
Abstract
This chapter details Toll-like receptors (TLRs) and the tools available to study their biology in vitro. Key parameters to consider before exploring TLR action such as receptor localization, signaling pathways, nature of ligands and cellular expression are introduced. Cellular models (i.e., host cells and readouts) based on the use of cell lines, primary cells, or whole blood are presented. The use of modified TLRs to circumvent some technical problems is also discussed.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Toll-Like Receptors
Toll-like receptors (TLRs) represent a family of Pattern Recognition Receptors (PRRs) that play a critical role in early defence against invading pathogens and responses to endogenous danger signals. To date ten human TLRs have been classified (TLR1-TLR10) and 12 in the mouse (TLR1-9, TLR11-13) [1]. From a technical point of view, TLRs are distinguished by ligand specificity, signal transduction, expression patterns, and cellular localization.
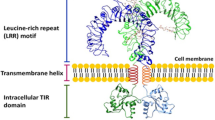
TLRs are type I transmembrane receptors and form part of the Toll/interleukin-1 (TIR) superfamily that includes the IL-1 receptors (IL-1Rs) because of the shared homology of their cytoplasmic regions [2]. In contrast, their extracellular regions (ectodomains) are considerably different. TLR ectodomains contain tandem repeats of leucine-rich regions termed leucine-rich repeats (LRRs), while IL-1Rs have three immunoglobulin (Ig)-like domains. The arrangement of LRR side chains confers a unique combinatorial code to each TLR enabling it to bind a specific ligand. In addition detection of ligand is also dependent on the cellular localization of the TLR in question [3, 4]. The distinct cellular location of different TLRs and the unique combinatorial code of their LRRs afford them the ability to interact with structurally unrelated ligands of endogenous and exogenous origin.
1.1 TLR Localization
TLRs are localized to the cell surface (plasma membrane) or intracellular compartments [5]. The location of any given TLR is related to the origin of the ligand it recognizes. TLR1, TLR2, TLR4, TLR5, and TLR6 are expressed on the plasma membrane and are largely involved in the detection of bacterial products in the extracellular space. On the other hand, TLR3, TLR7, TLR8, and TLR9 are located within endocytic compartments that present nucleic acids of viral origin to these TLRs [6, 7]. Localization is also important for the discrimination between “self” and “non-self.” For example, in contrast to most TLR ligands nucleic acids can be of self and foreign origin. Studies have demonstrated that a chimeric TLR9 consisting of a transmembrane and cytoplasmic domain of other TLRs localizes to the plasma membrane [8]. Here it is able to detect and respond to mammalian DNA yet remain unresponsive to viral nucleic acids, highlighting the importance of TLR location. Endogenous TLR9 is not exposed to mammalian DNA and can only be activated by viral DNA ingested and acidified within endosomes. As a consequence of their localization, activation of intracellular TLRs in in vitro experiments requires the use of cell permeable ligands or ligands complexed with cationic lipids to facilitate their uptake.
1.2 TLR Signaling
Knowledge of the different signaling pathways mobilized by TLRs is essential for the selection of appropriate readouts or reporter genes in cell-based assays (see Part III). TLR signaling is initiated by the binding of a TLR with its respective ligand, be it exogenous or endogenous. Invading microbes are detected by means of recognizing specific “pathogen-associated molecular patterns” or PAMPs. TLRs have also evolved to detect molecules derived from damaged cells referred to as endogenous “danger-associated molecular patterns” or DAMPs. Recognition of PAMPs or DAMPs by TLRs results in the activation of signaling pathways that induce the upregulation of cytokines, chemokines, and co-stimulatory molecules.
The initial step in signal transduction following the binding of ligand is dimerization of two TLR receptor chains. In the case of TLR4 a homodimer is induced by the binding of MD-2 (Myeloid differentiation protein-2) to the lipid A moiety of Lipopolysaccharide (LPS) [9]. To date the crystal structures of several TLR dimers have been elucidated including dimers of TLR3 [10–12], TLR2/1 [13] and TLR4 [14], TLR5 [15], TLR8 [16], and TLR10 [17].
Following dimerization conformational changes in the receptor leads to the association of the two cytoplasmic receptor TIR domains [4, 18]. It is believed the overall structure of the TLR ectodomain, transmembrane, and cytoplasmic regions in turn constitutes a molecular switch “turned-on” by ligand binding. Ultimately, the association of TLR cytoplasmic TIR domains produces two symmetrically related binding sites for the recruitment of specific adaptor molecules which also contain TIR domains [19]. The result is a post-receptor signaling complex associating relevant adaptor molecules to active TIR domains of TLR dimers.
Subsequently, signaling cascades are activated via recruitment of such adaptors. These include MyD88, Mal/TIRAP, TRIF/TICAM-1, TRAM/TICAM-2, and SARM [3, 20, 21]. The proximal events of ligand binding and adaptor recruitment to the active TIR domains of TLRs can result in the activation of two major signaling cascades, namely the MyD88-dependant and MyD88-independent pathways [22] (see Fig. 1).

TLR localization and activation of MyD88-dependant and -independent pathways
The MyD88-dependent pathway results in nuclear translocation of Nuclear Factor-kappaB (NF-κB) and induction of pro-inflammatory cytokines, while the MyD88-independent pathway mediates induction of Type I interferons and interferon-inducible genes via Interferon Response Factors (e.g., IRF3/7) [21]. All TLRs with the exception of TLR3 are known to recruit MyD88 and activate the MyD88-dependent pathway activating mitogen activated protein kinases (MAPKs) and NF-κB [6]. In addition to MyD88, TLR2 and TLR4 require Mal/TIRAP to activate the MyD88-dependant pathway [23]. TLR3 typically activates IRFs and expression of IFNs from its endocytic compartments via TRIF [24]. Signaling via TLR4 is unique in that it activates both the MyD88-dependant pathway via MyD88 and Mal/TIRAP to activate NF-κB and the MyD88-independent pathway via TRAM and TRIF to activate IRF3. It remains unclear as to whether an activated TLR4 dimer can stimulate Mal and TRAM directed pathways simultaneously or whether the engagement of each adaptor is mutually exclusive [19]. Of particular interest is the fact that TLR4 signaling via TRIF and TRAM also induces a late phase of NF-κB activation [25]. A role for TRAM in the induction of Type I IFN via TLR2 endosomal signaling has also recently been described [26]. TLR7, TLR8, and TLR9 act through MyD88 to induce pro-inflammatory cytokine secretion and the IFNs. A recent study has defined a role for TRIF in TLR9-induced IFNs in response to high doses of CpG [27]. Other than MyD88, the signaling proteins employed by TLR7-8 to activate IFNs remain unidentified.
2 Ligands
Knowledge about how TLRs recognize pathogenic ligands is critical to understand how these receptors are activated and important for designing therapeutic compounds that can target this family of receptors for inflammatory diseases. TLRs are activated by microorganisms such as bacteria, viruses, or fungi and by endogenous ligands. Most of these ligands are by nature complex and undefined. From a technical point of view, defined, specific ligands are needed. This section will focus on commercially available ligands purified for the purpose of TLR study and on the most recent TLR ligands described in the literature. Suppliers offering a large range of TLR-related products are Enzo Life Sciences, Hycult Biotech, Bio-Techne (formally Imgenex, Novus Biologicals), and InvivoGen; and most of the TLR ligands discussed in this chapter are commercially available from them (see Table 1). Different formats are proposed including 96-well plates precoated with TLR ligands (InvivoGen), and labeled ligands for staining purposes (e.g., flow cytometry). In addition, several pharmaceutical companies have developed new synthetic TLR ligands and TLR inhibitors (mainly for TLR7/8/9) [28].
The potency of TLR ligands relies on their ability to induce homo- or heterodimerization and/or conformational change of receptor chains [29]. Direct binding of several TLRs to their known ligands has been experimentally demonstrated for TLR9 [30], TLR1/2 [13], TLR4 [14] and TLR3 [10–12], TLR5 [15], TLR8 [16], and TLR13 [31].
2.1 TLR1
TLR1 forms functional heterodimers with TLR2. TLR1/2 heterodimers are receptors for triacyl lipopeptides found in bacteria and mycobacteria [32]. An artificial model suggests that signaling through TLR1 homodimers would trigger a weak signal characterized by the activation of the TNF promoter [33]. The ligand of choice for TLR1/2 is the synthetic molecule Pam3CSK4 and it is active when used at 10 ng/ml.
