
Summary
Due to their endosymbiotic origin, expression of plastid and mitochondrial genes retains several features of prokaryotes. Nevertheless, plant organelles acquired novel specific traits during evolution. Furthermore, due to the migration of many genes to the nucleus of the host cell, complex anterograde and retrograde signalling pathways evolved to coordinate gene expression in different subcellular compartments. Control of gene expression in plant organelles occurs at the transcriptional and posttranscriptional levels. In this chapter, we analyze the available data concerning the variability shown by both organelle genomes for different steps of gene expression in various genotypes or after environmental and developmental cues. Genotypic variability for the extent of RNA editing or transcript processing and stability in cytoplasmic organelles has been observed in natural populations at the interspecific and intraspecific level or in artificial CMS lines. The role of various plastid genes in global genome expression and chloroplast development has been highlighted in knock-out lines produced by plastid transformation. Significant differences in the transcriptome, editome and translatome have also been found comparing different plastid types in diverse organs or tissues. Similar differences have been found for mitochondrial genomes during the diurnal cycle or between cell suspensions and differentiated leaves. However, the precise level and mechanisms at which these changes are achieved and the signals necessary for their installation are barely understood.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
I. Introduction
According to the endosymbiotic theory, plastids and mitochondria derive from early prokaryotic organisms engulfed by a proto-eukaryotic cell (Buchanan et al. 2000). The origin and the following evolution of both plant organelles have had important implications not only for the structure of their genomes, but also for the expression of their genes.
Expression of plastid and mitochondrial genes retain several features of prokaryotes, e.g. the common, albeit not exclusive, organization in operons, implying co-transcription of individual genes, similar RNA polymerases and promoters (in plastids), similar structure of mature mRNAs, the presence of 70S-type ribosomes, and others. Nevertheless, different from bacteria and other prokaryotes, plant organelles show novel traits, such as uncoupled transcription and translation, phage-type RNA polymerases, and frequent RNA editing and splicing of transcript precursors. Furthermore, during evolution, many genes originally present in the endosymbionts’ genomes migrated to the nucleus of the host cell. Since in many cases nuclear and organelle genes encode subunits of the same protein complexes, their expression needs to be somehow co-regulated, implying complex anterograde and retrograde signalling pathways between different subcellular compartments (Bräutigam et al. 2007).
Control of gene expression in plant organelles occurs at the transcriptional and posttranscriptional levels, the latter including regulation of transcript maturation and stability, translation, protein stability and activity (Bollenbach et al. 2007; Liere and Börner 2007; Peled-Zehavi and Danon 2007; Schmitz-Linneweber and Barkan 2007). The recent development of novel technologies, in particular DNA arrays, allowed the genome-wide analyses of gene expression in different genotypes, tissues, and environmental conditions. In several cases, the concomitant analysis of nuclear genes involved in organelle gene expression allowed to dissect interorganellar regulatory pathways (Biehl et al. 2005).
In this chapter, after reviewing basic aspects of gene expression in plastids and plant mitochondria, we analyze the available data concerning the variability shown by both organelle genomes for different steps of gene expression in various genotypes or in response to environmental and developmental cues.
II. Regulation of Gene Expression in Plant Organelles
A.Transcription
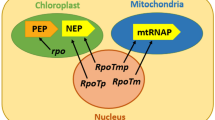
In plastids, the RNA-synthesizing activity is carried out by two enzymes of different evolutionary origins. A plastid-encoded RNA polymerase (PEP) is homologous to the eubacterial RNA polymerase also found in cyanobacteria, the closest extant bacterial relatives of plastids (Mereschkowsky 1905; Kaneko et al. 1996). The plastid genome encodes the core subunits of the bacterial-type RNA polymerase, consisting of the four proteins RpoA, RpoB, RpoC1 and RpoC2. The corresponding genes were identified in the first completely sequenced plastomes of Marchantia polymorpha and Nicotiana tabacum (Ohyama et al. 1986; Shinozaki et al. 1986). A second RNA-synthesizing activity is carried out through a nuclear-encoded RNA polymerase (NEP) with homology to phage-type RNA polymerases (Lerbs-Mache 1993). The genome of Arabidopsis thaliana contains three copies of RpoT genes designated as RpoTm, RpoTmp, and RpoTp, indicating sub-cellular localization in mitochondria (m) and/or plastids (p) (Hedtke et al. 1997, 2000; Chang and Stern 1999; Kobayashi et al. 2001). Although plastid genes encoding proteins involved in gene expression, including PEP, are preferentially transcribed by the NEP enzyme, several chloroplast genes are driven from promoters for both polymerases. The PEP enzyme transcribes predominantly photosynthesis-related genes (Allison et al. 1996; Hajdukiewicz et al. 1997; Silhavy and Maliga 1998; Liere and Maliga 1999). It was hypothesized that the NEP polymerase is activated early in chloroplast development resulting in transcription of PEP which, in turn, activates photosynthesis-related genes (Mullet 1993). Although a recent study could show that both enzymes are already present in seeds (Demarsy et al. 2006), transcripts encoding the gene expression machinery, which are predominantly transcribed by NEP, peak in their maximal abundance earlier during chloroplast development than the predominantly PEP-transcribed photosynthesis genes (Baumgartner et al. 1993).
Although it is known that auxiliary factors are required for efficient transcription initiation in vivo in plastids (Kühn et al. 2007), experiments to identify and characterize factors involved in NEP promoter recognition and transcription initiation have failed so far. By contrast, plastids require, like bacteria, additional σ-like factors for correct PEP promoter recognition. Whereas just one σ-like factor is known in Chlamydomonas reinhardtii (Carter et al. 2004; Bohne et al. 2006), six σ-factors, designated as Sig1-6, are encoded in the nuclear genome of A. thaliana (Isono et al. 1997b; Tanaka et al. 1997; Kanamaru et al. 1999; Fujiwara et al. 2000). They have a general role in transcription, recognize certain promoters or respond to environmental stimuli (for review see Shiina et al. 2005; Liere and Börner 2007).
Unlike plastids, plant mitochondria rely entirely on nuclear encoded RNA polymerases for transcription. Recent studies have shown that RpoTmp could be involved in the transcription of specific genes in mitochondria (Kühn et al. 2009) and requires additional protein partners to recognise specifically promoter sequences, as observed in human and yeast mitochondria (Tracy and Stern 1995). Contrary to vertebrates, where transcription is initiated at a single site on each DNA strand, plant mitochondrial transcription is initiated at multiple sites. Moreover, transcription of single genes can be initiated by multiple promoters (Lupold et al. 1999; Kühn et al. 2005). Promoter sequences of the A. thaliana mitochondrial genome often contain the consensus motif YRTA, although transcription can also be initiated at non-canonical sites that lack any kind of recognizable consensus motif (Binder and Brennicke 1993; Kühn et al. 2005; Remacle and Maréchal-Drouard 1996; Fey and Maréchal-Drouard 1999).
Inverted repeat sequences forming stem-loop structures in 3′-UTR of transcripts are present both in plastids and mitochondria. However, they were rather found to be involved in RNA maturation than in transcription termination (Dombrowski et al. 1997; Hoffmann et al. 1999). Indeed, despite the complex transcription mechanisms, it appears that posttranscriptional processes have a major role in the regulation of gene expression in both organelles and often override changes at the transcriptional level (Giegé et al. 2000; Holec et al. 2006; Bollenbach et al. 2007; Liere and Börner 2007; Peled-Zehavi and Danon 2007; Schmitz-Linneweber and Barkan 2007).
B. RNA Editing
The term RNA editing comprises a variety of single nucleotide alterations which change the genetic information at the RNA level. Editing was initially described in trypanosome mitochondria (Benne et al. 1986) and includes alterations like nucleotide insertions or deletions as well as nucleotide modifications and replacements. In higher plant chloroplasts and mitochondria, RNA editing (see also Chap. 13) is restricted to C-to-U conversions (Covello and Gray 1989; Gualberto et al. 1989; Hiesel et al. 1989; Shikanai 2006; Liere and Börner 2007; Stern et al. 2010), while less frequent U-to-C conversions have been reported in fern, hornwort and lycophyte organelles (Malek et al. 1996; Knoop 2004; Wolf et al. 2004; Duff and Moore 2005; Shikanai 2006; Takenaka et al. 2008). Several common features of the editing process in chloroplasts and mitochondria suggest a common evolutionary origin of the two organelle editing systems (Freyer et al. 1997; Tillich et al. 2006).
To date, 34 editing sites are known in A. thaliana plastids (Chateigner-Boutin and Small 2007), representing a typical number of editing sites found in vascular plant chloroplasts. In comparison, RNA editing affects over 500 cytidines in mitochondria (Giegé and Brennicke 1999; Chateigner-Boutin and Small 2007; Zehrmann et al. 2008), although the number of editing sites per gene is highly variable. In Arabidopsis mitochondria, complex I and CCM (cytochrome c maturation) mRNAs have the highest RNA editing frequencies (Giegé and Brennicke 1999). There exist a few examples for editing occurring in non-coding regions such as introns (Chateigner-Boutin and Small 2007), but most editing events restore conserved amino acids and create start or stop codons (Chapdelaine and Bonen 1991; Hoch et al. 1991; Neckermann et al. 1994; Maier et al. 1995; Giegé et al. 2004; Miyata and Sugita 2004; Okuda et al. 2006). In addition, it often affects positions that appear to be essential for the respective protein functions (Bock et al. 1994b), although in 10% of cases RNA editing is “silent” since the third position of a codon is affected and the amino acid identity is unchanged (Giegé and Brennicke 1999). In maize mitochondria, the editing of a nad7 intron is required for its proper folding and thus for efficient splicing (Carrillo and Bonen 1997). Similarly, RNA editing is required for the proper folding of mitochondrial tRNAs, which is a prerequisite for their maturation (Maréchal-Drouard et al. 1996a, b; Kunzmann et al. 1998).
The partial RNA editing at some sites and the consequent heterogeneous populations of transcripts (Chateigner-Boutin and Hanson 2003; Bentolila et al. 2008) could lead to the synthesis of different forms of individual proteins. Although both the edited and unedited versions of the plastid ndhD transcript are associated with ribosomes, edited transcripts are highly enriched in the most actively translated polysome fractions (Zandueta-Criado and Bock 2004). Other studies have shown that only the proteins resulting from fully edited transcripts accumulate in mitochondria or plastids and no examples exist showing that an unedited protein has a function within organelles (Grohmann et al. 1994; Lu and Hanson 1994; Phreaner et al. 1996). This suggests that translation of partially edited transcripts could be inhibited and/or that proteins resulting from partially edited RNA are instable and rapidly degraded. Hence, it has been hypothesized that instead of a regulatory role, the primary function of RNA editing could have been to correct genomic mutations that appeared during the invasion of land by plants and thus to enable the translation of functional proteins (Shikanai 2006; Takenaka et al. 2008).
The exact editing mechanism still remains elusive. The analysis of the hundreds of editing sites present in the plant mitochondrial transcriptome (Giegé and Brennicke 1999; Bentolila et al. 2008) has not enabled to define specific consensus signals around editing sites. However, the distribution of nucleotides around the sites is not random because a strong preference for pyrimidines is observed for the two nucleotides immediately upstream of the sites (Giegé and Brennicke 1999). Both in plastids and mitochondria trans-acting factors are involved in recognition of the endogenous editing sites (Chaudhuri et al. 1995; Bock and Koop 1997). So far, several pentatricopeptide repeat (PPR) proteins, encoded by a gene family with more than 450 members in A. thaliana and characterized by repeated motifs of a degenerate 35 amino-acid consensus, were found to be involved in editing site recognition (Small and Peeters 2000; Kotera et al. 2005; Okuda et al. 2006, 2007; Kim et al. 2009; Zehrmann et al. 2009; Tasaki et al. 2010; Verbitskiy et al. 2010) although, so far, it could not be shown that these proteins actually carry out the deamination reaction, which converts the cytidine to a uridine (Hirose and Sugiura 2001).
C. RNA Processing
Plastid and mitochondrial genes are often, like in their bacterial ancestors, transcribed from operons resulting in polycistronic transcripts. Numerous rearrangements that occurred during the evolution of plant mitochondrial genomes have led to the loss of ancient syntenies of gene organisation into functional units (Schuster 1993; Giegé et al. 2000). As a consequence, co-transcription often does not involve genes of related function. This phenomenon is somewhat less widespread in plastids, where genes encoding subunits of the same protein complex are more frequently present in the same operon. Generally, prior to protein synthesis, transcripts are cleaved intercistronically and their 5′ and 3′ ends undergo maturation steps.
5′ ends of chloroplast transcripts are either unprocessed and then characterized by a 5′ di- or triphosphate or carry a 5′ hydroxyl group in the case of processed mRNAs. The enzymes catalyzing these processing steps are so far unknown (for review see Bollenbach et al. 2007). In contrast, the mechanisms of 3′ end maturation are known in much greater detail. Transcription termination is rather inefficient in plastids, resulting in the requirement of 3′ end processing. This involves the binding of a high molecular weight complex downstream of the stem-loop structures formed by inverted repeats (reviewed in Stern and Kindle 1993; Hayes et al. 1999). A second mechanism for mRNA stabilization is the binding of PPR proteins (Pfalz et al. 2009).
Similarly, the maturation of plant mitochondrial precursor transcripts involves 5′- and 3′-maturation steps. These maturations could be achieved through direct endoribonuclease activities and/or with 5′-to-3′ exoribonucleases and 3′-to-5′ exoribonucleases. Such enzymes are encoded in the nucleus and must be imported from the cytosol. In higher plant mitochondria, no 5′-to-3′ exoribonuclease has been identified yet. In contrast, two 3′-to-5′ exoribonucleases were characterised: RNase II is dually localised in mitochondria and plastids and a polynucleotide phosphorylase (PNPase) is found in mitochondria. Studies suggest that the 3′-processing of mitochondrial transcripts is at least a two-step phenomenon (Gagliardi et al. 2001; Perrin et al. 2004a, b). tRNAs are also transcribed as precursor molecules and have to be matured at their 5′- and 3′-ends. These maturation steps are performed by two ubiquitous endoribonuclease activities called RNase P and RNase Z, respectively (Vogel et al. 2005; Canino et al. 2009; Gobert et al. 2010; Jonietz et al. 2010).
Splicing is an essential process in RNA maturation in plant organelles as introns disrupt reading frames of important genes involved in photosynthesis or gene expression. Twenty out of the 21 plastid introns found in land plants belong to group II introns and the remaining intron in the trnL-UAA is a group I intron (reviewed in Saldanha et al. 1993). Factors involved in plastid intron splicing are mostly encoded in the nucleus. A single maturase-like protein, MatK, is encoded in the trnK gene intron and is potentially involved in splicing of several group II introns (Liere and Link 1995; Jenkins et al. 1997). A rather unusual intron is one of the two introns in the rps12 gene. This bipartite gene is encoded at two distant locations in the plastid genome, splitting this intron into two separate parts. Thus, two precursor mRNAs are generated which are joined together in a trans-splicing event (Hildebrand et al. 1988).
