
Abstract
Despite the agricultural importance of both potato and tomato, very little is known about their chloroplast genomes. Analysis of the complete sequences of tomato, potato, tobacco, and Atropa chloroplast genomes reveals significant insertions and deletions within certain coding regions or regulatory sequences (e.g., deletion of repeated sequences within 16S rRNA, ycf2 or ribosomal binding sites in ycf2). RNA, photosynthesis, and atp synthase genes are the least divergent and the most divergent genes are clpP, cemA, ccsA, and matK. Repeat analyses identified 33–45 direct and inverted repeats ≥30 bp with a sequence identity of at least 90%; all but five of the repeats shared by all four Solanaceae genomes are located in the same genes or intergenic regions, suggesting a functional role. A comprehensive genome-wide analysis of all coding sequences and intergenic spacer regions was done for the first time in chloroplast genomes. Only four spacer regions are fully conserved (100% sequence identity) among all genomes; deletions or insertions within some intergenic spacer regions result in less than 25% sequence identity, underscoring the importance of choosing appropriate intergenic spacers for plastid transformation and providing valuable new information for phylogenetic utility of the chloroplast intergenic spacer regions. Comparison of coding sequences with expressed sequence tags showed considerable amount of variation, resulting in amino acid changes; none of the C-to-U conversions observed in potato and tomato were conserved in tobacco and Atropa. It is possible that there has been a loss of conserved editing sites in potato and tomato.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The chloroplast is a plant organelle that contains the entire enzymatic machinery in the stroma and electron carriers within the thylakoid membranes for photosynthesis. In addition to photosynthesis, several other biochemical pathways are present within chloroplasts, including biosynthesis of fatty acids, amino acids, pigments, and vitamins. The chloroplast genome generally has a highly conserved organization (Palmer 1991; Raubeson and Jansen 2005), with most land plant genomes composed of a single circular chromosome with a quadripartite structure that includes two copies of an inverted repeat (IR) that separate the large and small single copy regions (LSC and SSC). Our knowledge of the organization and evolution of chloroplast genomes has been expanding rapidly because of the large numbers of completely sequenced genomes published in the past decade. Currently, there are 47 completely sequenced plastid genomes (Raubeson and Jansen 2005; Jansen et al. 2005; http://www.megasun.bch.umontreal.ca/ogmp/projects/other/cp_list.html), and 29 of these are from various land plant lineages, with the best representation (21) from flowering plants. Comparative studies indicate that chloroplast genomes of land plants are highly conserved in both gene order and gene content. Several lineages of land plants have chloroplast DNAs (cpDNAs) with multiple rearrangements, including Pinus (Wakasugi et al. 1994) and the angiosperm families Campanulaceae (Cosner et al. 1997), Fabaceae (Palmer et al. 1988; Milligan et al. 1989; Kato et al. 2000), Geraniaceae (Palmer et al. 1987), and Lobeliaceae (Knox and Palmer 1998). In most of these studies, comparisons of gene content and order have been made among distantly related taxa because only one genome sequence was available from groups with rearranged genomes. Two exceptions are: grasses with genomic data available for four genera of crop plants (corn, wheat, sugar cane, and rice; Maier et al. 1995; Matsuoka et al. 2002; Tang et al. 2004) and legumes with genome sequences completed for three genera (alfalfa, soybean, and Lotus; Kato et al. 2000; Saski et al. 2005).
Chloroplast genetic engineering offers a number of unique advantages, including a high-level of transgene expression (DeCosa et al. 2001), multi-gene engineering in a single transformation event (DeCosa et al. 2001; Ruiz et al. 2003; Lossl et al. 2003; Quesada-Vargas et al. 2005), transgene containment via maternal inheritance (Daniell et al. 1998; Scott and Wilkenson 1999; Daniell 2002; Hagemann 2004) or cytoplasmic male sterility (Ruiz and Daniell 2005), lack of gene silencing (DeCosa et al. 2001; Lee et al. 2003; Dhingra et al. 2004), position effect (Daniell et al. 2002), pleiotropic effects (Lee et al. 2003; Daniell et al. 2001; Leelavathi and Reddy 2003) and lack of transformation vector sequences or selectable marker genes (Daniell et al. 2004a).
Plastid genetic engineering has also become a powerful tool for basic research in plastid biogenesis and function. This approach has helped unveil a wealth of information about plastid DNA replication origins, intron maturases, translation elements and proteolysis, import of proteins and several other processes (Daniell et al. 2004b). Although many successful examples of plastid engineering have set a solid foundation for various future applications, this technology has not been extended to many of the major crops. However, plastid transformation has been recently accomplished via somatic embryogenesis using partially sequenced chloroplast genomes in soybean (Dufourmantel et al. 2004), carrot (Kumar et al. 2004a), and cotton (Kumar et al. 2004b; Daniell et al. 2005). Transgenic carrot plants were able to withstand salt concentrations that only halophytes could tolerate (Kumar et al. 2004a).
The lack of complete chloroplast genome sequences is still one of the major limitations to extending this technology to useful crops; prior to 2004 only seven published crop chloroplast genomes were available and this number has increased to 23 during the past 2 years (Table 1). Chloroplast genome sequences are necessary for identification of spacer regions for integration of transgenes at optimal sites via homologous recombination, as well as endogenous regulatory sequences for optimal expression of transgenes (Daniell et al. 2005; Maier and Schmitz-Linneweber 2004). In higher plants, about 40–50% of each chloroplast genome contains noncoding spacer and regulatory regions (Saski et al. 2005; Lee et al. 2006; Jansen et al. 2006).
Once thought to be poisonous, tomato (Solanum lycopersicum) has become the second most commonly grown vegetable crop in the world behind potato. The total traded value of tomatoes in the United States is about US $13,493,496,000. The fresh-market export of US tomatoes was estimated to be 325,000 lbs while export was 2,095,000 lbs. Similarly, the volume of processed tomatoes exported in 2005 was about 1,295,500 lbs and imported about 3,080,000 lbs. Countries that export tomatoes to the United States include Canada, Chile, Mexico, Italy, and Israel (http://www.ers.usda.gov/Briefing/Tomatoes/trade.htm#tradetables). Traditional plant breeding has resulted in great progress in increasing yield, disease and pest resistance, environmental stress resistance, and quality and processing attributes. However, tomato plant breeding programs still strive to generate a better product. To assist in this goal, some plant breeding programs have been expanded to include biotechnological techniques. Tomato has long been recognized as an excellent genetic model for molecular biology studies. This has resulted in a flood of information including markers and genetic maps, identification of individual chromosomes, promoters and other nuclear genome sequences, and identification of genes and their function. However, there is not much information about the tomato chloroplast genome. Because of this, segments of the tobacco chloroplast genome were used as flanking sequences to facilitate integration of transgenes into the tomato chloroplast genome by homologous recombination, without knowing exact sequence identity (Ruf et al. 2001).