2.2 TLR2
It is difficult to demonstrate ligand specificity for TLR2 as it forms heterodimers with TLR1, TLR6, and possibly TLR10 to recognize a variety of microorganisms [34]. Zymosan from yeast cell wall, lipoteichoic acid [35], lipoarabinomannan from bacteria and mycobacteria [36], and lipoproteins from mycoplasma or gram-negative bacteria [32] can all activate TLR2 in the absence of TLR1 or TLR6. In vitro, in the absence of TLR1 and TLR6, TLR2 can be activated with High-mobility group box (HMGB) 1 [37], heat-killed Listeria monocytogenes [38], Pam3CSK4 [32], or Staphylococcus aureus peptidoglycan [39]. With regard to peptidoglycan, it has been shown that this ligand is recognized by the cytosolic receptor NOD1 [40].
2.3 TLR3
TLR3 is an intracellular TLR localized in endosomes; it binds dsRNA of viral origin. In addition, two synthetic TLR3 ligands mimicking dsRNA, polyriboinosinic-polyribocytidylic acid (polyI:C) and polyadenylic-polyuridylic acid (polyA:U), have been described [41]. Both low molecular weight (LMW; 0.2–1 kb) and high molecular weight (HMW; 1.5–1 kb) Poly I:C fragments can activate TLR3. However, low molecular weight fragments are less potent than the large fragments. Polyinosinic acid (polyI) has been shown to activate TLR3 in mouse B cells, macrophages, and bone marrow derived dendritic cells [42].
TLR3 ligands are active when added to the medium at 10–50 μg/ml. Complexation with a lipid-based transfection reagent results in a lower effective concentration but such a delivery system might also activate cytoplasmic receptors such as MDA-5 independently of TLR3 [43].
Direct addition of both HMW- and LMW-poly I:C to the cultures of primary macrophages and a human neuroblastoma cell line (CHP212) activated TLR3 [44]. However, the transfection of poly I:C was necessary to induce TLR3 activation in other cell types studied. The activation efficiency of TLR3 by poly I:C is influenced by various factors, including size of the ligands, delivery methods, and cell types.
2.4 TLR4
TLR4 was the first TLR identified in mammals. It is one of the most studied TLRs [45]. First considered as the receptor for lipopolysaccharide (LPS) from Gram-negative bacteria, it has been later shown that TLR4 requires MD2, LPS binding protein (LBP), and CD14 as co-receptors to function. LPS consists of a polysaccharide moiety and the active component lipid A. Lipid A is composed of a glucosamine disaccharide linked to fatty acids [46]. The potency to activate TLR4 depends on the number of fatty acids. Lipid A from pathogenic bacteria (e.g., E. coli, Salmonella species) contains six fatty acids whereas lipid A containing 4 or 5 fatty acids is found in less pathogenic bacteria (mutated E. coli, Rhodobacter sphaeroides, P. gingivalis). The latter LPS are considered as antagonists because they can inhibit the activation of TLR4 induced by hexaacylated LPS [47]. Complete competitive inhibition of LPS activity is possible using a 100-fold excess of the antagonistic LPS from Rhodobacter sphaeroides (available from InvivoGen).
Agonistic LPS are divided in two categories based on the morphology of bacteria colonies: smooth or rough (S-LPS or R-LPS, respectively). S-LPS contains O-polysaccharide chains which are absent from R-LPS. Wild-type Gram-negative bacteria synthesize S-LPS which needs CD14 to signal through TLR4. Signaling by S-LPS through TLR4/CD14 activates both arms of the TLR4 pathway (i.e., MyD88-dependent and MyD88-independent; see Fig. 1). R-LPS can signal in the absence of CD14 but it does not activate the MyD88-independent pathway [48]. This property might be useful for in vitro experiments in the absence of TLR4 co-receptors but such activation of TLR4 is incomplete. Interestingly, it has been demonstrated that monophospholipid A, a candidate vaccine adjuvant, stimulates only the TRIF/TRAM arm of the TLR4 signaling [49]. This ligand is a promising tool to study the MyD88-independent response induced by TLR4 activation.
Basic purification protocols lead to LPS containing associated lipoproteins and thereby activation of TLR2 and TLR4. Phenol re-extraction of LPS is needed to retain only a TLR4 activity [50]. Purified LPS from different bacterial strains are commercially available and are active at 10 ng/ml.
Surprisingly, murine (but not human) TLR4 recognizes the antitumoral agent taxol [51]. Proteins of viral origin (e.g., respiratory syncytial virus, mouse mammary tumor virus) can also activate TLR4 [34]. Recently, heme has been shown to activate TLR4 in a CD14-dependent and MD2-independent manner [52].
TLR4 also recognizes endogenous ligands [53]; these include hyaluronate, fibrinogen, and HMGB1. These substances are generally released after tissue injury and might trigger a danger signal through TLR4 [54]. Activation of TLR4 by heat shock proteins is controversial as some authors have shown that this could be due to endotoxin contamination [55, 56].
2.5 TLR5
TLR5 recognizes monomeric flagellin, a constituent protein of bacterial flagella [57, 58]. Purified flagellin from Salmonella typhimurium or Bacillus subtilis is commercially available (InvivoGen, Bio-Techne). InvivoGen also proposes a recombinant S. typhimurium flagellin produced in mammalian cells that is devoid of TLR2/4 activity of bacterial origin. Flagellins are active at 0.1–1 μg/ml.
2.6 TLR6
TLR6 associates with TLR2 to recognize diacyl lipopeptides such as MALP-2 and FSL-1 [59]. Pam2CSK4 is a synthetic ligand for TLR2/6; it activates TLR6-expressing cells when used at 1–10 ng/ml. The level of activation is increased by the presence of CD14 and CD36 [60]. Along with the standard bacterial cell wall components available commercially, whole preparation of heat-killed bacteria (Gram-negative and Gram-positive) is available for TLR2/6 activation (InvivoGen). None of the available TLR ligands seem to activate TLR6 in the absence of TLR2. Nevertheless, artificial activation of a TLR6-specific signaling cascade showed that signaling through TLR6 homodimers is theoretically possible [34].
2.7 TLR7
TLR7 is expressed in endosomes; it is a receptor for ssRNA from viruses, especially U or GU-rich oligoribonucleotides such as RNA40 from the U5 region of HIV-1 RNA [61–63]. Activation of murine TLR7 but not human TLR7 can be achieved using complexed ssRNAs. Imidazoquinolines, which are synthetic compounds with antiviral activities, including R848 (or resiquimod) and its water soluble derivative CL097 (InvivoGen), imiquimod and gardiquimod (specific for TLR7 when used at less than 1 μg/ml) have also been identified as TLR7 ligands [64]. In addition, TLR7 is specifically activated by nucleoside analogs, e.g., 1 mM loxoribine, a guanosine analog [65], and the adenine analogs CL264 and CL307 from InvivoGen [66]. Notably, while looking for siRNA directed against human TLR9, Hornung et al. have discovered that siRNA can specifically activate TLR7 [67]. This activity was attributed to a nine-base motif within a 19 mer sequence. Immune complexes (ICX) formed of RNA-specific antibodies and self-RNA can activate TLR7; this participates in the development of autoimmune diseases [53]. As a result, small nuclear ribonucleoproteins purified from ICX can be used as TLR7 ligands [68].
2.8 TLR8
TLR8 shares high sequence homology with TLR7 and can recognize most of the TLR7 ligands [62]. As mentioned above, ssRNAs activate human TLR8. Technically, it is possible to discriminate between TLR7 and TLR8 activity by using different concentrations of nucleotide analogs. 3M-002 from 3M Pharmaceuticals (sold as CL075 by InvivoGen) can specifically activate human TLR8 when used at 0.1 μg/ml (0.4 μM) whereas higher concentrations of this ligand are required to activate TLR7. Initially, TLR8 was considered to be active only in human and not in mouse, but a combination of 10 μM 3M-002 and 3M-003 with 1–3 μM polyT oligodeoxyribonucleotide (ODN) can activate mouse TLR8 [69].
