
Abstract
Damage-associated molecular patterns (DAMPs) or alarmins are endogenous danger signals that are derived from damaged cells and extracellular matrix degradation, capable of triggering innate immune response to promote tissue damage repair. Hemolytic or hemorrhagic episodes are often associated with inflammation, even when infectious agents are absent, suggesting that damaged red blood cells (RBCs) release DAMPs.
Hemoglobin (Hb) composes 96% of the dry weight of RBCs; therefore upon hemolysis, tremendous amounts of Hb are released into the extracellular milieu. Hb oxidation occurs outside the protective environment of RBCs, leading to the formation of different Hb oxidation products and heme. Heme acts as a prototypic DAMP participating in toll-like receptor as well as intracellular nucleotide-binding oligomerization domain-like receptor signaling. Oxidized Hb forms also possess some inflammatory actions independently of their heme releasing capability. Non-Hb-derived DAMPs such as ATP, interleukin-33, heat shock protein 70, as well as RBC membrane-derived microparticles might also contribute to the innate immune response triggered by hemolysis/hemorrhage.
In this chapter we will discuss the inflammatory properties of RBC-derived DAMPs with a particular focus on Hb derivatives, as well as therapeutic potential of the endogenous Hb and heme-binding proteins haptoglobin and hemopexin in the prevention of hemolysis/hemorrhage-associated inflammation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
9.1 Introduction
Damage-associated molecular patterns (DAMPs) or alarmins are endogenous danger signals that are derived from damaged cells and extracellular matrix degradation capable of triggering and/or exacerbating innate immune responses to promote tissue damage repair (Matzinger 1994). Hemolytic or hemorrhagic episodes are often associated with inflammation even when infectious agents are absent (Arruda et al. 2005), suggesting that damaged red blood cells (RBCs) release DAMPs (Mendonca et al. 2016).
The far most abundant protein in mature RBCs is hemoglobin (Hb) that composes 96% of the dry weight of RBCs; therefore upon hemolysis, tremendous amounts of Hb are released into the extracellular milieu. Once outside the protective environment of RBCs, Hb is prone to oxidation, in which process different Hb oxidation products form with diverse biological activities toward immune and nonimmune cells. Heme, the prosthetic group of Hb, is promptly released from oxidized Hb species and is the most studied RBC-derived alarmin (Soares and Bozza 2016). Heme is a strong prooxidant and is involved in toll-like receptor (TLR) as well as intracellular nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) signaling [reviewed in Dutra and Bozza (2014), Soares and Bozza (2016)]. Besides heme, oxidized Hb forms also possess some inflammatory actions independently of their heme releasing capability [reviewed in Jeney et al. (2014)]. Non-Hb-derived DAMPs such as adenosine triphosphate (ATP), interleukin (IL)-33, heat shock protein (Hsp) 70, as well as RBC membrane-derived microparticles (MPs) might also contribute to the innate immune response triggered by hemolysis/hemorrhage.
Deleterious effects of extracellular Hb and heme are controlled by haptoglobin (Hp) and hemopexin (Hx), respectively. These acute phase proteins bind extracellular Hb and heme avidly and facilitate their removal from circulation through receptor-mediated endocytotic routes. Upon massive intravascular hemolysis, the scavenging capacities of Hp and Hx are overwhelmed. Along with this notion, Hp- and Hx-based therapeutic interventions could be beneficial in pathologies associated with hemolysis/hemorrhage.
9.1.1 Physiology of RBCs
RBCs are the most prevalent cells in the human body, structurally and functionally dedicated to transport oxygen and carbon dioxide throughout the organism. RBCs are formed in the bone marrow from pluripotent hematopoietic stem cells in the process of erythropoiesis. Differentiation takes place mainly in the bone marrow, until reticulocytes released into the bloodstream where they mature further 1–2 days into terminally differentiated RBCs. During differentiation RBCs loose nuclei and cytoplasmic organelles including mitochondria and ribosomes. The advantage of not having nuclei in mature RBCs is twofold: first, anucleated cells are more flexible assuring that they can squeeze through small blood capillaries; second, there is more space for Hb resulting in increased oxygen-binding capacity. On the other side of this trade-off, mature anucleated RBCs are unable to divide, and their rescuing mechanisms are limited. This explains the relatively short life-span (100–120 days) of RBCs in the circulation, and the enormous turnover of making and breaking RBCs (200 billion RBCs/day).
Circulating RBCs are continuously exposed to high levels of reactive oxygen species (ROS) of both endogenous and exogenous origin [reviewed in Mohanty et al. (2014)]. Each ml of blood contains 0.3 g of Hb, and auto-oxidation (Table 9.1, equation #1) of Hb is the major source of endogenous ROS in RBCs. Besides Hb auto-oxidation, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases also contribute to endogenous ROS production in RBCs (George et al. 2013). To cope with this challenge, RBCs are equipped with a highly effective antioxidant defense system which includes enzymes such as Cu/Zn superoxide dismutase that convert superoxide anion to hydrogen peroxide (H2O2), catalase, glutathione peroxidase, and peroxiredoxins which decompose H2O2 to H2O [reviewed in Siems et al. (2000), Jeney et al. (2013), Mohanty et al. (2014)]. Nonenzymatic low-molecular-weight scavengers such as glutathione and ascorbic acid also contribute to this protection. Incomplete neutralization of ROS triggers RBC membrane damage and subsequent impairment of oxygen delivery to the tissues which eventually leads to tissue damage and inflammation.
Circulating RBCs lose 20% of their Hb content during their life-span via vesiculation (Willekens et al. 2003). Vesiculation is considered to be a self-protective mechanism of RBCs via which RBCs release membrane patches containing removal molecules including phosphatidylserine, immunoglobulin G, and senescent cell antigens (Willekens et al. 2008). Additionally, RBCs are able to get rid of intracellular inclusions, e.g., Heinz bodies via this mechanism (de Back et al. 2014), thereby postponing the premature loss of otherwise healthy RBCs from the circulation. RBC-derived vesicles are rapidly removed from the circulation by the mononuclear phagocyte system (Willekens et al. 2003).
At the end of their life-span, senescent RBCs are removed from the circulation by hemophagocytic macrophages, mainly in the spleen (Bratosin et al. 1998; de Back et al. 2014). Aged RBCs are smaller and denser because of the permanent loss of Hb and cell membrane via vesiculation and also characterized by decreased metabolic activity (Piomelli and Seaman 1993). At the terminal stage of RBC aging, “eat me” signals appear, and “don’t eat me” signals disappear on the surface of senescent RBCs, and shortly after they are internalized by macrophages [reviewed in de Back et al. (2014)].
Different theories exist about the entity of the removal surface markers of senescent RBCs [reviewed in de Back et al. (2014)]. Phosphatidylserine, a phospholipid normally found in the inner membrane of RBCs, is a very likely candidate of being a removal signal, when it appears in the outer membrane of the RBCs (Boas et al. 1998). Phosphatidylserine is a general marker for apoptotic cells (Fernandez-Boyanapalli et al. 2009), and although RBCs cannot undergo a classical apoptosis because of the lack of nucleus and other cellular organelles, evidence suggest that aged or damaged RBCs can undergo a regulated process called eryptosis that is in many terms resembles to that of programmed cell death (Lang et al. 2005). Eryptosis is characterized by cell shrinkage, membrane blebbing, activation of proteases, and exposition of phosphatidylserine at the outer membrane leaflet of RBCs. Importantly, the removal of these phosphatidylserine-positive senescent, or terminally damaged RBCs by macrophages, is a non-inflammatory process and allows efficient and safe recycling of the RBC components, particularly the heme iron (Muckenthaler et al. 2017).
9.1.2 Hemolysis and Hemorrhage
Numerous pathologies are associated with hemolysis or hemorrhage characterized by uncontrolled destruction of RBCs. Hemolysis can occur in the vasculature but also in the extravascular space. Inherited or acquired conditions can cause hemolysis as listed in Table 9.2. Inherited hemolytic diseases are caused by mutations in genes encoding Hb, RBC membrane components, or certain enzymes in RBCs. The repertoire of acquired conditions associated with hemolysis is quite wide. Auto- and alloimmune reactions, mechanical, physical, or chemical stress, and diverse infections can trigger substantial RBC lysis. RBCs outside the vasculature tend to lyse quickly; therefore hemorrhages are also associated with RBC lysis.
