
Abstract
Purpose
To create improved pharmaceutical formulations for nasal and sublingual administration of desmopressin and investigate their pharmacokinetic profiles in comparison with a commercial nasal liquid spray and finally to evaluate the volunteers’ opinions on the different dosage forms.
Methods
Both formulations were based on the characteristics of interactive mixtures. The nasal powder spray was produced by a rotary evaporator technique with sodium starch glycolate as carrier material and the sublingual tablet by direct compression after dry mixing with mannitol as carrier. The clinical study was an open-label, randomised cross-over pharmacokinetic study in healthy volunteers.
Results
The nasal powder formulation gave a threefold increase in the absorption, unaltered time to maximum plasma concentration and a tendency to lower variability in the amount absorbed compared with the liquid spray. The powder was reported to be more irritating than the liquid but was still well accepted by the volunteers. The tablet did not improve the uptake of desmopressin, likely because of a poor disintegration sublingually.
Conclusions
The nasal powder formulation is a promising new dosage form for the delivery of desmopressin and other compounds. The sublingual tablet has a beneficial means of production and may be further developed by decreasing its disintegration time.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The nasal route of administration is often applied for peptide drug delivery. The relatively large surface area, thin epithelium and rich vascularisation enable absorption of even large and hydrophilic compounds without first-pass intestinal and hepatic metabolism. Another plausible way of delivering peptide drugs is by sublingual administration. Also in this case, a direct systemic absorption is achieved, but normally to a lower degree than after nasal administration as a consequence of the smaller absorptive surface area and multiple epithelial cell layers (1,2). It is important for both administration routes that the drug remains at the site of absorption instead of being lost to the stomach, which would lead to much lower bioavailability because of chemical and enzymatic degradation.
The vasopressin analogue desmopressin is today administered as oral and orally disintegrating tablets and as a nasal liquid spray. The differences in the absorption capacities of the three administration routes are reflected in the bioavailability of the peptide that, according to the manufacturer’s summary of product characteristics (SPC), is 3–5% for the nasal spray, 0.25% for the sublingual freeze-dried tablet and 0.08–0.16% for the oral tablet (Ferring Pharmaceuticals. SPC: Minirin nasal spray, Minirin Freeze-dried tables and Minirin tablet). The variability in the amount absorbed is extensive and an improvement thereof would be most valuable. Especially nasal administration of desmopressin has been associated with high variability (3,4), although pharmacokinetic parameters with relative standard deviations of the same magnitude or higher have also been obtained after per oral administration (5). The variability in the bioavailability after nasal administration could be explained by differences in the amount of liquid that is actually deposited in the nasal cavity. Harris et al. (6) have, for example, previously shown that the absorption of desmopressin was significantly affected by the volume of the liquid spray dose. In addition to the spray volume, the shape of the nasal cavity and the administration could also affect the amount of liquid that runs straight down the oesophagus; if the total amount of the drug is instead retained in the nasal cavity it should lead to both an increased bioavailability and a reduced variability.
Attempts have been made to increase the bioavailability after nasal and sublingual administration by the use of mucoadhesive dosage forms. A mucoadhesive sublingual tablet has been developed for the sublingual administration of fentanyl (7), resulting in extensive and rapid absorption. The concentrated deposition and reduced risk of swallowing may also be beneficial for sublingual absorption of other drugs. Carbomer is a well-known mucoadhesive agent that has also been shown to have a positive effect on the paracellular absorption of desmopressin (8). The sublingual absorption of the peptide may be further increased by the inhibitory effect on the enzymatic degradation of carbomer (9), wherefore it should be a suitable mucoadhesive additive in a sublingual peptide formulation.
The nasal route of administration may be especially well suited for mucoadhesive dosage forms since the mucosa has an air interface that enables prolonged residence time with as simple means as swelling dry particles that create a viscous gel upon contact with the moist nasal mucosa (10,11). Such administration systems can also have an absorption enhancing effect by widening of the tight junctions as a result of a temporary dehydration of the nasal epithelium (12), which would be beneficial for the absorption of large and hydrophilic peptides. Several studies have been performed on different mucoadhesive particle systems since Nagai et al. (13) showed an increased uptake of insulin in comparison with both a liquid and water soluble powder formulation. Promising results have often been obtained, both concerning the absorption (e.g. 14,15), residence time (e.g. 10,16) and safety (e.g. 17,18) of the systems. However, literature comparing the bioavailability from dry powder formulations and liquid nasal sprays in humans is scarce.
