
Abstract
Purpose of the Review
Most of the primary vasculitis in children and adults has different clinical manifestations for the same disease, which suggests that they might not be part of the same clinical spectrum and requires a different approach in order to reduce the morbidity and mortality of these patients. In this work, we review the most recent literature and the most important studies that describe and compare adult and children primary vasculitides pathogenesis, clinical presentation, and treatment approach. Accordingly, we discuss recent research involving clinical trials, comparison studies, and pathogeny for these vasculitides.
Recent Findings
Clinical manifestations in the different primary vasculitis change in predominance from adults to children. There is a female sex predominance for the ANCA vasculitides in children compared with adults, but the same treatment works in most cases for both groups.
Summary
Identifying the diverse clinical spectrum in both adults and children primary vasculitides will reduce the need to extrapolate the diagnostic criteria from one group to another and individualize it, which will allow the clinician to establish a better approach.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vasculitides are a rare group of disorders characterized by inflammation of the blood vessels. The classification of vasculitis depends on the size of the vessel involved which will give diverse clinical features. Depending on the pathogeny, they could be described as primary vasculitides and secondary vasculitides [1, 2].
Although most forms of primary vasculitis are more common in adults than in children, some types of vasculitis such as Henoch-Schonlein purpura (HSP)/immunoglobulin A vasculitis (IgAV) and Kawasaki disease (KD) tend to occur predominantly in childhood [1].
During the last few years, rheumatologists and pediatricians have tried to standardize the management of some pediatric primary vasculitides by classifying and treating them the same way as in adults [3]. However, only the classification criteria for the most common vasculitides in childhood such as IgAV/HSP, KD, polyarteritis nodosa (PAN), granulomatous polyangiitis/Wegener granulomatosis (GPA/WG), and Takayasu arteritis (TA) have been developed and validated [4, 5].
Some of the pediatric vasculitis could behave clinically more aggressive than in the adult population, requiring a different approach in order to reduce the morbidity and mortality of these patients.
In this work, we review the current literature describing and comparing adult and children primary vasculitides pathogenesis, clinical presentation, and treatment approach. Accordingly, we discuss recent research involving clinical trials, comparison studies, and pathogeny for these vasculitides. The most relevant differences are described in Table 1.
Ultimately, similarities in children and adults vasculitides may define more precise diagnosis criteria and a better management, as well as differences that may represent an opportunity to improve classification criteria.
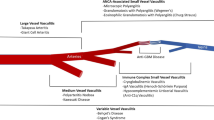
Large Vessel Vasculitis
This type of vasculitis causes chronic granulomatous inflammation predominantly of the aorta and its major branches.
Giant Cell Arteritis
Although giant cell arteritis (GCA) is exclusive in adults over 50 years, a similar pathology with a name that may cause confusion is worth mentioning: juvenile temporal arteritis (JTA), a rare and little-known local inflammatory disease of the temporal arteries that rarely occurs in young adults and children [6].
Patients may have a tender or painful palpable lump in the forehead area of the temporal artery; however, unlike GCA, the inflammation of the temporal arteries may be due to trauma. Another major difference is that GCA is a systemic disease that also involves high inflammatory markers and response to steroids [6].
Takayasu Arteritis
Takayasu Arteritis is characterized by an idiopathic chronic granulomatous vasculitis of the aorta and/or its main branches. Sometimes it can affect also coronary and pulmonary arteries.
TA usually manifests in women at their third or fourth decade, but children and adolescents can also develop the disease, with an incidence in all ages estimated at 2.6/100,000. The exact incidence and prevalence of TA for children only are not known, and girls and young women are more frequently affected when compared with boys [4, 7, 8••].
First described in 1830 and later described with its unique clinical features in 1951. It was not until 2008, when the European League Against Rheumatology (EULAR)/Pediatric Rheumatology European Society (PReS) working group developed classification criteria for it [9•, 10].
The molecular pathophysiology of TA remains unclear: genetic factors, humoral, autoimmunological factors, as well as infections have been discussed as contributors. It has been accepted that the inflammatory process begins at the vasa vasorum where T-cells and macrophages invade the outer layer of the media, progressing from the adventitial side to the intimal side of the vessel generating fibrosis, resulting in thickening of all three layers. The chronic process causes vessel stenosis, occlusion, and aneurysmal formation [4, 7, 10].
Regarding the clinical differences between adults and children, hypertension is the most common symptom in both groups. In children, hypertension could be the only symptom, but the absence of peripheral pulses can be commonly seen. Other symptoms in children are headaches, fever, dyspnea, weight loss, vomiting, and arthralgia-myalgia [4, 7, 11••].
In contrast, adults more commonly present vascular involvement of the arteries, bruits, claudication, and decreased pulses most commonly of the upper extremities, as well as arthritis/arthralgia [12].
Treatment regimens for initial induction of remission were different between children and adults: while adults were most often induced with systemic glucocorticoids alone, children received a combination of systemic glucocorticoids with Methotrexate or with Cyclophosphamide. In addition, anti-TNF therapy at follow-up was more commonly used in children than adults [12,13,14].
Medium Vessel Vasculitis
This type of vasculitis affects medium and small-sized vessels of all organs but do not involve capillaries, venules, or arterioles.
Kawasaki Disease
KD also called mucocutaneous lymph node syndrome, is an acute, self-limited, medium vessel-size vasculitis that predominantly affects children under 5 years of age and rarely occurs in adults [14].