9.1.3 The Fate of Extracellular Hemoglobin
Hb is released in large amounts from lysing RBCs. Extracellular Hb exerts diverse unfavorable vasoactive effects. For example, extracellular Hb scavenges nitric oxide, an important vasodilator and signaling molecule in the vasculature [reviewed in Rother et al. (2005)]. Furthermore, once outside the protective environment of RBCs, Hb tends to oxidize. Auto-oxidation of Hb occurs resulting in metHb generation meanwhile superoxide anions are formed (Table 9.1, equation 1). Peroxides, such as H2O2 or lipid hydroperoxides, induce a two-electron oxidation of Hb leading to the formation of ferryl (Fe4+ = O2−) Hb (Table 9.1, equation 2), whereas the reaction of metHb with H2O2 results in ferrylHb radical (Hb•+(Fe4+ = O2−)) in which the unpaired electron is located at either the globin chain or at the porphyrin ring (Table 9.1, equation 3) (Harel and Kanner 1988; Patel et al. 1996; Jia et al. 2007; Alayash et al. 2001). These high-valence iron compounds, i.e., ferrylHb and ferrylHb radical, are highly reactive intermediates that can decay by several ways (Reeder et al. 2008). FerrylHb induces additional production of globin radicals via an intramolecular electron transfer between the ferryl iron and specific amino acid residues of the globin chains such as 𝛼Tyr-24, 𝛼Tyr-42, 𝛼His-20, 𝛽Tyr-35, 𝛽Tyr-130, and 𝛽Cys-93 leading to the formation of metHb globin radical (Table 9.1, equation 4) (Deterding et al. 2004; Ramirez et al. 2003; Jeney et al. 2013). Termination reactions of globin- and porphyrin-centered radicals lead to the formation of globin-globin (Table 9.1, equation 5) or porphyrin-globin crosslinks.
To prevent the deleterious effects of extracellular Hb, efficient mechanisms have evolved for its removal from the circulation. Hp, an acute-phase protein, is present in plasma in high amounts (0.41−1.65 mg/ml) with the special recognized function of capturing cell-free Hb [reviewed in Alayash (2011)]. The formation of the Hp-Hb complex is virtually irreversible, and Hp binding has multiple beneficial effects. First of all, Hp binding facilitates the removal of Hb from circulation through the CD163 macrophage scavenger receptor-mediated endocytosis (Kristiansen et al. 2001). Besides this effect, studies showed that Hb bound to Hp is less prone to H2O2-mediated oxidation than free Hb (Buehler et al. 2009; Banerjee et al. 2012; Miller et al. 1997). In fact, the Hb-Hp complex acts as a fairly efficient peroxidase (Kapralov et al. 2009). Further studies proved that Hp prevents H2O2-induced oxidation of amino acids in critical regions of Hb chains—i.e., α-Tyr42, β-Tyr145, and β-Cys93—and polymerization of Hb (Pimenova et al. 2010). The recent determination of the crystal structure of the porcine Hp-Hb complex revealed that Hb residues known to be prone to oxidative modifications are buried in the Hp-Hb interface, thereby explaining this direct protective role of Hp against H2O2-induced oxidation (Andersen et al. 2012).
Although the Hb/Hp/CD163 system is highly efficient in removing intravascular free Hb, it has some limitations. Plasma Hp can bind and clear approximately 3 g of Hb from the circulation which is less than 1% of the total amount of circulating Hb. In case of pronounced hemolysis, when more than 1% of RBCs disrupt, Hp is depleted from the circulation in which case free Hb is cleared (rather inefficiently) via a low-affinity pathway through CD163 (Schaer et al. 2006) and/or by renal excretion (Schaer et al. 2013; Murray et al. 1961). This latter is accompanied by generation of free iron and organ damage.
Another limitation of the Hp/CD163 system is that Hp and CD163 have decreased affinity for structurally altered (e.g., covalently cross-linked) Hb species that might form upon Hb oxidation. Recent studies have revealed that elimination of oxidized Hb species via both high-affinity and low-affinity pathways can be severely compromised (Schaer et al. 2006; Vallelian et al. 2008).
Upon massive hemolysis Hp is consumed, causing accumulation and oxidation of cell-free Hb that eventually lead to the release of the prosthetic heme group. Hx is an acute-phase plasma protein that binds heme with the highest affinity of any known heme-binding proteins (Hrkal et al. 1974). Hx-heme complexes are internalized via the scavenger receptor LDL receptor-related protein 1/CD91 (Hvidberg et al. 2005) mainly by hepatocytes and macrophages (Herz and Strickland 2001).
Following internalization of Hb or heme, cells and tissues upregulate heme oxygenase-1 (HO-1) and ferritin. HO-1 catabolizes free heme into equimolar amounts of Fe2+, carbon monoxide (CO), and biliverdin (Tenhunen et al. 1968). Liberated iron drives the upregulation of ferritin that is the main intracellular iron storage protein (Eisenstein et al. 1991).
9.1.4 Pro-inflammatory Actions of Hb-Derived Species
Massive intravascular hemolysis or hemorrhage result in the exhaustion of the endogenous defense system leading to the accumulation of oxidized Hb forms and free heme in the plasma or in the extravascular space (Pamplona et al. 2007; Larsen et al. 2010; Nagy et al. 2010). These Hb derivatives, particularly free heme, exert prooxidant activities [reviewed in Immenschuh et al. (2017), Jeney et al. (2013)]. Moreover, hemolytic or hemorrhagic episodes are often associated with inflammation even when infectious agents are absent (Arruda et al. 2005). Considerable effort has been made to define the mediators and the target cells involved in the hemolysis-/hemorrhage-induced inflammatory response. Accumulating evidence suggest that Hb-derived oxidized species possess diverse pro-inflammatory actions targeting different immune and non-immune cells (Table 9.3).
9.1.4.1 Macrophage Activation
Macrophages, the frontline cells of innate immunity, respond to a variety of pathogen-associated molecular patterns (PAMPs) and DAMPs. Lysis of RBCs leads to the release of different RBC components that can potentially behave as DAMPs and induce a sterile inflammatory response dependently of receptors such as TLRs or NOD-like receptors (Table 9.3).
Accumulating evidence suggests that heme that is released from oxidized Hb forms modulate macrophage phenotype. Bozza et al. showed that heme triggers tumor necrosis factor-alpha (TNF-α) secretion by macrophages in a TLR4-dependent manner (Figueiredo et al. 2007). The activation of TLR4 by heme is strictly dependent on its coordinated iron and the vinyl groups of the porphyrin ring (Figueiredo et al. 2007). Sustained exposure of macrophages to free heme triggers programmed necrosis that is dependent on autocrine production of TNF-α and ROS (Fortes et al. 2012). The pathogenic role of heme-mediated TLR4 activation was investigated in a murine model of intracerebral hemorrhage (ICH)-induced neuro-inflammation. In comparison to wild-type mice, TLR4−/− mice exhibited less inflammation, reduced cerebral edema, and lower neurological deficit scores, suggesting that heme-mediated TLR4 activation plays a critical role in ICH-associated neuro-inflammation (Lin et al. 2012). Gram et al. showed that after intraventricular hemorrhage, metHb forms and its level correlates to the expression of TNF-α (Gram et al. 2013). In agreement with this finding, a recent study of Kwon et al. revealed that metHb is an important endogenous activator of TLR4 that promotes widespread TLR4-mediated neuro-inflammation upon subarachnoid hemorrhage (Kwon et al. 2015).
Activation of the cytosolic NOD-like receptors results in the assembly of a caspase-1-activating scaffold. Active caspase-1 subsequently cleaves the pro-inflammatory IL-1 family of cytokines into their bioactive forms, IL-1β and IL-18, those can trigger pyroptosis, a type of inflammatory cell death [reviewed in Guo et al. (2015)]. The NLR family pyrin domain containing 3 (NLRP3) inflammasome, which belongs to the NOD-like receptor family, is the most extensively studied inflammasome, that is formed after the oligomerization of NLRP3, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and pro-caspase-1 (Schroder and Tschopp 2010).
Besides PAMPs, the NLRP3 inflammasome is activated in response to a wide variety of DAMPs including extracellular ATP, crystals of monosodium urate or cholesterol, β-amyloid fibers, the degradation of extracellular matrix components, and environmental or industrial particles and nanoparticles (Martinon et al. 2006; Mariathasan et al. 2006; Duewell et al. 2010; Halle et al. 2008; Babelova et al. 2009; Yazdi et al. 2010; Hornung et al. 2008).