In seed plant mitochondria, group II introns are found in several genes (Unseld et al. 1997; Bonen and Vogel 2001), while only one example of a recently acquired group I intron has been found in the cox1 genes of Peperomia and of some other plants (Vaughn et al. 1995; Cho et al. 1998; Grewe et al. 2009). The genes encoded in the Arabidopsis mitochondrial genome are interrupted by altogether 23 group II introns with sizes varying from 485 to about 4,000 nucleotides (Unseld et al. 1997). Some genes are interrupted by more than one intron, e.g. nad7 has four introns. Trans-splicing is found in plant mitochondria in several instances (e.g. in the nad1, nad2 and nad5 genes), (Chapdelaine and Bonen 1991; Knoop et al. 1991; Wissinger et al. 1991; Binder et al. 1992; Glanz and Kück 2009). The highly conserved structure of the group II introns and specific protein factors, called maturases, are essential for splicing activity (Wank et al. 1999; Lambowitz and Zimmerly 2004; Meng et al. 2005; Fedorova and Zingler 2007). In plant mitochondria, one conserved gene encoding such a maturase, MatR, is located in the terminal nad1 intron. Moreover, several nuclear genes and nucleus-encoded proteins, involved in splicing in chloroplasts and, putatively, in mitochondria were identified (Mohr and Lambowitz 2003; Nakagawa and Sakurai 2006; Keren et al. 2008).
Total RNA abundance also depends on the rate of transcript degradation. It has been shown in spinach and barley that plastid mRNA stability can highly vary during leaf development and therefore also accounts for transcript abundance (Klaff and Gruissem 1991; Kim et al. 1993). Lack of ribosome association can result in mRNA degradation which has been shown for the rbcL transcript, but this mechanism cannot be generalized as many other transcripts remain unaffected despite decreased ribosome association (Barkan 1993). The actual RNA degradation pathway in plastids involves polyadenylation (Kudla et al. 1996), a process which is known to be a stabilizing signal for nuclear mRNAs (for review see Dreyfus and Régnier 2002), but also acts as RNA instability signal in prokaryotes.
D. Translation
Plastid translation is related to translation in eubacteria. Both systems share homologous compounds, like initiation factors, rRNAs, tRNAs and 70S-type ribosomes (for review see Peled-Zehavi and Danon 2007). The tRNAs, rRNAs and some ribosomal proteins are encoded by the plastid genome, the remaining components are encoded in the nucleus and imported from the cytosol. Plant mitochondria also require a fully functional translation machinery to express the about 30 mRNAs encoded in the mitochondrial genome. Since only a few ribosomal proteins, rRNAs and an incomplete set of tRNAs are encoded by the mtDNA (Unseld et al. 1997), plant mitochondria must import most of the components of their translational apparatus, e.g. several tRNAs (Salinas et al. 2008) and all the required aminoacyl-tRNA synthetases (Duchêne et al. 2005).
Plastid ribosomes were characterized in proteomics studies in C. reinhardtii and spinach, which has led to the identification of 59 proteins. While 53 ribosomal proteins share homologues with Escherichia coli, six are specific to plastids and termed PRSP1-6 (Yamaguchi and Subramanian 2000; Yamaguchi et al. 2000, 2002, 2003; Yamaguchi and Subramanian 2003). In eubacteria, the Shine-Dalgarno (SD) sequence plays a crucial role in the correct positioning of the ribosome during translation initiation (reviewed in Kozak 2005). In most plastid mRNAs, the SD-like sequence has a similar role, but the distance to the initiation codon is not as conserved as it is in E. coli. In addition, 30 of the 79 protein-coding genes in tobacco do not contain a SD-like sequence at all, indicating that alternative cis-elements and trans-acting factors may be responsible for correct translation initiation (Sugiura et al. 1998). In plant mitochondria, sequences resembling SD sequences are very rare and in the absence of an in vitro translation system, the function of these sequences in translation initiation could not be determined (Pring et al. 1992). Thus, the mechanism controlling translation initiation remains completely elusive in plant mitochondria. Potential translation regulation systems are also unknown. However, the function of PPR proteins might well be connected to plant mitochondrial translation as suggested by the involvement of CRP1 as a chloroplast translation regulator (Schmitz-Linneweber et al. 2005), by the requirement of Pet309 for translation in yeast mitochondria (Tavares-Carreón et al. 2008) and by the association of PPR336 to polysomes in plant mitochondria (Uyttewaal et al. 2008).
Sequence analysis has shown that translation is usually, but not always, initiated with an AUG codon in plant organelles. Alternative codons were found to be possible additional translation initiator triplets in mitochondria and plastids (Bock et al. 1994a; Unseld et al. 1997; Dong et al. 1998; Zandueta-Criado and Bock 2004). Moreover, plant mitochondrial genes can be expressed from mRNAs lacking canonical termination codons with no evidence that alternative termination codons had been created posttranscriptionally by either RNA editing or polyadenylation (Raczynska et al. 2006).
While in bacteria nascent transcripts are directly translated into proteins, the uncoupling of these two processes introduces a new level of regulation in organelles (Mayfield et al. 1995; Danon 1997; Zerges 2000; Peled-Zehavi and Danon 2007).
III. Technological Developments for the Expression Profiling of Organellar Genes
Northern blot analysis, in which a labeled probe is hybridized to a RNA target, was the first and most widely used technology to confirm and quantify gene expression. However, it can only be used to analyze the expression pattern of a limited number of genes under few experimental conditions. The recent development of “-omics” technologies enables researchers to carry out a genome-wide expression profiling, analyzing simultaneously up to thousands of genes. Most of these methods rely on the use of DNA arrays (macro- or micro-), although alternative methods such as differential display, expressed sequence tags (ESTs), serial analysis of gene expression (SAGE), quantitative RT-PCR (qRT-PCR), and others, can alternatively be used for such purposes. The above-mentioned transcriptional profiling technologies allow the analysis of complex RNA populations from different cells or tissues. Although DNA arrays produced a real advance in large-scale expression analysis and are currently widely used for transcript profiling, only limited datasets are as yet available for plant organelles and most of them are related to chloroplast genes or nuclear genes with chloroplast functions (Kurth et al. 2002; Legen et al. 2002; Richly et al. 2003; Kahlau and Bock 2008; Valkov et al. 2009).
In DNA arrays, DNA fragments or oligonucleotides corresponding to different genes or cDNAs are immobilized on a solid support (nylon membranes for macroarrays and glass slides for microarrays), and hybridized as probes to total RNA pools extracted from cells, tissues, whole organisms, etc. The hybridization signal detected for each spot can then be measured giving the relative abundance of the corresponding mRNA (Bouchez and Höfte 1998; Meyers et al. 2004). The simplest and cheapest array systems use nylon membranes in combination with labeled (radioactive) cDNA probes, detected by Phosphorimager instruments (Kurth et al. 2002; Legen et al. 2002; Richly et al. 2003; Geimer et al. 2009). This method allowed to study, on a genome scale, the expression of the entire plastid chromosome of tobacco wild-type and mutant (PEP-deficient) plants (Legen et al. 2002) and Euglena gracilis under different culture conditions (Geimer et al. 2009), or nuclear genes related to chloroplast functions in A. thaliana under different environmental and genetic conditions (Kurth et al. 2002; Richly et al. 2003).
Initially, DNA microarrays were produced with cDNA fragments immobilized on microscope slides, but a competing approach, based on DNA oligonucleotides, has recently become the most widely used system (Bouchez and Höfte 1998; Stears et al. 2003; Meyers et al. 2004). Different fluorescent labeling and detection techniques are used to produce graphical images and numerical data corresponding to the measurement of spot intensities (Schulze and Downward 2001; Stears et al. 2003). The source of variation needs to be carefully controlled by replicating experiments at technical and biological levels (Schulze and Downward 2001; Meyers et al. 2004). In addition, it is often necessary to verify a subset of array results by alternative techniques, generally northern blot analysis and/or qRT-PCR (Schulze and Downward 2001). Several tools for array data analyses have been developed by both commercial and public suppliers (Schulze and Downward 2001; Stears et al. 2003).
Compared to cDNA arrays, arrays based on oligonucleotides offer several advantages: they can be synthesized either in plates or directly on solid surfaces, produce strong hybridization signals of superior specificity, also in the case of individual transcripts of multigene families that share sequence homology (by synthesizing oligonucleotides corresponding to regions of non-identity; Lemieux et al. 1998; Stears et al. 2003). Obviously, this method requires the availability of genome sequence for the organism under study, but this is usually no limitation in the case of organelles, considering the number of genomes continuosly released and the high degree of sequence conservation (http://megasun.bch.umontreal.ca/ogmp/projects/other/all_list.html). A plastome microarray, useable for different Solanaceae species, was recently developed (Kahlau and Bock 2008). This array is based on long (68–71 nucleotides) oligonucleotides and contains all genes and conserved open reading frames present in Solanaceae plastomes. It was designed using the complete tobacco, tomato and potato plastid genomes (Gargano et al. 2005; Yukawa et al. 2005; Daniell et al. 2006; Kahlau et al. 2006), and used to analyze the expression of different tomato and potato plastid genomes and identify regulatory expression patterns in different tissues and plastid types (Kahlau and Bock 2008; Valkov et al. 2009).
As an alternative to the array-based approach, a sequenced-based transcriptomic approach has been recently developed, with significant advantages, such as the potential to quantify the abundance of any transcript and the independence of the availability of a sequenced genome (Wang et al. 2010). Although these technologies have great potential, expression profiling studies based on Ultra High-Throughput Sequencing (UHTS) methods are still limited in plants (Cheung et al. 2006; Emrich et al. 2007; Weber et al. 2007; Schnable et al. 2009) and, so far, none of them has been applied to organelle transcriptomes.
IV. Expression Profiling in Plastids
A. Genotype-Specific Variation
Nuclear and plastid mutants have been used to study the effect of genotypic variability on differential plastid gene expression. Early studies about the profiling of gene expression in mutant genotypes involved the striped and albino mutants of maize and barley (Han et al. 1993; Hess et al. 1993), which show altered chloroplasts in mutated tissues. In maize, the striped iojap 1 (ij1) and albino white 1 (w1) and white 2 (w2) mutants displayed alterations in the levels and sizes of several photosynthesis-related plastid transcripts. Furthermore, reduced protein accumulation was observed in ij1-affected plastids. Unchanged ptDNA content compared to wild-type plastids, and several other observations, suggested that ij1 and w1 mutants might have not only altered transcription rate, but also alterations in transcript processing and stability. On the other hand, the severe reduction of plastome copy number per plastid was likely responsible for the general reduction of transcripts in the w2 mutant (Han et al. 1993). In the ribosome and plastid protein biosynthesis-deficient albostrians mutant of barley (Hess et al. 1993), the accumulation of transcripts for the photosynthesis genes psbA, atpH, atpI, and rbcL was strongly reduced, due to differential transcription rates and transcript stabilities in mutant and wild type plastids. In contrast, transcript accumulation for rpo and rps genes, encoding some subunits of the plastid-encoded RNA polymerase and small ribosome proteins, respectively, was enhanced, suggesting the involvement of NEP in their synthesis.
More recently, macro- and microarray analyses were carried out with mutants of the alga C. reinhardtii and the model Brassicaceae A. thaliana (Erickson et al. 2005; Cho et al. 2009). In the former case, using two RNA stability mutants (mcd1-1 and mcd1-2), such analyses not only confirmed the gene-specific substrate (petD) of the nuclear Mcd1 gene, encoding an mRNA stability factor, but also allowed the discovery of an additional unlinked mutation (mda1-2) affecting accumulation of atpA mRNAs (Erickson et al. 2005). In Arabidopsis, the expression of 94 plastid genes was analyzed in a large set (75) of genotypes including albino mutants arrested at an early stage of chloroplast development, “high chlorophyll fluorescence” (hcf) mutants with impaired photosynthetic electron transport capacity and yellow/pale-green lines with altered, and often unknown, chloroplast functions (Cho et al. 2009). Transcriptomes were clustered in two main groups. Group I, including the majority of albino mutants, displayed up-regulation of non-photosynthetic genes transcribed by NEP and down-regulation of genes transcribed by PEP and encoding photosynthetic proteins. On the other hand, group II showed less evident expression changes and included all hcf, pale-green and the remaining albino mutants. Deviations from the expected profiles in the two groups allowed to identify 14 mutants specifically involved in plastid RNA metabolism.
Knock-out lines of Arabidopsis for the nuclear genes encoding PEP σ-like factors were used to analyze changes in global plastid gene expression and switch in promoter usage (Kanamaru et al. 2001; Nagashima et al. 2004; Schweer et al. 2006). In early experiments (Kanamaru et al. 2001), sig2-1 mutants, showed reduced accumulation of chlorophyll and photosynthesis-related proteins, without significant reduction of the respective mRNAs. On the other hand, the observed phenotypic and biochemical defects were likely related to reduction in synthesis of some tRNAs encoded by genes (trnE-UUC, trnV-UAC, trnM-CAU, trnQ-UUG) with conserved eubacteria-type promoter sequences. Later, microarray analyses carried out on the same mutant line (Nagashima et al. 2004) showed that out of 79 protein coding genes, only the psaJ transcript was reduced in the mutant, whereas transcripts of 47 genes, many under the control of NEP, were increased, suggesting increase of NEP activity in the sig2-1 genotype. In another σ-like factor knock-out line (sig6-2), the appearance of an unusually long transcript was observed in the atpB-E operon (Schweer et al. 2006), suggesting either a role of SIG6 in chloroplast RNA maturation or a differential usage of promoter sequences. Indeed, the authors detected motifs for NEP recognition far upstream of the atpB gene, between the accD and rbcL genes and thus devised a model for the developmentally regulated use of alternative σ-like factors and promoter sequences.
Transgenic tobacco plants overexpressing a plastid-targeted bacteriophage T7 RNA polymerase (T7RNAP) were analyzed for their effects on plastid gene expression (Magee and Kavanagh 2002; Magee et al. 2007). In the former study, northern analyses showed an increase in transcript accumulation for several genes generally transcribed by NEP, but no variation in other genes (psbD, ndhA, rrn16) transcribed exclusively or predominantly by PEP in chloroplasts. These results could be explained by: (a) T7RNAP-mediated transcription from NEP promoters, (b) increased activity of NEP triggered by the presence of T7RNAP activity in chloroplasts of transgenic plants, and/or (c) differential increase in stability of some plastid mRNAs in transgenic plants (Magee and Kavanagh 2002). When the presence of the nuclear-encoded T7RNAP was associated with that of plastid transgenes driven by T7G10 5′ regulatory sequences, hybrid transplastomic plants showed reduced growth and altered expression of both plastid and nuclear genes (Magee et al. 2004). The profile of gene expression in mutant plants was analyzed using a customized array containing target sequences from all 124 tobacco plastid genes and 61 nuclear genes coding for photosynthetic proteins and components of the plastid translational apparatus. Significantly down-regulated plastid genes included those encoding subunits of the PSII, NADH dehydrogenase, ATP synthase, and cytochrome b6f complexes as well as ribosomal RNAs and proteins. Genes co-transcribed in the same polycistronic operons were usually down-regulated similarly. At the same time, up-regulation was observed for the accD gene and the ndhCKJ operon, but while in the former case it was due to read-through transcription from the upstream promoters present in the construct used for transformation, in the latter case it was, as previously shown, likely due to non-specific recognition of a NEP promoter-like sequence by the plastid-directed T7RNAP. Furthermore, a reduction in the amount of the mature clpP mRNA was observed in mutant plants due to altered processing of the primary transcript (Magee et al. 2007).
