
Abstract
Diabetes mellitus, a metabolic disorder characterized by high blood glucose, is one of the main risk factors in the development of vascular complications affecting both large and small blood vessels. A major challenge is the discovery of new mediators and biomarkers for diabetes-related vascular complications. In this regard, accumulating evidence indicate that endothelial progenitor cells (EPCs), derived from the bone marrow and peripheral blood, are critical for the maintenance and regeneration of endothelial cells contributing to repair and restoration of vascular wall integrity. The studies reveal that the reduced number of circulating EPCs under diabetic conditions can predict cardiovascular outcomes, and EPC dysfunction could contribute to the pathogenesis of diabetes – associated vascular disease.
This chapter discusses the EPC dysfunction in relationship to vascular complications of diabetes, highlighting the pathophysiology of diabetic vascular complications, mechanisms leading to EPC dysfunction in diabetes and diabetic vascular complications, significance of EPCs in the pathogenesis of vascular complications of diabetes and potential therapeutic implications of EPCs in diabetes-associated vascular complications. In particular, to understand the EPC significance in diabetes, the effects of hyperglycaemia, insulin resistance, insulin like growth factor 1, nitric oxide, oxidative stress, PI3K/Akt signaling pathway, inflammation, and of altered microRNA expression on the EPC dysfunctionality have been considered.
A comprehensive knowledge of EPC role in all diabetic complications may help to develop new research strategies to demonstrate and consolidate their clinical relevance so that they become diagnostic biomarkers and pharmacological targets to prevent and treat diabetes-related vascular complications. Increasing the number and functional capacity of EPCs by different approaches may favorably modify the risk for cardiovascular complications and survival for people suffering from diabetes.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Endothelial progenitor cells
- Diabetes
- Cardiovascular disease
- Cerebrovascular disease
- Vascular complications
- Endothelial dysfunction
- Hyperglycemia
- Insulin resistance
1 Introduction
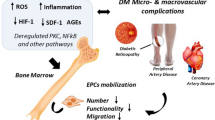
Diabetes mellitus represents a very serious issue in every public health system. Its worldwide prevalence is continuously increasing; recent statistics released by the International Diabetes Federation reveal that 1 in 11 adults suffer from diabetes (415 millions) and those numbers will increase to 1 in 10 adults (642 millions) by the year 2040 [1]. The global rise in diabetes occurs due to population growth and ageing, genetic susceptibility and to increasing trends towards an unhealthy diet, obesity, and sedentary lifestyle. The vascular complications of diabetes are among the most serious manifestations of the disease. Patients with type 2 diabetes (T2DM) represent about 85–95% of the people with diabetes in developed countries and an even higher percentage in developing countries [1]. The microvascular complications , like nephropathy , retinopathy or neuropathy , as well as the macrovascular ones – atherosclerotic disease in all its forms: ischaemic heart disease, cerebrovascular disease , or peripheral arterial disease (PAD) are usually irreversible and lead to a decrease in life expectancy and to a higher mortality rate in these patients.
Despite the progress made in the last few years, vascular complications due to diabetes mellitus still remain a huge problem, and identifying new mechanisms involved in their development, like dysfunction of endothelial progenitor cells (EPCs), could lead to new curative and preventive therapeutic options.
2 Pathophysiology of Diabetic Vascular Complications
2.1 Diabetes and Vascular Risk Factors
It is well known that diabetic patients are more frequently affected by cardiovascular disease (CVD) compared with those without diabetes. CVD increases the rate of all-cause death nearly threefold and the rate of cardiovascular death nearly fivefold in subjects with diabetes [2]. Most of this excess risk is associated with an increased prevalence of well-known traditional risk factors such as hypertension , dyslipidaemia, obesity (generalised or visceral), and smoking in these subjects. Hypertension is more than twice as common in people with diabetes as in people with normal blood glucose levels [3]. Premenopausal women who have diabetes have an increased risk of heart disease because diabetes cancels out the protective effects of estrogen. Nevertheless, these established risk factors do not fully explain the excess risk for CVD associated with diabetes.
Therefore, other non-traditional risk factors may be important in people with diabetes: insulin resistance and hyperinsulinemia; postprandial hyperglycaemia and glucose variability; microalbuminuria ; haematological and thrombogenic factors; inflammation assessed by high-sensitivity C-reactive protein; homocysteine and vitamins; genetics and epigenetics [4, 5].
Large clinical trials in type I diabetes mellitus (T1DM) and type II diabetes mellitus (T2DM) have demonstrated that hyperglycaemia plays an important role in the pathogenesis of microvascular complications [6]. Although diabetic patients with the most severe hyperglycaemia have the highest risk of microangiopathy , hyperglycaemia, however, is a necessary, but not sufficient, cause of clinically important microangiopathy . Hypertension, smoking , hypercholesterolaemia, dyslipidaemia, obesity and hyperhomocysteinaemia are additional major causes of microangiopathy. The risk of macroangiopathy does not appear to be strongly related to hyperglycaemia, but is related to general risk factors for atherothrombosis, such as age, smoking , hypertension , hypercholesterolaemia, dyslipidaemia, obesity and hyperhomocysteinaemia . Cardiovascular risk factors such as hypertension , dyslipidaemia, obesity, insulin resistance, hyperinsulinaemia and impaired fibrinolysis cluster in the metabolic syndrome [7]. All of the above-mentioned factors create a state of constant and progressive damage to the vascular wall, manifested by a low-grade inflammatory process and endothelial dysfunction [8].
2.2 Diabetes and Vascular Complications
2.2.1 Microvascular Complications
Diabetic Retinopathy
This is one of the most important microvascular complications in diabetes mellitus and is a leading cause of visual impairment in working-age adults [9]. Development of diabetic retinopathy in patients with T2DM was found to be related to the severity of hyperglycemia , duration of diabetes, and presence of hypertension [10].
Retinopathy is classified as nonproliferative (background) or proliferative. The most common early clinically visible manifestations of diabetic retinopathy include microaneurysm formation and intraretinal hemorrhages. Microvascular damage leads to retinal capillary nonperfusion, cotton wool spots , increased number of hemorrhages, venous abnormalities, and intraretinal microvascular abnormalities . During this stage, increased vasopermeability can result in retinal thickening (edema) and/or exudates that may lead to a loss in central visual acuity. Proliferative retinopathy is characterized by the formation of new blood vessels on the surface of the retina and can lead to vitreous hemorrhage. White areas on the retina (“cotton wool spots ”) can be a sign of impending proliferative retinopathy . These new vessels then lead to traction retinal detachments and neovascular glaucoma , respectively. Vision can be lost in this stage as a result of capillary nonperfusion or edema in the macula, vitreous hemorrhage, and distortion or traction retinal detachment [11].
Diabetic Nephropathy
It is one of the most common complications of diabetes mellitus. Among patients with T1DM, the incidence of diabetic nephropathy has decreased to 10–15% in more recent cohorts [12]. However, due to the increase in T2DM, the absolute prevalence of diabetic nephropathy has increased over the past two decades; in 2015, diabetic nephropathy was reported to be the cause of 43.9% of all cases of end-stage renal disease (ESRD) in the United States [13].
Diabetic nephropathy is characterized by an expanded mesangial volume, changes in the physical and biochemical properties of the glomerular basement membrane, and a decreased glomerular filtration rate . Diabetic nephropathy is a clinical syndrome characterized by the following: persistent albuminuria (>300 mg/day or >200 μg/min) that is confirmed on at least two occasions, 3–6 months apart; progressive decline in the glomerular filtration rate ; elevated arterial blood pressure [14]. It is preceded by lower degrees of proteinuria , or “microalbuminuria ” defined as albumin excretion of 30–299 mg/24 h. In the absence of an intervention , diabetic patients with microalbuminuria typically progress to proteinuria and overt diabetic nephropathy . This progression occurs in both T1DM and T2DM. As many as 7% of patients with T2DM may already have microalbuminuria at the time they are diagnosed with diabetes [15]. The evidence suggests that early treatment delays or prevents the onset of diabetic nephropathy or diabetic kidney disease.
The exact cause of diabetic nephropathy is unknown, but the main mechanisms are: hyperglycemia (causing hyperfiltration and renal injury), advanced glycation end-products (AGEs), and activation of cytokines . More recent research highlights the role of toll-like receptors, regulatory T-cells (Treg), and increased expression of transforming growth factor β (TGF-β) in the glomeruli [16]. TGF-β and vascular endothelial growth factor (VEGF) may contribute to the cellular hypertrophy and collagen synthesis and may induce the vascular changes observed in persons with diabetic nephropathy . Hyperglycemia also may activate protein kinase C (PKC), which may contribute to renal disease and other vascular complications of diabetes. Moreover, hypergycemia was shown to induce renal artery dysfunction in streptozotocin-induced diabetic mice [17]. This study has reported that the renal artery dysfunction is the result of the reduction of nitric oxide (NO ) bioavailability, endothelial nitric oxide synthase (eNOS ) expression, phospholipase C activity, and intracellular free calcium concentrations [17].
Diabetic Neuropathy
It has become the most common complication of diabetes, affecting as many as 50% of patients with T1DM and T2DM [18]. In T1DM, distal polyneuropathy typically becomes symptomatic after many years of chronic prolonged hyperglycemia , whereas in T2DM, it may be apparent after only a few years of known poor glycemic control or even at diagnosis. Chronic sensori-motor distal symmetric polyneuropathy is the most common form of neuropathy in diabetes. Diabetic autonomic neuropathy also causes significant morbidity in patients with diabetes. Neurological dysfunction may occur in most organ systems and can manifest by gastroparesis, constipation, diarrhea, anhidrosis, bladder dysfunction, erectile dysfunction, exercise intolerance, resting tachycardia, silent ischemia, and even sudden cardiac death [19].
Development of symptoms depends on many factors, such as total hyperglycemic exposure and other risk factors such as elevated lipids , blood pressure, smoking , increased height, and high exposure to other potentially neurotoxic agents such as ethanol. Genetic factors may also play a role. Important contributing biochemical mechanisms in the development of the more common symmetrical forms of diabetic polyneuropathy likely include the polyol pathway , AGEs, and oxidative stress [20].
2.2.2 Macrovascular Complications
Atherosclerosis
This is the central pathological mechanism in diabetic macrovascular disease. CVD is the primary cause of death in people with either T1DM or T2DM. T2DM is one of the components of metabolic syndrome which also includes abdominal obesity, hypertension , hyperlipidemia and increased coagulability; these factors act together to promote CVD.
Atherosclerosis results from chronic inflammation and injury to the arterial wall in the peripheral or coronary vascular system. The result of the process is the formation of a lipid-rich atherosclerotic lesion with a fibrous cap . The rupture of this lesion leads to acute vascular infarction [21]. Other mechanisms involved in macrovascular disease are: increased platelet adhesion and hypercoagulability, impaired NO generation, increased free radical formation in platelets and increased levels of plasminogen activator inhibitor type 1 (PAI-1) [22, 23].
Coronary Heart Disease
Coronary heart disease (CHD) has been associated with diabetes in numerous studies beginning with the Framingham study [24]. Other studies have shown that the risk of myocardial infarction (MI) in people with diabetes is equivalent to the risk in nondiabetic patients with a history of previous MI [25]. These results have lead to the recommendations of the American Diabetes Association and American Heart Association that diabetes should be considered a coronary artery disease (CAD) risk equivalent rather than a risk factor [26].
Stroke and Cerebrovascular Disease
Stroke and cerebrovascular disease have a higher incidence in patients with diabetes, the later being a strong independent predictor factor for these conditions [27]. Risk of stroke -related dementia and recurrence, as well as stroke-related mortality , is elevated in patients with diabetes [22].
Various subtypes of cerebrovascular diseases have been defined in T2DM. Lacunar strokes or the occlusion of the penetrating arteries that provide blood to the brain deep structures are the main subtypes of cerebrovascular disease in diabetic patients. It is considered that 28–43% of lacunar strokes are due to diabetes [28]. Ischemic stroke , caused by occlusion of the large cerebral vessels, and transient ischemic attacks are found in a smaller percentage compared to lacunar strokes and are mainly due to the strong association between diabetes mellitus and other cardiovascular risk factors [29]. Hemorrhagic stroke is also frequent in diabetic patients as several studies have assigned a relative risk for hemorrhagic stroke of 2.4 in diabetic patients [30].
Diabetes is an independent predictor of poor outcomes [31]. Various studies have highlighted the impact of hyperglycemia during the post-stroke phase. Apparently, hyperglycemia ≥155 mg/dL in patients with stroke, with or without diabetes, is associated with a higher risk of short-term mortality and a reduced chance of recovery [32].
Diabetes contributes significantly and increasingly to the burden of stroke [33]. In the INTERSTROKE case–control study, diabetes increased the rate of stroke by 35% when comparing the top to the bottom tertile, and has been associated with 5% of the population attributable risk for stroke [34]. The Emerging Risk Factors Collaboration analysed 698 782 people from 102 prospective studies, finding that diabetes was associated with a 2.27-fold increase in the risk of ischaemic stroke and 56% excess rate of haemorrhagic stroke [35]. Following stroke, diabetes attenuates cognitive recovery, limits functional outcome, and increases mortality . Diabetes increases the risk of recurrent stroke as well. In the Life Long After Cerebral ischemia (LiLAC) cohort study, diabetes increased the risk of recurrent fatal and non-fatal stroke more than two-fold [36].
Peripheral arterial disease Peripheral arterial disease (PAD) is another macrovascular complication in diabetic patients. Compared with patients without diabetes, patients with diabetes had a higher prevalence of PAD (26.3 vs. 15.3%) and intermittent claudication (5.1% vs. 2.1%) [37]. The rate of PAD in patients with diabetes also increases with age, as it does in non-diabetic persons. The PAD occurs earlier and is often more severe and diffuse [38]. In a multicentre cross-sectional study of patients older than 70 years with diabetes, 71% had PAD when detected by abnormal ankle–brachial index [39]. Diabetes increases the incidence of critical limb ischaemia (CLI) four-fold in patients with peripheral artery disease; moreover, in diabetic patients with CLI, 50% will develop CLI in the contralateral limb within 5 years [40].
Intermittent claudication occurs three times more often in men with diabetes and almost nine times more often in women with diabetes than in their counterparts without diabetes [41]. It is also important to note that diabetes is most strongly associated with femoral-popliteal and tibial PAD, whereas other risk factors (e.g. smoking and hypertension ) are associated with more proximal disease in the aorto-ilio-femoral vessels [33].
The true prevalence of PAD in people with diabetes has been difficult to determine, as most patients are asymptomatic, many do not report their symptoms as pain perception may be blunted by the presence of peripheral neuropathy [42]. Given the inconsistencies of clinical findings in the diagnosis of PAD in the diabetic patient, the measurement of ankle-brachial pressure index (ABI) has emerged as the relatively simple, non-invasive and inexpensive diagnostic tool of choice. An ABI smaller than 0.9 is not only diagnostic of PAD in the asymptomatic patients, but it is also an independent marker of increased morbidity and mortality from CVD [43].
2.3 Molecular Basis of the Vascular Dysfunction in Diabetes and Diabetic Vascular Complications
A better understanding of the mechanisms underlying diabetic vascular disease is mandatory because it may provide novel approaches to prevent or delay the development of its complications . The common etiology link for the different types of diabetes-associated vascular diseases is chronic hyperglycemia that evokes pathologic responses in the vasculature, which finally cause constitutive NO inhibition, smooth muscle cell dysfunction, overproduction of vascular endothelial growth factor , chronic inflammatio n, hemodynamic dysregulation, impaired fibrinolytic ability and enhanced platelet aggregation [44].
2.3.1 Hyperglycemia , Oxidative Stress and Vascular Disease in Diabetes
Vascular dysfunction in diabetes is based upon endothelial and smooth muscle cells dysfunction which eventually leads to atherothrombosis. Micro- and macrovascular complications are mainly due to prolonged exposure to hyperglycemia and its frequent association with other risk factors and genetic susceptibility [45]. Interestingly, the endothelial, mesangial and retinal cells are equipped to handle high sugar levels when compared with other cell types [46]. The detrimental effects of glucose already occur with glycemic levels below the threshold for the diagnosis of diabetes; this is explained by the concept of ‘glycemic continuum ’ across the spectrum of prediabetes, diabetes and cardiovascular risk [45, 47]. There is a strong relationship between dysglycemia, obesity-related insulin resistance and impaired insulin secretion that will determine functional and structural alterations of the vessel wall. Endothelial dysfunction occurs as a consequence of the imbalance between the accumulation of reactive oxygen species (ROS) and NO bioavailability, a decrease in the latter being a strong predictor of cardiovascular events [48]. The overproduction of ROS by the mitochondria is considered one of the key triggers of vascular complications in diabetes [49].