Recently heme was added to the long list of NLRP3 activating danger signals. Dutra et al. showed that heme triggers active IL-1β production in lipopolysaccharide (LPS)-primed macrophages in an NLRP3- and caspase-1-dependent manner (Dutra et al. 2014). They also investigated the structural requirements of heme-mediated NLRP3 inflammasome activation. Heme analogs such as protoporphyrin IX (PPIX) that lacks the central iron atom or metal substitution derivatives such as CoPPIX and SnPPIX were unable to induce IL-1β secretion in LPS-primed macrophages (Dutra et al. 2014). Based on these observations, they came to the conclusion that NLRP3 activation by heme is strictly dependent on its coordinated iron, which is in conflict with the findings of Li et al. who reported that PPIX is as efficient in inducing IL-1β maturation and secretion as heme (Li et al. 2014).
9.1.4.2 Neutrophil Activation
Polymorphonuclear neutrophils are the first leukocytes migrating from the blood into injured or infected tissues. Neutrophils kill pathogens via various cytotoxic mechanisms and clear cellular debris; therefore they play a fundamental role in innate and adaptive immunity (Rosales et al. 2016). In the recent years, it has become evident that neutrophils not only sense PAMPs but can recognize and respond to endogenous DAMPs as well. In line of this notion, heme triggers neutrophil chemotaxis and activation, characterized by elevated ROS production and increased expression of the pro-inflammatory cytokine IL-8 (Graca-Souza et al. 2002). Heme-induced neutrophil recruitment is regulated through signaling pathways that are characteristic of chemoattractant molecules (Porto et al. 2007) but independent of TLR4-mediated signaling (Figueiredo et al. 2007). Besides heme, oxidized Hb (ferrylHb) is a very potent trigger of neutrophil infiltration in mice independently of TLR4 signaling (Silva et al. 2009). Additionally, Kono et al. showed that PPIX was as efficient as heme in inducing neutrophil ROS production, pointing out that this effect is independent of the coordinated iron present in heme (Kono et al. 2013). Protoporphyrin ring-induced neutrophil activation was suggested to play a role in transfusion-related acute lung injury (Kono et al. 2013).
Additionally of ROS generation and the release of microbicidal molecules, neutrophils can release extracellular traps—a meshwork of chromatin fibers decorated by granular proteins—that represent an important strategy to immobilize and kill invading microorganisms (Brinkmann et al. 2004). Recently Chen et al. reported that heme is able to induce the formation of neutrophil extracellular traps and suggested that this mechanism contributes to vaso-occlusion crises in sickle cell disease (Chen et al. 2014).
9.1.4.3 Endothelial Cell Activation
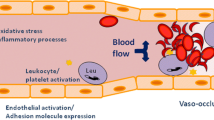
Endothelium, the interface between blood and tissue, has a pivotal role in the inflammatory response mainly through the induction of the leukocyte adhesion cascade to facilitate transmigration of inflammatory cells to the inflamed tissue. Accordingly, inflammatory stimuli, such as IL-1, TNF-α, or LPS, upregulate cellular adhesion molecules including intracellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E selectin, in endothelial cells (Bevilacqua et al. 1985; Pohlman et al. 1986). Wagener et al. found that exposure of endothelial cells to heme upregulated the expressions of ICAM-1, VCAM-1, and E selectin, in a similar manner to that of IL-1, TNF-α, or LPS (Wagener et al. 1997). Recently Belcher et al. showed that heme activates endothelial cells in a TLR4-dependent manner and that this heme-mediated TLR4-dependent endothelial activation plays a pathogenic role in vaso-occlusion in a murine model of sickle cell disease (Belcher et al. 2014).
While searching for other mediators of hemolysis-associated inflammation, Silva et al. reported that ferrylHb but not native Hb or metHb triggers upregulation of the pro-inflammatory adhesion molecules ICAM-1, VCAM-1, and E-selectin (Silva et al. 2009). FerrylHb induced rearrangement of actin cytoskeleton in endothelial cells leading to the disruption of the endothelial monolayer integrity (Silva et al. 2009). FerrylHb-induced inflammatory response was dependent on actin polymerization and the activation of the c-Jun N-terminal kinase and the p38 mitogen-activated protein kinase signal transduction pathways (Silva et al. 2009). Silva et al. showed that induction of endothelial inflammatory response is a unique property of ferrylHb because neither Hb nor metHb triggered these effects (Silva et al. 2009). FerrylHb can release its prosthetic heme group (Potor et al. 2013), and one can ask whether ferrylHb-mediated inflammatory response is mediated by the released heme. Many lines of evidence suggest that in fact this is not the case. First of all, metHb, that can also release heme in a similar manner as ferrylHb, does not induce inflammatory response in endothelial cells (Silva et al. 2009). Second, ferrylHb-induced inflammatory response is not dependent on TLR4 signaling (Silva et al. 2009). These results suggest that heme and ferrylHb are two Hb-derived pro-inflammatory agonists that trigger endothelial activation via different signaling mechanisms.
9.1.5 Cytoprotective and Anti-inflammatory Actions of Hb-Derived Species
Interestingly enough, besides its prooxidant and pro-inflammatory actions, under special circumstances heme can induce cytoprotective and anti-inflammatory responses. These protective mechanisms largely rely on the heme-mediated upregulation of the HO-1/ferritin system [reviewed in Gozzelino et al. (2010)], and it mostly relies on the ability of HO-1 to degrade heme into CO, iron, and biliverdin, in which the latter is promptly converted to bilirubin. The subsequent upregulation of ferritin is essential to obtain the protective effect, as it can store the released iron in a catalytically inactive form (Balla et al. 1992). Additionally, the side products of heme degradation, i.e., bilirubin and CO, exert diverse antioxidant and anti-inflammatory actions (Gozzelino et al. 2010).
Along with these notions, a subset of macrophages, called hemorrhage-associated or Mhem macrophages with anti-inflammatory properties, were identified in atherosclerotic plaques with intraplaque hemorrhage (Boyle et al. 2009). Mhem macrophages are characterized by facilitated iron sequestration assured by elevated expressions of HO-1 and CD163 and at the same time protection from foam cell formation secured by induction of genes central to cholesterol efflux (Boyle et al. 2009, 2012). Boyle et al. also showed that Mhem macrophage polarization is driven by heme and identified two key transcription factors nuclear factor erythroid 2-related factor 2 (NRF2) and activating transcription factor 1 involved in this process (Boyle et al. 2011, 2012).
Endothelial cells can also benefit from the cytoprotective mechanism provided by the HO-1/ferritin system. In the early 1990s, Balla et al. showed that a brief exposure of sublethal concentration of heme made endothelial cells highly resistant to subsequent oxidant-mediated killing in which cytoprotection was relied on the upregulation of the HO-1/ferritin system (Balla et al. 1992). Since that initial work, many investigations targeted the multifunctional role and therapeutic potential of HO-1 in the vascular endothelium [reviewed in Calay and Mason (2014)].
9.1.6 Non-Hb-Derived RBC DAMPs
Although Hb is the far more abundant molecule in RBCs, there are other components in RBCs that can potentially become DAMPs following RBC lysis. For example ATP, a universal energy source, is present in RBCs in high concentration (~1.6 mmol/L). When present in the extracellular milieu, ATP becomes a signaling molecule that activates P2 receptors in diverse cells (Dubyak 1991). It has been shown that hypoxia, elevated shear stress, and reduced pH lead to ATP release from RBCs, although it is still a matter of debate whether it occurs via an active or passive process. Bergfeld et al. showed that under hypoxic conditions, RBCs release ATP in a regulated way through the plasma membrane protein band 4.5 (Bergfeld and Forrester 1992). Recently Sridharan et al. proposed that pannexin 1, a channel-forming glycoprotein, is involved in hypoxia-mediated ATP release from RBCs (Sridharan et al. 2010). Regarding shear stress-induced ATP release, Wan et al. suggested that mechanosensitive ATP release is triggered by retraction of the spectrin-actin cytoskeleton network and influenced by membrane viscosity (Wan et al. 2008). Recently, Piezo1, a mechanically activated cation channel involved in physiological responses to touch, pressure, and stretch, was shown to regulate mechanosensitive release of ATP from RBCs via controlling the shear-induced calcium influx (Cinar et al. 2015). Contrary to the active process, Sikora et al. reported that hemolysis is the primary mechanism via which RBCs release ATP in response to hypoxia or mechanical stress (Sikora et al. 2014). Nevertheless, RBC-derived ATP can activate P2 purinergic receptors on vascular endothelial cells, resulting in the synthesis of powerful vasodilators such as nitric oxide and prostaglandins (Burnstock 2017). Via this mechanism RBCs actively participate in the regulation of microvascular blood flow and contribute to match oxygen delivery and local needs (Ellsworth et al. 1995).