The development of dry particle delivery systems for nasal administration may have been stalled by difficulties in their production. The particles are normally obtained after emulsification followed by lyophilisation or spray drying, which could make it difficult to achieve a large scale production of particles in a reproducible size range. Particles below 10 μm should be avoided to rule out pulmonary deposition, but they should also not be too large to enable a favourable deposition in the nasal cavity (19). Interactive mixtures with mucoadhesive carrier particles that are available in a suitable size range, such as sodium starch glycolate (SSG), represent an alternative strategy. SSG is commonly used as a superdisintegrant in tablet formulations and owing to its extensive capacity for absorbing water of up to 300 times its volume (20) it also possesses mucoadhesive characteristics (21). It has been shown that interactive mixtures can be created down to a carrier particle size of approximately 30 μm simply by dry mixing the micronized active component with SSG as carrier material (22). The drug is homogeneously deposited on the surface of the carrier in such formulations and the particle size of the formulation is determined by the size of the carrier particles. Surface deposition can also be favourable for the release and absorption of the active component (23), because complete hydration of the carrier particle is not necessary for drug release.
The aim of this study was to improve the bioavailability of desmopressin in humans by developing new and simple delivery systems for nasal and sublingual administration and comparing their efficiency with a commercial nasal liquid spray in a cross-over clinical study. A final objective was to evaluate the volunteers’ views on the different administration forms.
MATERIALS AND METHODS
Materials
Desmopressin acetate (MW 1069 Da, anhydrous free base) was purchased from Bachem California, USA. Sodium starch glycolate (Primojel®) was purchased from DMV International, The Netherlands. The tablet ingredients were obtained as follows: carbomer (Carbopol 974) from Noveon Inc., USA, crospovidone (Polyplasdone XL10) from ISP Technologies, USA, magnesium stearate from Peter Greven, The Netherlands, mannitol (Pearlitol 200 SD) from Roquette Pharma, France and silica colloidal anhydrous (Aerosil) from Evonik, Germany. The commercial liquid nasal spray (Desmopressin Alpharma) was purchased from Apoteket AB, Sweden and had a concentration of 100 μg/mL desmopressin acetate, corresponding to an amount of 10 μg desmopressin acetate per spray actuation. All materials and chemicals were of GMP-quality.
Nasal Powder Spray
Preparation
SSG particles between 10 and 50 μm were obtained by air-classification under GMP-conditions at Micron Technologies, UK. An amount of 24 g of the SSG powder was weighed into a 250 mL rotary evaporator (Büchi Rotavapor, Switzerland) and desmopressin acetate, dissolved in 99.5% ethanol, was added to give a final concentration of 2 μg desmopressin acetate per mg SSG. Ethanol was used instead of water to prevent SSG from swelling so that the compound would be deposited on the surface of the carrier as is the case in interactive mixtures (22,24). The ethanol was carefully evaporated at 35°C and below 100 mBar until the powder was completely dry. The desmopressin content was evaluated with isocratic HPLC on a Reprosil Pur C18 AQ column with acetonitrile in 20 mM phosphate buffer pH 7.0 (1:4) as the mobile phase and UV-detection at 220 nm. The uniformity of content, expressed as the coefficient of variation (CV), was determined with ten repeated measurements where the powder in one device was dissolved in mobile phase and the amount of desmopressin was measured. After determination of the final desmopressin content in the dry powder, it was manually dispensed in UniDose DP® (Bespak, UK) devices so that each contained a dose of 20 μg desmopressin acetate. The individual fill weights were noted for the devices that were to be used in the dose delivery studies. Finally, the UniDose DP® devices were individually packaged in sealed aluminium sachets.
Particle Size and Dose Delivery
The particle size of the powder actuated from the devices was measured with Malvern dry laser diffraction analysis at Bespak, UK. The devices were mechanically actuated while held in an angle to prevent particles from crossing the laser twice when falling down. The dose delivery efficiency was calculated by subtracting the device weight post actuation from the pre actuation weight and dividing the difference by the original fill weight. Devices were stored in desiccators at different temperature and relative humidity (25°C/60% RH and 40°C/75% RH) for 4 or 10 weeks to investigate the effect of storage condition on the delivered dose and particle size. The particle size and dose delivery efficiency were determined from 12 repeated measurements.
