
Abstract
Intranasal administration of topical drugs has always been used for symptomatic relief or treatment of local nasal dysfunctions. However, over the last years, the use of the nasal route for drug delivery as a viable and promising alternative to conventional oral and parenteral routes has tremendously increased. Indeed, owing to the high vascularisation and large absorptive surface, the nasal respiratory mucosa is recognised as an appropriate site for systemic entry of drugs, circumventing the gastrointestinal and hepatic first-pass metabolism. Accordingly, the extent of absorption and bioavailability of several therapeutic compounds have been improved with intranasal delivery, particularly those that are poorly permeable and/or highly susceptible to enzymatic degradation, such as small polar molecules, peptides and proteins. On the other hand, the intranasal route has also demonstrated to have potential for targeting central nervous system-acting drugs since nasal olfactory region allows the direct connection between the nose and the brain.
In this chapter, the most relevant in vivo and in vitro findings that support the advantages of the intranasal delivery of drugs with topical, systemic and central nervous system action are critically discussed as well as alternative approaches frequently adopted to overcome the high mucociliary clearance, the reduced residence time and the poor nasal permeability of some compounds. Throughout this overview, nasal therapeutic compounds already marketed and those currently under investigation are also highlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Nasal route
- Nasal drug delivery
- Intranasal administration
- Nasal therapeutic agents
- Intranasal delivery
- Small-molecule drugs
- Topical drugs
- Systemic drugs
- Central nervous system-acting drugs
- Biomolecular drugs
IN administration of topical drugs (decongestants, antihistamines, corticosteroids or antimicrobials) has been widely used for symptomatic relief and prevention/treatment of nasal dysfunctions, such as nasal congestion and acute or chronic rhinosinusitis.
IN administration is now recognised as a therapeutically viable way for delivery of systemic drugs as alternative to the parenteral and oral routes.
Over the last years, new pharmaceutical formulations and novel delivery strategies have been developed offering promising opportunities to expand the delivery of small-molecule drugs and biomacromolecular drugs by the nasal route.
Nasal drug delivery is particularly interesting for compounds such as polar small drugs, and therapeutic peptides and proteins.
IN drug delivery is a patient-friendly administration route avoiding the pain associated with parenteral administration and that enables to circumvent poor stability in gastrointestinal fluids, poor intestinal absorption and/or hepatic first-pass metabolism related with oral route.
A wide variety of IN drugs exhibit plasma concentrations and systemic bioavailability frequently higher than those obtained for oral administration. Sometimes they are even comparable to those obtained after IV administration.
The potential of the nasal route for administration of drugs into systemic circulation has been remarkably evidenced for a wide variety of drugs and it is particularly interesting when a rapid onset of action is a key requirement.
IN administration is currently emerging as a promising way for direct delivery of drugs to the brain, which may be extremely useful for treatment of neurological conditions such as epilepsy, Alzheimer’s disease and Parkinson’s disease.
IN delivery of some CNS-acting drugs has afforded higher concentrations in the brain than those reached after IV administration, probably due to readily access to the brain, avoiding the blood–brain barrier.
Non-invasive mucosal routes, with less importance for drug delivery in the past, have now assumed a greater interest for delivery of peptide-, protein- and nucleic acid-based drugs or vaccines, particularly the nasal route.
In the pharmaceutical formulation of peptide-, protein- and other biomacromolecular-based drugs intended for IN delivery, the use of suitable vehicles, enzyme inhibitors and/or penetration enhancers is of paramount importance.
The use of suitable vehicles, enzyme inhibitors and/or penetration enhancers is of paramount importance during the development of IN pharmaceutical formulations of peptide-, protein- and other biomacromolecular drugs.
The successful IN administration of some polypeptide drugs (desmopressin, calcitonin, buserelin, nafarelin and oxytocin) has promoted an extensive evaluation of the nasal route for delivery of many other protein and peptide drugs currently used as injectables.
The immunisation through the nasal route is an interesting opportunity that has been increasingly explored over the last years. The recent developments lead us to believe that the availability of new vaccines for IN delivery will be greatly expanded in the near future.
15.1 Introduction
Nasal drug delivery is now well recognised as a useful alternative to oral and parenteral routes. Undoubtedly, the intranasal (IN) administration of medicinal products for the symptomatic relief and prevention or treatment of topical nasal conditions has been widely used for a long period of time. However, recently, the nasal mucosa has seriously emerged as a therapeutically viable route for the systemic drug delivery. Among the primary target drugs for IN administration are those compounds with poor stability in gastrointestinal fluids, poor intestinal absorption and/or extensive hepatic first-pass elimination, such as polar small drugs, peptides and proteins. The nasal drug delivery seems to be also an encouraging way to circumvent the blood–brain barrier (BBB) enabling direct nose-to-brain delivery of central nervous system (CNS)-active agents.
Over the last years, several comprehensive reviews have been published discussing in some detail particular aspects of drug delivery through the nasal route. Therefore, this chapter is built based on this information and focuses on recent developments in the area, discussing the major factors affecting nasal drug delivery and highlighting nasal therapeutic agents currently available on the market and also some candidates for IN administration.
15.2 Intranasal Delivery of Topical Drugs
IN route has been widely used for a long time as an attractive option for local (or topical) drug delivery. Typical examples of locally acting intranasally administered drugs are decongestants, antihistamines, corticosteroids and antimicrobials. They are mainly indicated in the treatment of nasal congestion, rhinitis and sinusitis (rhinosinusitis), inflammation and infection. Topical therapies enable direct drug delivery to the target organ (biophase) and the use of lower effective doses which minimises the potential for systemic adverse effects that may occur with oral and parenteral therapy. The choice between topical and systemic therapy depends on spectrum disease and on the efficacy to safety ratio of each therapy (Bitter et al. 2011; Costantino et al. 2007; Salib and Howarth 2003).
15.2.1 Decongestants
Topical nasal decongestants are widely prescribed for the symptomatic relief of nasal congestion in common cold, allergic and nonallergic/vasomotor rhinitis, acute and chronic rhinosinusitis (CRS) and nasal polyposis (Meltzer et al. 2010).
The most common nasal topical decongestants are phenylephrine, pseudoephedrine, oxymetazoline and xylometazoline. The first two are sympathomimetic amines while the others are imidazoline derivatives. Their pharmacologic effect results from direct or indirect activation of postsynaptic α-adrenergic receptors of the nasal mucosa vasculature; this produces vasoconstriction and subsequent decrease of mucosa swelling and nasal resistance to airflow which leads to decongestion (Corboz et al. 2008). Both groups are α-adrenoceptors agonists; however, sympathomimetic amines preferentially bind to α1-adrenoceptors while imidazolines predominantly address α2-adrenoceptors. Moreover, imidazoline derivatives also cause a reduction in the nasal mucosal blood flow due to their activity on the resistance vessels (α2-adrenoceptors) which contributes to nasal decongestion (Caenen et al. 2005; Hochban et al. 1999). The effect of topical decongestants has already been studied in the past by several authors using different methods (Bende and Löth 1986; Maranta and Simmen 1996). The most common problem associated with overuse of topical decongestants is rebound nasal congestion, reduction in efficacy (tachyphylaxis) and nonspecific nasal hyperreactivity. This clinical condition is defined as rhinitis medicamentosa and it limits the practical utility of topical decongestants to short-term therapies. The treatment options for rhinitis medicamentosa include the immediate suspension of nasal decongestant and some authors suggest the use of corticosteroids (Akpinar et al. 2012; Graf 1997; Vaidyanathan et al. 2010). This has recently been the motivation for a novel approach consisting in the simultaneous use of nasal decongestants and corticosteroids to overcome the limitation of the long-term use. Vaidyanathan et al. (2010) evaluated the effect of combining IN fluticasone propionate with oxymetazoline after 14 days of treatment with IN oxymetazoline alone. After 3 days of this combined administration (day 17), tachyphylaxis of response and rebound congestion, induced by the prolonged use of IN oxymetazoline, were reduced by IN fluticasone propionate concomitant administration. This finding along with other studies may open new perspectives for the prolonged use of topical decongestants in clinical practice (Akpinar et al. 2012; Vaidyanathan et al. 2010).
15.2.2 Antihistamines and Corticosteroids
IN antihistamines and corticosteroids are efficacious topical drugs used in the treatment of allergic rhinitis (AR). This pathology is defined as an inflammation of the nasal mucosa caused by a hyperactive immune system response to benign, non-infectious environmental aeroallergens (e.g. pollens, mites, animal danders) (Dykewicz and Hamilos 2010).
Antihistamines, the commonly known H1 receptor antagonists, are particularly effective at reducing the symptoms of sneezing, nasal itching and rhinorrhoea in AR. Interestingly, as reviewed by Howarth in 2000, in vitro investigations revealed that some antihistamines have also the potential to modify the inflammatory process, in addition to their H1 histamine receptor blocking action. However, for these effects to be fully evident, in vivo antihistamine doses must be higher than those usually tolerated, leading to sedative adverse effects. Thus, the topical IN delivery of antihistamines appears as an advantageous strategy to directly target the organ with therapeutic drug concentrations, minimising the risk of systemic adverse effects (Howarth 2000; Meltzer et al. 2010; Salib and Howarth 2003).
Topical nasal antihistamines represent the latest therapeutic option added to the armamentarium of AR management. IN antihistamines include levocabastine, azelastine and olopatadine and their efficacy is equal or superior to that of second-generation oral antihistamines. These topical agents have a rapid onset of action, which makes them an appropriate “as required” therapy for episodic AR symptoms relief (Dykewicz and Hamilos 2010; Kaliner et al. 2009; Sur and Scandale 2010).
Although levocabastine is a second-generation antihistamine, it causes some sedation when administered orally. For this reason and because of its remarkable potency, levocabastine was subsequently developed for IN delivery. Topical levocabastine has an onset of action of 10–15 min and is effective for up to 12 h (Salib and Howarth 2003).
Topical azelastine is another second-generation antihistamine that has been developed to overcome the sedation effects of oral administration. This drug offers an onset of action of 15 min and has a systemic bioavailability of 40 % following IN administration. The estimated systemic exposure of topical azelastine is six to eightfold lower than that observed for the oral drug. IN azelastine exhibits superior efficacy compared to IN levocabastine. In a double-blind parallel group study, levocabastine and azelastine nasal sprays provided a good symptomatic treatment of seasonal AR; however, azelastine was statistically more effective and safer than levocabastine (Falser et al. 2001).
Olopatadine is the most recent topical nasal antihistamine introduced in the market. Firstly, it was approved as an ophthalmic solution, but in 2008, olopatadine appeared as a nasal spray indicated for the treatment of seasonal AR. Clinical trials of olopatadine nasal spray have shown an onset of action within 30 min and a significant efficacy in relieving nasal allergy symptoms, including nasal congestion (Kaliner et al. 2009; Roland et al. 2010).
The AR and its impact on asthma guidelines recommend the use of IN antihistamine in mild persistent disease or in occasional symptoms for intermittent disease. In the case of IN corticosteroids, they should be regarded as first-line therapy for moderate to severe persistent disease (Salib and Howarth 2003).
IN corticosteroids are recognised as “the gold standard” of therapeutic choice in AR. Compared with oral or local antihistamines, IN corticosteroids are more effective in what concerns the relief of nasal congestion symptom. There are a wide variety of IN corticosteroid molecules; they include beclomethasone, budesonide, triamcinolone acetonide, flunisolide, fluticasone propionate, mometasone furoate, ciclesonide and fluticasone furoate (Salib and Howarth 2003; Sastre and Mosges 2012). This pharmacological class acts very early in the inflammatory pathway, modifying the ability of pro-inflammatory transcription factors to up-regulate gene expression (Howarth 2000). This mechanism of action implies a time delay between administration and clinical activity. Hence, IN corticosteroids have a slower onset of action (several hours) than IN antihistamines with maximum efficacy developing over a period of days and weeks. In most cases, a once-daily regimen is sufficient and compatible with patient compliance; in severe cases and during exacerbation, twice-daily administration is indicated (Salib and Howarth 2003).