Solanum tuberosum (Irish or white potato) is the most economically significant crop in the US produce industry. With an annual farm value of US $2.5 billion and per capita use of 140 pounds in 2003, potato ranks first in value and consumption among all vegetables produced and consumed in the United States. Additionally, potato products such as french fries and potato chips generate billions more in revenue for the food-processing and food service industries. Currently exports account for 11% of US potato production in the form of fresh, seed, frozen and dehydrated potatoes (http://www.ers.usda.gov/Briefing/Potatoes). However, there is not much information on the potato chloroplast genome. When potato plastid genome was transformed, only the tobacco plastid genome flanking sequence was used to facilitate transgene integration by homologous recombination (Sidorov et al. 1999).
In this article we present the complete sequence of the chloroplast genomes of tomato and potato. One goal of this paper is to compare the genome organization of potato and tomato with the other two completely sequenced Solanaceae chloroplast genomes (tobacco and Atropa). In addition to examining gene content and gene order, we determine the distribution and location of repeated sequences among members of the Solanaceae. A second goal is to compare levels of DNA sequence divergence between chloroplast coding and noncoding regions. Intergenic spacer regions have been examined to identify ideal insertion sites for transgene integration and they are commonly used by plant systematists for resolving phylogenetic relationships among closely related species (Kelchner 2002; Shaw et al. 2005). A final goal of this paper is to examine the extent of RNA editing in Solanaceae chloroplast genomes by comparing the DNA sequences with available expressed sequence tags (EST) sequences. RNA editing is known to play an important role in several lineages of plants (Wolf et al. 2004; Kugita et al. 2003), but most of our knowledge about the frequency of this process in crop plants comes from studies in maize (Maier et al. 1995) and tobacco (Hirose et al. 1999).
Materials and methods
DNA sources
The bacterial artificial chromosome (BAC) libraries of potato and tomato were constructed by ligating size fractionated partial HindIII digests of total cellular high molecular weight DNA with the pINDIGOBAC vector. The average insert size of the potato and tomato libraries is 177 and 155 kb, respectively. BAC related resources for these public libraries can be obtained from the Clemson University Genomics Institute BAC/EST Resource Center (http://www.genome.clemson.edu).
BAC clones containing the chloroplast genome inserts were isolated by screening the library with a soybean chloroplast probe. The first 96 positive clones from screening were pulled from the library, arrayed in a 96-well microtiter plate, copied, and archived. Selected clones were then subjected to HindIII fingerprinting and NotI digests. End-sequences were determined and localized on the chloroplast genome of Arabidopsis thaliana to deduce the relative positions of the clones, then clones that covered the entire chloroplast genomes of potato and tomato were chosen for sequencing.
DNA sequencing and genome assembly
The nucleotide sequences of the BAC clones were determined by the bridging shotgun method. The purified BAC DNA was subjected to hydroshearing, end repair, and then size-fractionated by agarose gel electrophoresis. Fractions of approximately 3.0–5.0 kb were eluted and ligated into the vector pBLUESCRIPT IIKS+. The libraries were plated and arrayed into 40 96-well microtiter plates, respectively, for the sequencing reactions.
Sequencing was performed using the Dye-terminator cycle sequencing kit (Perkin Elmer Applied Biosystems, USA). Sequence data from the forward and reverse priming sites of the shotgun clones were accumulated. Sequence data equivalent to eight times the size of the genome was assembled using Phred-Phrap programs (Ewing and Green 1998).
Gene annotation
Annotation of the potato and tomato chloroplast genomes was performed using DOGMA (Dual Organellar GenoMe Annotator; Wyman et al. 2004; http://www.evogen.jgi-psf.org/dogma). This program uses a FASTA-formatted input file of the complete genomic sequences and identifies putative protein-coding genes by performing BLASTX searches against a custom database of previously published chloroplast genomes. The user must select putative start and stop codons for each protein coding gene and intron and exon boundaries for intron-containing genes. Both tRNAs and rRNAs are identified by BLASTN searches against the same database of chloroplast genomes.
Molecular evolutionary comparisons
Comparisons of gene content and gene order
Gene content comparisons were performed with Multipipmaker (Schwartz et al. 2003). Comparisons included four genomes: tobacco (NC_001879), potato (DQ 347958), tomato (DQ 347959), and Atropa (NC_004561) using tobacco as the reference genome. Gene orders were examined by pair-wise comparisons between the tobacco, potato, tomato, and Atropa genomes using PipMaker (Elnitski et al. 2002).
Examination of repeat structure
The repeat structure of the chloroplast genomes was examined in two stages. First, REPuter (Kurtz et al. 2001) was used to identify the number and location of direct and inverted (palindromic) repeats in the species of Solanaceae using a minimum repeat size of 30 bp and a Hamming distance of 3 (i.e., a sequence identity of ≥90%). Second, the repeats identified for tobacco were blasted against the complete chloroplast genomes of all four Solanaceae genomes. Blast hits of size 30 bp and longer with a sequence identity of ≥90% were identified to determine the shared repeats among the four genomes examined.
Comparisons of DNA sequence divergence
An aligned data set of all of the shared genes among the four Solanaceae chloroplast genomes was constructed by extracting these sequences from the annotated genomes either using DOGMA (Wyman et al. 2004) or the Chloroplast Genome Database (Cui et al. 2006; http://www.cbio.psu.edu/chloroplast/index.html). The sequences were aligned using ClustalX (Higgins et al. 1996) followed by manual adjustments using Seq Ap.
Molecular evolutionary analyses were then performed on the aligned data matrix using MEGA2 (Molecular Evolutionary Genetics Analysis; Kumar et al. 2001). Estimates of sequence divergence were based on the Kimura 2-parameter distance correction (Kimura 1980).