The incidence of KD is as high as 240 cases per 100,000 in children of Japanese origin, while the incidence is around 9 to 17 cases per 100,000 in children from other ethnicities with a tendency to increase [15, 16]. It is considered the second most common vasculitis in children and the leading cause of acquired heart disease in childhood [17, 18]. Data in adults is limited since it is often misdiagnosed, and the differential diagnosis is broad: drug hypersensitivity reactions, toxic shock syndrome, erythema multiforme, scarlet fever, measles, rubella, parvovirus, infectious mononucleosis, leptospirosis, rocky mountain, syphilis, endocarditis, rheumatic fever, palmoplantar psoriasis, Behçet disease, and polyarteritis nodosa [19•].
This disease was first described in Japan in 1967, but it occurs in children of all races and rarely in adults [15, 20]. A genetic susceptibility has been identified by genome-wide association study (GWAS): the ITPKC gene implicated in negative regulation of T-cells via and the FcRg2a gene associated with susceptibility to KD and is also linked to IVIg response. This GWAS could explain why adults develop Kawasaki disease, but to date, a satisfactory explanation of the pathophysiology remains unclear [15, 17].
The clinical differences between adults and children are not remarkable. In general, any patient is classified as having KD if she/he has fever persisting for at least 5 days (mandatory criterion) and four of the following five criteria: changes in the peripheral extremities and perianal area, polymorphous exanthema, bilateral conjunctival injection, changes of lips, and cardiac abnormalities. In incomplete cases, most of them were diagnosed by the presence of coronary vasculitis [20].
In the case of the adults, it was reported by Fraison JB et al. [19•] that the main symptoms were fever, exanthema, changes in the extremities, conjunctivitis (77%), oral cavity changes (89%), cervical adenitis (55%), and cardiac abnormalities (45%). Overall, 35% of patients showed coronary vasculitis (26%) and coronary aneurysm (19%). In addition, there was a marked increase in acute-phase reactants.
Given the rarity of adult patients with KD, treatment basically remains the same for children and adults with a focus for cardiovascular complications in the long term [19•, 21•]. As awareness of this diagnosis increases, more data and more genetic studies will be made and will give more clues as to whether the adult and children Kawasaki disease are part of the same clinical spectrum.
Polyarteritis Nodosa
PAN is a necrotizing arteritis that affects medium- and small-sized vessels of all organs except the lungs [22]. It does not involve capillaries, venules, or arterioles, and patients have a negative result for antineutrophil cytoplasmic antibodies (ANCA) [23].
The incidence of PAN can vary from 2 to 9/million in adults, but epidemiological studies in childhood are scarce since this disease is less frequent in children [23]. The age at disease onset is between 25 and 50 years in adults while it is around 9–10 years of age in children [23, 24]. It is interesting to note that this data should be reviewed since PAN incidence has decreased due to:
A better distinction between similar diseases like Microscopic Polyangiitis, adenosine deaminase-2(ADA2) deficiency and infectious diseases that may cause identical symptoms.
Increase in hygiene measures, increase in blood-transfusion safety and anti-HBV vaccination (especially in children) have markedly decreased the PAN incidence specially in developed countries.
First described by Küssmaul and Maier in 1866, PAN has changed its diagnostic criteria, with the last update in 2012 by the Chapel Hill consensus [2]. The majority of PAN cases reflect a variety of phenotypes and a lack of a clear systemic subclassification. Currently, PAN could be divided in the following: classic PAN, cutaneous PAN, MPA, HBV-related PAN, and ADA2 deficiency [25].
For this part, we will focus only in the classical PAN. Although, it is worth mentioning that patients with cutaneous PAN, which is mostly seen by dermatologists, can also have systemic complaints and occasionally elevated levels of acute-phase reactants, making obligatory a complete assessment to rule out classic PAN.
Regarding clinical characteristics in adults, in one of the biggest series of patients with PAN from the French Vasculitis Study Group Database [26], the characteristics of 348 adult PAN patients (225 PAN and 123 HBV-PAN) was reviewed. In this study, the most frequent findings were constitutional symptoms such as fever, fatigue, and weight loss (93.1%), neurologic findings (79%), urologic and renal manifestations (50.6%), skin involvement (49.7%), and gastrointestinal symptoms (37.9%). In another major study from Turkey [23], among 133 adult PAN patients (108 PAN, 13 HBV-PAN, 12 not known), the most frequent findings were skin involvement (81.2%), constitutional symptoms (64.7%), neurological involvement (32.3%), and GI involvement (24.1%).
In the case of children, in the largest multicenter study [27] including 110 children with PAN, the most common symptom was constitutional features (86.4%), followed by skin involvement in 74.5%, myalgia in 33.6%, and gastrointestinal symptoms in 17.3%.
There are not significant clinical differences between children and adults; childhood-onset PAN has a slightly higher proportion of patients who are female compared with adult PAN. Another important difference is that children with PAN have better prognosis [28, 29].
Interestingly, PAN could be seen as part of the spectrum of KD (or vice-versa). PAN with aneurysmal involvement of major coronary arteries, and KD are clinically and pathologically indistinguishable [22, 30]. PAN is usually diagnosed when other organs, rarely affected by Kawasaki disease, are involved. In these cases, the major distinction between KD and PAN is that the diagnosis of KD is based entirely on clinical criteria, while the diagnosis of PAN is based on histologic findings [31].