Since oral and some high-dose inhaled corticosteroids have systemic adverse effects, IN administration of corticosteroids emerges as a promising alternative route to enhance the safety profile of these agents. Nevertheless, one should be aware of the possibility of these topical agents reaching the systemic circulation in sufficient concentration to produce adverse effects (Salib and Howarth 2003; Sastre and Mosges 2012). The newer IN corticosteroid agents (e.g. fluticasone propionate, mometasone furoate, ciclesonide, fluticasone furoate) have pharmacokinetic properties that minimise their systemic bioavailability compared to older IN corticosteroids (e.g. beclomethasone, flunisolide, triamcinolone acetonide, budesonide) and oral agents (e.g. methylprednisolone). The systemic bioavailability of the newer IN corticosteroids drugs is negligible (<1 %) which contributes for minimal risk of systemic adverse effects (Sastre and Mosges 2012). Drug lipophilicity plays an important role in pharmacokinetic profile and pharmacodynamic action of IN corticosteroids. Increased lipophilicity is associated with greater deposition and slower release from the nasal respiratory tissue, greater binding affinity for corticosteroid receptor and consequently less free drug is available for systemic absorption, which results in fewer systemic adverse effects. Fluticasone furoate is a novel-enhanced affinity corticosteroid recently approved by the Food and Drug Administration (FDA) in 2007; experimental studies have shown that it has the most potent and fastest anti-inflammatory activity (Giavina-Bianchi et al. 2008). The clinical efficacy of IN corticosteroids does not depend only on the relative affinity for corticosteroid receptor but also on the drug retention in the nasal mucosa. This may be primarily attributed to lipophilicity as in the case of fluticasone propionate; however, for budesonide, an additional contribution is provided by its ability to reversibly form fatty acid esters in the mucosa that may hold and release the regenerated budesonide locally. This reversible nasal metabolism adds to lipophilicity to originate the higher retention of budesonide when compared to fluticasone propionate (Petersen et al. 2001).
15.2.3 Antimicrobials
Since the early 1990s, topical antimicrobial treatment of CRS has attracted increasing attention. Although the exact aetiology and pathophysiology of CRS are still unknown, bacteria and fungi appear to be implicated in the development of this disease. Furthermore, bacterial and fungal biofilms, which are microcolonies embedded in an extracellular polysaccharide matrix with greater antimicrobial resistance than planktonic bacteria, have been associated with chronic infections. The aims of CRS treatment are reduction of nasal and paranasal mucosal inflammation, control of infection and re-establishment of mucociliary clearance (MCC) (Dykewicz and Hamilos 2010; Foreman et al. 2012; Suh and Kennedy 2011). The mainstay of CRS treatment is topical corticosteroids and oral antibiotics; the efficacy of topical antibiotics is still under investigation. These have the theoretical advantage of achieving higher concentrations of antibiotics at the target site which has been shown to be effective against bacteria in biofilm form. Moreover, topical usage is less liable to produce systemic adverse effects (Lim et al. 2008).
A systematic review of IN antimicrobials in the management of CRS was presented by Lim et al. (2008). Antimicrobials investigated included topical tobramycin, mupirocin, N-chlorotaurine, fosfomycin, ceftazidime, cefmenoxime and amphotericin. The purpose of this study was to identify evidence for the benefit of topical antimicrobials in several CRS subgroups, classified according to method of delivery, culture-directed or empiric therapy, presence or absence of previous surgery, stable or acute exacerbations of CRS and type of antimicrobial (antibiotics and antifungals). The authors concluded that a low level of evidence points to the efficacy of topical antibiotics in both stable and acute exacerbations of CRS and no definite conclusion could be made regarding the use of antifungals (Adappa et al. 2012; Lim et al. 2008). In what concerns the mode of delivery, there was evidence for the use of nasal irrigation or nebulisation rather than delivery by nasal spray. Nebulisers and nasal irrigations have advantages over the nasal sprays for the successful delivery of topical drugs. In fact, nasal sprays achieve a smaller deposition surface area than that covered by nebulisation and their drug distribution effect depends on MCC which is impaired in CRS. Although non-aerosol based, nasal irrigations may be beneficial; their efficacy may arise from removal of inflammatory cells and excess mucus with consequent improvement of sinus drainage, rather than from direct action on sinus pathology (Adappa et al. 2012; Lim et al. 2008). Lim et al. (2008) also reported that culture-directed bacterial studies present a higher level of evidence than empiric treatment. The American Academy of Otolaryngology – Head and Neck Surgery recommends irrigation or nebulisation with ceftazidime, aminoglycoside (e.g. tobramycin) and quinolones (e.g. ciprofloxacin, levofloxacin) when cultures present pseudomonas and the use of amphotericin B irrigation in cases of proven fungal infections. The highest level of evidence was found for studies with postsurgical patients. Topical antibiotics may play a unique role in CRS patients with post-surgery infections with Streptococcus aureus and pseudomonas. In summary, topical antibiotics should not be first-line management but may be successfully used in refractory patients to the recommended topical corticosteroids and oral antibiotics. However, a full evaluation of this emergent modality of CRS treatment requires more investigation (Adappa et al. 2012; Lim et al. 2008).
15.3 Intranasal Delivery of Systemic Drugs
As the market for the IN delivery of topical drugs matures, the potential of IN route for administration of drugs acting systemically has been investigated at a remarkably fast rate. Indeed, the IN administration is today regarded as a potential alternative route for systemic delivery of drugs that are conventionally administered by intravenous (IV) route or that undergo extensive first-pass metabolism after oral administration (Bitter et al. 2011; Illum 2012). Nevertheless, the nasal route is less suitable for chronic drugs that must be frequently administered daily, and drugs that require sustained blood levels should not be considered for nasal delivery unless they are included in sustained-release type dosage forms for nasal administration (Atluri et al. 2005). This is mainly due to the anatomic and physiological characteristics of the nose, transport mechanisms involved throughout nasal systemic absorption and physicochemical properties of the drugs.
Due to its anatomical localisation, high vascularisation and permeability, the respiratory mucosa around the turbinates is recognised as the main site for systemic entry of drugs. Generally, lipophilic drugs easily diffuse through nasal mucosa by the transcellular route (Illum 2002), while the polar drugs are mostly transported through small connections between epithelial adjacent cells, called tight junctions (Arora et al. 2002). However, only polar compounds with molecular weight lower than 1,000 Da can cross this semipermeable membrane as the normal diameter of tight junctions is 3.9–8.4 Å (Alsarra et al. 2010).
15.3.1 Intranasal Systemic Delivery of Small Molecules
The awareness that drugs may reach widespread circulation in few minutes after nasal administration expanded remarkably the number of systemically acting drugs marketed as nasal formulations (Table 15.1). Furthermore, the number of investigations regarding the feasibility of IN route for delivering many other small compounds to the systemic circulation is also continuously emerging (Table 15.1).
Underlying this wide interest on exploiting nasal cavity for systemic delivery is the rapid and direct systemic absorption of compounds, the circumvention of gastrointestinal and hepatic first-pass metabolism and, consequently, the achievement of higher drug plasma levels and higher bioavailability through nasal route than oral administration. IN drug administration may enable the reduction of the dose administered, a quick onset of pharmacological activity and fewer side effects.
Among the several alternative formulations currently developed and under development, solution-based formulations are the most frequent because they are the easiest to administer and they have the greatest chance for systemic drug delivery across the nasal mucosa. Moreover, systems incorporating mucoadhesive excipients and/or enzyme inhibitors and/or nasal permeation enhancers have been developed in order to improve the therapeutic efficacy once they enhance drug nasal residence time, prolong duration of action and increase the absorption extent of drugs (Grassin-Delyle et al. 2012; Jiang et al. 2010; Pires et al. 2009).
15.3.1.1 Analgesic Drugs
Opioids are considered the cornerstone of an analgesic regimen and are indicated for the treatment of breakthrough pain and acute, moderate to severe and chronic pain. Ideally, they must exhibit a rapid onset time and a prolonged duration of action that coincides with the episode’s time course. Although oral and parenteral solutions are generally used for treatment of the breakthrough pain, the onset effect is not achieved before 30–45 min and the maximal effect within 1 h (Tveita et al. 2008). IN administration of opioids arises hence as a hope to easily and quickly achieve pain relief and improvement of the life quality of patients.
Indeed, a wide variety of opioid drugs have been under investigation, including morphine, fentanyl and buprenorphine. Although recognised as the standard opioid for cancer pain relief, morphine has a significant intestinal and hepatic first-pass metabolism that limits its bioavailability, which is around 20–32 % (Fitzgibbon et al. 2003). Similarly, the bioavailability of morphine solutions administered intranasally rounds only 10–22 % in humans and sheep (Illum et al. 2002) probably due to its low lipophilicity. In order to increase the nasal residence time, the bioavailability and the elimination half-life time of morphine after its IN administration, a wide variety of formulations have been currently under development, including formulations containing chitosan as microspheres or in solution (formulations based on starch microspheres coupled with lysophosphatidylcholine) (Illum et al. 2002) and solutions added of oleic acid as absorption promoter (Fitzgibbon et al. 2003). One of the most relevant clinical studies consisted in assessing the pharmacokinetic profile and tolerability of Rylominetm composed by morphine mesylate and chitosan in 13 subjects (Stoker et al. 2008). Based on the area under the concentration-time curve (AUC) values, bioavailability of IN morphine was considerably higher when compared to the other administration routes.
In opposition to morphine, fentanyl and butorphanol can be effectively and quickly absorbed at nasal cavity without using absorption promoters due to their relative high lipophilicity and low molecular weight. Particularly, IN fentanyl is currently marketed (Table 15.1) as two distinct forms: the aqueous solution Instanyl® and the pectin-based mucoadhesive formulation PecFent®. In a pharmacokinetic study in 19 cancer patients with breakthrough pain, nasal spray fentanyl was quickly absorbed through the nasal mucosa, attaining peak plasma concentrations within 12–15 min when administered at 50, 100 and 200 μg (Kaasa et al. 2010). One of the most important in vivo studies within this framework consisted in a balanced, randomised, double-blind, two-way crossover study in which patients received the same fentanyl dose by IN and IV administration (Christrup et al. 2008). The time to onset of action of around 10 min and the onset and duration of analgesia were not significantly different between single doses of IN and IV fentanyl in these adults. Recent researches have also shown an improvement of the bioavailability of fentanyl when administered as IN mucoadhesive formulations (Fisher et al. 2010; Kaasa et al. 2010).
Sumatriptan and zolmitriptan are analgesic drugs particularly used for migraine and cluster headaches. They are currently available as nasal formulations that provide onset times significantly quicker than those obtained after oral dosing (Dodick et al. 2005; Gawel et al. 2005) (Table 15.1). This success is due to the high lipophilicity of sumatriptan and zolmitriptan that facilitate their systemic absorption through nasal respiratory mucosa (Uemura et al. 2005) but also due to their direct access to CNS as it is referred in Sect. 15.4.1.5.
15.3.1.2 Cardiovascular Drugs
For a long time, nasal administration has been investigated as an attractive route for administration of cardiovascular drugs such as propranolol, nifedipine, nitroglycerin and carvedilol (Costantino et al. 2007).
The IN dosing of propranolol provides a pharmacokinetic profile that is very similar to that of IV administration, specifically when regarding the onset time and bioavailability (Ahn et al. 1995).