The availability of rpo deletion mutants, derived by plastid transformation, and the comparative analysis of expression profiles in wild-type and mutant tobacco plants allowed to establish the existence of the nuclear-encoded RNA polymerase in higher plant plastids and to investigate the interplay of the two polymerase types (PEP and NEP) at several levels of gene expression (Allison et al. 1996; Hajdukiewicz et al. 1997; Serino and Maliga 1998; De Santis-Maciossek et al. 1999; Krause et al. 2000; Legen et al. 2002). Based on results of northern analyses and mapping of transcription initiation sites, early seminal studies (Allison et al. 1996; Hajdukiewicz et al. 1997) showed that, while some mRNA accumulation was always detected for all genes analyzed, plastid genes could be grouped in three classes on the basis of relative transcript accumulation levels in wild-type and mutant plants: (1) genes with high accumulation levels in wild-type leaves, but negligible levels in leaves of ΔrpoB plants; (2) genes with similar mRNA leaves in wild-type and mutant plants; (3) genes with significantly more transcript accumulation in mutant than in wild-type plants. Genes or operons with different physiological functions (e.g. photosynthesis or transcription/translation) belonged to different classes and displayed upstream sequences for recognition of either one polymerase type or both. It was thus proposed that transcription by NEP or PEP through differential promoter recognition could determine the pattern of group-specific gene regulation in plastids (Hajdukiewicz et al. 1997). Later studies, however, indicated a more complex picture, likely not based only on differential promoter usage but also on differential posttranscriptional mechanisms (Krause et al. 2000; Legen et al. 2002). In fact, the entire plastome was found to be transcribed in both wild-type and PEP-deficient plastids and, in genes specifying different functions, no correlation was found between transcription rates, transcript levels, transcript patterns, and accumulation levels of derived polypeptides. A predominance of posttranscriptional regulation mechanisms over transcriptional ones was also found in a tobacco psbA gene deletion mutant showing changes in PSII protein accumulation levels and upregulation of the alternative electron transport pathways (Baena-González et al. 2003).
A microarray analysis of the expression profile of 108 plastid DNA fragments carried out in a chloroplast trnR-CCG gene knock-out mutant of the moss Physcomitrella patens indicated that most plastid genes were expressed at comparable levels in mutant and wild-type genotypes, suggesting that the arginine trnR-CCG gene is not essential for plastid gene expression in P. patens (Nakamura et al. 2005).
As previously discussed, the RNA editing process occurs in all major lineages of land plants and only marchantiid liverworts do not seem to edit plastid transcripts (Table 14.1, Freyer et al. 1997; Duff and Moore 2005). By investigating editing patterns of the ndhB and rbcL transcripts, it could be observed that neither plastid editing frequencies nor the editing patterns of a specific transcript correlated with the phylogenetic tree of the plant kingdom (Freyer et al. 1997). Later genome-wide comparisons of the editotypes of one hornwort (Anthoceros formosae), one fern (Adiantum capillus-veneris) and five seed plants (Arabidopsis thaliana, Nicotiana tabacum, Atropa belladonna, Zea mays and Pinus thunbergii) showed that only 18 of the total 85 chloroplast editing sites in seed plants were present also in either one or both other taxa, while the latter shared 53 sites (Tillich et al. 2006). Tsudzuki et al. (2001) compared the conservation of editing sites among several higher plant species. The dicotyledonous plant tobacco and the monocots rice and maize have 12 editing sites in common, which may already have been present before divergence of both taxa (Hirose et al. 1999; Tsudzuki et al. 2001). The conservation of editing sites between gymnosperms and angiosperms is lower. Just five sites out of 26 are shared by the gymnosperm black pine (Wakasugi et al. 1996) and the so far analyzed angiosperms (Tsudzuki et al. 2001). The editing sites were also compared within more closely related taxa. The three Solanaceae tobacco (N. tabacum), tomato (Solanum lycopersicum) and deadly nightshade (A. belladonna) have 30 of the so far known sites in common. While 2–3 sites are always shared by two species, 1–2 appear to be species-specific (Schmitz-Linneweber et al. 2002; Kahlau et al. 2006). Differences in editing can even be observed at the sub-species level. The editotypes of three different ecotypes of A. thaliana, Columbia (Col-0), Cape Verde Islands (Cvi-0) and Wassilewskija (Ws-2) were characterized. One non-synonymous point mutation was detected in Cvi-0 at the first position of codon 17 of the ndhG gene. This codon is usually edited in all three ecotypes with a C-to-U transition at the second position. This mutation changes a serine codon in Col-0 and Ws-2 into an alanine codon in Cvi-0 at the DNA level, resulting at the RNA level in a phenylalanine or valine codon, respectively (Tillich et al. 2005).
By analyzing differences among various species, it is interesting to note how fast the capability to edit certain sites was lost during evolution. In A. thaliana, the cis-elements of matK(2) and ndhB(11) editing sites show striking similarities, suggesting that they share the same trans-acting factor, a common mechanism which has been experimentally proven for other sites (Chateigner-Boutin et al. 2008; Hammani et al. 2009). However, while the matK(2) editing event restores a conserved tyrosine, the ndhB(11) does eliminate an evolutionary conserved serine. These results indicate that the matK(2) editing is the primary target whereas ndhB(11) editing might be secondary (Tillich et al. 2005). The potential of one trans-acting factor to recognize several similar cis-elemets might explain the capability of some species to edit foreign sites. The site rps12(74) is edited in tomato, but absent from tobacco, as the ‘T’ is already encoded at the DNA level (Kahlau et al. 2006). After introduction of the tomato editing site into the tobacco plastid genome, the transplastomic tobacco plant is able to edit this site with high efficiency. There are two possible explanations for this scenario: (1) either the responsible trans-acting factor is still present in the tobacco nuclear genome and was not lost during evolution or (2) the pre-existence of the rpoB(667) site facilitated the evolution of the rps12 site, as both cis-elements share high similarity (Karcher et al. 2008). Other attempts to edit heterologous editing sites in vivo have not been successful so far. In contrast to the tomato rps12 site, tobacco is not able to edit a foreign psbF site introduced from spinach (Spinacia oleracea, Bock et al. 1994b). But also more closely related species show nuclear-plastidial incompatibilities. By creating cybrids and introducing tobacco chloroplasts into deadly nightshade nuclear backgrounds, it became evident that the nuclear genome of nightshade is not able to support editing at all tobacco editing sites. The editing at site atpA(264) is absent, resulting in an albino phenotype. The most likely explanation is the absence of a nuclear-encoded editing factor in the nightshade which is responsible for correct processing of the site in tobacco (Schmitz-Linneweber et al. 2005).
B. Variation Due to Developmental and Environmental Cues
Most plastids are able to interconvert into other types following developmental and environmental cues (for review see Pyke 2007). Plastid gene expression and its regulation have been extensively studied in chloroplasts, which are present in photosynthetically active green tissues and generally develop from proplastids in meristems or etioplasts after illumination of dark-grown tissues. Non-green plastids, such as amyloplasts, chromoplasts and others, fulfill important functions in storage and pigmentation and are also the place of important metabolic pathways. The knowledge on gene expression in such plastid types, however, is still very limited.
Monocots are good model organisms to study changes in gene expression during chloroplast development. Cells at the leaf base contain proplastids which develop eventually into mature chloroplasts, present in the tip of the leaves. Using a custom maize chloroplast biogenesis cDNA microarray, it was shown that the abundance of most plastid transcripts in maize leaves (52 out of 63 analyzed) increases more than twofold during development (Cahoon et al. 2008). Ten transcripts which are present at similar levels in both plastid types are mostly involved in plastid gene expression and are transcribed by NEP (Cahoon et al. 2008). Since, in maize, transcription activity increases in developing chloroplasts, the latter transcripts are likely less stable in mature chloroplasts compared to proplastids at the leaf base (Cahoon et al. 2004, 2008). In the same study, a coordinated gene expression pattern in the nucleus and the plastids, likely based on a combination of anterograde and retrograde signalling between the two organelles, was found (Cahoon et al. 2008). Barley, another monocot, shows differences in transcript patterns during proplastid-to-chloroplast conversion compared to maize. At the leaf base, plastid transcriptional activity and transcript abundance are low. An increase in both can be seen in etioplasts, present in cells which already stopped dividing and entered the cell elongation phase. In contrast to maize, after illumination and further chloroplast maturation, transcript abundance and transcriptional activity decline again (Baumgartner et al. 1989, 1993). Another plastome-wide study in barley could not detect global quantitative changes in gene expression. During the de-etiolation process, no changes were found between etioplasts and chloroplasts, neither in relative transcription rates nor in transcript stability (Krupinska and Apel 1989). Differences between maize and barley could be caused by the differences in CO2 fixation mechanisms. Barley uses the C3 fixation mechanism while maize is a C4 plant showing the typical Kranz anatomy. Maize bundle sheath and mesophyll cells have very different tasks during CO2 fixation and also differ in their plastid transcript profiles. Transcripts for subunits of photosystem II are more abundant in mesophyll cells while rbcL is more abundant in bundle-sheath cells (Kubicki et al. 1994), the cell type in which concentrated CO2 is fixed by Rubisco. It is possible that these two cell types also differ in their transcript patterns during proplastid-to-chloroplast development. In another monocot study based on macroarrays for studying chloroplast gene expression profiles, changes in gene expression levels were monitored using RNA isolated from germinating wheat seeds and seedlings at different stages of development (Siniauskaya et al. 2008). While transcript levels for PSI and PSII genes increased after imbibition until 1 week of development, the levels of other transcripts (e.g. those of ndh and atp genes) either did not change or decreased.
In the dicot N. tabacum, dark-grown etioplast-containing and illuminated chloroplast-containing seedlings were compared by using a microarray with 220 ptDNA fragments, each corresponding to a single known gene or an intergenic region and altogether covering the whole plastome (Nakamura et al. 2003). A clear trend of gene expression within the two functional groups of plastid-encoded genes was evident. The majority of photosynthesis-related genes increased in their abundance in illuminated plants. On the other hand, the majority of genes involved in gene expression were expressed at similar levels in both plastid types. Furthermore, unexpected signals were found in several intergenic regions, suggesting the existence of novel transcripts (Nakamura et al. 2003). Similar studies conducted in the unicellular red alga Cyanidioschyzon merolae with a microarray containing almost all plastid protein coding genes, northern blot analyses and run-on transcription assays, showed differential activation of gene transcription by illumination (Minoda et al. 2005).
Results from microarray analyses in tobacco and Arabidopsis (MacLean et al. 2008) showed coordinated expression of nuclear and plastid genes encoding ribosomal proteins during seedling development. Transcript accumulation responded similarly to light and inhibitors of plastid signaling. In another study comprising the same two species, the effect of green light on seedling development and plastid gene expression was analyzed by using genome microarrays and RNA gel blot experiments (Dhingra et al. 2006). In both species, etiolated seedlings subjected to a short, dim, single pulse of green light showed stem elongation and concomitant decrease in a sub-set of plastid-encoded transcripts, including several ones known to be light inducible. The majority of plastid transcripts did not vary, while only three increased in abundance, indicating that the effect of green light on plastid gene expression is gene-specific.
As a representative for eukaryotes carrying secondary endosymbionts, plastid gene expression was analyzed in the protist Euglena gracilis. Similar to primary endosymbionts, E. gracilis plastids encode mainly genes involved in transcription, translation and photosynthesis (Hallick et al. 1993). Early work on E. gracilis using RNA-DNA hybridizations had already shown that plastid genes are transcribed in proplastids of dark-grown cells (Chelm and Hallick 1976; Rawson and Boerma 1976; Chelm et al. 1979). Although Dix and Rawson (1983) could not identify individual genes, they could distinguish between two major groups: (1) genes which are constitutively transcribed and (2) genes encoding transcripts which increase in their abundance during the greening process, as e.g. psbA, encoding a core subunit of photosystem II (Hollingsworth et al. 1984). A recent study analyzed the complete E. gracilis transcriptome under 12 different growth and stress conditions using a macroarray-based approach (Geimer et al. 2009). Overall, the organelle transcriptome showed pronounced global quantitative changes, but qualitative changes were negligible. After growth in darkness, the overall transcript abundance was much lower than in light-grown cells, but psbA transcription increased drastically. The trnI-CAU gene, involved in gene expression and an example for genes identified by Dix and Rawson (1983) as being constitutively transcribed, did not change in abundance (Geimer et al. 2009). As plastid gene expression patterns in E. gracilis remain more or less constant with quantitative changes on a global scale, these results suggest that fine-tuning of protein production might be regulated posttranscriptionally. In contrast to the limited global transcriptional changes happening in E. gracilis (Geimer et al. 2009), translational regulation is much more pronounced (Miller et al. 1983). Although this study did not identify single plastid-encoded proteins, it could clearly show the gap between transcriptional changes, which are just up to threefold, and the total rates of protein synthesis, which were increased to up to 100-fold. Different sets of proteins were expressed at different time points during proplastid-to-chloroplast development (Miller et al. 1983). Analyzing and comparing psbA transcription with protein accumulation during de-etiolation identified similar patterns in barley. Without noticeable changes in psbA mRNA, the encoded protein increased drastically once the plants were illuminated (Klein and Mullet 1987).
Eberhard and coworkers (2002) analyzed the ability of plastids to override transcriptional changes at the translational level more systematically. C. reinhardtii cells were treated with rifampicin causing depletion of plastid transcripts by binding to and inhibiting the eubacterial-type RNA polymerase. Most of the analyzed transcripts dropped in their abundance to 10% compared to prior to the treatment. Despite these significant changes in RNA levels, the rate of protein synthesis measured with pulse-chase labeling experiments did not drop during the treatment (Eberhard et al. 2002).
Amyloplasts are present in storage organs like tubers or seed endosperm as well as in columella cells of root tips (for review see Pyke 2007). Several genes (16S rRNA, atpB/E, psbA, rbcL) were analyzed with respect to their abundance in A. thaliana and spinach root amyloplasts (Deng and Gruissem 1988; Isono et al. 1997a). All analyzed transcripts could be detected, but their abundance was highly decreased compared to leaf chloroplasts. Although plastid DNA levels were lower in Arabidopsis roots than in leaves, these differences could not explain the large differences in transcript abundances (Isono et al. 1997a). Similar observations were made by analyzing amyloplasts of potato tubers (Brosch et al. 2007; Valkov et al. 2009). The rather small reduction in plastid DNA content of approximately two- to threefold could not account for the reduction in transcript accumulation (Valkov et al. 2009). Run-on assays showed that the decrease in steady-state RNA levels was largely due to a decrease in transcriptional activity in amyloplasts (Sakai et al. 1992; Brosch et al. 2007; Valkov et al. 2009), although differences in stability could explain variable transcript accumulation levels. To get a more detailed insight into tissue versus plastid specificity, bell pepper (Capsicum annuum) and tomato (Solanum lycopersicum) fruits, which convert chloroplasts (highly transcriptionally active in leaves) into red chromoplasts during fruit ripening, were analyzed. Surprisingly, chloroplasts in green fruits already show reduced transcript abundance and differ from their counterparts in leaves, pointing towards a developmental regulation of plastid transcription. Bell pepper fruit plastids show reduction in steady-state RNA levels which is due to a reduced transcriptional activity. However, no significant changes during ripening and chloroplast-to-chromoplast conversion in transcriptional activity could be detected (Kuntz et al. 1989). Similarly, in tomato and pumpkin (Cucurbita pepo), RNAs are present at lower levels in fruits compared to leaves, while transcriptional activity is already downregulated in green fruits and does not change significantly during ripening (Piechulla et al. 1985; Marano and Carrillo 1992; Obukosia et al. 2003; Kahlau and Bock 2008).
