
Abstract
Sensitization is a prerequisite for the development of allergic contact dermatitis. Its initiation, however, is—as mistakenly assumed for quite a while—not just dependent on an adaptive immune response, but also requires a supplementary signal that activates the innate immune system. Many metal allergens are capable of delivering both signals—the mandatory antigenic stimulus and an innate proinflammatory signal. In recent years, the molecular mechanisms of metal-induced innate immune activation have at least in part been deciphered. It turned out that nickel, cobalt and presumably palladium directly bind to Toll-like receptor 4 (TLR4) in humans, resulting in receptor dimerization, activation and subsequent gene transcription. On the other hand, metal compounds may injure cells and tissue leading to alteration of extracellular matrix molecules such as hyaluronan, which can serve as endogenous ligands for TLRs. Additionally, metal haptens may induce reactive oxygen species (ROS) that result in activation of the inflammasome, a multimeric protein platform that controls release of the proinflammatory cytokines IL-1β and IL-18. A recently identified powerful activator of the inflammasome is the widespread contact hapten dichromate. Importantly, TLR and inflammasome activation may occur at the same time and collaborate in delivering innate immune signals. Finally, the coincident presence of microbial pathogens that can activate TLRs may support the development of contact allergies to metals. A better understanding of the mechanisms by which metal allergens are sensed by the innate immune system may contribute to the design of novel therapeutic approaches for this common allergic disease.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

1 The Significance of Innate Immune Activation in Metal Allergy
Contact allergies to normally harmless metal ions in our environment are prototypic of T cell-mediated delayed-type hypersensitivity (type IV) [1]. The capability to mount a T cell-dependent adaptive response is the key determinant governing the clinical manifestation of metal allergies. However, it is frequently neglected that de novo formation of metal-reactive T cells crucially requires innate immune activation [2]. In fact, the initial step in sensitization to metals is the proinflammatory activation of dendritic cells (DCs), which take up and process the metal allergen and carry it to the regional lymph node. There, they present it to naïve T cells, which become primed, proliferate and re-enter the circulation as metal-reactive effector and memory T cells (Fig. 8.1). One has to bear in mind that it is this first direct encounter between an activated DC and naïve T cells which paves the way for sensitization to metal hypersensitivity, as this interaction is a conditio sine qua non for de novo generation of metal-specific T cells. Of note, the degree of DC activation is considered to be of imminent importance for the fate of the resulting metal-responsive T cell as ineffective or partial DC activation is believed to result in generation of regulatory T cells and tolerance [3]. Thus, development of a T effector response requires robust DC activation. This may either result from direct stimulation or secondary activation due to the release of proinflammatory cytokines or damage-associated molecular patterns (DAMPs) by other skin-resident cells [4].

Requirement of innate immune activation in the different phases of allergic contact dermatitis. During sensitization innate immune activation is necessary to activate and mobilize skin-resident dendritic cells, which migrate to the regional lymph node to present the metal allergen (red) to naïve T cells, which become primed in response to productive DC interaction. Primed metal-specific T cells subsequently proliferate and differentiate into metal-specific effector and memory T cells released to the circulation. In the elicitation phase, innate immune activation is required to locally activate endothelial cells, which in turn upregulate leucocyte adhesion receptors and cytokines to recruit metal-specific effector T cells to the site of exposure and to allow their attachment and transmigration through the endothelial barrier into the skin where they initiate the adaptive immune response by interaction with various skin-resident cells including hapten-presenting DCs
Besides its role during sensitization, innate immune activation is also relevant for endothelial activation at the elicitation phase of metal-induced allergic contact dermatitis. Once activated, endothelial cells induce expression of cytokines and cell adhesion receptors, which is necessary for recruitment of metal-specific effector T cells and other leukocytes from the circulation and their transmigration through the endothelial barrier to the site of challenge [5] (Fig. 8.1). The importance of endothelial activation is also evident by the observation that lack of the endothelial-specific leukocyte interaction molecule E-selectin in combination with P-selectin deficiency interfered with experimental contact hypersensitivity in mice [6]. Thus, innate immune activation is not only essential for initial production of metal-specific T cells but also crucial to recall and instruct T cell effector activity upon repeated or continuous challenge with the metal hapten.
2 Initiation of Innate Immune Activation by Metal Allergens: The Example of Nickel
It has long been a mystery how metals initiate the required innate immune response to trigger metal allergy. For some metal allergens, the underlying mechanisms now have started to emerge. The best example is nickel, for which the mechanism of proinflammatory activation has recently been solved [7, 8].