Schematically (Fig. 8.1), high concentrations of intracellular glucose determine [45]:
-
PKC activation, followed by:
-
increased nicotinamide adenine dinucleotide phosphate (NADPH ) oxidase levels [50], phosphorylate p66Shc at serine 36 [51], and oxidative stress and ROS generation; all of which quickly inactivate NO and facilitate peroxynitrite (ONOO−) formation, a pro-oxidant compound responsible for protein nitrosylation;
-
eNOS deregulation with decreased activity, further reduction of NO availability, and accumulation of free radicals [52]; furthermore, hyperglycemia reduces eNOS activity by blunting activatory phosphorylation at Ser1177;
-
increased synthesis of ET-1, favouring vasoconstriction and platelet aggregation [53];
-
increased synthesis of vasoconstrictors and prostanoids by up-regulation of cyclooxygenase-2 (COX-2) associated with increased thromboxane A2 (TXA2) synthesis and decreased prostacyclin (PGI2) release [54];
-
structural and functional changes in the vasculature: alterations in cellular permeability, inflammation , angiogenesis , cell growth, extracellular matrix expansion and apoptosis [53].
-
-
Overproduction of ROS by mitochondria is involved in:
-
decreased NO bioavailability;
-
up-regulation of proinflammatory genes encoding for monocyte chemo-attractant protein-1 (MCP-1 ), selectins, vascular cell adhesion molecule-1 (VCAM-1) and intracellular cell adhesion molecule-1 (ICAM-1), via activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) subunit p65 signalling; these factors cause monocyte adhesion, rolling and diapedesis with foam cells formation in the sub-endothelial layer, thus accelerating the atherosclerotic process [55];
-
increased synthesis of methylglyoxal (a glucose metabolite) leading to AGE synthesis, accumulation and ultimately to endothelial dysfunction [56]; generation of AGEs leads to cellular dysfunction by activation of AGEs receptors (RAGE); AGE-RAGE signalling activates ROS-sensitive biochemical pathways such as the pro-oxidant hexosamine flux [57];
-
activation of the polyol pathway flux involved in vascular redox stress [49].
-

Mechanisms of hyperglycemia -induced vascular damage
2.3.2 Insulin Resistance and Atherothrombosis
The main feature of T2DM, insulin resistance, often precedes its onset by many years. Insulin resistance is critically involved in vascular dysfunction in subjects with T2DM [58] and is strongly related with obesity, since the adipose tissue is the main source for inflammatory mediators and free fatty acids (FFAs). Increased levels of FFAs stimulate toll-like receptors (TLR) that cause, on one hand, the activation of NF-kB nuclear translocation, with subsequent up-regulation of inflammatory genes interleukin-6 (IL-6) and tumor necrosis factor (TNF-α), and, on the other hand, the activation of c-Jun amino-terminal kinase (JNK) and PKC, phosphorylation of insulin receptor substrate-1 (IRS-1) , thus blunting its downstream targets phosphatidylinositol 3-kinase (PI3K) and Akt (a serine/threonine kinase also known as protein kinase B). These results in down-regulation of glucose transporter GLUT-4 and, hence, insulin resistance [45, 59]. In the vascular endothelium, a decrease in PI3K/Akt levels leads to increased FFA oxidation and subsequent ROS generation, with the aforementioned consequences: PKC activation, AGE synthesis, reduced PGI2 synthase activity and protein glycosylation; as a result, NO levels decrease dramatically and endothelial dysfunction ensues [60]. The blood coagulation system is also affected by insulin resistance, through alterations in IRS1/PI3K pathway leading to Ca2+ accumulation and increased platelet aggregation. Furthermore, insulin resistance facilitates atherothrombosis through increased cellular synthesis of PAI-1 and fibrinogen and reduced production of tissue plasminogen activator (tPA) [61].
The tight bond between insulin resistance and atherosclerosis is further established by the alterations in the lipid profile, such as high triglycerides, low HDL cholesterol, increased remnant lipoproteins, elevated apolipoprotein B (ApoB) as well as small and dense LDL cholesterol [62]. Accordingly, the experimental association of hyperlipemia with diabetes diminished the relaxation of the resistance arteries to bradykinin by an NO -dependent and an NO -independent mechanism (mediated via Ca2+ activated K+ channels) [63]. Moreover, the simultaneous insult of hyperlipemia-hyperglycemia has been associated with the highest contractility of the resistance arteries to prostaglandin F2a and the highest circulating glucose and cholesterol levels; the activation of PKC pathway, the alteration of cyclooxigenase and the Ca2+ dependent K+ channels generate the augmented contractility [64].
2.3.3 Micro RNA and Diabetic Vascular Disease
MicroRNAs (miRNAs) are a newly identified class of small non-coding ribonucleic acids (RNAs); they regulate gene expression at the post-transcriptional level. Alterations in miRNA expression occurring in T2DM play an important role in hyperglicemia-induced vascular damage pathogenesis [65]. Thus, in endothelial cells exposed to hyperglicemia, miR-320, miR-221, miR-503 are highly expressed, while miR-222 and miR-126 are submitted to down-regulation. The alterations in miRNA expression lead to decreased angiogenesis , generation of AGEs, decreased EPC proliferation, migration and homing, endothelial dysfunction and impaired vascular repair [45].
There is evidence that suggest that reduced miR-126 expression levels are partially responsible for impaired vascular repair capacities in diabetes; in contrast, restored expression of this miRNA promotes EPCs-related repair capacities and inhibits apoptosis [66].
2.3.4 Thrombosis and Coagulation
Both diabetes and insulin resistance are associated with a prothrombotic status, as a result of the alterations in clotting factors and platelet aggregation [67]. The most frequent alterations consist of: increased PAI-1 and fibrinogen, reduced tPA levels, increased expression of tissue factor (TF) with procoagulant activity and thrombin generation, platelet hyperreactivity, up-regulation of glycoproteins Ib and IIb/IIIa, increased levels of microparticles (MPs) released in the circulation [45]. Platelet hyperactivity and hyperaggregability in T2DM is induced by several factors including oxidative stress , abnormal intracellular Ca2+ homeostasis and hyperhomocysteinaemia . It has been showed that the endogenous production of ROS, Ca2+ mobilization and platelet aggregation are significantly greater in platelets from diabetic patients than in controls, even though they have been exposed to the same concentrations of homocysteine (Hcy), indicating that platelets from diabetic donors are more sensitive to plasma Hcy levels [68]. Besides, the exogenous oxidative stress, thrombin activation, and ageing lead to protein carbonyl formation in platelets from diabetic patients [69]. Moreover, it has been shown that MPs from patients with T2DM increase coagulation activity in endothelial cells. MPs carrying TF promote thrombus formation at the sites of injury, representing a novel and additional mechanism of coronary thrombosis in diabetes [70]. On the other hand, it has been reported that enoxaparin – a low molecular weight heparin, restores the altered vascular reactivity of resistance arteries in aged and aged-diabetic hamsters [71]. The author concludes that, these pharmacological effects supplement the anticoagulant properties of enoxaparin and may be of relevance for improving perfusion/circulation in the microvasculature of aged and of aged–diabetic persons [71].
2.3.5 Vascular Hyperglycemic Memory
The “hyperglycemic memory” concept derived from large observational studies, where adequate control of patients’ glucose blood levels acquired years after disease onset, failed to result in a lower cardiovascular risk [72]. However, in patients with early-onset therapy , well-established benefits were obtained [73]. The persistence of hyperglycemic stress despite blood glucose normalization has been defined as “hyperglycemic memory” [45]. Transitory episodes of hyperglycemia activate NF-kB, with a lingering effect even after blood glucose level become optimal. Hyperglycemia induces endothelial dysfunction , vascular inflammation and apoptosis through Sirtuin 1 (SIRT1) downregulation, p53 and p66Shc activation, PKCβII activation, inhibition of eNOS activity, expression of inflammatory genes and mitochondrial ROS accumulation , thus perpetuating a vicious circle that maintains the vascular lesional status in patients with diabetes despite optimal glycemic control [74].
3 Endothelial Progenitor Cell Biology
3.1 Definition of Endothelial Progenitor Cells
EPCs are a heterogeneous population of cells that reside in the bone marrow (BM) in close association with hematopoietic stem cells (HSCs) and the stroma [75]. These cells can be found (circulate) in the peripheral and umbilical cord blood and have been first isolated using magnetic micro beads by Asahara et al. (1997) [76]. EPCs represent between 1 and 5% of the total BM cells and less than 0.0001–0.01% of peripheral circulating mononuclear cells [77]. EPCs are involved in the maintenance of endothelial regeneration, vascular repair and in angiogenesis processes [78].
3.2 Ontogeny of Endothelial Progenitor Cells
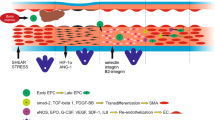
In circulation two categories of EPCs can be found: a population with hematopoietic origin, and another population named non-hematopoietic EPCs [79]. It is well known that hematopoietic EPCs arise from a progenitor cell of mesodermal origin, defined as hemangioblast [76, 80, 81]. This cell type is rare, slowly proliferating and is described as a precursor for hematopoietic cells (myeloid and lymphocytic lineages), and also for a part of EPCs [82, 83]. The angioblast (immature stage of EPCs) and primitive HSCs present common hematopoietic stem cell markers as: CD133, CD34, CD45 or Flk-1/KDR [80, 84,85,86,87] (Fig. 8.2). During the differentiation process the angioblasts start to express new cell surface markers (CD) and become primitive EPCs, an immature population of cells (Fig. 8.2). Some markers (CD14 and CD45) are common with myeloid lineage suggesting the hematopoietic origin of these EPCs [88]. In BM or in circulation, the hematopoietic EPCs begin to express specific markers for endothelial cells (ECs): vascular endothelial growth factor receptor 2 (VEGFR-2) and von Willebrand factor (vWf), in addition to CD133 and CD34 [80, 82, 85]. Regarding CD133, its expression is downregulated in non-hematopoietic cells and absent in mature ECs [85, 89, 90].

The origin and differentiation of EPCs from hematopoietic and non hematopoietic stem cells: the profile of cell surface markers (+: positive cells, –: negative cells)
In vitro, hematopoietic EPCs generate the endothelial cell colony forming units (CFU-ECs) [91] with spindle shape and low proliferative capacity, named also “early endothelial colonies” or early EPCs (Table 8.1). These cells are able to incorporate acetylated LDL (AcLDL) and to bind specific lectins (BS-1 and ulex europaeus) which are usually considered endothelial specific [92, 93]. They are also characterized by the expression of vWf, VEGFR-2 and CD31 [88, 94]. However, early EPCs do not generate vascular tubes in vitro, but they can induce the angiogenesis indirectly by producing angiogenic factors and inflammatory cytokines /chemokines permitting new vessels to form and to extend [95,96,97,98,99]) (Table 8.1).
Another EPC subtype is known as non-hematopoietic EPCs or late EPCs or outgrowth endothelial cells (OECs), because in the culture they generate the endothelial colony forming cell (ECFC) that develop into monolayers with a typical “cobblestone” morphology [79, 85]. OECs have a higher proliferative potential and they easily form tube-like structures in vitro [79]. These cells are present in peripheral and cord-blood and non-hematopoietic tissues [79]. Late EPCs do not express hematopoietic marker CD45 or the monocyte markers CD14 and CD115, but they express many EC antigens CD31, CD105, CD144, CD146, vWF, KDR, and UEA-1 [100] (Table 8.1 and Fig. 8.2). It has been also observed that, in vivo, these cells continue to differentiate and incorporate into the endothelium, and the expression of CD31 and vWF increases [91].
4 Endothelial Progenitor Cell Dysfunction, a Link Between Diabetes and Vascular Disruption
Cardiovascular risk factors induce endothelial injury. The occurring damages represent a balance between the degree of injury and the capacity of various complex mechanisms of repairing it. Diabetes mellitus is considered to be a clinical condition characterized by early and extended endothelial dysfunction . Hyperglycemia impairs vascular endothelial function and contributes to the vascular damage in diabetic patients [107]. Current studies suggest there is a negative correlation between the severity of diabetes and EPC count and function [108].
The complex pathophysiology of vascular damage in diabetes is not fully comprehended. Oxidative stress plays a crucial role in the pathogenesis of late diabetic complications . EPC dysfunction in diabetic patients has been correlated to oxidative stress and the generation of ROS [109]. Reduced extracellular superoxide dismutase (SOD) activity, the major antioxidant enzyme system of the vessel wall, has been associated with increased vascular oxidative stress and has been implicated in the endothelial dysfunction . In patients with CAD, SOD activity was substantially reduced [110].
NO is a biologically active unstable radical that is synthesized in vascular endothelial cells by eNOS . EPC mobilization from bone marrow to the peripheral blood and function requires NO [111]. Endothelial dysfunction is characterized by low biovailability of endothelium-derived NO, witch is itself an independent predictor of future cardiovascular events.
Chen et al. [112] have reported that prolonged exposure of early or late EPCs to high glucose concentrations reduces their number and proliferative ability, NO bioavailability, and the extent of phosphorylation of eNOS [112]. Exposure of EPC to high glucose concentrations has increased NADPH oxidase activity which results in increased O2- generation and reduced NO bioavailability because O2- inactivates NO and uncouples eNOS [113]. Therefore, decreased NO bioavailability is one of the determinants of vascular damage in diabetes.
On the other hand, ischemia induces neovascularization in diabetic patients. The oxygen deficit is considered the strongest stimulus for EPC mobilization from the bone marrow, through the up regulation of VEGF. It seems that EPC recruitment in regenerating tissues is mediated by a hypoxic gradient by Hypoxic Inducible Factor -1 (HIF-1 ) [114]. The expression of angiogenic factors, VEGF and HIF-1 , has been reduced in the hearts of diabetic patients during acute coronary syndromes (ACS). In rats, myocardial infarct size has increased in hyperglycemic conditions and has been associated with a reduced expression of the HIF-1 gene [115]. Lambiase et al. (2004) have shown that modest coronary collateral vessels development, which is typical for diabetes, may be related to low levels of circulating EPCs [116]. Diabetic EPCs have not been able to stimulate vascularization, even becoming anti-angiogenic. Gill et al. [117] have reported that coronary artery bypass grafting is followed by a marked increase in circulating EPCs that peaks at 6–12 h, resembling very closely to VEGF increase effects [117].
Nondiabetic patients with PAD alone and patients with uncomplicated diabetes have had similar EPC reduction versus control subjects [118]. Patients with diabetes and PAD have had a further significant decrease in circulating EPC levels, especially in the presence of ischemic foot lesions. EPC levels have been strongly correlated with the ankle-brachial index, the most objective diagnostic and prognostic test for lower-extremity arterial disease.
In addition, hyperglycemia induces retinal ischemia and the release of angiogenic factors that stimulate the proliferation of microvessels, leading to proliferative retinopathy . EPCs may be involved in the development of proliferative retinopathy . This is a paradox as, in diabetic patients, the vascular ischemia may coexist with a condition of pathological neovascularization . Interestingly, the pericyte loss is an early and selective event leading to endothelial activation and proliferation in the retina, and CD34+ progenitors of perivascular cells have been demonstrated in peripheral blood [119]. Thus, depletion of generic CD34+ progenitor cells may be one cause of pericyte loss.
Another possible link between diabetes and EPC alterations is the effect of insulin resistance per se. It has been demonstrated that patients with metabolic syndrome have decreased levels of CD34+KDR+EPCs compared with patients without the syndrome [120].
Given the EPC effects revealed by ongoing clinical studies we may consider new pathways of understanding and treatment of diabetic complications .
5 Mechanisms Leading to Endothelial Progenitor Cell Dysfunction in Diabetes and Diabetic Vascular Complications
EPCs from humans and animals with T2DM have multiple functional defects in vitro, with biologically relevance in vivo, including decreased migration to chemotactic stimuli, reduced proliferative potential and differentiation, diminished ability to form vascular-like structures, which limit their regenerative capacity [121, 122].
In the following sections, we highlight the putative mechanisms by which metabolic features of diabetes impair EPC functions.
5.1 Effect of Hyperglycaemia
The abnormalities of glucose regulation are associated with changes in EPC biology, including reduced circulating EPC numbers, incorrect mobilization from bone marrow, decreased functional properties, lowered capacity to mediate endothelial repair, and altered differentiation propensity. These alterations of EPCs reduce their potential to generate vascular regenerative cells favouring the development of proinflammatory cells [123,124,125].
It has been shown that in both patients with T2DM or pre-diabetic states (meaning impaired fasting glucose and reduced glucose tolerance) and animal models of diabetes, the function and number of circulating EPCs are decreased compared to normoglycemic conditions and these are correlated with disease severity [118, 125,126,127,128,129,130]. EPCs have been negatively associated with glucose levels after a glucose challenge, in individuals with impaired glucose tolerance [131], and also with serum glucose and glycated haemoglobin A1c levels, in patients with T2DM [132].