Although the functions and structure of the two plastid types are very different, amyloplasts and chromoplasts share striking similarities in their gene expression profiles (Fig. 14.1). In both plastid types, transcript abundance was highly reduced compared to leaf chloroplasts, but in both cases the differences could not be attributed to the absence of one of the two RNA polymerase activities present in plastids (Kahlau and Bock 2008; Valkov et al. 2009). In fact, although differences in promoter utilization were observed by comparing amyloplasts or chromoplasts to chloroplasts, both the nuclear-encoded and plastid-encoded RNA polymerases are active in non-green plastids. Plastome-wide expression profiling showed that two genes, clpP and accD, are expressed at similar high levels in leaf chloroplasts and amyloplasts or chromoplasts (Kahlau and Bock 2008; Valkov et al. 2009). clpP, a subunit of a protease, is essential in tobacco and important for plant development (Shikanai et al. 2001; Kuroda and Maliga 2003; Clarke et al. 2005; Adam 2007). Many nuclear-encoded proteins are imported into all plastid types (Baginsky et al. 2004; Siddique et al. 2006; Bancel et al. 2010; Barsan et al. 2010; Daher et al. 2010), indicating that the Clp protease is probably needed for the removal of damaged proteins (Zybailov et al. 2009). The accD gene is also essential and cannot be deleted from the plastid genome (Kode et al. 2005). The encoded protein is part of the plastid-localized Acetyl-CoA carboxylase (ACCase), catalyzing the first committed step in fatty acid biosynthesis. Residual expression of the plastid gene expression machinery may be necessary to produce the ACCase and secure therefore further production of lipids needed in all cell membranes (Kahlau and Bock 2008; Valkov et al. 2009).

Genome-wide analysis of total (a) and polysomal (b) RNA accumulation in potato tuber amyloplasts (Log2 T/L) and tomato red fruit chromoplasts (Log2 RF/L) compared to leaf chloroplasts (Kahlau and Bock 2008; Valkov et al. 2009). Based on their function, genes analyzed were grouped in three classes. The accD and clpP genes are highlighted.
Regulation of plastid translation at the level of polysome formation was investigated in various species and plastid types. In spinach root amyloplasts, representative transcripts involved in photosynthesis were detectable, but specifically depleted from polysomal fractions (Deng and Gruissem 1988). The situation is similar in potato tuber amyloplasts as well as in tomato fruit chromoplasts (Brosch et al. 2007; Kahlau and Bock 2008; Valkov et al. 2009). Both plastid types show a large reduction in polysome-associated mRNAs. In addition to the constantly low mRNAs levels in tomato fruit plastid-types, translation is increasingly down-regulated during chloroplast-to-chromoplast conversion for almost all mRNAs. The only genes which showed potentially similar translation levels in leaves and non-green plastids were genetic system genes like those encoding the subunits of the plastid-encoded RNA polymerase, ycf1 and ycf2 (open reading frames of unknown function) and, interestingly, clpP and accD (Kahlau and Bock 2008; Valkov et al. 2009). Hence, also at the translational level, transcripts of the latter two genes differ in their regulation pattern from almost all other plastid mRNAs and the low level of plastid gene expression is probably maintained to secure the production of the Clp protease and the ACCase for fatty acid biosynthesis (Kahlau and Bock 2008; Valkov et al. 2009).
Both in higher and lower plant plastids, the RNA editing process was likewise shown to be affected by changes in the environmental conditions as well as the organ and plastid type analyzed (Bock et al. 1993; Hirose et al. 1996; Hirose and Sugiura 1997; Ruf and Kössel 1997; Karcher and Bock 1998; Hirose et al. 1999; Karcher and Bock 2002a, b; Peeters and Hanson 2002; Chateigner-Boutin and Hanson 2003; Miyata and Sugita 2004; Kahlau and Bock 2008; Valkov et al. 2009). However, results of studies analyzing individual sites as well as those of a comprehensive study in maize, involving 27 editing sites in 15 genes and 10 different tissues (Peeters and Hanson 2002), demonstrate that environmental and developmental effects on RNA editing efficiency are not consistent in different genes and/or editing sites. Furthermore, developmental co-variation of RNA editing extent in some editing sites was shown by surveying 34 editing sites in 15 tobacco genes (Chateigner-Boutin and Hanson 2003). In bell pepper chromoplasts, the psbL initiation codon is still edited although the product is obviously not needed in the non-photosynthesizing ripe fruits. These results suggest that editing is in this case not responsible for the regulation of PsbL protein expression (Kuntz et al. 1992). psbL and psbF editing were also analyzed in illuminated and etiolated leaf tissue as well as in roots and seeds of spinach (Bock et al. 1993). Editing was complete in leaf etioplasts as well as chloroplasts, indicating that light had no influence on editing extent in these two plastid-types. However, editing of these two sites in proplastids (seeds) and amyloplasts (roots) was incomplete. As unedited start codons render transcripts probably untranslatable, editing might be one mechanism controlling plastid gene expression (Bock et al. 1993). Several editing sites in the ndhA, ndhB and ndhF transcripts, encoding subunits of the NAD(P)H dehydrogenase complex, show incomplete editing in A. thaliana roots (Chateigner-Boutin and Hanson 2003). However, a functional significance of incomplete editing for regulation of gene expression is questionable in this case as the NdhD protein is completely absent in roots. The ndhD start codon is only partially edited in tobacco, tomato, potato and Arabidopsis leaf tissue. The incomplete editing in leaves is conserved across several species, but the editing extent in non-green plastid types varies considerably. Editing of ndhD(1) is completely absent in Arabidopsis roots and potato tubers, but is partial in tobacco and spinach roots as well as tomato fruits (Chateigner-Boutin and Hanson 2003; Kahlau and Bock 2008; Valkov et al. 2009). All available results suggest the importance of selective activation/inhibition of site-specific nuclear-encoded trans-factors (sometimes able to recognize more than one editing site) in explaining developmental and/or environmental differences in RNA editing efficiency. By comparing different species and plastid-types, however, no common pattern can be identified which could hint towards a role of editing in the regulation of plastid gene expression. In addition, no editing sites specific for non-green plastid types have been identified so far.
Although genome-wide studies on the effect of environment and/or development on transcript processing are missing, available data for a number of genes showed at least in some cases a reduction of transcript splicing in non-green plastids compared to leaf chloroplasts, suggesting a possible link with limited expression of the plastid genome in some tissues and plastid types (Barkan 1989; Kahlau and Bock 2008; Valkov et al. 2009).
V. Expression Profiling in Mitochondria
A. Genotype-Specific Variation
Only few studies investigated the mitochondrial expression profiling of natural and mutant plant populations. The albostrians mutant of barley, characterized by a very low expression level of photosynthesis-related plastid and nuclear genes, was used to study the influence of impaired chloroplast development on mitochondrial gene and transcript levels (Hedtke et al. 1999). The analysis of mitochondrial steady-state RNA levels in different tissues showed an enhanced transcript accumulation of all mitochondrial genes tested in white leaves, due to a threefold higher mitochondrial gene copy number. Further, because the increased transcript levels in mitochondria of white leaves could be caused by either the differentiation state of plastids or the direct action of the mutated nuclear albostrians allele, plants derived by reciprocal crosses between a green wild-type and a white (striped) albostrians parents were analyzed, showing that the enhanced transcript levels were a consequence of the impaired plastids and not of the nuclear mutant allele. These results highlight the crucial importance of inter-organellar crosstalk in plant cells.
In order to gain more knowledge about species-specific regulation of plant mitochondrial gene expression, Leino et al. (2005) compared transcriptional activity and RNA turnover in a cytoplasmic male-sterile (CMS) Brassica napus line, the corresponding male-fertile progenitors (A. thaliana and B. napus), and a fertility-restored line. The alloplasmic CMS line was obtained by protoplast fusion between A. thaliana and B. napus and contained mitochondrial DNA (mtDNA) mostly inherited from A. thaliana with some mtDNA fragments from B. napus, whereas the nucleus contained pure B. napus DNA (Leino et al. 2003). The fertility-restored line was isogenic for its mtDNA but had an additional pair of A. thaliana chromosome III in the nuclear genome (Leino et al. 2004). The analysis of transcriptional activities by run-on assays revealed a high variability between the parental species, with a higher transcript activity in B. napus than in A. thaliana for the atp8, ccmB, rps7 and rrn5 genes, and an opposite relationship for the nad4L, nad9 and cox1 genes. By contrast, the values obtained for the CMS and restored lines were very similar for all tested genes. The authors suggested that the differences observed in transcription activity could be due to differences in promoter strength, as already found in other species (Muise and Hauswirth 1992; Giegé et al. 2000). In comparison with transcription activities, the transcript steady-state levels were more homogeneous demonstrating that RNA turnover might act as a compensating mechanism.
In another study, the major transcript ends of all mitochondrial protein-genes were compared in three A. thaliana accessions (Forner et al. 2008). Authors identified mRNA polymorphisms for several genes (nad4, nad9, ccmB, ccmC, rpl5-cob), and linked them to variations at the 5′ ends that were conserved in all tissues analyzed. Since the polymorphisms observed could be caused by mitochondrial sequences or by differences in nuclear genes, they analyzed the inheritance of polymorphic mRNAs in reciprocal F1 hybrids. These analyses showed a maternal (ccmC) or biparental (nad4, nad9, ccmB and rpl5) inheritance for polymorphic transcripts, suggesting that they could arise from differences in mtDNA or nuclear-encoded trans-factors, respectively. Despite intensive research in the past years, most of the cis-acting sequence elements and trans-factors required to generate mature 5′ and 3′ ends of mtRNA of higher plants are still unknown, Forner et al. (2008) suggested that the analysis of reciprocal F1 hybrids is a promising approach to identify mitochondrial cis-elements and nuclear-encoded trans-factors involved in 5′ end formation or mRNA stability.
Comprehensive studies were carried out on RNA editing in different ecotypes and tissues of Arabidopsis mitochondria (Table 14.1, Giegé and Brennicke 1999; Bentolila et al. 2008; Zehrmann et al. 2008). Giegé and Brennicke (1999) identified a total of 456 C-to-U conversions in suspension cultures of A. thaliana, of which 441 reside in open reading frames (orfs). Differences among Arabidopsis ecotypes both for the extent of RNA editing and accession-specific editing sites were found (Bentolila et al. 2008; Zehrmann et al. 2008). Dominance relationships and maternal effects were assessed for the most polymorphic sites by evaluating the degree of editing in reciprocal hybrids. Dominance was more common in non-silent than in silent sites, while additivity was observed only in silent sites. For more than half of the inspected sites, a significant difference depending on the direction of the cross was found (Bentolila et al. 2008). Quantitative variations among ecotypes suggested that the extent of editing can evolve more rapidly than the species (Zehrmann et al. 2008).
A comparative analysis of the mitochondrial genes and RNA editing sites of B. napus L. and A. thaliana was carried out by Handa (2003), identifying 427 editing sites in genes and orfs of B. napus compared with 441 sites in A. thaliana (Table 14.1, Giegé and Brennicke 1999). The number of editing sites shared by both plant mitochondria was 358, which correspond to 83.8% and 81.2% of the total editing sites in B. napus L. and A. thaliana transcripts, respectively. These percentages seem to be low considering that mitochondrial DNA nucleotide identity (for protein coding regions) between the two species was 99.2%. This means that, as already found in plastids, RNA editing variations in plant mitochondria evolve more rapidly than coding sequences. By contrast, in the moss model system Physcomitrella patens, only 11 editing sites in 9 mitochondrial genes (atp9, cox1, cox2, cox3, nad3, nad4, nad5, rps14 and ccmFC) were found, and only the codon positions reconstituting highly conserved amino acids in the encoded proteins were subjected to C-to-U conversions (Rüdinger et al. 2009).
B. Variation Due to Developmental and Environmental Cues
The plant mitochondrial genome is far from being able to express all the required proteins for mitochondrial respiration and translation (Unseld et al. 1997). Various and precise communication mechanisms must be necessary for the biogenesis of mitochondrial protein complexes and especially for the modulation of this biogenesis. A number of studies have established that mitochondrial respiration can be modulated in the plant cell in response to environmental stimuli, at some particular developmental stages or in response to stress (Wood et al. 1996; Svensson and Rasmusson 2001; Giegé et al. 2005; Ribas-Carbo et al. 2005). If this modulation of respiration is due to changes in the number of respiratory complexes per cell, it means that the biogenesis of respiratory complexes can be adjustable as well. A coordinated expression must exist between mitochondrial and nuclear genes, between nuclear genes and between mitochondrial genes encoding subunits of the same respiratory complexes (Giegé et al. 2005; Welchen and Gonzalez 2006; Gonzalez et al. 2007). While many nuclear genes are clearly (co-)regulated at the transcriptional level, the mechanisms regulating coordination of mitochondrial gene expression are less clear.
A global study of the Arabidopsis mitochondrial transcriptome had shown that individual genes or transcription units are transcribed with distinct rates even if they encode components of the same multi-subunit complexes. These differences are at least partially counterbalanced at the steady-state RNA level by posttranscriptional processes and different RNA stabilities (Giegé et al. 2000). Are the steady-state RNA levels obtained invariable or can they be regulated, e.g. during changing developmental stages? To address this question, Li-Pook-Than and colleagues (2004) examined RNA levels of wheat mitochondrial genes during the developmental period when seeds leave dormancy, germinate and develop into seedlings. Mitochondrial transcript levels from 0 h to 6 days post-imbibition were analysed. Stable and edited messengers were observed in dormant seeds and precursor RNAs were subsequently detected early in embryo germination. Respiratory chain genes showed mRNA profiles comparable to those of ribosomal RNAs, whereas ribosomal protein genes had proportionately lower steady-state mRNA levels in later stages of seedling development. The relative levels of precursors compared with the respective mRNAs decreased during development, consistent with transcription outpacing RNA processing in early stages of development. However, coordination was more effective several days after imbibition. In the case of multiply split genes containing group II introns, complex patterns of splicing intermediates were observed. This suggested an absence of strict polarity for splicing. Spliced introns were typically more abundant in embryos than in seedlings. These observations suggest a transient delay of the RNA processing mechanisms at the beginning of seed germination, a period where mitochondrial biogenesis is rapid and apparently demanding for the posttranscriptional machinery (Li-Pook-Than et al. 2004). In another global study, Howell and colleagues (2006) described mitochondrial biogenesis during imbibition of rice embryos both at the morphological and the molecular levels. For a subset of mitochondrial encoded subunits of the respiratory chain genes, they observed two different transcript expression profiles. While complex V atp1 and complex IV cox2 transcripts reached maximum levels at 48 h after imbibition, complex I nad9 and complex III cob message levels peaked much earlier at 8 h (Howell et al. 2006). Similar to the previous case, this showed that gene expression does not seem to be synchronized in early developmental stages and could suggest that mitochondrial transcripts rather follow a defined expression pattern in early development for the biogenesis of mitochondrial complexes. A more comprehensive investigation of mitochondrial transcript profiles during germination and early seedling development in wheat gave similar results (Khanam et al. 2007). In this study, the mitochondrial transcripts were present in the initial dry embryo at variable levels. During early development, gene expression levels of individual genes were very variable. However, genes could be classified into four categories according to their expression patterns. Most mitochondrial respiratory genes were found in two categories. For one category, the timing of RNA accumulation corresponded to the activation of respiration, but not for the other one. Altogether, this work suggested that the initial respiratory burst during early development is supported by stored preexisting respiratory components, whereas de novo mitochondrial gene expression rather supports the subsequent seedling growth (Khanam et al. 2007). It also suggested that the availability of substrates might be a regulatory factor or a signal for the initiation of gene expression in plant mitochondria. Gene expression profiles were also monitored for later developmental stages. Mitochondrial encoded transcript levels, together with chloroplast and nuclear RNA levels, were followed along a maize leaf developmental gradient (Cahoon et al. 2008). Twenty-five out of the 27 mitochondrial transcripts investigated had at least twofold higher steady-state levels in the leaf base than in the rest of the leaf. This mitochondrial gene expression pattern is not surprising because the actively dividing and expanding base of maize leaves has high energy demands and is expected to contain highly active mitochondria (Cahoon et al. 2008). However, from this particular study, it is difficult to conclude whether mitochondrial gene expression had been up-regulated in response to a developmental signal or whether the transcript level differences observed were due to an enriched content in mitochondria per cell at the maize leaf base.