Bioadhesive sodium alginate microspheres of metoprolol tartrate for IN systemic delivery were also investigated as an alternative therapy for the treatment of hypertension and angina pectoris (Rajinikanth et al. 2003). Promising results were found in rabbits and rats, with maximum plasma drug concentrations (C max) clearly higher after IN administration than those after oral administration.
Nifedipine is a calcium channel blocking agent frequently used for the treatment of angina pectoris and hypertension. Kubota et al. (2001) performed a crossover clinical study in order to investigate the optimal administration method of nifedipine for rapid management of hypertension. It is interesting to highlight that although the value of C max was clearly lower after IN administration of nifedipine than that obtained for oral administration, the mean serum concentration of nifedipine 5 min after IN administration was higher (and remained higher until after 15 min). These results sustain that IN administration of nifedipine guarantees the fastest increase of drug plasma concentrations and the most significant effect on blood pressure reduction.
More recently, IN administration of carvedilol, a non-selective β-adrenergic antagonist also used in the treatment of hypertension and stable angina pectoris (Packer et al. 2002), has been under investigation due to its significant hepatic first-pass metabolism and low absolute bioavailability (25 %). Recent investigations reported that when administered by IN route to rabbits, sodium alginate microspheres and mucoadhesive chitosan microspheres containing carvedilol, the mean residence and half-life times of the drug were at least twice of those observed after IV administration. Furthermore, the high absolute bioavailability and the low t max achieved for carvedilol sustain that both pharmaceutical formulations are promissory to prolong the therapeutic effect of carvedilol (Patil et al. 2010, 2012).
15.3.1.3 Antiviral Drugs
The antiviral acyclovir is currently available as several dosage forms that present limitations. Firstly, the intestinal absorption of acyclovir is slow, variable and incomplete, with an absolute bioavailability of approximately 15–20 % which requires a frequent oral dose regimen. On the other hand, its low solubility in water and lipids hamper the administration of acyclovir by intramuscular route (Shao et al. 1994). Even when intravenously administered, acyclovir is mainly excreted unchanged through urine by glomerular filtration and tubular secretion, demanding a high dose to be administered in order to attain therapeutic drug concentrations.
Hence, the IN administration of acyclovir emerged recently as an innovative strategy that could maintain the drug for a longer time in systemic circulation within effective and non-toxic concentration ranges (Alsarra et al. 2008). Since acyclovir is also practically impermeable through the nasal mucosa, neutral mucoadhesive liposomes were formulated in order to enhance the nasal penetration and systemic absorption. In a study performed in rabbits, the absolute bioavailability of nasal liposomes with acyclovir was 60.7 % while that of free acyclovir was only around 5 %. This discrepancy was also observed for AUC values, clearly demonstrating that liposomes pass directly into systemic circulation, resulting in a considerable systemic concentration of acyclovir (Alsarra et al. 2008).
Similarly, zanamivir is another antiviral drug which, although presenting higher bioavailability when administered by IN route than orally (Cass et al. 1999), is poorly absorbed at nasal level especially due to its high hydrophilicity. Thus, similar investigations to those executed for acyclovir are expected to be soon performed for zanamivir.
15.3.1.4 Antiemetic and Motion Sickness Drugs
The nasal delivery of drugs for the treatment of nausea and motion sickness is steadily appearing as a desirable alternative to parenteral and oral medications especially because a rapid onset of action is required in acute situations. Moreover, the gastric dysmotility associated to the pathological situation is probable to affect the intestinal drug absorption and the drug fraction that is absorbed after oral administration.
For instance, when orally administered, metoclopramide bioavailability is highly variable (32–98 %) and it has a short half-life (3–4 h) that demands an oral administration three to four times daily. The IN administration of metoclopramide is identified as a good alternative (Mahajan and Gattani 2010). There are, however, limitations related to the low permeability across the nasal mucosa and the rapid MCC of metoclopramide, and in order to overcome these features, new nasal formulations have been developed and are under investigation. They consist on aqueous solutions added of absorption enhancers to increase nasal permeability (Zaki et al. 2006) or on gel and mucoadhesive formulations to prolong the residence time at the nasal absorption local and facilitate the drug uptake (Mahajan and Gattani 2010; Tas et al. 2009). Zaki et al. (2006) demonstrated that when nasal spray solution was administered to humans, the C max of metoclopramide was significantly higher than that observed after oral administration while values of t max and half-life time were significantly lower (Zaki et al. 2006). However, no statistical differences were observed for the mean residence times of metoclopramide, and therefore, the same research group developed and administered gel and mucoadhesive formulations composed by gellan gum (0.4 %, w/v) and Carbopol (0.15 %, w/v) to rabbits. The superior absolute bioavailability of the nasal gel compared to the oral solution clearly indicated higher absorption of metoclopramide when administered intranasally. Favourable results were also found for gel dosage forms based on mucoadhesive polymer sodium carboxymethylcellulose for IN administration of metoclopramide to sheep (Tas et al. 2009).
Ondansetron has also been under investigation to be administered by IN route, although it is currently available in IV solutions and oral dosage forms. The low oral bioavailability of ondansetron in humans (60 %) and its administration at least 30 min prior to chemotherapy sessions (Gungor et al. 2010) propelled Hussain and collaborators (2000) to investigate for the first time the feasibility of ondansetron IN administration to rats. The plasma concentration-time profiles for IN administration were comparable to that of IV administration and the rapid absorption through the nasal mucosa allowed ondansetron to reach systemic circulation almost instantaneously. Equivalent results were also reported by Gungor et al. (2010). Nevertheless, several ondansetron formulations have been developed and demonstrated to enhance drug delivery, reduce the onset time and prolong drug effect duration in relation to the oral administration (Cho et al. 2008; Gungor et al. 2010)
Scopolamine, an antimuscarinic agent indicated for motion sickness, is another example of a drug in this area that is suitable for IN dosing as depicted by human pharmacokinetic studies developed by Ahmed et al. 2000.
15.3.1.5 Erectile Dysfunction Drugs
Sildenafil citrate is considered a standard treatment for erectile dysfunction. It is rapidly absorbed after oral administration but only with an absolute bioavailability of 40 %, an onset of action time within 15.5 min and effect duration of approximately 40 min (Deveci et al. 2004). Recently, Elshafeey et al. (2009) attempted to take advantage of nasal administration to improve these limitations and developed a new microemulsion of sildenafil citrate composed of oleic acid/Labrasol/Transcutol/water. The research group achieved drug concentrations that were nearly twofold higher than those obtained for oral tablets. A higher bioavailability and faster onset systemic levels were also observed for IN formulation probably due to the fact that liver metabolism was bypassed.
15.4 Intranasal Delivery of CNS-Acting Drugs
The brain is a delicate organ that plays a set of vital functions to maintain convenient body homeostasis; therefore, its integrity is ensured by specific physiological barriers and mechanisms of defence which efficiently protect and isolate the CNS from harmful endogenous substances and external insults (e.g. xenobiotics and virus).
The BBB represents one of the strictest structural and functional barriers in segregating the brain from the systemic circulation. It is characterised by the presence of non-fenestrated capillary endothelial cells with intercellular tight junctions, a very high transendothelial electric resistance (Misra et al. 2003; Vyas et al. 2005a) and a high metabolic activity associated to the expression of numerous carrier-mediated efflux transporters (Anderson 1996; Rautio et al. 2008) that regulate the influx and efflux of a variety of compounds. Unfortunately, the CNS delivery of proficuous therapeutic agents is also frequently prevented. In the last decades, several different approaches have been attempted in order to circumvent the BBB and to deliver drugs efficiently to the brain for therapeutic or diagnostic applications (Illum 2000). For example, recent developments have generated much interest in the possibility of exploiting the IN administration as a non-invasive alternative route for delivery of drugs to the CNS. In fact, assuming the olfactory region as a unique direct connection between the nose and the brain, the IN administration has emerged as a promising approach for the delivery of therapeutic agents to the CNS bypassing the BBB (Hanson and Frey 2008; Illum 2004; Vyas et al. 2005a).
In many CNS disorders, a rapid and/or specific targeting of drugs to the brain would be beneficial. Therefore, valuable efforts have been conducted to improve brain delivery of various therapeutic agents via the IN route, in order to provide higher drug bioavailability at the biophase and consequently better therapeutic efficacy.
15.4.1 Nose-to-Brain Drug Delivery
IN drug administration provides a promising method to deliver therapeutics from the nasal cavity directly to the CNS, bypassing the BBB. Indeed, IN delivery represents an attractive alternative to oral and parenteral routes since, in addition to being non-invasive, it also allows the avoidance of gastrointestinal destruction and hepatic first-pass metabolism. Direct transport of drugs to the brain may lead to reducing systemic exposure and peripheral side effects, which allows the decrease of the dose and frequency of dosing as well as minimises toxicity and improves therapeutic efficacy by achieving desired drug concentrations at the biophase (Kumar et al. 2008; Seju et al. 2011). In addition, the rapid onset delivery of drugs to the CNS and the higher brain uptake congregate the essential conditions for the application of the IN route in the management of emergency situations (Florence et al. 2011; Li et al. 2002; Vyas et al. 2006a; Wolfe and Bernstone 2004).
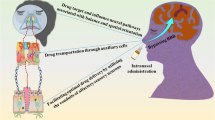
The possible transport pathways by which a drug can be delivered to the CNS after IN administration are schematically depicted in Fig. 15.1. In general, therapeutic agents can travel from the nasal cavity to the brain via the olfactory route by two possible mechanisms: the olfactory epithelial pathway and the olfactory neural pathway (Merkus and Van den Berg 2007). Similar to drug absorption through nasal respiratory mucosa, in the olfactory epithelial pathway, drugs can be absorbed across the olfactory epithelium either by transcellular or paracellular transport.

Schematic representation of the possible pathways involved in the transport of drugs from nose to brain
In the olfactory neural pathway, drugs can be transferred via axonal internalisation with subsequent transport along the olfactory sensory nerves directly to the brain. Nevertheless, it is believed that such transport is slow, taking hours or even days for drugs to reach the brain parenchymal tissue (Dhuria et al. 2010; Thorne and Frey 2001). As an alternative, it was suggested that drugs after traversing the olfactory epithelium could make their way by paracellularly entering into the perineuronal channels that surround the olfactory nerves, requiring only few minutes (<30 min) to travel along the olfactory axon up to the cerebral spinal fluid (CSF) (Dhuria et al. 2010). Recently, trigeminal nerve pathway has also been advocated as another and additional valid route for the transport of molecules directly from the nasal cavity to the brain (Dhuria et al. 2010; Ross et al. 2004; Thorne et al. 2004).
The hypothetic mechanisms of direct delivery of drugs from nasal passages to the CNS were described; notwithstanding, the contributions underlying each one are not yet clearly elucidated. Generally, the rapid appearance of a drug in the brain and CSF indicates preferential involvement of extracellular transport pathways rather than the olfactory neural route. However, the possibility of occurring later axonal drug internalisation cannot be entirely ruled out. Nasally applied drugs could reach the CNS by means of one or a combination of various transport pathways (Fig. 15.1).
15.4.1.1 Alzheimer’s Disease Drugs
Several oral acetylcholinesterase inhibitors including rivastigmine, donepezil, galantamine and tacrine have been used for the treatment of Alzheimer’s disease symptoms. Notwithstanding, oral administration of such molecules has often been associated with low bioavailability, extensive first-pass metabolism, short elimination half-life, hepatotoxicity and severe gastrointestinal side effects (Costantino et al. 2008).
The potential of the IN delivery route for targeting acetylcholinesterase inhibitors to the brain seems to provide valuable benefits and has been investigated in animal models. The uptake of NXX-066 (a physostigmine analogue) in the CSF after nasal and IV administration to rats was investigated in order to assess whether a direct nose-to-brain pathway is involved (Dahlin and Björk 2001). Study results demonstrated that only low concentrations of NXX-066 were detected in the CSF following both routes of administration. However, nasal administration resulted in extremely rapid and complete absorption of NXX-066 into the systemic circulation exhibiting an absolute bioavailability near to 100 %. The high values of nasal bioavailability suggest that this route could be a suitable alternative to oral and parenteral administrations.
