take the form of vaccines or monoclonal antibody therapy. Vaccines may sensitise an individual's immune system to resist potential malignancies [66]. One of the most critical elements in the success of a cancer vaccine is the choice of appropriate antigen target(s) that will allow for identification and elimination of tumour cells with minimum undesired autoimmune toxicity [128]. It is possible that analysis of gene expression in CSCs could identify tumour antigens whose expression is limited to CSCs. Such tumour antigens would be desirable targets in immunotherapy [66, 128]. For example, a study has shown that the adhesion receptor, CD44, has splice variants that are differentially expressed by CSCs and normal stem cells, making it a suitable new target for antibody-based therapy [151]. Through a similar approach, Dick et al. have proposed a new therapeutic strategy for the treatment of AML, based on the use of anti-CD44 Abs to selectively target leukaemic stem cells, while sparing the normal counterpart [152]. The interleukin-3 receptor alpha (IL-3Rα) chain (CD123) is reported to be overexpressed in AML cells [153] and in leukaemic stem cells, but not in normal HSCs [154, 155]. A neutralising monoclonal antibody against CD123 inhibited the IL-3-mediated survival of leukaemic stem cells in vitro as well as homing, engraftment, expansion, and serial transplantation of AML cells in immunodeficient mice, with lower effects on normal hematopoietic cells [156]. In human hepatocellular carcinoma, CD90+/CD44+ cells—a subpopulation of CSCs—indicated a phenotype more aggressive than that of CD90+/CD44– cells, characterised by formation of metastatic lesions in the lungs of immunodeficient mice. Systemic administration of a human CD44–-specific mAb at the time of subcutaneous CD90+/CD44+ carcinoma cell injection markedly inhibited tumour initiation and growth compared with controls. Quantification of apoptosis in CD90+/CD44+ tumour cells after in vitro treatment with the human CD44–-specific mAb indicated that mAb induced CSC apoptosis as a possible mechanism underlying the observed inhibition of tumour xenograft formation [14]. Most antibody therapies that have been successful, whether by passive or active immunotherapy, have targeted those molecules that are essential for the survival of a cancer cell [128].
Genes expressed in ESCs, encoding proteins involved in phosphatase and tensin homologue (PTEN), Wnt, Hh and Notch signalling pathways, are key factors in regulating self-renewal. Although these genes are expressed in normal stem cells, they are frequently mutated or aberrantly activated in cancers, thus making them potential therapeutic targets. The specific therapies targeting these signal pathways in CSCs will be briefly illustrated here [157]. Conditional gene deletion studies in murine HSCs provided evidence about a differential role of PTEN in normal HSCs versus leukaemic stem cells. PTEN deficiency caused an initial expansion of normal HSCs due to their cycling, followed by their exhaustion. In contrast to this requirement for PTEN in the maintenance of HSCs, LSCs arose and expanded in numbers following PTEN deletion. The observation that PTEN deletion had opposite effects on normal HSCs compared to leukaemic stem cells raised the possibility for therapeutic targeting of this pathway to eliminate leukaemic stem cells, without affecting normal HSCs. Since PTEN deletion caused increased AKT and mammalian target of rapamycin (mTOR) activation, it seemed logical that mTOR targeting by pharmacological agents, such as rapamycin, could represent an interesting option for AML treatment [68]. On the other hand, it has been shown that upon BMP activation, the mTOR kinase mediates glial differentiation in neural stem cells [158], and it has subsequently been shown that BMP reduces the tumourigenic potential of a putative glioma stem cell population [136]. In fact, rapamycin is used in the clinic in some treatment regimens for glioma [159]. One may therefore consider the mTOR kinase and possibly members of this kinase family to be potential targets for selective manipulation of some CSCs [128].