Sublingual Tablet
Preparation
The sublingual tablet formulation was based on an interactive mixture with mannitol as carrier material (7); carbomer (2%) was added for mucoadhesion and absorption enhancement. Mannitol was mixed with desmopressin in a stainless steel jar (500 mL) in a Turbula mixer T2F (WA Bachofen AG, Switzerland) for 65 h after which the other tablet ingredients, except magnesium stearate, were added and mixed for a further 30 min; magnesium stearate was finally added and co-mixed for 2 min. The tablets were made by direct compression in a Korsch XPI tablet press (Korsch AG, Germany) using 6 mm punches to provide tablets that were small enough for sublingual administration. Each tablet was to contain 240 μg desmopressin, corresponding to 253 μg desmopressin acetate. The amount of 240 μg desmopressin base is equal to the composition of the commercial orally disintegrating tablet, Minirin®. The tablet homogeneity (CV) was determined from ten repeated measurements where one tablet was dissolved in mobile phase and the desmopressin content was determined with the same HPLC method as for the nasal powder formulation. The disintegration time was determined according to the European Pharmacopoeia (2005) method 2.9.1 as a mean of six measurements on individual tablets in a DISI-1M dissolution bath (Charles Ischi AG, Germany). The disintegration evaluation was performed in 37°C water and the gel forming tablets were fastened with a plastic disc to prevent them from floating.
Clinical Study
The clinical study was performed during 1 month as an open-label, randomised, three-period crossover pharmacokinetic study in healthy female and male volunteers. The study was approved by the Swedish medical products agency and the regional independent ethics committee in Linköping (EudraCT number 2006-006774-25). The study was performed at the Berzelius Clinical Research Center, Linköping, Sweden, in accordance with the Declaration of Helsinki, Good Clinical Practice and applicable regulatory requirements. Subjects were enrolled in the study after having given their written, informed consent.
Study Population
Healthy male and female volunteers underwent a screening procedure including medical and physical examination, where tests were made to ensure that none of the exclusion criteria were fulfilled. Owing to the nature of the study drugs, the exclusion criteria contained restrictions regarding use of nicotine products within 6 months, use of nasally administered medications within 24 h, ongoing upper respiratory tract infection or allergic rhinitis and previously known hypersensitivity to desmopressin. The intake of fluid was restricted to a maximum of 500 mL from 1 h before until 8 h after drug administration to minimise the risk of hyponatremia. The occurrence of adverse events was recorded throughout the study. A total number of 13 volunteers, six male and seven female, were included in the study. They had a mean age of 26 ± 6 years, mean weight of 70.9 ± 11.0 kg and a mean Body Mass Index of 23.6 ± 2.7 kg/m2.
Drug Administration and Blood Sampling
Each subject received the nasal liquid spray, the nasal powder spray and the sublingual tablet once in a randomised order. The three study periods were separated by a wash-out period of at least 3 days. All drug formulations were administered by the trained study personnel, who were provided with an instruction sheet to ensure that the nasal spray devices for powder and liquid administration were held at a constant angle to give an accurate dose straight into the nasal cavity. The powder formulation was sprayed into one nostril, whereas the commercial liquid formulation (Desmopressin Alpharma) was sprayed into both nostrils with a minimal delay between doses. The nasal liquid formulation was primed by spraying it five times into the air before dosing. The volunteers were instructed to blow their noses before administration and were asked not to inhale or exhale during the actuation of the spray device. They were also asked to try to avoid sneezing or blowing the nose and to remain seated in an upright position for at least 15 min after the administration.
The sublingual tablet was placed under the tongue in the deepest part of the oral cavity and the patients were instructed to keep their mouths shut without moving their tongues for 15 min after dosage; the tablet was to dissolve under the tongue without chewing or sucking. There was no water intake permitted from 1 h before until 1 h after the tablet administration.
Venous blood samples of 10 mL were collected through an indwelling catheter no more than 15 min prior to dosage and at 5, 10, 15, 30, 45, 60, 75, 90, 120, 180, 240, 360, 480 and 720 min after nasal administration and at 15, 30, 45, 60, 75, 90, 120, 180, 240, 360, 480 and 720 min after sublingual administration. The samples were collected in K2 EDTA plastic tubes and were centrifuged at 2 700 g for 10 min within 1 h after sampling. Approximately 4 mL of the supernatant was transferred to a new plastic tube and stored at −20°C until analysis.
Sample Analysis
The plasma concentrations of desmopressin were determined with a validated radioimmunoassay, based on a method previously described by Lundin et al. (25). Each sample was analysed in duplicate by MDS Pharma Sevices Switzerland AG, Switzerland. The lower limit of quantification (LLOQ) was 2.5 pg/mL and concentrations below the LLOQ were set to 0.