Nickel is by far the most relevant contact allergen in humans. Still, it has never been popular as a model allergen to study contact hypersensitivity (CHS) in mice. The reason is simple: Wild-type mice are highly resistant to metal-induced contact hypersensitivity [9], making native mouse models unattractive for investigation of nickel allergy. Hence, most early studies analysing the effect of nickel on innate immunity focused on patient studies or human in vitro systems. Below we will briefly summarize some of the key findings that helped to shed light on the mechanism by which nickel initiates its allergic response.
An important finding was the discovery that nickel could directly trigger proinflammatory gene expression in human primary endothelial cells in vitro [10, 11], which later greatly facilitated identification of the responsible receptor since those cells only express a limited number of dedicated innate immune receptors. Endothelial activation was subsequently confirmed as a physiological response to nickel exposure, as it could be demonstrated that epicutaneous nickel administration to the skin of sensitized patients resulted in strong mRNA expression of endothelial-specific inflammation markers in skin sections [12]. Intriguingly, proinflammatory activation of dermal endothelial cells as well as unidentified cells in the vicinity or within the keratinocyte basal cell layer of the epidermis was initiated as early as 6 h after nickel application [12]. At this early time point, no infiltration of T cells or other leukocytes was observed, excluding an activation mode via infiltrating leukocytes. The early activation kinetics of skin-resident cells also argued against other indirect activation mechanisms, for instance, via DAMP release that may secondarily activate innate immune receptors. This corroborated previous in vitro findings that stimulation of primary human endothelial cells with nickel or cobalt triggered a rapid activation of the proinflammatory IKK2/NFκB pathway and induction of NFκB-dependent gene expression [11]. Importantly, the observed inflammatory response of human primary endothelial cells did not depend on release of TNF or IL-1, which are well-known NFκB-activating cytokines since neither interfering antibodies to TNF or IL-1 [11] nor treatment with the IL-1 receptor antagonist anakinra [8] was able to block the inflammatory response to nickel. This supported the notion that nickel could directly trigger activation of an IKK2-/NFκB-inducing innate immune receptor.
3 Identification of TLR4 as Direct Mediator of Nickel-Induced Innate Immune Activation
Innate immune responses critically depend on activation of a relatively small number of pattern recognition receptors (PRRs) that are dedicated to the broad detection of evolutionarily conserved pathogen-associated molecular patterns (PAMPs) rather than to the detection of single pathogenic structures [13, 14]. Prominent examples are Toll-like receptors (TLRs), but PRRs additionally involve Nod-like receptors (NLRs), Rig-like receptors (RLRs) and DNA sensory proteins such as AIM2.
Among those, TLRs such as TLR2 and TLR4 qualified best as potential nickel sensors: For one, TLR2 and TLR4 had previously been shown to be important for contact hypersensitivity responses as TLR2/TLR4 double-deficient mice were resistant to contact hypersensitivity induced by the model hapten 2,4,6-trinitro-1-chlorobenzene (TNCB) [15]. Secondly, TLR4 was also shown to be involved in unrelated allergic responses such as innate immune activation by the dust mite allergen Der p2 [16] or the cat dander protein Fel D1 [17]. Finally, TLRs evolved to detect a large variety of different molecular patterns ranging from PAMPs such as bacterial lipids, glycoproteins, peptidoglycans or nucleic acids [18] to endogenous DAMPs such as hyaluronic acid [19], heat shock proteins [20], S100 proteins [21], or the extracellular matrix protein biglycan [22].