The concentrations of tacrine in blood and brain after IN and IV administration to mice were also evaluated by Jogani et al. (2007). Pharmacokinetic data revealed that drug concentrations in brain tissue were found to be significantly higher for IN administration and the delivery of nasal tacrine to the brain showed to be much quicker than given via the IV route. These findings demonstrated that after IN delivery, a preferential nose-to-brain transport is implied in the selective distribution of tacrine to the brain.
15.4.1.2 Parkinson’s Disease Drugs
Until now, there is no cure for Parkinson’s disease but its symptoms can be attenuated by the replacement of the dopamine basal levels at the brain. However, dopamine is unable to cross the BBB in appreciable amounts making its administration via oral and parenteral routes not feasible. Therefore, levodopa (L-dopa) is currently the gold standard treatment in Parkinson’s disease, since it easily penetrates the BBB and is rapidly converted to dopamine within the brain. Unfortunately, the clinical response to oral L-dopa is commonly variable and unreliable, due to its erratic absorption and first-pass metabolism (Kao et al. 2000). Additionally, about 95 % of the drug undergoes decarboxylation to dopamine in the peripheral tissues (Dahlin et al. 2000), compromising the amount of unchanged drug available to reach the brain and enhancing the occurrence of adverse effects. In this context, the transfer of dopamine along the olfactory pathway to the CNS following nasal administration has been assessed in rodents (Dahlin et al. 2000, 2001). The experimental results showed that there was an effective transport of dopamine from the nasal cavity into the CNS, since concentration levels after nasal administration were, in comparison to IV injection, 2.3 and 6.8 times higher in the CSF and olfactory bulb, respectively (Dahlin et al. 2000). Nevertheless, the fraction of the nasally administered drug that reached the brain tissue was only 0.12 % of the total dose, suggesting that higher doses of dopamine may be required to guarantee therapeutic efficacy (Dahlin et al. 2000).
The potential of direct nose-to-brain transport of L-dopa was also investigated in rats. Although the AUC values of nasal L-dopa were more than two times higher in plasma and brain comparatively to oral administration, a large fraction of drug was systemically absorbed via the nasal route, and therefore, the fraction of drug transported by the direct nose-to-brain pathway was minimal (Kim et al. 2009). More promising results were achieved by Kao et al. (2000) using the prodrug approach. Following IN administration of the butyl ester prodrug of L-dopa, CNS bioavailability of L-dopa was improved comparing to an equivalent dose given intravenously.
15.4.1.3 Anticonvulsant and Antiepileptic Drugs
Oral administration of anticonvulsant drugs has generally been associated with high systemic distribution into nontargeted tissues, peripheral adverse effects and limited brain uptake. Moreover, patient’s physical condition immediately after a convulsive episode is incompatible with the oral ingestion of a tablet dosage form. Apart from its advantages on the clinical emergencies in acute seizure situations, nasally administered anticonvulsant drugs may represent a valuable approach for the long-term treatment of epilepsy by providing the decrease of the dose, frequency of dosing and related side effects thus improving therapeutic efficacy and tolerability.
IV benzodiazepines, such as diazepam, lorazepam, midazolam and clonazepam, have been used as the first-line therapy for the termination of seizure activity in status epilepticus. However, benzodiazepines IV dosing may unleash hypotension, cardiac dysrhythmia and respiratory failure (Li et al. 2000). Aiming to minimise the disadvantages and potentiate the therapeutic index of such drugs, several studies were carried out on the subject of IN delivery. A comparative study between IV injection and three nasal formulations of clobazam (solution, microemulsion and mucoadhesive microemulsion) was performed in mice in order to assess and characterise its pharmacokinetic profile and pharmacodynamic performance (Florence et al. 2011). The pharmacokinetic results revealed that the systemic blood distribution of the drug was significantly lower with IN-administered formulations comparatively to IV injection, thus ensuring drug targeting at the site of action and minimising the possibility of systemic side effects. Furthermore, higher brain AUC and C max for microemulsion formulations reflect an enhanced CNS uptake, indicating that a preferential nose-to-brain transport may be involved, revealing consistency with similar previous studies with clonazepam (Vyas et al. 2006a). By virtue of their lipophilic nature and lower interfacial tension, microemulsions heighten the drug permeability across the nasal mucosa. On the other hand, the incorporation of a mucoadhesive agent (Carbopol) improves drug uptake by opening tight junctions, increasing paracellular transport of the molecules.
To investigate brain targeting via nasal administration, the antiepileptic drug carbamazepine (CBZ) was chosen as a model. Taking into account that CBZ is absorbed slowly and erratically after oral administration, displays a bioavailability of less than 50 % and usually attains peak plasma concentration 4–8 h after oral ingestion (Barakat et al. 2006), a direct delivery of this drug to the brain circumventing the BBB would be highly beneficial. In this context, a CBZ gel formulation composed by hypromellose and Carbopol 974P (3:1) was nasally administered to rats, aiming to compare CBZ concentrations in blood and brain tissue samples with other conventional routes, such as oral and IV administration (Barakat et al. 2006). Experimental data revealed that IN CBZ concentrations were greater in brain than in plasma, also achieving remarkably higher levels in CNS compared to oral or IV administration. A direct transport pathway from nose to brain was demonstrated since peak brain concentration after nasal administration was attained in only 5 min and CBZ absorption from the nasal cavity into the brain was rapid and complete.
15.4.1.4 Analgesic Drugs
Nasal administration of morphine is currently under development in order to overcome its extensive hepatic first-pass effect, affording a more rapid drug absorption and faster onset of action. Indeed, systemic absorption of morphine after nasal administration undoubtedly contributes to achieve these goals as already stated in Sect. 15.3.1.1 . However, taking into account that morphine is a small hydrophilic molecule with limited BBB permeability, direct transport of the drug along the olfactory pathway from nose to the brain would be advantageous for pain relief. For these reasons, some investigations have been carried out in order to evaluate the direct access of morphine to the brain. Following IN administration of morphine to rodents, Westin and collaborators (2005) found that the drug was rapidly transferred via the olfactory epithelium to the CNS, reaching the highest concentration in the olfactory bulb after 15 and 60 min in rats and mice, respectively. Upon these facts, the same research group intended to quantify the olfactory transfer of morphine to the brain by comparing drug levels in brain and plasma after both IN and IV administration (Westin et al. 2006). The results showed that after nasal and IV administration of the same dose (1 mg/kg body weight), equal morphine concentrations were obtained in the brain at 5 and 15 min. However, brain to plasma AUC ratio from 0 to 5 min was substantially higher for nasal delivery compared to IV infusion, proving an early distribution of morphine to the CNS via the nasal route.
15.4.1.5 Migraine and Cluster Headaches Drugs
Sumatriptan and zolmitriptan are the drugs most commonly used in the effective treatment of migraine and cluster headaches (Jain et al. 2010). Although these drugs present potent analgesic activity on acute migraine pain relief, current oral therapies are commonly associated with a slow onset of action and significant hepatic first-pass metabolism which results in low absolute plasma bioavailability (Jain et al. 2010; Vyas et al. 2005b). Furthermore, the majority of migraine patients experience several gastrointestinal disturbances during the attacks making the intake of oral tablets often inappropriate (Yates et al. 2005).
Although systemic absorption of IN sumatriptan and zolmitriptan is undeniable, their eventual transport from the nasal cavity directly to the brain may also have an important contribution for the treatment of migraine and cluster headaches. Therefore, the assessment of nose-to-brain delivery of IN mucoadhesive microemulsions of both sumatriptan and zolmitriptan has been investigated in rats (Vyas et al. 2005b, 2006b). The mucoadhesive microemulsions showed better results than microemulsions or drug solutions given nasally. Superior pharmacokinetic results were even attained for the developed sumatriptan microemulsions compared to an already marketed nasal product (Vyas et al. 2006b). Comparatively to IV administration, higher C max and AUC values were found in the brain at all sampling time points for nasally administered formulations, suggesting that preferential nose-to-brain transport may be attributed to both drugs. These findings were also sustained by Jain et al. (2010) who demonstrated that zolmitriptan is predominantly transported to the brain via the olfactory and trigeminal pathways.
15.4.1.6 Antipsychotic and Antidepressant Drugs
Atypical antipsychotic drugs are currently the first choice for the treatment of schizophrenia, and they are available in the market predominantly under oral dosage forms. Oral formulations are, however, related to low plasma drug bioavailability which frequently demands the increase of dose and frequency of dosing. As a consequence, the occurrence of adverse effects is also often potentiated. In this context, the development of IN delivery systems of several antipsychotic agents like risperidone and olanzapine has been attempted considering the potential of this route for direct brain targeting (Kumar et al. 2008; Seju et al. 2011). Promising results were obtained for the IN delivery experiments of a mucoadhesive nanoemulsion of risperidone- and olanzapine-loaded poly(lactic-co-glycolic acid) (PLGA) nanoparticles using animal models. Higher drug concentrations were observed in the brain for the developed formulations compared to the plain solution of the drug given either nasally or intravenously. Pharmacokinetic data of olanzapine-loaded PLGA nanoparticles even showed an additional therapeutic gain by providing sustained drug delivery to the brain (Seju et al. 2011). In fact, the nanoparticle strategy offers an improvement in nose-to-brain delivery since, in addition to protecting the encapsulated drug from biological or chemical degradation and efflux P-glycoprotein (P-gp) transport, it also enables the increase of the drug residence time within the nasal cavity. As a result, the opportunity to provide sustained delivery of olanzapine is increased, allowing the enhancement of brain drug concentrations.
An IN delivery system of milnacipran was also investigated for the treatment of depression (Uchida et al. 2011). A pharmacokinetic assessment of plasma and CSF milnacipran concentrations following nasal drug delivery to rats revealed that, in comparison to intraduodenal administration, higher C max and lower t max were observed for both matrices. These pharmacokinetic data were in agreement with the results obtained for the pharmacodynamic evaluations in which the antidepressant effect after IN administration of milnacipran was higher and quicker than after oral dosing. The impact of the co-administration of IN milnacipran with 0.5 % chitosan was also addressed in this study. The incorporation of this polysaccharide into the nasal formulation led to an even greater antidepressant effect since it provided a long residence time of milnacipran within the nasal cavity, thus resulting in the increase of the systemic absorption as well as direct transport of the drug to the CNS.
15.4.1.7 Antiviral Drugs
The efficacy of antiviral therapy in the treatment of neuroinfections is often limited due to reduced drug uptake into the CNS as a consequence of its poor permeation across the BBB (Colombo et al. 2011). Indeed, most of the antiviral agents are highly hydrophilic compounds and therefore cannot passively diffuse through the BBB easily. Moreover, it is estimated that a huge part of them are also substrates of the P-gp efflux pump (Hanson and Frey 2007) which has a markedly role on CNS protection by hindering the access of a wide variety of substances to the brain.
Several studies have recently investigated the pharmacokinetics and brain distribution profiles of some antiviral agents after nasal and IV administration to animal models. A preferential transfer of zidovudine, a reverse transcriptase inhibitor, into the CSF and brain tissues following IN administration to rabbits was successfully demonstrated, providing a promising therapeutic option for the treatment of CNS dysfunctions caused by human immunodeficiency virus (HIV) (Ved and Kim 2011). By using a thermo-reversible gelling system comprising Poloxamer 407 as a mucoadhesive polymer and n-tridecyl-β-D-maltoside as a permeation enhancer, the authors guaranteed a larger increase of zidovudine brain bioavailability relatively to solutions given both nasally and intravenously. The existence of a direct nose-to-brain pathway to transport zidovudine from the nasal cavity to the CNS was also strongly proven. According to Ved and Kim (2011), approximately 99 % of zidovudine content was directly transferred to the brain via the olfactory route.