The mechanisms by which hyperglycaemia influences EPC function involve the formation of AGEs and oxidative stress with augmentation of ROS production through the activation of NAPDH oxidase in mitochondrion, with role in EPC apoptosis [133]. Increased ROS generation could also stimulate the AGE production, which further triggers ROS production. These activate nuclear factor-kappa B (NF-B) and subsequently the target genes that encode inflammatory proteins inducing interleukin 1𝛽 (IL-1𝛽) and tumor necrosis factor-𝛼 (TNF-𝛼). In parallel, NF-𝜅B transcription factor activates p53 accelerating cell senescence and inducible NOS (iNOS) that further potentiates the ROS production [134,135,136]. Hyperglycaemia causes also endoplasmic reticulum (ER) stress and excessive autophagy which further facilitate EPC death and reduce their migration [137, 138]. Apart from that, high glucose concentrations influence the proliferative capacity of EPCs either via inhibition of Akt phosphorylation followed by NOS activation or via activation of C-JunN-terminal kinase (JNK) pathway [139,140,141]. It has been demonstrated that the exposure to high levels of glucose, in vitro, induces decreased early and late EPC number and activity by downregulation of eNOS expression and phosphorylation, suggesting that eNOS is an important target for high glucose adverse effects [112]. However, it is still unclear whether high glucose-associated eNOS damage causes oxidative stress or if oxidative stress associated with high glucose causes eNOS deactivation [142]. Hamed et al. (2009) showed that in patients with T2DM an inverse relationship between plasma glucose and reduced NO bioavailability in EPCs can be found, due to enhanced oxidative stress which damages the protein signaling pathways that lead to diminished NO generation [143]. The relationship between the NO signaling pathway and EPC dysfunction will be discussed in detail below. High glucose levels also induce EPC senescence by one of NF-𝜅B target genes, p53, and by the activation of the p38 mitogen-activated protein kinase (MAPK ) pathway [144] (Fig. 8.3).

The mechanisms by which diabetic hallmarks induce EPC dysfunction
A very recent study has shown that the main factors (AGE, oxidative stress ) for EPC apoptosis and dysfunction induced by hyperglycaemia are also potent inducers for epigenetic changes in EPCs [145]. For example, ROS has been associated with a series of histone changes in the promoter and enhancer of superoxide dismutase (SOD) 2 gene in retinal endothelial cells isolated from diabetic rats with retinopathy [146]. In human microvascular endothelial cells, hyperglycaemia has induced the increase of H3K4mel expression and decreases of H3K9me2 and H3K9me3 levels on the of NF-𝜅B promoter leading to NF-𝜅B activation [147]. Moreover, the histone codes H3K9ac, H3K12ac, H3K4me2, and H3K4me3 suppress the eNOS transcription conducting to decreased NO [148].
Taken together, these studies demonstrate the obvious and complex influence of hyperglycaemia on impaired EPC levels and function .
5.2 Effect of Insulin Resistance and Insulin Like Growth Factor 1
Insulin resistance, a key feature of T2DM and the metabolic syndrome , results in a variety of metabolic and vascular phenomena such as dyslipidaemia, inflammation and a pro-thrombotic tendency, which eventually promote the development of atherosclerosis . Insulin resistance has been correlated with impaired downstream insulin signal transduction that reduces the glucose uptake in metabolic tissues [124].
The homoeostasis model assessment (HOMA) of insulin resistance (a method used to quantify insulin resistance and beta-cell function), has been found to be negatively correlated with EPCs, in patients with cardiovascular risk [120]. In addition, it has been shown that healthy men of South Asian descent, that are more insulin resistant than Caucasian peoples, present a reduced EPCs number and function [149]. Also, in insulin receptor (IR)-null mice, the number of circulating EPCs has been decreased [150]. Flow cytometric and cell culture analyses have revealed lower levels of circulating EPCs across the spectrum of insulin-resistant states [124]. Furthermore, the treatment with an insulin sensitizer, metformin , or thiazolidinediones, such as rosiglitazone, restored circulating EPC levels in diabetes [151,152,153]. The reduction of circulating EPC levels could be the result of a number of factors, such as defective mobilization, diminished proliferation and shortened survival into the circulation [94, 154].
However, the direct effect of insulin on the mobilization and differentiation of EPCs remains underexplored [155]. On this line, it has been shown that insulin resistance is closely associated with abnormalities in NO bioavailability and PI3K/Akt signaling, both playing an essential role in EPC mobilization from the bone marrow [94, 156,157,158,159]. Furthermore, in diabetic patients, EPCs have reduced clonogenicity and uncoupled eNOS mediated by ROS, which additionally contribute to augmented oxidative stress and impaired vascular repair [113] (Fig. 8.3). In one study on patients with poorly controlled T2DM, insulin significantly enhanced EPC mobilization in subjects with the stromal cell-derived factor 1 (SDF-1)-3′-A/G allele, a polymorphism known to be correlated with increased EPC mobilization, suggesting that this peptide plays a role in this EPC function [160, 161].
The mechanism by which insulin stimulates the in vitro outgrowth of EPCs from patients with T2DM involved the insulin-like growth factor (IGF-I) receptor, the stimulation of MAPKs and extracellular-signal-regulated kinase (ERK1/2) signaling pathways, but not IR [162]. IGF-I has complementary activity to insulin, and low IGF-I levels are recognized as an independent risk factors for CVD [163]. Treatment with growth hormone, which increases circulating IGF-I levels induced, in middle-aged humans, both the enhancement of circulating EPC levels and their incorporation into tube-like structures, and eNOS expression followed by the improvement of EPC colony forming and migratory capacity [157]. In vitro, IGF-I stimulates via the IGF-I receptor the EPC differentiation, migratory capacity and ability to incorporate into vascular networks [157]. Furthermore, it has been demonstrated that haploinsufficiency of the IGF1-receptor increases endothelial repair and favorably modifies the angiogenic progenitor cell phenotype . This angiogenic trait accelerated the endothelial regeneration in vivo, and increased the tube formation ability and adhesion potential of progenitor cells in vitro, and in general enhanced vascular repair [164]. It should be noted that a study has shown that IGF-I increases the eNOS expression, phosphorylation and activity in a PI3K/Akt-dependent manner in EPCs [157] (Fig. 8.3).
The heterozygous mouse models for IR knockout (IRKO), although non-diabetic, have revealed the presence of endothelial dysfunction and reduced EPC number and function. The descendants of IRKO mice crossed with transgenic mice with Tie-2-driven human IR expression in endothelial cells (HIRECO), have presented restored insulin signaling in endothelial cells through IR, and improved blood pressure, endothelial function, NO bioavailability, and vascular repair in the setting of global IR. This has not been related to glucoregulation or EPC abundance [165]. Insulin resistant, non-diabetic hemizygous mice for IRKO have presented a lower number of circulating EPCs in peripheral blood, but not in bone marrow and decreased EPC mobilization compared to wild-type mice [150]. Moreover, in IRKO mice, after arterial injury, the endothelial regeneration was delayed, but it has been restored after the transfusion of mononuclear cells or c-kit+bone marrow cells from wild-type mice [150].
All these studies demonstrate that both insulin and IGF-I significantly influence the EPC function, but more investigations are needed to understand their mode of action.
5.3 Nitric Oxide as a Key Factor of Endothelial Progenitor Cell Dysfunction
NO , a biologically active unstable free radical is synthesized from L-arginine in vascular endothelial cells by eNOS , an enzyme which is constitutively expressed in these cells. NO bioavailability depends on the balance between the rate of its generation and its inactivation, particularly by ROS [166, 167]. Moreover, NO and eNOS play an important role in mobilization of EPCs from bone marrow stem cell niches to the peripheral circulation [11, 168, 169]. NO bioavailability in sites of active vascularization seems to be crucial for EPC biology and function. The administration of endogenous NOS inhibitors, such as asymmetric dimethylarginine (ADMA), induces decreased EPC mobilization, differentiation, and proliferation in patients with CVD, suggesting the essential role of this enzyme in EPC function regulation [170].
Impaired NO bioavailability, the hallmark of endothelial dysfunction , is one of the contributing factors to the vascular damage in T2DM. NO bioavailability may be diminished either due to the lower overall systemic fraction of L-arginine that is converted to NO , or due to the reduction of essential eNOS cofactor and (6R)-5,6,7,8-tetrahydro-L-biopterin (BH4) [171, 172]. Reduced NO concentration contributes to defective migratory activity in diabetic EPCs. It has been demonstrated that EPCs isolated from diabetic patients have an impaired migration to stimulation with SDF-1 due to defective cell deformability, and the NO treatment improves deformability and normalizes the migration of these diabetic cells [173]. The EPC dysfunction in T2DM has been reported to be restored through NO -dependent mechanisms by various ways: (i) treatment with a NO donor drug which normalized their migration [173]; (ii) treating wounds with SDF-1α which reestablished their homing [140]; (iii) inactivation of NADPH oxidase which improved their reendothelialization capacity, in vivo [174]; (iv) preservation of the NO bioavailability with SOD which restored EPC proliferation [169]. Furthermore, since it has been demonstrated that prostacyclin (PGI2), an vasorelaxant prostanoid, has a direct effect on EPC functions and number in an autocrine or paracrine manner through an NO -dependent mechanism [175,176,177], it has been considered that PGI2 may have a substantial therapeutic role in diabetes as well [142].
5.4 PI3K/Akt Signaling Pathway and Endothelial Progenitor Cell Dysfunction
The phosphatidylinositol triphosphate kinase/protein kinase B (PI3K/Akt) pathway has been suggested to be involved in the regulation of EPC recruitment, mobilization, and proliferation [178]. Well-known activators of the PI3K/Akt pathway such as hydroxymethylglutaryl-coenzyme A reductase inhibitors (statins ), erythropoietin, estrogens, and VEGF, are able to increase circulating EPC levels, proliferation and migration [156, 179]. Pharmacological inhibition of PI3K and the overexpression of a dominant-negative Akt construct have been shown to abolish EPC proliferation and differentiation induced by statin and VEGF, in vitro and in vivo [156]. Moreover, Akt is an upstream enzyme of the eNOS signaling pathway which, as we mentioned above, is essential for EPC mobilization. Thus, perturbations in the PI3K/Akt/eNOS/NO signaling pathway or in one of its members may result in EPC dysfunction [168].
5.5 Oxidative Stress Impairs the Function of Endothelial Progenitor Cells
Oxidative stress is defined as an imbalance between ROS production and antioxidant defences. ROS generation is promoted by the p66shc, an adaptor protein [180, 181], while the antioxidant protection is provided by catalase, SOD, and glutathione peroxidase (GPx), which scavenge the excess of oxygen-free radicals and reduce ROS action. Previous reports have shown that the oxidative stress has a pivotal damaging effect on EPC functions [155, 182]. Thus, enhanced superoxide generation reduces the EPC levels and impairs EPC function [113]. Dysregulations of p66shc expression and SOD activity have been detected in AGEs-stimulated late EPCs , changes that are mediated by high mobility group box-1 (HMGB-1), a nonchromosomal nuclear protein [183, 184]. However, in the early-stage of diabetic EPCs, increased levels of ROS are not found, owing to the enhanced expression of antioxidant enzymes such as catalase [185].
Additionally to the indirect effects of ROS on EPCs it has been suggested that ROS exert direct effects on EPCs. Hydrogen peroxide (H2O2) induces in EPCs the increase of Forkhead box O3 (FOXO3a) protein expression, in a dose-dependent manner, and thereafter the activation of pro-apoptotic protein, Bim, that leads to the following effects: decreased viability, increased apoptosis, and the impairment of tube formation [186]. Also, H2O2 stimulates EPC apoptosis by the activation of apoptosis signal-regulating kinase 1 (ASK1), due to the oxidation of sulfhydryl groups of multiple anti-oxidant proteins such as glutaredoxin and thioredoxin [187, 188]. Moreover, H2O2 produces the oxidation of important EPC proteins such as the T-complex protein 1 subunit α, cofilin-1, peroxiredoxin-4, isoform A of prelamin-A/C, and actin [189].
Under diabetic conditions, enhanced oxidative stress induces the excessive generation of oxidized low density lipoprotein (oxLDL) [142]. It has been shown that the oxLDL reduces the number of viable EPCs in culture and induces the dysfunction of cultured EPCs isolated from healthy subjects [190,191,192]. These effects are mediated by NADPH oxidase, NF-𝜅B activation, or LOX-1 activation that subsequently inhibit the Akt/eNOS pathway [190, 192]. In T2DM patients, elevated levels of circulating oxLDL contribute to cardiovascular symptoms [193]. OxLDL accelerates the EPC senescence by the activation of the Akt/p53/p21 signaling pathway [144, 190] and inhibits VEGF-mediated differentiation via LOX-1 receptors, increasing the LOX-1 mRNA expression [194].
High-density lipoproteins (HDL), particles with antioxidant and anti-inflammatory properties, have a positive impact on EPC physiology [195, 196]. In T2DM patients, the HDL particles are dysfunctional, and the serum levels of oxidized HDL (oxHDL) and myeloperoxidase (MPO) enzyme have been found to be elevated as well [197, 198]. The administration of reconstituted HDL to T2DM patients has improved circulating EPC functions [199], while the treatment with HDL of cultured EPCs has induced the intensifications in their proliferation, migration, adhesion, and tube formation and also protected them from apoptosis [200]. In addition, HDL protects EPCs from the deleterious effects of ox-LDL. On the other hand, high concentrations of HDL (>400 μg/ml) seem to induce EPC senescence and to decrease their tube formation ability via the activation of Rho kinase that inhibits the Akt and p38 MAPK signaling pathways [201]. Conversely, ox-HDL stimulates EPC apoptosis in a dose-dependent manner, via the CD36 pathway. Interaction of ox-HDL with CD36 also enhances the NADPH oxidase activity, upregulates Nox2 mRNA (NADPH oxidase subunit), and activates the MAPK /NF-𝜅B pathway [202].
Other data have revealed that ROS induce the impairment of EPC function in diabetes, but the mechanisms that explain this phenomenon have not yet been studied by these authors [155, 203]. One of the mechanisms of diabetes-induced oxidative stress action has been recently investigated by Wu et al. (2016). This study has indicated that HMGB-1 has a significantly involvement via a positive feedback loop including the AGE/ROS/HMGB-1 pathway [203].
Regarding the antioxidant protection, it has been shown that EPCs from healthy humans contain high intracellular expression levels of manganese SOD (MnSOD) [204, 205], while EPCs from T2DM patients have increased SOD activity that neutralizes the high levels of superoxide anions [142]. Moreover, it has been reported that the antioxidant therapy with SOD in diabetic mice has reduced oxidative stress and improved EPC levels and differentiation capacity [206]. The treatment with SOD of glucose-stressed EPCs has restored their proliferation through an NO -dependent mechanism suggesting that the interaction between NO and superoxide anions has an important role in the development of EPC dysfunction and subsequently in CVD development in T2DM patients [169]. The augmentation of SOD expression in human EPCs by shear stress can accelerate the neutralization of superoxide anions, preventing the peroxynitrite formation, and thus increasing NO bioavailability in EPCs [207]. Likewise, the MnSOD overexpression effectively reversed the diabetic EPC dysfunction including tube formation, migration, while the transplantation with MnSOD-overexpressed diabetic EPCs improved in vivo wound healing ability [208]
5.6 Inflammation and Endothelial Progenitor Cells Dysfunction
Inflammation affects both EPC number and function, and EPCs react in two different ways to an inflammatory environment [208]: (1) at low concentrations of inflammatory cytokines , the number and function of EPCs are positively regulated, meaning that the increased number of circulating EPCs adheres and is recruited to the injured area; (2) at high concentrations of inflammatory cytokines , in a severe and chronic inflammatory environment such as diabetes, EPC functions (mobilization, adhesive capacity and proliferation) are impaired and the EPC number is reduced, leading to deficiency in angiogenesis . Subclinical inflammation has been shown to be a powerful predictor of cardiovascular events and T2DM [155]. In these conditions, the systemic inflammation is characterized by elevated levels of C-reactive protein (CRP), TNF-α, and many cytokines , such as interleukins (ILs): IL-1, IL-6, IL-10 and IL-18 [209, 210]. The interaction of these factors with different receptors results in the increase of oxidative stress and activation of NF-𝜅B in EPCs, which lead to their dysfunction (Fig. 8.3).
CRP has been reported to have the following effects, mediated through receptors for AGE, on EPCs: (i) significantly disturbs migration, adhesion and proliferation; (ii) reduces eNOS expression, increases apoptosis and necrosis [211, 212]. In addition, CRP increases mitochondrial ROS production, modulating the expression of anti-oxidant enzymes, such as GPx and MnSOD [212]. There was no association found between plasma levels of CRP and EPCs [213]. Regarding the effect of ILs, it has been shown that IL-1β: (i) induces murine EPC viability, proliferation, and migration both in vivo and in vitro, via ERK1/2 pathway activation [214]; (ii) increases mRNA and protein levels of VEGF-A in EPCs, via the PI3K/Akt signaling pathway [215]; (iii) reduces the number and proliferation of pig EPCs, and also EPC migration, adhesion, and angiogenesis , through p38 MAPK pathway activation [216]. Also, IL-18 reduces the ability of EPCs from healthy individuals to differentiate into mature endothelial cells [217] while IL-6 increases EPC migration, proliferation, and differentiation in cell culture, by activating both the JNK/STAT3 pathway and the ERK1/2 pathway [218]. Moreover, IL-10 alone has no effect on EPC migration and differentiation, although it did augmente significantly the expressions of VEGF and matrix metallopeptidase-9 (MMP-9) and potentiated the negative effects of TNF-α on EPCs [219].