Because of the rarity of childhood PAN, widely accepted and evidence-based treatment recommendations do not exist. Treatment is usually the same for adults and children, and includes Corticosteroids, Intravenous Immunoglobulin, or Cyclophosphamide and for maintenance, DMARDS. Because PAN does not often recur, its treatment duration may be shorter than for other systemic necrotizing vasculitides [22, 32, 33].
Small Cell Vasculitides
They can be divided in anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitides and immune complex vasculitides [34, 35]. Despite the existence of classification criteria for each ANCA-associated vasculitides, some of them cannot be classified, which will be briefly described in a later paragraph.
Henoch-Schonlein Purpura/IgA Vasculitis
HSP/IgAV is the most common vasculitis in children as well as the most studied. The incidence of IgA vasculitis has been reported for several areas over the past 25 years ranging from 6 to 26.7/100000 for children and 1 to 5/100000 in adults, with a medium age of 66 years and 6.5 years of age in adults and children respectively [36,37,38,39,40].
First described in 1837 by Dr. Johann Lukas Schonlein, it was reported as a purpura rheumatica in children [41]. More than a century later, the American College of Rheumatology (ACR) and Michel’s criteria in 1990 and 1992 respectively; specify that the diagnosis should be in patients under 20 years of age at disease onset, and no specific histopathology marker was proposed [41].
In 2008, the EULAR/PRINTO/PRES proposed other criteria for diagnosing HSP in children which had more sensitivity than the ACR [42]. However, this disease can rarely occur in adults in whom it is believed to be a more severe form with poor renal outcomes [39, 43]. Because of that, these criteria have been used since 2010 for adults as well, since they do not include the age criteria and have higher sensitivity than the one from the ACR [40].
In 2012, it was renamed as IgA vasculitis because of the presence of IgA1-dominant immune deposits that affect small vessels [2]. Because these IgA1 deposits are aberrantly glycosylated, they generate IgG autoantibodies and increase tissue levels of inflammatory cytokines mainly in dermal, gastrointestinal, and glomerular capillaries [3, 44]. However, the stimulus and primary site for production of the aberrant IgA in IgAV is unknown, but polymeric IgA1 dominates immune complexes, supporting a mucosal origin [5].
There is no clear reason why it affects mainly children, but it is hypothesized that an unknown infection and genetic susceptibility trigger the disease at different ages [36, 44, 45]. Nonetheless, underlying malignancy should be considered in adult patients with IgAV [5]. Although there are no any big current studies, besides report cases and abstracts, that show that there is no association, it is wise to look for malignancy in adults that develop IgA vasculitis [46, 47].
As far as the knowledge we have regarding pathogeny and genetics in this disease, we could say they are part of the same clinical spectrum. However, the clinical manifestations vary in children and in adults. In adulthood, IgAV tends to have a more severe course with purpura as the first clinical manifestation as well as joint involvement. Children more frequently present/show abdominal pain before the appearance of purpuric rash. However, there are no prominent differences regarding laboratory findings [3]. Renal involvement severity between adults and children varies from study to study [48, 49].
For treatment, there was no difference in the frequency of immunosuppressive treatment required by the two groups [48,49,50].
Granulomatosis with Polyangiitis
Previously known as Wegener’s granulomatosis is a necrotizing granulomatous inflammation which involves mainly the upper and lower respiratory tract and necrotizing vasculitis which predominantly affects small to medium-sized vessels [51].
The incidence of GPA in adults ranges from 0.2 to 1.2 per 100,000 persons per year with a median age of approximately 50 years GPA [53]. GPA is described as occurring at higher incidences in higher latitudes such as Norway and areas of the UK [34]. This disease is rare in children, its incidence is estimated to be around 1/1000,000 and the mean age at disease-onset is around 14 years of age [35, 52]. It is interesting to note that there is female predominance in GPA in children, but there is no gender difference in adults. Black and Hispanic children represent less than 5% of reported cases of ANCA vasculitides [51]. Because of disease rarity in children, most data on GPA is derived from the adult literature [34].
It is difficult to assess how old this disease is, because previously it was classified as part of PAN, but it was not until 1930s, when Wegener established the disease in adults in a short paper entitled “On Generalized Septic Vessel Diseases,” where he identified the major characteristics of the disease as a septic course, with extremely severe necrotizing granulomatous inflammation of the inner nose, pharynx, and larynx; localized glomerulonephritis; and generalized arteritis [53].
The cause of GPA is unknown. Causes like viral infections, exposure to silica, some medication, or nasal carriage of Staphylococcus aureus have been proposed [34], but like other polygenic systemic autoimmune diseases, it is likely a result of interactions between genetic factors, epigenetic factors, and environmental factors that predispose subjects to loss of self-tolerance [34, 51].
The association with autoantibodies against the cytoplasmic region of the neutrophil, namely proteinase 3 (PR3), and myeloperoxidase (MPO) gives ANCA a role in the pathogenesis and may also determine the extent or severity of disease manifestations, being more strongly associated with generalized versus limited disease [54]. ANCAs associated with GPA can have a cytoplasmic (c) or perinuclear (p) immunofluorescence pattern, with their primary antigenic targets being proteinase 3 (PR3) or myeloperoxidase (MPO), respectively [34, 55].