15.5 Intranasal Delivery of Biomacromolecular Drugs
IN administration represents a promising choice for delivery of a variety of high molecular weight therapeutic agents such as peptide-, protein- or nucleic acid-based drugs (Csaba et al. 2009; Singh et al. 2012). Because of the higher susceptibility of biological therapies to enzymatic degradation and due to their low permeability across the epithelium via transcellular and paracellular pathways, the absorption of these biomacromolecular drugs from mucosal sites is poor. Therefore, to increase their bioavailability, they are mostly administered by parenteral routes. Over the last years, new pharmaceutical formulations and novel delivery strategies have been developed offering promising opportunities to expand the IN delivery of biomacromolecules (Ozsoy et al. 2009; Singh et al. 2012).
As the nasal mucosa is one of the most permeable and highly vascularised tissues, also avoiding gastrointestinal and hepatic first-pass metabolism, the extent of absorption of biomacromolecules may be potentiated by IN administration comparatively to that achieved through oral route. Accordingly, the nasal route has gained a great interest as an alternative and non-invasive way for systemic and/or direct brain delivery of various classes of biological therapeutic agents (Ozsoy et al. 2009; Veronesi et al. 2011).
The recent advances in the field of biotechnology have promoted the emergence of a range of biodrugs. Besides therapeutic peptides and proteins, a broad variety of other biodrugs are coming into clinical practice or moving to a greater extent into clinical research, namely, vaccines, cell or gene therapies, cytokines, tissue growth factors and monoclonal antibodies (Csaba et al. 2009; Ozsoy et al. 2009; Singh et al. 2012). Hence, it is expected that the number of biomacromolecular drugs commercially available for administration via nasal route will progressively increase.
15.5.1 Peptides and Proteins
Peptides and proteins represent interesting targets for IN administration (Table 15.2). Nevertheless, as peptides and proteins are charged, hydrophilic and usually high molecular weight molecules, they are obviously poorly permeable across lipid biomembranes. Therefore, in the development of suitable protein- and peptide-based formulations intended for IN delivery, some chemical and pharmaceutical strategies need to be employed to overcome the physicochemical instability, enzymatic barrier of the nasal mucosa and low permeability, aiming to increase their bioavailability. Hence, in the formulation process of these medicinal products, the use of appropriate vehicles, enzyme inhibitors and/or penetration enhancers is of paramount importance (Bahadur and Pathak 2012; Mistry et al. 2009).
As previously referred, although the nasal mucosa poses a permeation barrier to high molecular weight therapeutics, the tight junctions between adjacent epithelial cells also limit the movement of molecules through the intercellular spaces, forming a barrier against paracellular drug delivery. However, the IN bioavailability of some macromolecules was considerably improved by using nasal permeation enhancers, which may affect the barrier function of the tight junctions (Costantino et al. 2007).
In spite of the advances progressively reached in the formulation of biological medicinal products, major hurdles remain to overcome the combined barriers of drug permeability, drug stability, pharmacokinetics and pharmacodynamics of peptide- and protein-based drugs (Gupta and Sharma 2009). Therefore, despite the hundreds of biological medicinal products already developed, these problems may explain why just a handful of non-injection biomacromolecular drugs have reached the market, mainly as IN formulations.
The success achieved with the IN administration of polypeptide drugs, such as desmopressin, calcitonin, buserelin, nafarelin and oxytocin, has promoted an extensive investigation of the viability of this route of administration for delivery of other protein and peptide drug candidates. Among them, human insulin represents perhaps the biomolecule most extensively assessed for systemic delivery by IN route (Benedict et al. 2011; Hallschmid et al. 2012; Jogani et al. 2008). In addition, many other protein and peptide drugs currently used as injectables have also been evaluated for nasal delivery (Table 15.2). Moreover, due to the increasing evidences on the possibility of direct nose-to-brain delivery of large-sized drugs, the research work targeting the IN delivery of neuropeptides has been largely potentiated in the last years (Lochhead and Thorne 2012; Veronesi et al. 2011).
The peptide- and protein-based drugs currently available in the market as IN formulations will be discussed below.
15.5.1.1 Salmon Calcitonin
Calcitonin is a polypeptide hormone of 32 amino acids (molecular weight of 3.4 kDa) and it has a physiological role in the regulation of calcium homeostasis. Calcitonin is produced in humans and other mammalian species, and also in birds and fish (du Plessis et al. 2010; Ozsoy et al. 2009).
Salmon calcitonin is more potent than natural human calcitonin at inhibiting osteoclast function. Therefore, salmon calcitonin is favoured comparatively to the human calcitonin, and the former is the only form of this peptide commercially available (Lee et al. 2011). Although calcitonin is available as several formulations, the IN formulations are the most widely used (Chesnut et al. 2008). The salmon calcitonin nasal spray has shown to be effective but, like other peptides, presents a low IN bioavailability (3 %) comparatively to those achieved by intramuscular or subcutaneous injections (Ozsoy et al. 2009). As a result, new pharmaceutical formulations have been progressively investigated to enhance the absorption of salmon calcitonin from nasal mucosa (Chen et al. 2009; du Plessis et al. 2010).
15.5.1.2 Desmopressin
Antidiuretic hormone (also called arginine-vasopressin) is produced in the hypothalamus and secreted by the neurohypophysis in conditions of increased plasma osmolality, decreased arterial pressure and cardiac volume reduction (Babey et al. 2011; Treschan and Peters 2006). A lack of arginine-vasopressin is the most common cause of diabetes insipidus (Babey et al. 2011). A dysfunction in the secretion of arginine-vasopressin may also induce the appearance of other clinical conditions (e.g. nocturnal enuresis) (Nevéus 2011). Thus, replacement therapy with the analogue desmopressin is justified in cases of insufficiency of arginine-vasopressin (Chanson and Salenave 2011).
Desmopressin (1-deamino-8-D-arginine-vasopressin; molecular weight of 1,069 Da) is a vasopressin analogue but retains the hormone’s antidiuretic effects and also exerts haemostatic effects. Therefore, despite its clinical use in diabetes insipidus and complex enuresis states, desmopressin is also useful for treating or preventing bleeding episodes (Ozgönenel et al. 2007; Ozsoy et al. 2009). Desmopressin has been used in clinical practice for more than 30 years and it is commercially available as IN solution, injectable solution, tablets and more recently also as oral lyophilisate (Van de Walle et al. 2007, 2010).
Usually, therapeutic peptides are highly potent and specific in their functions, but difficulties in their administration require parallel development of viable delivery systems to enhance their bioavailability. Indeed, the systemic absorption of desmopressin is very low from available formulations. Therefore, efforts have been made to develop improved pharmaceutical formulations (Fransén et al. 2009).
15.5.1.3 Gonadotropin-Releasing Hormone (GnRH) Analogues: Buserelin and Nafarelin
The GnRH neuronal system is the final common pathway for central regulation of fertility. GnRH is a single neuroendocrine decapeptide produced in the hypothalamus (Balasubramanian et al. 2010; Moenter 2010).
Over the times, an intensive research of potent GnRH agonist analogues with acceptable pharmacokinetics has been carried out. In fact, by specific amino acid substitutions in the structure of the natural GnRH, several GnRH agonists were developed and are now clinically available, such as histrelin acetate (Shore et al. 2012), goserelin acetate (Berglund et al. 2012), leuprolide acetate (Tunn 2011), buserelin acetate (Safdarian et al. 2007) and nafarelin acetate (Takeuchi et al. 2001). However, like other peptide-based drugs, the majority of these GnRH agonists are only marketed in parenteral formulations. Fortunately, buserelin acetate and nafarelin acetate are both commercially available as a spray formulation suitable for IN delivery (Franco et al. 2001; Tuvemo et al. 2002). Nevertheless, the absolute IN bioavailability of buserelin (6 %) and nafarelin (2.8 %) is very low (Costantino et al. 2007). Hence, it remains as a remarkable challenge in the development of new formulations of buserelin and nafarelin affording a greater drug bioavailability after IN delivery.
15.5.1.4 Oxytocin
Oxytocin, a neurohypophyseal nonapeptide hormone, is well known not only for its prominent role in parturition and lactation but also as a drug of choice for prevention of the postpartum haemorrhage (Anderson and Etches 2007; Lee et al. 2009; Wei et al. 2010). Oxytocin is a drug frequently used in the management of labour or for preventing postpartum haemorrhage, particularly through parenteral administration (Arnott et al. 2000; Bellad et al. 2012; Zhang et al. 2011). On the other hand, as a nasal spray formulation, oxytocin has been used to assist breast-feeding and milk expression (Fewtrell et al. 2006). Indeed, for a long time, those evidences exist about the effectiveness and safety of oxytocin IN spray as a means of enhancing lactation (Ruis et al. 1981).
More recently, many research works have focused on possible functions of oxytocin in the brain. As a result, oxytocin appears to be involved in learning, anxiety, feeding, sexual and maternal behaviour, aggression and pain perception, among others (Lee et al. 2009). Accordingly, the therapeutic spectrum for IN oxytocin delivery may be largely extended in the next years.
15.5.2 Vaccines
The majority of disease-causing viruses and bacteria reach the body through mucosal surfaces, including through the nasal mucosa (Chadwick et al. 2010). Immunisation by the nasal route is an interesting opportunity that has been increasingly explored. The nasal mucosa possesses many advantages for vaccine delivery; it is readily accessible (non-invasive needle-free option) and has a large surface area with a leaky and highly vascularised epithelium. In addition, and perhaps the most important aspect in this context, the nasal cavity is rich in nasal-associated lymphoid tissue (NALT) which is equivalent to that found in gut. The NALT is crucial to uptake the particulate carriers and is also an inductive and effective site of the immune system. NALT contains all the immunocompetent cells in the body that mediate the induction of mucosal immune responses to inhaled antigens. Moreover, IN vaccination becomes even more attractive because it is effective at inducing antigen-specific immune responses in both mucosal and systemic compartments (Kang et al. 2009; Zaman et al. 2010).
Despite the several well-recognised advantages of nasal vaccines, important limitations also exist. One of the most important limitations of nasal immunisation is the rapid clearance of the vaccine formulation from nasal mucosal surface owing to the MCC. Therefore, the use of mucoadhesive adjuvants to increase the residence time of vaccines in the nasal passages may be useful to improve their efficacy. Another limitation is the proteolytic activity of the nasal mucosal enzymatic barrier which, consequently, restricts the nasally delivered vaccines (Kang et al. 2009).
Actually, vaccines are based on protein antigens, or DNA (usually called DNA vaccines), and they are poorly permeable, unstable and susceptible to enzymatic degradation and, therefore, need to be protected. The advances in nanotechnology have brought the development of a great spectrum of nasal nanosystem carriers that provide protection against biological degradation and may facilitate the passage of the antigen across nasal barriers, leading to an efficient antigen presentation to the immune system. Some interesting reviews have been published focusing on the application of particulate systems as adjuvants and carriers for nasal vaccine delivery (Csaba et al. 2009; Köping-Höggård et al. 2005; Sharma et al. 2009). More recently, considerable advances have been made toward the development and testing of novel adjuvants and delivery vehicles to use in nasal vaccines, particularly Lactococcus lactis (Medina et al. 2010), adenoviral vectors encoding pathogen antigens (Tutykhina et al. 2011), live attenuated Bordetella pertussis BPZE1 strain (Li et al. 2011) and several strains of Lactobacillus (Wells 2011).