Besides its vasoactive effects, activation of P2 purinergic receptors by ATP can trigger inflammatory responses in various immune and nonimmune cells (Idzko et al. 2014). For example, ATP activates P2X purinoceptor 7 (P2X7) and promotes IL-1β and IL-18 secretion in LPS-primed macrophages (Perregaux et al. 2000). Activation of P2X7 receptors by ATP on endothelial cells leads to nuclear factor kappa B (NF-κB) activation and subsequent upregulation of its target genes such as E-selectin (von Albertini et al. 1998). Extracellular ATP induces deterioration of endothelial barrier function and may trigger apoptotic cell death (McClenahan et al. 2009). ATP can induce activation of the NLRP3 inflammasome and subsequent release of low levels of IL-1β in endothelial cells primed with LPS or TNF-α (Huck et al. 2015; Champaiboon et al. 2014). Furthermore, both progenitor and mature RBCs express P2 purinergic receptors, and accumulating evidence suggest that extracellular ATP exerts various biological effects on these cells (Burnstock 2015; Sluyter 2015). ATP induces the release of MPs, ROS formation and apoptotic cell death in erythroid progenitor cells (Chahwala and Cantley 1984; Constantinescu et al. 2010; Wang and Sluyter 2013). Activation of P2 purinergic receptors in mature RBCs triggers eicosanoid release and phosphatidylserine exposure and eventually leads to hemolysis (Jiang et al. 2006; Sluyter et al. 2007a, b).
IL-33, the member of the IL-1 cytokine superfamily, is a well-known alarmin that is released upon stress and contributes to the pathogenesis of diverse inflammatory diseases through the activation of innate immune cells (Rider et al. 2017). Recently Wei et al. showed that RBCs contain IL-33 and that IL-33 is released in large amounts upon RBC lysis (Wei et al. 2015). They found association between plasma IL-33 levels and the degree of hemolysis in sickle cell disease patients with intravascular hemolysis (Wei et al. 2015). Similar association between plasma IL-33 concentration and hemolysis was reported in patients with autoimmune hemolytic anemia (Bu et al. 2015). Released IL-33 signals through ST2 receptors and enhances the functions of diverse lymphoid and myeloid immune cells [reviewed in Griesenauer and Paczesny (2017)].
Hsps are ubiquitously expressed proteins exerting diverse protective mechanisms during cellular stress. For example, both constitutive and inducible forms of the 70 kDa Hsp, Hsc70, and Hsp70, respectively, function as cytosolic chaperons during erythrocyte maturation. Although the expressions of Hsc70 and Hsp70 decrease significantly at the terminal stage of erythroid progenitor cell differentiation (Patterson et al. 2009), they are still present in mature RBCs (Gromov and Celis 1991). Vabulas et al. showed that extracellular Hsp70 activates macrophage IL-12 and E-selectin production via CD14/TLR2 and CD14/TLR4 receptor complex-mediated signal transduction pathways (Vabulas et al. 2002). However, recent evidence suggests that the reported cytokine effects of Hsp70 and other Hsps may be due to the contaminating LPS (Tsan and Gao 2004).
Microparticles (MPs) are small membrane-encapsulated vesicles present in body fluids. Blood MPs can originate from platelets, RBCs, leukocytes, or endothelial cells. They are shed from cells in response to cell activation, cell stress, or apoptosis, and besides the phospholipid bilayer, they contain cytosolic components of their parental cells. RBCs release MPs during their normal lifetime in which process they lose a substantial amount of Hb content and surface area (Willekens et al. 2003). Hemoglobinopathies, characterized by shortened life-span of RBCs, such as sickle cell disease and thalassemia major, are associated with accelerated formation of RBC-derived MPs (Tantawy et al. 2013a, b). Interestingly increased levels of RBC-derived MPs are present in patients with metabolic syndrome (Helal et al. 2011). Recently RBC-derived MPs attracted attention in transfusion medicine as well. For therapeutic interventions, packed RBCs are stored in the blood bank for up to 42 days. Storage is associated with diverse morphological and biochemical alterations of RBCs including reduced integrity of the RBC membrane and the formation of RBC-derived MPs (Kim-Shapiro et al. 2011; D’Alessandro et al. 2015). RBC-derived MPs exert diverse biological actions. For example, RBC-derived MPs scavenge nitric oxide (Donadee et al. 2011; Liu et al. 2013) and amplify systemic inflammation via thrombin-dependent activation of complement system (Zecher et al. 2014). Moreover, RBC-derived MPs enhance coagulation activation (van Beers et al. 2008) and are involved in endothelial activation via heme transfer (Camus et al. 2015). RBC-derived MPs are internalized by myeloid cells and induce pro-inflammatory cytokine secretion (Awojoodu et al. 2014). These mechanisms contribute significantly to sickle cell disease-associated vascular dysfunction and cardiovascular complications (Tantawy et al. 2013b) and involved in transfusion-induced inflammatory responses (Cognasse et al. 2015).
9.1.7 Therapeutic Interventions
Different therapeutic approaches were designed and investigated to limit the pathological consequences of massive hemolysis or hemorrhages. Some strategies are focusing on limiting the formation or fostering the elimination of RBC-derived prooxidant and pro-inflammatory molecules. For example, Pamplona et al. showed that CO—the product of heme catabolism—suppress the pathogenesis of experimental cerebral malaria. The effect is mediated by the binding of CO to Hb, preventing Hb oxidation and the generation of free heme, a molecule that plays a critical role in the pathogenesis of cerebral malaria (Pamplona et al. 2007). Recently the therapeutic potential of the natural plasma Hb and heme scavenger proteins, Hp and Hx, have been tested in preclinical animal studies and in small-scale human studies [reviewed in Schaer et al. (2013), Smith and McCulloh (2015)]. In humans Hp supplementation prevented hemoglobinuria or the development of acute kidney injury in a variety of hemolytic conditions [reviewed in Schaer et al. (2013)]. Vinchi et al. showed that Hx therapy improves cardiovascular function in mouse models of sickle cell anemia and β-thalassemia by preventing endothelial dysfunction (Vinchi et al. 2013) and inhibits heme-induced pro-inflammatory phenotypic change of macrophages in a mouse model of sickle cell disease (Vinchi et al. 2016).
Other therapeutic approaches against hemolysis-/hemorrhage-associated adverse effects rely on the induction of the natural antioxidant response. For example upregulation of the NRF2/HO-1 system suppresses the pathogenesis of severe malaria in mice, a pathology driven by RBC-derived heme (Pamplona et al. 2007; Ferreira et al. 2008; Seixas et al. 2009; Jeney et al. 2014). The protective mechanism provided by the NRF2/HO-1 system is very complex and relies on the effective removal of heme, the cytoprotective and anti-inflammatory actions of heme degradation products (bilirubin and CO), and the upregulation of the iron-sequestering protein, ferritin (Gozzelino et al. 2010).
9.2 Conclusions
The RBC is usually a blessing but sometimes a curse. It is a blessing, when it functions properly: circulates throughout the body about 170,000 times during its lifetime to deliver oxygen and remove carbon dioxide from cells and phagocytosed unperceivably at the end of its life-span by macrophages, and curse, when it is involved in pathophysiologic mischief upon hemorrhage or intravascular hemolysis.
Since the dogma breaking “danger model” introduced by Polly Matzinger in 1994 our understanding of how the immune system discriminates between dangerous and safe by recognition of pathogens or alarmins released by injured or stressed cells, underwent a fundamental revision. Diverse endogenous DAMPs were identified and their critical contributions were unquestionably verified in different pathologies. In the last decade, it became evident that upon hemolysis or hemorrhage RBCs release DAMPs that can activate immune and nonimmune cells via diverse signaling mechanisms. A lot of work needs to be done in the future to complete the colorful picture of RBC-derived DAMPs, their targeted cells, and the mechanisms of their actions. Fuller understanding of hemolysis/hemorrhage-associated inflammation could contribute to the development of novel therapeutics intended to interrupt these pathological events.