2.9 TLR9
TLR9 is an intracellular TLR involved in the recognition of DNA from bacterial and viral origin but also self-DNA in ICX [8]. Non-self-DNA is detected by the presence of unmethylated CpG motifs. Extensive research has been done to characterize immunostimulatory CpG DNA motifs [70]. Two main classes have been described. Class A CpG is active on plasmacytoid dendritic cells (pDC). These reagents contain polyG motifs and a palindromic sequence on a mixed phosphodiester/phosphorothioate backbone and can multimerize to form large structures. Class B CpG contains one or more CpG and no polyG motifs on a phosphorothioate backbone and they activate B cells. Class C CpG shares class A and class B characteristics and properties. Despite the endosomal location of TLR9, CpG ODNs are usually added to the culture medium to activate TLR9+ cells. The mechanism of activation by CpG has thus been challenging to understand. Tian et al. showed that class A CpG interacts with HMGB1 and the resulting complex is efficiently internalized by a mechanism involving the receptor for advanced glycation end products (RAGE) [71]. This RAGE-dependent mechanism is thought to facilitate the delivery of class A CpG to TLR9.
In vitro, the optimal concentration for immunostimulatory CpG ODN is 1 μM. Higher concentrations are usually less effective. It should be noted that some CpG ODNs show species specificities (human vs. mouse). The specificity of the activation by CpG ODN can be confirmed by the use of control CpG ODN which contains the same sequence as immunostimulatory CpG ODN but in which CpG dinucleotides have been replaced by GpC dinucleotides. Purified endotoxin-free E. coli DNA at 50 μg/ml can also be used as a TLR9 ligand. Natural CpG sequences from Gram-negative bacteria termed repetitive extragenic palindromics (REPs) have been isolated and shown to induce innate responses via TLR9 [72]. Immune complexes present in serum from SLE patients activate TLR9 by a mechanism involving the cell surface receptor FcγR (CD32) [73]. In vitro, patients’ serum can activate TLR9-expressing cells.
Besides immunostimulatory ODNs acting through TLR9, inhibitory ODNs have been identified. The latter bind TLR9 but fail to induce the switch to an active conformation of TLR9. Inhibitory ODNs block signaling by competing with immunostimulatory ODNs for the binding site on TLR9 [74]. Two types of inhibitory ODNs have been identified: repeated TTAGGG motifs found in telomeres or ODN containing either unmethylated GC or methylated CG [75].
Hemozoin from Plasmodium falciparum, which was initially identified as a TLR9 ligand, seems to actually help bringing parasite DNA in close proximity with TLR9 to induce a response [76].
2.10 TLR10
TLR10 is expressed on B cells; it is closely related to TLR1 and TLR6 (48 and 46 % amino acid sequence identity, respectively) and may interact with TLR2 [77]. The ligand for TLR10 is still unknown. By use of homology modeling recent studies have shown the binding orientation of a human TLR10—TLR2 heterodimer to be similar with that of TLR2/1 and TLR2/6. Moreover, the study revealed that Pam3CSK4 might be the ligand for a human TLR10/2 complex and PamCysPamSK4 might activate a human TLR10/1 hetero- and TLR10 homodimer [78]. Using chimeric CD4TLR10 (see Fig. 2 and Subheading 5), Hasan et al. showed that promoters activated by signaling through TLR10 include CXCL5, IL-4, NF-κB and to a lesser extent IL-2, TNF, and AP-1 [33]. Most recently, Oosting et al. demonstrate that TLR10 acts as an inhibitory receptor, with suppressive effects. The use of specific antibodies to block TLR10 significantly upregulated TLR2-mediated cytokine production [79].

Example of engineered TLRs (a, b)
2.11 TLR11 and TLR12
It is worth pointing out that while the mouse genome encodes 13 TLRs, the human genome lacks functional TLR11, TLR12, and TLR13. TLR11 is an endosomal TLR only functional in mice. It was first described as a receptor for uropathogenic bacteria [80] and studies have shown that it is activated by a protein resembling Toxoplasma gondii profilin [81]. TLR12 is expressed in mice and rats [82]. TLR12 has been shown to form heterodimers with TLR11 for sensing and responding to Toxoplasma gondii profilin [83]. The expression and distribution of TLRs 11–13 in normal and parasite infected mouse brains has been demonstrated suggesting a role for them in central nervous system (CNS) infections [84].
2.12 TLR13
TLR13 is an endosomal murine receptor; the exact role and ligand for which remains unclear. However, several groups have identified the bacterial 23S ribosomal RNA (rRNA) sequence as a ligand for TLR13. Specifically, the conserved 23S ribosomal sequence “CGGAAAGACC,” has been shown to induce cytokine production in a TLR13-, MyD88-, and UNC93B-dependent manner. (UNC93B is an accessory protein involved in the trafficking of intracellular TLRs including, TLR3/7/9). The conserved 23S rRNA sequence is the binding site of the macrolide-lincosamide-streptogramin (MLS) antibiotics, and 23S rRNA from bacteria resistant to these antibiotics is not recognized by TLR13. ORN Sa19 is a 19 mer Staphylococcus aureus rRNA derived oligoribonucleotide stabilized by a phosphorothioate modification available commercially and found to be highly stimulatory in murine TLR13-expressing cells (InvivoGen).
3 Host Cells
When it comes to choosing a relevant host cell to study TLR biology in vitro, several options are possible. Firstly, cell lines lacking TLR expression can be transfected with one or two TLRs with or without accessory molecules. The second option is to select cell lines naturally expressing TLR(s). Primary cells are another option as they might be more physiologically relevant (i.e., presence of normal expression level of TLRs and presence of crosstalk between signaling pathways) but they are technically more difficult to handle than cell lines.
3.1 Cell Lines Lacking TLR Expression
Cells devoid of endogenous active TLR are a valuable tool to study individual TLRs. The most commonly used cell lines are of epithelial origin; they express all the signaling components needed downstream of TLR activation. Stable or transient transfection of one or two TLRs with or without accessory molecules renders them responsive to the corresponding ligand(s). This has been demonstrated in the following cell lines: HEK293 [85, 86], HeLa [87], COS7 [88], or CHO [89]. It should be noted that HEK293 cells, unlike the variant HEK293T, might have endogenous TLR3 and TLR5 activity [90].
Cells lacking endogenous TLR activity have been used to study TLR signaling, interactions between TLRs [91], requirement for accessory molecules [60], intracellular trafficking [85], or localization. The transfection efficiency in these epithelial cells is very high and the experiments described above can be easily done in transiently transfected cells. Alternatively, stable cell lines expressing TLR, accessory molecules, and even a reporter gene can be generated.
HEK293 cells stably expressing functional human or mouse TLR stably transfected or not with a reporter gene are commercially available from InvivoGen (HEK Blue™) and Bio-Techne (SEAPorter™ and LUCPorter™). The one concern about the use of cells overexpressing a TLR is that overexpression could result in mislocalization, i.e., an intracellular TLR might be found on the cell surface when overexpressed [92]. For that reason, cell lines expressing normal levels of TLRs are often the preferred model.
3.2 TLR Expressing Cell Lines
Cell lines expressing TLRs endogenously may also be used to study TLR function. However, they may require transfection of accessory molecules or co-receptors to increase the responsiveness of the cell. It is important to profile cell lines for TLR and accessory molecules expression before using them in an assay. This can be done by RT-PCR and confirmed by flow cytometry or western blot.
The following cell lines have been used in numerous TLR studies: RAW264.7 (mouse macrophage), THP-1 (human acute monocytic leukemia), and U937 (human promonocytic leukemia) and all belong to the monocyte/macrophage lineage. U373 are epithelial human glioblastoma astrocytoma cells and A549 are human alveolar basal epithelial cells. Cell lines having characteristics similar to human pDC have been described: PMDC05 [93], GEN2.2 [94], and CAL-1 [95]. These cell lines could be a good alternative to the use of primary pDC which are present in very low numbers among circulating blood cells. CD14 expression is needed to render U373 cells responsive to LPS [96] or to increase the responsiveness of THP-1 cells to several TLR2/4/5 ligands [97]. Differentiation induced by phorbol myristate acetate (PMA) or LPS also increases expression of TLRs and hence responsiveness of THP-1 cells to TLR ligands [98–100]. LPS responsiveness of A549 cells through TLR4 is controversial [101]. Table 2 shows the TLR profile of some of these cell lines. In addition, human myeloma cell lines have been shown to express a variety of TLR genes [102].