Despite the large number of vaccines commercially available for prevention of numerous infectious diseases, the majority is formulated for parenteral administration. Actually, in spite of the intensive research presently ongoing for developing nasal vaccines, it seems there is only currently authorised by FDA and/or European Medicines Agency (EMA) vaccines against influenza for IN administration in humans (FluMist®, FluMist® Quadrivalent and Fluenz®) (Chadwick et al. 2010; EMA 2012; FDAb 2012). Nevertheless, many other vaccines for IN delivery are under investigation, for instance, against measles (Simon et al. 2011), HIV infection (Hinkula et al. 2008), hepatitis B (Tiwari et al. 2011), Mycobacterium tuberculosis (Lorenzi et al. 2010), Bacillus anthracis (Wang et al. 2012), H5N1 influenza (Wu et al. 2012), Streptococcus pneumoniae (Xu et al. 2011), norovirus infections (Velasquez et al. 2011) and shigellosis (Tribble et al. 2010).
Although traditional vaccines have comprised subunit proteins, live attenuated viruses or killed bacteria, much attention has recently focused on non-replicating DNA or RNA vaccine delivery systems (Goodsell et al. 2008). Hence, as a result of the huge development achieved during the last years in the field of genetic engineering, and perhaps motivated by the successful clinical introduction of the first gene-therapy medicinal product (Gendicine®) (Wilson 2005), the investigation of DNA- and RNA-based vaccines has significantly enhanced, targeting even the IN delivery of the therapeutic genes or oligonucleotides encoding antigens for specific pathogens.
Therefore, the most recent developments on IN delivery of vaccines lead us to believe that the nasal route is a viable option for effective immunisation and the clinical introduction of new nasal vaccines is expected in the next years.
15.6 Conclusions
Nowadays, the most part of IN medicinal products available in clinical practice are targeted toward local (or topical) relief or prevention of nasal symptoms usually associated to acute or chronic diseases affecting the upper respiratory tract, such as common cold, rhinitis and sinusitis. Over the last few years, some small-molecule drugs also reached the market for acute or chronic pain management, smoking cessation and hormone replacement therapy, but many others are currently under clinical or preclinical development. In addition to small-molecule drugs, the nasal route has also attracted the interest of scientific community for the delivery of therapeutic macromolecules such as proteins, peptides and nucleic acids; these biomolecules are highly susceptible to enzymatic or acidic degradation and, therefore, they are typically administered by parenteral routes; thus, in these cases, the IN delivery represents a viable alternative to oral route and enables to overcome the problems associated to parenteral drug delivery.
Hence, taking into account all the intrinsic advantages of the nasal route, and considering that it has become one of the most explored ways for non-invasive drug delivery, in the future, it will be certainly possible to routinely use a broad spectrum of nasal products for the pharmacological management of multiple clinical conditions.
References
Adappa ND, Wei CC, Palmer J. Nasal irrigation with or without drugs: the evidence. Curr Opin Otolaryngol Head Neck Surg. 2012;20:23–57.
Ahmed S, Sileno AP, deMeireles JC, et al. Effects of pH and dose on nasal absorption of scopolamine hydrobromide in human subjects. Pharm Res. 2000;17:974–7.
Ahn B-N, Kim S-K, Shim C-K. Proliposomes as an intranasal dosage form for the sustained delivery of propranolol. J Contol Release. 1995; 34:203–10.
Akpinar ME, Yigit O, Akakin D, et al. Topical glucocorticoid reduces the topical decongestant-induced histologic changes in an animal model nasal mucosa. Laryngoscope. 2012;122:741–6.
Alsarra IA, Hamed AY, Alanazi FK. Acyclovir liposomes for intranasal systemic delivery: development and pharmacokinetics evaluation. Drug Deliv. 2008;15:313–21.
Alsarra IA, Hamed AY, Alanazi FK, et al. Vesicular systems for intranasal drug delivery. In: Jain KK, editor. Drug delivery to the central nervous system. New York: Humana; 2010. doi:10.1007/978-1-60761-529-3_8.
Anderson BD. Prodrugs for improved CNS delivery. Adv Drug Deliv Rev. 1996;19:171–202.
Anderson JM, Etches D. Prevention and management of postpartum hemorrhage. Am Fam Physician. 2007;75:875–82.
Arnott N, Harrold AJ, Lynch P. Variations in oxytocin regimes in Scottish labour wards in 1998. J Obstet Gynaecol. 2000;20:235–8.
Arora P, Sharma S, Garg S. Permeability issues in nasal drug delivery. Drug Discov Today. 2002;7:967–75.
Atluri H, Tirucherai GS, Dias CS, et al. Ocular, nasal, pulmonary and otic routes of drug delivery. In: Jasti BR, Ghosh TK, editors. Theory and practice of contemporary pharmaceutics. New York: CRC Press; 2005. doi:10.1201/9780203644478.ch16.
Babey M, Kopp P, Robertson GL. Familial forms of diabetes insipidus: clinical and molecular characteristics. Nat Rev Endocrinol. 2011;7:701–14.
Bahadur S, Pathak K. Physicochemical and physiological considerations for efficient nose-to-brain targeting. Expert Opin Drug Deliv. 2012;9:19–31.
Balasubramanian R, Dwyer A, Seminara SB, et al. Human GnRH deficiency: a unique disease model to unravel the ontogeny of GnRH neurons. Neuroendocrinology. 2010;92:81–99.
Barakat NS, Omar SA, Ahmed AA. Carbamazepine uptake into rat brain following intra-olfactory transport. J Pharm Pharmacol. 2006;58:63–72.
Behl CR, Pimplaskar HK, Sileno AP, et al. Effects of physicochemical properties and other factors on systemic nasal drug delivery. Adv Drug Deliv Rev. 1998;29:89–116.
Bellad M, Tara D, Ganachari M, et al. Prevention of postpartum haemorrhage with sublingual misoprostol or oxytocin: a double-blind randomised controlled trial. BJOG. 2012;119:975–86.
Bende M, Löth S. Vascular effects of topical oxymetazoline on human nasal mucosa. J Laryngol Otol. 1986;100:285–8.
Benedict C, Brede S, Schiöth HB, et al. Intranasal insulin enhances postprandial thermogenesis and lowers postprandial serum insulin levels in healthy men. Diabetes. 2011;60:114–8.
Berglund RK, Tangen CM, Powell IJ, et al. Ten-year follow-up of neoadjuvant therapy with goserelin acetate and flutamide before radical prostatectomy for clinical T3 and T4 prostate cancer: update on Southwest Oncology Group Study 9109. Urology. 2012;79:633–7.
Bitter C, Suter-Zimmermann K, Surber C. Nasal drug delivery in humans. Curr Probl Dermatol. 2011;40:20–35.
Caenen M, Hamels K, Deron P, et al. Comparison of decongestive capacity of xylometazoline and pseudoephedrine with rhinomanometry and MRI. Rhinology. 2005;43:205–9.
Cass LM, Efthymiopoulos C, Bye A. Pharmacokinetics of zanamivir after intravenous, oral, inhaled or intranasal administration to healthy volunteers. Clin Pharmacokinet. 1999;36:1–11.
Chadwick S, Kriegel C, Amiji M. Nanotechnology solutions for mucosal immunization. Adv Drug Deliv Rev. 2010;62:394–407.
Chanson P, Salenave S. Treatment of neurogenic diabetes insipidus. Ann Endocrinol (Paris). 2011;72:496–9.
Chen M, Li XR, Zhou YX, et al. Improved absorption of salmon calcitonin by ultraflexible liposomes through intranasal delivery. Peptides. 2009;30:1288–95.
Chesnut 3rd CH, Azria M, Silverman S, et al. Salmon calcitonin: a review of current and future therapeutic indications. Osteoporos Int. 2008;19:479–91.
Cho E, Gwak H, Chun I. Formulation and evaluation of ondansetron nasal delivery systems. Int J Pharm. 2008;349:101–7.
Christrup LL, Foster D, Popper LD, et al. Pharmacokinetics, efficacy, and tolerability of fentanyl following intranasal versus intravenous administration in adults undergoing third-molar extraction: a randomized, double-blind, double-dummy, two-way, crossover study. Clin Ther. 2008;30:469–81.
Colombo G, Lorenzini L, Zironi E, et al. Brain distribution of ribavirin after intranasal administration. Antiviral Res. 2011;92:408–14.
Corboz MR, Rivelli MA, Mingo GG, et al. Mechanism of decongestant activity of alpha 2-adrenoceptor agonists. Pulm Pharmacol Ther. 2008;21:449–54.
Costantino HR, Illum L, Brandt G, et al. Intranasal delivery: physicochemical and therapeutic aspects. Int J Pharm. 2007;337:1–24.
Costantino HR, Leonard AK, Brandt G, et al. Intranasal administration of acetylcholinesterase inhibitors. BMC Neurosci. 2008;9 Suppl 3:S6.
Csaba N, Gracia-Fuentes M, Alonso MJ. Nanoparticles for nasal vaccination. Adv Drug Deliv Rev. 2009;61:140–57.
Dahlin M, Björk E. Nasal administration of a physostigmine analogue (NXX-066) for Alzheimer’s disease to rats. Int J Pharm. 2001;212:267–74.
Dahlin M, Bergman U, Jansson B, et al. Transfer of dopamine in the olfactory pathway following nasal administration in mice. Pharm Res. 2000;17:737–42.
Dahlin M, Jansson B, Björk E. Levels of dopamine in blood and brain following nasal administration to rats. Eur J Pharm Sci. 2001;14:75–80.
Deveci S, Peşkircioğlu L, Aygün C, et al. Sublingual sildenafil in the treatment of erectile dysfunction: faster onset of action with less dose. Int J Urol. 2004;11:989–92.
Devogelaer JP, Boutsen Y, Manicourt DH. Biologicals in osteoporosis: teriparatide and parathyroid hormone in women and men. Curr Osteoporos Rep. 2010;8:154–61.
Dhuria SV, Hanson LR, Frey 2nd WH. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99:1654–73.
Dodick D, Brandes J, Elkind A, et al. Speed of onset, efficacy and tolerability of zolmitriptan nasal spray in the acute treatment of migraine: a randomised, double-blind, placebo-controlled study. CNS Drugs. 2005;19:125–36.
du Plessis LH, Lubbe J, Strauss T, et al. Enhancement of nasal and intestinal calcitonin delivery by the novel PheroidTM fatty acid based delivery system, and by N-trimethyl chitosan chloride. Int J Pharm. 2010;385:181–6.
Dykewicz MS, Hamilos DL. Rhinitis and sinusitis. J Allergy Clin Immunol. 2010;125 Suppl 2:S103–15.
Elshafeey AH, Bendas ER, Mohamed OH. Intranasal microemulsion of sildenafil citrate: in vitro evaluation and in vivo pharmacokinetic study in rabbits. AAPS PharmSciTech. 2009;10:361–7.
EMA, European Medicines Agency. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/001101/human_med_001405.jsp&mid=WC0b01ac058001d124 (2012). Accessed 13 July 2012.
Falser N, Wober W, Rahlfs VW, et al. Comparative efficacy and safety of azelastine and levocabastine nasal sprays in patients with seasonal allergic rhinitis. Arzneimittelforschung. 2001;51:387–93.
FDAa – U.S. Food and Drug Administration. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm (2012). Accessed 13 July 2012.
FDAb – U.S. Food and Drug Administration. http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm093833.htm (2012). Accessed 13 July 2012.
Fewtrell MS, Loh KL, Blake A, et al. Randomised, double blind trial of oxytocin nasal spray in mothers expressing breast milk for preterm infants. Arch Dis Child Fetal Neonatal Ed. 2006;91:F169–74.
Fisher A, Watling M, Smith A, et al. Pharmacokinetic comparisons of three nasal fentanyl formulations; pectin, chitosan and chitosan-poloxamer 188. Int J Clin Pharmacol Ther. 2010;48:138–45.