GPA as well as other ANCA-associated vasculitides (AAV) is chronic and often relapsing diseases that can be organ or life-threatening [55], so it is vital to identify early-onset manifestations in children and in adults. Regarding the clinical differences between children and adults, children experience the following symptoms more frequently: ischemic abdominal pain, kidney involvement, nose deformities, and subglottic stenosis [52, 55,56,57]. In the case of adults, they have significantly more mononeuritis multiplex than children [58••].
Inflammatory markers do not show significant differences when compared in both groups and regarding ANCA, its positivity is higher in children compared with adults [59,60,61].
When it is not treated, GPA has mortality close to 100% within the first year, and regardless of treatment, up to 60% of patients experience subsequent disease flares [52]. The treatment has been adapted from the experience in adult GPA, so there are no differences when treating children: combined aggressive use of glucocorticoids and cyclophosphamide/rituximab for the treatment of GPA has led to marked improvement [62].
Although there are some interesting differences in GPA between children and adults as depicted, there is not enough information yet to confirm or deny that they share the same biology. The information gathered so far shows that clinically this does not matter, since treatment will mostly be equal for both groups.
Eosinophilic Granulomatosis with Polyangiitis
EGPA formerly known as Churg-Strauss vasculitis is a necrotizing granulomatosis vasculitis characterized by pulmonary, systemic small-vessel vasculitis, and hypereosinophilia infiltrates in medium- and small-sized vessels.
The incidence of EGPA in adults in the USA and Europe ranges from 0.5 to 6.8 cases per 1000,000 adults and the median age of 50 [63, 64]. In children, a reliable demographic information does not exist; in the work of Fina et al. [65], she described the largest case series with 14 patients with a median age, at time of diagnosis, of 12.3 [9•, 10, 11••, 12,13,14], and a systematic review of literature from Zwerina et al. [66] described 33 pediatric EGPA cases with a median age of 12.
It was first described in 1951 by Jacob Churg and Lotte Strauss as a triad of asthma, periarteritis, and eosinophilia in their monograph entitled “Allergic Granulomatosis, Allergic angiitis, and Periarteritis nodosa” [67].
Genetic association studies identified several loci associated with increased EGPA risk. The putative pathogenesis of EGPA involves a Th2-mediated inflammatory response. IL-10 is an important anti-inflammatory cytokine and inhibits Th1-type T cell responses. Increased levels may influence the dysregulated Th2 inflammatory response in EGPA [67]. It has been suggested the existence of different disease subsets in EGPA; for example, ANCA-positive patients have more clinical and histopathological features of small-vessel vasculitis, whereas ANCA-negative patients show tissue infiltration. However, since the Chapel Hill Consensus in 2012, the classification criteria have not been modified [2, 64].
Children had significantly more cardiac, ear, nose, throat, cutaneous, and gastrointestinal symptoms. Adult EGPA cases showed more neurological manifestations, such as peripheral neuropathy and mononeuritis multiplex. Adults had a higher rate of vasculitis on biopsy sample, with no differences for granuloma or eosinophilic infiltrate. Relapse rates were higher in the pediatric cohort, but no difference was found between the mortality rates [63, 65].
ANCA testing does not vary for adults and children with about 31% for each group. Children with positive ANCA had more neurological symptoms and more myalgia than children with negative ANCA testing [65, 68, 69].
For treatment, few randomized controlled trials have been conducted in EGPA. In general, treatment for adults and children has often been extrapolated from data regarding the other ANCA vasculitides [66, 67, 70]. Treatment with corticosteroids is a cornerstone in EGPA treatment and when the disease is severe, Cyclophosphamide is added. For maintenance, it is recommended to use immunosuppressant like methotrexate, leflunomide, mycophenolate mofetil, and azathioprine. A fairly new medication, mepolizumab, an anti-IL-5, is being used for refractory cases, but more studies are needed [66, 67].
We could hypothesize that EGPA in children is a subset of EGPA, since clinical manifestations are statistically different for adults and children. Nonetheless, the same treatment approach seems to work well for both groups. Future studies should aim to establish a marker that could help identify the ANCA-negative cases sooner.
Microscopic Polyangiitis
MPA in contrast to GPA and EGPA is a non-granulomatous necrotizing vasculitis of small vessels where immune complex deposition is limited or absent (pauci-immune vasculitis). Although characterized by the nearly constant positivity of circulating ANCAs, this disease is defined by histopathological and clinical criteria [71]. The patients usually develop pulmonic hemorrhage and glomerulonephritis.
The annual incidence of the disease in adults has been estimated at 5.9–11.3 in 1,000,000 with an average age of onset between 50 and 60 years [71, 72]. In children, there is scarce data to estimate an incidence, but case series have found that the mean age for MPA in children is 12 years [35, 52, 61, 73].
Initially confused with PAN, MPA is distinctly different in terms of clinical manifestations and a high prevalence of ANCA that is instead consistently absent in patients with PAN [72]. It was not until 1985, when Savage and colleagues described the presentation, pathology, and prognosis of what they termed microscopic polyarteritis, based on studies of 34 adult patients (ANCA). The authors emphasized the necessity of considering this entity in patients with focal embolic nephritis. Systemic arteritis without significant glomerular change or lung lesions would correspond to PAN [53].
As with the other ANCA vasculitides, what triggers the disease is unknown. In adults, up to 95% of patients have detectable peri-nuclear MPO-ANCA; PR3-ANCA is found in less than 5% [45, 74]. It is important to advise that a negative ANCA test does not exclude MPA [75, 76]. A meta-analysis made by Iudici et al. [61] in children with MPA, describes c-ANCA in 4% of patients and p-ANCA in 94% of patients with a prevalence of anti-MPO antibodies was 93%.