Abbreviations
- ASC:
-
Apoptosis-associated speck-like protein containing a caspase recruitment domain
- ATP:
-
Adenosine triphosphate
- CO:
-
Carbon monoxide
- Cys:
-
Cysteine
- DAMPs:
-
Damage-associated molecular patterns
- FerrylHb:
-
Ferrylhemoglobin
- Hb:
-
Hemoglobin
- HO-1:
-
Heme oxygenase-1
- H2O2:
-
Hydrogen-peroxide
- Hp:
-
Haptoglobin
- Hsp:
-
Heat shock protein
- Hx:
-
Hemopexin
- ICAM-1:
-
Intracellular adhesion molecule-1
- ICH:
-
Intracerebral hemorrhage
- IL:
-
Interleukin
- LPS:
-
Lipopolysaccharide
- MetHb:
-
Met(ferric) hemoglobin
- Mhem macrophage:
-
Hemorrhage-associated macrophage
- MPs:
-
Microparticles
- MyD88:
-
Myeloid differentiation primary response gene 88
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NF-κB:
-
Nuclear factor kappa B
- NLR:
-
NOD-like receptor
- NLRP3:
-
NLR family pyrin domain containing 3
- NOD:
-
Nucleotide-binding oligomerization domain
- NRF2:
-
Nuclear factor erythroid 2-related factor 2
- PAMPs:
-
Pathogen-associated molecular patterns
- PPIX:
-
Protoporphyrin IX
- RBC:
-
red blood cell
- P2X7:
-
P2X purinoceptor 7
- TLR:
-
Toll-like receptor
- ROS:
-
Reactive oxygen species
- TNF-α:
-
Tumor necrosis factor-alpha
- TRIF:
-
TIR-domain-containing adapter-inducing interferon-β
- Tyr:
-
Tyrosine
- VCAM-1:
-
Vascular cell adhesion molecule-1
References
Alayash AI (2011) Haptoglobin: old protein with new functions. Clin Chim Acta 412(7–8):493–498. https://doi.org/10.1016/j.cca.2010.12.011
Alayash AI, Patel RP, Cashon RE (2001) Redox reactions of hemoglobin and myoglobin: biological and toxicological implications. Antioxid Redox Signal 3(2):313–327. https://doi.org/10.1089/152308601300185250
Andersen CB, Torvund-Jensen M, Nielsen MJ, de Oliveira CL, Hersleth HP, Andersen NH, Pedersen JS, Andersen GR, Moestrup SK (2012) Structure of the haptoglobin-haemoglobin complex. Nature 489(7416):456–459. https://doi.org/10.1038/nature11369
Arruda MA, Graca-Souza AV, Barja-Fidalgo C (2005) Heme and innate immunity: new insights for an old molecule. Mem Inst Oswaldo Cruz 100(7):799–803. doi:/S0074-02762005000700022
Awojoodu AO, Keegan PM, Lane AR, Zhang YY, Lynch KR, Platt MO, Botchwey EA (2014) Acid sphingomyelinase is activated in sickle cell erythrocytes and contributes to inflammatory microparticle generation in SCD. Blood 124(12):1941–1950. https://doi.org/10.1182/blood-2014-01-543652
Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, Bruckner P, Pfeilschifter J, Schaefer RM, Grone HJ, Schaefer L (2009) Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem 284(36):24035–24048. https://doi.org/10.1074/jbc.M109.014266
Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW, Vercellotti GM (1992) Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem 267(25):18148–18153
Banerjee S, Jia Y, Siburt CJ, Abraham B, Wood F, Bonaventura C, Henkens R, Crumbliss AL, Alayash AI (2012) Haptoglobin alters oxygenation and oxidation of hemoglobin and decreases propagation of peroxide-induced oxidative reactions. Free Radic Biol Med 53(6):1317–1326. https://doi.org/10.1016/j.freeradbiomed.2012.07.023
Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, Smith A, Nath KA, Hebbel RP, Vercellotti GM (2014) Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123(3):377–390. https://doi.org/10.1182/blood-2013-04-495887
Bergfeld GR, Forrester T (1992) Release of Atp from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovasc Res 26(1):40–47. https://doi.org/10.1093/Cvr/26.1.40
Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA Jr (1985) Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J Clin Invest 76(5):2003–2011. https://doi.org/10.1172/JCI112200
Boas FE, Forman L, Beutler E (1998) Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc Natl Acad Sci U S A 95(6):3077–3081
Boyle JJ, Harrington HA, Piper E, Elderfield K, Stark J, Landis RC, Haskard DO (2009) Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol 174(3):1097–1108. https://doi.org/10.2353/ajpath.2009.080431
Boyle JJ, Johns M, Lo J, Chiodini A, Ambrose N, Evans PC, Mason JC, Haskard DO (2011) Heme induces heme oxygenase 1 via Nrf2: role in the homeostatic macrophage response to intraplaque hemorrhage. Arterioscler Thromb Vasc Biol 31(11):2685–2691. https://doi.org/10.1161/ATVBAHA.111.225813
Boyle JJ, Johns M, Kampfer T, Nguyen AT, Game L, Schaer DJ, Mason JC, Haskard DO (2012) Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res 110(1):20–33. https://doi.org/10.1161/CIRCRESAHA.111.247577
Bratosin D, Mazurier J, Tissier JP, Estaquier J, Huart JJ, Ameisen JC, Aminoff D, Montreuil J (1998) Cellular and molecular mechanisms of senescent erythrocyte phagocytosis by macrophages. A review. Biochimie 80(2):173–195
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535. https://doi.org/10.1126/science.1092385
Bu XM, Zhang TL, Wang CH, Ren T, Wen ZK (2015) IL-33 reflects dynamics of disease activity in patients with autoimmune hemolytic anemia by regulating autoantibody production. J Transl Med 13:381. https://doi.org/10.1186/S12967-015-0745-0
Buehler PW, Abraham B, Vallelian F, Linnemayr C, Pereira CP, Cipollo JF, Jia Y, Mikolajczyk M, Boretti FS, Schoedon G, Alayash AI, Schaer DJ (2009) Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood 113(11):2578–2586. https://doi.org/10.1182/blood-2008-08-174466
Burnstock G (2015) Blood cells: an historical account of the roles of purinergic signalling. Purinergic Signal 11(4):411–434. https://doi.org/10.1007/s11302-015-9462-7
Burnstock G (2017) Purinergic signaling in the cardiovascular system. Circ Res 120(1):207–228. https://doi.org/10.1161/Circresaha.116.309726
Calay D, Mason JC (2014) The multifunctional role and therapeutic potential of HO-1 in the vascular endothelium. Antioxid Redox Signal 20(11):1789–1809. https://doi.org/10.1089/ars.2013.5659
Camus SM, De Moraes JA, Bonnin P, Abbyad P, Le Jeune S, Lionnet F, Loufrani L, Grimaud L, Lambry JC, Charue D, Kiger L, Renard JM, Larroque C, Le Clesiau H, Tedgui A, Bruneval P, Barja-Fidalgo C, Alexandrou A, Tharaux PL, Boulanger CM, Blanc-Brude OP (2015) Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 125(24):3805–3814. https://doi.org/10.1182/blood-2014-07-589283
Cerqueira BA, Boas WV, Zanette AD, Reis MG, Goncalves MS (2011) Increased concentrations of IL-18 and uric acid in sickle cell anemia: contribution of hemolysis, endothelial activation and the inflammasome. Cytokine 56(2):471–476. https://doi.org/10.1016/j.cyto.2011.08.013
Chahwala SB, Cantley LC (1984) Extracellular ATP induces ion fluxes and inhibits growth of Friend erythroleukemia cells. J Biol Chem 259(22):13717–13722
Champaiboon C, Poolgesorn M, Wisitrasameewong W, Sa-Ard-Iam N, Rerkyen P, Mahanonda R (2014) Differential inflammasome activation by Porphyromonas gingivalis and cholesterol crystals in human macrophages and coronary artery endothelial cells. Atherosclerosis 235(1):38–44. https://doi.org/10.1016/j.atherosclerosis.2014.04.007
Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS (2014) Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 123(24):3818–3827. https://doi.org/10.1182/blood-2013-10-529982
Cinar E, Zhou S, DeCourcey J, Wang YX, Waugh RE, Wan J (2015) Piezo1 regulates mechanotransductive release of ATP from human RBCs. Proc Natl Acad Sci U S A 112(38):11783–11788. https://doi.org/10.1073/pnas.1507309112