The examination of plant mitochondrial transcript profiling studies has shown that specific transcript profiles emerge during development. Is this also the case in response to external stimuli? Variations for plant mitochondrial transcript profiles have also been observed during the day and night cycle (Okada and Brennicke 2006). In Arabidopsis mitochondria, these authors found that the transcription activity (measured by run-on RNA assays) varied during the diurnal cycle. In contrast, the steady-state transcript levels did not vary between light and dark phases and were stable throughout the diurnal as well as the circadian time course. From this, the authors concluded that the steady-state transcript levels available in plant mitochondria are sufficient to provide sufficient translation capacity at any time during the diurnal cycle (Okada and Brennicke 2006). This, together with previous work (Giegé et al. 2000), also illustrates that, in mitochondria, transcriptional variations are buffered at the level of posttranscriptional processes.
In a global study, where coordination of gene expression between the nucleus and mitochondria was investigated, authors have applied sugar starvation to Arabidopsis cells (Giegé et al. 2005). In this study, the overall mitochondrial transcript levels appeared to increase when sucrose was removed from the growth medium. On the other hand, the levels of mitochondrial transcripts drastically decreased when sugar was added back to the medium. These variations of RNA levels did not necessarily reflect adjustments in mitochondrial gene expression. The authors rather concluded from their results that the relative increase of mitochondrial transcript levels was due to an overall decrease of nuclear transcript levels in response to stress. Thus, after starvation, the proportion of mitochondrial RNA had increased among total RNA and vice versa, when sugar was added back, nuclear RNA expression had increased again and the proportion of mitochondrial RNA decreased among total RNA. Therefore, at least in this particular case, it appears that mitochondrial transcript levels had not been regulated in response to environmental demands. The required adjustment had rather been achieved by changes in nuclear gene expression and was reflected at the level of mitochondrial protein complexes assembly (Giegé et al. 2005). In another study, the effect of antimycin A treatment on mitochondrial function in wheat embryos was described. The transcript levels of five mitochondrial genes and two nuclear genes encoding mitochondrial proteins decreased in response to stress whereas the alternative oxidase (AOX) level increased (Naydenov et al. 2008). Although this study had not been conducted on a global scale, it suggested that in this case, antimycin A treatment had indeed been reflected at the level of mitochondrial gene expression. Finally, in a recent and very comprehensive study, the effects of low temperature, high salinity and high osmotic potential on the mitochondrial transcriptome have been monitored in wheat embryos (Naydenov et al. 2010). Most of the transcript level variations were stress specific. However, groups of genes could be defined with common responses to different stresses (Fig. 14.2). The authors predict from these results that common regulatory mechanisms must be active in response to some conditions whereas other regulatory processes appear to be active to specifically regulate the mitochondrial transcriptome in response to a particular situation (Naydenov et al. 2010).

Mitochondrial transcriptome variations in wheat embryos in response to stresses, as modified from Naydenov et al. (2010). Stresses were applied for 3 days. Up- and down-regulated genes (>1.5-fold) are shown in bold or plain font, respectively. Stress-specific responses are observed for some genes; however, other genes show common response patterns to two or three different stresses.
The extent of editing in plant mitochondria was also found to be affected by developmentally-related effects. In fact, 67 new editing sites not previously observed in A. thaliana Col-0 cell-suspension cultures (Giegé and Brennicke 1999), were detected in rosette leaves (Bentolila et al. 2008). In contrast, 37 of the 441 editing events reported in suspension cultures were not observed in rosette leaves (Table 14.1). The proportion of silent sites in the two classes showing differential editing in the two tissues was similar: 48% (32/67) and 43% (16/37). These percentages were significantly higher than the proportion of silent sites found in the whole population of sites edited in either tissue (20%, Bentolila et al. 2008).
VI. Conclusions
Gene expression in plant organelles can be controlled either at the transcriptional or posttranscriptional level. The former is based on the differential use of multiple promoters and RNA polymerases (PEP in plastids, different NEP isoforms in plastids and mitochondria), and the action of various auxiliary factors. At the RNA level, the posttranscriptional regulation relates to differential editing, processing, stability and translatability of transcripts. Although the investigation of transcript profiling in plant organelles does not enable to draw general conclusions, available studies suggests that transcription itself is not highly regulated both in plastids and mitochondria, and that the steady-state levels of transcripts rather appear to be predominantly obtained through posttranscriptional processes.
Various steps of gene expression in plant organelles have been analyzed at a genome-wide scale by using DNA array-based technologies or others. Genotypic variability for the extent of RNA editing or transcript processing and stability in cytoplasmic organelles has been observed in natural populations at the interspecific and intraspecific level or in artificial CMS lines. The possibility to produce knock-out lines by plastid transformation has been particularly useful to highlight the role of various plastid genes on global genome expression and chloroplast development.
Specific transcript profiles can clearly be achieved also in response to developmental signals and environmental stimuli. Significant differences in the transcriptome, editome and translatome have been found comparing different plastid types in diverse organs or tissues. Similar differences have been found for mitochondrial genomes during the diurnal cycle or between cell suspensions and differentiated leaves. However, the precise levels and mechanisms at which these changes are achieved and the signals necessary to trigger them are barely understood.
Acknowledgments
Work in authors’ labs was supported by the European Union (FP6 Plastomics Project grant no. LSHG–CT–2003–503238) to TC and by research grant 07-JCJC-0123 from the Agence Nationale de la Recherche (ANR) to PG. IGV publication no. 357.
Abbreviations
- ACCase:
-
Acetyl-CoA carboxylase;
- AOX:
-
Alternative oxidase;
- CMS:
-
Cytoplasmic male sterility;
- NEP:
-
Nuclear encoded polymerase;
- PEP:
-
Plastid encoded polymerase;
- PPR:
-
Pentatricopeptide repeat;
- PSI:
-
Photosystem I;
- PSII:
-
Photosystem II;
- RNAP:
-
RNA polymerase
References
Adam Z (2007) Protein stability and degradation in plastids. Top Curr Genet 19:315–338
Allison LA, Simon LD, Maliga P (1996) Deletion of rpoB reveals a second distinct transcription system in plastids of higher plants. EMBO J 15:2802–2809
Baena-González E, Allahverdiyeva Y, Svab Z, Maliga P, Josse EM, Kuntz M, Mäenpää P, Aro EM (2003) Deletion of the tobacco plastid psbA gene triggers an upregulation of the thylakoid-associated NAD(P)H dehydrogenase complex and the plastid terminal oxidase (PTOX). Plant J 35:704–716
Baginsky S, Siddique A, Gruissem W (2004) Proteome analysis of tobacco bright yellow-2 (BY-2) cell culture plastids as a model for undifferentiated heterotrophic plastids. J Proteome Res 3:1128–1137
Bancel E, Rogniaux H, Debiton C, Chambon C, Branlard G (2010) Extraction and proteome analysis of starch granule-associated proteins in mature wheat kernel (Triticum aestivum L.). J Proteome Res 9:3299–3310
Barkan A (1989) Tissue-dependent plastid RNA splicing in maize: transcripts from four plastid genes are predominantly unspliced in leaf meristems and roots. Plant Cell 1:437–445
Barkan A (1993) Nuclear mutants of maize with defects in chloroplast polysome assembly have altered chloroplast RNA metabolism. Plant Cell 5:389–402
Barsan C, Sanchez-Bel P, Rombaldi C, Egea I, Rossignol M, Kuntz M, Zouine M, Latché A, Bouzayen M, Pech JC (2010) Characteristics of the tomato chromoplast revealed by proteomic analysis. J Exp Bot 61:2413–2431
Baumgartner BJ, Rapp JC, Mullet JE (1989) Plastid transcription activity and DNA copy number increase early in barley chloroplast development. Plant Physiol 89:1011–1018
Baumgartner BJ, Rapp JC, Mullet JE (1993) Plastid genes encoding the transcription/translation apparatus are differentially transcribed early in barley (Hordeum vulgare) chloroplast development (evidence for selective stabilization of psbA mRNA). Plant Physiol 101:781–791
Benne R, Van den Burg J, Brakenhoff J, Sloof P, Van Boom JH, Tromp MC (1986) Major transcript of the frameshift coxII from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell 46:819–826
Bentolila S, Elliott LE, Hanson MR (2008) Genetic architecture of mitochondrial editing in Arabidopsis thaliana. Genetics 178:1693–1708
Biehl A, Richly E, Noutsos C, Salamini F, Leister D (2005) Analysis of 101 nuclear transcriptomes reveals 23 distinct regulons and their relationship to metabolism, chromosomal gene distribution and co-ordination of nuclear and plastid gene expression. Gene 344:33–41
Binder S, Brennicke A (1993) A transfer RNA gene transcription initiation site is similar to messenger RNA and rRNA promoters in plant mitochondria. Nucleic Acids Res 21:5012–5019
Binder S, Marchfelder A, Brennicke A, Wissinger B (1992) RNA editing in trans-splicing intron sequences of nad2 mRNAs in Oenothera mitochondria. J Biol Chem 267:7615–7623
Bock R, Koop HU (1997) Extraplastidic site-specific factors mediate RNA editing in chloroplasts. EMBO J 16:3282–3288
Bock R, Hagemann R, Kössel H, Kudla J (1993) Tissue- and stage-specific modulation of RNA editing of the psbF and psbL transcript from spinach plastids–a new regulatory mechanism? Mol Gen Genet 240:238–244
Bock H, Brennicke A, Schuster W (1994a) Rps3 and rpl16 genes do not overlap in Oenothera mitochondria: GTG as a potential translation initiation codon in plant mitochondria? Plant Mol Biol 24:811–818
Bock R, Kössel H, Maliga P (1994b) Introduction of a heterologous editing site into the tobacco plastid genome: the lack of RNA editing leads to a mutant phenotype. EMBO J 13:4623–4628
Bohne AV, Irihimovitch V, Weihe A, Stern DB (2006) Chlamydomonas reinhardtii encodes a single sigma70-like factor which likely functions in chloroplast transcription. Curr Genet 49:333–340
Bollenbach T, Schuster G, Portnoy V, Stern D (2007) Processing, degradation, and polyadenylation of chloroplast transcripts. Top Curr Genet 19:175–211
Bonen L, Vogel J (2001) The ins and outs of group II introns. Trends Genet 17:322–331
Bouchez D, Höfte H (1998) Functional genomics in plants. Plant Physiol 118:725–732
Bräutigam K, Dietzel L, Pfannschmidt T (2007) Plastid-nucleus communication: anterograde and retrograde signalling in the development and function of plastids. Top Curr Genet 19:409–456
Brosch M, Krause K, Falk J, Krupinska K (2007) Analysis of gene expression in amyloplasts of potato tubers. Planta 227:91–99
Buchanan BB, Gruissem W, Jones RL (eds) (2000) Biochemistry & molecular biology of plants. American Society of Plant Physiologists, Rockwille
Cahoon AB, Harris FM, Stern DB (2004) Analysis of developing maize plastids reveals two mRNA stability classes correlating with RNA polymerase type. EMBO Rep 5:801–806
Cahoon AB, Takacs EM, Sharpe RM, Stern DB (2008) Nuclear, chloroplast, and mitochondrial transcript abundance along a maize leaf developmental gradient. Plant Mol Biol 66:33–46
Canino G, Bocian E, Barbezier N, Echeverría M, Forner J, Binder S, Marchfelder A (2009) Arabidopsis encodes four tRNase Z enzymes. Plant Physiol 150:1494–1502
Carrillo C, Bonen L (1997) RNA editing status of nad7 intron domains in wheat mitochondria. Nucleic Acids Res 25:403–409
Carter ML, Smith AC, Kobayashi H, Purton S, Herrin DL (2004) Structure, circadian regulation and bioinformatic analysis of the unique sigma factor gene in Chlamydomonas reinhardtii. Photosynth Res 82:339–349
Chang CC, Stern DB (1999) DNA-binding factors assemble in a sequence-specific manner on the maize mitochondrial atpA promoter. Curr Genet 35:506–511
Chapdelaine Y, Bonen L (1991) The wheat mitochondrial gene for subunit-I of the NADH dehydrogenase complex – a trans-splicing model for this gene-in-pieces. Cell 65:465–472
Chateigner-Boutin AL, Hanson MR (2003) Developmental co-variation of RNA editing extent of plastid editing sites exhibiting similar cis-elements. Nucleic Acids Res 31:2586–2594
Chateigner-Boutin AL, Small I (2007) A rapid high-throughput method for the detection and quantification of RNA editing based on high-resolution melting of amplicons. Nucleic Acids Res 35:e114
Chateigner-Boutin AL, Ramos-Vega M, Guevara-García A, Andrés C, de la Luz Gutiérrez-Nava M, Cantero A, Delannoy E, Jiménez LF, Lurin C, Small I, León P (2008) CLB19, a pentatricopeptide repeat protein required for editing of rpoA and clpP chloroplast transcripts. Plant J 56:590–602
Chaudhuri S, Carrer H, Maliga P (1995) Site-specific factor involved in the editing of the psbL mRNA in tobacco plastids. EMBO J 14:2951–2957
Chelm BK, Hallick RB (1976) Changes in the expression of the chloroplast genome of Euglena gracilis during chloroplast development. Biochemistry 15:593–599
Chelm BK, Hallick RB, Gray PW (1979) Transcription program of the chloroplast genome of Euglena gracilis during chloroplast development. Proc Natl Acad Sci USA 76:2258–2262