TNF-α serum levels are higher in diabetes and have been associated with various complications of this disease [220, 221]. It has been shown that TNF-α influences the EPC function by different ways: (i) induces IL-18 expression that has negative effects on EPC differentiation; (ii) decreases Akt phosphorylation mediated by insulin and increases apoptosis through NF-𝜅B pathway activation [222]; (iii) inhibits migration and proliferation in a dose and time-dependent manner; (iv) mediates downexpressions of VEGFR-1 and SDF-1 as well as of the iNOS and eNOS [223]. On the contrary, in another study it has been reported that TNF-α enhances EPC migration, adhesion, and tube formation [219].
Regarding the effect of NF-𝜅B, it has been indicated that its overexpression: (i) improves EPC adherence to the endothelium by increasing the expressions of E-selectin and P-selectin glycoprotein ligand-1 [224]; (ii) does not impair the migration or vasculogenesis, in murine embryonic EPCs. In addition, simultaneous stimulation with TNF-α and NF-𝜅B of EPCs isolated from insulin resistant ZO rats induces apoptosis via caspase-3 [222]. The activation of NF-𝜅B can mediate the damage induced by Benzo[a]pyrene, an environmental toxin, on EPCs by increasing ROS production, thus impairing their migration, proliferation, and vasculogenesis [225].
5.7 Altered Micro RNA Expression and Dysfunctionality of Endothelial Progenitor Cells
The small noncoding molecules, microRNAs (miRNAs), are key regulators of diverse cellular processes, and their expression reflects the disease pathology [226]. The miRNAs in the body fluid seem promising to be used as biomarkers to monitor diabetes onset, and their number has been found to play a significant physiological role in tissues where diabetes complications occur.
Regarding the involvement of miRNAs in diabetic EPC dysfunctions, there are several data sustaining this aspect. For example, it has been shown that in T2DM, the miRNA-126 expression has been downregulated in EPCs, inhibited EPC proliferation/migration ability, and induced apoptosis, leading to diabetes-mediated CVD [227]. The altered expressions of miRNA-126 as well as of miRNA-130a have been involved in EPC dysfunction through extracellular signal–regulated kinase, Ras/ERK/VEGF, and the PI3K/Akt/eNOS signaling pathway [227, 228]. In addition, dysregulated miR-130a has impaired EPC function by directly targeting MAP3K12, a newly identified target gene of the JNK signaling pathway [141]. Alternatively, in T1DM patients the expression of miR-126 in EPCs has increased compared to control subjects [229]. In primary cultured EPCs from diabetic patients, an increased expression of miR-21 has been detected compared to that from control individuals, and it was suggested that elevated levels of muR21 protect EPCs from apoptosis via the regulation of downstream target DAXX [230]. Moreover, the overexpression of miR-34a in EPCs results in an increase in EPC senescence with impaired angiogenesis and SIRT1 expression [231] (Fig. 8.3). Also, augmented levels of miR-34a and miR-217 have induced the downregulation of some important targets of SIRT1, such as FOXO1 and eNOS, thereby leading to premature endothelial cell senescence and apoptosis [231, 232]. More recently, it was demonstrated that in T1DM patients with diabetic retinopathy , the miR-221 expression in EPCs has been significantly higher than in T1DM patients without diabetic retinopathy and control subjects [229]. Thus, it was hypothesized that when retinal damage is widespread with chronic hypoxia and nonperfusion, the EPCs would respond by increases of miR-221 expression and specific chemokines , a process not activated in earlier stages in noncomplicated diabetic patients.
The identification of miRNAs as diabetic biomarkers and pathogenic factors would not only contribute to the detection of early complications and progressive changes of diabetes, but also would provide targets for strategic therapeutic approaches in diabetes mellitus.
6 Significance of Endothelial Progenitor Cells in the Pathogenesis of Vascular Complications of Diabetes
Several studies have revealed the innate complex mechanisms underlying changes that occur in the vasculature during diabetes and lead to the cardiovascular risk associated with macrovascular and microvascular complications of diabetes [233]. It is well known that EPCs play an essential role in endothelial repair, angiogeneosis, neovascularization and attenuation of vascular dysfunction. Therefore, alterations in EPC number and functions are considered markers of cardiovascular risk in the general population and in diabetic patients, as well as a cause of diabetic vascular complications [120, 234, 235].
6.1 Endothelial Progenitor Cell Dysfunction and Macrovascular Complications in Diabetes
The linkage between diabetes mellitus and macrovascular disease has been very well established in many scientific studies [236]. It has been reported that diabetic patients have a two to fourfold increased risk of developing CAD and PAD compared with non-diabetic individuals [22]. Also, the severity of macrovascular complications in diabetes has been attributed to a profoundly impaired collateralization of vascular ischemic beds [237]. In addition, EPCs have been found to be involved into the mechanisms that delay ischemia-induced neovascularization in diabetes. In animal models of diabetic vasculopathy, it has been shown that diabetic EPCs are not able to promote vascularization, becoming antiangiogenic [238, 239], while the administration of EPCs from control animals has reduced defective collateralization. Consequently, a referenced study has established that EPCs play an important role in the vascularization and also, in healing of diabetic wounds [240]. Additionally, it has been demonstrated that the EPC reduction in diabetes is strongly correlated with the severity of both carotid and lower-limb atherosclerosis , suggesting that EPC number can be a valuable marker of atherosclerotic involvement [115]. In agreement with these findings, other studies have indicated that the lower circulating EPC number reflects the evolution of atherosclerotic disease both in animal models [241,242,243] and in patients [244]. These papers have used for EPC analyzing and quantification the flow cytometry technique. Furthermore, it has been reported that the determination of EPC number, using flow cytometry, is sufficiently reproducible to be used in the clinical practice, providing additional information over the classical risk factor analysis. This EPC measuring reflects not only vascular function and atherosclerotic changes, but also the endogenous vasculoregenerative potential [120, 245, 246]. The CD34+ KDR+ EPC count has been showed to predict the cardiovascular events independently of risk factors and hard indexes, such as left ventricular ejection fraction [244, 245, 247].
These findings have indicated that both decreased levels and dysfunction of EPCs play a significant role in enhanced cardiovascular risk and diabetes-related complications .
6.1.1 Endothelial Progenitor Cells and Diabetic Coronary Artery Disease
It is well known that diabetic patients die from CVD, diabetes representing the major cause of death among this population and contributing to a shortening of average life span by 5–10 years in these patients [248]. Diabetes increases the risk of future MI more than any other risk factors , and the consequences of MI are greater in these patients compared to the patients without diabetes mellitus [236].
It has been shown that EPCs isolated from the peripheral blood (PB-EPCs) of subjects with cardiovascular risk factors and previously diagnosed diabetic CAD, have altered phenotypes [247, 249], while in patients with known CAD, these cells have exhibited a reduced migratory capacity and weak proliferative response [250]. Additionally, lower levels of EPCs have been found in patients with severe atherosclerosis or diabetes-related vasculopathy [251, 252], and it was concluded that the circulating EPC levels predict cardiovascular events in patients with CAD [245, 253].
Most importantly, due to the EPC heterogeneity and the variable changes in the EPC phenotype at different stages of CAD and diabetes development, there are some limitations in establishing the predictive value of the number and functionality of EPCs in cardiovascular risk calculation [233].
Moreover, modulating EPC levels in T2DM with known CAD using different drugs is still under study. Regarding this aspect, it was found that valsartan, an angiotensin-2 receptor blocker, in high doses, has a positive influence on bone marrow-derived EPCs phenotyped as CD14+ CD309+ and CD14+ CD309+ Tie2+ in T2DM patients with known asymptomatic CAD [254]. Additionally, strong evidence has been provided to support that statins (atorvastatin and pravastatin) have a favourable in vitro effect on functional parameters of EPCs derived from diabetic patients with acute ST segment elevation MI (STEMI) [248]. These data indicate that treatment with statins may be beneficial for EPC-driven vascular repair after an acute MI (AMI) and may improve the cardiovascular outcome of diabetic patients.
6.1.2 Endothelial Progenitor Cells and Diabetic Peripheral Arterial Disease
PAD is a common vascular complication in the diabetic population , diabetes increasing the risk of developing PAD at least two-fold [255, 256]. Patients suffering from both diabetes and PAD present poor lower extremity function and are at risk of developing critical limb ischaemia and ulceration, potentially requiring limb amputation [257, 258]. Moreover, these patients respond poorly to the treatment of PAD and exhibit a higher mortality [245, 246].
Regarding EPC involvement in this pathology , it has been shown that patients with PAD alone and patients with uncomplicated diabetes had similar EPC decrease versus control subjects, while patients with PAD and diabetes had a more significantly reduction in circulating EPC levels, mainly in the presence of ischemic foot lesions [115]. EPC levels are strongly correlated with the ankle brachial index, the most objective diagnostic and prognostic test for lower extremity arterial disease [118]. A recent study has demonstrated that ankle-brachial index is the determinant of EPC population state in disease-affected groups, and EPCs could predict the prevalence and severity of symptomatic PAD [259]. Moreover, EPCs isolated from diabetic patients with PAD have exhibited impaired proliferation and adhesion capacity to mature endothelium [260], while EPCs isolated from diabetic mice had suppressed EPC mobilization following hindlimb ischaemia [261,262,263,264,265]. In ischaemic tissue the existence of an inverse relationship was proven between diabetes duration and EPC number [266]. Furthermore, it has been reported that the administration of: (i) non-diabetic EPCs into diabetic hindlimbs, following ischaemia, have accelerated the blood flow restoration [238]; (ii) vitamin B1 analogue, benfotiamine or statins , have prevented the diabetes-induced reduction in circulating EPCs in mice subjected to limb ischaemia [265, 267]; (iii) insulin and G-CSF (granulocyte colony stimulating factor) have partially restored the deficient EPC mobilization in diabetic rats after ischaemia/reperfusion injury [268].
6.1.3 Endothelial Progenitor Cells and Diabetic Cerebrovascular Disease
In diabetic patients, ischemic cerebral damage is exacerbated, and the outcome is poor, but the responsible mechanisms are not well known. Likewise, there is less information regarding the correlation of circulating EPCs with cerebral vascular density (as an index of angiogenesis ) and ischemic injury [269]. Information on ischemic stroke in diabetic animal models is also lacking. In a study using db/db mice as a T2DM animal model for in vivo ischemic stroke it has been shown that impaired circulating EPC number, reduced EPC production/function, and increased generation of microparticles (MPs) might be the mechanisms responsible for increased ischemic damage [269]. Moreover, these data suggest that circulating EPCs and MPs could be used as predictive biomarkers for ischemic stroke complications in diabetes and might be thus targeted, offering new therapeutic possibilities for diabetes and ischemic stroke. In another study it has been reported that EPC transplantation alone had a modest effect on stroke recovery in diabetic mice in terms of angiogenesis , neurogenesis, axonal remodeling, and neurological behavior. These phenomena may be explained by the fact that only a small number of transplanted cells survived and successfully homed to the ischemic brain in these diabetic animals [270]. Recently, the same group has reported that EPC transplantation combined with p38 mitogen-activated protein kinase inhibitor administration into db/db diabetic mice, after ischemic stroke induction, have accelerated recovery, by increasing levels of proangiogenic and neurotrophic factors [271].
As a result, EPC dysfunction is perhaps a promising target for diabetes treatment strategies . Indeed, the improvement of EPC number and functionality seems to reduce cardiovascular risk and diabetes-related macrovascular complications , but the mechanisms underlying these outcomes are not fully clear, requiring more investigations.
6.2 Endothelial Progenitor Cell Dysfunction and Microvascular Complications in Diabetes
Patients with diabetes mellitus are at high risk for the development of microvascular complications and major adverse cardiovascular events. The EPC dysfunction related to the three manifestations of microvascular disease in diabetes: retinopathy , nephropathy , and neuropathy , will be discussed in further detail below.
6.2.1 Endothelial Progenitor Cells and Diabetic Retinopathy
Diabetic retinopathy represents an important cause of visual deficiency in the Western world [9]. In the United States this disease has been responsible for ~8% of cases of legal blindness and ~12% of all new cases of blindness in each year in the last decade of the twentieth century [236]. The majority of T1DM patients and more than 60% of patients with T2DM develop background retinopathy . The severity of hyperglycemia , duration of diabetes mellitus, insulin resistance and additionally, hypertension , dyslipidemia , inflammation and smoking are important factors that contribute to the development of microvascular disease [272, 273].
The role of EPC in the development of diabetic retinopathy is controversial [145]. EPC number has been reported as either decreased, increased or unchanged in diabetic patients with severe retinopathy when compared to diabetic patients with or without mild retinopathy , or to healthy subjects [229, 274,275,276,277]. Additionally, there are studies showing that in patients with nonproliferative diabetic retinopathy the circulating EPC number is reduced [127] compared to proliferative diabetic retinopathy patients which have increased EPC levels [278]. In T1DM and T2DM patients with diabetic retinopathy , it was found that although the EPC number is increased, their functions, such as migration, mobilization and homing, are often impaired [277, 279]. Intravitreal delivery of cartilage oligomeric matrix protein-angiopoietin 1 (COMP-Ang1) recovers the endothelial integrity and ameliorates the vascular leakage by promoting incorporation of endothelial colony-forming cells into retinal vasculature [280] in diabetic mice, and this way reverses diabetic retinopathy . Moreover, it has been demonstrated in culture studies that the early EPC (eEPCs) are responsible for ‘provisional repair’, first homing at the lesion and attracting the CD34+ cells, and later on attracting late outgrowth endothelial progenitor cells (late EPCs ) [281]. In nonproliferative diabetic retinopathy , eEPCs are dysfunctional and they can not recruit late EPCs into the retina to repair the acellular capillaries, while in proliferative diabetic retinopathy the eEPCs take a proinflammatory phenotype and recruit too many late EPCs leading to pathological neovascularization . Correcting these dysfunctions may allow the use of a diabetic patient’s own EPCs to repair their injured retinal and systemic vasculature, in both the early and intermediate phase of vasodegeneration, to enhance vessel repair, reverse ischemia, and prevent progression to the late stages of diabetic retinopathy [281]. Thus, for durable repair and sustained correction of retinal ischemia the use of these expanded in vitro cells (eEPCs and late EPCs ) has been proposed as being better than the use of the freshly isolated ones [282,283,284]. Nevertheless, more rigorous investigations are needed to solve this problem.
6.2.2 Endothelial Progenitor Cells and Diabetic Nephropathy
Diabetic nephropathy is found at a rate of ~7% of patients already diagnosed with T2DM. It occurs in less than 12% of patients with T1DM at 7 years after the diagnosis has been made, and in ~25% of patients with T2DM at 10 years after diagnosis [236]. Diabetic nephropathy is characterized in the early stages by hyperperfusion and hyperfiltration, due to the endothelial cell damage and abnormal angiogenesis , and in the late stages by the development of glomeruli fibrosis that results in renal failure. However, the exact mechanisms of nephropathy are not fully elucidated. At the present time, it has been reported that AGEs, oxidative stress , and the activation of the renin-angiotensin-aldosterone system (RAAS) are involved in these changes partially through the activation of TGF-1 signaling and increased VEGF expression in the kidney [285,286,287]. The negative correlation between EPC number and microalbuminuria or albumin excretion rate reported in both T1DM and T2DM patients, has suggested that EPCs have a protective effect in the structure and function of glomeruli [179, 288]. The involvement of dysfunctional EPC has been described in both endothelial damage and microcirculatory impairment that occurs in the early pathogenetic events in diabetic nephropathy and also in defective glomerular repair and renal disease progression in diabetes [115]. Moreover, it has been suggested that EPCs, being pluripotent, have the ability to transdifferentiate into different phenotypes . Due to the kidney-derived hormone, erythropoietin, that has a major role in the regulation of EPC mobilization and differentiation, the relations between EPCs and renal function are more complicated [179]. In diabetes, the oxygen-erythropoietin feedback that depends on the hypoxia-sensing system, hypoxia-inducible factor 1-alpha (HIF-1 α), is dysregulated. The erythropoietin response is affected by microangiopathy and progressive tubulointerstitial fibrosis which increase the latency of the erythropoietin system, and by ROS production and hyperglycemia which themselves stabilize HIF-1 α [289]. It has been demonstrated that HIF-1 α downregulation had a negative impact on EPC mobilization in diabetes [268]. Another factor that has complicated the relationship between EPCs and renal function is represented by ADMA. This endogenous NO inhibitor that is accumulated in patients with chronic kidney disease (CKD) [290] and diabetes [291], is also a potent inhibitor of EPC mobilization and function [170]. Thus, the disrupted erythropoietin system and an excess of ADMA in CKD seem to inhibit EPC mobilization, differentiation, and homing, while EPC alterations that occur in diabetes impair the renal microvasculature. Due to this vicious circle, diabetic nephropathy can be associated with a deficiency of EPCs rather than with CKD in general, which would represent an additional risk for CVD and death [268].