The first phytochemical shown to inhibit the Hh pathway was cyclopamine, a natural compound found in the plant Veratrum californicum [160]. Cyclopamine treatment of a murine medulloblastoma inhibited proliferation and induced neuronal differentiation, effectively depleting the CSC population [161]. In breast cancer and multiple myeloma stem cells, cyclopamine decreased mammosphere formation and stem cell proliferation, respectively [162, 163]. Several phytochemicals such as selenium, EGCG (one of the polyphenol compounds in green tea) and vitamin D, were indicated to inhibit Wnt signalling in cancers and could potentially be excellent candidates for targeting CSCs [164]. Furthermore, inhibiting of Notch pathway with specific gamma secretase inhibitors could also inhibit CSC self-renewal and decrease tumour growth [165]. Recently, Harrison et al. showed that Notch4 activity was increased in breast CSCs, and that inhibition of Notch4 signalling reduced breast CSCs and completely inhibited tumour-initiation [166]. Interestingly, Notch1 activity was lower in breast CSCs compared to more differentiated progenitor cells. This suggests that there is specificity for different Notch receptors in the regulation of breast stem and progenitor cells. If this is the case, then selective inhibition of Notch4 may be more effective and potentially less toxic than Notch1 inhibitors or secretase inhibitors that inhibit all Notch receptors [167]. Phytochemical resveratrol (found in grapes, berries and peanuts) shows anti-cancer properties [168, 169]. Acute lymphoblastic leukaemia cells treated with resveratrol resulted in reduced Notch expression [170].
As mentioned, current opinion is that CSCs are either dormant or in a proliferative phase. Dormant CSCs may confer drug resistance [37]; therefore, a main hurdle in designing strategies targeting CSCs would be the targeting of a quiescent cell among the dividing population of normal and transformed cells. One method to address this is to target molecules, which sustain the quiescence and stemness of the CSCs [128]. If quiescence is actively maintained by some factors in the milieu of the niche, interfering with their function could force the CSCs to undergo apoptosis, differentiation or division [128]. For instance, maintenance of AML stem cells in a quiescent state at the level of the bone marrow had important implications for their response to therapy. Interestingly, it was recently shown that LSCs can be triggered to enter cell cycle by treatment with granulocyte colony-stimulating factor, and this strategy conjugated with cell cycle-dependent chemotherapy significantly induces the apoptosis and elimination of primary AML stem cells [171].
Some strategies for cancer therapy by targeting the CSCs have been discussed so far. In addition to CSCs, there are also non-CSCs in tumours which can give rise to CSCs at a low but significant rate. It is also possible that the elimination of the CSCs within a tumour may not result in its complete regression, since non-CSCs, while less aggressive, may be capable of maintaining an already-established tumour for an extended period of time. Either of these possibilities would compromise the therapeutic utility of agents that exclusively target CSCs. One strategy to address this concern would be to look for agents that target both CSCs and non-CSCs within tumours. Alternatively, it may be better to develop combination therapies that use agents with specific toxicity for CSCs together with agents that specifically target non-CSC populations within tumours [138]. For example, it was shown that treatment of prostate tumour xenografts in mice with a combination of a standard chemotherapeutic drug, targeting non-CSCs and NVP-BEZ235 targeting CD133+/CD44+ tumour progenitors, leads to near-complete tumour regression. In contrast, the use of cytotoxic drugs such as Taxotere or 5-fluorouracil alone results in a decrease in tumour mass (non-CSCs) but leads to an overall increase in the relative size of the TICs population—the source of tumour relapse and resistance [172]. Therefore, it is suggested that the use of combination therapy directed against TICs together with non-CSCs is more effective in eradicating tumours and may provide a better strategy for cancer treatment.
Conclusion and perspective
According to current knowledge, initiation, recurrence and metastasis of cancers may be explained, at least in part by the presence of CSCs. In general, CSC model predicts that cancer therapies must destroy CSCs in order to be effective, therefore the development of CSC-targeted therapies able to preserve the normal stem cell compartment is currently a golden approach in cancer research [1]. For highly effective cancer treatments, we need to meticulously identify CSCs in the various forms of tumours and perform gene and protein profiling studies to determine how these cells differ from normal stem cells and other cancerous cells within the same tissue. Understanding these different expression patterns will be important in designing more effective and more specific treatments and also will aid in the development of novel treatments that destroy CSCs without adversely affecting self-renewal of normal stem cells. For example, the analysis of the gene expression profile has provided helpful information to specifically target the leukemogenic population without affecting normal stem cells [173]. Further studies are necessary for identification of CSC-specific surface markers for antibody therapy, elucidation of CSC-specific pathways that can be pharmacologically targeted and evaluation of agents that promote the differentiation of CSCs into progenitors that do not self-renew. We also speculate that a vaccine approach to CSCs will allow sensitising of the immune system and thus prevent some critical signalling pathways that could initiate stem cell transformation.