TLRs are integral glycoproteins, consisting of a cytoplasmic or luminal leucine-rich receptor domain, an anchoring transmembrane domain and an intracellular Toll-/interleukin 1-receptor (TIR) domain that mediates intracellular signal transduction in response to pathogen challenge [18]. In humans ten different functional TLRs have been described, which differ both in PAMP specificity, cellular localization and downstream signalling. They are subcategorized into cell surface TLRs, including TLR1, TLR2, TLR4, TLR5 and TLR6 due to their localization within the cytoplasmic membrane, and intracellular TLRs comprising TLR3, TLR7, TLR8 and TLR9, which reside in the membrane of endosomes, the endoplasmatic reticulum, lysosomes and endolysosomes [18]. Their different localization reflects the distinct core function of those two TLR subtypes, with surface TLRs mainly being involved in the defence of extracellular bacterial or fungal pathogens by recognition of microbial membrane components such as proteins, lipoproteins and lipids, and intracellular TLRs mediating host responses to intracellular microbes and viruses by detection of nucleic acids. Despite their fundamental differences in core functionality, the two TLR subgroups share central downstream signalling components required for proinflammatory activation. TLR activation invariably results in activation of the IKK2/NFκB transcription factor pathway [18], which is essential for transcriptional induction of key proinflammatory cytokines such as TNF or IL-8 [23]. Except for TLR3, the capacity of TLRs to stimulate NFκB activity critically relies on recruitment of the TIR domain-containing adaptor protein, MyD88 and members of the IL-1 receptor-associated kinases (IRAKs) such as IRAK1 and IRAK4, which also are essential for mediation of signal responses downstream of the IL-1 receptor. It was particularly this critical dependency of TLR signalling on MyD88 and IRAK1 that finally implicated TLRs as mediators of nickel-induced innate immune activation since siRNA-mediated knockdown of MyD88 or IRAK1 could abrogate nickel-induced proinflammatory gene expression in primary human endothelial cells [24]. As those cells only express a limited number of TLRs including TLR4 and TLR3 [25], which triggers NFκB activation in a MyD88-independent manner via the TIR-domain-containing adapter TRIF [26], the candidate list of TLR mediators of nickel-induced NFκB activation quickly boiled down to TLR4, which physiologically detects bacterial lipopolysaccharide (LPS) [27]. Indeed, siRNA-mediated depletion of either TLR4 or its co-receptor MD2 abolished nickel-induced NFκB activity in human primary endothelial cells [24]. Conversely, reconstitution of human TLR4 along with its co-receptor MD2 but not expression of either receptor component alone or other TLRs restored the missing innate immune response of TLR-deficient HEK293 cells to nickel [24], confirming the human TLR4/MD2 complex as a nickel-responsive innate immune receptor. Surprisingly, only in human cells, TLR4/MD2 positivity turned out to be a reliable predictor of nickel-induced proinflammatory activation, whereas nickel failed to trigger proinflammatory gene expression in TLR4-/MD2-positive murine cells [24]. Consistently, expression of murine TLR4 in HEK293 cells stably expressing either murine or human MD2 was unable to restore nickel-induced proinflammatory gene expression [24]. This also explains the aforementioned insensitivity of mice to nickel-induced contact hypersensitivity since murine TLR4 was unable to initiate an innate immune signal in response to nickel stimulation. Accordingly, only transgenic expression of human TLR4 but not of murine TLR4 in a TLR4−/− background was capable of restoring innate immune activation by nickel and allowed for contact hypersensitivity induction by nickel in mice [24].
This peculiar species dependency of nickel-induced proinflammatory activation and contact hypersensitivity finally turned out to be due to the presence of two non-conserved histidines, i.e. H456 and H458, at the dimerization interface of human TLR4, which are missing in murine TLR4. Accordingly, mutation of the equivalent sites of murine TLR4 to histidines restored its capacity to induce nickel-dependent proinflammatory gene expression in reconstitution experiments in MD2-expressing HEK293 cells [24]. Structural modelling of dimeric human TLR4 further revealed that the non-conserved histidines at position H456 and H458 in combination with a conserved histidine at position H431 provided by the opposing TLR4 monomer form two putative metal binding sites in the assembled TLR4 dimer [24], suggesting that nickel may initiate proinflammatory signalling by cross-linking two TLR4 molecules. Indeed, this concept was validated when it was demonstrated that nickel stimulation promoted co-immunoprecipitation of differently tagged TLR4 variants co-expressed in HEK293 cells [28]. Direct interaction studies using in vitro-synthesized peptides comprising the predicted metal-binding region of human TLR4 later on confirmed H456 and H458 as genuine metal-binding residues [29].
Of note, the above-mentioned studies revealed important differences in nickel- and LPS-induced TLR4 activation. First, H456 and H458 were found to be dispensable for LPS-induced proinflammatory activation [24, 28]. Second, LPS required the presence of the TLR4 co-receptor MD2 to trigger TLR4 dimerization, whereas in the case of nickel, MD2 expression was not obligatory for TLR4 dimerization per se [28] but strictly required for subsequent signal initiation [24], most likely by stabilizing the nickel-bound TLR4 dimer [30]. Accordingly, administration of a MD2-free soluble ectodomain of human TLR4 could reduce nickel-induced proinflammatory expression in human primary endothelial cells but left the LPS-induced response unaffected [28]. This inhibitory effect of soluble TLR4 required the dimerization capacity of the TLR4 ectodomain since neither alanine mutation of asparagine 433, which interferes with TLR4 dimerization, nor double mutation of H456/H458 of the soluble TLR4 ectodomain were able to impair nickel-induced proinflammatory activation in such transfer experiments [28]. These observations indicate that it should principally be possible to therapeutically interfere with nickel-induced proinflammatory activation without touching the innate immune system’s important function in host defence against LPS-releasing gram-negative bacteria [8].