It has been recently suggested that for treating diabetic nephropathy the endothelial colony-forming cells (ECFCs) could be a promising and complimentary therapeutic target [145]. Another promising idea is to apply ECFC with higher level of NO or angiopoietin 1 (Ang1) that will be favorable for stabilizing capillaries by reversing ‘uncoupled VEGF with NO ’ balancing ‘Ang1/Ang2 competition’ and ‘rendering Ang1/VEGF’. Alternatively, induced pluripotent stem cells (iPSC)-based ECFCs would be one of the major strategies for diabetic microvascular abnormality treatment. In this direction it has been disclosed that the endothelial progenitors generated from human iPSCs derived from cord blood have a greater capacity for homing and long term incorporation into injured retinal vessels [292, 293]. To improve endothelial function and protect vessel from retinopathy as well as nephropathy , ECFC administration has been proposed in the early stage of diabetes for better efficacy [145].
6.2.3 Endothelial Progenitor Cells and Diabetic Neuropathy
The development of diabetic neuropathy is associated with vascular and nonvascular abnormalities. The neuropathy is characterized by basement membrane thickening, pericyte loss, reduced capillary blood flow to C fibers, resulting in attenuated nerve perfusion and attendant endoneurial hypoxia, axonal thickening and eventual loss of neurons [294]. There are two major types of clinical manifestations: (1) chronic, symmetrical, length-dependent sensorimotor polyneuropathy, that is associated with the severity and duration of hyperglycemia [295]; (2) asymmetrical polyneuropathies that develops at more unpredictable times during the development of diabetes [296].
In the experimental diabetic neuropathy , the reduction of vasa nervorum is an obvious characteristic of peripheral nerves , and decreased blood supply to peripheral nerves can accelerate disease progression [297]. It was hypothesized that EPCs may have a crucial role in the homeostasis of the nutritive microvasculature, their dysfunction contributing to the acceleration of disease. Due to the ability of these cells to differentiate also toward the neural phenotype [298], it is possible that the imbalance of immature circulating cells in diabetes influences this chronic complication, downregulating both endothelial and neuronal progenitors [268]. To support this hypothesis it has been reported that the EPC intramuscular administration can reverse the impairment of sciatic nerve conduction velocity and nerve blood flow in diabetic rats [299]. Chavez et al. (2005) have demonstrated that the EPC dysregulation in diabetic neuropathy may be attributed to a defective HIF-1 α activation [300]. Other groups have shown that diabetic neuropathy can by delayed by the administration of some EPC-modulating agents, such as erythropoietin and statins [301]. Consequently, the EPC alterations have contributed to the pathogenesis of diabetic neuropathy , but future studies are needed to elucidate the involved mechanisms .
Taken together, these findings indicate that, although very important, the role of EPCs in the pathogenesis of diabetic microvascular diseases is still uncertain and future investigations are necessary to reveal the EPC mysterious nature for therapeutic applications.
7 Potential Therapeutic Implications of Endothelial Progenitor Cells in Diabetes -Associated Vascular Complications
7.1 Prognostic Value of Endothelial Progenitor Cells
In the recent years many studies have focused on an attempt to define the role of EPCs in identifying patients with increased cardiovascular risk. Clinical studies have demonstrated a correlation between the levels of circulating EPCs and the increasing cardiovascular risk profile [250, 302]. Thus, the adjuvant potential of EPCs as a cardiovascular risk biomarker has been proposed, based on the inverse link between EPC number, their migratory/proliferative potential and risk factors for CVD. Thereby, it has been demonstrated that the number of circulating EPCs and their migratory activity are reduced in the presence of classic cardiovascular risk factors such as smoking [94, 303,304,305], hypertension [306,307,308], hypercholesterolemia [250, 309], obesity [310, 311], T1DM and T2DM [115, 121, 128, 235] (Fig. 8.4). These effects could be possibly explained by three different mechanisms, either separate or in combination: (a) an impaired mobilization of EPCs from the bone marrow, (b) an increased uptake of EPCs at sites of vascular injury to induce the endothelial repair; and (c) a decreased half-life of circulating EPCs by accelerated senescence and apoptosis of the remaining cells [94, 312]. In this way the reduction in mobilization, homing, and differentiation/survival of EPCs may limit their ability to repair injured tissues. The endothelial dysfunction and alteration have also determined the higher tissue request for EPCs and their increased turnover [305]. On the other hand, with ageing there is a decrease in the production of EPCs in BM [313].

The influence of physiological and pathological factors on EPC number and function
In contrast, some pathologies such as ACS and acute myocardial infarction (AMI) cause hypoxia and vascular injury determining increased levels of inflammatory and hematopoietic cytokines , which induced a rapid mobilization of EPCs in the circulation [314, 315] (Fig. 8.4). Also, it is well known that physical exercises , hypoxia and some chemokines and growth factors (VEGF, SDF-1, angiogenin and colony-stimulating factor-CSF) increase EPC number and improve their function [106, 315, 316] (Fig. 8.4).
7.2 Pharmacological Manipulation of Endothelial Progenitor Cells
Besides their role as diagnostic and prognostic biomarkers, EPCs may be important targets in the CVD therapy . Thereby, many cardiovascular pharmacotherapies have been used to improve the number and function of EPCs in patients with cardiovascular risk (Table 8.2).
8 Conclusions
In diabetes mellitus, the hyperglycemia has profound detrimental effects on the vascular endothelial cells, due to their anatomical location in the blood vessel, leading to the emergence of endothelial dysfunction . The vascular complications , particularly macrovascular ( coronary artery disease, peripheral arterial disease, cerebrovascular diseas e) and microvascular (retinopathy , nephropathy , neuropathy ), are principal causes of disability and death in patients suffering from diabetes mellitus.
Accumulating data evoke that the mechanisms which are involved in the pathogenesis of vascular complications in diabetes have a well-defined role in the mobilization and function of EPCs. Thus, hyperglycaemia, insulin resistance, insulin like growth factor 1, nitric oxide, oxidative stress , PI3K/Akt signaling pathway, inflammation , and altered microRNA expression can contribute to decreasing of circulating EPC levels and to EPC dysfunctionality in diabetes. Many studies have shown that, in patients with diabetes and CVD, the number of EPCs from peripheral blood is reduced and EPC function is impaired. On the other hand, the alterations in EPC number and function may have a relevant role in the development of diabetes-related vascular complications .
A better understanding of the mechanisms leading to impairment of EPC mobilization and function in diabetes can further help in identifying the targets to prevent or reduce the risk of disease progression towards vascular complications .
It is currently hoped that addressing EPCs as targets for diagnostic and therapy in diabetes will favourably modify the risk for cardiovascular complications and survival. The drug therapy on EPC number and function can enhance the protection against vascular complications during diabetes. Therefore, EPCs could represent a diagnostic biomarker and pharmacological target to conduct the preventive or therapeutic interventions in diabetes. Nevertheless, further studies need to elucidate the exact role of EPCs in the pathogenesis of vascular complications in diabetes and their potential therapeutic implications.
References
International Diabetes Federation. Atlas7thEdition. Available online: http://www.diabetesatlas.org/key-messages.html
Barkoudah E, Skali H, Uno H et al (2012) Mortality rates in trials of subjects with type 2 diabetes. J Am Heart Assoc 1:8–15
Jenkins AJ, Januszewski AS, O’Neal DN (2015) Addressing vascular risk factors in diabetes. Endocrinology Today 4:35–38
Fonseca V, Desouza C, Asnani S et al (2004) Nontraditional risk factors for cardiovascular disease in diabetes. Endocrine Reviews 25:153–175
Saito I, Folsom AR, Brancati FL et al (2000) Non-traditional risk factors for coronary heart disease incidence among persons with diabetes: the Atherosclerosis Risk in Communities (ARIC) Study. Ann Intern Med 133:81–91
United Kingdom Prospective Diabetes Study (UKPDS) (1995) Relative efficacy of randomly allocated diet, sulphonylurea, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. Br Med J 310:83–88
Martín-Timón I, Sevillano-Collantes C, Segura-Galindo A et al (2014) Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J Diabetes 5:444–470
Caballero AE (2003) Endothelial dysfunction in obesity and insulin resistance: a road to diabetes and heart disease. Obes Res 11:1278–1289
Fong DS, Aiello LP, Ferris FL et al (2004) Diabetic retinopathy. Diabetes Care 27:2540–2553
United Kingdom Prospective Diabetes Study Group (UKPDS) (1998) Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. Lancet 352:837–853
American Academy of Ophtalmology. Diabetic retinopathy preffered practice pattern guidelines- Updated-2016. Available online: http://www.aao.org/preferred-practice-pattern/diabetic-retinopathy-ppp-updated-2016
Hovind P, Tarnow L, Rossing K et al (2003) Decreasing incidence of severe diabetic microangiopathy in type 1 diabetes. Diabetes Care 26:1258–1264
United States Renal Data System. Available online: www.usrds.org 2015 Annual Data Report
Tang SC, Chan GC, Lai KN (2016) Recent advances in managing and understanding diabetic nephropathy Version 1. F1000 Res. 5: F1000 Faculty Rev-1044. doi:10.12688/f1000research.7693.1
Gross JL, de Azevedo MJ, Silveiro SP et al (2005) Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care 28:164–176
Odegaard JI, Chawla A (2012) Connecting type 1 and type 2 diabetes through innate immunity. Cold Spring Harb Perspect Med 2:a007724
Georgescu A, Popov D, Dragan E et al (2007) Protective effects of nebivolol and reversal of endothelial dysfunction in diabetes associated with hypertension. Eur J Pharmacol 570:149–158
Diabetic Neuropathy. Available online: http://emedicine.medscape.com/article/1170337
Boulton AJ, Vinik AI, Arezzo JC et al (2005) Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 28:956–962
Zochodne DW (2008) Diabetic polyneuropathy: an update. Curr Opin Neurol 5:527–533
Boyle PJ (2007) Diabetes mellitus and macrovascular disease: mechanisms and mediators. Am J Med 120:S12–S17
Beckman JA, Creager MA, Libby P (2002) Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA 287:2570–2581
Fowler MJ (2008) Microvascular and macrovascular complications of diabetes. Clinical Diabetes 26:77–82
Kannel WB, McGee DL (1979) Diabetes and cardiovascular disease: the Framingham study. JAMA 241:2035–2038
Haffner SM, Lehto S, Ronnemaa T et al (1998) Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 339:229–234
Buse JB, Ginsberg HN, Bakris GL et al (2007) Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Diabetes Care 30:162–172
Lehto S, Ronnemaa T, Pyorala K et al (1996) Predictors of stroke in middle-aged patients with non-insulin-dependent diabetes. Stroke 27:63–68
Ohira T, Shahar E, Chambless LE et al (2006) Risk factors for ischemic stroke subtypes: the Atherosclerosis Risk in Communities Study. Stroke 37(10):2493–2498
Muntean C, Mitrea A, Mota M et al (2011) Type 2 diabetes and its implications in cerebrovascular disease. Rom J Diabetes Nutr Metab Dis 19:81–88
Feldmann E, Broderick JP, Kernan WN et al (2005) Major risk factors for intracerebral hemorrhage in the young are modifiable. Stroke 36:1881–1885
Jia Q, Zhao X, Wang C et al (2011) Diabetes and poor outcomes within 6 months after acute ischemic stroke: the China National Stroke Registry. Stroke 42:2758–2762
Fuentes B, Ortega-Casarrubios M, SanJosé B et al (2010) Persistent Hyperglycemia >155 mg/dL in acute ischemic stroke patients: how well are we correcting it? Stroke 41:2362–2365
Beckman JA, Paneni F, Cosentino F et al (2013) Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part II. Eur Heart J 34:2444–2456
O’Donnell MJ, Xavier D, Liu L et al (2010) Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case control study. Lancet 376:112–123
Sarwar N, Gao P, Seshasai SR et al (2010) Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet 375:2215–2222
van Wijk I, Kappelle LJ, van Gijn J et al (2005) Long-term survival and vascular event risk after transient ischaemic attack or minor ischaemic stroke: a cohort study. Lancet 365:2098–2104
Lange S, Diehm C, Darius H et al (2003) High prevalence of peripheral arterial disease but low antiplatelet treatment rates in elderly primary care patients with diabetes. Diabetes Care 26:3357–3358
Jude EB, Oyibo SO, Chalmers N et al (2001) Peripheral arterial disease in diabetic and nondiabetic patients. Diabetes Care 24:1433–1437
Escobar C, Blanes I, Ruiz A et al (2011) Prevalence and clinical profile and management of peripheral arterial disease in elderly patients with diabetes. Eur J Intern Med 22:275–281
Faglia E, Clerici G, Mantero M et al (2007) Incidence of critical limb ischemia and amputation outcome in contralateral limb in diabetic patients hospitalized for unilateral critical limb ischemia during 1999–2003 and followed-up until 2005. Diabetes Res Clin Pract 77:445–450
World Heart Federation. Available online: www.world-heart-federation.org/cardiovascular-health/cardiovascular-disease-risk-factors/diabetes
Jude EB (2004) Intermittent claudication in the patient with diabetes. British Journal of Diabetes & Vascular Disease 4:238–242
Fowkes FG, Murray GD, Butcher I et al (2008) Ankle brachial index combined with Framingham risk score to predict cardiovascular events and mortality. JAMA 300:197e208
Wang F, Guo X, Shen X et al (2014) Vascular dysfunction associated with type 2 diabetes and Alzheimer’s disease: a potential etiological linkage. Med Sci Monit Basic Res 20:118–129
Paneni F, Beckman JA, Creager MA et al (2013) Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy. Eur Heart J 34:2436–2446
Naudi A, Jove M, Ayala V, et al (2012) Cellular dysfunction in diabetes as maladaptive response to mitochondrial oxidative stress. Exp Diabetes Res 2012:696215
Bartnik M, Cosentino F (2009) Dysglycaemia, cardiovascular outcome and treatment. Is the jury still out? Eur Heart J 30:1301–1304
Lerman A, Zeiher AM (2005) Endothelial function: cardiac events. Circulation 111:363–368
Nishikawa T, Edelstein D, Du XL et al (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Inoguchi T, Li P, Umeda F et al (2000) High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49:1939–1945
Cosentino F, Francia P, Camici GG et al (2008) Final common molecular pathways of aging and cardiovascular disease: role of the p66shc protein. Arterioscler Thromb Vasc Biol 28:622–628
Du XL, Edelstein D, Dimmeler S et al (2001) Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the akt site. J Clin Invest 108:1341–1348
Geraldes P, King GL (2010) Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 106:1319–1331
Cosentino F, Eto M, De Paolis P et al (2003) High glucose causes up-regulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation 107:1017–1023
Hink U, Li H, Mollnau H et al (2001) Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 88:E14–E22
Sena CM, Matafome P, Crisostomo J et al (2012) Methylglyoxal promotes oxidative stress and endothelial dysfunction. Pharmacol Res 65:497–506
Giacco F, Brownlee M (2010) Oxidative stress and diabetic complications. Circ Res 107:1058–1070
Kim JA, Montagnani M, Koh KK et al (2006) Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113:1888–1904
Kim JK (2012) Endothelial nuclear factor kB in obesity and aging: is endothelial nuclear factor kappaB a master regulator of inflammation and insulin resistance? Circulation 125:1081–1083
Du X, Edelstein D, Obici S et al (2006) Insulin resistance reduces arterial prostacyclin synthase and eNOs activities by increasing endothelial fatty acid oxidation. J Clin Invest 116:1071–1080
Vinik AI, Erbas T, Park TS et al (2001) Platelet dysfunction in type 2 diabetes. Diabetes Care 24:1476–1485
Zhang H, Dellsperger KC, Zhang C (2012) The link between metabolic abnormalities and endothelial dysfunction in type 2 diabetes: an update. Basic Res Cardiol 107:237