Calculations and Statistical Evaluation
The pharmacokinetic parameters of interest were the area under the plasma concentration–time curve until the last plasma sample (AUC0–t ) or infinity (AUC∞), the time to reach maximum plasma concentration (t max), the maximum plasma concentration (C max) and the plasma half-life (t 1/2). The pharmacokinetic parameters were calculated with non-compartmental analysis using WinNonlin® 4.0 (Pharsight Corp., CA, USA). The AUC was calculated with the linear/logarithmic trapezoidal method. All statistical analyses were performed in Minitab® Release 15. A p value of less than 0.05 was regarded as significant. Differences in t max were evaluated with the nonparametric Kruskal–Wallis test and all other statistical comparisons were performed with general linear models (GLM). Treatment, dosing sequence and period were included as fixed parameters and subject number within sequence as a random effect in the GLM for statistical evaluation of the pharmacokinetic parameters. A logarithmic transformation of the AUC and C max values was performed to ensure normal distribution. The homogeneity of the variances for logarithmically transformed AUC and C max was determined with Bartlett’s test to evaluate if the dosage form had an effect on the absorption variability. The standard deviations of logarithmically transformed data can be used instead of the coefficient of variation (CV) to evaluate the homoscedasticity of data in different size ranges (26), which should be especially suitable when the data is normally distributed after transformation.
Evaluation of the Volunteers’ Views on the Formulations
The volunteers were asked to fill out an evaluation form after each administration to look into their opinions on using the different formulations. The questionnaire was divided into four parts: one for each dosage form (nasal powder, sublingual tablet and commercial nasal liquid spray) and one with final comparisons. The final comparative part was completed after all doses had been received. The volunteers were asked if they had previously used nasal sprays or sublingual tablets and after each administration they were also asked if they felt the administration, taste or smell of the dosage form on a scale from 1 to 5, where one was not at all and 5 very much, and they were also requested to grade this sensation on the same scale, where 1 was very pleasant and 5 very unpleasant. Furthermore, they were requested to state approximately how long the sensation lasted and were given the opportunity to write down any specific comments regarding the administrations. In the final part, the subjects were asked which one of the three formulations they preferred and why.
RESULTS
Nasal Powder Spray
The homogeneity evaluation showed that desmopressin was uniformly distributed in the nasal powder bulk with a CV of 2.4%. Each UniDose DP® device for use in the clinical trial was filled with 11.76 mg powder containing 20 μg desmopressin; a variation of ±0.59 mg, corresponding to 1 μg desmopressin acetate, was allowed. The individually weighed devices, for investigation of the delivery efficiency and particle size, contained 11.8 ± 0.2 mg powder.
The particle size of the powder was suitable for nasal administration with an initial median particle size of 33.4 μm. The particle size was not significantly affected by storage at high humidity (Table I) and the fraction of the particles below 10 μm was low (<1.5%) and did not increase with storage. However, the dose delivery efficiency was affected by high moisture and temperature and was significantly lower after 10 weeks storage at 40°C and 75% RH (91.7%) compared with the initial delivery (99.5%) (p < 0.01). In the case of desmopressin delivery, a dose delivery efficiency of 91.7% would correspond to a decrease in the delivered dose of 1.7 μg. The delivery efficiency was not significantly affected by storage at 25°C and 60% RH for 10 weeks.
Sublingual Tablet
Carbomer containing tablets were successfully created in a small size suitable for sublingual administration (ϕ 6 mm). The dry mixing with mannitol as carrier material led to a homogeneous desmopressin content (CV 1.25%) after direct compression of the tablets, less cost efficient methods such as granulation or lyophilisation were thus superfluous. The tablets contained an amount of 237 μg desmopressin, corresponding to 250 μg desmopressin acetate, and the disintegration time was determined to 46.2 ± 3.8 s.
Clinical Study
The data from the nasal liquid spray and sublingual tablet were obtained from 13 volunteers, whereas the results from the powder spray were obtained from 11 subjects; one of the volunteers dropped out of the study before receiving the powder spray and one of the nasal powder administrations failed and leading to plasma concentrations too low for pharmacokinetic analysis. There were no serious adverse events nor cases of sneezing reported after drug administration.
Pharmacokinetics of the Dosage Forms
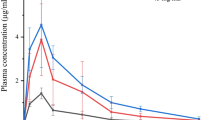
The plasma desmopressin concentration–time profiles are presented in Fig. 1 and other pharmacokinetic parameters are given in Table II. The nasal liquid spray resulted in an AUC0–12 h of 125.6 ± 71.7 pg h/mL, a C max of 34.1 ± 20. pg/mL and a plasma half-life of 162.1 ± 31.3 min (Table II). The plasma concentration–time profile of the commercial nasal liquid spray is well in accordance with previously published bioequivalence studies (3,4). The median time to reach maximum plasma concentration (t max) of 45 min was typical for the absorption of desmopressin, but comparatively long for nasal delivery.

Plasma pharmacokinetics after administration of the commercial nasal liquid spray (empty square, n = 13), the nasal powder spray (filled circle, n = 11) or the sublingual tablet (filled triangle, n = 13). Mean values and standard deviations.