Comparison of intergenic spacer regions
Intergenic regions from four Solanaceae chloroplast genomes were compared using MultiPipMaker (Schwartz et al. 2003; http://www.pipmaker.bx.psu.edu/pipmaker/tools.html). MultiPipMaker offers a suite of software tools to analyze relationships among more than two sequences. In the current study, we used a program known as ‘all_bz’ that iteratively compares a pair of nucleotide sequences at a time until all possible pairs from all species have been compared. However, this program processes only one set of intergenic regions at a time. For genome-wide comparisons of corresponding intergenic regions from all species, we developed two programs written in PERL. The first program iteratively creates a set of input files containing corresponding intergenic regions from each species and compares them using ‘all_bz’ program, until all the intergenic regions in the chloroplast genome are processed. The second program parses the output from the above comparisons, calculates percent identity by using the number of identities over the length of the longer sequence and generates results in tab-delimited tabular format.
Variation between coding sequences and cDNAs
Each of the gene sequences from the potato chloroplast genome was used to perform a BLAST search of expressed sequence tags (ESTs) from Genbank. The retrieved EST sequences from potato, tomato, and tobacco were then aligned with the corresponding gene for each species separately, using Clustal X. In the case of Atropa, no sequences were retrieved from the Genbank even though its chloroplast sequence has been completed and studies of RNA editing have been previously performed (Schmitz-Linneweber et al. 2002). To maintain consistency in this study, only EST sequences were used and no other genomic sequences were considered. The aligned sequences were then screened and nucleotide and amino acid changes were detected using the Megalign software. The following criteria were used for comparisons of the DNA and EST sequences: (1) when more than one EST sequence was retrieved using BLAST, a change was recorded only if all sequences had the same change (substitution); (2) changes were recorded based on the base substitutions, that is, if there was an indel that affected the DNA sequence, it was not considered; and (3) if a retrieved EST sequence was too different (more than three consecutive nucleotide substitutions in a given sequence), it was not used for the analysis. In most cases, EST sequences were not of the same length as that of the corresponding gene, so the length of the analyzed sequence was recorded. Once a variable site was detected, the sequence was translated using the Megalign program using the plastid/bacterial genetic code and differences in the amino acid sequence were recorded.
Results
Size, gene content and organization of the tomato and potato chloroplast genomes
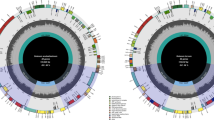
The complete sizes of the tomato and potato chloroplast genomes are 155,461 and 155,371 bp (Fig. 1), respectively. The genomes include a pair of inverted repeats of 25,611 bp (tomato) and 25,588 bp (potato), separated by a small single copy region of 18,363 bp (tomato) and 18,381 bp (potato) and a large single copy region of 85,876 bp (tomato) and 85,814 bp (potato). The difference in size of the two genomes is due partly to a slight expansion of the IR in tomato resulting in a partial duplication rps19, a phenomenon that is quite common in chloroplast genomes (Goulding et al. 1996).

Gene map of tomato and potato chloroplast genomes. The thick lines indicate the extent of the inverted repeats (IRa and IRb), which separate the genome into small (SSC) and large (LSC) single copy regions. Genes on the outside of the map are transcribed in the clockwise direction and genes on the inside of the map are transcribed in the counterclockwise direction. Numbers around the map indicate the location of repeated sequences found in Solanaceae genomes (see Table 2 for details). Lines with asterisks indicate the five groups of repeats that are shared by all four Solanaceae genomes: *tobacco and tomato, **tobacco and Atropa, ***tobacco
The potato and tomato chloroplast genomes contain 113 unique genes, and 20 of these are duplicated in the IR, giving a total of 133 genes (Fig. 1). There are 30 distinct tRNAs, and seven of these are duplicated in the IR. Seventeen genes contain one or two introns, and five of these are in tRNAs. The genomes consist of 58.3% (tomato), 59.6% (potato) coding regions that includes 50.7% (tomato), 52.0% (potato) protein coding genes and 7.6% (tomato and potato) RNA genes and 41.7% (tomato), 40.4% (potato) noncoding regions, containing both intergenic spacers and introns. The overall GC and AT content of the potato and tomato chloroplast genomes are 37.86% (tomato), 37.88% (potato) and 62.14% (tomato), 62.12% (potato), respectively.
Gene content and gene order
Gene content of the four sequenced species of Solanaceae (potato [DQ347958] & tomato [DQ347959] published here; tobacco [NC_001879] and Atropa [NC_004561]) is identical. Similarly, the gene order is identical among all four sequenced Solanaceae genomes. However, there are significant additions or deletions of nucleotides within certain coding sequences. For example, ACACGGGAAAC sequence is uniquely present within the 16S rRNA gene of potato, tomato, and Atropa but absent in tobacco or any other sequenced chloroplast genome (Fig. 2). Several deletions also occur within the coding sequence of ycf2 in Atropa, tomato, potato, and tobacco (Fig. 3). It should be noted that deleted nucleotides within the 16S rRNA and ycf2 are repeated sequences. In tomato ycf2 has three ribosome binding sites (GGAGG), whereas there is only one in all other Solanaceae members sequenced so far (Fig. 3).

Alignment of a portion of the 5′ end of the 16S ribosomal RNA showing a 9 bp insertion in Atropa, potato, and tomato. Nucleotides shown in red indicate base substitutions, yellow indicate the repeated sequence. Nucleotides shown are for the 16S rRNA gene, from nucleotides 46 to 96 or 105

Alignment of four regions of the ycf2 gene among the four Solanaceae chloroplast genomes showing insertion and deletion events. Nucleotides shown in red indicate base substitutions, yellow indicate the repeated sequence, and green indicate the start codon
Repeat structure
REPuter found 33–45 direct and inverted repeats 30 bp or longer with a sequence identity of at least 90% among the four chloroplast genomes examined (Fig. 4; see Supplemental Table 1 for a list of all repeats in all four genomes). The majority of the repeats in all four genomes are between 30 and 40 bp in length. The longest repeats other than the inverted repeats are found in tomato and consist of four 57 bp repeats not found in any of the other three genomes. Both tobacco and potato share a 50 and 56 bp repeat, whereas Atropa does not have a single repeat in the 50+ bp size range (excluding the IR).

