
Abstract
Primary systemic vasculitides of adolescents and young adults AYA are relatively rare diseases, but may be associated with significant morbidity and mortality. We provide an overview of systemic vasculitides and emphasise the problems to be anticipated in adolescence and young adulthood.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Key Management Points-
1.
Systemic vasculitides are a group of disorders, characterized by inflammation of vessel walls.
-
2.
Some type of vasculitides are more common or more severe in adolescence and young adulthood.
-
3.
For professionals caring for AYA with vasculitis, stage and developmental considerations of adolescence and young adulthood must be considered.
-
4.
Effective transition programs are needed for AYA with vasculitides for a better transfer of AYAs to adult clinics.
Introduction
Systemic vasculitides are a group of disorders, characterized by inflammation of vessel walls. The estimated incidence of pediatric vasculitis is approximately 50 cases per 100,000 children per year [1]. However, the incidence and prevalence of vasculitis among adolescents are unknown.
Adolescence is a unique period of life (see Chaps. 1, 2 and 3) and poor health status during adolescence may lead to lifelong persistent effects [2]. Since vasculitides have chronic and devastating features, it is important to be aware of the problems to be anticipated at adolescence and take a multidisciplinary approach with an AYA perspective in caring for AYA with vasculitis.
Definitions and Classifications
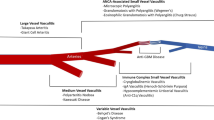
The vasculitides are mainly classified according to predominant size of involved vessels. In 2012, the International Chapel Hill Consensus Conference (CHCC 2012) updated the definition of vasculitides (Fig. 10.1) [3]. Pediatricians have established the Ankara classification criteria which includes criteria for the main vasculitides of childhood and was endorsed by EULAR/PReS/PRINTO [4, 5] (Table 10.1).

Chapel Hill Consensus Conferences 2012 classification criteria for systemic vasculitis [3]
Clinical and Laboratory Findings of Systemic Vasculitides
Clinical features of the systemic vasculitides may vary according to predominant size of involved vessels. Constitutional symptoms such as malaise, fever, weight loss, myalgia, and arthralgia may accompany their course.
There is no diagnostic laboratory test for vasculitides except for Antineutrophil cytoplasmic antibody (ANCA – see below). Elevated acute phase reactants such as C-reactive protein (CRP) , erythrocyte sedimentation rate (ESR) are common. Urinalysis and renal function tests should be performed to monitor renal involvement routinely. Renal and liver function tests, echocardiogram, slit lamp examination of eyes and appropriate evaluation of the lung should also be a part of the work-up in the assessment of vasculitis (Fig. 10.2).

Clinical and laboratory finds that suggest a systemic vasculitis
The main features and treatment approaches for vasculitides will be summarized, emphasizing issues related to adolescence and young adulthood.
Predominantly Small Vessel Disease
Immunoglobulin A Vasculitis (IgAV)/Henoch-Schönlein Purpura (HSP)
Immunoglobulin A vasculitis (IgAV)/Henoch-Schönlein purpura (HSP) is the one of the most common vasculitides during childhood with an annual incidence of 13–20/100,000 [1]. It occurs most frequently between the age of 3 and 15 with a male predominance [1].
In adolescent male patients the gastrointestinal involvement is more severe; furthermore previous studies have demonstrated that older children have a more severe course with worse renal outcomes and relapsing course than younger children [1].
There is no specific test for IgAV/HSP. Urinalysis should be performed to monitor renal involvement routinely for 1 year [1]. Ultrasound studies may help to detect specific gastrointestinal abnormalities in children with abdominal complaints such as dilated loops of bowel or intussusception.
The main therapy of IgAV/HSP depends on a conservative approach pain relief and hydration. Corticosteroids are used for severe GI involvement. Routine steroid treatment does not protect from renal involvement. Whilst we lack high evidence data for the best management of renal involvement, corticosteroids combined with other immunosuppressive agents are often used [1].
Antineutrophil Cytoplasmic Antibody (ANCA) Associated Vasculitis (AAA)
Antineutrophil cytoplasmic antibody (ANCA) associated vasculitis (AAV) is a group of vasculitides, including granulomatosis with polyangiitis (GPA) (formerly known as Wegeners Granulomatosis), eosinophilic granulomatosis with polyangiitis (EGPA) (previously known as Churg-Strauss syndrome), microscopic polyangiitis (MPA ), and single organ disease (renal-limited vasculitis) . AAV mainly is characterized with predominantly small size vessels inflammation, and commonly the presence of ANCA [3].