The plasma exposure (AUC0–12 h, AUC∞ and C max) after administering the nasal powder formulation was significantly higher than after the liquid formulation (p < 0.0001); the arithmetic means being 353.9 ± 113.3 pg h/mL and 103.3 ± 30.8 pg/mL for AUC0–12 h and C max, respectively (Table II). No significant differences in t max or plasma half-life were shown. The nasal powder spray gave 3.06 and 3.26 times higher AUC0–12 h and C max than the nasal liquid spray when comparing the geometric means of the log-normally distributed parameters (Table III). The sublingual tablet led to significantly lower plasma concentrations (AUC0–12 h, AUC∞ and C max) than the nasal liquid spray (p < 0.001) despite the higher dose; the arithmetic means being 60.7 ± 27.4 pg h/mL and 17.9 ± 7.5 pg/mL for AUC0–12 h and C max, respectively (Table II). The relative bioavailability was approximately 25 times lower from the sublingual tablet than from the nasal liquid spray (Table III). The t max was significantly delayed after sublingual administration (p < 0.001), which is a natural result considering the differences in the mucosal constitution. A significantly shorter half-life of desmopressin was also detected after sublingual administration (p < 0.01), but this value was probably underestimated due to the low plasma concentrations and delayed t max that led to fewer concentrations above LLOQ in the elimination phase.
The variability in the absorption of desmopressin is usually expressed as the CV of the pharmacokinetic parameters despite their log-normal distribution. In this study, the CV tended to be lower after administration of the nasal powder spray (32.0 and 29.8% for AUC0–12 h and C max, respectively), both in comparison with the nasal liquid spray (57.1% and 58.5%) and the sublingual tablet (45.1% and 42.0%). However, it was not possible to detect a significant difference in the variation when comparing the standard deviations of the logarithmic values (Fig. 2).

Standard deviations of the logarithmically transformed values of AUC0–12 h (circles) and C max (squares) after administration of the intranasal (I.N.) liquid or powder spray or the sublingual (S.L.) tablet. Error bars represent the 95% confidence interval.
Volunteers’ Views on the Formulations
As many as 12 of the volunteers reported that they had used nasal sprays previously and two that they had used sublingual tablets before. One of the subjects dropped out of the study before receiving the nasal powder spray and was thus not included in the evaluation thereof or in the final comparative part. Furthermore, one of the volunteers did not feel the powder administration, indicating that it was probably not delivered properly, especially since there was no significant increase in the plasma concentration of desmopressin in this person. This was taken into account when evaluating the questionnaires and does not have an impact on the presented data.
There was no prevalence of taste or smell from the two nasal formulations. The administration of the nasal powder spray caused more discomfort than the nasal liquid spray; 15% and 73% of the volunteers reported that they sensed the spray dose much or very much after administration of the liquid or powder, respectively (failed administration not included). Some of the volunteers described a stinging sensation from the powder spray dose. Correspondingly, 8% and 36% graded the subjective sensation associated with the two formulations, respectively, as unpleasant and 0% and 9% as very unpleasant. The sensation was temporary and said to last for approximately 3 min for both dosage forms.
All volunteers experienced the administration of the sublingual tablet as simple or very simple and reported that they could feel the tablet under the tongue to some extent. The sensation of the tablet remained for an average of 15 min, but was not reported as uncomfortable more than in one case. Most volunteers reported that they could taste the tablet, but were not considerably disturbed by this. The main objective against the tablet was that it disintegrated too slowly.
The order of the preferred administration forms are displayed in Fig. 3. One volunteer did not choose a specific alternative, but said that all formulations were conceivable. This was not included in Fig. 3 as it was not an alternative given in the questionnaire. The preferred administration was in four cases said to be nasal spray, but without specifying if it should be in liquid or powder form. It was also not possible to determine the preferred alternative from the reasons given for this choice, wherefore these four answers are gathered under the collective term “Intranasal spray”. The predominant reasons for choosing a nasal spray seemed to be that the administration was the fastest and simplest. The volunteers, who preferred the powder spray, also all stated that a reason for this was that it was not runny. Administration of the sublingual tablet was perceived as simple, but its major drawback was that it disintegrated too slowly.

Percentage distribution of the first hand choices among the different formulations as given by the volunteers after sublingual (S.L.) or intranasal (I.N.) administration (n = 11). A third unspecified I.N. alternative was included in the figure because I.N. spray was chosen in some cases without specifying if it should be in liquid or powder form.
DISCUSSION
The plasma concentration–time profile after administration of the nasal liquid spray is in accordance with previously published data (3,4). According to the SPC, the bioavailability for the investigated commercial liquid spray should be 3–5%, which is in agreement with values for nasal sprays in scientific literature (27,28). However, the nasal uptake has also been reported to be higher with, for example, 11.3% absorbed (29). If comparing these values with the AUC ratios in Table III, the bioavailabilities would be in the order of 9–34% and 0.11–0.44% for the nasal powder spray and the sublingual tablet, respectively.