Histogram showing the number and type (direct or inverted) of repeated sequences ≥30 bp long with a sequence identity ≥90% in the four Solanaceae chloroplast genomes using REPuter (Kurtz et al. 2001)
BlastN comparisons of the tobacco repeats (excluding the inverted repeat) against the chloroplast genomes of Atropa, potato, and tomato identified 42 repeats that show a sequence identity ≥90% with sequences ≥30 bp and a bit score greater than 40 (Table 2, Fig. 1). Thirty-seven of the 42 repeats are found in all four Solanaceae chloroplast genomes and all of these are located in the same genes or intergenic regions.
Intergenic spacer regions
All intergenic spacer regions except those less than 11 bp across the four Solanaceae chloroplast genomes were compared (Fig. 5a, Table 3; see Supplemental Table 2 for a list of sequence identities for all intergenic spacers). Only four spacer regions (rps11 - rpl 36, rps 7 - rps 12 3′ end, trnI-GAU - trnA-UGC, ycf 2 - ycf 15) have 100% sequence identity among all genomes (~2.5% of the spacer regions) and three of these regions are in the inverted repeat. Between tomato and potato 21 intergenic spacer regions have 100% sequence identity, whereas only eight regions have 100% sequence identity between tomato and Atropa, tobacco and potato, Atropa and potato, nine regions between tobacco and tomato and ten regions between tobacco and Atropa. The number of intergenic spacer regions with 100% sequence identity reflects the close phylogenetic relationship among the four Solanaceae genomes (Bohs and Olmstead 1997; Olmstead et al. 1999). It is noteworthy that one of the intergenic spacer regions that has 100% sequence identity between Atropa and potato (trnI-CAU - ycf 2) has only 66–69% sequence identity among the other Solanaceae species examined. Similarly, ycf4 - cemA has only 27% identity between tobacco and Atropa, potato and tomato, whereas it has greater than 90% identity between other Solanaceae species examined. There are several deletions or insertions in the intergenic spacer regions between trnQ - rps16, trnE - trnT, trnK - rps16, trnT - ycf 15, trnS - trnG, ycf2 - trnI, ycf 4 - cemA, ycf15 - trnL.

Histogram showing sequence divergence in pairwise comparisons among four Solanaceae chloroplast genomes for intergenic spacers (a) and coding regions (b). Pot potato, Tom tomato, Atr Atropa, and Tob tobacco. a Comparisons of 21 of the most variable intergenic regions. *, **, and *** indicate the tier 1, tier 2, and tier 3 regions reported in Shaw et al. (2005). The values plotted in this histogram come from Supplemental Table 2, which showed percent sequence identities for all intergenic spacers. The plotted values were converted from percent identity to sequence divergence on a scale from 0 to 1 and included on the Y-axis. b Sequence divergence of coding regions for the 11 different functional groups (Table 3)
Sequence divergence
We classified the chloroplast genes into 11 functional groups for comparisons of sequence divergence among coding regions (Table 4; Fig. 5b). Sequence divergence, which represents the proportion of nucleotide sites that differ, were estimated for all genes using the Kimura 2-parameter method (Kimura 1980). Overall, sequence divergence corresponds to the phylogenetic relationships among the four species of Solanaceae examined (Bohs and Olmstead 1997; Olmstead et al. 1999; Spooner et al. 1993). For example, the two most closely related species, potato and tomato, have the lowest divergence values for all classes of genes. Comparisons of sequence divergence among functional groups indicates that the RNA, photosynthesis, and atp synthase genes are the least divergent and that the most divergent genes are cemA, clpP, matK, and ccsA. Our comparisons of the levels of sequence divergence between noncoding and coding regions (Fig. 5a, b) indicate that the noncoding regions are more divergent than coding regions.
RNA variable sites in tomato and potato chloroplast transcripts
Based on the alignment of EST sequences retrieved from Genbank, 53 nucleotide substitution differences were observed in the tomato sequence (Table 5) and 47 were observed in potato (Table 6). However, with the exception of rpl23, all nucleotide substitutions occurred in different positions among both species. Of these substitutions, 11 were synonymous and 42 were nonsynonymous in tomato, whereas potato had 19 synonymous and 24 nonsynonymous substitutions. Potato had nine C-to-U conversions, five of which resulted in amino acid changes (Table 6). In tomato, seven C-to-U conversions were observed, all of which resulted in an amino acid change (Table 5). Although most genes in both species experienced one and three nucleotide substitutions, four genes had more than five variable sites. These were rpl36 and rpoC2 in tomato, with 7 and 10 nucleotide substitutions, respectively (Table 5), and rpl16 and ycf1 in potato, with 5 and 7 substitutions, respectively (Table 6). In addition, an amino acid alteration was observed in the tomato ycf1 gene that results in a stop codon at position 604. There is a complete copy of ycf1 and the truncated copy is at the IR/SSC boundary. It is the truncated copy that has the stop codon due to RNA editing. Thus there is still a full, functional copy of ycf1. Although there is evidence that ycf1 is a necessary chloroplast gene, it is missing from all grass genomes (Maier et al. 1995).
Discussion
Implications for integration of transgenes
Several intergenic spacer regions have been used to integrate foreign genes into the tomato and potato plastid genomes. These spacer regions are located between the following genes: trnfM and trnG, rbcL and accD, trnV and 3′-rps 12, and 16S rRNA and orf 70B (Ruf et al. 2001; Sidorov et al. 1999; Nguyen et al. 2005). Unfortunately, none of these regions have 100% sequence identity to the tobacco flanking sequence used in plastid transformation vectors. Potato plastid transformants were generated at 10–30 times lower frequencies than tobacco (Nguyen et al. 2005) and the intergenic spacer region between rbcL and accD shows only 94% identity. Similarly, the trnfM and trnG intergenic spacer region used for tomato plastid transformation has only 82% sequence identity, resulting in inefficient transgene integration. There are major deletions in the tomato chloroplast genome in this intergenic spacer region when compared to tobacco, which was used for plastid transformation (Ruf et al. 2001). These studies point out the importance of choosing appropriate intergenic spacers for plastid transformation. The use of one of the regions between tobacco and tomato or potato with 100% sequence identity (Table 3) might have enhanced recombination efficiency and thereby increased the efficiency of plastid transformation. Alternatively, if species-specific vectors are used, then one could use any of the intergenic spacer regions for transgene integration.