There are several TLR expressing cells stably transfected with a reporter gene available on the market which can be used to measure TLR activity: THP1-Blue™, THP1-Lucia™ (human monocytes), Ramos-Blue™ (B lymphocytes), Jurkat-Dual™ (T lymphocytes, TLR3+), Raw-Blue™, Raw-Lucia™ (mouse macrophages) from InvivoGen (San Diego, CA), CellSensor™ (Life Technologies) derived from THP1 or RAW264.7 cells, and SEAPorter™ and LUCPorter™ RAW264.7 from Bio-Techne.
3.3 Isolated Primary Cells
Ideally, cell line studies should be complemented by studies in primary cells. The main problem with the use of primary cells is to choose the right cell type as there are discrepancies in the literature regarding the expression and functionality of TLRs.
It was initially thought that TLRs are restricted to cells of the immune system. However, TLRs have been found in other cell types such as epithelial cells (TLR2, 3, 4, 5, 9) [103], fibroblasts, and even cells from the nervous system [104]. Hornung et al. [105] and Applequist et al. [106] have measured the expression of TLR1-10 mRNA in different types of human or murine immune cells, but the presence of mRNA for a given TLR does not mean that this TLR is active. For example, eosinophils express TLR1, TLR4, TLR7, TLR8, TLR9, and TLR10 mRNA and only respond to stimulation with a TLR7 agonist [107]. Also worth noting is that while naive human CD4+ T cells express significant levels of intracellular TLR2 and TLR4 protein, cell surface expression of TLR2 and TLR4 is found only in activated CD4+ T cells [108]. Expression of TLRs in T cells remains controversial. Hemont et al. have investigated TLR responsiveness of human mDC subsets using a whole blood assay followed by flow cytometry analysis [109]. Table 3 summarizes the panel of functional TLRs expressed by human immune cells. Expression levels are altered by pro-inflammatory signals or type I IFN treatment [99, 110]. In practice, the most commonly used primary cells are non-sorted peripheral blood mononuclear cells (PBMCs), monocytes, or B cells. Other cell types which are very valuable for the study of TLR biology (e.g., pDC) are more difficult to obtain in sufficient quantity.
Interestingly, mouse cells display a TLR profile similar to the corresponding human cells. One important difference is the LPS responsiveness of murine but not human naïve B cells through TLR4 [111]. Among primary cells, murine embryonic fibroblasts (MEF) isolated from mouse embryos are a very good model to study TLRs as they express all known TLRs [112]. Ex vivo, MEFs can be grown for over ten passages, after which their proliferation rate then declines.
3.4 Whole Blood
Whole blood assays emerged as a powerful way to investigate innate immune responses mediated by TLRs [113, 114]. They allow evaluation of TLR responsiveness in the absence of exogenous additives such as animal serum and antibiotics. In this type of assay, anticoagulated whole blood is incubated 3–48 h in the presence of TLR ligand(s) to activate different populations of blood cells resulting in the release of chemokines, cytokines, and interferons in the supernatant (i.e., plasma). From a practical point of view, several parameters have to be controlled to ensure the robustness of this type of assay: freshness of the blood, choice of anticoagulant, dilution of blood, time of sampling [115, 116], and medical treatment as immunosuppressive treatment will result in decreased ex vivo responses. It has been shown that a few hours after sampling the responsiveness of the blood is reduced possibly due to the short half-life of some blood cell populations such as neutrophils [114]. Nevertheless, it is still possible to induce a TLR-mediated response in whole blood stored at room temperature for up to 48 h [114]. For whole blood assays, the preferred anticoagulant is heparin over EDTA which might interfere with TLR function. Frozen whole blood (stored at −80 °C) has been proposed as an alternative to the use of fresh blood [117]. Upon thawing, red blood cells are lyzed and hemoglobin is released in the supernatant which can interfere with some detection techniques. According to a supplier of frozen whole blood (e.g., sold as part of the pyrogen detection kit PyroDetect, Merck Millipore), storage at temperatures above −80 °C quickly reduces the responsiveness of frozen blood which limits its use in most of the labs.
Whole blood assays performed in 96- or 384-well plates allows the testing of many different stimuli with a small volume of blood as starting material. Blood from several donors/patients or even from newborns [118–121] can be processed in parallel. Of note, this type of assay is transposable to animal blood which allows the assessment of cross-species TLR responses [122–126].
Whole blood assays reflect donor to donor variability. Hence, in blood from normal donors, the stimulation by TLR ligands often results in secretion of cytokines over a wide range of concentrations. Great efforts are needed to normalize these assays. This process is ongoing in order to be able to use whole blood assays as a diagnostic test to identify immune disorders [127].
The use of cell lines or isolated primary cells remains a technique of choice when high throughput is required. In combination with this technique, whole blood assays bring additional information about the bioactivity of TLRs, their ligands and compounds interacting with them. As a consequence, whole blood assays have also been used in preclinical studies to characterize TLR modulators whether they are small molecules or biologics.
4 Readouts
As mentioned previously, signaling through the different TLRs involves mainly two families of transcription factors: NF-κB via a MyD88-dependent pathway (all TLRs except TLR3) and the IRFs via a MyD88-independent TRIF-dependent pathway (TLR3) or a MyD88-dependent pathway (TLR7/8/9). NF-κB and IRFs induce the expression of different sets of cytokines, chemokines, and interferons [128].
In order to determine TLR activity in each of the cell models outlined in Subheading 3, the following readouts can be used: measurement of cytokines, chemokines, and/or interferons secretion by simplex or multiplex immunoassays (ELISA, AlphaLISA, HTRF®, immunoPCR, Luminex); detection of activation markers by western blot, RT-PCR, or flow cytometry; phosphorylation of signaling proteins; and nuclear translocation of transcription factors and/or reporter gene assays.
4.1 Protein Secretion
The simplest method to assay for the presence of functional TLR is to measure the production of pro-inflammatory cytokines and/or chemokines after TLR stimulation using specific ligands. ELISA is a widely used technique and validated kits to detect most of the biomarkers are available from several suppliers. ELISA results can be sufficient to distinguish between two signaling pathways, e.g., TRIF vs. MyD88 after TLR4 activation. TRIF activation will result in the production of G-CSF, CXCL10, CCL2, and CCL5 while MyD88 activation induces the secretion of IFNβ, IL-1β, IL-6, and CCL3 [20].
Multiple cytokines and chemokines can be detected simultaneously in a single small volume sample using multiplex techniques. Cytometric Bead Array (CBA; BD Biosciences) is based on the use of beads coated with an antibody recognizing a specific analyte. This system requires the use of a flow cytometer. xMAP technology (Luminex®) also uses beads available from Affymetrix eBioscience (ProcartaPlex™), Bio-Rad (Bio-Plex®), Merck Millipore (Milliplex®), and Bio-Techne but in a 96-well plate format. A compatible microplate reader is needed to process the samples. The multi-array technology by Meso Scale Discovery (MSD) uses electroluminescence detection. Wells of dedicated microplates (96 or 384 well) are coated with specific antibodies directed against cytokines, chemokines, or signaling proteins allowing the detection of up to ten different targets. Secondary antibodies are labeled with the SULFO-TAGTM reagent which emits light upon electrochemical stimulation. Specific readers (SECTOR) are required to process MSD microplates.
Immuno-PCR is a combination of ELISA and PCR [129]. Antibodies specific for the target(s) of interest are conjugated with a short DNA sequence. After the antibodies recognize their target, the DNA tag is amplified by real-time PCR. Compared to ELISA, immuno-PCR is more sensitive (down to fg/ml). Multiplexing and conjugation of DNA tags remain challenging and limit the generalization of this technique. Specialized suppliers are Innova Biosciences, Chimera Biotec, and Olink Bioscience.
Biomarker quantification by HTRF® (CisBio Bioassays) is based on time-resolved fluorescence resonance energy transfer (FRET). Two antibodies specific for the target are labeled with two different fluorophores: an acceptor and a donor. When the two antibodies are in close proximity, i.e., when they recognize their target, the excitation of the donor by a laser will result in energy transfer to the donor which then emits fluorescence. Unlike ELISA which requires several incubation/wash steps, HTRF® is a homogenous technique. HTRF® measurement is typically done in small volume 384-well plates with only 10 μl sample. The detection range is wider with HTRF® compared to ELISA. There are much less HTRF® than ELISA kits available on the market and HTRF® requires the use of a microplate reader capable of time-resolved FRET measurement.