Assuming that CSCs represent only a small proportion of the entire tumour, killing them in the short term, might have little impact on the size of the tumour as a whole. However, over time the tumour would be expected to exhaust itself and wither away, because it has lost the capacity for long-term self-renewal. From a clinical point, it remains to be seen whether such therapies are effective on their own; it is possible that, for some cancers, continued proliferation of transit-amplifying cells that make up the bulk of the tumour may be sufficient to cause irreversible histological and physiological damage. Therefore, combination therapies that target both CSCs and bulk cancer populations are likely to emerge as particularly effective clinical strategies. Furthermore, the potentially severe side effects of CSC-targeted therapy still have to be evaluated in animal models before we can suggest it for clinical trials (an important question, however, is how realistically tumour xenograft models in immunodeficient mice recapitulate what is happening in human patients). Therefore, more studies are needed to improve therapeutic approaches and outcomes in patients with cancer disease.
References
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer and cancer stem cells. Nature. 2001;414:105–11.
Tu SM, Lin SH, Logothetis CJ. Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet Oncol. 2002;3:508–13.
Passegue E, Jamieson CH, Ailles LE, Weissman IL. Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics. Proc Natl Acad Sci USA. 2003;100:11842–9.
Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–99.
Virchow R. Editorial. Arch Pathol Anat Physiol Klin Med. 1855;8:23.
Sell S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol. 2004;51:1–28.
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumourigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401.
Fang B, Zheng C, Liao L, Han Q, Sun Z, Jiang X, et al. Identification of human chronic myelogenous leukemia progenitor cells with hemangioblastic characteristics. Blood. 2005;105:2733–40.
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7.
Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007;104:973–8.
Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–9.
Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–66.
Suvà ML, Riggi N, Stehle JC, Baumer K, Tercier S, Joseph JM, et al. Identification of cancer stem cells in Ewing's sarcoma. Cancer Res. 2009;69:1776–81.
Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133–7.
Rasheed ZA, Yang J, Wang Q, Kowalski J, Freed I, Murter C, et al. Prognostic significance of tumourigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst. 2010;102:340–51.
Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumour growth need not be driven by rare cancer stem cells. Science. 2007;317:337.
Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–8.
Ishizawa K, Rasheed ZA, Karisch R, Wang Q, Kowalski J, Susky E, et al. Tumour initiating cells are rare in many human tomour. Cell Stem Cell. 2010;7:279–82.
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7.
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumours. Cancer Res. 2003;63:5821–8.
Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–35.
Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, et al. Highly purified CD44+ prostate cancer cells from xenograft human tumours are enriched in tumourigenic and metastatic progenitor cells. Oncogene. 2006;25:1696–708.
Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumourigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–51.
Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian inhibiting substance responsiveness. Proc Natl Acad Sci USA. 2006;103:11154–9.
O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10.
Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon cancer-initiating cells. Nature. 2007;445:111–5.
Taylor G, Lehrer MS, Jensen PJ, Sun TT, Lavker RM. Involvement of follicular stem cells in forming not only the follicle but also the epidermis. Cell. 2000;102:451–61.
Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 2004;118:635–48.
Andrews PW, Matin M, Bahrami AR, Damjanov I, Gokhale P, Draper JS. Embryonic stem (ES) cells and embryonal carcinoma (EC) cells: opposite sides of the same coin. Biochem Soc Trans. 2005;33:1526–30.
Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902.
Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–60.
Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–31.
Al-Hajj M, Berker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14:43–7.
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–83.
Ishii H, Iwatsuki M, Ieta K, Ohta D, Haraguchi N, Mimori K, et al. Cancer stem cells and chemoradiation resistance. Cancer Sci. 2008;99:1871–7.
Terpstra W, Ploemacher RE, Prins A, van Lom K, Pouwels K, Wognum AW, et al. Fluorouracil selectively spares acute myeloid leukemia cells with long-term growth abilities in immunodeficient mice and in culture. Blood. 1996;88:1944–50.
Ohyashiki JH, Sashida G, Tauchi T, Ohyashiki K. Telomeres and telomerase in hematologic neoplasia. Oncogene. 2002;21:680–7.
Ju Z, Rudolph KL. Telomeres and telomerase in stem cells during aging and disease. Genome Dyn. 2006;1:84–103.
Rosner MH, Vigano MA, Ozato K, Timmons PM, Poirier F, Rigby PW, et al. A POU-domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature. 1990;345:686–92.