Fitzgibbon D, Morgan D, Dockter D, et al. Initial pharmacokinetic, safety and efficacy evaluation of nasal morphine gluconate for breakthrough pain in cancer patients. Pain. 2003;106:309–15.
Florence K, Manisha L, Kumar BA, et al. Intranasal clobazam delivery in the treatment of status epilepticus. J Pharm Sci. 2011;100:692–703.
Foreman A, Boase S, Psaltis A, et al. Role of bacterial and fungal biofilms in chronic rhinosinusitis. Curr Allergy Asthma Rep. 2012;12:127–35.
Franco Jr JG, Baruffi RL, Mauri AL, et al. Prospective randomized comparison of ovarian blockade with nafarelin versus leuprolide during ovarian stimulation with recombinant FSH in an ICSI program. J Assist Reprod Genet. 2001;18:593–7.
Fransén N, Bredenberg S, Björk E. Clinical study shows improved absorption of desmopressin with novel formulation. Pharm Res. 2009;26:1618–25.
Gao L, Yu S, Chen Q, et al. A randomized controlled trial of low-dose recombinant human interferons alpha-2b nasal spray to prevent acute viral respiratory infections in military recruits. Vaccine. 2010;28:4445–51.
Gawel M, Aschoff J, May A, et al. Zolmitriptan 5 mg nasal spray: efficacy and onset of action in the acute treatment of migraine–results from phase 1 of the REALIZE Study. Headache. 2005;45:7–16.
Giavina-Bianchi P, Agondi R, Stelmach R, et al. Fluticasone furoate nasal spray in the treatment of allergic rhinitis. Ther Clin Risk Manag. 2008;4:465–72.
Goodsell A, Zhou F, Gupta S, et al. Beta7-integrin-independent enhancement of mucosal and systemic anti-HIV antibody responses following combined mucosal and systemic gene delivery. Immunology. 2008;123:378–89.
Graf P. Rhinitis medicamentosa: aspects of pathophysiology and treatment. Allergy. 1997;52 Suppl 4:28–34.
Grassin-Delyle S, Buenestado A, Naline E, et al. Intranasal drug delivery: an efficient and non-invasive route for systemic administration: focus on opioids. Pharmacol Ther. 2012;134:366–79.
Gungor S, Okyar A, Erturk-Toker S, et al. Ondansetron-loaded chitosan microspheres for nasal antiemetic drug delivery: an alternative approach to oral and parenteral routes. Drug Dev Ind Pharm. 2010;36:806–13.
Gupta H, Sharma A. Recent trends in protein and peptide drug delivery systems. Asian J Pharm. 2009;3:69–75. Available from: http://www.asiapharmaceutics.info/text.asp?2009/3/2/69/55041. Accessed 13 July 2012.
Hallschmid M, Higgs S, Thienel M, et al. Postprandial administration of intranasal insulin intensifies satiety and reduces intake of palatable snacks in women. Diabetes. 2012;61:782–9.
Hanson LR, Frey 2nd WH. Strategies for intranasal delivery of therapeutics for the prevention and treatment of neuroAIDS. J Neuroimmune Pharmacol. 2007;2:81–6.
Hanson LR, Frey 2nd WH. Intranasal delivery bypasses the blood–brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008;9 Suppl 3:S5.
Hinkula J, Hagbom M, Wahren B, et al. Safety and immunogenicity, after nasal application of HIV-1 DNA gagp37 plasmid vaccine in young mice. Vaccine. 2008;26:5101–6.
Hochban W, Althoff H, Ziegler A. Nasal decongestion with imidazoline derivatives: acoustic rhinometry measurements. Eur J Clin Pharmacol. 1999;55:7–12.
Howarth PH. A comparison of the anti-inflammatory properties of intranasal corticosteroids and antihistamines in allergic rhinitis. Allergy. 2000;62:6–11.
Hussain AA, Dakkuri A, Itoh S. Nasal absorption of ondansetron in rats: an alternative route of drug delivery. Cancer Chemother Pharmacol. 2000;45:432–4.
Illum L. Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci. 2000;11:1–18.
Illum L. Nasal drug delivery: new developments and strategies. Drug Discov Today. 2002;7:1184–9.
Illum L. Is nose-to-brain transport of drugs in man a reality? J Pharm Pharmacol. 2004;56:3–17.
Illum L. Nasal drug delivery – recent developments and future prospects. J Control Release. 2012;161:254–63.
Illum L, Watts P, Fisher AN, et al. Intranasal delivery of morphine. J Pharmacol Exp Ther. 2002;301:391–400.
Jain R, Nabar S, Dandekar P, et al. Micellar nanocarriers: potential nose-to-brain delivery of zolmitriptan as novel migraine therapy. Pharm Res. 2010;27:655–64.
Jiang L, Gao L, Wang X, et al. The application of mucoadhesive polymers in nasal drug delivery. Drug Dev Ind Pharm. 2010;36:323–36.
Jogani VV, Shah PJ, Mishra P, et al. Nose-to-brain delivery of tacrine. J Pharm Pharmacol. 2007;59:1199–205.
Jogani V, Jinturkar K, Vyas T, et al. Recent patents review on intranasal administration for CNS drug delivery. Recent Pat Drug Deliv Formul. 2008;2:25–40.
Kaasa S, Moksnes K, Nolte T, et al. Pharmacokinetics of intranasal fentanyl spray in patients with cancer and breakthrough pain. J Opioid Manag. 2010;6:17–26.
Kaliner MA, Storms W, Tilles S, et al. Comparison of olopatadine 0.6% nasal spray versus fluticasone propionate 50 microg in the treatment of seasonal allergic rhinitis. Allergy Asthma Proc. 2009;30:255–62.
Kang ML, Cho CS, Yoo HS. Application of chitosan microspheres for nasal delivery of vaccines. Biotechnol Adv. 2009;27:857–65.
Kao HD, Traboulsi A, Itoh S, et al. Enhancement of the systemic and CNS specific delivery of L-dopa by the nasal administration of its water soluble prodrugs. Pharm Res. 2000;17:978–84.
Kim TK, Kang W, Chun IK, et al. Pharmacokinetic evaluation and modeling of formulated levodopa intranasal delivery systems. Eur J Pharm Sci. 2009;38:525–32.
Köping-Höggård M, Sánchez A, Alonso MJ. Nanoparticles as carriers for nasal vaccine delivery. Expert Rev Vaccines. 2005;4:185–96.
Kubota R, Komiyama T, Shimada H. Evaluation of the method for nifedipine administration for a rapid onset of clinical effect: a clinical study in normal volunteers. Yakugaku Zasshi. 2001;121:355–64.
Kumar M, Misra A, Babbar AK, et al. Intranasal nanoemulsion based brain targeting drug delivery system of risperidone. Int J Pharm. 2008;358:285–91.
Lee HJ, Macbeth AH, Pagani JH, et al. Oxytocin: the great facilitator of life. Prog Neurobiol. 2009;88:127–51.
Lee SL, Yu LX, Cai B, et al. Scientific considerations for generic synthetic salmon calcitonin nasal spray products. AAPS J. 2011;13:14–9.
Lerner EN, van Zanten EH, Stewart GR. Enhanced delivery of octreotide to the brain via transnasal iontophoretic administration. J Drug Target. 2004;12:273–80.
Li L, Gorukanti S, Choi YM, et al. Rapid-onset intranasal delivery of anticonvulsants: pharmacokinetic and pharmacodynamic evaluation in rabbits. Int J Pharm. 2000;199:65–76.
Li L, Nandi I, Kim KH. Development of an ethyl laurate-based microemulsion for rapid-onset intranasal delivery of diazepam. Int J Pharm. 2002;237:77–85.
Li R, Lim A, Alonso S. Attenuated Bordetella pertussis BPZE1 as a live vehicle for heterologous vaccine antigens delivery through the nasal route. Bioeng Bugs. 2011;2:315–9.
Lim M, Citardi MJ, Leong JL. Topical antimicrobials in the management of chronic rhinosinusitis: a systematic review. Am J Rhinol. 2008;22:381–9.
Lochhead JJ, Thorne RG. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev. 2012;64:614–28.
Lorenzi JC, Trombone AP, Rocha CD, et al. Intranasal vaccination with messenger RNA as a new approach in gene therapy: use against tuberculosis. BMC Biotechnol. 2010;10:77.
Mahajan HS, Gattani S. In situ gels of metoclopramide hydrochloride for intranasal delivery: in vitro evaluation and in vivo pharmacokinetic study in rabbits. Drug Deliv. 2010;17:19–27.
Maranta CA, Simmen D. Decongestant nasal spray. Results of a rhinomanometric double-blind study. Schweiz Med Wochenschr. 1996;126:1875–80.
Mathias NR, Hussain MA. Non-invasive systemic drug delivery: developability considerations for alternate routes of administration. J Pharm Sci. 2010;99:1–20.
Medina M, Vintiñi E, Villena J. Lactococcus lactis as an adjuvant and delivery vehicle of antigens against pneumococcal respiratory infections. Bioeng Bugs. 2010;1:313–25.
Meltzer EO, Caballero F, Fromer LM, et al. Treatment of congestion in upper respiratory diseases. Int J Gen Med. 2010;3:69–91.
Merkus FW, Van den Berg MP. Can nasal drug delivery bypass the blood–brain barrier? Questioning the direct transport theory. Drugs R D. 2007;8:133–44.
Misra A, Ganesh S, Shahiwala A, et al. Drug delivery to the central nervous system: a review. J Pharm Pharm Sci. 2003;6:252–73.
Mistry A, Stolnik S, Illum L. Nanoparticles for direct nose-to-brain delivery of drugs. Int J Pharm. 2009;379:146–57.
Moenter SM. Identified GnRH neuron electrophysiology: a decade of study. Brain Res. 2010;1364:10–24.
Nevéus T. Nocturnal enuresis – theoretic background and practical guidelines. Pediatr Nephrol. 2011;26:1207–14.
Ozgönenel B, Rajpurkar M, Lusher JM. How do you treat bleeding disorders with desmopressin? Postgrad Med J. 2007;83:159–63.
Ozsoy Y, Gungor S, Cevher E. Nasal delivery of high molecular weight drugs. Molecules. 2009;14:3754–79.
Packer M, Fowler MB, Roecker EB, et al. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation. 2002;106:2194–9.
Parra AL, Rodriguez JC. Nasal neuro EPO could be a reliable choice for neuroprotective stroke treatment. Cent Nerv Syst Agents Med Chem. 2012;12:60–8.
Patil S, Babbar A, Mathur R, et al. Mucoadhesive chitosan microspheres of carvedilol for nasal administration. J Drug Target. 2010;18:321–31.
Patil SB, Kaul A, Babbar A, et al. In vivo evaluation of alginate microspheres of carvedilol for nasal delivery. J Biomed Mater Res B Appl Biomater. 2012;100:249–55.
Petersen H, Kullberg A, Edsbäcker S, et al. Nasal retention of budesonide and fluticasone in man: formation of airway mucosal budesonide-esters in vivo. Br J Clin Pharmacol. 2001;51:159–63.
Pires A, Fortuna A, Alves G, et al. Intranasal drug delivery: how, why and what for? J Pharm Pharm Sci. 2009;12:288–311.
Rajinikanth PS, Sankar C, Mishra B. Sodium alginate microspheres of metoprolol tartrate for intranasal systemic delivery: development and evaluation. Drug Deliv. 2003;10:21–8.
Rautio J, Laine K, Gynther M, et al. Prodrug approaches for CNS delivery. AAPS J. 2008;10:92–102.
Roland PS, Marple BF, Wall GM. Olopatadine nasal spray for the treatment of allergic rhinitis. Expert Rev Clin Immunol. 2010;6:197–204.