Cognasse F, Hamzeh-Cognasse H, Laradi S, Chou ML, Seghatchian J, Burnouf T, Boulanger C, Garraud O, Amabile N (2015) The role of microparticles in inflammation and transfusion: A concise review. Transfus Apher Sci 53(2):159–167. https://doi.org/10.1016/j.transci.2015.10.013
Constantinescu P, Wang B, Kovacevic K, Jalilian I, Bosman GJ, Wiley JS, Sluyter R (2010) P2X7 receptor activation induces cell death and microparticle release in murine erythroleukemia cells. Biochim Biophys Acta 1798(9):1797–1804. https://doi.org/10.1016/j.bbamem.2010.06.002
D’Alessandro A, Kriebardis AG, Rinalducci S, Antonelou MH, Hansen KC, Papassideri IS, Zolla L (2015) An update on red blood cell storage lesions, as gleaned through biochemistry and omics technologies. Transfusion 55(1):205–219. https://doi.org/10.1111/trf.12804
de Back DZ, Kostova EB, van Kraaij M, van den Berg TK, van Bruggen R (2014) Of macrophages and red blood cells; a complex love story. Front Physiol 5:9. https://doi.org/10.3389/fphys.2014.00009
Deterding LJ, Ramirez DC, Dubin JR, Mason RP, Tomer KB (2004) Identification of free radicals on hemoglobin from its self-peroxidation using mass spectrometry and immuno-spin trapping: observation of a histidinyl radical. J Biol Chem 279(12):11600–11607. https://doi.org/10.1074/jbc.M310704200
Donadee C, Raat NJH, Kanias T, Tejero J, Lee JS, Kelley EE, Zhao XJ, Liu C, Reynolds H, Azarov I, Frizzell S, Meyer EM, Donnenberg AD, Qu LR, Triulzi D, Kim-Shapiro DB, Gladwin MT (2011) Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation 124(4):465–U294. https://doi.org/10.1161/Circulationaha.110.008698
Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, Suva ML, Stehle JC, Kopf M, Stamenkovic I, Corradin G, Tschopp J (2009) Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One 4(8):e6510. https://doi.org/10.1371/journal.pone.0006510
Dubyak GR (1991) Signal transduction by P2-purinergic receptors for extracellular ATP. Am J Respir Cell Mol Biol 4(4):295–300. https://doi.org/10.1165/ajrcmb/4.4.295
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464(7293):1357–1361. https://doi.org/10.1038/nature08938
Dutra FF, Bozza MT (2014) Heme on innate immunity and inflammation. Front Pharmacol 5:115. https://doi.org/10.3389/fphar.2014.00115
Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, Zamboni DS, Bozza MT (2014) Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A 111(39):E4110–E4118. https://doi.org/10.1073/pnas.1405023111
Eisenstein RS, Garcia-Mayol D, Pettingell W, Munro HN (1991) Regulation of ferritin and heme oxygenase synthesis in rat fibroblasts by different forms of iron. Proc Natl Acad Sci U S A 88(3):688–692
Ellsworth ML, Forrester T, Ellis CG, Dietrich HH (1995) The erythrocyte as a regulator of vascular tone. Am J Physiol Heart C 269(6):H2155–H2161
Fernandez-Boyanapalli RF, Frasch SC, McPhillips K, Vandivier RW, Harry BL, Riches DW, Henson PM, Bratton DL (2009) Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood 113(9):2047–2055. https://doi.org/10.1182/blood-2008-05-160564
Ferreira A, Balla J, Jeney V, Balla G, Soares MP (2008) A central role for free heme in the pathogenesis of severe malaria: the missing link? J Mol Med 86(10):1097–1111. https://doi.org/10.1007/s00109-008-0368-5
Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, Bozza MT (2007) Characterization of heme as activator of Toll-like receptor 4. J Biol Chem 282(28):20221–20229. https://doi.org/10.1074/jbc.M610737200
Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, Souto-Padron T, De Rosa MJ, Kelliher M, Golenbock D, Chan FK, Bozza MT (2012) Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 119(10):2368–2375. https://doi.org/10.1182/blood-2011-08-375303
George A, Pushkaran S, Konstantinidis DG, Koochaki S, Malik P, Mohandas N, Zheng Y, Joiner CH, Kalfa TA (2013) Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 121(11):2099–2107. https://doi.org/10.1182/blood-2012-07-441188
Gibb DR, Calabro S, Liu D, Tormey CA, Spitalnik SL, Zimring JC, Hendrickson JE, Hod EA, Eisenbarth SC (2016) The Nlrp3 inflammasome does not regulate alloimmunization to transfused red blood cells in mice. EBioMedicine 9:77–86. https://doi.org/10.1016/j.ebiom.2016.06.008
Gozzelino R, Jeney V, Soares MP (2010) Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 50:323–354. https://doi.org/10.1146/annurev.pharmtox.010909.105600
Graca-Souza AV, Arruda MAB, de Freitas MS, Barja-Fidalgo C, Oliveira PL (2002) Neutrophil activation by heme: implications for inflammatory processes. Blood 99(11):4160–4165. https://doi.org/10.1182/blood.V99.11.4160
Gram M, Sveinsdottir S, Ruscher K, Hansson SR, Cinthio M, Akerstrom B, Ley D (2013) Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J Neuroinflammation 10:100. https://doi.org/10.1186/1742-2094-10-100
Griesenauer B, Paczesny S (2017) The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol 8:475. https://doi.org/10.3389/Fimmu.2017.00475
Gromov PS, Celis JE (1991) Identification of two molecular chaperons (HSX70, HSC70) in mature human erythrocytes. Exp Cell Res 195(2):556–559
Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21(7):677–687. https://doi.org/10.1038/nm.3893
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9(8):857–865. https://doi.org/10.1038/ni.1636
Harel S, Kanner J (1988) The generation of ferryl or hydroxyl radicals during interaction of haemproteins with hydrogen peroxide. Free Radic Res Commun 5(1):21–33
Helal O, Defoort C, Robert S, Marin C, Lesavre N, Lopez-Miranda J, Riserus U, Basu S, Lovegrove J, McMonagle J, Roche HM, Dignat-George F, Lairon D (2011) Increased levels of microparticles originating from endothelial cells, platelets and erythrocytes in subjects with metabolic syndrome: relationship with oxidative stress. Nutr Metab Cardiovasc Dis 21(9):665–671. https://doi.org/10.1016/j.numecd.2010.01.004
Herz J, Strickland DK (2001) LRP: a multifunctional scavenger and signaling receptor. J Clin Invest 108(6):779–784. https://doi.org/10.1172/JCI13992
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9(8):847–856. https://doi.org/10.1038/ni.1631
Hrkal Z, Vodrazka Z, Kalousek I (1974) Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. Eur J Biochem/FEBS 43(1):73–78
Huck O, Elkaim R, Davideau JL, Tenenbaum H (2015) Porphyromonas gingivalis-impaired innate immune response via NLRP3 proteolysis in endothelial cells. Innate Immun 21(1):65–72. https://doi.org/10.1177/1753425914523459
Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK (2005) Identification of the receptor scavenging hemopexin-heme complexes. Blood 106(7):2572–2579. https://doi.org/10.1182/blood-2005-03-1185
Idzko M, Ferrari D, Eltzschig HK (2014) Nucleotide signalling during inflammation. Nature 509(7500):310–317. https://doi.org/10.1038/nature13085
Immenschuh S, Vijayan V, Janciauskiene S, Gueler F (2017) Heme as a target for therapeutic interventions. Front Pharmacol 8:146. https://doi.org/10.3389/Fphar.2017.00146
Jeney V, Eaton JW, Balla G, Balla J (2013) Natural history of the bruise: formation, elimination, and biological effects of oxidized hemoglobin. Oxidative Med Cell Longev 2013:703571. https://doi.org/10.1155/2013/703571
Jeney V, Balla G, Balla J (2014) Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front Physiol 5:379. https://doi.org/10.3389/fphys.2014.00379
Jia Y, Buehler PW, Boykins RA, Venable RM, Alayash AI (2007) Structural basis of peroxide-mediated changes in human hemoglobin: a novel oxidative pathway. J Biol Chem 282(7):4894–4907. https://doi.org/10.1074/jbc.M609955200