Granulomatosis with polyangiitis (GPA) presents mainly with granulomatous inflammation of upper and lower respiratory tracts, necrotizing, pauci-immune glomerulonephritis, but may also present with multi-systemic involvements. In the pediatric population, GPA occurs around adolescence, with a median disease onset is 11.6 years old in pediatric age and an estimated incidence of 0.1 per 100,000 [6].
Meta-analysis data shows [7], about 69% of pediatric GPA patients were anti-PR3 ANCA (c-ANCA) positive, while presence of anti-MPO ANCA (p-ANCA) was approximately 21% among the pediatric GPA patients [7]. The characteristic histopathological findings are granulomatous inflammation of predominantly small vessels or pauci-immune glomerulonephritis [7]. Chest X-rays may show pulmonary nodules, infiltrations, cavitations, pleural effusions. High resonance computed tomography is more sensitive to detect pulmonary abnormalities, and magnetic resonance imaging is highly helpful to determine ENT involvement.
Microscopic polyangiitis (MPA) has limited epidemiological data in childhood and adolescent. A meta-analysis in adults showed that 94% had renal involvement, 79% had systemic symptoms, 57% musculoskeletal involvement, 44% cutaneous findings, 37% pulmonary involvement (pulmonary hemorrhage), and 28% GI involvement [7]. Almost all of these adult patients were anti-MPO ANCA (93%) positive whereas only 5% patients were anti-PR3 ANCA positive [7].
Eosinophilic granulomatosis with polyangiitis (EGPA) formally known as Churg-Strauss syndrome , is a rare necrotizing vasculitis, characterized by severe asthma or allergic rhinitis, along with skin, cardiovascular, renal, nervous system, GI system involvements. EGPA is a very rare disease in children. There are no validated, standard classification criteria for pediatric EGPA patients. According to the American College of Rheumatology (ACR) criteria for classification of EGPA, patients can be classified as EGPA in presence of at least four of the following criteria: (1) asthma, (2) eosinophilia (eosinophil counts >10% of differential white blood cell count), (3) history of allergy, (4) mononeuropathy or polyneuropathy, (5) pulmonary infiltrates, (6) paranasal sinus abnormality, (7) extravascular eosinophilia [8].
Elevation of serum immunoglobulin E and peripheral blood eosinophilia are common. Approximately 25% of pediatric EGPA patients are MPO-ANCA positive [9]. The main findings in pathology specimens of EGPA patients are angiitis and extravascular necrotizing granulomas accompanied by eosinophilic infiltration.
The treatment of AAV includes remission induction and maintenance therapy. In case of new onset organ-threating or life-threating AAV, treatment with a combination of glucocorticoids and either cyclophosphamide or rituximab is recommended for the remission-induction. However, in patients with non-organ threating AAV, combination of glucocorticoids and either methotrexate or mycophenolate mofetil may be preferred for remission-induction [10]. Combination of low-dose glucocorticoids and either azathioprine, rituximab, methotrexate or mycophenolate mofetil are used as maintenance treatments [10].
Predominantly Medium Vessel Disease
Polyarteritis Nodosa (PAN)
Polyarteritis nodosa (PAN) is a primary systemic necrotizing vasculitis, characterized by inflammatory lesions of medium size vessels [11]. It is estimated to be the third most common vasculitis among the children [12].
According to largest multicenter study including 110 pediatric PAN patients (63 with systemic PAN), 74.5% had cutaneous lesions, 17.3% had GI involvement, 14.5% had nervous system symptoms, 11.8% had renal involvement, and 5.5% had cardiac findings [13]. Laboratory work-up usually reveals leukocytosis, thrombocytosis and elevated acute phase reactants. ANCA is negative. The typical histopathological finding in PAN is fibrinoid necrosis of the predominantly medium size arteries walls with an inflammatory response surrounding the vessel wall [11]. Characteristic features in angiography are aneurysms in renal, celiac, mesenteric, or other arteries. Although conventional angiography is accepted as a gold standard, computerized tomography angiography has emerged an alternative, non-invasive technique to detect vasculitic lesions [14].