Possible reasons for the significantly improved absorption from the powder formulation could be facilitated absorption of the peptide and/or better deposition and residence time in the nasal cavity owing to its mucoadhesive characteristics (21). The latter assumption is supported by the volunteers reporting that one of the advantages with the powder formulation was that it did not run down the throat. Desmopressin has previously been administered as a viscous solution with the objective of increasing the residence time and thus the bioavailability of the substance (30,31). The viscous dosage form was indeed found to give a prolonged residence time, but did still not lead to an improved bioavailability. A decreased clearance alone should thus not be enough to increase the bioavailability of desmopressin and it is probable that the nasal powder spray also had an absorption enhancing effect, possibly caused by an increased paracellular uptake as a result of a temporary opening of the tight junctions (12). An optimised uptake from the nasal cavity, for whichever reason, should lead to a higher bioavailability and a lower variability as was also the case after administration of the nasal powder formulation. Hence, it is likely that it was the formulation, and not the administration route itself, that previously resulted in a high variability after nasal administration (3,4). The promising effect on the variability of the absorption from the powder formulation would thus be worth investigating in a larger study population.
The nasal powder formulation should also be applicable to other drugs. An advantage, in addition to the simple and reproducible production, is that the carrier particles are not swollen when adding the drug. A fast absorption is thus favoured because the release of the drug will not be dependent on total hydration of the particles and subsequent diffusion from the core of the particle to the epithelium. This was also seen in the current study where the more extensive plasma concentrations after administration of the powder formulation were not combined with a prolonged absorption phase.
SSG is a promising material for nasal powder systems as it is a well-known tablet excipient that is easily obtained in the appropriate particle size range. It should, however, also be possible to use other dry powders as carriers with this technique. The nasal powder was reported to be sensed to a greater extent than the liquid spray, but despite this it was chosen as the preferred dosage form more often than the sublingual tablet. The sensation will vary depending on the carrier powder and amount thereof and may even be less irritating than the corresponding liquid spray (32). The fact that the powder spray did not run down the throat was acknowledged by the volunteers and it is possible that the formulation would have seemed even more advantageous, had it been compared with a liquid spray containing a drug such as sumatriptan, which is known to have a bad taste (33).
The sublingual tablet did not give plasma concentrations comparable to any of the nasal sprays, likely due to the inferior paracellular absorption from this administration route compared with the nasal one. Although the non-keratinised sublingual mucosa should be the most permeable in the oral cavity (1), it is not comparable to the nasal mucosa (2). Previously published studies on a sublingual spray and an instantly dissolving tablet (34,35) have shown plasma desmopressin concentrations in the same range as were also seen herein after sublingual administration. A prolonged residence time sublingually facilitates both a site-specific absorption and possibilities for absorption enhancement and should therefore be more beneficial for the bioavailability than a formulation that is instantly dissolved and swallowed; yet, this requires that the drug is dissolved and accessible for absorption. It is thus reasonable to believe that the potentially enhancing effect of carbomer was counteracted by a too long disintegration time of the tablet, which was also supported by the volunteers’ comments on the formulation. The benefit of the investigated tablet is instead that it can be produced by direct compression since sufficient homogeneity is obtained by dry mixing of the peptide with coarse mannitol particles as carrier material. An improvement of the bioavailability may well be reached after further development of the tablet to decrease its disintegration time.
CONCLUSIONS
This is one of few published studies that show improved nasal absorption of a peptide drug with a powder formulation in human volunteers. The significantly increased absorption and the strong tendency to lower variability compared with the liquid spray were likely achieved by a better deposition in the nasal cavity and increased site-specific uptake. Furthermore, the production of the novel powder formulation is favourable for a large scale manufacturing. This makes the nasal powder formulation a promising new dosage form for the delivery of desmopressin and other compounds. The desmopressin containing sublingual tablet could be produced by direct compression, which makes it interesting to evaluate further after decreasing its disintegration time. Both study formulations were well accepted by the volunteers.
References
D. Harris, and J. R. Robinson. Drug delivery via the mucous membranes of the oral cavity. J. Pharm. Sci. 81:1–10 (1992) doi:10.1002/jps.2600810102.
A. Yamamoto, T. Iseki, M. Ochi-Sugiyama, N. Okada, T. Fujita, and S. Muranishi. Absorption of water-soluble compounds with different molecular weights and [Asu1.7]-eel calcitonin from various mucosal administration sites. J. Control Release. 76:363–374 (2001) doi:10.1016/S0168-3659(01)00454-0.