4 TLR-Dependent Innate Immune Activation by Other Metals
The discovery of human TLR4 as a direct mediator of nickel-induced innate immune responses raised the question whether other metal allergens might use similar mechanisms of innate immune activation. Indeed, in the meantime also other metal allergens were shown to induce proinflammatory gene expression via TLR4 or other TLRs. The first one was cobalt, which could trigger proinflammatory activation by a very similar if not identical TLR4-dependent mechanism [28, 30]. In analogy to nickel, cobalt-induced proinflammatory gene expression was similarly found to rely on human TLR4 and required MD2, TLR4 dimerization and the presence of the two non-conserved histidines H456/H458 [28, 30,31,32,33]. Analogous to nickel, cobalt further was able to trigger a type I interferon response downstream of TLR4 [30]. This most likely relied on TRIF-dependent activation of the transcription factor interferon response factor 3 (IRF3) that physiologically is initiated by endocytosis of activated TLR4, as cobalt could transcriptionally induce expression of typical TRIF-dependent cytokines such as CCL5/RANTES or CXCL10 [28] and was able to stimulate an IRF-responsive reporter construct [30].
More recently, palladium was reported as a third metal allergen to trigger TLR4-dependent proinflammatory gene expression as shown by differential responsiveness of wild-type and human TLR4-/MD2-positive HEK293 cells to palladium stimulation [34]. Consistently, other TLR4-positive cells such as primary human monocyte-derived DCs could also initiate proinflammatory gene expression in response to palladium stimulation [34]. However, the response to palladium was substantially weaker than with nickel and cobalt, and it is currently unclear whether the observed palladium response was likewise species-dependent and requiring MD2 and histidines at positions H456 and H458 of TLR4.
Apart from TLR4, TLR3 has recently not only been implicated in the elicitation phase of CHS to TNCB [35] but also the innate immune response to gold particles used in dental applications. It was demonstrated that gold thiosulphate dose-dependently triggered proinflammatory activation in TLR3-positive primary human monocyte-derived DCs and TLR3-supplemented HEK293 cells [36]. The exact mechanism of TLR3 activation, however, is currently unclear. Considering that TLR3 belongs to the group of intracellular TLRs and has been shown to serve as an endogenous sensor for RNA species formed during necrosis [37], it is conceivable that DAMP release in response to cellular damage triggers this response. Alternatively, phagocytosed gold particles may trigger nonphysiological signalling by directly influencing TLR3 activity independently of its natural ligand.
5 Indirect Roles of TLRs in Metal Allergy
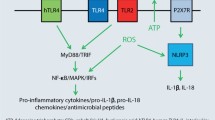
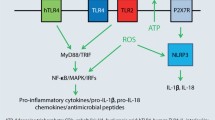
Besides their role as direct mediators of metal-induced proinflammatory signalling (Fig. 8.2a), TLRs may exert important indirect roles in metal-induced innate immune activation (Fig. 8.2b, c). For instance, TLR activation critically contributes to inflammasome activation [38] (Fig. 8.2b), another important innate immune mechanism known to contribute to contact hypersensitivity [39]. The inflammasome, in its core composed of a NLR (in most cases NLRP3), the adaptor protein ASC and Caspase-1, regulates maturation of IL-1β and IL-18 via Caspase-1-mediated proteolytic cleavage [38]. Importantly, inflammasome activation commonly requires a TLR-dependent priming signal that triggers pro-IL-1β and pro-IL-18 mRNA expression (signal 1) before Caspase-1-mediated release can be initiated by a distinct NLR-inducing signal (signal 2) [38] (Fig. 8.2b). Additionally, TLR-mediated priming was also found to be essential for transcriptional and post-transcriptional activation of NLRP3 [40, 41], without which no effective inflammasome activation can occur. Thus, TLR activation is considered an important prerequisite for inflammasome activation.