Burdon T, Smith A, Savatier P. Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 2002;12:432–8.
Boiani M, Scholer HR. Regulatory networks in embryo-derived pluripotent stem cells. Nat Rev Mol Cell Biol. 2005;6:872–84.
Matin MM, Walsh JR, Gokhale PJ, Draper JS, Bahrami AR, Morton I, et al. Specific knockdown of Oct4 and beta2-microglobulin expression by RNA interference in human embryonic stem cells and embryonic carcinoma cells. Stem Cells. 2004;22:659–68.
Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumourigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–11.
Chen YC, Hsu HS, Chen YW, Tsai TH, How CK, Wang CY, et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133+ cells. PLoS One. 2008;3:e2637.
Schoenhals M, Kassambara A, De Vos J, Hose D, Moreaux J, Klein B. Embryonic stem cell markers expression in cancers. Biochem Biophys Res Commun. 2009;383:157–62.
Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–50.
Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–34.
Van Stijn A, Van der Pol MA, Kok A, Bontje PM, Roemen GM, Beelen RH, et al. Differences between the CD34+ and CD34− blast compartments in apoptosis resistance in acute myeloid leukemia. Haematologica. 2003;88:497–508.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60.
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–3.
Raguz S, Yague E. Resistance to chemotherapy: new treatments and novel insights into an old problem. Br J Cancer. 2008;99:387–91.
Fuchs E, Segre JA. Stem cells: a new lease on life. Cell. 2000;100:143–55.
Morrison SJ, Qian D, Jerebek L, Thiel BA, Park IK, Ford PS, et al. A genetic determinant that specifically regulates the frequency of hematopoietic stem cells. J Immunol. 2002;168:635–42.
Shackleton M. Normal stem cells and cancer stem cells: similar and different. Semin Cancer Biol. 2010;20:85–92.
Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–14.
Knudson Jr AG, Strong LC, Anderson DE. Heredity and cancer in man. Prog Med Genet. 1973;9:113–58.
Jaiswal S, Traver D, Miyamoto T, Akashi K, Lagasse E, Weissman IL. Expression of BCR/ABL and BCL-2 in myeloid progenitors leads to myeloid leukemia. Proc Natl Acad Sci USA. 2003;100:10002–7.
Shiras A, Chettiar ST, Shepal V, Rajendran G, Prasad GR, Shastry P. Spontaneous transformation of human adult nontumourigenic stem cells to cancer stem cells is driven by genomic instability in a human model of glioblastoma. Stem Cells. 2007;25:1478–89.
Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403–17.
Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, et al. Non-stem cell origin for oligodendroglioma. Cancer Cell. 2010;18:669–82.
Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-ssociated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–35.
Sell S, Pierce G. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest. 1994;70:6–22.
Ratajczak MZ. Cancer stem cells-normal stem cells "Jedi" that went over to the "dark side". Folia Histochem Cytobiol. 2005;43:175–81.
Zhao RC, Zhu Y, Shi Y. New hope for cancer treatment: exploring the distinction between normal adult stem cells and cancer stem cells. Pharmacol Ther. 2008;119:74–82.
Spandidos DA. Oncogenes and tumour suppressor genes as paradigms in oncogenesis. J BUON. 2007;12:S9–12.
Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–82.
He XC, Yin T, Grindley JC, Tian Q, Sato T, Tao WA, et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet. 2007;39:189–98.
Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–5.
Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005;19:1432–7.
Gil J, Stembalska A, Pesz KA, Sasiadek MM. Cancer stem cells: the theory and perspectives in cancer therapy. J Appl Genet. 2008;49:193–9.
Kléber M, Sommer L. Wnt signaling and the regulation of stem cell function. Curr Opin Cell Biol. 2004;16:681–7.
Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;14:843–50.
Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signaling. Nature. 2008;452:650–3.
Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, et al. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell. 2010;18:606–18.
Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–87.
Pepinsky RB, Rayhorn P, Day ES, Dergay A, Williams KP, Galdes A, et al. Mapping sonic hedgehog–receptor interactions by steric interference. J Biol Chem. 2000;275(10):10995–1001.
Murone M, Rosenthal A, de Sauvage FJ. Sonic hedgehog signaling by the patched-smoothened receptor complex. Curr Biol. 1999;9:76–84.
Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–21.