In addition to providing insight into genome organization and evolution, availability of complete DNA sequence of chloroplast genomes should facilitate plastid genetic engineering. Thus far, transgenes have been stably integrated and expressed via the tobacco chloroplast genome to confer several useful agronomic traits, including insect resistance (DeCosa et al. 2001; McBride et al. 1995; Kota et al. 1999), herbicide resistance (Daniell et al. 1998; Iamtham and Day 2000), disease resistance (DeGray et al. 2001), drought tolerance (Lee et al. 2003), salt tolerance (Kumar et al. 2004a), phytoremediation (Ruiz et al. 2003), and cytoplasmic male sterility (Ruiz and Daniell 2005). The chloroplast has been used as a bioreactor to produce vaccine antigens (Daniell et al. 2001; Molina et al. 2004; Tregoning et al. 2003; Watson et al. 2004; Koya et al. 2005), human therapeutic proteins (Daniell et al. 2004a; Staub et al. 2000; Fernandez-San Millan et al. 2003; Grevich and Daniell 2005), industrial enzymes (Leelavathi et al. 2003), and biomaterials (Lossl et al. 2003; Guda et al. 2000; Vitanen et al. 2004). Although many successful examples of plastid engineering in tobacco have set a solid foundation for various future applications, this technology has not been extended to many of the major crops. Complete chloroplast genome sequences should provide valuable information on spacer regions for integration of transgenes at optimal sites via homologous recombination, as well as endogenous regulatory sequences for optimal expression of transgenes and should help in extending this technology to other useful crops.
Evolutionary implications
Our comparisons of chloroplast genome organization between tomato and potato parallel earlier mapping studies of the nuclear genome of these important crop plants. Gene order of tomato and potato chloroplast genomes is identical, and this conservation extends to more distantly related genera (tobacco and Atropa) of Solanaceae. This is in contrast to the syntenic differences in the nuclear chromosomes of tomato and potato, which can be explained by three paracentric and two pericentric inversions (Bonierbale et al. 1988; Tanksley et al. 1992).
The analysis of repeated sequences in Solanaceae chloroplast genomes revealed 42 groups of repeats shared among various members of the family (Table 2, Fig. 1). Both direct and inverted repeats were identified. The origin of the repeats in the Solanaceae is not known, although replication slippage could be responsible for generating direct repeats. This mechanism has been suggested for chloroplast DNA (Palmer 1991) and evidence for replication slippage has been reported in the Oenothera chloroplast genome (Sears et al. 1996).
The fact that 37 of these 42 repeats are found in all four genomes examined suggests a high level of conservation of repeat structure. Furthermore, examination of the location of these repeats in the four genomes suggests that all of them occur in the same location, either in genes, introns or within intergenic spacers. This high level of conservation of both sequence identity and location suggests that these elements may play a functional role in the genome.
Except for the large inverted repeat, repeated sequences have generally been considered to be relatively uncommon in chloroplast genomes (Palmer 1991). One extraordinary exception is Chlamydomonas, which was estimated to have a genome comprised of more than 20% dispersed repeats (Maul et al. 2002). Dispersed repeats have also been identified in several families of flowering plants, including Trachelium (Cosner et al. 1997) (Campanulaceae), Trifolium (Milligan et al. 1989) (Fabaceae), wheat (Bowman and Dyer 1986; Howe 1985) (Poaceae), and Oenothera (Hupfer et al. 2000; Sears et al. 1996; Vomstein and Hachtel 1988) (Onagraceae). All of these genomes have gene order changes, suggesting that the repeats may have played a role in these changes. The chloroplast genomes of Solanaceae are not rearranged yet they still have a substantial number of repeats. A similar comparison of repeat structure among three legume chloroplast genomes (Saski et al. 2005) also identified a substantial number of repeat elements. Thus, it is becoming evident that chloroplast genomes contain a substantial number of repeated sequences other than the inverted repeat. Additional studies are needed to assess the possible functional role of these repeat elements.
Intergenic spacer regions are the most widely used chloroplast markers for phylogenetic investigations at lower taxonomic levels in plants (Kelchner 2002; Raubeson and Jansen 2005; Shaw et al. 2005). Plant phylogeneticists have utilized these markers because IGS regions are considered more variable and therefore should provide more characters. Several early studies support this contention; however, other studies questioned the systematic utility of chloroplast intergenic spacer regions (see references in Kelchner 2002). Our first genome-wide comparisons of the levels of sequence conservation in the intergenic spacer regions of four Solanaceae chloroplast genomes (Table 3, Fig. 5a, and Supplemental Table 2) demonstrate a wide range of sequence divergence in different regions. Furthermore, comparisons of coding (Fig. 5b) and noncoding (Fig. 5a) regions generally support the contention that intergenic spacer regions are more variable and could provide more phylogenetically informative characters for phylogenetic studies at lower taxonomic levels. Shaw et al. (2005) recently compared the phylogenetic utility of 21 noncoding chloroplast DNA regions. In their study, they ranked these 21 regions into three tiers based on their phylogenetic utility with tier one being the most useful by calculating the number of potentially informative characters. Although our genome-wide comparisons are based on sequence divergence, our results agree with the relative ranking of these regions in the Solanaceae (Fig. 5a; number of asterisks by gene names indicate Shaw et al.’s tiers). However, our comparisons have identified several intergenic regions that have higher sequence divergence than the most variable tier 1 regions identified by Shaw et al. (2005). Thus, our genome-wide comparisons provide valuable new information for the plant systematics community about the potential phylogenetic utility of the chloroplast intergenic spacer regions.
Our comparisons of DNA and EST sequences identified a substantial number of differences. Many of these differences are not likely due to RNA editing because previous studies of both Atropa (Schmitz-Linneweber et al. 2002) and tobacco (Hirose et al. 1999) have indicated that these types of events are exclusively C-to-U edits. Our analyses of both potato and tomato sequences (Tables 5, 6) showed a lower number of C-to-U changes than previously observed for these species (Hirose et al. 1999; Schmitz-Linneweber et al. 2002). In addition, none of the C-to-U conversions observed in potato and tomato were conserved with respect to the previous observations in tobacco and Atropa. It is more likely that the differences observed between the DNA and EST sequences are due to polymorphisms within these species, or even errors in the EST sequences. However, if future studies in the Solanaceae confirm that these differences are real and due to RNA editing, then it is possible that there has been a loss of conserved editing sites in potato and tomato. Evolutionary loss of RNA editing sites has been previously observed and could possibly be due to a decrease in the effect of RNA-editing enzymes (Wolf et al. 2004). Additionally, a considerable number of variable sites other than C-to-U conversions were observed in tomato and potato, suggesting that these chloroplast genomes may be accumulating considerable amounts of nucleotide substitutions, and some of the genes accumulate more variable sites than others. This has been previously observed in several chloroplast genes, such as petL and ndh genes, which have a high frequency of RNA editing (Fiebig et al. 2004). This suggests that, even though the chloroplast genome is relatively highly conserved among species, much of its variability could also be accounted for at the transcript level.