Roles of TLRs in metal-induced innate immune activation. TLRs can either directly mediate metal-induced innate immune activation as in case of TLR4 activation by nickel (Ni2+), cobalt (Co2+) and Pd2+ or TLR3 stimulation in response to exposure with gold (Au3+) particles, resulting in NFκB-dependent proinflammatory gene expression (a), or indirectly contribute to metal-induced innate immune responses, e.g. by priming of inflammasome activation as shown for hexavalent chromium (Cr6+) (b) or by triggering proinflammatory gene expression in response to hypothetical DAMP release by tissue damage, for instance, by oxidative degradation of extracellular matrix components due to metal-induced production of reactive oxygen species (ROS) (c). HA hyaluronic acid, mROS mitochondrial reactive oxygen species
An intriguing example illustrating the importance of TLR signalling for metal-induced inflammasome activation is the innate immune response to chromium (Fig. 8.2b). Recent studies from our lab have shown that the major allergenic oxidation form of chromium, chromium (VI), unlike nickel or cobalt, fails to trigger proinflammatory gene expression in various cells including THP-1 cells [42], a human monocytic cell line expressing multiple TLRs including TLR4 [43] that frequently is employed as a model system for studying inflammasome activation. This suggested that chromium (VI) is unable to trigger a direct TLR4-dependent innate immune signal, confirming previous observations that chromium (VI) failed to trigger innate immune activation in TLR4-positive human primary endothelial cells [11]. However, when THP1 were primed by TLR stimulation, they strongly initiated IL-1β cleavage, Caspase-1 activation and IL-1β release, indicative of inflammasome activation [42]. Knockdown of NLRP3 abolished this response, identifying NLRP3 as the responsible NLR for inflammasome activation. Chromium (VI)-induced inflammasome activation subsequently was also shown to occur in other cells, including murine bone marrow-derived DCs and human primary keratinocytes. Remarkably, the latter cells still required priming by TNF or TLR stimulation to trigger a significant inflammasome response to chromium (VI) albeit they constitutively express pro-IL-1β [42], suggesting that even in cells with basal pro-IL-1β expression, TLR-mediated priming is important to allow chromium (VI)-induced innate immune activation. Of note, only hexavalent chromium, but not trivalent chromium that is substantially less allergenic [44,45,46], was able to trigger NLRP3 activation [42]. While evidence is still pending to prove that inflammasome activation indeed contributes to chromium (VI)-induced contact hypersensitivity and may account for the divergent allergenicity of the different chromium compounds, these data strongly suggest that some metal allergens may critically rely on additional signals such as TLR activation present at sensitization to trigger sufficient innate immune activation to launch an adaptive immune response.
While in the case of chromium (VI), TLR activation and inflammasome activation cooperate to trigger sufficient innate immune activation, experimental data in mice also suggest that TLR stimulation can fully replace a missing innate immune signal. For instance, it has been shown that naturally nickel-resistant mice can be sensitized to nickel by co-treatment with the TLR4 agonist LPS [9] or the TLR2 agonist Pam3CSK4 [47]. This suggests that in the presence of sufficient TLR activation, for instance, by a coincidental infection, even metals normally incapable of initiating an innate immune response by themselves may efficiently trigger an adaptive response as long as they are efficiently haptenized. It is further conceivable that even in the case of metals that efficiently can activate the innate immune system such as nickel, a supporting unrelated TLR activation may be required for efficient sensitization as local nickel concentrations in the skin might not reach sufficient levels for effective TLR4 activation. On the other hand, a strong TLR4 activator such as nickel may act as an adjuvant for sensitization to another metal hapten, e.g. to cobalt [48].
Another yet unproven possibility by which TLRs indirectly may foster metal allergy is their secondary activation by DAMPs (Fig. 8.2c) [49]. For instance, it has been shown that some DAMPs produced via oxidative degradation of the extracellular matrix such as low molecular weight hyaluronic acid can act as ligands for TLR2 and TLR4 [19]. Notably, several metals including nickel and chromium (VI) trigger the release of ROS in the mitochondria [42, 50] so that in vivo a secondary TLR2/TLR4 activation via DAMPs produced after oxidative degradation of the extracellular matrix is at least conceivable. DAMP-mediated inflammasome activation that occurs independently of TLR4-mediated innate immune activation has recently been reported in cobalt-induced implant-related inflammation [51]. Such mechanisms may perhaps also explain why some groups, in contrast to our own experience, were able to induce contact hypersensitivity to nickel in mice, even in TLR4-deficent animals, when high doses of nickel were epicutaneously and repeatedly applied to the skin [52]. In combination with its known capacity to trigger species-independent inflammasome activation [50], this may result in sufficient innate immune activation to allow for adjuvant-free induction of contact hypersensitivity in this particular model.