Diagnostic or classification criteria for children with MPA do not exist [77]. Its diagnosis has been adapted from the Chapel Hill Consensus Conference [2]. Thus, MPA remains a diagnosis of exclusion. There is a female predominance of MPA in children; about 80% of the MPA patients were girls. This finding contrasts with those for studies in adults, which have reported a clear absence of difference in frequency between the sexes. There are not significant differences regarding clinical presentation in adults and children [51, 61, 71].
Regarding treatment for MPA, when it is compared with the treatment for GPA, there are no major differences in therapeutic implications [34, 78]. As in the other ANCA vasculitides, the treatment for adults has been adapted for children. Prompt and intensive treatment with the combination of corticosteroids and cyclophosphamide is the usual first-line approach and maintenance therapy includes lower doses of corticosteroids, azathioprine, and methotrexate. Rituximab has been shown to be effective in resistant and relapsing patients [72].
Adult and children patients with MPA are probably the most similar when it comes to pathogeny, clinical characteristics, and treatment. In other words, the clinical spectrum is similar. More genetic studies, especially in children, have to be done.
Other Vasculitides
There are some cases in children and adults where some vasculitides cannot be categorized with the current classification criteria. In those cases, regular close follow-ups may elucidate a new symptom. More reported cases may give a better classification and better approach for this subset of patients [51, 79, 80].
Conclusion
Primary vasculitis are rare diseases; this situation has been a major problem for establishing adequate classification and diagnosis criteria for adults and children due to the lack of big population studies. From a practical view, although clinical characteristics in almost every primary vasculitis between adults and children differ, the usual treatment works in most cases for both groups.
Elucidating if adult and children primary vasculitides are part of the same clinical spectrum still requires many studies, but the main limitation is the difficulty of recruiting patients.
Identifying the diverse clinical spectrum in both adults and children primary vasculitides will reduce the need to extrapolate the diagnostic criteria from one group to another and to individualize it, which will allow the clinician to establish a more early diagnosis, since many of these patients require long periods of time to obtain a diagnosis from and, therefore, a treatment. In the end, this will permit an adequate treatment to reduce morbidity and incapacitating organ damage.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ozen S, Onmez HE, Demir S. Pediatric forms of vasculitis. Best Pract Res Clin Rheumatol. 2018;32:137–47. https://doi.org/10.1016/j.berh.2018.09.007.
Jennette JC. Overview of the 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Clin Exp Nephrol. 2013;17:603–6. https://doi.org/10.1007/s10157-013-0869-6.
Demir S et al. Vasculitis: decade in review. Current Rheumatology Reviews, 2018, 14, 00–00. This study Doi: https://doi.org/10.2174/1573397114666180726093731, Vasculitis: Decade in Review.
Sag E, et al. Childhood systemic vasculitis. Best practice & research clinical rheumatology. 2017;31(4):558–75. https://doi.org/10.1016/j.berh.2017.11.009.
Moran S, et al. IgA vasculitis in adults. Curr Treat Options in Rheum. 2018;4:119–32. https://doi.org/10.1007/s40674-018-0088-0.
Journeau L, Pistorius MA, et al. Juvenile temporal arteritis: a clinicopathological multicentric experience. Autoimmun Rev. 2019;18(5):476–83. https://doi.org/10.1016/j.autrev.2019.03.007.
Di Santo M, Stelmaszewski EV, Villa A. Takayasu arteritis in paediatrics. Cardiol Young. 2018;28:354–61. https://doi.org/10.1017/S1047951117001998.
Panupattanapong S. Epidemiology and outcomes of granulomatosis with polyangiitis in pediatric and working-age adult populations in the United States: analysis of a large National Claims Database. Arthritis Rheumatol. 2018;70(12):2067–76. https://doi.org/10.1002/art.40577. This is one of the biggest and most descriptive works about GPA.
Watts R. Evolving concepts in classification of systemic vasculitis: where are we and what is the way forward? International Journal of Rheumatic Diseases 2018. 22 Suppl 1:21–27. This article discusses the current classification and the future of it. doi: https://doi.org/10.1111/1756-185X.13304.
Johnston SL, Lock RJ, Gompels MM. Takayasu arteritis: a review. J Clin Pathol. 2002;55:481–6. https://doi.org/10.1136/jcp.55.7.481.
Mathew AJ, et al. Childhood-onset Takayasu arteritis: an update. International Journal of Rheumatic Diseases. 2016;19:116–26. This article describes the epidemiology and the biggest works on childhood Takayasu Vasculitis. https://doi.org/10.1111/1756-185X.12718.
Eleftheriou D, et al. Takayasu arteritis in childhood: retrospective experience from a tertiary referral centre in the United Kingdom. Arthritis Research & Therapy. 2015:17–36. https://doi.org/10.1186/s13075-015-0545-1.
Aeschlimann FA, Eng SWM, Sheikh S, Laxer RM, Hebert D, Noone D, et al. Diagnosis, treatment, and long-term management of Kawasaki disease a Statement for health professionals from the committee on rheumatic fever, endocarditis and Kawasaki disease, council on cardiovascular disease in the young, American Heart Association endorsed by the American Academy of Pediatrics Childhood Takayasu arteritis: disease course and response to therapy. Circulation. 2017;19:255. https://doi.org/10.1186/s13075-017-1452-4.