References
Asano T, Tsudzuki T, Takahashi S, Shimada H, Kadowaki K (2004) Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: a comparative analysis of four monocot chloroplast genomes. DNA Res 11:93–99
Bausher MG, Singh ND, Mozoru J, Lee S-B, Jansen RK, Daniell H (2006) The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var. ‘Ridge Pineapple’: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol (in review)
Bonierbale MW, Plaisted RL, Tanksley SD (1988) RFLP maps based on a common set of clones reveal modes of chromosomal evolution in potato and tomato. Genetics 120:1095–1103
Bohs L, Olmstead RG (1997) Phylogenetic relationships in Solanum (Solanaceae) based on ndhF sequences. Syst Bot 22:5–17
Bowman CM, Dyer T (1986) The location and possible evolutionary significance of small dispersed repeats in wheat ctDNA. Curr Genet 10:931–941
Cosner ME, Jansen RK, Palmer JD, Downie SR (1997) The highly rearranged chloroplast genome of Trachelium caeruleum (Campanulaceae): Multiple inversions, inverted repeat expansion and contraction, transposition, insertions/deletions, and several repeat families. Curr Genet 31:419–429
Cui L, Veeraraghavan N, Wall K, Jansen RK, Leebens-Mack J, Makalowska I, dePamphilis CW (2006) ChloroplastDB: the chloroplast genome database. Nucleic Acids Res 34:D692–D696
Daniell H (2002) Molecular strategies for gene containment in transgenic crops. Nat Biotechnol 20:581–586
Daniell H, Datta R, Varma S, Gray S, Lee S-B (1998) Containment of herbicide resistance through genetic engineering of the chloroplast genome. Nat Biotechnol 16:345–348
Daniell H, Lee S-B, Panchal T, Wiebe PO (2001) Expression of cholera toxin B subunit gene and assembly as functional oligomers in transgenic tobacco chloroplasts. J Mol Biol 311:1001–1009
Daniell H, Khan M, Allison L (2002) Milestones in chloroplast genetic engineering: an environmentally friendly era in biotechnology. Trends Plant Sci 7:84–91
Daniell H, Carmona-Sanchez O, Burns BB (2004a) Chloroplast-derived vaccine antibodies, biopharmaceuticals, and edible vaccines in transgenic plants engineered via the chloroplast genome. In: Schillberg S (ed) Molecular farming. Wiley, Germany, Chapter 8 pp 113–133
Daniell H, Cohill PR, Kumar S, Dufourmantel N (2004b) Chloroplast genetic engineering In: Daniell H, Chase CD (eds) molecular biology and biotechnology of plant organelles. Springer Publishers, Netherlands, pp 443–490
Daniell H, Kumar S, Duformantel N (2005) Breakthrough in chloroplast genetic engineering of agronomically important crops. Trends Biotechnol 23(5):238–245
DeCosa B, Moar W, Lee S-B, Miller M, Daniell H (2001) Overexpression of the Bt cry2Aa2 operon in chloroplasts leads to formation of insecticidal crystals. Nat Biotechnol 9:71–74
DeGray G, Rajasekaran K, Smith F, Sanford J, Daniell H (2001) Expression of an antimicrobial peptide via the chloroplast genome to control phytopathogenic bacteria and fungi. Plant Physiol 127:852–862
Dhingra A, Portis AR, Daniell H (2004) Enhanced translation of a chloroplast expressed rbcS gene restores SSU levels and photosynthesis in nuclear antisense RbcS plants. Proc Natl Acad Sci USA 101:6315–6320
Dufourmantel N, Pelissier B, Garçon F, Peltier JM, Tissot G (2004) Generation of fertile transplastomic soybean. Plant Mol Biol 55(4):479–89
Elnitski L, Riemer C, Petrykowska H et al (2002) PipTools: a computational toolkit to annotate and analyze pairwise comparisons of genomic sequences. Genomics 80:681–690
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred II error probabilities. Genome Res 8:186–194
Fernandez-San Millan A, Mingo-Castel A, Miller M, Daniell H (2003) A chloroplast transgenic approach to hyper express and purify human serum albumin, a protein highly susceptible to proteolytic degradation. Plant Biotechnol J 1:71–79
Fiebig A, Stegemann S, Bock R (2004) Rapid evolution of RNA editing sites in a small non-essential plastid gene. Nucleic Acids Res 32:3615–3622
Goulding SE, Olmstead RG, Morden CW, Wolfe KH (1996) Ebb and flow of the chloroplast inverted repeat. Mol Gen Genet 252:195–206
Grevich JJ, Daniell H (2005) Chloroplast genetic engineering: Recent advances and future perspectives. Crit Rev Plant Sci 24:83–108
Guda C, Lee S-B, Daniell H (2000) Stable expression of biodegradable protein based polymer in tobacco chloroplasts. Plant Cell Rep 19:257–262
Hagemann R (2004) The sexual inheritance of plant organelles. In: Daniell H, Chase C (eds) Molecular biology and biotechnology of plant organelles. Springer Publishers, Dordrecht, pp 93–113
Higgins DG, Thompson JD, Gibson TJ (1996) Using CLUSTAL for multiple sequence alignments. Meth Enzymol 266:383–402
Hiratsuka J, Shimada H, Whittier R et al (1989) The complete sequence of rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol Gen Genet 217:185–194
Hirose T, Kusumegi T, Tsudzuki T, Sugiura M (1999) RNA editing sites in tobacco chloroplast transcripts: editing as a possible regulator of chloroplast RNA polymerase activity. Mol Gen Genet 262:462–467
Howe CJ (1985) The endpoints of an inversion in wheat chloroplast DNA are associated with short repeated sequences containing homology to att-lambda. Curr Genet 10:139–145
Hupfer H, Swaitek M, Hornung S et al. (2000) Complete nucleotide sequence of the Oenothera elata plastid chromosome, representing plastome 1 of the five distinguishable Euoenthera plastomes. Mol Gen Genet 263:581–585
Iamtham S, Day A (2000) Removal of antibiotic resistance genes from transgenic tobacco plastids. Nat Biotechnol 18:1172–1176
Jansen RK, Raubeson LA, Boore JL et al (2005) Methods for obtaining and analyzing chloroplast genome sequences. Meth Enzym 395:348–384