Cheung F, Haas BJ, Goldberg SM, May GD, Xiao Y, Town CD (2006) Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology. BMC Genomics 7:272
Cho Y, Qiu Y-L, Kuhlman P, Palmer JD (1998) Explosive invasion of plant mitochondria by a group I intron. Proc Natl Acad Sci USA 95:14244–14249
Cho WK, Geimer S, Meurer J (2009) Cluster analysis and comparison of various chloroplast transcriptomes and genes in Arabidopsis thaliana. DNA Res 16:31–44
Clarke AK, MacDonald TM, Sjögren LLE (2005) The ATP-dependent Clp protease in chloroplasts of higher plants. Physiol Plant 123:406–412
Corneille S, Lutz K, Maliga P (2000) Conservation of RNA editing between rice and maize plastids: are most editing events dispensable? Mol Gen Genet 264:419–424
Covello PS, Gray MW (1989) RNA editing in plant mitochondria. Nature 341:662–666
Daher Z, Recorbet G, Valot B, Robert F, Balliau T, Potin S, Schoefs B, Dumas-Gaudot E (2010) Proteomic analysis of Medicago truncatula root plastids. Proteomics 10:2123–2137
Daniell H, Lee S-B, Grevich J, Saski C, Quesada-Vargas T, Guda C, Tomkins J, Jansen R (2006) Complete chloroplast genome sequences of Solanum bulbocastanum, Solanum lycopersicum and comparative analyses with other Solanaceae genomes. Theor Appl Genet 112:1503–1518
Danon A (1997) Translational regulation in the chloroplast. Plant Physiol 115:1293–1298
De Santis-Maciossek G, Kofer W, Bock A, Schoch S, Maier RM, Wanner G, Rüdiger W, Koop HU, Herrmann RG (1999) Targeted disruption of the plastid RNA polymerase genes rpoA, B and C1: molecular biology, biochemistry and ultrastructure. Plant J 18:477–489
Demarsy E, Courtois F, Azevedo J, Buhot L, Lerbs-Mache S (2006) Building up of the plastid transcriptional machinery during germination and early plant development. Plant Physiol 142:993–1003
Deng XW, Gruissem W (1988) Constitutive transcription and regulation of gene expression in non-photosynthetic plastids of higher plants. EMBO J 7:3301–3308
Dhingra A, Bies DH, Lehner KR, Folta KM (2006) Green light adjusts the plastid transcriptome during early photomorphogenic development. Plant Physiol 142:1256–1266
Dix KP, Rawson JRY (1983) In vivo transcriptional products of the chloroplast DNA of Euglena gracilis. Curr Genet 7:265–272
Dombrowski S, Brennicke A, Binder S (1997) 3′-Inverted repeats in plant mitochondrial mRNAs are processing signals rather than transcription terminators. EMBO J 16:5069–5076
Dong FG, Wilson KG, Makaroff CA (1998) The radish (Raphanus sativus L.) mitochondrial cox2 gene contains an ACG at the predicted translation initiation site. Curr Genet 34:79–87
Dreyfus M, Régnier P (2002) The poly(A) tail of mRNAs: bodyguard in eukaryotes, scavenger in bacteria. Cell 111:611–613
Duchêne AM, Giritch A, Hoffmann B, Cognat V, Lancelin D, Peeters NM, Zaepfel M, Maréchal-Drouard L, Small ID (2005) Dual targeting is the rule for organellar aminoacyl-tRNA synthetases in Arabidopsis thaliana. Proc Natl Acad Sci USA 102:16484–16489
Duff RJ, Moore FB (2005) Pervasive RNA editing among hornwort rbcL transcripts except Leiosporoceros. J Mol Evol 61:571–578
Eberhard S, Drapier D, Wollman FA (2002) Searching limiting steps in the expression of chloroplast-encoded proteins: relations between gene copy number, transcription, transcript abundance and translation rate in the chloroplast of Chlamydomonas reinhardtii. Plant J 31:149–160
Emrich SJ, Barbazuk WB, Li L, Schnable PS (2007) Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res 17:69–73
Erickson B, Stern DB, Higgs DC (2005) Microarray analysis confirms the specificity of a Chlamydomonas reinhardtii chloroplast RNA stability mutant. Plant Physiol 137:534–544
Fedorova O, Zingler N (2007) Group II introns: structure, folding and splicing mechanism. Biol Chem 388:665–678
Fey J, Maréchal-Drouard L (1999) Compilation and analysis of plant mitochondrial promoter sequences: an illustration of a divergent evolution between monocot and dicot mitochondria. Biochem Biophys Res Commun 256:409–414
Forner J, Hölzle A, Jonietz C, Thuss S, Schwarzländer M, Weber B, Meyer RC, Binder S (2008) Mitochondrial mRNA polymorphisms in different Arabidopsis accessions. Plant Physiol 148:1106–1116
Freyer R, Kiefer-Meyer MC, Kössel H (1997) Occurrence of plastid RNA editing in all major lineages of land plants. Proc Natl Acad Sci USA 94:6285–6290
Fujiwara M, Nagashima A, Kanamaru K, Tanaka K, Takahashi H (2000) Three new nuclear genes, sigD, sigE and sigF, encoding putative plastid RNA polymerase sigma factors in Arabidopsis thaliana. FEBS Lett 481:47–52
Gagliardi D, Perrin R, Maréchal-Drouard L, Grienenberger JM, Leaver CJ (2001) Plant mitochondrial polyadenylated mRNAs are degraded by a 3′- to 5′- exoribonuclease activity, which proceeds unimpeded by stable secondary structures. J Biol Chem 276:43541–43547
Gargano D, Vezzi A, Scotti N, Gray JC, Valle G, Grillo S, Cardi T (2005) The complete nucleotide sequence genome of potato (Solanum tuberosum cv Désirée) chloroplast DNA. In: Proceedings of the 2nd Solanaceae Genome Workshop 2005, Ischia, 25–29 Sept 2005
Geimer S, Belicová A, Legen J, Sláviková S, Herrmann RG, Krajcovic J (2009) Transcriptome analysis of the Euglena gracilis plastid chromosome. Curr Genet 55:425–438
Giegé P, Brennicke A (1999) RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc Natl Acad Sci USA 96:15324–15329
Giegé P, Hoffmann M, Binder S, Brennicke A (2000) RNA degradation buffers asymmetries of transcription in Arabidopsis mitochondria. EMBO Rep 1:164–170
Giegé P, Rayapuram N, Meyer EH, Grienenberger JM, Bonnard G (2004) CcmF(C) involved in cytochrome c maturation is present in a large sized complex in wheat mitochondria. FEBS Lett 563:165–169
Giegé P, Sweetlove LJ, Cognat V, Leaver CJ (2005) Coordination of nuclear and mitochondrial genome expression during mitochondrial biogenesis in Arabidopsis. Plant Cell 17:1497–1512
Glanz S, Kück U (2009) Trans-splicing of organelle introns–a detour to continuous RNAs. Bioessays 31:921–934
Gobert A, Gutmann B, Taschner A, Gössringer M, Holzmann J, Hartmann RK, Rossmanith W, Giegé P (2010) A single Arabidopsis organellar protein has RNase P activity. Nat Struct Mol Biol 17:740–744
Gonzalez DH, Welchen E, Attallah CV, Comelli RN, Mufarrege EF (2007) Transcriptional coordination of the biogenesis of the oxidative phosphorylation machinery in plants. Plant J 51:105–116
Grewe F, Viehoever P, Weisshaar B, Knoop V (2009) A trans-splicing group I intron and tRNA-hyperediting in the mitochondrial genome of the lycophyte Isoetes engelmannii. Nucleic Acids Res 37:5093–5104
Grewe F, Herres S, Viehover P, Polsakiewicz M, Weisshaar B, Knoop V (2010) A unique transcriptome: 1782 positions of RNA editing alter 1406 codon identities in mitochondrial mRNAs of the lycophyte Isoetes engelmannii. Nucleic Acids Res 39:2890–2902
Grohmann L, Thieck O, Herz U, Schröder W, Brennicke A (1994) Translation of nad9 mRNAs in mitochondria from Solanum tuberosum is restricted to completely edited transcripts. Nucleic Acids Res 22:3304–3311
Gualberto JM, Lamattina L, Bonnard G, Weil JH, Grienenberger JM (1989) RNA editing in wheat mitochondria results in the conservation of protein sequences. Nature 341:660–662
Hajdukiewicz PT, Allison LA, Maliga P (1997) The two RNA polymerases encoded by the nuclear and the plastid compartments transcribe distinct groups of genes in tobacco plastids. EMBO J 16:4041–4048
Hallick RB, Hong L, Drager RG, Favreau MR, Monfort A, Orsat B, Spielmann A, Stutz E (1993) Complete sequence of Euglena gracilis chloroplast DNA. Nucleic Acids Res 21:3537–3544
Hammani K, Okuda K, Tanz SK, Chateigner-Boutin AL, Shikanai T, Small I (2009) A study of new Arabidopsis chloroplast RNA editing mutants reveals general features of editing factors and their target sites. Plant Cell 21:3686–3699
Han C-d, Patrie W, Polacco M, Coe EHJ (1993) Aberrations in plastid transcripts and deficiency of plastid DNA in striped and albino mutants in maize. Planta 191:552–563
Handa H (2003) The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res 31:5907–5916
Hayes R, Kudla J, Gruissem W (1999) Degrading chloroplast mRNA: the role of polyadenylation. Trends Biochem Sci 24:199–202
Hedtke B, Börner T, Weihe A (1997) Mitochondrial and chloroplast phage-type RNA polymerases in Arabidopsis. Science 277:809–811
Hedtke B, Wagner I, Börner T, Hess WR (1999) Inter-organellar crosstalk in higher plants: impaired chloroplast development affects mitochondrial gene and transcript levels. Plant J 19:635–643
Hedtke B, Börner T, Weihe A (2000) One RNA polymerase serving two genomes. EMBO Rep 1:435–440
Hess WR, Prombona A, Fieder B, Subramanian AR, Börner T (1993) Chloroplast rps15 and the rpoB/C1/C2 gene cluster are strongly transcribed in ribosome-deficient plastids: evidence for a functioning non-chloroplast-encoded RNA polymerase. EMBO J 12:563–571
Hiesel R, Wissinger B, Schuster W, Brennicke A (1989) RNA editing in plant mitochondria. Science 246:1632–1634
Hildebrand M, Hallick RB, Passavant CW, Bourque DP (1988) Trans-splicing in chloroplasts: the rps12 loci of Nicotiana tabacum. Proc Natl Acad Sci USA 85:372–376
Hirose T, Sugiura M (1997) Both RNA editing and RNA cleavage are required for translation of tobacco chloroplast ndhD mRNA: a possible regulatory mechanism for the expression of a chloroplast operon consisting of functionally unrelated genes. EMBO J 16:6804–6811
Hirose T, Sugiura M (2001) Involvement of a site-specific trans-acting factor and a common RNA-binding protein in the editing of chloroplast mRNAs: development of a chloroplast in vitro RNA editing system. EMBO J 20:1144–1152
Hirose T, Fan H, Suzuki JY, Wakasugi T, Tsudzuki T, Kössel H, Sugiura M (1996) Occurrence of silent RNA editing in chloroplasts: its species specificity and the influence of environmental and developmental conditions. Plant Mol Biol 30:667–672
Hirose T, Kusumegi T, Tsudzuki T, Sugiura M (1999) RNA editing sites in tobacco chloroplast transcripts: editing as a possible regulator of chloroplast RNA polymerase activity. Mol Gen Genet 262:462–467
Hoch B, Maier RM, Appel K, Igloi GL, Kössel H (1991) Editing of a chloroplast mRNA by creation of an initiation codon. Nature 353:178–180
Hoffmann M, Dombrowski S, Guha C, Binder S (1999) Cotranscription of the rpl5-rps14-cob gene cluster in pea mitochondria. Mol Gen Genet 261:537–545
Holec S, Lange H, Kühn K, Alioua M, Börner T, Gagliardi D (2006) Relaxed transcription in Arabidopsis mitochondria is counterbalanced by RNA stability control mediated by polyadenylation and polynucleotide phosphorylase. Mol Cell Biol 26:2869–2876
Hollingsworth MJ, Johanningmeier U, Karabin GD, Stiegler GL, Hallick RB (1984) Detection of multiple, unspliced precursor mRNA transcripts for the Mr 32,000 thylakoid membrane protein from Euglena gracilis chloroplasts. Nucleic Acids Res 12:2001–2017
Howell KA, Millar AH, Whelan J (2006) Ordered assembly of mitochondria during rice germination begins with promitochondrial structures rich in components of the protein import apparatus. Plant Mol Biol 60:201–223
Inada M, Sasaki T, Yukawa M, Tsudzuki T, Sugiura M (2004) A systematic search for RNA editing sites in pea chloroplasts: an editing event causes diversification from the evolutionarily conserved amino acid sequence. Plant Cell Physiol 45:1615–1622
Isono K, Niwa Y, Satoh K, Kobayashi H (1997a) Evidence for transcriptional regulation of plastid photosynthesis genes in Arabidopsis thaliana roots. Plant Physiol 114:623–630
Isono K, Shimizu M, Yoshimoto K, Niwa Y, Satoh K, Yokota A, Kobayashi H (1997b) Leaf-specifically expressed genes for polypeptides destined for chloroplasts with domains of sigma70 factors of bacterial RNA polymerases in Arabidopsis thaliana. Proc Natl Acad Sci USA 94:14948–14953
Jenkins BD, Kulhanek DJ, Barkan A (1997) Nuclear mutations that block group II RNA splicing in maize chloroplasts reveal several intron classes with distinct requirements for splicing factors. Plant Cell 9:283–296
Jonietz C, Forner J, Holzle A, Thuss S, Binder S (2010) RNA PROCESSING FACTOR2 is required for 5′ end processing of nad9 and cox3 mRNAs in mitochondria of Arabidopsis thaliana. Plant Cell 22:443–453
Kahlau S, Bock R (2008) Plastid transcriptomics and translatomics of tomato fruit development and chloroplast-to-chromoplast differentiation: chromoplast gene expression largely serves the production of a single protein. Plant Cell 20:856–874
Kahlau S, Aspinall S, Gray JC, Bock R (2006) Sequence of the tomato chloroplast DNA and evolutionary comparison of solanaceous plastid genomes. J Mol Evol 63:194–207
Kanamaru K, Fujiwara M, Seki M, Katagiri T, Nakamura M, Mochizuki N, Nagatani A, Shinozaki K, Tanaka K, Takahashi H (1999) Plastidic RNA polymerase sigma factors in Arabidopsis. Plant Cell Physiol 40:832–842
Kanamaru K, Nagashima A, Fujiwara M, Shimada H, Shirano Y, Nakabayashi K, Shibata D, Tanaka K, Takahashi H (2001) An Arabidopsis sigma factor (SIG2)-dependent expression of plastid-encoded tRNAs in chloroplasts. Plant Cell Physiol 42:1034–1043
Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, Nakamura Y, Miyajima N, Hirosawa M, Sugiura M, Sasamoto S, Kimura T, Hosouchi T, Matsuno A, Muraki A, Nakazaki N, Naruo K, Okumura S, Shimpo S, Takeuchi C, Wada T, Watanabe A, Yamada M, Yasuda M, Tabata S (1996) Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res 3:109–136
Karcher D, Bock R (1998) Site-selective inhibition of plastid RNA editing by heat shock and antibiotics: a role for plastid translation in RNA editing. Nucleic Acids Res 26:1185–1190
Karcher D, Bock R (2002a) The amino acid sequence of a plastid protein is developmentally regulated by RNA editing. J Biol Chem 277:5570–5574
Karcher D, Bock R (2002b) Temperature sensitivity of RNA editing and intron splicing reactions in the plastid ndhB transcript. Curr Genet 41:48–52
Karcher D, Kahlau S, Bock R (2008) Faithful editing of a tomato-specific mRNA editing site in transgenic tobacco chloroplasts. RNA 14:217–224