Georgescu A, Popov D, Simionescu M (2001) Mechanisms of impeded bradykinin-induced vasodilation in experimental hyperlipemia-hyperglycemia: contribution of nitric oxide and Ca2+ activated K+ channels. Fund Clin Pharmacol 15:335–342
Georgescu A, Popov D (2003) The contractile response of the mesenteric resistance arteries to prostaglandin F2α; effects of simultaneous hyperlipemia-diabetes. Fund Clin Pharmacol 17:683–689
Shantikumar S, Caporali A, Emanueli C (2012) Role of microRNAs in diabetes and its cardiovascular complications. Cardiovasc Res 93:583–593
Wang DE (2009) MicroRNA regulation and its biological significance in personalized medicine and aging. Curr Genomics 10:143
Vazzana N, Ranalli P, Cuccurullo C et al (2012) Diabetes mellitus and thrombosis. Thromb Res 129:371–377
Alexandru N, Jardín I, Popov D et al (2008) Effect of homocysteine on calcium mobilisation and platelet function in type 2 diabetes mellitus. J Cell Mol Med 12:2015–2026
Alexandru N, Constantin A, Popov D (2008) Carbonylation of platelet proteins occurs as consequence of oxidative stress and thrombin activation, and is stimulated by ageing and type 2 diabetes. Clin Chem Lab Med 46:528–536
Tsimerman G, Roguin A, Bachar A et al (2011) Involvement of microparticles in diabetic vascular complications. Thromb Haemost 106:310–321
Georgescu A, Popov D, Capraru M et al (2003) Enoxaparin-a low molecular weight heparin, restores the altered vascular reactivity of resistance arteries in aged and aged-diabetic hamsters. Vasc Pharmacol 40:167–174
Duckworth W, Abraira C, Moritz T et al (2009) Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 360:129–139
Holman RR, Paul SK, Bethel MA et al (2008) 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 359:1577–1589
Schisano B, Tripathi G, McGee K et al (2011) Glucose oscillations, more than constant high glucose, induce p53 activation and a metabolic memory in human endothelial cells. Diabetologia 54:1219–1226
Ribatti D, Nico B, Crivellato E et al (2005) Endothelial progenitor cells in health and disease. Histol Histopathol 20:1351–1358
Asahara T, Murohara T, Sullivan A et al (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science 275:964–967
Khan SS, Solomon MA, McCoy JP Jr (2005) Detection of circulating endothelial cells and endothelial progenitor cells by flow cytometry. Cytometry B 64:1–8
Zhang M, Rehman J, Malik AB (2014) Endothelial progenitor cells and vascular repair. Curr Opin Hematol 21:224–228
Asahara T, Kawamoto A, Masuda H (2011) Concise review: circulating endothelial progenitor cells for vascular medicine. Stem Cells 29:1650–1655
Urbich C, Dimmeler S (2004) Endothelial progenitor cells: characterization and role in vascular biology. Circ Res 95:343–353
Barber CL, Iruela-Arispe ML (2006) The ever-elusive endothelial progenitor cell: identities, functions and clinical implications. Pediatr Res 59:26R–32R
Schatteman GC, Awad O (2004) Hemangioblasts, angioblasts, and adult endothelial cell progenitors. Anat Rec A Discov Mol Cell Evol Biol 276:13–21
Cao N, Yao Z-X (2011) The hemangioblast: from concept to authentication. Anat Rec (Hoboken) 294:580–588
Weiss MJ, Orkin SH (1996) In vitro differentiation of murine embryonic stem cells. New approaches to old problems. J Clin Invest 97:591–595
Hristov M, Weber C (2004) Endothelial progenitor cells: characterization, pathophysiology, and possible clinical relevance. J Cell Mol Med 8:498–508
Yoder MC (2012) Human endothelial progenitor cells. Cold Spring Harb Perspect Med 2:a006692
Reale A, Melaccio A, Lamanuzzi A et al (2016) Functional and biological role of endothelial precursor cells in tumour progression: a new potential therapeutic target in haematological malignancies. Stem Cells Int 2016:7954580
Salingova B, Madarasova M, Stejskal S et al (2014) From endothelial progenitor cells to tissue engineering: how fare have we come? J Stem Cell Res Ther 4:1000185 o
Kakiuchi-Kiyota S, Crabbs TA, Arnold LL et al (2013) Evaluatin of expression profiles of hematopoietic stem cell, endothelial cell, and myeloid cell antigens in spontaneous and chemically induced hemangiosarcomas and hemangiomas in mice. Toxicologic Pathology 41:709–721
Handgretinger R, Gordon PR, Leimig T et al (2003) Biology and plasticity of CD133+ hematopoietic stem cells. Ann N Y Acad Sci 996:141–151
Shantsila E, Watson T, Tse H-F et al (2008) New insights on endothelial progenitor cell subpopulations and their angiogenic properties. J Am Coll Cardiol 51:669–671
Kaushal S, Amiel GE, Guleserian KJ et al (2001) Functional small diameter neovessels created using endothelial progenitor cells expanded ex vivo. Nat Med 7:1035–1040
Reyes M, Dudek A, Jahagirdar B et al (2002) Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest 109:337–346
Hristov M, Erl W, Weber PC (2003) Endothelial progenitor cells: mobilization, differentiation, and homing. Arterioscler Thromb Vasc Biol 23:1185–1189
Rehman J, Li J, Orschell CM et al (2003) Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation 107:1164–1169
Mukai N, Akahori T, Komaki M et al (2008) A comparison of the tube forming potentials of early and late endothelial progenitor cells. Exp Cell Res 314:430–440
Sieveking DP, Buckle A, Celermajer DS et al (2008) Strikingly different angiogenic properties of endothelial progenitor cell subpopulations: insights from a novel human angiogenesis assay. J Am Coll Cardiol 51:660–668
Williamson K, Stringer SE, Alexander EY (2012) Endothelial progenitor cells enter the aging arena. Frontiers in Physiology 3:1–7
Cheng CC, Chang SJ, Chueh YN et al (2013) Distinct angiogenesis roles and surface markers of early and late endothelial progenitor cells revealed by functional group analyses. BMC Genomics 14:182
Bouvard C, Gafsou B, Dizier B et al (2010) ɑ6 Integrin subunit plays a major role in the proangiogenic properties of endothelial progenitor cells. Arterioscler Thromb Vasc Biol 30:1569–1575
Hill JM, Zalos G, Halcox JP et al (2003) Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med 348:593–600
Lin Y, Weisdorf DJ, Solovey A et al (2000) Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest 105:71–77
Prater DN, Case J, Ingram DA et al (2007) Working hypothesis to redefine endothelial progenitor cells. Leukemia 21:1141–1149
Padfield GJ, Newby DE, Mills NL (2010) Understanding the role of endothelial progenitor cells in percutaneous coronary intervention. J Am Coll Cardiol 55:1553–1565
Fadini GP, Losordo D, Dimmeler S (2012) Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ Res 110:624–637
Mao M, Xu X, Zhang Y et al (2013) Endothelial progenitor cells: the promise of cell-based therapies for acute lung injury. Inflamm Res 62:3–8
Schaumberg DA, Glynn RJ, Jenkins AJ et al (2005) Effect of intensive glycemic control on levels of markers of inflammation in type 1 diabetes mellitus in the diabetes control and complications trial. Circulation 111:2446–2453
Fadini GP, Agostini C, Avogaro A (2005) Endothelial progenitor cells and vascular biology in diabetes mellitus: current knowledge and future perspectives. Curr Diabetes Rev 1:41–58
Galasso G, Schiekofer S, Sato K et al (2006) Impaired angiogenesis in glutathione peroxidase-1-deficient mice is associated with endothelial progenitor cell dysfunction. Circ Res 98:254–261
Landmesser U, Merten R, Spiekermann S et al (2000) Vascular extracellular superoxide dismutase activity in patients with coronary artery disease: relation to endothelium-dependent vasodilation. Circulation 101:2264–2270
Ozuyaman B, Ebner P, Niesler U et al (2005) Nitric oxide differentially regulates proliferation and mobilization of endothelial progenitor cells but not of hematopoietic stem cells. Thromb Haemost 94:770–772
Chen YH, Lin SJ, Lin FY et al (2007) High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes 56:1559–1568
Thum T, Fraccarollo D, Schultheiss M et al (2007) Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes 56:666–674
Ceradini DJ, Yao D, Grogan RH et al (2008) Decreasing intracellular superoxide corrects defective ischemia-induced new vessel formation in diabetic mice. J Biol Chem 283:10930–10938
Fadini GP, Sartore S, Agostini C et al (2007) Significance of endothelial progenitor cells in subjects with diabetes. Diabetes Care 30:1305–1313
Lambiase PD, Edwards RJ, Anthopoulos P et al (2004) Circulating humoral factors and endorhelial progenitor cell in patients with differing coronary collateral support. Circulation 109:2993–2999
Gill M, Dias S, Hattori K et al (2001) Vascular trauma induces rapid but transient mobilization of VEGF endothelial precursor cells. Circ Res 88:167–7416
Fadini GP, Miorin M, Facco M et al (2005) Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol 45:1449–1457
Hammes HP (2005) Pericytes and the pathogenesis of diabetic retinopathy. Horm Metab Res 37:39–43
Fadini GP, de Kreutzenberg SV, Coracina A et al (2006a) Circulating CD34+ cells, metabolic syndrome, and cardiovascular risk. Eur Heart J 27:2247–2255
Tepper OM, Galiano RD, Capla JM et al (2002) Human endothelial progenitor cells from type 2 diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 106:2781–2786
Ii M, Takenaka H, Asai J et al (2006) Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ Res 98:697–704
Loomans CJ, van Haperen R, Duijis JM et al (2009) Differentiation of bone marrow-derived endothelialprogenitor cells is shifted into a proinflammatory phenotype byhyperglycemia. Mol Med 15:152–159
Cubbon RM, Kahn MB, Wheatcroft SB (2009) Effects of insulin resistance on endothelial progenitor cells and vascular repair. Clin Sci (Lond) 117:173–190
Altabas V (2015) Diabetes, endothelial dysfunction, and vascular repair: what should a diabetologist keep his eye on? Int J Endocrinol 2015:848272
Fadini GP, Boscaro E, de Kreutzenberg S et al (2010) Time course and mechanisms of circulating progenitor cell reduction in the natural history of type 2 diabetes. Diabetes Care 33:1097–1102
Kusuyama T, Omura T, Nishiya D et al (2006) Effects of treatment for diabetes mellitus on circulating vascular progenitor cells. J Pharmacol Sci 102:96–102
Egan CG, Lavery R, Caporali F et al (2008) Generalised reduction of putative endothelial progenitors and CXCR4-positive peripheral blood cells in type 2 diabetes. Diabetologia 51:1296–1305
De Falco E, Avitabile D, Totta P et al (2009) Altered SDF-1-mediated differentiation of bone marrow-derived endothelial progenitor cells in diabetes mellitus. J Cell Mol Med 13:3405–3414
Barthelmes D, Irhimeh MR, Gillies MC (2013) Diabetes impairs mobilization of mouse bone marrow-derivedLin(−)/VEGF-R2(+) progenitor cells. Blood Cells Mol Dis 51:163–173
Fadini GP, Pucci L, Vanacore R et al (2007) Glucose tolerance is negatively associated with circulating progenitor cell levels. Diabetologia 50:2156–2163
Churdchomjan W, Kheolamai P, Manochantr S et al (2010) Comparison of endothelial progenitor cell function in type 2 diabetes with good and poor glycemic control. BMC Endocr Disord 10:5
Kränkel N, Adams V, Linke A et al (2005) Hyperglycemia reduces survival and impairs function of circulating blood-derived progenitor cells. Arterioscler Thrombos Vasc Biol 25:698–703
Park J, Min J-S, Kim B et al (2015) Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-𝜅B pathways. Neurosci Lett 584:191–196
Yang H-L, Huang P-J, Liu Y-R et al (2014) Toona sinensis inhibits LPS-induced inflammation and migration in vascular smooth muscle cells via suppression of reactive oxygen species and NF-𝜅B signaling pathway. Ox Med Cell Longev 2014:901315
Zhen J, Lu H, Wang XQ et al (2008) Upregulation of endothelial and inducible nitric oxide synthase expression by reactive oxygen species. Am J Hypertens 21:28–34
Kim K-A, Shin Y-J, Akram M et al (2014) High glucose condition induces autophagy in endothelial progenitor cells contributing to angiogenic impairment. Biol Pharm Bull 37:1248–1252
Bhatta M, Ma JH, Wang JJ et al (2015) Enhanced endoplasmic reticulum stress in bone marrow angiogenic progenitor cells in a mouse model of long-term experimental type 2 diabetes. Diabetologia 58:2181–2190
Sollier CBD, Berge N, Boval B et al (2009) Functional variability of platelet response to clopidogrel correlates with P2Y12 receptor occupancy. Thromb Haemost 101:116–122
Gallagher KA, Liu ZJ, Xiao M et al (2007) Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest 117:1249–1259
Ye M, Li D, Yang J et al (2015) MicroRNA-130a targets MAP3K12 to modulate diabetic endothelial progenitor cell function. Cell Physiol Biochem 36:712–726
Hamed S, Brenner B, Roguin A (2011) Nitric oxide: a key factor behind the dysfunctionality of endothelial progenitor cells in diabetes mellitus type-2. Cardiovasc Res 91:9–15
Cohen RA, Tong X (2010) Vascular oxidative stress: the common link in hypertensive and diabetic vascular disease. J Cardiovasc Pharmacol 55:308–316
Rosso A, Balsamo A, Gambino R et al (2006) p53 mediates the accelerated onset of senescence of endothelial progenitor cells in diabetes. J Biol Chem 281:4339–4347
Yu CG, Zhang N, Yuan SS et al (2016) Endothelial progenitor cells in diabetic microvascular complications: friends or foes? Stem Cells 2016:1803989
Zhong Q, Kowluru RA (2013) Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: role of histone methylation. Invest Ophth Vis Sci 54:244–250
Li Y-D, Ye B-Q, Zheng S-X et al (2009) NF-𝜅B transcription factor p50 critically regulates tissue factor in deep vein thrombosis. J Biol Chem 284:4473–4483
Fish JE, Matouk CC, Rachlis A et al (2005) The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem 280:24824–24838
Murphy C, Kanaganayagam GS, Jiang B et al (2007) Vascular dysfunction and reduced circulating endothelial progenitor cells in young healthy UK South Asian men. Arterioscler Thromb Vasc Biol 27:936–942
Kahn MB, Yuldasheva NY, Cubbon RM et al (2011) Insulin resistance impairs circulating angiogenic progenitor cell function and delays endothelial regeneration. Diabetes 60:1295–1303
Chen LL, Liao YF, Zeng TS et al (2010) Effects of metformin plus gliclazide compared with metformin alone on circulating endothelial progenitor cell in Type 2 diabetic patients. Endocrine 38:266–275
Liao YF, Chen LL, Zeng TS et al (2010) Number of circulating endothelial progenitor cells as a marker of vascular endothelial function for Type 2 diabetes. Vasc Med 15:279–285
Pistrosch F, Herbrig K, Oelschlaegel U et al (2005) PPAR-agonist rosiglitazone increases number and migratory activity of cultured endothelial progenitor cells. Atherosclerosis 183:163–167
Aicher A, Heeschen C, Dimmeler S (2004) The role of NOS3 in stem cell mobilization. Trends Mol Med 10:421–425
Saad MI, Abdelkhalek TM, Salleh MM et al (2015) Insights into the molecular mechanisms of diabetes-induced endothelial dysfunction: focus on oxidative stress and endothelial progenitor cells. Endocrine 50:537–567
Dimmeler S, Aicher A, Vasa M et al (2001) HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI3-kinase/Akt pathway. J Clin Invest 108:391–397
Thum T, Hoeber S, Froese S et al (2007b) Age-dependent impairment of endothelial progenitor cells is corrected by growth-hormone-mediated increase of insulin-like growth-factor-1. Circ Res 100:434–443
Werner C, Kamani CH, Gensch C et al (2007) The peroxisome proliferator-activated receptor-gamma agonist pioglitazone increases number and function of endothelial progenitor cells in patients with coronary artery disease and normal glucose tolerance. Diabetes 56:2609–2615
Urao N, Okigaki M, Yamada H et al (2006) Erythropoietin-mobilized endothelial progenitors enhance reendothelialization via Akt-endothelial nitric oxide synthase activation and prevent neointimal hyperplasia. Circ Res 98:1405–1413
Humpert PM, Neuwirth R, Battista MJ et al (2005) SDF-1 genotype influences insulin-dependent mobilization of adult progenitor cells in type 2 diabetes. Diabetes Care 28:934–936
Benboubker L, Watier H, Carion A et al (2001) Association between the SDF1-SDF3’A allele and high levels of CD34+ progenitor cells mobilized into peripheral blood in humans. Br J Haematol 113:247–250
Humpert PM, Djuric Z, Zeuge U et al (2008) Insulin stimulates the clonogenic potential of angiogenic endothelial progenitor cells by IGF-1 receptor-dependent signaling. Mol Med 14:301–308
Juul A, Scheike T, Davidsen M et al (2002) Low serum insulin-like growth factor I is associated with increased risk of ischemic heart disease: a population-based case-control study. Circulation 106:939–944
Yuldasheva NY, Rashid ST, Haywood NJ et al (2014) Haploinsufficiency of the insulin-like growth factor-1 receptor enhances endothelial repair and favorably modifies angiogenic progenitor cell phenotype. Arterioscler Thrombos Vasc Biol 34:2051–2058