Jansen RK, Kaittanis C, Saski C, Lee S-B, Tompkins J, Alverson AJ, Daniell H (2006) Phylogenetic analyses of Vitis (Vitaceae) based on complete chloroplast genome sequences: effects of taxon sampling and phylogenetic methods on resolving relationships among rosids. BMC Evol Biol (in press)
Kato T, Kaneko T, Sato S, Nakamura Y, Tabata S (2000) Complete structure of the chloroplast genome of a legume, Lotus japonicus. DNA Res 7:323–330
Kelchner SA (2002) The evolution of non-coding chloroplast DNA and its application in plant systematics. Ann Missouri Bot Gard 87:482–498
Kim K-J, Lee H-L (2004) Complete chloroplast genome sequence from Korean Ginseng (Panax schiseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res 11:247–261
Kim J-S, Jung JD, Lee J-A et al. (2006) Complete sequence and organization of the cucumber (Cucumis sativus L. cv. Baekmibaekdadagi) chloroplast genome. Plant Cell Rep, online
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Knox EB, Palmer JD (1998) Chloroplast DNA evidence on the origin and radiation of the giant lobelias in eastern Africa. Syst Bot 23:109–149
Kota M, Daniel H, Varma S, Garczynski SF, Gould F, William MJ (1999) Overexpression of the Bacillus thuringiensis (Bt) Cry2Aa2 protein in chloroplasts confers resistance to plants against susceptible and Bt-resistant insects. Proc Natl Acad Sci USA 96:1840–1845
Koya V, Moayeri M, Leppla SH, Daniell H (2005) Plant based vaccine: mice immunized with chloroplast-derived anthrax protective antigen survive anthrax lethal toxin challenge. Infect Immun 73:8266–8274
Kugita M, Yamamoto Y, Fujikawa T, Matsumoto T, Yoshinaga K (2003) RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucleic Acids Res 31:2417–2423
Kumar S, Koichiro T, Jakobsen IB, Nei M (2001) MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244–1245
Kumar S, Dhingra A, Daniell H (2004a) Plastid expressed betaine aldehyde dehydrogenase gene in carrot cultured cells, roots and leaves confers enhanced salt tolerance. Plant Physiol 136:2843–2854
Kumar S, Dhingra A, Daniell H (2004b) Manipulation of gene expression facilitates plastid transformation of cotton by somatic embryogenesis and maternal inheritance of transgenes. Plant Mol Biol 56:203–216
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29:4633–4642
Lee S-B, Kwon H-B, Kwon S-J et al. (2003) Accumulation of trehalose within transgenic chloroplasts confers drought tolerance. Mol Breed 11:1–13
Lee S-B, Kaittanis C, Hostetler J, Town C, Jansen RK, Daniell H (2006) The complete chloroplast genome sequence of Gossypium hirsutum: organization and phylogenetic relationships to other angiosperms. BMC Genomics (in press)
Leelavathi S, Reddy VS (2003) Chloroplast expression of His-tagged GUS-fusions: a general strategy to overproduce and purify foreign proteins using transplastomic plants as bioreactors. Mol Breed 11:49–58
Leelavathi S, Gupta N, Maiti S, Ghosh A, Reddy VS (2003) Overproduction of an alkali-and thermo-stable xylanase in tobacco chloroplasts and efficient recovery of the enzyme. Mol Breed 11:59–67
Lossl A, Eibl C, Harloff HJ, Jung C, Koop HU (2003) Polyester synthesis in transplastomic tobacco (Nicotiana tabacum L): significant contents of polyhydroxybutyrate are associated with growth reduction. Plant Cell Rep 21:891–899
Maier RM, Schmitz-Linneweber (2004) Plastid genomes. In: Daniell H Chase CD (eds) Molecular biology and biotechnology of plant organelles. Springer publishers, Netherlands, pp 115–150
Maier RM, Neckermann K, lgloi GL, Kossel H (1995) Complete sequence of the maize chloroplast genome: Gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J Mol Biol 251:614–628
Masood MS, Nishikawa T, Fukuoka S, Njenga PK, Tsudzuki T, Kadowaki K (2004) The complete nucleotide sequence of wild rice (Oryza nivara) chloroplast genome: first genome wide comparative sequence analysis of wild and cultivated rice. Gene 340:133–139
Matsuoka Y, Yamazaki Y, Ogihara Y, Tsunewaki K (2002) Whole chloroplast genome comparison of rice, maize, and wheat: implications for chloroplast gene diversification and phylogeny of cereals. Mol Biol Evol 19:2084–2091
Maul JE, Lilly JW, Cui L et al. (2002) The Chlamydomonas reinhardtii plastid chromosome: Islands of genes in a sea of repeats. The plant Cell 14:1–22
McBride KE, Svab Z, Schaaf DJ, Hogan PS, Stalker DM, Maliga P (1995) Amplification of a chimeric Bacillus gene in chloroplasts leads to an extraordinary level of an insecticidal protein in tobacco. Bio Technol 13:362–365
Milligan BG, Hampton JN, Palmer JD (1989) Dispersed repeats and structural reorganization in subclover chloroplast DNA. Mol Biol Evol 6:355–368
Molina A, Herva-Stubbs S, Daniell H, Mingo-Castel AM, Veramendi J (2004) High yield expression of a viral peptide animal vaccine in transgenic tobacco chloroplasts. Plant Biotechnol J 2:141–153
Nguyen TT, Nugent G, Cardi T, Dix PJ (2005) Generation of homoplasmic plastid transformants of a commercial cultivar of potato (Solanum tuberosum L). Plant Sci 168:1495–1500
Ogihara Y, Isono K, Kojima T et al. (2000) Chinese spring wheat (Triticum aestivum L.) chloroplast genome: complete sequence and contig clones. Plant Mol Biol Rep 18:243–253
Olmstead RG, Sweere JA, Spangler RE, Bohs L, Palmer JD (1999) Phylogeny and provisional classification of the Solanaceae based on chloroplast DNA. In: Nee M, Symon DE, Jessup JP, Hawkes JG (eds) Solanaceae IV, advances in biology and utilization. Royal Botanic Gardens, Kew, pp 111–137
Palmer JD (1991) Plastid chromosomes: structure and evolution. In: Hermann RG (ed) The molecular biology of plastids. Cell culture and somatic cell genetics of plants, vol 7A. Springer-Verlag, Vienna, pp 5–53