Keren I, Klipcan L, Bezawork-Geleta A, Kolton M, Shaya F, Ostersetzer-Biran O (2008) Characterization of the molecular basis of group II intron RNA recognition by CRS1-CRM domains. J Biol Chem 283:23333–23342
Khanam SM, Naydenov NG, Kadowaki K, Nakamura C (2007) Mitochondrial biogenesis as revealed by mitochondrial transcript profiles during germination and early seedling growth in wheat. Genes Genet Syst 82:409–420
Kim M, Christopher DA, Mullet JE (1993) Direct evidence for selective modulation of psbA, rpoA, rbcL and 16S RNA stability during barley chloroplast development. Plant Mol Biol 22:447–463
Kim SR, Yang JI, Moon S, Ryu CH, An K, Kim KM, Yim J, An G (2009) Rice OGR1 encodes a pentatricopeptide repeat-DYW protein and is essential for RNA editing in mitochondria. Plant J 59:738–749
Klaff P, Gruissem W (1991) Changes in chloroplast mRNA stability during leaf development. Plant Cell 3:517–529
Klein RR, Mullet JE (1987) Control of gene expression during higher plant chloroplast biogenesis. Protein synthesis and transcript levels of psbA, psaA-psaB, and rbcL in dark-grown and illuminated barley seedlings. J Biol Chem 262:4341–4348
Knoop V (2004) The mitochondrial DNA of land plants: peculiarities in phylogenetic perspective. Curr Genet 46:123–139
Knoop V, Schuster W, Wissinger B, Brennicke A (1991) Trans-splicing integrates an exon of 22 nucleotides into the nad5 messenger RNA in higher plant mitochondria. EMBO J 10:3483–3493
Kobayashi Y, Dokiya Y, Sugita M (2001) Dual targeting of phage-type RNA polymerase to both mitochondria and plastids is due to alternative translation initiation in single transcripts. Biochem Biophys Res Commun 289:1106–1113
Kode V, Mudd EA, Iamtham S, Day A (2005) The tobacco plastid accD gene is essential and is required for leaf development. Plant J 44:237–244
Kotera E, Tasaka M, Shikanai T (2005) A pentatricopeptide repeat protein is essential for RNA editing in chloroplasts. Nature 433:326–330
Kozak M (2005) Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361:13–37
Krause K, Maier RM, Kofer W, Krupinska K, Herrmann RG (2000) Disruption of plastid-encoded RNA polymerase genes in tobacco: expression of only a distinct set of genes is not based on selective transcription of the plastid chromosome. Mol Gen Genet 263:1022–1030
Krupinska K, Apel K (1989) Light-induced transformation of etioplasts to chloroplasts of barley without transcriptional control of plastid gene expression. Mol Gen Genet 219:467–473
Kubicki A, Steinmüller K, Westhoff P (1994) Differential transcription of plastome-encoded genes in the mesophyll and bundle-sheath chloroplasts of the monocotyledonous NADP-malic enzyme-type C4 plants maize and sorghum. Plant Mol Biol 25:669–679
Kudla J, Hayes R, Gruissem W (1996) Polyadenylation accelerates degradation of chloroplast mRNA. EMBO J 15:7137–7146
Kugita M, Yamamoto Y, Fujikawa T, Matsumoto T, Yoshinaga K (2003) RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucleic Acids Res 31:2417–2423
Kühn K, Weihe A, Börner T (2005) Multiple promoters are a common feature of mitochondrial genes in Arabidopsis. Nucleic Acids Res 33:337–346
Kühn K, Bohne AV, Liere K, Weihe A, Börner T (2007) Arabidopsis phage-type RNA polymerases: accurate in vitro transcription of organellar genes. Plant Cell 19:959–971
Kühn K, Richter U, Meyer EH, Delannoy E, de Longevialle AF, O’Toole N, Börner T, Millar AH, Small ID, Whelan J (2009) Phage-type RNA polymerase RPOTmp performs gene-specific transcription in mitochondria of Arabidopsis thaliana. Plant Cell 21:2762–2779
Kuntz M, Evrard J-L, d’Harlingue A, Weil JH, Camara B (1989) Expression of plastid and nuclear genes during chromoplast differentiation in bell pepper (Capsicum annuum) and sunflower (Helianthus annuus). Mol Gen Genet 216:156–163
Kuntz M, Camara B, Weil JH, Schantz R (1992) The psbL gene from bell pepper (Capsicum annuum): plastid RNA editing also occurs in non-photosynthetic chromoplasts. Plant Mol Biol 20:1185–1188
Kunzmann A, Brennicke A, Marchfelder A (1998) 5′ end maturation and RNA editing have to precede tRNA 3′ processing in plant mitochondria. Proc Natl Acad Sci USA 95:108–113
Kuroda H, Maliga P (2003) The plastid clpP1 protease gene is essential for plant development. Nature 425:86–89
Kurth J, Varotto C, Pesaresi P, Biehl A, Richly E, Salamini F, Leister D (2002) Gene-sequence-tag expression analyses of 1,800 genes related to chloroplast functions. Planta 215:101–109
Lambowitz AM, Zimmerly S (2004) Mobile group II introns. Annu Rev Genet 38:1–35
Legen J, Kemp S, Krause K, Profanter B, Herrmann RG, Maier RM (2002) Comparative analysis of plastid transcription profiles of entire plastid chromosomes from tobacco attributed to wild-type and PEP-deficient transcription machineries. Plant J 31:171–188
Leino M, Teixeira R, Landgren M, Glimelius K (2003) Brassica napus lines with rearranged Arabidopsis mitochondria display CMS and a range of developmental aberrations. Theor Appl Genet 106:1156–1163
Leino M, Thyselius S, Landgren M, Glimelius K (2004) Arabidopsis thaliana chromosome III restores fertility in a cytoplasmic male-sterile Brassica napus line with A. thaliana mitochondrial DNA. Theor Appl Genet 109:272–279
Leino M, Landgren M, Glimelius K (2005) Alloplasmic effects on mitochondrial transcriptional activity and RNA turnover result in accumulated transcripts of Arabidopsis orfs in cytoplasmic male-sterile Brassica napus. Plant J 42:469–480
Lemieux B, Aharoni A, Schena M (1998) Overview of DNA chip technology. Mol Breed 4:277–289
Lerbs-Mache S (1993) The 110-kDa polypeptide of spinach plastid DNA-dependent RNA polymerase: single-subunit enzyme or catalytic core of multimeric enzyme complexes? Proc Natl Acad Sci USA 90:5509–5513
Liere K, Börner T (2007) Transcription and transcriptional regulation in plastids. Top Curr Genet 19:121–174
Liere K, Link G (1995) RNA-binding activity of the matK protein encoded by the chloroplast trnK intron from mustard (Sinapis alba L.). Nucleic Acids Res 23:917–921
Liere K, Maliga P (1999) In vitro characterization of the tobacco rpoB promoter reveals a core sequence motif conserved between phage-type plastid and plant mitochondrial promoters. EMBO J 18:249–257
Li-Pook-Than J, Carrillo C, Bonen L (2004) Variation in mitochondrial transcript profiles of protein-coding genes during early germination and seedling development in wheat. Curr Genet 46:374–380
Lu B, Hanson MR (1994) A single homogeneous form of ATP6 protein accumulates in petunia mitochondria despite the presence of differentially edited atp6 transcripts. Plant Cell 6:1955–1968
Lupold DS, Caoile AG, Stern DB (1999) Genomic context influences the activity of maize mitochondrial cox2 promoters. Proc Natl Acad Sci USA 96:11670–11675
MacLean D, Jerome CA, Brown AP, Gray JC (2008) Co-regulation of nuclear genes encoding plastid ribosomal proteins by light and plastid signals during seedling development in tobacco and Arabidopsis. Plant Mol Biol 66:475–490
Magee AM, Kavanagh TA (2002) Plastid genes transcribed by the nucleus-encoded plastid RNA polymerase show increased transcript accumulation in transgenic plants expressing a chloroplast-localized phage T7 RNA polymerase. J Exp Bot 53:2341–2349
Magee AM, Coyne S, Murphy D, Horvath EM, Medgyesy P, Kavanagh TA (2004) T7 RNA polymerase-directed expression of an antibody fragment transgene in plastids causes a semi-lethal pale-green seedling phenotype. Transgenic Res 13:325–337
Magee AM, MacLean D, Gray JC, Kavanagh TA (2007) Disruption of essential plastid gene expression caused by T7 RNA polymerase-mediated transcription of plastid transgenes during early seedling development. Transgenic Res 16:415–428
Maier RM, Neckermann K, Igloi GL, Kössel H (1995) Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J Mol Biol 251:614–628
Malek O, Lättig K, Hiesel R, Brennicke A, Knoop V (1996) RNA editing in bryophytes and a molecular phylogeny of land plants. EMBO J 15:1403–1411
Marano MR, Carrillo N (1992) Constitutive transcription and stable RNA accumulation in plastids during the conversion of chloroplasts to chromoplasts in ripening tomato fruits. Plant Physiol 100:1103–1113
Maréchal-Drouard L, Kumar R, Remacle C, Small I (1996a) RNA editing of larch mitochondrial tRNA(His) precursors is a prerequisite for processing. Nucleic Acids Res 24:3229–3234
Maréchal-Drouard L, Cosset A, Remacle C, Ramamonjisoa D, Dietrich A (1996b) A single editing event is a prerequisite for efficient processing of potato mitochondrial phenylalanine tRNA. Mol Cell Biol 16:3504–3510
Mayfield SP, Yohn CB, Cohen A, Danon A (1995) Regulation of chloroplast gene expression. Annu Rev Plant Physiol Plant Mol Biol 46:147–166
Meng Q, Wang Y, Liu XQ (2005) An intron-encoded protein assists RNA splicing of multiple similar introns of different bacterial genes. J Biol Chem 280:35085–35088
Mereschkowsky C (1905) Ueber Natur und Ursprung der Chromatophoren im Pflanzenreiche. Biol Centralbl 25:593–604
Meyers BC, Galbraith DW, Nelson T, Agrawal V (2004) Methods for transcriptional profiling in plants. Be fruitful and replicate. Plant Physiol 135:637–652
Miller ME, Jurgenson JE, Reardon EM, Price CA (1983) Plastid translation in organello and in vitro during light-induced development in Euglena. J Biol Chem 258:14478–14484
Minoda A, Nagasawa K, Hanaoka M, Horiuchi M, Takahashi H, Tanaka K (2005) Microarray profiling of plastid gene expression in a unicellular red alga, Cyanidioschyzon merolae. Plant Mol Biol 59:375–385
Miyata Y, Sugita M (2004) Tissue- and stage-specific RNA editing of rps14 transcripts in moss (Physcomitrella patens) chloroplasts. J Plant Physiol 161:113–115
Mohr G, Lambowitz AM (2003) Putative proteins related to group II intron reverse transcriptase/maturases are encoded by nuclear genes in higher plants. Nucleic Acids Res 31:647–652
Mower J, Palmer J (2006) Patterns of partial RNA editing in mitochondrial genes of Beta vulgaris. Mol Genet Genomics 276:285–293
Muise RC, Hauswirth WW (1992) Transcription in maize mitochondria: effects of tissue and mitochondrial genotype. Curr Genet 22:235–242
Mullet JE (1993) Dynamic regulation of chloroplast transcription. Plant Physiol 103:309–313
Nagashima A, Hanaoka M, Motohashi R, Seki M, Shinozaki K, Kanamaru K, Takahashi H, Tanaka K (2004) DNA microarray analysis of plastid gene expression in an Arabidopsis mutant deficient in a plastid transcription factor sigma, SIG2. Biosci Biotechnol Biochem 68:694–704
Nakagawa N, Sakurai N (2006) A mutation in At-nMat1a, which encodes a nuclear gene having high similarity to group II intron maturase, causes impaired splicing of mitochondrial NAD4 transcript and altered carbon metabolism in Arabidopsis thaliana. Plant Cell Physiol 47:772–783
Nakamura T, Furuhashi Y, Hasegawa K, Hashimoto H, Watanabe K, Obokata J, Sugita M, Sugiura M (2003) Array-based analysis on tobacco plastid transcripts: preparation of a genomic microarray containing all genes and all intergenic regions. Plant Cell Physiol 44:861–867
Nakamura T, Sugiura C, Kobayashi Y, Sugita M (2005) Transcript profiling in plastid arginine tRNA-CCG gene knockout moss: construction of Physcomitrella patens plastid DNA microarray. Plant Biol 7:258–265
Naydenov NG, Khanam SM, Atanassov A, Nakamura C (2008) Expression profiles of respiratory components associated with mitochondrial biogenesis during germination and seedling growth under normal and restricted conditions in wheat. Genes Genet Syst 83:31–41
Naydenov NG, Khanam S, Siniauskaya M, Nakamura C (2010) Profiling of mitochondrial transcriptome in germinating wheat embryos and seedlings subjected to cold, salinity and osmotic stresses. Genes Genet Syst 85:31–42
Neckermann K, Zeltz P, Igloi GL, Kössel H, Maier RM (1994) The role of RNA editing in conservation of start codons in chloroplast genomes. Gene 146:177–182
Notsu Y, Masood S, Nishikawa T, Kubo N, Akiduki G, Nakazono M, Hirai A, Kadowaki K (2002) The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol Genet Genomics 268:434–445
Obukosia SD, Richards CM, Boyer CD (2003) Expression of plastid-encoded photosynthetic genes during chloroplast or chromoplast differentiation in Cucurbitae pepo L. fruits. Phytochemistry 64:1213–1221
Ohyama K, Fukuzawa H, Kohchi T, Shirai H, Sano T, Sano S, Umesono K, Shiki Y, Takeuchi M, Chang Z, Aota S, Inokuchi H, Ozeki H (1986) Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 322:572–574
Okada S, Brennicke A (2006) Transcript levels in plant mitochondria show a tight homeostasis during day and night. Mol Genet Genomics 276:71–78
Okuda K, Nakamura T, Sugita M, Shimizu T, Shikanai T (2006) A pentatricopeptide repeat protein is a site recognition factor in chloroplast RNA editing. J Biol Chem 281:37661–37667
Okuda K, Myouga F, Motohashi R, Shinozaki K, Shikanai T (2007) Conserved domain structure of pentatricopeptide repeat proteins involved in chloroplast RNA editing. Proc Natl Acad Sci USA 104:8178–8183
Peeters NM, Hanson MR (2002) Transcript abundance supercedes editing efficiency as a factor in developmental variation of chloroplast gene expression. RNA 8:497–511
Peled-Zehavi H, Danon A (2007) Translation and translational regulation in chloroplasts. Top Curr Genet 19:249–281
Perrin R, Lange H, Grienenberger JM, Gagliardi D (2004a) AtmtPNPase is required for multiple aspects of the 18S rRNA metabolism in Arabidopsis thaliana mitochondria. Nucleic Acids Res 32:5174–5182
Perrin R, Meyer EH, Zaepfel M, Kim YJ, Mache R, Grienenberger JM, Gualberto JM, Gagliardi D (2004b) Two exoribonucleases act sequentially to process mature 3′-ends of atp9 mRNAs in Arabidopsis mitochondria. J Biol Chem 279:25440–25446
Pfalz J, Bayraktar OA, Prikryl J, Barkan A (2009) Site-specific binding of a PPR protein defines and stabilizes 5′ and 3′ mRNA termini in chloroplasts. EMBO J 28:2042–2052
Phreaner CG, Williams MA, Mulligan RM (1996) Incomplete editing of rps12 transcripts results in the synthesis of polymorphic polypeptides in plant mitochondria. Plant Cell 8:107–117
Piechulla B, Imlay KRC, Gruissem W (1985) Plastid gene expression during fruit ripening in tomato (Lycopersicon esculentum). Plant Mol Biol 5:373–384