C. Joukhadar, B. Schenk, S. T. Kaehler, C. J. Kollenz, P. Bauer, M. Muller, and H. G. Eichler. A replicate study design for testing bioequivalence: a case study on two desmopressin nasal spray preparations. Eur. J. Clin. Pharmacol. 59:631–636 (2003) doi:10.1007/s00228-003-0682-3.
N. Eller, C. J. Kollenz, and G. Hitzenberger. A comparative study of pharmacodynamics and bioavailability of 2 different desmopressin nasal sprays. Int. J. Clin. Pharmacol. Ther. 36:139–145 (1998).
S. T. Kaehler, I. M. Steiner, R. Sauermann, H. Scheidl, M. Mueller, and C. Joukhadar. A bioequivalence study of two oral desmopressin tablet formulations. Pharmacology. 77:46–52 (2006) doi:10.1159/000092625.
A. S. Harris, M. Ohlin, S. Lethagen, and I. M. Nilsson. Effects of concentration and volume on nasal bioavailability and biological response to desmopressin. J. Pharm. Sci. 77:337–339 (1988) doi:10.1002/jps.2600770412.
S. Bredenberg, M. Duberg, B. Lennernas, H. Lennernas, A. Pettersson, M. Westerberg, and C. Nystrom. In vitro and in vivo evaluation of a new sublingual tablet system for rapid oromucosal absorption using fentanyl citrate as the active substance. Eur. J. Pharm. Sci. 20:327–334 (2003) doi:10.1016/j.ejps.2003.07.002.
L. Li, N. R. Mathias, C. L. Heran, P. Moench, D. A. Wall, and R. L. Smith. Carbopol-mediated paracellular transport enhancement in Calu-3 cell layers. J. Pharm. Sci. 95:326–335 (2006) doi:10.1002/jps.20541.
H. L. Luessen, B. J. de Leeuw, M. W. Langemeyer, A. B. de Boer, J. C. Verhoef, and H. E. Junginger. Mucoadhesive polymers in peroral peptide drug delivery. VI. Carbomer and chitosan improve the intestinal absorption of the peptide drug buserelin in vivo. Pharm. Res. 13:1668–1672 (1996) doi:10.1023/A:1016488623022.
L. Illum, H. Jorgensen, H. Bisgaard, O. Krogsgaard, and N. Rossing. Bioadhesive microspheres as a potential nasal drug delivery system. Int. J. Pharm. 39:189–199 (1987) doi:10.1016/0378-5173(87)90216-X.
C. Callens, and J. P. Remon. Evaluation of starch–maltodextrin–Carbopol 974 P mixtures for the nasal delivery of insulin in rabbits. J. Control Release. 66:215–220 (2000) doi:10.1016/S0168-3659(99)00271-0.
E. Björk, U. Isaksson, P. Edman, and P. Artursson. Starch microspheres induce pulsatile delivery of drugs and peptides across the epithelial barrier by reversible separation of the tight junctions. J. Drug Target. 2:501–507 (1995) doi:10.3109/10611869509015920.
T. Nagai, Y. Nishimoto, N. Nambu, Y. Suzuki, and K. Sekine. Powder dosage form of insulin for nasal administration. J. Control Release. 1:15–22 (1984) doi:10.1016/0168-3659(84)90017-8.
A. M. Dyer, M. Hinchcliffe, P. Watts, J. Castile, I. Jabbal-Gill, R. Nankervis, A. Smith, and L. Illum. Nasal delivery of insulin using novel chitosan based formulations: a comparative study in two animal models between simple chitosan formulations and chitosan nanoparticles. Pharm. Res. 19:998–1008 (2002) doi:10.1023/A:1016418523014.
N. Vivien, P. Buri, L. Balant, and S. Lacroix. Nasal absorption of metoclopramide administered to man. Eur. J. Pharm. Biopharm. 40:228–231 (1994).
R. J. Soane, M. Frier, A. C. Perkins, N. S. Jones, S. S. Davis, and L. Illum. Evaluation of the clearance characteristics of bioadhesive systems in humans. Int. J. Pharm. 178:55–65 (1999) doi:10.1016/S0378-5173(98)00367-6.
F. W. Merkus, J. C. Verhoef, N. G. Schipper, and E. Marttin. Nasal mucociliary clearance as a factor in nasal drug delivery. Adv. Drug Deliv. Rev. 29:13–38 (1998) doi:10.1016/S0169-409X(97)00059-8.
C. Callens, E. Adriaens, K. Dierckens, and J. P. Remon. Toxicological evaluation of a bioadhesive nasal powder containing a starch and Carbopol 974 P on rabbit nasal mucosa and slug mucosa. J. Control Release. 76:81–91 (2001) doi:10.1016/S0168-3659(01)00419-9.