Finally, in a polymorphic population of cells such as whole blood, intracellular staining of cytokines by flow cytometry combined with cell surface staining allows the identification of cell types responsible for each cytokine produced in response to TLR stimulation [118].
4.2 Phosphorylation
The phosphorylation of signaling components, such as the MAPK p38 or the NF-κB subunit p65, occurs usually within minutes after TLR stimulation. There are several techniques to monitor protein phosphorylation, all of them rely on the availability of phospho-specific antibodies. Flow cytometry is performed on permeabilized cells whereas western blots and immunoassays (e.g., TransAM® from Active Motif and CASE™ from SABiosciences™, HTRF and AlphaScreen® (Perkin Elmer)) are performed using cell lysates. AlphaScreen® is interesting as it is designed to be used in high-throughput screening. This technique is a bead-based luminescent assay used to measure interactions between molecules. In the case of protein phosphorylation, two sets of beads containing different dyes are needed: one set of beads is coated with an antibody against the nonphosphorylated form of the target protein and another set of beads is coated with an antibody against the phosphorylated form of the protein. Beads are mixed with cell lysates and upon excitation light is emitted if both sets of beads are in close proximity, i.e., when the phosphorylated protein is present in the lysates. HTRF allows quantification of protein phosphorylation without the need for beads as antibodies are directly coupled with FRET-compatible fluorophores.
4.3 Translocation of Transcription Factors
Another way to monitor TLR activation is to look at the change in localization of transcription factors (TF) activated after ligand binding, e.g., NF-κB, IRFs. This can be assayed using high content screening (HCS) or high content analysis (HCA) assays. The TF is detected using immunochemistry techniques before and after stimulation and the intensity of the signal in the nucleus vs. cytoplasm is measured. Alternatively, cells can be transfected to express a fluorescent fusion protein with the TF (e.g., GFP-TF) and the change in localization can be observed in live cells [130]. HCS/HCA techniques require using adherent cells having a low nucleus/cytoplasm ratio. Finally, preparation of subcellular fractions using specific lysis buffers followed by protein detection by western blot is another method of choice.
4.4 Reporter Genes
Reporter gene assays are also used to monitor TLR activity. The most widely used promoter is derived from the ELAM-1 gene promoter modified to contain additional NF-κB motifs [131]. An artificial promoter containing five NF-κB binding sites is another option [132]. The reporter gene itself can code for a secreted protein (e.g., human placental alkaline phosphatase (SEAP) [133] or secreted luciferase [134]) or an intracytoplasmic protein like firefly luciferase [135]. Cells can be transiently or stably transfected with the reporter gene construct and a construct coding for a TLR. TLR activation induces NF-κB-dependent production of the reporter gene. SEAP is secreted in the supernatants of cells and can be detected using colorimetric (e.g., Quantiblue™ from InvivoGen) or chemiluminescent (Phosphalight™ from Applied Biosystems) techniques. With a secreted reporter protein, it is possible to measure TLR activation over the time simply by sampling the supernatants. It should be noted that serum (human or animal) often used in culture media contains heat labile alkaline phosphatase activity. Heating of samples at 65 °C for 10 min is sufficient to inactivate endogenous alkaline phosphatase activity prior to the measurement of SEAP activity which is not altered by heating.
Secreted luciferase systems are available from Clontech (Ready-To-Glow™), New England Biolabs (Gaussia Luciferase), and InvivoGen. Unlike techniques based on the detection of a secreted protein, “classical” luciferase measurement does not allow to do time course experiments as it requires lysing the cells. Luciferase detection products are available from several suppliers. In reporter gene assays, IRF-dependent and NF-κB-independent promoters are needed to cover the full range of signaling pathways activated by the TLRs. IRF-specific reporter gene assays can be designed based on specific binding sites described for certain cytokines, chemokines, or interferon promoters [33, 136, 137].
5 Engineered TLRs
The study of TLR biology is sometimes rendered difficult by the lack of a known ligand (e.g., for TLR10) or the accessibility of the receptor to its ligand(s) (e.g., TLR3/7/8/9). To overcome these technical problems, two techniques based on the use of engineered TLRs have been described.
In the first technique the extracellular part of a TLR is fused with the transmembrane (TM) and intracellular (IC) parts of hCD32a [41] (see Fig. 2a). The resulting chimeric protein can be expressed in HEK293 cells where it localizes on the cell surface. The engagement of this TLR by its ligand(s) results in a Ca2+ response/influx mediated by CD32a. The two main advantages of this technique are (1) intracellular TLRs become accessible to their ligands added exogenously in the culture medium thereby facilitating the identification of new ligands and (2) one single readout (Ca2+) can be used to monitor the activity of any TLR. Ca2+ influx is a very fast response that has been widely used as a readout in other domains (e.g., 7-TM receptors) and a lot of screening tools are available [138].
Using this technique, De Bouteiller et al. [41] have demonstrated the need for an acidic pH (~5.7) for TLR3 activity and the minimal requirements for TLR3 ligands (see TLR3 above). Nevertheless, when these chimeras are used to study intracellular TLRs, one should keep in mind that, in normal cells, a ligand identified with this technique will need access to the TLR. In addition, the amount of ligand needed to induce Ca2+ mobilization was about 50 times less than the amount needed to induce cytokine secretion by TLR3-expressing cells.
The second technique involves another fusion protein using the extracellular part of mouse CD4 fused with the TM and IC part of a TLR (see Fig. 2b) [39, 45]. These chimeric proteins (CD4TLRx) are expressed on the cell surface when transfected in HEK293T cells. The association of extracellular CD4 domains triggers the signaling cascade corresponding to the TM + IC of the TLR. Hence, the ligand for this TLR can remain unknown to study the signaling. This technique can also be used to select the best readout for a given TLR. For example, besides repeated κB binding sites which are the most used promoter for reporter gene assays, the promoters for the chemokines IL-8 and CXCL5 were shown to be strongly activated by several TLRs. Focusing on TLR10, which has no identified ligand yet, Hasan et al. [33] show that CD4TLR10 specifically activated IL-4, CXCL5, and NF-κB promoters after 48 h stimulation. This could be the basis to design an assay to identify TLR10 ligands. These results stress the importance of looking at more than one readout (usually NF-κB) when one studies TLR activity.
A last example of modified TLRs is the dominant negative (DN) forms. These mutants can be obtained by inserting a point mutation in the TIR domain preventing the binding of adaptor proteins or by deleting the intracellular TIR domain. Such mutated TLRs can bind their ligands but they cannot trigger the signaling cascade. Hence, overexpression of one mutated TLR specifically inhibits the activity of the endogenous TLR by competition [36, 39]. This technique can be used to show the involvement of each individual TLR in cells expressing several TLRs.
With new ligands discovered regularly, novel accessory molecules identified, and the potential for more TLRs to be discovered, there is a need for robust cell-based assays to monitor TLR activity. To keep up to date with latest developments, a rich database of bioassays is maintained by NCBI and hundreds of them are related to TLRs [139]. While “empty shells” such as HEK293 transformed to express one TLR are still a very convenient tool, the future will probably see the rise of HCS and HTS assays in primary cells as the preferred techniques. Finally, the normalization of TLR assays using whole blood could be the basis for new diagnosis tools.
TLRs have the ability to harness great immunostimulatory signals and are therefore tightly regulated and activated. Incorrect and/or overactivation of these pathways can lead to autoimmune disease [140] and fatal sepsis [141]. Many questions remain surrounding the exact interaction of TLRs and their ligands, the complexities of which remain under constant investigation. TLR signaling and the responses they control continue to challenge thinking regarding the pathogenesis and treatment of cancers, immune and infectious diseases (see Part V, Chapters 21–25).