Jiang HL, Zhu AG, Wong PYK, McGiff JC (2006) Stimulation of rat erythrocyte P2X7 receptor induces the release of epoxyeicosatrienoic acids. FASEB J 20(4):A483–A483
Kapralov A, Vlasova II, Feng W, Maeda A, Walson K, Tyurin VA, Huang Z, Aneja RK, Carcillo J, Bayir H, Kagan VE (2009) Peroxidase activity of hemoglobin-haptoglobin complexes: covalent aggregation and oxidative stress in plasma and macrophages. J Biol Chem 284(44):30395–30407. https://doi.org/10.1074/jbc.M109.045567
Kim-Shapiro DB, Lee J, Gladwin MT (2011) Storage lesion: role of red blood cell breakdown. Transfusion 51(4):844–851. https://doi.org/10.1111/j.1537-2995.2011.03100.x
Kono M, Saigo K, Takagi Y, Kawauchi S, Wada A, Hashimoto M, Sugimoto T, Takenokuchi M, Morikawa T, Funakoshi K (2013) Morphological and flow-cytometric analysis of haemin-induced human neutrophil activation: implications for transfusion-related acute lung injury. Blood transfus 11(1):53–60. https://doi.org/10.2450/2012.0141-11
Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, Moestrup SK (2001) Identification of the haemoglobin scavenger receptor. Nature 409(6817):198–201. https://doi.org/10.1038/35051594
Kwon MS, Woo SK, Kurland DB, Yoon SH, Palmer AF, Banerjee U, Iqbal S, Ivanova S, Gerzanich V, Simard JM (2015) Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int J Mol Sci 16(3):5028–5046. https://doi.org/10.3390/ijms16035028
Land WG (2013) Transfusion-related acute lung injury: the work of DAMPs. Transfus Med Hemother 40(1):3–13. https://doi.org/10.1159/000345688
Lang KS, Lang PA, Bauer C, Duranton C, Wieder T, Huber SM, Lang F (2005) Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem 15(5):195–202. https://doi.org/10.1159/000086406
Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A, Marguti I, Cardoso S, Sepulveda N, Smith A, Soares MP (2010) A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med 2(51):51ra71. https://doi.org/10.1126/scitranslmed.3001118
Li Q, Fu W, Yao J, Ji Z, Wang Y, Zhou Z, Yan J, Li W (2014) Heme induces IL-1beta secretion through activating NLRP3 in kidney inflammation. Cell Biochem Biophys 69(3):495–502. https://doi.org/10.1007/s12013-014-9823-9
Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, Wang JZ, Su BY, Yang QW (2012) Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation 9:46. https://doi.org/10.1186/1742-2094-9-46
Liu C, Zhao WX, Christ GJ, Gladwin MT, Kim-Shapiro DB (2013) Nitric oxide scavenging by red cell microparticles. Free Radic Biol Med 65:1164–1173. https://doi.org/10.1016/j.freeradbiomed.2013.09.002
Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440(7081):228–232. https://doi.org/10.1038/nature04515
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440(7081):237–241. https://doi.org/10.1038/nature04516
Matzinger P (1994) Tolerance, danger, and the extended family. Annu Rev Immunol 12:991–1045. https://doi.org/10.1146/annurev.iy.12.040194.005015
McClenahan D, Hillenbrand K, Kapur A, Carlton D, Czuprynski C (2009) Effects of extracellular ATP on bovine lung endothelial and epithelial cell monolayer morphologies, apoptoses, and permeabilities. Clin Vaccine Immunol 16(1):43–48. https://doi.org/10.1128/Cvi.00282-08
Mendonca R, Silveira AA, Conran N (2016) Red cell DAMPs and inflammation. Inflamm Res 65(9):665–678. https://doi.org/10.1007/s00011-016-0955-9
Miller YI, Altamentova SM, Shaklai N (1997) Oxidation of low-density lipoprotein by hemoglobin stems from a heme-initiated globin radical: antioxidant role of haptoglobin. Biochemistry 36(40):12189–12198. https://doi.org/10.1021/bi970258a
Mohanty JG, Nagababu E, Rifkind JM (2014) Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front Physiol 5:84. https://doi.org/10.3389/fphys.2014.00084
Muckenthaler MU, Rivella S, Hentze MW, Galy B (2017) A red carpet for iron metabolism. Cell 168(3):344–361. https://doi.org/10.1016/j.cell.2016.12.034
Murray RK, Connell GE, Pert JH (1961) The role of haptoglobin in the clearance and distribution of extracorpuscular hemoglobin. Blood 17:45–53
Nagy E, Eaton JW, Jeney V, Soares MP, Varga Z, Galajda Z, Szentmiklosi J, Mehes G, Csonka T, Smith A, Vercellotti GM, Balla G, Balla J (2010) Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler Thromb Vasc Biol 30(7):1347–1353. https://doi.org/10.1161/ATVBAHA.110.206433
Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha-Rodrigues M, Portugal S, Soares MP, Mota MM (2007) Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med 13(6):703–710. https://doi.org/10.1038/nm1586
Patel RP, Svistunenko DA, Darley-Usmar VM, Symons MC, Wilson MT (1996) Redox cycling of human methaemoglobin by H2O2 yields persistent ferryl iron and protein based radicals. Free Radic Res 25(2):117–123
Patterson ST, Li J, Kang JA, Wickrema A, Williams DB, Reithmeier RAF (2009) Loss of specific chaperones involved in membrane glycoprotein biosynthesis during the maturation of human erythroid progenitor cells. J Biol Chem 284(21):14547–14557. https://doi.org/10.1074/jbc.M809076200
Perregaux DG, McNiff P, Laliberte R, Conklyn M, Gabel CA (2000) ATP acts as an agonist to promote stimulus-induced secretion of IL-1 beta and IL-18 in human blood. J Immunol 165(8):4615–4623
Pimenova T, Pereira CP, Gehrig P, Buehler PW, Schaer DJ, Zenobi R (2010) Quantitative mass spectrometry defines an oxidative hotspot in hemoglobin that is specifically protected by haptoglobin. J Proteome Res 9(8):4061–4070. https://doi.org/10.1021/pr100252e
Piomelli S, Seaman C (1993) Mechanism of red blood cell aging: relationship of cell density and cell age. Am J Hematol 42(1):46–52
Pohlman TH, Stanness KA, Beatty PG, Ochs HD, Harlan JM (1986) An endothelial cell surface factor(s) induced in vitro by lipopolysaccharide, interleukin 1, and tumor necrosis factor-alpha increases neutrophil adherence by a CDw18-dependent mechanism. J Immunol 136(12):4548–4553
Porto BN, Alves LS, Fernandez PL, Dutra TP, Figueiredo RT, Graca-Souza AV, Bozza MT (2007) Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J Biol Chem 282(33):24430–24436. https://doi.org/10.1074/jbc.M703570200
Potor L, Banyai E, Becs G, Soares MP, Balla G, Balla J, Jeney V (2013) Atherogenesis may involve the prooxidant and proinflammatory effects of ferryl hemoglobin. Oxidative Med Cell Longev 2013:676425. https://doi.org/10.1155/2013/676425
Ramirez DC, Chen YR, Mason RP (2003) Immunochemical detection of hemoglobin-derived radicals formed by reaction with hydrogen peroxide: involvement of a protein-tyrosyl radical. Free Radic Biol Med 34(7):830–839
Reeder BJ, Cutruzzola F, Bigotti MG, Hider RC, Wilson MT (2008) Tyrosine as a redox-active center in electron transfer to ferryl heme in globins. Free Radic Biol Med 44(3):274–283. https://doi.org/10.1016/j.freeradbiomed.2007.06.030
Reimer T, Shaw MH, Franchi L, Coban C, Ishii KJ, Akira S, Horii T, Rodriguez A, Nunez G (2010) Experimental cerebral malaria progresses independently of the Nlrp3 inflammasome. Eur J Immunol 40(3):764–769. https://doi.org/10.1002/eji.200939996
Rider P, Voronov E, Dinarello CA, Apte RN, Cohen I (2017) Alarmins: feel the stress. J Immunol 198(4):1395–1402. https://doi.org/10.4049/jimmunol.1601342
Rosales C, Demaurex N, Lowell CA, Uribe-Querol E (2016) Neutrophils: their role in innate and adaptive immunity. J Immunol Res 2016:1469780. https://doi.org/10.1155/2016/1469780
Rother RP, Bell L, Hillmen P, Gladwin MT (2005) The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA 293(13):1653–1662. https://doi.org/10.1001/jama.293.13.1653