Pasca di Magliano M, Hebrok M. Hedgehog signaling in cancer formation and maintenance. Nat Rev Cancer. 2003;3:903–11.
Medina V, Calvo MB, Díaz-Prado S, Espada J. Hedgehog signalling as a target in cancer stem cells. Clin Transl Oncol. 2009;11:199–207.
Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6.
Bray S. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–89.
Katoh M, Katoh M. Notch signaling in gastrointestinal tract. Int J Oncol. 2007;30:247–51.
Fox V, Gokhale PJ, Walsh JR, Matin M, Jones M, Andrews PW. Cell-cell signaling through NOTCH regulates human embryonic stem cell proliferation. Stem Cells. 2008;26:715–23.
Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–54.
Radtke F, Raj K. The role of Notch in tumourigenesis: oncogene or tumour suppressor? Nat Rev Cancer. 2003;3:756–67.
Boulay JL, Miserez AR, Zweifel C, Sivasankaran B, Kana V, Ghaffari A, et al. Loss of NOTCH2 positively predicts survival in subgroups of human glial brain tumours. PLoS One. 2007;2:e576.
Lee SY, Kumano K, Nakazaki K, Sanada M, Matsumoto A, Yamamoto G, et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci. 2009;100:920–6.
Lino MM, Merlo A, Boulay JL. Notch signaling in glioblastoma: a developmental drug target? BMC Med. 2010;8:72.
Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58–62.
Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci USA. 1997;94:5320–5.
Hogan CJ, Shpall EJ, Keller G. Differential long-term and multilineage engraftment potential from subfractions of human CD34+ cord blood cells transplanted into NOD/SCID mice. Proc Natl Acad Sci USA. 2002;99:413–8.
Kimura T, Asada R, Wang J, Kimura T, Morioka M, Matsui K. Identification of long-term repopulating potential of human cord blood-derived CD34-flt3- severe combined immunodeficiency-repopulating cells by intra-bone marrow injection. Stem Cells. 2007;25:1348–55.
Kitamura T, Matsuoka Y, Kimura T, Takahashi M, Nakamoto T, Yosuda K, et al. In vivo dynamic of human cord-blood derived CD34−SCID-repopulating cells using intra-bone marrow injection. Leukemia. 2010;24:162–8.
Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–75.
Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood. 2010;115:1976–84.
Schatton T, Frank NY, Frank MH. Identification and targeting of cancer stem cells. Bioessays. 2009;31:1038–49.
Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, et al. Identification and characterization of tumourigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–56.
Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumourigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–14.
Cui F, Wang J, Chen D, Chen YJ. CD133 is a temporary marker of cancer stem cells in small cell lung cancer, but not in non-small cell lung cancer. Oncol Rep. 2010. doi:10.3892/or.2010.1115.
Chu P, Clanton DJ, Snipas TS, Lee J, Mitchell E, Nguyen M-L, et al. Characterization of a subpopulation of colon cancer cells with stem cell-like properties. Int J Cancer. 2009;124:1312–21.
Shi MF, Jiao J, Lu WG, Ye F, Ma D, Dong QG, et al. Identification of cancer stem cell-like cells from human epithelial ovarian carcinoma cell line. Cell Mol Life Sci. 2010;67:3915–25.
Baba T, Convery PA, Matsumura N, Whitaker RS, Kondoh E, Perry T, et al. Epigenetic regulation of CD133 and tumourigenicity of CD133C ovarian cancer cells. Oncogene. 2009;28:209–18.
Dontu G, Al-Hajj M, Abdullah W, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003;36:59–72.
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–67.
Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumourigenesis. Cancer Res. 2009;69:3382–9.
Su Y, Qiu Q, Zhang X, Jiang Z, Leng Q, Liu Z, et al. Aldehyde dehydrogenase 1 A1-positive cell population is enriched in tumour-initiating cells and associated with progression of bladder cancer. Cancer Epidemiol Biomarkers Prev. 2010;19:327–37.
Penumatsa K, Edassery SL, Barua A, Bradaric MJ, Luborsky JL. Differential expression of aldehyde dehydrogenase 1A1 (ALDH1) in normal ovary and serous ovarian tumours. J Ovarian Res. 2010;3:28.
Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–5.
Joo KM, Kim SY, Jin X, Song SY, Kong DS, Lee JI, et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab Invest. 2008;88:808–15.
Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133 metastatic colon cancer cells initiate tumours. J Clin Invest. 2008;118:2111–20.