Y. W. Chien, K. S. E. Su, and S.-F. Chang. Nasal systemic drug delivery. Marcel Dekker, New York, 1989.
R. C. Rowe, P. J. Sheskey, and S. C. Owen. Handbook of pharmaceutical excipients. 5Pharmaceutical Press, Somerset, UK, 2006, pp. 701–704.
N. Fransén, E. Björk, and K. Edsman. Changes in the mucoadhesion of powder formulations after drug application investigated with a simplified method. J. Pharm. Sci.(2008).
N. Fransén, E. Björk, and C. Nyström. Development and characterisation of interactive mixtures with a fine-particulate mucoadhesive carrier for nasal drug delivery. Eur. J. Pharm. Biopharm. 67:370–376 (2007) doi:10.1016/j.ejpb.2007.03.006.
L. Pereswetoff-Morath, and P. Edman. Dextran microspheres as a potential nasal drug delivery system for insulin—in vitro and in vivo properties. Int. J. Pharm. 124:37–44 (1995) doi:10.1016/0378-5173(95)00070-Y.
J. A. Hersey. Ordered mixing: a new concept in powder mixing practice. Powder Technol. 11:41–44 (1975) doi:10.1016/0032-5910(75)80021-0.
S. Lundin, P. Melin, and H. Vilhardt. Plasma concentrations of 1-deamino-8-D-arginine vasopressin after intragastric administration in the rat. Acta Endocrinol. (Copenh). 108:179–183 (1985).
R. C. Lewontin. On the measurement of relative variability. Syst. Zool. 15:141–142 (1966) doi:10.2307/2411632.
M. Kohler, and A. Harris. Pharmacokinetics and haematological effects of desmopressin. Eur. J. Clin. Pharmacol. 35:281–285 (1988) doi:10.1007/BF00558266.
A. Fjellestad-Paulsen, L. d'Agay-Abensour, P. Hoglund, and J. C. Rambaud. Bioavailability of 1-deamino-8-D-arginine vasopressin with an enzyme inhibitor (aprotinin) from the small intestine in healthy volunteers. Eur. J. Clin. Pharmacol. 50:491–495 (1996) doi:10.1007/s002280050146.
H. Vilhardt, and S. Lundin. Biological effect and plasma concentrations of DDAVP after intranasal and peroral administration to humans. Gen. Pharmacol. 17:481–483 (1986) doi:10.1016/0306-3623(86)90198-9.
A. S. Harris, M. Ohlin, E. Svensson, S. Lethagen, and I. M. Nilsson. Effect of viscosity on the pharmacokinetics and biological response to intranasal desmopressin. J. Pharm. Sci. 78:470–471 (1989) doi:10.1002/jps.2600780610.
A. S. Harris, E. Svensson, Z. G. Wagner, S. Lethagen, and I. M. Nilsson. Effect of viscosity on particle size, deposition, and clearance of nasal delivery systems containing desmopressin. J. Pharm. Sci. 77:405–408 (1988) doi:10.1002/jps.2600770510.
D. Teshima, A. Yamauchi, K. Makino, Y. Kataoka, Y. Arita, H. Nawata, and R. Oishi. Nasal glucagon delivery using microcrystalline cellulose in healthy volunteers. Int. J. Pharm. 233:61–66 (2002) doi:10.1016/S0378-5173(01)00930-9.
R. Ryan, A. Elkind, C. C. Baker, W. Mullican, S. DeBussey, and M. Asgharnejad. Sumatriptan nasal spray for the acute treatment of migraine. Results of two clinical studies. Neurology. 49:1225–1230 (1997).
I. M. Steiner, S. T. Kaehler, R. Sauermann, H. Rinosl, M. Muller, and C. Joukhadar. Plasma pharmacokinetics of desmopressin following sublingual administration: an exploratory dose-escalation study in healthy male volunteers. Int. J. Clin. Pharmacol. Ther. 44:172–179 (2006).
O. Osterberg, R. M. Savic, M. O. Karlsson, U. S. Simonsson, J. P. Norgaard, J. V. Walle, and H. Agerso. Pharmacokinetics of desmopressin administrated as an oral lyophilisate dosage form in children with primary nocturnal enuresis and healthy adults. J. Clin. Pharmacol. 46:1204–1211 (2006) doi:10.1177/0091270006291838.
Acknowledgements
Orexo AB is gratefully acknowledged for financial support. The devoted work of the whole project team is thankfully recognised.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fransén, N., Bredenberg, S. & Björk, E. Clinical Study Shows Improved Absorption of Desmopressin with Novel Formulation. Pharm Res 26, 1618–1625 (2009). https://doi.org/10.1007/s11095-009-9871-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-009-9871-9