Classic treatment of PAN is glucocorticoids and cyclophosphamide as remission induction and either azathioprine or methotrexate combination with low-dose steroid as maintenance treatment [15].
Kawasaki Disease (KD)
Kawasaki disease is an acute vasculitis of childhood, resulting in coronary artery aneurysms in around 25% of the untreated cases. It is predominantly a disease of young children and is very rare in adolescence, although it may have important cardiovascular sequelae for AYA (see below). A patient may be classified as having KD when they have fever for more than 5 days plus at least four following clinical findings: (1) bilateral conjunctival injection, (2) changes in oropharyngeal mucous (fissured lips, strawberry tongue, injected pharynx), (3) changes in peripheral extremities (erythema, edema of hands and feet, desquamation), (4) polymorphous rash, (5) cervical lymphadenopathy. A single administration of 2 g/kg intravenous immunoglobulin (IVIG) plus acetylsalicylic acid (ASA) (with a total daily dose of 80–100 mg/kg/day in the United States and 30–50 mg/kg/day in Japan and Western Europe) is the still standard therapy of KD [16]. In the presence of coronary abnormalities, low dose ASA should be continued indefinitely [16]. In IVIG resistance cases, high-dose pulse steroids, infliximab, cyclosporine, and anakinra are indicated [16].
Predominantly Large Vessel Disease
Takayasu Arteritis (TA)
Takayasu arteritis (TA) is a chronic, granulomatous vasculitis of large vessels, involving aorta and its major branches. TA is rare in children and the incidence is unknown. TA is mostly seen among adolescents with a median age of diagnosis of 11.8 years among children [17].
The most common findings at diagnosis are arterial hypertension (82%), headache (31%), fever (29%), dyspnea (23%) and weight loss. Conventional angiography, MR angiography and CT angiography are used for diagnosis and assessing the extent of involvement.
Steroids are still the mainstay of therapy in TA. Immunosuppressive agents or biologics may be administered for remission induction and maintenance treatment may continue with lower dose of steroids and methotrexate [17, 18]. Surgical procedures may be required for treatment due to the destructive effect of the disease on the large vessels [19].
Variable Vessel Disease
Behçet’s Disease (BD)
Behçet’s disease (BD) was first described as a clinical triad of aphthous stomatitis, genital ulceration, and uveitis although it is now known that BD is a variable vessel vasculitis, characterized by multi-system involvement. Roughly, 5–8% of BD patients have disease onset in childhood and the usual presentation is in adolescence [20, 21]. An international expert consensus group (the pediatric BD [PEDBD] group) has established a new set of classification criteria for pediatric Behçet’s Disease (BD) [22]. By the PEDBD criteria patients are classified as having BD when they have three or more of the following criteria [22]:
-
1.
oral aphthosis (≥3 attacks per year)
-
2.
genital aphthosis (typical with scars)
-
3.
skin involvement (necrotic folliculitis, acneiform lesions, erythema nodosum)
-
4.
neurologic involvement (except isolated headaches)
-
5.
ocular manifestations (anterior uveitis, posterior uveitis, retinal vasculitis)
-
6.
vascular signs (venous thrombosis, arterial thrombosis, arterial aneurysms).
BD may affect all vessel sizes and involves both arteries and venules. Central nervous system, musculoskeletal, gastrointestinal, cardiac, pulmonary involvements, orchitis, renal vasculitis, and glomerulonephritis may accompany the disease [20, 23]. Angiography and magnetic resonance imaging are used to assess vascular and central nervous system involvement [20, 23].
Oral aphthosis is common in young people, the majority of whom who will not have BD. Furthermore, sexually transmitted infections should also be considered in the assessment of genital aphthosis in this age group (see below).
The treatment of BD usually depends on the site and severity of involvement. Mild oral or genital ulcers are usually treated with topical therapies (corticosteroid- or lidocaine-containing creams, sucralfate suspension) and colchicine. Azathioprine, interferon alfa, and TNF-α antagonists are recommended for refractory cases. Vascular involvement is a serious complication in BD. Corticosteroids and azathioprine , treatments are recommended for acute deep vein thrombosis, while cyclophosphamide and corticosteroids are recommended for pulmonary and peripheral artery aneurysm. Corticosteroids, IFN-alpha, azathioprine, cyclophosphamide, methotrexate and anti-tumor necrosis factor-alpha may be used for central nervous system involvement [24].