References
Takeda K, Akira S (2005) Toll-like receptors in innate immunity. Int Immunol 17:1–14
Gay NJ, Keith FJ (1991) Drosophila Toll and IL-1 receptor. Nature 351:355–356
Brikos C, O'Neill LA (2008) Signalling of Toll-like receptors. Handb Exp Pharmacol 183:21–50
Gay NJ, Gangloff M, Weber AN (2006) Toll-like receptors as molecular switches. Nat Rev Immunol 6:693–698
Kawai T, Akira S (2006) TLR signaling. Cell Death Differ 13:816–825
Boehme KW, Compton T (2004) Innate sensing of viruses by toll-like receptors. J Virol 78:7867–7873
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4:499–511
Barton GM, Kagan JC, Medzhitov R (2006) Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol 7:49–56
Saitoh S, Akashi S, Yamada T, Tanimura N, Kobayashi M, Konno K, Matsumoto F, Fukase K, Kusumoto S, Nagai Y, Kusumoto Y, Kosugi A, Miyake K (2004) Lipid A antagonist, lipid IVa, is distinct from lipid A in interaction with Toll-like receptor 4 (TLR4)-MD-2 and ligand-induced TLR4 oligomerization. Int Immunol 16:961–969
Botos I, Liu L, Wang Y, Segal DM, Davies DR (2009) The toll-like receptor 3:dsRNA signaling complex. Biochim Biophys Acta 1789:667–674
Wang Y, Liu L, Davies DR, Segal DM (2010) Dimerization of Toll-like receptor 3 (TLR3) is required for ligand binding. J Biol Chem 285:36836–36841
Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR (2008) Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 320:379–381
Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO (2007) Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130:1071–1082
Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO (2007) Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130:906–917
Yoon SI, Kurnasov O, Natarajan V, Hong M, Gudkov AV, Osterman AL, Wilson IA (2012) Structural basis of TLR5-flagellin recognition and signaling. Science 335:859–864
Tanji H, Ohto U, Shibata T, Miyake K, Shimizu T (2013) Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 339:1426–1429
Nyman T, Stenmark P, Flodin S, Johansson I, Hammarstrom M, Nordlund P (2008) The crystal structure of the human toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J Biol Chem 283:11861–11865
Gangloff M, Weber AN, Gay NJ (2005) Conserved mechanisms of signal transduction by Toll and Toll-like receptors. J Endotoxin Res 11:294–298
Nunez Miguel R, Wong J, Westoll JF, Brooks HJ, O'Neill LA, Gay NJ, Bryant CE, Monie TP (2007) A dimer of the Toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signalling adaptor proteins. PLoS One 2, e788
Akira S, Yamamoto M, Takeda K (2003) Role of adapters in Toll-like receptor signalling. Biochem Soc Trans 31:637–642
Lu YC, Yeh WC, Ohashi PS (2008) LPS/TLR4 signal transduction pathway. Cytokine 42:145–151
Kaisho T, Akira S (2006) Toll-like receptor function and signaling. J Allergy Clin Immunol 117:979–987, quiz 988
Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, Takeda K, Akira S (2002) Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 420:324–329
Takeda K, Akira S (2004) TLR signaling pathways. Semin Immunol 16:3–9
Kawai T, Adachi O, Ogawa T, Takeda K, Akira S (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115–122
Stack J, Doyle SL, Connolly DJ, Reinert LS, O'Keeffe KM, McLoughlin RM, Paludan SR, Bowie AG (2014) TRAM is required for TLR2 endosomal signaling to type I IFN induction. J Immunol 193(12):6090–6102
Volpi C, Fallarino F, Pallotta MT, Bianchi R, Vacca C, Belladonna ML, Orabona C, De Luca A, Boon L, Romani L, Grohmann U, Puccetti P (2013) High doses of CpG oligodeoxynucleotides stimulate a tolerogenic TLR9-TRIF pathway. Nat Commun 4:1852
Hennessy EJ, Parker AE, O'Neill LA (2010) Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov 9:293–307
Gay NJ, Gangloff M (2007) Structure and function of Toll receptors and their ligands. Annu Rev Biochem 76:141–165
Rutz M, Metzger J, Gellert T, Luppa P, Lipford GB, Wagner H, Bauer S (2004) Toll-like receptor 9 binds single-stranded CpG-DNA in a sequence- and pH-dependent manner. Eur J Immunol 34:2541–2550
Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, Bathke B, Lauterbach H, Suter M, Dreher S, Koedel U, Akira S, Kawai T, Buer J, Wagner H, Bauer S, Hochrein H, Kirschning CJ (2012) TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337:1111–1115
Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S (2002) Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol 169:10–14
Hasan UA, Dollet S, Vlach J (2004) Differential induction of gene promoter constructs by constitutively active human TLRs. Biochem Biophys Res Commun 321:124–131
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ (1999) Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J Biol Chem 274:17406–17409
Sandor F, Latz E, Re F, Mandell L, Repik G, Golenbock DT, Espevik T, Kurt-Jones EA, Finberg RW (2003) Importance of extra- and intracellular domains of TLR1 and TLR2 in NFkappa B signaling. J Cell Biol 162:1099–1110
Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E (2006) High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol 290:C917–C924
Flo TH, Halaas O, Lien E, Ryan L, Teti G, Golenbock DT, Sundan A, Espevik T (2000) Human toll-like receptor 2 mediates monocyte activation by Listeria monocytogenes but not by group B streptococci or lipopolysaccharide. J Immunol 164(4):2064–2069
Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A (2000) The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A 97:13766–13771
Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ (2003) Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300:1584–1587
de Bouteiller O, Merck E, Hasan UA, Hubac S, Benguigui B, Trinchieri G, Bates EE, Caux C (2005) Recognition of double-stranded RNA by human toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem 280(46):38133–38145
Marshall-Clarke S, Downes JE, Haga IR, Bowie AG, Borrow P, Pennock JE, Grencis RK, Rothwell P (2007) Polyinosinic acid is a ligand for toll-like receptor 3. J Biol Chem 282(34):24759–24766
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita C, Akira S (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105
Zhou Y, Guo M, Wang X, Li J, Wang Y, Ye L, Dai M, Zhou L, Persidsky Y, Ho W (2013) TLR3 activation efficiency by high or low molecular mass poly I:C. Innate Immun 19:184–192
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397
Caroff M, Karibian D (2003) Structure of bacterial lipopolysaccharides. Carbohydr Res 338:2431–2447
Coats SR, Reife RA, Bainbridge BW, Pham TT, Darveau RP (2003) Porphyromonas gingivalis lipopolysaccharide antagonizes Escherichia coli lipopolysaccharide at toll-like receptor 4 in human endothelial cells. Infect Immun 71:6799–6807
Huber M, Kalis C, Keck S, Jiang ZF, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Beutler B, Galanos C, Freudenberg MA (2006) R-form LPS, the master key to the activation of TLR4/MD-2-positive cells. Eur J Immunol 36:701–711
Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC (2007) The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 316:1628–1632
Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ (2000) Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J Immunol 165:618–622
Kawasaki K, Akashi S, Shimazu R, Yoshida T, Miyake K, Nishijima M (2001) Involvement of TLR4/MD-2 complex in species-specific lipopolysaccharide-mimetic signal transduction by Taxol. J Endotoxin Res 7:232–236
Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, Bozza MT (2007) Characterization of heme as activator of toll-like receptor 4. J Biol Chem 282:20221–20229
Marshak-Rothstein A (2006) Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol 6:823–835
Matzinger P (2002) The danger model: a renewed sense of self. Science 296:301–305
Gao B, Tsan MF (2003) Recombinant human heat shock protein 60 does not induce the release of tumor necrosis factor alpha from murine macrophages. J Biol Chem 278:22523–22529
Ohashi K, Burkart V, Flohe S, Kolb H (2000) Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol 164:558–561
Smith KD, Andersen-Nissen E, Hayashi F, Strobe K, Bergman MA, Barrett SL, Cookson BT, Aderem A (2003) Toll-like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nat Immunol 4:1247–1253
Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–1103
Okusawa T, Fujita M, Nakamura J, Into T, Yasuda M, Yoshimura A, Hara Y, Hasebe A, Golenbock DT, Morita M, Kuroki Y, Ogawa T, Shibata K (2004) Relationship between structures and biological activities of mycoplasmal diacylated lipopeptides and their recognition by toll-like receptors 2 and 6. Infect Immun 72:1657–1665