Newburger JW, Takahashi M., Gerber MA, Gewitz MH, Tani LY, Burns JC, Shulman ST, Bolger AF, Ferrieri P, Baltimore RS, Wilson WR, Baddour LM, Levison ME, Pallasch TJ, Falace DA, Taubert KA, Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease., Council on Cardiovascular Disease in the Young., American Heart Association., American Academy of Pediatrics. . 2004;110:2747–71. Doi:https://doi.org/10.1161/01.CIR.0000145143.19711.78.
Uehara R. Recent topics in the Epidemiology of Kawasaki disease. In: Tsutomu-Saji B, Newburger JW, Burns JC, Takahashi M, editors. Kawasaki Disease: Current Understanding of the Mechanism and Evidence-Based Treatment. Springer; 2017. 91–94.
Tai-Lin M, Wu MH. The global epidemiology of Kawasaki disease: review and future perspectives. Global Cardiology Science and Practice 2017:20. Doi: https://doi.org/10.21542/gcsp.2017.20
Newburger JW, Takahashi M, Burns JC. Kawasaki Disease Journal of the American College of Cardiology. 2016;67(14). https://doi.org/10.1016/j.jacc.2015.12.073.
Mossberg M, Segelmark M, Kahn R, Englund M, Mohammad AJ. Epidemiology of primary systemic vasculitis in children: a population-based study from southern Sweden. Scand J Rheumatol. 2018;47(4):295–302. https://doi.org/10.1080/03009742.2017.1412497.
Fraison JB, et al. Kawasaki disease in adults: observations in France and literature review. Autoimmun Rev. 2016;(3):242–9. This article has a extended description and revision of adult Kawasaki. https://doi.org/10.1016/j.autrev.2015.11.010.
Burns JC, Kushner HI, Bastian JF, Shike H, Shimizu C, Matsubara T, et al. Kawasaki disease: a brief history. Pediatrics. 2000;106(2):E27. https://doi.org/10.1542/peds.106.2.e27.
Denby KJ, Clark DE, Markham LW Management of Kawasaki disease in adults. Heart. 2017 0:1–9. Doi: https://doi.org/10.1136/heartjnl-2017-311774, 103.
Guillevin L. Polyarteritis Nodosa. In Nussinovitch U, editor. The heart in rheumatic, autoimmune and inflammatory diseases pathophysiology, clinical aspects and therapeutic approaches. Elsevier. 2017. Chapter 18: 419–427.
Sönmez H, et al. Polyarteritis nodosa: lessons from 25 years of experience. Clin Exp Rheumatol. 2019;37(Suppl. 117):S52–6.
Jayne DJ, Karadag O. Polyarteritis nodosa revisited: a review of historical approaches, subphenotypes and a research agenda. Clin Exp Rheumatol 2018. s135-s142.
Ozen S. The changing face of polyarteritis nodosa and necrotizing vasculitis. Nat Rev Rheumatol. 2017;13(6):381–6. https://doi.org/10.1038/nrrheum.2017.68.
Pagnoux C, Seror R, Henegar C, Mahr A, Cohen P, le Guern V, et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis study group database. Arthritis & Rheumatism. 2010;62(2):616–26. https://doi.org/10.1002/art.27240.
Ozen S. Juvenile polyarteritis: results of a multicenter survey of 110 children. J Pediatr. 2004;145:517–22. https://doi.org/10.1016/j.jpeds.2004.06.046.
De Virgilio A, et al. Polyarteritis nodosa: a contemporary overview. Autoimmunity Reviews. 15(6):564–70. https://doi.org/10.1016/j.autrev.2016.02.015.
Erden A. Comparing polyarteritis nodosa in children and adults: a single center study. Int J Rheum Dis. 2017;20(8):1016–22. https://doi.org/10.1111/1756-185X.13120.
Mahr A, de Menthon M. Classification and classification criteria for vasculitis. Curr Opin Rheumatol. 2015;27(1):1–9. https://doi.org/10.1097/bor.0000000000000134.
Lahiry S, et al. Childhood polyarteritis nodosa: a rare presentation. Asian Journal of Medical Sciences. 2016;7(6). https://doi.org/10.3126/ajms.v7i6.15724.
Iudici M. Childhood- versus adult-onset polyarteritis nodosa results from the French Vasculitis Study Group Registry. Autoimmunity Reviews. 2018;17:984–9. https://doi.org/10.1016/j.autrev.2018.08.001. This article is the biggest study that compares adult and child PAN.
Barut K, Sahin S, Kasapcopur O. Pediatric vasculitis. Curr Opin Rheumatol. 2016;28:29–38. https://doi.org/10.1097/BOR.0000000000000236.
Cabral DA, Morishita K. Granulomatosis with polyangiitis in children. In S. Sawhney, editor. Pediatric rheumatology: a clinical viewpoint. Springer. 2017. Chapter 36:461–479.
Bingham D, et al. A10: younger age and severity of renal presentation distinguishes microscopic polyangiitis from granulomatosis with polyangiitis in children: an ARChiVe study. Arthritis & Rheumatology. 2014;66(S3):S15–6. https://doi.org/10.1002/art.38421.
Piram M, Mahr A. Epidemiology of immunoglobulin a vasculitis(Henoch–Schonlein): current state of knowledge. Curr Opin Rheumatol. 2013;25:171–8. https://doi.org/10.1097/BOR.0b013e32835d8e2a.