Differential Diagnosis of Vasculitis in Adolescence and Young Adulthood
Treatment and Follow-up of AYA with Systemic Vasculitis
Treatment goals are both to control disease, but also help AYA with vasculitis to achieve a good quality of life. Thus, patients should be evaluated not only for disease activity but also for the burden of the disease (see Chaps. 2 and 3) and specific treatment side effects. Success in treatment mainly depends on the adherence to therapies which for AYA needs a multidisciplinary approach, including the psychologist (see Chap. 19).
Glucocorticoids remain the main treatment for remission induction and maintenance in systemic vasculitis. Patients, treated with long term corticosteroids, still are at risk of serious side effects such as Cushing syndrome , growth suppression, osteoporosis (see Chap. 16), metabolic effects, immunosuppression, glaucoma, hypertension, and mood disorders.
Gonadal toxicity is a common long-term consequence of cyclophosphamide. The risk of gonadal toxicity is increased after puberty. Administration of intravenous bolus cyclophosphamide has lower total cumulative dose than daily oral administration [26]. Luteinizing hormone–releasing hormones may protect the ovaries against cyclophosphamide-induced damage [27].
Kawasaki disease (KD) is the most common cause of acquired heart disease during childhood in developed countries. Furthermore, KD is becoming an important cause of myocardial ischemia in young adults. Patients should be followed by cardiologists routinely [16]. Apart from KD the association between CAD and vasculitis is scare in childhood, although adult studies have demonstrated an increased risk of coronary artery disease (CAD) among the patients with rheumatic disease.
Transition to the adult clinics is another sensitive issue to be dealt with in this period (see Chap. 21) and is often more complex for AYA with vasculitis in view of the number of different disciplines involved.
According to the World Health Organization (WHO) about 16 million girls aged 15–19 give birth every year, mostly those living in low- and middle-income countries. Chronic health problems such as vasculitis may further threaten the health of the baby and the mother. From adult studies, it is well known that pregnancy complications, including pregnancy loss and preterm birth, are higher among women with vasculitis [28]. For those conditions with a thrombotic tendency eg BD, oestrogen containing contraceptives are contraindicated. Promotion of cardiovascular health is similarly important with advice and support regarding the importance of physical activity, weight management and avoidance or nicotine and/or other drug use is integral to the management of such young people.
Summary
In conclusion, certain vasculitides are more common or more severe in adolescence and young adulthood. The burden of the disease and side effects of treatment may have a more pronounced effect for AYA. An effective, multidisciplinary and interdisciplinary transition program is required for an effective and holistic transfer of adolescents to adult clinics.
References
Eleftheriou D, Batu ED, Ozen S, Brogan PA. Vasculitis in children. Nephrol Dial Transpl Off Publ Eur Dial Transpl Assoc Eur Renal Assoc. 2015;30(Suppl 1):i94–103.
Donaldson L, Banatvala N. Health is global: proposals for a UK Government-wide strategy. Lancet. 2007;369(9564):857–61.
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65(1):1–11.
Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, et al. EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis. 2010;69(5):798–806.
Ruperto N, Ozen S, Pistorio A, Dolezalova P, Brogan P, Cabral DA, et al. EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part I: overall methodology and clinical characterisation. Ann Rheum Dis. 2010;69(5):790–7.
Bohm M, Gonzalez Fernandez MI, Ozen S, Pistorio A, Dolezalova P, Brogan P, et al. Clinical features of childhood granulomatosis with polyangiitis (wegener’s granulomatosis). Pediatr Rheumatol Online J. 2014;12:18.
Iudici M, Quartier P, Terrier B, Mouthon L, Guillevin L, Puechal X. Childhood-onset granulomatosis with polyangiitis and microscopic polyangiitis: systematic review and meta-analysis. Orphanet J Rare Dis. 2016;11(1):141.
Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990;33(8):1094–100.
Zwerina J, Eger G, Englbrecht M, Manger B, Schett G. Churg-Strauss syndrome in childhood: a systematic literature review and clinical comparison with adult patients. Semin Arthritis Rheum. 2009;39(2):108–15.
Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, Hauser T, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann Rheum Dis. 2016;75(9):1583–94.
Eleftheriou D, Dillon MJ, Tullus K, Marks SD, Pilkington CA, Roebuck DJ, et al. Systemic polyarteritis nodosa in the young: a single-center experience over thirty-two years. Arthritis Rheum. 2013;65(9):2476–85.
Ozen S, Bakkaloglu A, Dusunsel R, Soylemezoglu O, Ozaltin F, Poyrazoglu H, et al. Childhood vasculitides in Turkey: a nationwide survey. Clin Rheumatol. 2007;26(2):196–200.
Ozen S, Anton J, Arisoy N, Bakkaloglu A, Besbas N, Brogan P, et al. Juvenile polyarteritis: results of a multicenter survey of 110 children. J Pediatr. 2004;145(4):517–22.
Ozcakar ZB, Yalcinkaya F, Fitoz S, Yuksel S, Acar B, Caltik A, et al. Polyarteritis nodosa: successful diagnostic imaging utilizing pulsed and color Doppler ultrasonography and computed tomography angiography. Eur J Pediatr. 2006;165(2):120–3.
Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009;68(3):310–7.
McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term Management of Kawasaki Disease: a scientific statement for health professionals from the American Heart Association. Circulation. 2017;135(17):e927–e99.
Eleftheriou D, Varnier G, Dolezalova P, McMahon AM, Al-Obaidi M, Brogan PA. Takayasu arteritis in childhood: retrospective experience from a tertiary referral Centre in the United Kingdom. Arthritis Res Ther. 2015;17:36.
Ozen S, Duzova A, Bakkaloglu A, Bilginer Y, Cil BE, Demircin M, et al. Takayasu arteritis in children: preliminary experience with cyclophosphamide induction and corticosteroids followed by methotrexate. J Pediatr. 2007;150(1):72–6.
Keser G, Aksu K. What is new in management of Takayasu arteritis? Presse Med. 2017;46(7–8 Pt 2):e229–e35.
Sarica R, Azizlerli G, Kose A, Disci R, Ovul C, Kural Z. Juvenile Behcet’s disease among 1784 Turkish Behcet’s patients. Int J Dermatol. 1996;35(2):109–11.
Zouboulis CC, Kotter I, Djawari D, Kirch W, Kohl PK, Ochsendorf FR, et al. Epidemiological features of Adamantiades-Behcet’s disease in Germany and in Europe. Yonsei Med J. 1997;38(6):411–22.
Kone-Paut I, Shahram F, Darce-Bello M, Cantarini L, Cimaz R, Gattorno M, et al. Consensus classification criteria for paediatric Behcet’s disease from a prospective observational cohort: PEDBD. Ann Rheum Dis. 2016;75(6):958–64.
Ozen S, Eroglu FK. Pediatric-onset Behcet disease. Curr Opin Rheumatol. 2013;25(5):636–42.
Hatemi G, Silman A, Bang D, Bodaghi B, Chamberlain AM, Gul A, et al. EULAR recommendations for the management of Behcet disease. Ann Rheum Dis. 2008;67(12):1656–62.
Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International league of associations for rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31(2):390–2.
Miller JJ 3rd, Williams GF, Leissring JC. Multiple late complications of therapy with cyclophosphamide, including ovarian destruction. Am J Med. 1971;50(4):530–5.
Clowse ME, Behera MA, Anders CK, Copland S, Coffman CJ, Leppert PC, et al. Ovarian preservation by GnRH agonists during chemotherapy: a meta-analysis. J Women’s Health. 2009;18(3):311–9.
Machen L, Clowse ME. Vasculitis and pregnancy. Rheum Dis Clin N Am. 2017;43(2):239–47.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sönmez, H.E., Bilginer, Y., Özen, S. (2019). Systemic Vasculitis in Adolescence and Young Adulthood. In: McDonagh, J., Tattersall, R. (eds) Adolescent and Young Adult Rheumatology In Clinical Practice. In Clinical Practice. Springer, Cham. https://doi.org/10.1007/978-3-319-95519-3_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-95519-3_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95518-6
Online ISBN: 978-3-319-95519-3
eBook Packages: MedicineMedicine (R0)