Triantafilou M, Gamper FG, Haston RM, Mouratis MA, Morath S, Hartung T, Triantafilou K (2006) Membrane sorting of toll-like receptor (TLR) -2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem 281:31002–31011
Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529–1531
Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S (2004) Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303:1526–1529
Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA (2004) Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A 101
Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S (2002) Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol 3:196–200
Heil F, Ahmad-Nejad P, Hemmi H, Hochrein H, Ampenberger F, Gellert T, Dietrich H, Lipford G, Takeda K, Akira S, Wagner H, Bauer S (2003) The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol 33:2987–2997
Kobold S, Wiedemann G, Rothenfusser S, Endres S (2014) Modes of action of TLR7 agonists in cancer therapy. Immunotherapy 6:1085–1095
Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G (2005) Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med 11:263–270
Savarese E, Chae OW, Trowitzsch S, Weber G, Kastner B, Akira S, Wagner H, Schmid RM, Bauer S, Krug A (2006) U1 small nuclear ribonucleoprotein immune complexes induce type I interferon in plasmacytoid dendritic cells through TLR7. Blood 107:3229–3234
Gorden KK, Qiu XX, Binsfeld CC, Vasilakos JP, Alkan SS (2006) Cutting edge: activation of murine TLR8 by a combination of imidazoquinoline immune response modifiers and polyT oligodeoxynucleotides. J Immunol 177:6584–6587
Krieg AM (2006) Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov 5:471–484
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G et al (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8(5):487–496
Magnusson M, Tobes R, Sancho J, Pareja E (2007) Cutting edge: natural DNA repetitive extragenic sequences from gram-negative pathogens strongly stimulate TLR9. J Immunol 179:31–35
Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR (2004) Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J Exp Med 199:1631–1640
Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DC, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, Espevik T, Golenbock DT (2007) Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol 8:772–779
Lenert P (2005) Inhibitory oligodeoxynucleotides – therapeutic promise for systemic autoimmune diseases? Clin Exp Immunol 140:1–10
Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, Latz E, Gazzinelli RT, Golenbock DT, Fitzgerald KA (2014) Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. Cell Rep 6:196–210
Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, Guiet C, Briere F, Vlach J, Lebecque S, Trinchieri G, Bates EE (2005) Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through {MyD} 88. J Immunol 174:2942–2950
Govindaraj RG, Manavalan B, Lee G, Choi S (2010) Molecular modeling-based evaluation of hTLR10 and identification of potential ligands in Toll-like receptor signaling. PLoS One 5, e12713
Oosting M, Cheng SC, Bolscher JM, Vestering-Stenger R, Plantinga TS, Verschueren IC, Arts P, Garritsen A, van Eenennaam H, Sturm P, Kullberg BJ, Hoischen A, Adema GJ, van der Meer JW, Netea MG, Joosten LA (2014) Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc Natl Acad Sci U S A 111:E4478–E4484
Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, Ghosh S (2004) A toll-like receptor that prevents infection by uropathogenic bacteria. Science 303:1522–1526
Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A (2005) TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308:1626–1629
Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A (2005) The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci U S A 102:9577–9582
Andrade WA, Souza Mdo C, Ramos-Martinez E, Nagpal K, Dutra MS, Melo MB, Bartholomeu DC, Ghosh S, Golenbock DT, Gazzinelli RT (2013) Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe 13:42–53
Mishra BB, Gundra UM, Teale JM (2008) Expression and distribution of Toll-like receptors 11–13 in the brain during murine neurocysticercosis. J Neuroinflammation 5:53
Latz E, Visintin A, Lien E, Fitzgerald KA, Monks BG, Kurt-Jones EA, Golenbock DT, Espevik T (2002) Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J Biol Chem 277:47834–47843
Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, Goddard A, Wood WI, Gurney AL, Godowski PJ (1998) Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature 395:284–288
Pridmore AC, Wyllie DH, Abdillahi F, Steeghs L, van der Ley P, Dower SK, Read RC (2001) A lipopolysaccharide-deficient mutant of Neisseria meningitidis elicits attenuated cytokine release by human macrophages and signals via toll-like receptor (TLR) 2 but not via TLR4/MD2. J Infect Dis 183:89–96
Matsuguchi T, Takagi K, Musikacharoen T, Yoshikai Y (1999) Gene expressions of lipopolysaccharide receptors, toll-like receptors 2 and 4, are differently regulated in mouse T lymphocytes. Blood 95(56):1378–1385
Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D (1999) Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol 163:1–5
Hasan UA, Trinchieri G, Vlach J (2005) Toll-like receptor signaling stimulates cell cycle entry and progression in fibroblasts. J Biol Chem 280:20620–20627
Wang J, Shao Y, Bennett TA, Shankar RA, Wightman PD, Reddy LG (2006) The functional effects of physical interactions among Toll-like receptors 7, 8, and 9. J Biol Chem 281:37427–37434
Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, Klinman DM (2001) Cutting edge: role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. J Immunol 167:3555–3558
Watanabe N, Narita M, Yamahira A, Nakamura T, Tochiki N, Saitoh A, Kaji M, Hashimoto S, Furukawa T, Toba K, Fuse I, Aizawa Y, Takahashi M (2010) Transformation of dendritic cells from plasmacytoid to myeloid in a leukemic plasmacytoid dendritic cell line (PMDC05). Leuk Res 34:1517–1524
Chaperot L, Blum A, Manches O, Lui G, Angel J, Molens JP, Plumas J (2006) Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J Immunol 176:248–255
Maeda T, Murata K, Fukushima T, Sugahara K, Tsuruda K, Anami M, Onimaru Y, Tsukasaki K, Tomonaga M, Moriuchi R, Hasegawa H, Yamada Y, Kamihira S (2005) A novel plasmacytoid dendritic cell line, CAL-1 established from a patient with blastic natural killer, cell lymphoma. Int J Hematol 81:148–154
Tapping RI, Orr SL, Lawson EM, Soldau K, Tobias PS (1999) Membrane-anchored forms of lipopolysaccharide (LPS)-binding protein do not mediate cellular responses to LPS independently of CD14. J Immunol 162:5483–5487
InvivoGen (2006) InvivoGen Insight Newsletter: TLR7 and TLR8. http://www.InvivoGen.com/docs/Insight200609.pdf
Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS (2007) Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm Res 56:45–50
Zarember KA, Godowski PJ (2002) Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol 168:554–561
Chanput W, Mes JJ, Wichers HJ (2014) THP-1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol 23:37–45
Hippenstiel S, Opitz B, Schmeck B, Suttorp N (2006) Lung epithelium as a sentinel and effector system in pneumonia—molecular mechanisms of pathogen recognition and signal transduction. Respir Res 7:97
Jego G, Bataille R, Geffroy-Luseau A, Descamps G, Pellat-Deceunynck C (2006) Pathogen-associated molecular patterns are growth and survival factors for human myeloma cells through Toll-like receptors. Leukemia 20:1130–1137
Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP (2004) Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol 31:358–364
Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lu J, Kosaras B, Sidman RL, Volpe JJ, Vartanian T (2006) Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol 175:209–215
Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G (2002) Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol 168:4531–4537
Applequist SE, Wallin RP, Ljunggren HG (2002) Variable expression of Toll-like receptor in murine innate and adaptive immune cell lines. Int Immunol 14:1065–1074
Nagase H, Okugawa S, Ota Y, Yamaguchi M, Tomizawa H, Matsushima K, Ohta K, Yamamoto K, Hirai K (2003) Expression and function of Toll-like receptors in eosinophils: activation by Toll-like receptor 7 ligand. J Immunol 171:3977–3982
Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY (2004) TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A 101:3029–3034