Palmer JD, Nugent JM, Herbon LA (1987) Unusual structure of Geranium chloroplast DNA—a triple-sized inverted repeat, extensive gene duplications, multiple inversions, and two repeat families. Proc Natl Acad Sci USA 84:769–773
Palmer JD, Osorio B, Thompson WF (1988) Evolutionary significance of inversions in Legume chloroplast DNAs. Curr Genet 14:65–74
Quesada-Vargas T, Ruiz ON, Daniell H (2005) Characterization of heterologous multigene operons in transgenic chloroplasts: transcription, processing, translation. Plant Physiol 138:1746–1762
Raubeson LA, Jansen RK (2005) Chloroplast genomes of plants. In: Henry R (ed) Diversity and evolution of plants-genotypic and phenotypic variation in higher plants. CABI Publishing, Wallingford, pp 45–68
Ruf S, Hermann M, Berger I, Carrer H, Bock R (2001) Stable genetic transformation of tomato plastids and expression of a foreign protein in fruit. Nat Biotechnol 19:870–875
Ruiz ON, Daniell H (2005) Engineering. cytoplasmic male sterility via the chloroplast genome. Plant Phys 138:1232–1246
Ruiz ON, Hussein H, Terry N, Daniell H (2003) Phytoremediation of organomercurial compounds via chloroplast genetic engineering. Plant Phys 32:1344–1352
Saski C, Lee S-B, Daniell HT et al. (2005) Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol Biol 59:309–322
Schmitz-Linneweber C, Maier RM, Alcaraz JP, Cottet A, Herrmann RG, Mache R (2001) The plastid chromosome of spinach (Spinacia oleracea): complete nucleotide sequence and gene organization. Plant Mol Biol 45:307–315
Schmitz-Linneweber C, Regel R, Du TG, Hupfer H, Herrmann RG, Maier RM (2002) The plastid chromosome of Atropa belladonna and its comparison with that of Nicotiana tabacum: the role of RNA editing in generating divergence in the process of plant speciation. Mol Biol Evol 19:1602–1612
Schwartz S, Elnitski L, Li M et al. (2003) MultiPipMaker and supporting tools: alignments and analysis of multiple genomic DNA sequences. Nucleic Acids Res 31:3518–3524
Scott SE, Wilkenson MJ (1999) Low probability of chloroplast movement from oilseed rape (Brassica napus) into wild Brassica rapa. Nat Biotechnol 17:390–392
Sears BB, Stoike LL, Chiu WL (1996) Proliferation of direct repeats near the Oenothera chloroplast DNA origin of replication. Mol Biol Evol 13:850–863
Shaw J, Lickey EB, Beck JT et al. (2005) The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analyses. Am J Bot 92:142–166
Shinozaki K, Ohme M, Tanaka et al. (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. The EMBO J 5:2043–2049
Sidorov VA, Kasten D, Pang SZ, Hajdukiewicz PT, Staub JM, Nehra NS (1999) Technical advance: stable chloroplast transformation in potato: use of green fluorescent protein as a plastid marker. Plant J 19:209–216
Spooner DM, Anderson GJ, Jansen RK (1993) Chloroplast DNA evidence for the interrelationships of Tomatoes, Potatoes, and Pepinos. Am J Bot 8:676–688
Staub JM, Garcia B, Graves J, Hajdukiewicz PTJ, Hunter P, Nehra N (2000) High-yield production of a human therapeutic protein in tobacco chloroplasts. Nat Biotechnol 18:333–338
Steane DA (2005) Complete nucleotide sequence of the chloroplast genome from the Tasmanian Blue Gum, Eucalyptus globules (Myrtaceae). DNA Res 12:215–220
Tang J, Xia H, Cao M (2004) A comparison of rice chloroplast genomes. Plant Phys 135:412–420
Tanksley SD, Ganal MW, Prince JP et al. (1992) High density molecular linkage maps of tomato and potato genomes. Genetics 132:1141–1160
Timme RE, Kuehl JV, Boore JL, Jansen RK (2006) A comparison of the first two sequenced chloroplast genomes in Asteraceae: Lettuce and Sunflower. BMC Evol Biol (in review)
Tregoning JS, Nixon P, Kuroda H et al. (2003) Expression of tetanus toxin Fragment C in tobacco chloroplasts. Nucleic Acids Res 31(4):1174–1179
Vitanen PV, Devine AL, Kahn S, Deuel DL, Van-Dyk DE, Daniell H (2004) Metabolic engineering of the chloroplast genome using the E coli ubiC gene reveals that corismate is a readily abundant precursor for 4-hydroxybenzoic acid synthesis in plants. Plant Phys 136:4048–4060
Vomstein J, Hachtel W (1988) Deletions, insertions, short inverted repeats, sequences resembling att-lambda, and frame shift mutated open reading frames are involved in chloroplast DNA differences in the genus Oenothera subsection Munzia. Mol Gen Genet 213:513–518
Wakasugi T, Tsudzuki J, Ito S, Nakashima K, Tsudzuki T, Sugiura M (1994) Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc Natl Acad Sci USA 91:9794–9798
Watson J, Koya V, Leppla SH, Daniell H (2004) Expression of Bacillus anthracis protective antigen in transgenic chloroplasts of tobacco, a non-food/feed crop. Vaccine 22:4374–4384
Wolf PG, Rowe CA, Hasebe M (2004) High levels of RNA editing in a vascular plant chloroplast genome: analysis of transcripts from the fern Adiantum capillus-veneris. Gene 339:89–97
Wyman SK, Boore JL, Jansen RK (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20:3252–3255
Acknowledgments
Investigations reported in this article were supported in part by grants from USDA 3611-21000-017-00D to Henry Daniell and from NSF DEB 0120709 to Robert K. Jansen.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Hagemann
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Daniell, H., Lee, SB., Grevich, J. et al. Complete chloroplast genome sequences of Solanum bulbocastanum, Solanum lycopersicum and comparative analyses with other Solanaceae genomes. Theor Appl Genet 112, 1503–1518 (2006). https://doi.org/10.1007/s00122-006-0254-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-006-0254-x