Schaer DJ, Schaer CA, Buehler PW, Boykins RA, Schoedon G, Alayash AI, Schaffner A (2006) CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood 107(1):373–380. https://doi.org/10.1182/blood-2005-03-1014
Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM (2013) Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121(8):1276–1284. https://doi.org/10.1182/blood-2012-11-451229
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140(6):821–832. https://doi.org/10.1016/j.cell.2010.01.040
Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, Rebelo S, Penido C, Smith NR, Coutinho A, Soares MP (2009) Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci U S A 106(37):15837–15842. https://doi.org/10.1073/pnas.0903419106
Siems WG, Sommerburg O, Grune T (2000) Erythrocyte free radical and energy metabolism. Clin Nephrol 53(1 Suppl):S9–S17
Sikora J, Orlov SN, Furuya K, Grygorczyk R (2014) Hemolysis is a primary ATP-release mechanism in human erythrocytes. Blood 124(13):2150–2157. https://doi.org/10.1182/blood-2014-05-572024
Silva G, Jeney V, Chora A, Larsen R, Balla J, Soares MP (2009) Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J Biol Chem 284(43):29582–29595. https://doi.org/10.1074/jbc.M109.045344
Sluyter R (2015) P2X and P2Y receptor signaling in red blood cells. Front Mol Biosci 2:60. https://doi.org/10.3389/fmolb.2015.00060
Sluyter R, Shemon AN, Hughes WE, Stevenson RO, Georgiou JG, Eslick GD, Taylor RM, Wiley JS (2007a) Canine erythrocytes express the P2X(7) receptor: greatly increased function compared with human erythrocytes. Am J Physiol Reg I 293(5):R2090–R2098. https://doi.org/10.1152/ajpregu.00166.2007
Sluyter R, Shemon AN, Wiley JS (2007b) P2X(7) receptor activation causes phosphatidylserine exposure in human erythrocytes. Biochem Biophys Res Commun 355(1):169–173. https://doi.org/10.1016/j.bbrc.2007.01.124
Smith A, McCulloh RJ (2015) Hemopexin and haptoglobin: allies against heme toxicity from hemoglobin not contenders. Front Physiol 6:187. https://doi.org/10.3389/fphys.2015.00187
Soares MP, Bozza MT (2016) Red alert: labile heme is an alarmin. Curr Opin Immunol 38:94–100. https://doi.org/10.1016/j.coi.2015.11.006
Sridharan M, Adderley SP, Bowles EA, Egan TM, Stephenson AH, Ellsworth ML, Sprague RS (2010) Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am J Physiol Heart C 299(4):H1146–H1152. https://doi.org/10.1152/ajpheart.00301.2010
Stanojcic M, Chen P, Harrison RA, Wang V, Antonyshyn J, Zuniga-Pflucker JC, Jeschke MG (2014) Leukocyte infiltration and activation of the NLRP3 inflammasome in white adipose tissue following thermal injury. Crit Care Med 42(6):1357–1364. https://doi.org/10.1097/CCM.0000000000000209
Tantawy AA, Adly AA, Ismail EA, Habeeb NM (2013a) Flow cytometric assessment of circulating platelet and erythrocytes microparticles in young thalassemia major patients: relation to pulmonary hypertension and aortic wall stiffness. Eur J Haematol 90(6):508–518. https://doi.org/10.1111/ejh.12108
Tantawy AA, Adly AA, Ismail EA, Habeeb NM, Farouk A (2013b) Circulating platelet and erythrocyte microparticles in young children and adolescents with sickle cell disease: relation to cardiovascular complications. Platelets 24(8):605–614. https://doi.org/10.3109/09537104.2012.749397
Tenhunen R, Marver HS, Schmid R (1968) The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci U S A 61(2):748–755
Tsan MF, Gao BC (2004) Cytokine function of heat shock proteins. Am J Physiol Cell Physiol 286(4):C739–C744. https://doi.org/10.1152/ajpcell.00364.2003
Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H (2002) HSP70 as endogenous stimulus of the toll/interleukin-1 receptor signal pathway. J Biol Chem 277(17):15107–15112. https://doi.org/10.1074/jbc.M111204200
Vallelian F, Pimenova T, Pereira CP, Abraham B, Mikolajczyk MG, Schoedon G, Zenobi R, Alayash AI, Buehler PW, Schaer DJ (2008) The reaction of hydrogen peroxide with hemoglobin induces extensive alpha-globin crosslinking and impairs the interaction of hemoglobin with endogenous scavenger pathways. Free Radic Biol Med 45(8):1150–1158. https://doi.org/10.1016/j.freeradbiomed.2008.07.013
van Beers EJ, Schaap MC, Berckmans RJ, Nieuwland R, Sturk A, Meijers JCM, Biemond BJ (2008) Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Blood 112(11):52–53
Vinchi F, De Franceschi L, Ghigo A, Townes T, Cimino J, Silengo L, Hirsch E, Altruda F, Tolosano E (2013) Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 127(12):1317–1329. https://doi.org/10.1161/CIRCULATIONAHA.112.130179
Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, Cerwenka A, Tolosano E, Muckenthaler MU (2016) Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 127(4):473–486. https://doi.org/10.1182/blood-2015-08-663245
von Albertini M, Palmetshofer A, Kaczmarek E, Koziak K, Stroka D, Grey ST, Stuhlmeier KM, Robson SC (1998) Extracellular ATP and ADP activate transcription factor NF-kappa B and induce endothelial cell apoptosis. Biochem Biophys Res Commun 248(3):822–829. https://doi.org/10.1006/bbrc.1998.9055
Wagener FA, Feldman E, de Witte T, Abraham NG (1997) Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc Soc Exp Biol Med 216(3):456–463
Wan JD, Ristenpart WD, Stone HA (2008) Dynamics of shear-induced ATP release from red blood cells. Proc Natl Acad Sci U S A 105(43):16432–16437. https://doi.org/10.1073/pnas.0805779105
Wang B, Sluyter R (2013) P2X7 receptor activation induces reactive oxygen species formation in erythroid cells. Purinergic Signal 9(1):101–112. https://doi.org/10.1007/s11302-012-9335-2
Wei JX, Zhao J, Schrott V, Zhang YZ, Gladwin M, Bullock G, Zhao YT (2015) Red blood cells store and release interleukin-33. J Investig Med 63(6):806–810. https://doi.org/10.1097/Jim.0000000000000213
Willekens FL, Roerdinkholder-Stoelwinder B, Groenen-Dopp YA, Bos HJ, Bosman GJ, van den Bos AG, Verkleij AJ, Werre JM (2003) Hemoglobin loss from erythrocytes in vivo results from spleen-facilitated vesiculation. Blood 101(2):747–751. https://doi.org/10.1182/blood-2002-02-0500
Willekens FL, Werre JM, Groenen-Dopp YA, Roerdinkholder-Stoelwinder B, de Pauw B, Bosman GJ (2008) Erythrocyte vesiculation: a self-protective mechanism? Br J Haematol 141(4):549–556. https://doi.org/10.1111/j.1365-2141.2008.07055.x
Yazdi AS, Guarda G, Riteau N, Drexler SK, Tardivel A, Couillin I, Tschopp J (2010) Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1 alpha and IL-1 beta. Proc Natl Acad Sci U S A 107(45):19449–19454. https://doi.org/10.1073/pnas.1008155107
Zecher D, Cumpelik A, Schifferli JA (2014) Erythrocyte-derived microvesicles amplify systemic inflammation by thrombin-dependent activation of complement. Arterioscler Thromb Vasc 34(2):313–320. https://doi.org/10.1161/Atvbaha.113.302378
Acknowledgments
This work was supported by grant from the National Research, Development and Innovation Office (NKFIH grant number: K116024).
Conflict of Interest
The author has no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Jeney, V. (2018). Pro-Inflammatory Actions of Red Blood Cell-Derived DAMPs. In: Cordero, M., Alcocer-Gómez, E. (eds) Inflammasomes: Clinical and Therapeutic Implications. Experientia Supplementum, vol 108. Springer, Cham. https://doi.org/10.1007/978-3-319-89390-7_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-89390-7_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-89389-1
Online ISBN: 978-3-319-89390-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)