Hemont C, Neel A, Heslan M, Braudeau C, Josien R (2013) Human blood mDC subsets exhibit distinct TLR repertoire and responsiveness. J Leukoc Biol 93:599–609
Siren J, Pirhonen J, Julkunen I, Matikainen S (2005) IFN-alpha regulates TLR-dependent gene expression of IFN-alpha, IFN-beta, IL-28, and IL-29. J Immunol 174:1932
Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S (1999) Cutting edge: Toll-like receptor 4 (TLR4) -deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162:6686–6687
Kurt-Jones EA, Sandor F, Ortiz Y, Bowen GN, Counter SL, Wang TC, Finberg RW (2004) Use of murine embryonic fibroblasts to define Toll-like receptor activation and specificity. J Endotoxin Res 10:419–424
Thurm CW, Halsey JF (2005) Measurement of cytokine production using whole blood. Curr Protoc Immunol Unit 7:18B
Blimkie D, Fortuno ES III, Yan H, Cho P, Ho K, Turvey SE, Marchant A, Goriely S, Kollmann TR (2011) Variables to be controlled in the assessment of blood innate immune responses to Toll-like receptor stimulation. J Immunol Methods 366:89–99
Hermann C, von Aulock S, Dehus O, Keller M, Okigami H, Gantner F, Wendel A, Hartung T (2006) Endogenous cortisol determines the circadian rhythm of lipopolysaccharide—but not lipoteichoic acid—inducible cytokine release. Eur J Immunol 36:371–379
Petrovsky N, Harrison LC (1995) Cytokine-based human whole blood assay for the detection of antigen-reactive T cells. J Immunol Methods 186:37–46
Schindler S, Asmus S, von Aulock S, Wendel A, Hartung T, Fennrich S (2004) Cryopreservation of human whole blood for pyrogenicity testing. J Immunol Methods 294:89–100
Smolen KK, Cai B, Gelinas L, Fortuno ES III, Larsen M, Speert DP, Chamekh M, Cooper PJ, Esser M, Marchant A, Kollmann TR (2014) Single-cell analysis of innate cytokine responses to pattern recognition receptor stimulation in children across four continents. J Immunol 193:3003–3012
Burl S, Townend J, Njie-Jobe J, Cox M, Adetifa UJ, Touray E, Philbin VJ, Mancuso C, Kampmann B, Whittle H, Jaye A, Flanagan KL, Levy O (2011) Age-dependent maturation of Toll-like receptor-mediated cytokine responses in Gambian infants. PLoS One 6, e18185
Levy O, Zarember KA, Roy RM, Cywes C, Godowski PJ, Wessels MR (2004) Selective impairment of TLR-mediated innate immunity in human newborns: neonatal blood plasma reduces monocyte TNF-alpha induction by bacterial lipopeptides, lipopolysaccharide, and imiquimod, but preserves the response to R-848. J Immunol 173:4627–4634
Labuda LA, de Jong SE, Meurs L, Amoah AS, Mbow M, Ateba-Ngoa U, van der Ham AJ, Knulst AC, Yazdanbakhsh M, Adegnika AA (2014) Differences in innate cytokine responses between European and African children. PLoS One 9, e95241
Deitschel SJ, Kerl ME, Chang CH, DeClue AE (2010) Age-associated changes to pathogen-associated molecular pattern-induced inflammatory mediator production in dogs. J Vet Emerg Crit Care (San Antonio) 20:494–502
Figueiredo MD, Moore JN, Vandenplas ML, Sun WC, Murray TF (2008) Effects of the second-generation synthetic lipid A analogue E5564 on responses to endotoxin in [corrected] equine whole blood and monocytes. Am J Vet Res 69:796–803
Karper JC, Ewing MM, de Vries MR, de Jager SC, Peters EA, de Boer HC, van Zonneveld AJ, Kuiper J, Huizinga EG, Brondijk TH, Jukema JW, Quax PH (2013) TLR accessory molecule RP105 (CD180) is involved in post-interventional vascular remodeling and soluble RP105 modulates neointima formation. PLoS One 8, e67923
Chen YW, Smith ML, Sheets MP, Ballaron SJ, Trevillyan JM, Fey TA, Gauvin DM, Kolano R, Pong MS, Hsieh GC, Bauch J, Marsh K, Carter G, Luly J, Djuric S, Mollison KW (1999) Ex vivo assessment of immunosuppression in undiluted whole blood from pigs dosed with tacrolimus (FK506). Clin Immunol 90:133–140
Dietsch GN, Dipalma CR, Eyre RJ, Pham TQ, Poole KM, Pefaur NB, Welch WD, Trueblood E, Kerns WD, Kanaly ST (2006) Characterization of the inflammatory response to a highly selective PDE4 inhibitor in the rat and the identification of biomarkers that correlate with toxicity. Toxicol Pathol 34:39–51
Duffy D, Rouilly V, Libri V, Hasan M, Beitz B, David M, Urrutia A, Bisiaux A, Labrie ST, Dubois A, Boneca IG, Delval C, Thomas S, Rogge L, Schmolz M, Quintana-Murci L, Albert ML, Milieu Interieur Consortium (2014) Functional analysis via standardized whole-blood stimulation systems defines the boundaries of a healthy immune response to complex stimuli. Immunity 40:436–450
O'Neill LA, Bowie AG (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7:353–364
Hansen MC, Nederby L, Henriksen MO, Hansen M, Nyvold CG (2014) Sensitive ligand-based protein quantification using immuno-PCR: a critical review of single-probe and proximity ligation assays. Biotechniques 56:217–228
Pranada AL, Metz S, Herrmann A, Heinrich PC, Muller-Newen G (2004) Real time analysis of STAT3 nucleocytoplasmic shuttling. J Biol Chem 279:15114–15123
Schindler U, Baichwal VR (1994) Three NF-kappa B binding sites in the human E-selectin gene required for maximal tumor necrosis factor alpha-induced expression. Mol Cell Biol 14:5820–5831
Johnson CM, Tapping RI (2007) Microbial products stimulate human Toll-like receptor 2 expression through histone modification surrounding a proximal NF-kappaB-binding site. J Biol Chem 282:31197–31205
Berger J, Hauber J, Hauber R, Geiger R, Cullen BR (1988) Secreted placental alkaline phosphatase: a powerful new quantitative indicator of gene expression in eukaryotic cells. Gene 66:1–10
Markova SV, Golz S, Frank LA, Kalthof B, Vysotski ES (2004) Cloning and expression of cDNA for a luciferase from the marine copepod Metridia longa. A novel secreted bioluminescent reporter enzyme. J Biol Chem 279:3212–3217
Zhou S, Cerny AM, Bowen G, Chan M, Knipe DM, Kurt-Jones EA, Finberg RW (2010) Discovery of a novel TLR2 signaling inhibitor with anti-viral activity. Antiviral Res 87:295–306
Ehrhardt C, Kardinal C, Wurzer WJ, Wolff T, von Eichel-Streiber C, Pleschka S, Planz O, Ludwig S (2004) Rac1 and PAK1 are upstream of IKK-epsilon and TBK-1 in the viral activation of interferon regulatory factor-3. FEBS Lett 567:230–238
Civas A, Genin P, Morin P, Lin R, Hiscott J (2006) Promoter organization of the interferon-A genes differentially affects virus-induced expression and responsiveness to TBK1 and IKKepsilon. J Biol Chem 281:4856–4866
Monteith GR, Bird GS (2005) Techniques: high-throughput measurement of intracellular Ca(2+)—back to basics. Trends Pharmacol Sci 26:218–223
National Center for Biotechnology Information (NCBI) (2015) http://www.ncbi.nlm.nih.gov/pcassay/?term = tlr
Reindl M, Lutterotti A, Ingram J, Schanda K, Gassner C, Deisenhammer F, Berger T, Lorenz E (2003) Mutations in the gene for toll-like receptor 4 and multiple sclerosis. Tissue Antigens 61:85–88
Karima R, Matsumoto S, Higashi H, Matsushima K (1999) The molecular pathogenesis of endotoxic shock and organ failure. Mol Med Today 5:123–132
Mansson A, Adner M, Hockerfelt U, Cardell LO (2006) A distinct Toll-like receptor repertoire in human tonsillar B cells, directly activated by PamCSK R-837 and CpG-2006 stimulation. Immunology 118:539–548
Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H, Delneste Y (2005) Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol 175:1551–1557
Tabiasco J, Devevre E, Rufer N, Salaun B, Cerottini JC, Speiser D, Romero P (2006) Human effector CD8+ T lymphocytes express TLR3 as a functional coreceptor. J Immunol 177:8708–8713
Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, Liu YJ (2001) Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med 194:863–869
Hart OM, Athie-Morales V, O'Connor GM, Gardiner CM (2005) TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J Immunol 175:1636–1642
Ito T, Wang YH, Liu YJ (2005) Plasmacytoid dendritic cell precursors/type I interferon-producing cells sense viral infection by Toll-like receptor (TLR) 7 and TLR9. Springer Semin Immunopathol 26:221–229
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Dowling, J.K., Dellacasagrande, J. (2016). Toll-Like Receptors: Ligands, Cell-Based Models, and Readouts for Receptor Action. In: McCoy, C. (eds) Toll-Like Receptors. Methods in Molecular Biology, vol 1390. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3335-8_1
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3335-8_1
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3333-4
Online ISBN: 978-1-4939-3335-8
eBook Packages: Springer Protocols