Sengupta A, Viswambharan H, Yuldasheva N et al (2014) Endothelial insulin sensitization enhances vascular repair in systemic insulin resistance and improves endothelial function by restoring nitric oxide bioavailability. Circulation 130:A13829
Wattanapitayakul SK, Weinstein DM, Holycross BJ et al (2000) Endothelial dysfunction and peroxynitrite formation are early events in angiotensin-induced cardiovascular disorders. FASEB J 14:271–278
Versari D, Daghini E, Virdis A et al (2009) Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care 32:S314–S321
Aicher A, Heeschen C, Mildner-Rihm C et al (2003) Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med 9:1370–1376
Hamed S, Brenner B, Aharon A et al (2009) Nitric oxide and superoxide dismutase modulate endothelial progenitor cell function in type 2 diabetes mellitus. Cardiovasc Diabetol 8:56
Thum T, Tsikas D, Stein S et al (2005) Suppression of endothelial progenitor cells in human coronary artery disease by the endogenous nitric oxide synthase inhibitor asymmetric dimethylarginine. J Am Coll Cardiol 46:1693–1701
Avogaro A, Toffolo G, Kiwanuka E et al (2003) L-Arginine-nitric oxide kinetics in normal and type 2 diabetic subjects: a stable-labelled 15N arginine approach. Diabetes 52:795–802
Bauersachs J, Schäfer A (2005) Tetrahydrobiopterin and eNOS dimer/monomer ratio-a clue to eNOS uncoupling in diabetes? Cardiovasc Res 65:768–769
Segal MS, Shah R, Afzal A et al (2006) Nitric oxide cytoskeletal-induced alterations reverse the endothelial progenitor cell migratory defect associated with diabetes. Diabetes 55:102–109
Sorrentino SA, Bahlmann FH, Besler C et al (2007) Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus. Circulation 116:163–173
Miyahara Y, Ohnishi S, Obata H et al (2006) Beraprost sodium enhances neovascularization in ischemic myocardium by mobilizing bone marrow cells in rats. Biochem Biophys Res Commun 349:1242–1249
Di Stefano R, Barsotti MC, Melillo E et al (2008) The prostacyclin analogue iloprost increases circulating endothelial progenitor cells in patients with critical limb ischemia. Thromb Haemost 100:871–877
Kawabe J, Yuhki K, Okada M et al (2010) Prostaglandin I2 promotes recruitment of endothelial progenitor cells and limits vascular remodeling. Arterioscler Thromb Vasc Biol 30:464–470
Morello F, Perino A, Hirsch E (2009) Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc Res 82:261–271
Bahlmann FH, De Groot K, Spandau J-M et al (2004) Erythropoietin regulates endothelial progenitor cells. Blood 103:921–926
Vikram A, Kim Y-R, Kumar S et al (2014) Canonical Wnt signaling induces vascular endothelial dysfunction via p66Shc-regulated reactive oxygen species. Arterioscler Thrombos Vasc Biol 34:2301–2309
Zhou S, Chen H-Z, Wan Y-Z et al (2011) Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res 109:639–648
Case J, Ingram DA, Haneline LS (2008) Oxidative stress impairs endothelial progenitor cell function. Antioxid Redox Signal 10:1895–1907
Di Stefano V, Cencioni C, Zaccagnini G et al (2009) P66ShcA modulates oxidative stress and survival of endothelial progenitor cells in response to high glucose. Cardiovasc Res 82:421–429
Li H, Zhang X, Guan X et al (2012) Advanced glycation end products impair the migration, adhesion and secretion potentials of late endothelial progenitor cells. Cardiovasc Diabetol 11:46
Sukmawati D, Fujimura S, Jitsukawa S et al (2015) Oxidative stress tolerance of early stage diabetic endothelial progenitor cell. Regener Therapy 1:38–44
Wang F, Wang Y-Q, Cao Q et al (2013) Hydrogen peroxide induced impairment of endothelial progenitor cell viability is mediated through a FoxO3a dependant mechanism. Microvasc Res 90:48–54
Song JJ, Lee YJ (2003) Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem J 373:845–853
Ingram TR, Krier LE, Mead C et al (2007) Clonogenic endothelial progenitor cells are sensitive to oxidative stress. Stem Cells 25:297–304
Wei J, Liu Y, Chang M et al (2012) Proteomic analysis of oxidative modification in endothelial colony-forming cells treated by hydrogen peroxide. Int J Mol Med 29:1099–1105
Ma FX, Zhou B, Chen Z et al (2006) Oxidized low density lipoprotein impairs endothelial progenitor cells by regulation of endothelial nitric oxide synthase. J Lipid Res 47:1227–1237
Hamed S, Brenner B, Abassi Z et al (2010) Hyperglycemia and oxidized-LDL exert a deleterious effect on endothelial progenitor cell migration in type 2 diabetes mellitus. Thromb Res 126:166–174
Ji K-T, Qian L, Nan JL et al (2015) Ox-LDL induces dysfunction of endothelial progenitor cells via activation of NF-κB. Biomed Res Int 2015:175291
Shimada K, Mokuno H, Matsunaga E et al (2004) Predictive value of circulating oxidized LDL for cardiac events in type 2 diabetic patients with coronary artery disease. Diabetes Care 27:843–844
Imanishi T, Hano T, Sawamura T et al (2004) Oxidized lowdensity lipoprotein induces endothelial progenitor cell senescence, leading to cellular dysfunction. Clin Exp Pharmacol Physiol 31:407–413
Tso C, Martinic G, Fan WH et al (2006) High-density lipoproteins enhance progenitormediated endothelium repair in mice. Arteriosclerosis, Trombosis, and Vascular Biology 26:1144–1149
Zhang Q, Yin H, Liu P et al (2010) Essential role of HDL on endothelial progenitor cell proliferation with PI3K/Akt/cyclin D1 as the signal pathway. Exp Biol Med 235:1082–1092
Pan B, Ma Y, Ren H et al (2012) Diabetic HDL is dysfunctional in stimulating endothelial cell migration and proliferation due to down regulation of SR-BI expression. PLoS ONE 7:e48530
Marin MT, Dasari PS, Tryggestad JB et al (2015) Oxidized HDL and LDL in adolescents with type 2 diabetes compared to normal weight and obese peers. J Diabetes Complic 29:679–685
Van Oostrom O, Nieuwdorp M, Westerweel P et al (2007) Reconstituted HDL increases circulating endothelial progenitor cells in patients with type 2 diabetes. Arterioscler Thrombos Vasc Biol 27:1864–1865
Petoumenos V, Nickenig G, Werner N (2009) High-density lipoprotein exerts vasculoprotection via endothelial progenitor cells. J Cell Mol Med 13:4623–4635
Huang C-Y, Lin F-Y, Shih C-M et al (2012) Moderate to high concentrations of high-density lipoprotein from healthy subjects paradoxically impair human endothelial progenitor cells and related angiogenesis by activating Rho-associated kinase pathways. Arterioscler Thrombos Vasc Biol 32:2405–2417
Wu J, He Z, Gao X et al (2015) Oxidized highdensity lipoprotein impairs endothelial progenitor cells’ function by activation of CD36-MAPK-TSP-1 pathways. Antioxid Redox Signal 22:308–324
Wu H, Li R, Wei ZH et al (2016) Diabetes-induced oxidative stress in endothelial progenitor cells may be sustained by a positive feedback loop involving high mobility group box-1. Oxid Med Cell Longev 2016:1943918
Dernbach E, Urbich C, Brandes RP et al (2004) Antioxidative stress-associated genes in circulating progenitor cells: evidence for enhanced resistance against oxidative stress. Blood 104:3591–3597
He T, Peterson TE, Holmuhamedov EL et al (2004) Human endothelial progenitor cells tolerate oxidative stress due to intrinsically high expression of manganese superoxide dismutase. Arterioscler Thromb Vasc Biol 24:2021–2027
Ohshima M, Li TS, Kubo M et al (2009) Antioxidant therapy attenuates diabetes-related impairment of bone marrow stem cells. Circ J 73:162–166
Tao J, Yang Z, Wang JM et al (2007) Shear stress increases Cu/ Zn SOD activity and mRNA expression in human endothelial progenitor cells. J Hum Hypertens 21:353–358
Kim KA, Shin YJ, Kim JH et al (2012) Dysfunction of endothelial progenitor cells under diabetic conditions and its underlying mechanisms. Arch Pharm Res 35:223–234
Hartge MM, Unger T, Kintscher U (2007) The endothelium and vascular inflammation in diabetes. Diabetes Vasc Dis Res 4:84–88
Goldberg RB (2009) Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J Clin Endocrinol Metab 94:3171–3182
Chen J, Jin J, Song M et al (2012) C-reactive protein down-regulates endothelial nitric oxide synthase expression and promotes apoptosis in endothelial progenitor cells through receptor for advanced glycation end-products. Gene 496:128–135
Fujii H, Li S-H, Szmitko PE et al (2006) C-reactive protein alters antioxidant defenses and promotes apoptosis in endothelial progenitor cells. Arterioscler Thrombos Vasc Biol 26:2476–2482
Fasing KA, Nissan BJ, Greiner JJ et al (2014) Influence of elevated levels of C-reactive protein on circulating endothelial progenitor cell function. Clin Transl Sci 7:137–140
Rosell A, Arai K, Lok J et al (2009) Interleukin-1β augments angiogenic responses of murine endothelial progenitor cells in vitro. J Cereb Blood Flow Metab 29:933–943
Yang L, Guo X-G, Du C-Q et al (2012) Interleukin-1 beta increases activity of human endothelial progenitor cells: involvement of PI3K-Akt signaling pathway. Inflammation 35:1242–1250
Mao A, Liu C, Guo Y et al (2012) Modulation of the number and functions of endothelial progenitor cells by interleukin 1β in the peripheral blood of pigs: involvement of p38 mitogen-activated protein kinase signaling in vitro. J Trauma Acute Care Surg 73:1145–1151
Kahlenberg JM, Thacker SG, Berthier CC et al (2011) Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol 187:6143–6156
Fan Y, Ye J, Shen F et al (2008) Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J Cereb Blood Flow Metab 28:90–98
Wang Y, Chen Q, Zhang Z et al (2015) Yan, Interleukin-10 overexpression improves the function of endothelial progenitor cells stimulated with TNF-α through the activation of the STAT3 signaling pathway. Int J Mol Med 35:471–477
Makino N, Maeda T, Sugano M et al (2005) High serum TNF-α level in Type 2 diabetic patients with microangiopathy is associated with eNOS down-regulation and apoptosis in endothelial cells. J Diabetes Complic 19:347–355
Zorena K, Mysliwska J, Mysliwiec M et al (2007) Relationship between serum levels of tumor necrosis factor-alpha and interleukin-6 in diabetes mellitus type 1 children. Central Eur J Immunol 32:124
Desouza CV, Hamel FG, Bidasee K, O’Connell K (2011) Role of inflammation and insulin resistance in endothelial progenitor cell dysfunction. Diabetes 60:1286–1294
Chen T-G, Zhong Z-Y, Sun G-F et al (2011) Effects of tumour necrosis factor-alpha on activity and nitric oxide synthase of endothelial progenitor cells from peripheral blood. Cell Prolif 44:352–359
Pfosser A, El-Aouni C, Pfisterer I et al (2010) NF-κB activation in embryonic endothelial progenitor cells enhances neovascularization via PSGL-1 mediated recruitment: novel role for LL37. Stem Cells 28:376–385
Ji K, Xing C, Jiang F et al (2013) Benzo [a] pyrene induces oxidative stress and endothelial progenitor cell dysfunction via the activation of the NF-κB pathway. Int J Mol Med 31:922–930
Chien HY, Lee TP, Chen CY et al (2015) Circulating microRNA as a diagnostic marker in populations with type 2 diabetes mellitus and diabetic complications. J Chin Med Assoc 78:204–211
Meng S, Cao JT, Zhang B et al (2012) Downregulation of microRNA-126 in endothelial progenitor cells from diabetes patients, impairs their functional properties, via target gene Spred-1. J Mol Cell Cardiol 53:64–72
Meng S, Cao J, Zhang X et al (2013) Downregulation of microRNA-130a contributes to endothelial progenitor cell dysfunction in diabetic patients via its target Runx3. PLoS One 8:e68611
de la Torre NG, Fernández-Durango R, Gómez R et al (2015) Expression of angiogenic microRNAs in endothelial progenitor cells from Type 1 diabetic patients with and without diabetic retinopathy. Invest Ophthalmol Vis Sci 56:4090–4098
Zeng J, Xiong Y, Li G et al (2013) MiR-21 is overexpressed in response to high glucose and protects endothelial cells from apoptosis. Exp Clin Endocrinol Diabetes 121:425e30
Zhao T, Li J, Chen AF (2010) MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am Physiol Endocrinol Metab 299:E110–E116
Menghini R, Casagrande V, Cardellini M et al (2009) MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 120:1524–1532
Berezin AE (2016) Endothelial progenitor cells dysfunction and impaired tissue reparation: the missed link in diabetes mellitus development. Diabetes Metab Syndr. pii: S1871-4021(16)30145-X
Georgescu A (2011) Vascular dysfunction in diabetes: the endothelial progenitor cells as new therapeutic strategy. World J Diabetes 2:92–97
Georgescu A, Alexandru N, Constantinescu A et al (2011) The promise of EPCs-based therapies on vascular dysfunction in diabetes. Eur J Pharmacol 669:1–6
Beckman JA, Creager MA (2016) Vascular complications of diabetes. Circ Res 118:1771–1785
Waltenberger J (2001) Impaired collateral vessel development in diabetes: potential cellular mechanisms and therapeutic implications. Cardiovasc Res 49:554–560
Schatteman GC, Hanlon HD, Jiao C et al (2000) Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J Clin Invest 106:571–578
Awad O, Jiao C, Ma N et al (2005) Obese diabetic mouse environment differentially affects primitive and monocytic endothelial cell progenitors. Stem Cells 23:575–583
Asai J, Takenaka H, Kusano KF et al (2006) Topical sonic hedgehog gene therapy accelerates wound healing in diabetes by enhancing endothelial progenitor cell mediated microvascular remodeling. Circulation 113:2413–2424
Georgescu A, Alexandru N, Andrei E et al (2012) Circulating microparticles and endothelial progenitor cells in atherosclerosis; pharmacological effects of irbesartan. J Thromb Haemost 10:680–691
Georgescu A, Alexandru N, Andrei E et al (2016) Effects of transplanted circulating endothelial progenitor cells and platelet microparticles in atherosclerosis development. Biol Cell 108:219–243
Andrei E, Alexandru N, Dragan E et al (2014) Flow cytometric analysis of circulating microparticles and endothelial progenitor cells in plasma; a research tool for atherosclerosis and therapy. Exp Clin Cardiol 20:1555–1563
Bădila E, Daraban AM, Ghiorghe S et al (2014) Rethinking cardiovascular therapy – the effect of irbesartan on circulating microparticles and endothelial progenitor cells in patients with hypertension and dyslipidemia. Farmacia 62:93–106
Werner N, Kosiol S, Schiegl T et al (2005) Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med 353:999–1007
Rustemeyer P, Wittkowski W, Jurk K et al (2006) Optimized flow cytometric analysis of endothelial progenitor cells in peripheral blood. J Immunoass Immunoch 27:77–88
Berezin AE, Kremzer AA, Samura TA et al (2015a) Impaired immune phenotype of circulating endothelial-derived microparticles in patients with metabolic syndrome and diabetes mellitus. J Endocrinol Invest 38:865–874
António N, Soares A, Fernandes R et al (2014) Endothelial progenitor cells in diabetic patients with myocardial infarction can statins improve their function? Eur J Pharmacol 741:25–36
Berezin AE (2014) Diabetes mellitus and cellular replacement therapy: expected clinical potential and perspectives. World J Diabetes 5:777–786
Vasa M, Fichtlscherer S, Aicher A et al (2001) Number and migratory capacity of circulating Endothelial Progenitor Cells inversely correlate with risk factors for coronary artery disease. Circ Res 89:1–7
Delva P, De Marchi S, Prior M et al (2008) Endothelial progenitor cells in patients with severe peripheral arterial disease. Endothelium 15:246–253
Lee LC, Chen CS, Choong PF et al (2010) Time-dependent dynamic mobilization of circulating progenitor cells during percutaneous coronary intervention in diabetics. Int J Cardiol 142:199–201
Schmidt-Lucke C, Rossig L, Fichtlscherer S et al (2005) Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation 111:2981–2987
Berezin AE, Kremzer AA, Martovitskaya YV et al (2015b) The effect of angiotensin-2 receptor blocker valsartan on circulating level of endothelial progenitor cells in diabetic patients with asymptomatic coronary artery disease. Diabetes Metab Syndr 9:305–309
Gregg EW, Sorlie P, Paulose-Ram R et al (2004) Prevalence of lower-extremity disease in the US adult population ≥40 years of age with and without diabetes: 1999–2000 national health and nutrition examination survey. Diabetes Care 27:1591–1597
Tapp RJ, Zimmet PZ, Harper CA et al (2004) Diabetes care in an Australian population: frequency of screening examinations for eye and foot complications of diabetes. Diabetes Care 27:688–693
Dolan NC, Liu K, Criqui MH et al (2002) Peripheral artery disease, diabetes, and reduced lower extremity functioning. Diabetes Care 25:113–120
Prompers L, Schaper N, Apelqvist J et al (2008) Prediction of outcome in individuals with diabetic foot ulcers: focus on the differences between individuals with and without peripheral arterial disease. The EURODIALE Study. Diabetologia 51:747–755
Bitterli L, Afan S, Bühler S et al (2016) Endothelial progenitor cells as a biological marker of peripheral artery disease. Vasc Med 21:3–11