Pring DR, Mullen JA, Kempken F (1992) Conserved sequence blocks 5′ to start codons of plant mitochondrial genes. Plant Mol Biol 19:313–317
Pyke KA (2007) Plastid biogenesis and differentiation. Top Curr Genet 19:1–28
Raczynska KD, Le Ret M, Rurek M, Bonnard G, Augustyniak H, Gualberto JM (2006) Plant mitochondrial genes can be expressed from mRNAs lacking stop codons. FEBS Lett 580:5641–5646
Rawson JR, Boerma CL (1976) A measurement of the fraction of chloroplast DNA transcribed during chloroplast development in Euglena gracilis. Biochemistry 15:588–592
Remacle C, Maréchal-Drouard L (1996) Characterization of the potato mitochondrial transcription unit containing ‘native’ trnS (GCU), trnF (GAA) and trnP (UGG). Plant Mol Biol 30:553–563
Ribas-Carbo M, Taylor NL, Giles L, Busquets S, Finnegan PM, Day DA, Lambers H, Medrano H, Berry JA, Flexas J (2005) Effects of water stress on respiration in soybean leaves. Plant Physiol 139:466–473
Richly E, Dietzmann A, Biehl A, Kurth J, Laloi C, Apel K, Salamini F, Leister D (2003) Covariations in the nuclear chloroplast transcriptome reveal a regulatory master-switch. EMBO Rep 4:491–498
Rüdinger M, Funk HT, Rensing SA, Maier UG, Knoop V (2009) RNA editing: only eleven sites are present in the Physcomitrella patens mitochondrial transcriptome and a universal nomenclature proposal. Mol Genet Genomics 281:473–481
Ruf S, Kössel H (1997) Tissue-specific and differential editing of the two ycf3 editing sites in maize plastids. Curr Genet 32:19–23
Sakai A, Kawano S, Kuroiwa T (1992) Conversion of proplastids to amyloplasts in tobacco cultured cells is accompanied by changes in the transcriptional activities of plastid genes. Plant Physiol 100:1062–1066
Saldanha R, Mohr G, Belfort M, Lambowitz AM (1993) Group I and group II introns. FASEB J 7:15–24
Salinas T, Duchêne AM, Maréchal-Drouard L (2008) Recent advances in tRNA mitochondrial import. Trends Biochem Sci 33:320–329
Schmitz-Linneweber C, Barkan A (2007) RNA splicing and RNA editing in chloroplasts. Top Curr Genet 19:213–248
Schmitz-Linneweber C, Regel R, Du TG, Hupfer H, Herrmann RG, Maier RM (2002) The plastid chromosome of Atropa belladonna and its comparison with that of Nicotiana tabacum: the role of RNA editing in generating divergence in the process of plant speciation. Mol Biol Evol 19:1602–1612
Schmitz-Linneweber C, Williams-Carrier R, Barkan A (2005) RNA Immunoprecipitation and microarray analysis show a chloroplast pentatricopeptide repeat protein to be associated with the 5′ region of mRNAs whose translation it activates. Plant Cell 17:2791–2804
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P, Reily AD, Courtney L, Kruchowski SS, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock SM, Belter E, Du F, Kim K, Abbott RM, Cotton M, Levy A, Marchetto P, Ochoa K, Jackson SM, Gillam B, Chen W, Yan L, Higginbotham J, Cardenas M, Waligorski J, Applebaum E, Phelps L, Falcone J, Kanchi K, Thane T, Scimone A, Thane N, Henke J, Wang T, Ruppert J, Shah N, Rotter K, Hodges J, Ingenthron E, Cordes M, Kohlberg S, Sgro J, Delgado B, Mead K, Chinwalla A, Leonard S, Crouse K, Collura K, Kudrna D, Currie J, He R, Angelova A, Rajasekar S, Mueller T, Lomeli R, Scara G, Ko A, Delaney K, Wissotski M, Lopez G, Campos D, Braidotti M, Ashley E, Golser W, Kim H, Lee S, Lin J, Dujmic Z, Kim W, Talag J, Zuccolo A, Fan C, Sebastian A, Kramer M, Spiegel L, Nascimento L, Zutavern T, Miller B, Ambroise C, Muller S, Spooner W, Narechania A, Ren L, Wei S, Kumari S, Faga B, Levy MJ, McMahan L, Van Buren P, Vaughn MW, Ying K, Yeh CT, Emrich SJ, Jia Y, Kalyanaraman A, Hsia AP, Barbazuk WB, Baucom RS, Brutnell TP, Carpita NC, Chaparro C, Chia JM, Deragon JM, Estill JC, Fu Y, Jeddeloh JA, Han Y, Lee H, Li P, Lisch DR, Liu S, Liu Z, Nagel DH, McCann MC, SanMiguel P, Myers AM, Nettleton D, Nguyen J, Penning BW, Ponnala L, Schneider KL, Schwartz DC, Sharma A, Soderlund C, Springer NM, Sun Q, Wang H, Waterman M, Westerman R, Wolfgruber TK, Yang L, Yu Y, Zhang L, Zhou S, Zhu Q, Bennetzen JL, Dawe RK, Jiang J, Jiang N, Presting GG, Wessler SR, Aluru S, Martienssen RA, Clifton SW, McCombie WR, Wing RA, Wilson RK (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326:1112–1115
Schulze A, Downward J (2001) Navigating gene expression using microarrays-a technology review. Nat Cell Biol 3:E190–E195
Schuster W (1993) Ribosomal protein gene rpl5 is cotranscribed with the nad3 gene in Oenothera mitochondria. Mol Gen Genet 240:445–449
Schweer J, Loschelder H, Link G (2006) A promoter switch that can rescue a plant sigma factor mutant. FEBS Lett 580:6617–6622
Serino G, Maliga P (1998) RNA polymerase subunits encoded by the plastid rpo genes are not shared with the nucleus-encoded plastid enzyme. Plant Physiol 117:1165–1170
Shiina T, Tsunoyama Y, Nakahira Y, Khan MS (2005) Plastid RNA polymerases, promoters, and transcription regulators in higher plants. In: Jeon KW (ed) International review of cytology, vol 244. Academic, Amsterdam, pp 1–68
Shikanai T (2006) RNA editing in plant organelles: machinery, physiological function and evolution. Cell Mol Life Sci 63:698–708
Shikanai T, Shimizu K, Ueda K, Nishimura Y, Kuroiwa T, Hashimoto T (2001) The chloroplast clpP gene, encoding a proteolytic subunit of ATP-dependent protease, is indispensable for chloroplast development in tobacco. Plant Cell Physiol 42:264–273
Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K, Ohto C, Torazawa K, Meng BY, Sugita M, Deno H, Kamogashira T, Yamada K, Kusuda J, Takaiwa F, Kato A, Tohdoh N, Shimida H, Sugiura M (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J 5:2043–2049
Siddique MA, Grossmann J, Gruissem W, Baginsky S (2006) Proteome analysis of bell pepper (Capsicum annuum L.) chromoplasts. Plant Cell Physiol 47:1663–1673
Silhavy D, Maliga P (1998) Mapping of promoters for the nucleus-encoded plastid RNA polymerase (NEP) in the iojap maize mutant. Curr Genet 33:340–344
Siniauskaya M, Naydenov N, Davydenko O, Nakamura C (2008) Macroarray for studying chloroplast gene expression profiles associated with the initial development of wheat. In: Proceedings of the 11th International Wheat Genetics Symposium, Sidney University Press, Sidney
Small ID, Peeters N (2000) The PPR motif – a TPR-related motif prevalent in plant organellar proteins. Trends Biochem Sci 25:46–47
Stears RL, Martinsky T, Schena M (2003) Trends in microarray analysis. Nat Med 9:140–145
Stern DB, Kindle KL (1993) 3′end maturation of the Chlamydomonas reinhardtii chloroplast atpB mRNA is a two-step process. Mol Cell Biol 13:2277–2285
Stern DB, Goldschmidt-Clermont M, Hanson MR (2010) Chloroplast RNA metabolism. Annu Rev Plant Biol 61:125–155
Sugiura M, Hirose T, Sugita M (1998) Evolution and mechanism of translation in chloroplasts. Annu Rev Genet 32:437–459
Svensson AS, Rasmusson AG (2001) Light-dependent gene expression for proteins in the respiratory chain of potato leaves. Plant J 28:73–82
Takenaka M, Verbitskiy D, van der Merwe JA, Zehrmann A, Brennicke A (2008) The process of RNA editing in plant mitochondria. Mitochondrion 8:35–46
Tanaka K, Tozawa Y, Mochizuki N, Shinozaki K, Nagatani A, Wakasa K, Takahashi H (1997) Characterization of three cDNA species encoding plastid RNA polymerase sigma factors in Arabidopsis thaliana: evidence for the sigma factor heterogeneity in higher plant plastids. FEBS Lett 413:309–313
Tasaki E, Hattori M, Sugita M (2010) The moss pentatricopeptide repeat protein with a DYW domain is responsible for RNA editing of mitochondrial ccmFc transcript. Plant J 62:560–570
Tavares-Carreón F, Camacho-Villasana Y, Zamudio-Ochoa A, Shingú-Vázquez M, Torres-Larios A, Pérez-Martínez X (2008) The pentatricopeptide repeats present in Pet309 are necessary for translation but not for stability of the mitochondrial cox1 mRNA in yeast. J Biol Chem 283:1472–1479
Tillich M, Funk HT, Schmitz-Linneweber C, Poltnigg P, Sabater B, Martin M, Maier RM (2005) Editing of plastid RNA in Arabidopsis thaliana ecotypes. Plant J 43:708–715
Tillich M, Lehwark P, Morton BR, Maier UG (2006) The evolution of chloroplast RNA editing. Mol Biol Evol 23:1912–1921
Tracy RL, Stern DB (1995) Mitochondrial transcription initiation: promoter structures and RNA polymerases. Curr Genet 28:205–216
Tsudzuki T, Wakasugi T, Sugiura M (2001) Comparative analysis of RNA editing sites in higher plant chloroplasts. J Mol Evol 53:327–332
Unseld M, Marienfeld JR, Brandt P, Brennicke A (1997) The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat Genet 15:57–61
Uyttewaal M, Mireau H, Rurek M, Hammani K, Arnal N, Quadrado M, Giegé P (2008) PPR336 is associated with polysomes in plant mitochondria. J Mol Biol 375:626–636
Valkov VT, Scotti N, Kahlau S, Maclean D, Grillo S, Gray JC, Bock R, Cardi T (2009) Genome-wide analysis of plastid gene expression in potato leaf chloroplasts and tuber amyloplasts: transcriptional and posttranscriptional control. Plant Physiol 150:2030–2044
Vaughn JC, Mason MT, Sper-Whitis GL, Kuhlman P, Palmer JD (1995) Fungal origin by horizontal transfer of a plant mitochondrial group I intron in the chimeric coxI gene of Peperomia. J Mol Evol 41:563–572
Verbitskiy D, Zehrmann A, Brennicke A, Takenaka M (2010) A truncated MEF11 protein shows site-specific effects on mitochondrial RNA editing. Plant Signal Behav 5:558–560
Vogel A, Schilling O, Späth B, Marchfelder A (2005) The tRNase Z family of proteins: physiological functions, substrate specificity and structural properties. Biol Chem 386:1253–1264
Wakasugi T, Hirose T, Horihata M, Tsudzuki T, Kössel H, Sugiura M (1996) Creation of a novel protein-coding region at the RNA level in black pine chloroplasts: the pattern of RNA editing in the gymnosperm chloroplast is different from that in angiosperms. Proc Natl Acad Sci USA 93:8766–8770
Wang L, Li P, Brutnell TP (2010) Exploring plant transcriptomes using ultra high-throughput sequencing. Brief Funct Genomics 9:118–128
Wank H, SanFilippo J, Singh RN, Matsuura M, Lambowitz AM (1999) A reverse transcriptase/maturase promotes splicing by binding at its own coding segment in a group II intron RNA. Mol Cell 4:239–250
Weber AP, Weber KL, Carr K, Wilkerson C, Ohlrogge JB (2007) Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiol 144:32–42
Welchen E, Gonzalez DH (2006) Overrepresentation of elements recognized by TCP-domain transcription factors in the upstream regions of nuclear genes encoding components of the mitochondrial oxidative phosphorylation machinery. Plant Physiol 141:540–545
Wissinger B, Schuster W, Brennicke A (1991) Trans splicing in Oenothera mitochondria: nad1 mRNAs are edited in exon and trans-splicing group-II intron sequences. Cell 65:473–482
Wolf PG, Rowe CA, Hasebe M (2004) High levels of RNA editing in a vascular plant chloroplast genome: analysis of transcripts from the fern Adiantum capillus-veneris. Gene 339:89–97
Wood CK, Dudley P, Albury MS, Affourtit C, Leach GR, Pratt JR, Whitehouse DG, Moore AL (1996) Developmental regulation of respiratory activity and protein import in plant mitochondria. Biochem Soc Trans 24:746–749
Yamaguchi K, Subramanian AR (2000) The plastid ribosomal proteins. Identification of all the proteins in the 50 S subunit of an organelle ribosome (chloroplast). J Biol Chem 275:28466–28482
Yamaguchi K, Subramanian AR (2003) Proteomic identification of all plastid-specific ribosomal proteins in higher plant chloroplast 30S ribosomal subunit. Eur J Biochem 270:190–205
Yamaguchi K, von Knoblauch K, Subramanian AR (2000) The plastid ribosomal proteins. Identification of all the proteins in the 30 S subunit of an organelle ribosome (chloroplast). J Biol Chem 275:28455–28465
Yamaguchi K, Prieto S, Beligni MV, Haynes PA, McDonald WH, Yates JR 3rd, Mayfield SP (2002) Proteomic characterization of the small subunit of Chlamydomonas reinhardtii chloroplast ribosome: identification of a novel S1 domain-containing protein and unusually large orthologs of bacterial S2, S3, and S5. Plant Cell 14:2957–2974
Yamaguchi K, Beligni MV, Prieto S, Haynes PA, McDonald WH, Yates JR 3rd, Mayfield SP (2003) Proteomic characterization of the Chlamydomonas reinhardtii chloroplast ribosome. Identification of proteins unique to the 70S ribosome. J Biol Chem 278:33774–33785
Yukawa M, Tsudzuki T, Sugiura M (2005) The 2005 version of the chloroplast DNA sequence from tobacco (Nicotiana tabacum). Plant Mol Biol Rep 23:359–365
Zandueta-Criado A, Bock R (2004) Surprising features of plastid ndhD transcripts: addition of non-encoded nucleotides and polysome association of mRNAs with an unedited start codon. Nucleic Acids Res 32:542–550
Zehrmann A, van der Merwe JA, Verbitskiy D, Brennicke A, Takenaka M (2008) Seven large variations in the extent of RNA editing in plant mitochondria between three ecotypes of Arabidopsis thaliana. Mitochondrion 8:319–327
Zehrmann A, Verbitskiy D, van der Merwe JA, Brennicke A, Takenaka M (2009) A DYW domain-containing pentatricopeptide repeat protein is required for RNA editing at multiple sites in mitochondria of Arabidopsis thaliana. Plant Cell 21:558–567
Zerges W (2000) Translation in chloroplasts. Biochimie 82:583–601
Zybailov B, Friso G, Kim J, Rudella A, Rodriguez VR, Asakura Y, Sun Q, van Wijk KJ (2009) Large scale comparative proteomics of a chloroplast Clp protease mutant reveals folding stress, altered protein homeostasis, and feedback regulation of metabolism. Mol Cell Proteomics 8:1789–1810
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Cardi, T., Giegé, P., Kahlau, S., Scotti, N. (2012). Expression Profiling of Organellar Genes. In: Bock, R., Knoop, V. (eds) Genomics of Chloroplasts and Mitochondria. Advances in Photosynthesis and Respiration, vol 35. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-2920-9_14
Download citation
DOI: https://doi.org/10.1007/978-94-007-2920-9_14
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-2919-3
Online ISBN: 978-94-007-2920-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)