Cho RW, Clarke MF. Recent advances in cancer stem cells. Curr Opin Genet Dev. 2008;18:48–53.
Tan BT, Park CY, Ailles LE, Weissman IL. The cancer stem cell hypothesis: a work in progress. Lab Invest. 2006;86:1203–7.
Huang Y, Anderle P, Bussey KJ, Barbacioru C, Shankavaram U, Dai Z, et al. Membrane transporters and channels: role of the transportome in cancer chemosensitivity and chemoresistance. Cancer Res. 2004;64:4294–301.
Elliott A, Adams J, Al-Hajj M. The ABCs of cancer stem cell drug resistance. IDrugs. 2010;13:632–5.
Tan B, Piwnica-Worms D, Ratner L. Multidrug resistance transporters and modulation. Curr Opin Oncol. 2000;12:450–8.
Tamura K, Aoyagi M, Wakimoto H, Ando N, Nariai T, Yamamoto M, et al. Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. J Neurosurg. 2010;113:310–8.
Facchino S, Abdouh M, Chatoo W, Bernier G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci. 2010;30:10096–111.
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85.
Ropolo M, Daga A, Griffero F, Foresta M, Casartelli G, Zunino A, et al. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol Cancer Res. 2009;7:383–92.
Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature. 2009;457:51–6.
Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9.
Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–91.
Kai K, Arima Y, Kamiya T, Saya H. Breast cancer stem cells. Breast Cancer. 2010;17:80–5.
Massard C, Deutsch E, Soria JC. Tumour stem cell-targeted treatment: elimination or differentiation. Ann Oncol. 2006;17:1620–4.
Rajan P, Srinivasan R. Targeting cancer stem cells in cancer prevention and therapy. Stem Cell Rev. 2008;4:211–6.
Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells—old concepts, new insights. Cell Death Differ. 2008;15:947–58.
Spira AI, Carducci MA. Differentiation therapy. Curr Opin Pharmacol. 2003;3:338–43.
Campos B, Wan F, Farhadi M, Ernst A, Zeppernick F, Tagscherer KE, et al. Differentiation therapy exerts antitumour effects on stem-like glioma cells. Clin Cancer Res. 2010;16:2715–28.
Ohno R, Asou N, Ohnishi K. Treatment of acute promyelocytic leukemia: strategy toward further increase of cure rate. Leukemia. 2003;17:1454–63.
Clarke N, Germain P, Altucci L, Gronemeyer H. Retinoids: potential in cancer prevention and therapy. Expert Rev Mol Med. 2004;6:1–23.
Taddei A, Roche D, Bickmore WA, Almouzni G. The effects of histone deacetylase inhibitors on heterochromatin: implications for anticancer therapy? EMBO Rep. 2005;6:520–4.
Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci USA. 2002;99:11700–5.
Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, et al. Bone morpho-genetic proteins inhibit the tumourigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–5.
Schatzlein AG. Delivering cancer stem cell therapies—a role for nanomedicines? Eur J Cancer. 2006;42:1309–15.
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59.
Riccioni R, Dupuis ML, Bernabei M, Petrucci E, Pasquini L, Mariani G, et al. The cancer stem cell selective inhibitor salinomycin is a p-glycoprotein inhibitor. Blood Cells Mol Dis. 2010;45:86–92.
Jodoin J, Demeule M, Beliveau R. Inhibition of the multidrug resistance P-glycoprotein activity by green tea polyphenols. Biochim Biophys Acta. 2002;1542:149–59.
Harbottle A, Daly AK, Atherton K, Campbell FC. Role of glutathione S-transferase P1, P-glycoprotein and multidrug resistance associated protein 1 in acquired doxorubicin resistance. Int J Cancer. 2001;92:777–83.
Verma SP, Goldin BR, Lin PS. The inhibition of the estrogenic effects of pesticides and environmental chemicals by curcumin and isoflavonoids. Environ Health Perspect. 1998;106:807–12.
Behnam Rassouli F, Matin MM, Iranshahi M, Bahrami AR, Neshati V, Mollazadeha S, et al. Mogoltacin enhances vincristine cytotoxicity in human transitional cell carcinoma (TCC) cell line. Phytomedicine. 2009;16:181–7.