Fadini GP, Sartore S, Albiero M et al (2006b) Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler Thromb Vasc Biol 26: 2140–2146
Yan J, Tie G, Park B et al (2009) Recovery from hind limb ischemia is less effective in type 2 than in type 1 diabetic mice: roles of endothelial nitric oxide synthase and endothelial progenitor cells. J Vasc Surg 50:1412–1422
Spinetti G, Kraenkel N, Emanueli C et al (2008) Diabetes and vessel wall remodelling: from mechanistic insights to regenerative therapies. Cardiovasc Res 78:265–273
Kang LN, Chen Q, Wang L et al (2009) Decreased mobilization of endothelial progenitor cells contributes to impaired neovascularization in diabetes. Clin Exp Pharmacol Physiol 36:e47–e56
Avogaro A, Fadini GP, Gallo A et al (2006) Endothelial dysfunction in type 2 diabetes mellitus. Nutr Metab Cardiovasc Dis 16:S39–S45
Gadau S, Emanueli C, Van Linthout S et al (2006) Benfotiamine accelerates the healing of ischaemic diabetic limbs in mice through protein kinase B/Akt-mediated potentiation of angiogenesis and inhibition of apoptosis. Diabetologia 49:405–420
Zhou B, Bi YY, Han ZB et al (2006) G-CSF-mobilized peripheral blood mononuclear cells from diabetic patients augment neovascularization in ischemic limbs but with impaired capability. J Thromb Haemostasis 4:993–1002
Emanueli C, Monopoli A, Kraenkel N et al (2007) Nitropravastatin stimulates reparative neovascularization and improves recovery from limb ischaemia in type-1 diabetic mice. Br J Pharmacol 150:873–882
Fadini GP, Sartore S, Schiavon M et al (2006c) Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia 49:3075–3084
Chen J, Chen S, Chen Y et al (2011) Circulating endothelial progenitor cells and cellular membrane microparticles in db/db diabetic mouse: possible implications in cerebral ischemic damage. Am J Physiol Endocrinol Metab 301:E62–E71
Bai YY, Wang L, Peng XG et al (2015a) Noninvasive monitoring of transplanted endothelial progenitor cells in diabetic ischemic stroke models. Biomaterials 40:43–50
Bai YY, Wang L, Chang D et al (2015b) Synergistic effects of transplanted endothelial progenitor cells and RWJ 67657 in diabetic ischemic stroke models. Stroke 46:1938–1946
Nguyen TT, Wang JJ, Wong TY (2007) Retinal vascular changes in pre-diabetes and prehypertension: new findings and their research and clinical implications. Diabetes Care 30:2708–2715
Zhang X, Saaddine JB, Chou CF et al (2010) Prevalence of diabetic retinopathy in the United States, 2005-2008. JAMA 304:649–656
Busik JV, Tikhonenko M, Bhatwadekar A et al (2009) Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med 206:2897–2906
Hazra S, Jarajapu YPR, Stepps V et al (2013) Long-term type 1 diabetes influences haematopoietic stem cells by reducing vascular repair potential and increasing inflammatory monocyte generation in a murine model. Diabetologia 56:644–653
Hu P, Thinschmidt JS, Yan Y et al (2013) CNS inflammation and bone marrow neuropathy in type 1 diabetes. Am J Pathol 183:1608–1620
Tan K, Lessieur E, Cutler A et al (2010) Impaired function of circulating CD34(+) CD45(-) cells in patients with proliferative diabetic retinopathy. Exp Eye Res 91:229–237
Lee IG, Chae SL, Kim JC (2006) Involvement of circulating endothelial progenitor cells and vasculogenic factors in the pathogenesis of diabetic retinopathy. Eye (Lond) 20:546–552
Zerbini G, Maestroni A, Palini A et al (2012) Endothelial progenitor cells carrying monocyte markers are selectively abnormal in type 1 diabetic patients with early retinopathy. Diabetes 61:908–914
Cahoon JM, Rai RR, Carroll LS et al (2015) Intravitreal AAV2.COMP-ang1 prevents neurovascular degeneration in a murine model of diabetic retinopathy. Diabetes 64:4247–4259
Li Calzi S, Neu MB, Shaw LC et al (2010) Endothelial progenitor dysfunction in the pathogenesis of diabetic retinopathy: treatment concept to correct diabetes-associated deficits. EPMA J 1:88–100
Yoder MC, Mead LE, Prater D et al (2007) Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 109:1801–1809
Case J, Mead LE, Bessler WK et al (2007) Human CD34+ AC133+VEGFR-2+ cells are not endothelial progenitor cells but distinct, primitive hematopoietic progenitors. Exp Hematol 35:1109–1118
Yoon CH, Hur J, Park KW et al (2005) Synergistic neovascularization by mixed transplantation of early endothelial progenitor cells and late outgrowth endothelial cells: the role of angiogenic cytokines and matrix metalloproteinases. Circulation 112:1618–1627
Tsuchida K, Makita Z, Yamagishi S et al (1999) Suppression of transforming growth factor beta and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation end product inhibitor, OPB-9195. Diabetologia 42:579–588
Kanesaki Y, Suzuki D, Uehara G et al (2005) Vascular endothelial growth factor gene expression is correlated with glomerular neovascularization in human diabetic nephropathy. Am J Kidney Dis 45:288–294
Furukawa M, Gohda T, Tanimoto M et al (2013) Pathogenesis and novel treatment from the mouse model of type 2 diabetic nephropathy. Sci World J 2013:928197
Makino H, Okada S, Nagumo A et al (2009) Decreased circulating CD34+ cells are associated with progression of diabetic nephropathy: short report. Diabetic Med 26:171–173
Thomas MC, Cooper ME, Rossing K et al (2006) Anaemia in diabetes: is there a rationale to treat? Diabetologia 49:1151–1157
Vallance P, Leone A, Calver A et al (1992) Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 339:572–575
Krzyzanowska K, Mittermayer F, Krugluger W et al (2006) Asymmetric dimethylarginine is associated with macrovascular disease and total homocysteine in patients with type 2 diabetes. Atherosclerosis 189:236–240
Park TS, Bhutto I, Zimmerlin L et al (2014) Vascular progenitors from cord blood-derived induced pluripotent stem cells possess augmented capacity for regenerating ischemic retinal vasculature. Circulation 129:359–372
Chan XY, Black R, Dickerman K et al (2015) Three-dimensional vascular network assembly from diabetic patient-derived induced pluripotent stem cells. Arterioscler Thrombos Vasc Biol 35:2677–2685
Tavakoli M, Mojaddidi M, Fadavi H et al (2008) Pathophysiology and treatment of painful diabetic neuropathy. Curr Pain Headache Rep 12:192–197
Dyck PJ, Davies JL, Clark VM et al (2006) Modeling chronic glycemic exposure variables as correlates and predictors of microvascular complications of diabetes. Diabetes Care 29:2282–2288
Tesfaye S, Boulton AJ, Dyck PJ et al (2010) Toronto Diabetic Neuropathy Expert Group. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 33:2285–2293
Cameron N, Cotter M, Low P (1991) Nerve blood flow in early experimental diabetes in rats: relation to conduction deficits. Am J Physiol Endocrinol Metab 261:E1–E8
Porat Y, Porozov S, Belkin D et al (2006) Isolation of an adult blood-derived progenitor cell population capable of differentiation into angiogenic, myocardial and neural lineages. Br J Haematol 135:703–714
Naruse K, Hamada Y, Nakashima E et al (2005) Therapeutic neovascularization using cord blood-derived endothelial progenitor cells for diabetic neuropathy. Diabetes 54:1823–1828
Chavez JC, Almhanna K, Berti-Mattera LN (2005) Transient expression of hypoxia-inducible factor-1 alpha and target genes in peripheral nerves from diabetic rats. Neurosci Lett 374:179–182
Leiter LA (2005) The prevention of diabetic microvascular complications of diabetes: is there a role for lipid lowering? Diabetes Res Clin Pract 68:S3–S14
Lee PSS, Poh KK (2014) Endothelial progenitor cells in cardiovascular diseases. World J Stem Cells 6:355–366
Di Stefano R, Barsotti MC, Felice F et al (2010) Smoking and endothelial progenitor cells: a revision of literature. Curr Pharm Des 16:2559–2566
Ludwig A, Jochmann N, Kertesz A et al (2010) Smoking decreases the level of circulating CD34 progenitor cells in young healthy women–a pilot study. BMC Womens Health 10:20
Siddique A, Shantsila E, Lip Gregory YH et al (2010) Endothelial progenitor cells: what use for the cardiologist? Journal of Angiogenes Res 2:6
Giannotti G, Doerries C, Mocharla PS et al (2010) Impaired endothelial repair capacity of early endothelial progenitor cells in prehypertension: relation to endothelial dysfunction. Hypertension 55:1389–1397
Yang Z, Chen L, Su C et al (2010) Impaired endothelial progenitor cell activity is associated with reduced arterial elasticity in patients with essential hypertension. Clin Exp Hypertens 32:444–452
Lee CW, Huang PH, Huang SS et al (2011) Decreased circulating endothelial progenitor cell levels and function in essential hypertensive patients with electrocardiographic left ventricular hypertrophy. Hypertens Res 34:999–1003
Rossi F, Bertone C, Montanile F et al (2010) HDL cholesterol is a strong determinant of endothelial progenitor cells in hypercholesterolemic subjects. Microvasc Res 80:274–279
Heida NM, Muller JP, Cheng IF et al (2010) Effects of obesity and weight loss on the functional properties of early outgrowth endothelial progenitor cells. J Am Coll Cardiol 55:357–367
Tobler K, Freudenthaler A, Baumgartner-Parzer SM et al (2010) Reduction of both number and proliferative activity of human endothelial progenitor cells in obesity. Int J Obes (Lond) 34:687–700
Dzau VJ, Gnecchi M, Pachori AS et al (2005) Therapeutic potential of endothelial progenitor cells in cardiovascular diseases. Hypertension 46:7–18
Jie KE, Goossens MH, van Oostrom O et al (2009) Circulating endothelial progenitor cell levels are higher during childhood than in adult life. Atherosclerosis 202:345–347
Shintani S, Murohara T, Ikeda H et al (2001) Mobilization of endothelial progenitor cells in patients with acute myocardial infarction. Circulation 103:2776–2779
Leone AM, Valgimigli M, Giannico MB et al (2009) From bone marrow to the arterial wall: the ongoing tale of endothelial progenitor cells. European Heart Journal 30:890–899
Miller−Kasprzak E, Jagodziński PP (2007) Endothelial progenitor cells as a new agent contributing to vascular repair. Arch Immunol Ther Exp 55:247–259
Strehlow K, Werner N, Berweiler J et al (2003) Estrogen increases bone marrow-derived endothelial progenitor cell production and diminishes neointima formation. Circulation 107:3059–3065
Heeschen C, Aicher A, Lehmann R et al (2003) Erythropoietin is a potent physiological stimulus for endothelial progenitor cell mobilization. Blood 102:1340–1346
Tilling L, Chowienczyk P, Clapp B (2009) Progenitors in motion: mechanisms of mobilization of endothelial progenitor cells. Br J Clin Pharmacol 68:484–492
Yu H, Feng Y (2008) The potential of statin and stromal cell-derived factor-1 to promote angiogenesis. Cell Adh Migr 2:254–257
Steiner S, Niessner A, Ziegler S et al (2005) Endurance training increases the number of endothelial progenitor cells in patients with cardiovascular risk and coronary artery disease. Atherosclerosis 181:305–310
George J, Goldstein E, Abashidze S et al (2004) Circulating endothelial progenitor cells in patients with unstable angina: association with systemic inflammation. Eur Heart J 25:1003–1008
Shantsila E, Watson T, Lip GY (2007) Endothelial progenitor cells in cardiovascular disorders. J Am Coll Cardiol 49:741–752
Imanishi T, Moriwaki C, Hano T et al (2005) Endothelial progenitor cell senescence is accelerated in both experimental hypertensive rats and patients with essential hypertension. J Hypertens 23:1831–1837
Fadini GP, Albiero M, Vigili de Kreutzenberg S et al (2013) Diabetes impairs stem cell and proangiogenic cell mobilization in humans. Diabetes Care 36:943–949
Kissel CK, Lehmann R, Assmus B et al (2007) Selective functional exhaustion of hematopoietic progenitor cells in the bone marrow of patients with postinfarction heart failure. J Am Coll Cardiol 49:2341–2349
Massa M, Rosti V, Ferrario M et al (2005) Increased circulating hematopoietic and endothelial progenitor cells in the early phase of acute myocardial infarction. Blood 105:199–206
Yu Y, Fukuda N, Yao EH et al (2008) Effects of an ARB on endothelial progenitor cell function and cardiovascular oxidation in hypertension. Am J Hypertens 21:72–77
Honda A, Matsuura K, Fukushima N et al (2009) Telmisartan induces proliferation of human endothelial progenitor cells via PPARgamma-dependent PI3K/Akt pathway. Atherosclerosis 205:376–384
Pelliccia F, Pasceri V, Cianfrocca C et al (2010) Angiotensin II receptor antagonism with telmisartan increases number of endothelial progenitor cells in normotensive patients with coronary artery disease: a randomized, double-blind, placebocontrolled study. Atherosclerosis 210:510–515
Min TQ, Zhu CJ, Xiang WX et al (2004) Improvement in endothelial progenitor cells from peripheral blood by ramipril therapy in patients with stable coronary artery disease. Cardiovasc Drugs Ther 18:203–209
Cacciatore F, Bruzzese G, Vitale DF et al (2011) Effects of ACE inhibition on circulating endothelial progenitor cells, vascular damage, and oxidative stress in hypertensive patients. Eur J Clin Pharmacol 67:877–883
Sugiura T, Kondo T, Kureishi-Bando Y et al (2008) Nifedipine improves endothelial function: role of endothelial progenitor cells. Hypertension 52:491–498
de Ciuceis C, Pilu A, Rizzoni D et al (2011) Effect of antihypertensive treatment on circulating endothelial progenitor cells in patients with mild essential hypertension. Blood Press 20:77–83
DiFabio JM, Thomas GR, Zucco L et al (2006) Nitroglycerin attenuates human endothelial progenitor cell differentiation, function, and survival. J Pharmacol Exp Ther 318:117–123
Spadaccio C, Pollari F, Casacalenda A et al (2010) Atorvastatin increases the number of endothelial progenitor cells after cardiac surgery: a randomized control study. J Cardiovasc Pharmacol 55:30–38
Huang B, Cheng Y, Xie Q et al (2012) Effect of 40 mg versus 10 mg of atorvastatin on oxidized low-density lipoprotein, high-sensitivity C-reactive protein, circulating endothelial-derived microparticles, and endothelial progenitor cells in patients with ischemic cardiomyopathy. Clin Cardiol 35:125–130
Erbs S, Beck EB, Linke A et al (2011) High-dose rosuvastatin in chronic heart failure promotes vasculogenesis, corrects endothelial function, and improves cardiac remodeling – results from a randomized, double-blind, and placebo-controlled study. Int J Cardiol 146:56–63
Zhou LJ-Z, Wang L, Zhang T-X (2009) Effects of Xuezhikang and Pravastatin on circulating endothelial progenitor cells in patients with essential hypertension. Chin J Integr Med 15:266–271
Smadja DM, Godier A, Susen S et al (2009) Endothelial progenitor cells are selectively mobilised immediately after coronary artery bypass grafting or valve surgery. Thromb Haemost 101:983–985
Kondo T, Hayashi M, Takeshita K et al (2004) Smoking cessation rapidly increases circulating progenitor cells in peripheral blood in chronic smokers. Arterioscler Thromb Vasc Biol 24:1442–1447
Wang XX, Zhu JH, Chen JZ (2004) Effects of nicotine on the number and activity of circulating endothelial progenitor cells. J Clin Pharmacol 44:881–889
Makino H, Okada S, Nagumo A et al (2008) Pioglitazone treatment stimulates circulating CD34-positive cells in type 2 diabetes patients. Diabetes Res Clin Pract 81:327–330
Wang CH, Ting MK, Verma S et al (2006) Pioglitazone increases the numbers and improves the functional capacity of endothelial progenitor cells in patients with diabetes mellitus. Am Heart J 152:1051.e1–1051.e8
Acknowledgements
This work is supported by grants of the Romanian National Authority for Scientific Research, CNCS – UEFISCDI, project numbers: PN-II-RU-TE-2014-4-0525, PN-II-RU-TE-2014-4-0523 and PN-II-PT-PCCA-2013-4-0816.
Also, authors’ work is supported by: the Romanian Academy; the Competitiveness Operational Programme 2014-2020, Priority Axis 1/Action 1.1.4/Financing Contract no.115/13.09.2016/MySMIS:104362; the MODERNIZE project infrastructure, funded by the National Authority of Scientific Research and Innovation, in the name of the Ministry of European Funds, through the Operational Program Increase of Economic Competitiveness, Priority axis 2, Operation 2.2.1 (POSCCE-A2- 0.2.2.1- 2013-1), co-financed by the European Regional Development Fund.
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Alexandru, N., Titorencu, I., Frunzã, S., Weiss, E., Bãdilã, E., Georgescu, A. (2017). Endothelial Progenitor Cell Dysfunction in the Pathogenesis of Vascular Complications of Diabetes. In: Kartha, C., Ramachandran, S., Pillai, R. (eds) Mechanisms of Vascular Defects in Diabetes Mellitus. Advances in Biochemistry in Health and Disease, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-319-60324-7_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-60324-7_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-60323-0
Online ISBN: 978-3-319-60324-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)