6 Conclusion
Successful initiation of contact allergy to metals requires an antigen stimulus leading to an adaptive immune response, as well as a proinflammatory danger signal. Potent metal allergens are able to generate both at the same time. The innate signal is delivered by activation of TLRs and/or the inflammasome. Metal allergens may directly (e.g. nickel and cobalt, which directly bind to distinct histidines of TLR4 resulting in receptor dimerization and activation) or indirectly (e.g. by damaging extracellular matrix molecules such as hyaluronan that serve as TLR ligands) activate TLRs resulting in DC maturation and activation as well as expression of proinflammatory cytokines, chemokines and adhesion molecules. Moreover, as exemplified by dichromate, they may activate the inflammasome via mitochondrial ROS production, which leads to release of IL-1β and IL-18. Importantly, TLR activation may prime the inflammasome, making the latter susceptible for subsequent activation by distinct metal allergens. Finally, the coincident presence of microbial pathogens can contribute to (or even supplement for) the delivery of the required metal allergen innate immune signal. A deeper understanding of the sensing of metal allergens by the innate immune system will surely contribute to the development of novel therapeutic approaches for this common allergic skin disease.
References
McKee AS, Fontenot AP. Interplay of innate and adaptive immunity in metal-induced hypersensitivity. Curr Opin Immunol. 2016;42:25–30. doi:10.1016/j.coi.2016.05.001.
Schmidt M, Goebeler M. Immunology of metal allergies. J Dtsch Dermatol Ges. 2015;13(7):653–60. doi:10.1111/ddg.12673.
Mahnke K, Ring S, Enk AH. Antibody targeting of “steady-state” dendritic cells induces tolerance mediated by regulatory T cells. Front Immunol. 2016;7:63. doi:10.3389/fimmu.2016.00063.
Kaplan DH, Igyarto BZ, Gaspari AA. Early immune events in the induction of allergic contact dermatitis. Nat Rev Immunol. 2012;12(2):114–24. doi:10.1038/nri3150.
Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41(5):694–707. doi:10.1016/j.immuni.2014.10.008.
Staite ND, Justen JM, Sly LM, Beaudet AL, Bullard DC. Inhibition of delayed-type contact hypersensitivity in mice deficient in both E-selectin and P-selectin. Blood. 1996;88(8):2973–9.
Saito M, Arakaki R, Yamada A, Tsunematsu T, Kudo Y, Ishimaru N. Molecular mechanisms of nickel allergy. Int J Mol Sci. 2016;17(2). doi:10.3390/ijms17020202.
Schmidt M, Goebeler M. Nickel allergies: paying the Toll for innate immunity. J Mol Med (Berl). 2011;89(10):961–70. doi:10.1007/s00109-011-0780-0.
Sato N, Kinbara M, Kuroishi T, Kimura K, Iwakura Y, Ohtsu H, Sugawara S, Endo Y. Lipopolysaccharide promotes and augments metal allergies in mice, dependent on innate immunity and histidine decarboxylase. Clin Exp Allergy. 2007;37(5):743–51. doi:10.1111/j.1365-2222.2007.02705.x.
Goebeler M, Meinardus-Hager G, Roth J, Goerdt S, Sorg C. Nickel chloride and cobalt chloride, two common contact sensitizers, directly induce expression of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and endothelial leukocyte adhesion molecule (ELAM-1) by endothelial cells. J Invest Dermatol. 1993;100(6):759–65.
Goebeler M, Roth J, Brocker EB, Sorg C, Schulze-Osthoff K. Activation of nuclear factor-kappa B and gene expression in human endothelial cells by the common haptens nickel and cobalt. J Immunol. 1995;155(5):2459–67.
Goebeler M, Trautmann A, Voss A, Brocker EV, Toksoy A, Gillitzer R. Differential and sequential expression of multiple chemokines during elicitation of allergic contact hypersensitivity. Am J Pathol. 2001;158(2):431–40.
Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16(4):343–53. doi:10.1038/ni.3123.
Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–73. doi:10.1128/CMR.00046-08.
Martin SF, Dudda JC, Bachtanian E, Lembo A, Liller S, Durr C, Heimesaat MM, Bereswill S, Fejer G, Vassileva R, Jakob T, Freudenberg N, Termeer CC, Johner C, Galanos C, Freudenberg MA. Toll-like receptor and IL-12 signaling control susceptibility to contact hypersensitivity. J Exp Med. 2008;205(9):2151–62. doi:10.1084/jem.20070509.
Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, Thorne PS, Wills-Karp M, Gioannini TL, Weiss JP, Karp CL. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. 2009;457(7229):585–8. doi:10.1038/nature07548.
Herre J, Gronlund H, Brooks H, Hopkins L, Waggoner L, Murton B, Gangloff M, Opaleye O, Chilvers ER, Fitzgerald K, Gay N, Monie T, Bryant C. Allergens as immunomodulatory proteins: the cat dander protein Fel d 1 enhances TLR activation by lipid ligands. J Immunol. 2013;191(4):1529–35. doi:10.4049/jimmunol.1300284.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–84. doi:10.1038/ni.1863.
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11(11):1173–9. doi:10.1038/nm1315.
Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277(17):15028–34. doi:10.1074/jbc.M200497200.
Loser K, Vogl T, Voskort M, Lueken A, Kupas V, Nacken W, Klenner L, Kuhn A, Foell D, Sorokin L, Luger TA, Roth J, Beissert S. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat Med. 2010;16(6):713–7. doi:10.1038/nm.2150.
Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Gotte M, Malle E, Schaefer RM, Grone HJ. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115(8):2223–33. doi:10.1172/JCI23755.
Viemann D, Schmidt M, Tenbrock K, Schmid S, Muller V, Klimmek K, Ludwig S, Roth J, Goebeler M. The contact allergen nickel triggers a unique inflammatory and proangiogenic gene expression pattern via activation of NF-kappaB and hypoxia-inducible factor-1alpha. J Immunol. 2007;178(5):3198–207.
Schmidt M, Raghavan B, Muller V, Vogl T, Fejer G, Tchaptchet S, Keck S, Kalis C, Nielsen PJ, Galanos C, Roth J, Skerra A, Martin SF, Freudenberg MA, Goebeler M. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat Immunol. 2010;11(9):814–9. doi:10.1038/ni.1919.
Muller V, Viemann D, Schmidt M, Endres N, Ludwig S, Leverkus M, Roth J, Goebeler M. Candida albicans triggers activation of distinct signaling pathways to establish a proinflammatory gene expression program in primary human endothelial cells. J Immunol. 2007;179(12):8435–45.
Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169(12):6668–72.
Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–8.
Raghavan B, Martin SF, Esser PR, Goebeler M, Schmidt M. Metal allergens nickel and cobalt facilitate TLR4 homodimerization independently of MD2. EMBO Rep. 2012;13(12):1109–15. doi:10.1038/embor.2012.155.
Zoroddu MA, Peana M, Medici S, Potocki S, Kozlowski H. Ni(II) binding to the 429-460 peptide fragment from human Toll like receptor (hTLR4): a crucial role for nickel-induced contact allergy? Dalton Trans. 2014;43(7):2764–71. doi:10.1039/c3dt52187g.
Oblak A, Pohar J, Jerala R. MD-2 determinants of nickel and cobalt-mediated activation of human TLR4. PLoS One. 2015;10(3):e0120583. doi:10.1371/journal.pone.0120583.
Lawrence H, Mawdesley AE, Holland JP, Kirby JA, Deehan DJ, Tyson-Capper AJ. Targeting Toll-like receptor 4 prevents cobalt-mediated inflammation. Oncotarget. 2016;7(7):7578–85. doi:10.18632/oncotarget.7105.
Potnis PA, Dutta DK, Wood SC. Toll-like receptor 4 signaling pathway mediates proinflammatory immune response to cobalt-alloy particles. Cell Immunol. 2013;282(1):53–65. doi:10.1016/j.cellimm.2013.04.003.
Tyson-Capper AJ, Lawrence H, Holland JP, Deehan DJ, Kirby JA. Metal-on-metal hips: cobalt can induce an endotoxin-like response. Ann Rheum Dis. 2013;72(3):460–1. doi:10.1136/annrheumdis-2012-202468.
Rachmawati D, Bontkes HJ, Verstege MI, Muris J, von Blomberg BM, Scheper RJ, van Hoogstraten IM. Transition metal sensing by Toll-like receptor-4: next to nickel, cobalt and palladium are potent human dendritic cell stimulators. Contact Dermatitis. 2013;68(6):331–8. doi:10.1111/cod.12042.
Nakamura N, Tamagawa-Mineoka R, Ueta M, Kinoshita S, Katoh N. Toll-like receptor 3 increases allergic and irritant contact dermatitis. J Invest Dermatol. 2015;135(2):411–7. doi:10.1038/jid.2014.402.