Neshati V, Matin MM, Iranshahi M, Bahrami AR, Behravan J, Mollazadeh S, et al. Cytotoxicity of vincristine on the 5637 cell line is enhanced by combination with conferone. Z Naturforsch C. 2009;64:317–22.
Mollazadeh S, Matin MM, Iranshahi M, Bahrami AR, Neshati V, Behnam Rassouli F. The enhancement of vincristine cytotoxicity by combination with feselol. J Asian Nat Prod Res. 2010;12:569–75.
Le Cesne A, Blay JY, Judson I, Van Oosterom A, Verweij J, Radford J, et al. Phase II study of ET-743 in advanced soft tissue sarcomas: a European Organisation for the Research and Treatment of Cancer (EORTC) soft tissue and bone sarcoma group trial. J Clin Oncol. 2005;23:576–84.
Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–34.
Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, et al. Nuclear factor-κB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–7.
Griessinger E, Imbert V, Lagadec P, Gonthier N, Dubruil P, Romanelli A, et al. AS602868, a dual inhibitor of IKK2 and FLT3 to target AML cells. Leukemia. 2007;21:877–85.
Fuchs O. Transcription factor NF-κB inhibitors as a single therapeutic agent or in combination with classical chemotherapeutic agents for the treatment of hematologic malignancies. Curr Mol Pharmacol. 2010;3:98–122.
Miletti-González KE, Chen S, Muthukumaran N, Saglimbeni GN, Wu X, Yang J, et al. The CD44 receptor interacts with P-glycoprotein to promote cell migration and invasion in cancer. Cancer Res. 2005;65:6660–7.
Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–74.
Testa U, Riccioni R, Militi S, Coccia E, Stellacci E, Samoggia P, et al. Elevated expression of IL-3Rα in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100:2980–8.
Graf M, Hecht K, Reif S, Pelka-Fleischer R, Pfister K, Schmetzer H. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): implications for future therapeutical strategies. Eur J Haematol. 2004;72:89–106.
van Rhenen A, Moshaver B, Kelder A, Feller N, Nieuwint AW, Zweegman S, et al. Aberrant marker expression patterns on the CD34+CD38−-stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia. 2007;21:1700–7.
Jin L, Lee EM, Ramshaw HS, Busflield SJ, Peoppl AG, Wilkinson L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor α chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5:31–42.
Grinstein E, Wernet P. Cellular signaling in normal and cancerous stem cells. Cell Signal. 2007;19:2428–33.
Rajan P, Panchision DM, Newell LF, McKay RD. BMPs signal alternately through a SMAD or FRAP-STAT pathway to regulate fate choice in CNS stem cells. J Cell Biol. 2003;161:911–21.
Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, et al. Antitumour activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5(1):e8.
Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–7.
Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–61.
Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–71.
Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumour stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA. 2007;104:4048–53.
Byers S, Shah S. Vitamin D and the regulation of Wnt/beta-catenin signaling and innate immunity in colorectal cancer. Nutr Rev. 2007;65:S118–20.
Shih Ie M, Wang TL. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res. 2007;67:1879–82.
Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–18.
McDermott SP, Wicha MS. Targeting breast cancer stem cells. Mol Oncol. 2010;4:404–19.
Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24:2783–840.
Das DK, Maulik N. Resveratrol in cardioprotection: a therapeutic promise of alternative medicine. Mol Interv. 2006;6:36–47.
Cecchinato V, Chiaramonte R, Nizzardo M, Cristofaro B, Basile A, Sherbet GV, et al. Resveratrol-induced apoptosis in human T-cell acute lymphoblastic leukaemia MOLT-4 cells. Biochem Pharmacol. 2007;74:1568–74.
Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med. 2010;2:17ra9.
Dubrovska A, Elliott J, Salamone RJ, Kim S, Aimone LJ, Walker JR, et al. Combination therapy targeting both tumour-initiating and differentiated cell populations in prostate carcinoma. Clin Cancer Res. 2010;16:5692–702.
Qian Z, Fernald AA, Godley LA, Larson RA, Le Beau MM. Expression profiling of CD34+ haematopoietic stem/progenitor cells reveals distinct subtypes of therapy-related acute myeloid leukemia. Proc Natl Acad Sci USA. 2002;99:14925–30.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Soltanian, S., Matin, M.M. Cancer stem cells and cancer therapy. Tumor Biol. 32, 425–440 (2011). https://doi.org/10.1007/s13277-011-0155-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-011-0155-8