Piram M, Maldini C. Incidence of IgA vasculitis in children estimated by four-source capture_recapture analysis: a population-based study. Rheumatology. 2017;56(8):1358–66. https://doi.org/10.1093/rheumatology/kex158.
Chen JY, Mao JH. Henoch-Schönlein purpura nephritis in children: incidence, pathogenesis and management. World J Pediatr. 2015;11(1):29–34. https://doi.org/10.1007/s12519-014-0534-5.
Tracy A, Subramanian A, Adderley NJ, et al. Cardiovascular, thromboembolic and renal outcomes in IgA vasculitis (Henoch-Schönlein purpura): a retrospective cohort study using routinely collected primary care data. Ann Rheum Dis. 2019;78:261–9. https://doi.org/10.1136/annrheumdis-2018-214142.
Hocevar A, Rotar Z, Jurcic V. IgA vasculitis in adults: the performance of the EULAR/PRINTO/PRES classification criteria in adults. Arthritis Res Ther. 2016;18:58. https://doi.org/10.1186/s13075-016-0959-4.
Yao-Hsu Y, Hsin-hui Y. The diagnosis and classification of Henoch–Schönlein purpura: an updated review. Autoimmunity Reviews. 2014;13(4–5):355–8. https://doi.org/10.1016/j.autrev.2014.01.031.
EULAR/PRINTO/PRES criteria for Henoch–Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis 2010;69:798–806. doi: 10.1136/ard.2009.116657.
Gupta V, Aggarwal A, Gupta R. Differences between adult and pediatric onset. Henoch-Schonlein purpura from North India. Int J Rheum Dis. 2018;21:292–8. https://doi.org/10.1111/1756-185X.13221.
Vogler C, Eliason SC, Wood EG. Glomerular membranopathy in children with IgA nephropathy and Henoch Schonlein Purpura. Pediatr Dev Pathol. 1999;2:227–35.
Brogan P, Eleftheriou D. Vasculitis update: pathogenesis and biomarkers. Pediatr Nephrol. 2018;33(2):187–98. https://doi.org/10.1007/s00467-017-3597-4.
Pertuiset E. Liotd F. et al. Adult Henoch-SchSnlein purpura associated with malignancy. Seminars in arthritis and rheumatism. 2000. Vo1 29, 6: 360–367.
Ostrovršnik J, Rotar &, Ješe R, et al THU0448 Is there an association between adult iga vasculitis and cancer? Ann Rheum Dis 2018;77:436. https://doi.org/10.1136/annrheumdis-2018-eular.5359
Batu E, et al. Comparing immunoglobulin a vasculitis (Henoch–Schönlein purpura) in children and adults: a single-centre study from Turkey. Scand J Rheumatol. 2018:1–6. https://doi.org/10.1080/03009742.2018.144811150.
Audemard-Verger A, Terrier B, et al. Characteristics and management of IgA vasculitis (Henoch-Schonlein) in Adults. Arthritis & Rheumatology. 2017;69(9):1862–70. https://doi.org/10.1002/art.40178.
Selewski DT, Ambruzs JM, et al. Clinical characteristics and treatment patterns of children and adults with IgA nephropathy or IgA vasculitis: Findings from the CureGN study. Kidney Int Rep. 2018;3(6):1373–84. https://doi.org/10.1016/j.ekir.2018.07.021.
Gibson K, Glenn D. ANCA-associated vasculitis, Pediatric. In Gibson, Keisha & Glenn, Dorey, editor. Glomerulonephritis. 2019. Chapter 17: 419–427. Doi: https://doi.org/10.1007/978-3-319-49379-4_18
Cabral DA, et al. Comparing presenting clinical features in 48 children with microscopic polyangiitis to 183 children who have granulomatosis with polyangiitis (Wegener’s): an ARChiVe cohort study. Arthritis & Rheumatology. 2016;68(10):2514–26. https://doi.org/10.1002/art.39729.
Ball GV. The history of ANCA associated vasculitis. Rheum Dis Clin N Am. 2010;36:439–46. https://doi.org/10.1016/j.rdc.2010.05.004.
Jariwala MP, Laxer RM. Primary vasculitis in childhood: GPA and MPA in childhood. Front Pediatr. 2018;6:226. https://doi.org/10.3389/fped.2018.00226.
Plumb LA, Oni L, Marks SD, Tullus K. Paediatric anti-neutrophil cytoplasmic antibody (ANCA) -associated vasculitis: an update on renal management. Pediatr Nephrol. 2018;33:25–39.
Ozen S, Acar-Ozen NP. Recent advances in childhood vasculitis. Curr Opin Rheumatol 2017. 29:000–000. doi: https://doi.org/10.1097/BOR.0000000000000424, 534.
Berti A, Cornec D, Crowson CS, Specks U, Matteson EL. The epidemiology of ANCA associated vasculitis in Olmsted County, Minnesota (USA): a 20 year population-based study. Arthritis Rheumatol. 2017 Dec;69(12):2338–50. https://doi.org/10.1002/art.40313.
Iudici M, et al. Childhood- versus adult-onset ANCA-associated vasculitides: a nested, matched case-control study from the French Vasculitis Study Group Registry. Autoimmun Rev. 2018;17(2):108–14. https://doi.org/10.1016/j.autrev.2017.11.014. The French vasculitis group has described extensively ANCA vasculitis in children and adults.