Ross TM, Martinez PM, Renner JC, et al. Intranasal administration of interferon beta bypasses the blood–brain barrier to target the central nervous system and cervical lymph nodes: a non-invasive treatment strategy for multiple sclerosis. J Neuroimmunol. 2004;151:66–77.
Ruis H, Rolland R, Doesburg W, et al. Oxytocin enhances onset of lactation among mothers delivering prematurely. Br Med J (Clin Res Ed). 1981;283:340–2.
Safdarian L, Mohammadi FS, Alleyassin A, et al. Clinical outcome with half-dose depot triptorelin is the same as reduced-dose daily buserelin in a long protocol of controlled ovarian stimulation for ICSI/embryo transfer: a randomized double-blind clinical trial (NCT00461916). Hum Reprod. 2007;22:2449–54.
Salib RJ, Howarth PH. Safety and tolerability profiles of intranasal antihistamines and intranasal corticosteroids in the treatment of allergic rhinitis. Drug Saf. 2003;26:863–93.
Sastre J, Mosges R. Local and systemic safety of intranasal corticosteroids. J Investig Allergol Clin Immunol. 2012;22:1–12.
Schulz C, Paulus K, Jöhren O, et al. Intranasal leptin reduces appetite and induces weight loss in rats with diet-induced obesity (DIO). Endocrinology. 2012;153:143–53.
Seju U, Kumar A, Sawant KK. Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: in vitro and in vivo studies. Acta Biomater. 2011;7:4169–76.
Shao Z, Park GB, Krishnamoorthy R, et al. The physicochemical properties, plasma enzymatic hydrolysis, and nasal absorption of acyclovir and its 2′-ester prodrugs. Pharm Res. 1994;11:237–42.
Sharma S, Mukkur TK, Benson HA, et al. Pharmaceutical aspects of intranasal delivery of vaccines using particulate systems. J Pharm Sci. 2009;98:812–43.
Shore N, Cookson MS, Gittelman MC. Long-term efficacy and tolerability of once-yearly histrelin acetate subcutaneous implant in patients with advanced prostate cancer. BJU Int. 2012;109:226–32.
Simon JK, Ramirez K, Cuberos L, et al. Mucosal IgA responses in healthy adult volunteers following intranasal spray delivery of a live attenuated measles vaccine. Clin Vaccine Immunol. 2011;18:355–61.
Singh AK, Singh A, Madhav NV. Nasal cavity: a promising transmucosal platform for drug delivery and research approaches from nasal to brain targeting. J Drug Deliv Ther. 2012;2:22–33.
Steyn D, du Plessis L, Kotzé A. Nasal delivery of recombinant human growth hormone: in vivo evaluation with Pheroid technology and N-trimethyl chitosan chloride. J Pharm Pharm Sci. 2010;13:263–73.
Stoker DG, Reber KR, Waltzman LS, et al. Analgesic efficacy and safety of morphine-chitosan nasal solution in patients with moderate to severe pain following orthopedic surgery. Pain Med. 2008;9:3–12.
Suh JD, Kennedy DW. Treatment options for chronic rhinosinusitis. Proc Am Thorac Soc. 2011;8:132–40.
Sur D, Scandale S. Treatment of allergic rhinitis. Am Fam Physician. 2010;81:1440–6.
Takeuchi S, Minoura H, Shibahara T, et al. A prospective randomized comparison of routine buserelin acetate and a decreasing dosage of nafarelin acetate with a low-dose gonadotropin-releasing hormone agonist protocol for in vitro fertilization and intracytoplasmic sperm injection. Fertil Steril. 2001;76:532–7.
Tas C, Ozkan CK, Savaser A, et al. Nasal administration of metoclopramide from different dosage forms: in vitro, ex vivo, and in vivo evaluation. Drug Deliv. 2009;16:167–75.
Teshima D, Yamauchi A, Makino K, et al. Nasal glucagon delivery using microcrystalline cellulose in healthy volunteers. Int J Pharm. 2002;233:61–6.
Thorne RG, Frey 2nd WH. Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin Pharmacokinet. 2001;40:907–46.
Thorne RG, Pronk GJ, Padmanabhan V, et al. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127:481–96.
Thorne RG, Hanson LR, Ross TM, et al. Delivery of interferon-beta to the monkey nervous system following intranasal administration. Neuroscience. 2008;152:785–97.
Tiwari S, Verma SK, Agrawal GP, et al. Viral protein complexed liposomes for intranasal delivery of hepatitis B surface antigen. Int J Pharm. 2011;413:211–9.
Treschan TA, Peters J. The vasopressin system: physiology and clinical strategies. Anesthesiology. 2006;105:599–612.
Tribble D, Kaminski R, Cantrell J, et al. Safety and immunogenicity of a shigella flexneri 2a Invaplex 50 intranasal vaccine in adult volunteers. Vaccine. 2010;28:6076–85.
Tunn UW. A 6-month depot formulation of leuprolide acetate is safe and effective in daily clinical practice: a non-interventional prospective study in 1273 patients. BMC Urol. 2011;11:15.
Tutykhina IL, Logunov DY, Shcherbinin DN, et al. Development of adenoviral vector-based mucosal vaccine against influenza. J Mol Med (Berl). 2011;89:331–41.
Tuvemo T, Gustafsson J, Proos LA. Suppression of puberty in girls with short-acting intranasal versus subcutaneous depot GnRH agonist. Horm Res. 2002;57:27–31.
Tveita T, Thoner J, Klepstad P, et al. A controlled comparison between single doses of intravenous and intramuscular morphine with respect to analgesic effects and patient safety. Acta Anaesthesiol Scand. 2008;52:920–5.
Uchida M, Katoh T, Mori M, et al. Intranasal administration of milnacipran in rats: evaluation of the transport of drugs to the systemic circulation and central nervous system and the pharmacological effect. Biol Pharm Bull. 2011;34:740–7.
Uemura N, Onishi T, Mitaniyama A, et al. Bioequivalence and rapid absorption of zolmitriptan nasal spray compared with oral tablets in healthy Japanese subjects. Clin Drug Investig. 2005;25:199–208.
Vaidyanathan S, Williamson P, Clearie K, et al. Fluticasone reverses oxymetazoline-induced tachyphylaxis of response and rebound congestion. Am J Respir Crit Care Med. 2010;182:19–24.
Van de Walle J, Stockner M, Raes A, et al. Desmopressin 30 years in clinical use: a safety review. Curr Drug Saf. 2007;2:232–8.
Van de Walle J, Van Herzeele C, Raes A. Is there still a role for desmopressin in children with primary monosymptomatic nocturnal enuresis?: a focus on safety issues. Drug Saf. 2010;33:261–71.
Ved PM, Kim K. Poly(ethylene oxide/propylene oxide) copolymer thermo-reversible gelling system for the enhancement of intranasal zidovudine delivery to the brain. Int J Pharm. 2011;411:1–9.
Velasquez LS, Shira S, Berta AN, et al. Intranasal delivery of Norwalk virus-like particles formulated in an in situ gelling, dry powder vaccine. Vaccine. 2011;29:5221–31.
Veronesi MC, Kubek DJ, Kubek MJ. Intranasal delivery of neuropeptides. Methods Mol Biol. 2011;789:303–12.
Vyas TK, Shahiwala A, Marathe S, et al. Intranasal drug delivery for brain targeting. Curr Drug Deliv. 2005a;2:165–75.
Vyas TK, Babbar AK, Sharma RK, et al. Intranasal mucoadhesive microemulsions of zolmitriptan: preliminary studies on brain-targeting. J Drug Target. 2005b;13:317–24.
Vyas TK, Babbar AK, Sharma RK, et al. Intranasal mucoadhesive microemulsions of clonazepam: preliminary studies on brain targeting. J Pharm Sci. 2006a;95:570–80.
Vyas TK, Babbar AK, Sharma RK, et al. Preliminary brain-targeting studies on intranasal mucoadhesive microemulsions of sumatriptan. AAPS PharmSciTech. 2006b;7:E8.
Wang SH, Kirwan SM, Abraham SN, et al. Stable dry powder formulation for nasal delivery of anthrax vaccine. J Pharm Sci. 2012;101:31–47.
Wei SQ, Luo ZC, Qi HP, et al. High-dose vs low-dose oxytocin for labor augmentation: a systematic review. Am J Obstet Gynecol. 2010;203:296–304.
Wells J. Mucosal vaccination and therapy with genetically modified lactic acid bacteria. Annu Rev Food Sci Technol. 2011;2:423–45.
Westin U, Piras E, Jansson B, et al. Transfer of morphine along the olfactory pathway to the central nervous system after nasal administration to rodents. Eur J Pharm Sci. 2005;24:565–73.
Westin UE, Boström E, Gråsjö J, et al. Direct nose-to-brain transfer of morphine after nasal administration to rats. Pharm Res. 2006;23:565–72.
Wilson JM. Gendicine: the first commercial gene therapy product. Hum Gene Ther. 2005;16:1014–5.
Wolfe TR, Bernstone T. Intranasal drug delivery: an alternative to intravenous administration in selected emergency cases. J Emerg Nurs. 2004;30:141–7.
Wu Y, Wei W, Zhou M, et al. Thermal-sensitive hydrogel as adjuvant-free vaccine delivery system for H5N1 intranasal immunization. Biomaterials. 2012;33:2351–60.
Xu J, Dai W, Wang Z, et al. Intranasal vaccination with chitosan-DNA nanoparticles expressing pneumococcal surface antigen a protects mice against nasopharyngeal colonization by Streptococcus pneumoniae. Clin Vaccine Immunol. 2011;18:75–81.
Yang T, Hussain A, Bai S, et al. Positively charged polyethylenimines enhance nasal absorption of the negatively charged drug, low molecular weight heparin. J Control Release. 2006;115:289–97.
Yates R, Sörensen J, Bergström M, et al. Distribution of intranasal 11C-zolmitriptan assessed by positron emission tomography. Cephalalgia. 2005;25:1103–9.
Youn YS, Jeon JE, Chae SY, et al. PEGylation improves the hypoglycaemic efficacy of intranasally administered glucagon-like peptide-1 in type 2 diabetic db/db mice. Diabetes Obes Metab. 2008;10:343–6.
Zaki NM, Mortada ND, Awad GA, et al. Rapid-onset intranasal delivery of metoclopramide hydrochloride Part II: safety of various absorption enhancers and pharmacokinetic evaluation. Int J Pharm. 2006;327:97–103.
Zaman M, Simerska P, Toth I. Synthetic polyacrylate polymers as particulate intranasal vaccine delivery systems for the induction of mucosal immune response. Curr Drug Deliv. 2010;7:118124.
Zhang YJ, Ma CH, Lu WL, et al. Permeation-enhancing effects of chitosan formulations on recombinant hirudin-2 by nasal delivery in vitro and in vivo. Acta Pharmacol Sin. 2005;26:1402–8.
Zhang J, Branch DW, Ramirez MM, et al. Oxytocin regimen for labor augmentation, labor progression, and perinatal outcomes. Obstet Gynecol. 2011;118:249–56.
Acknowledgements
The authors thank to Fundação para a Ciência e a Tecnologia (Portugal) for the postdoctoral (SFRH/BPD/46826/2008) and doctoral grants (SFRH/BD/64895/2009; SFRH/BD/69378/2010). The authors also thank to POPH (Programa Operacional Potencial Humano) which is co-funded by FSE (Fundo Social Europeu), União Europeia.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Serralheiro, A., Alves, G., Sousa, J., Fortuna, A., Falcão, A. (2013). Nose as a Route for Drug Delivery. In: Önerci, T. (eds) Nasal Physiology and Pathophysiology of Nasal Disorders. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-37250-6_15
Download citation
DOI: https://doi.org/10.1007/978-3-642-37250-6_15
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-37249-0
Online ISBN: 978-3-642-37250-6
eBook Packages: MedicineMedicine (R0)