Rachmawati D, Alsalem IW, Bontkes HJ, Verstege MI, Gibbs S, von Blomberg BM, Scheper RJ, van Hoogstraten IM. Innate stimulatory capacity of high molecular weight transition metals Au (gold) and Hg (mercury). Toxicol In Vitro. 2015;29(2):363–9. doi:10.1016/j.tiv.2014.10.010.
Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205(11):2609–21. doi:10.1084/jem.20081370.
He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–21. doi:10.1016/j.tibs.2016.09.002.
Honda T, Egawa G, Grabbe S, Kabashima K. Update of immune events in the murine contact hypersensitivity model: toward the understanding of allergic contact dermatitis. J Invest Dermatol. 2013;133(2):303–15. doi:10.1038/jid.2012.284.
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–91. doi:10.4049/jimmunol.0901363.
Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287(43):36617–22. doi:10.1074/jbc.M112.407130.
Adam C, Wohlfarth J, Haußmann M, Sennefelder H, Rodin A, Maler M, Martin SF, Goebeler M, Schmidt M. Allergy-inducing chromium compounds trigger potent innate immune stimulation via ROS-dependent inflammasome activation. J Invest Dermatol. 2016;137(2):367–76. doi:10.1016/j.jid.2016.10.003.
Dowling JK, Dellacasagrande J. Toll-like receptors: ligands, cell-based models, and readouts for receptor action. Methods Mol Biol. 2016;1390:3–27. doi:10.1007/978-1-4939-3335-8_1.
Hansen MB, Johansen JD, Menne T. Chromium allergy: significance of both Cr(III) and Cr(VI). Contact Dermatitis. 2003;49(4):206–12.
Iyer VJ, Banerjee G, Govindram CB, Kamath V, Shinde S, Gaikwad A, Jerajani HR, Raman G, Cherian KM. Role of different valence states of chromium in the elicitation of allergic contact dermatitis. Contact Dermatitis. 2002;47(6):357–60.
Nethercott J, Paustenbach D, Adams R, Fowler J, Marks J, Morton C, Taylor J, Horowitz S, Finley B. A study of chromium induced allergic contact dermatitis with 54 volunteers: implications for environmental risk assessment. Occup Environ Med. 1994;51(6):371–80.
Takahashi H, Kinbara M, Sato N, Sasaki K, Sugawara S, Endo Y. Nickel allergy-promoting effects of microbial or inflammatory substances at the sensitization step in mice. Int Immunopharmacol. 2011;11(10):1534–40. doi:10.1016/j.intimp.2011.05.010.
Bonefeld CM, Nielsen MM, Vennegaard MT, Johansen JD, Geisler C, Thyssen JP. Nickel acts as an adjuvant during cobalt sensitization. Exp Dermatol. 2015;24(3):229–31. doi:10.1111/exd.12634.
Martin SF, Esser PR, Weber FC, Jakob T, Freudenberg MA, Schmidt M, Goebeler M. Mechanisms of chemical-induced innate immunity in allergic contact dermatitis. Allergy. 2011;66(9):1152–63. doi:10.1111/j.1398-9995.2011.02652.x.
Li X, Zhong F. Nickel induces interleukin-1beta secretion via the NLRP3-ASC-caspase-1 pathway. Inflammation. 2014;37(2):457–66. doi:10.1007/s10753-013-9759-z.
Samelko L, Landgraeber S, McAllister K, Jacobs J, Hallab NJ. Cobalt alloy implant debris induces inflammation and bone loss primarily through danger signaling, not TLR4 activation: implications for DAMP-ening implant related inflammation. PLoS One. 2016;11(7):e0160141. doi:10.1371/journal.pone.0160141.
Vennegaard MT, Dyring-Andersen B, Skov L, Nielsen MM, Schmidt JD, Bzorek M, Poulsen SS, Thomsen AR, Woetmann A, Thyssen JP, Johansen JD, Odum N, Menne T, Geisler C, Bonefeld CM. Epicutaneous exposure to nickel induces nickel allergy in mice via a MyD88-dependent and interleukin-1-dependent pathway. Contact Dermatitis. 2014;71(4):224–32. doi:10.1111/cod.12270.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Schmidt, M., Goebeler, M. (2018). Innate Immune System Response in Metal Allergy: Toll-Like Receptors. In: Chen, J., Thyssen, J. (eds) Metal Allergy. Springer, Cham. https://doi.org/10.1007/978-3-319-58503-1_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-58503-1_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-58502-4
Online ISBN: 978-3-319-58503-1
eBook Packages: MedicineMedicine (R0)