Noone D, Hebert D, Licht C. Pathogenesis and treatment of ANCA-associated vasculitis—a role for complement. Pediatr Nephrology. 2018;33(1):1–11. https://doi.org/10.1007/s00467-016-3475-5.
Morishita KA. Early outcomes in children with Antineutrophil cytoplasmic antibody-associated Vasculitis. Arthritis Rheumatol. 2017;69(7):1470–9. https://doi.org/10.1002/art.40112.
Iudici M, Quartier P, Terrier B, Mouthon L, Guillevin L, Puéchal X. Childhood-onset granulomatosis with polyangiitis and microscopic polyangiitis: systematic review and meta-analysis. Orphanet Journal of Rare Diseases. 2016;11:141. https://doi.org/10.1186/s13023-016-0523-y.
James KE, Xiao R, Merkel PA, Weiss PF. Variation in the treatment of children hospitalized with Antineutrophil cytoplasmic antibody-associated Vasculitis in the US. Arthritis Care Res (Hoboken). 2017;69(9):1377–83. https://doi.org/10.1002/acr.23142.
Comarmond C. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group Cohort. Arthritis & Rheumatism. 2013;65(1):270–81. https://doi.org/10.1002/art.37721.
Sada KE, Amano K, Uehara R, Yamamura M, Arimura Y, Nakamura Y, et al. Mod Rheumatol. 2014;24(4):640–4. https://doi.org/10.3109/14397595.2013.857582.
Fina A, Dubus JC, Tran A, Derelle J, Reix P, Fayon M, et al. Eosinophilic granulomatosis with polyangiitis in children: data from the French RespiRare® cohort. Pediatr Pulmonol. 2018;53:1–11. https://doi.org/10.1002/ppul.24089.
Zwerina J. Churg–Strauss syndrome in childhood: a systematic literature review and clinical comparison with adult patients. Semin Arthritis Rheum. 2009;39(2):108–15. https://doi.org/10.1016/j.semarthrit.2008.05.004.
Wu EY. Eosinophilic granulomatosis with polyangiitis: clinical pathology conference and review. The Journal of Allergy and Clinical Immunology: In Practice. 2018;6(5):1496–504. https://doi.org/10.1016/j.jaip.2018.07.001.
Alba MA, Jennette J, Falk R. Pathogenesis of ANCA-associated pulmonary vasculitis. Semin Respir Crit Care Med. 2018;39:413–24. https://doi.org/10.1055/s-0038-1673386.
Dumoitier N, Terrier B, London J, Lofek S, Mouthon L. Implication of B lymphocytes in the pathogenesis of ANCA-associated vasculitides. Autoimmun Rev. 2015;14(11):996–1004.
Hanna L, V G. Presentation and classification of rare primary vasculitides in children. Journal of Vasculitis. 2016;2:2. https://doi.org/10.4172/2471-9544.100107.
Karras A. Microscopic polyangiitis: new insights into pathogenesis, clinical features and therapy. Semin Respir Crit Care Med. 2018;39:459–64. https://doi.org/10.1055/s-0038-1673387.
Dammacco F, Vacca A. Microscopic Polyangiitis in Dammacco, F., Ribatti, D., & Vacca, A. (Eds.). Systemic vasculitides: current status and perspectives. 2016. Chapter 10: 109–118.
Wang H, Sun L, Tan W. Clinical features of children with pulmonary microscopic polyangiitis: report of 9 cases. PLoS One. 2015;10(4):e0124352. https://doi.org/10.1371/journal.pone.0124352.
Schirmer JH. Clinical presentation and long-term outcome of 144 patients with microscopic polyangiitis in a monocentric German cohort. Rheumatology (Oxford). 2016;55(1):71–9. https://doi.org/10.1093/rheumatology/kev286.
Microscopic polyangiitis: from pathogenesis to treatment. Urol Nephrol Open Access J 2017, 5(2): 00167. Doi: https://doi.org/10.15406/unoaj.2017.05.00167
Pagnoux C. Updates in ANCA-associated vasculitis. Eur J Rheumatol. 2016;3(3):122–33.
et al. Lythgoe H, Presentation and classification of rare primary vasculitides in children. J Vasc. 2016;2:2. https://doi.org/10.4172/2471-9544.100107.
Jarrot PA, Kaplanski G. Pathogenesis of ANCA-associated vasculitis: an update. Autoimmun Rev. 2016;15(7):704–13. https://doi.org/10.1016/j.autrev.2016.03.007.
Nakano N, Mori M, Umebayashi H, Iwata N, Kobayashi N, Masunaga K, et al. Characteristics and outcome of intractable vasculitis syndrome in children: nation-wide survey in Japan. Mod Rheumatol. 2017;28:1439–7595. https://doi.org/10.1080/14397595.2017.1404700.
Schnabel A, Hedrich CM. Childhood vasculitis. Front. Pediatr. 2019;6:421. https://doi.org/10.3389/fped.2018.00421.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors of this work do not have conflict of interest to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Vasculitis
Rights and permissions
About this article

Cite this article
Ferrandiz-Espadin, R., Ferrandiz-Zavaler, M. Childhood- Versus Adult-Onset Primary Vasculitides: Are They Part of the Same Clinical Spectrum?. Curr Rheumatol Rep 21, 51 (2019). https://doi.org/10.1007/s11926-019-0851-8
Published:
DOI: https://doi.org/10.1007/s11926-019-0851-8