
Abstract
In the early twentieth century, histopathological staining of Parkinson’s and Alzheimer’s disease patient brains and later other neurodegenerative disorders revealed large intracellular and extracellular protein aggregates. Despite these pathological findings, 100 years later, scientists are yet to fully unravel disease pathogenesis sufficiently enough to allow early disease detection and development of therapeutic agents. While genetic and environmental factors are known to cause protein self-aggregation, recent discoveries have shown that protein intermediates are more toxic than the protein aggregates, serving as invaluable biomarkers for early disease detection. Mutations in alpha-synuclein, amyloid precursor, tau, and huntingtin proteins are known to cause self-aggregation and, via a series of conformational changes, produce oligomers and fibrils before being deposited extra- or intracellularly. In Alzheimer’s and Parkinson’s diseases, intermediate oligomeric forms of alpha-synuclein and beta-amyloid have been described not only to alter calcium homeostasis, which is vital to neuronal signalling, but also to interact with and alter mitochondrial function and therefore the energetic status of neurons. Research into the effects on the energy status of brain cells is vital to understanding the process of neurotoxicity observed in neurodegenerative diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Alzheimer’s disease
- Parkinson’s disease
- Huntington’s disease
- Mitochondria
- Oligomers
- Protein aggregation
- Tau
- Alpha-synuclein
- Huntingtin
1 Introduction
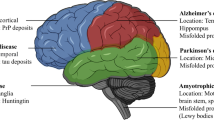
Most neurodegenerative diseases have been linked to the deposition of misfolded proteins in the brain. These deposits can be either extra- or intracellularly located and are associated with an altered biochemistry, often leading to neuronal loss. Extracellular deposits of beta-amyloid in the form of amyloid plaques and intracellular tau fibrils are histopathological hallmarks in Alzheimer’s disease (AD) [1, 2]. In Parkinson’s disease (PD), misfolded alpha-synuclein aggregates into intracellular Lewy bodies, while intracellular aggregates of nuclear-encoded mutant huntingtin protein (Htt) are found in the brain of Huntington’s disease patients. These deposits consist predominantly of protein fibrils and have long been thought to be the trigger of cellular dysregulations leading to the pathologies seen in these neurodegenerative diseases. However, insights gained more recently have revealed that in fact oligomeric forms of these proteins are more toxic than monomeric or fibrillary forms [3, 4]. Oligomers are cell permeable, allowing them to pass through plasma membranes and propagate throughout the brain. The propagation hypothesis by which proteins can spread progressively between cells and interconnecting brain regions in neurodegenerative diseases found support when oligomeric forms of alpha-synuclein were found in the cerebrospinal fluid (CSF) of affected individuals and in neuronal graft studies [5–7]. Furthermore, beta-amyloid, tau, mutant Htt, and alpha-synuclein have been shown to directly interact with mitochondria which makes bioenergetic disturbances in the cell likely and increases neuronal vulnerability [3, 8, 9].
2 Alpha-Synuclein
The exact physiological function of alpha-synuclein is yet to be unravelled despite a presynaptic localisation long being established [10]. However, studies of alpha-synuclein knockout mice and alpha-synuclein overexpressing models have proposed a physiological function related to neurotransmitter release [11, 12]. The fact that genome-wide association studies looking for risk factors for the development of idiopathic PD revealed the alpha-synuclein gene as the biggest risk locus makes research into this protein and its cellular function invaluable [13].
Alpha-synuclein is an intrinsically disordered protein and contains seven 11-amino acid repeats in the N-terminal that are predicted to form amphipathic alpha-helices which are highly conserved in vertebrates. Normal, soluble alpha-synuclein aggregates to form insoluble fibrils via a series of conformational changes and oligomeric intermediates. Native monomeric forms of this protein are α-helical structures, whereas misfolded polymers form more highly ordered β-sheet structures. Mounting evidence suggests that the soluble oligomeric forms generated in this misfolding process are the most toxic, and these have been directly linked to neurodegeneration [3, 14]. Interestingly, alpha-synuclein mutations associated with early-onset, autosomal dominant PD such as A30P (age of onset 54–79 years) and A53T (average age of onset 46 years) are all located in the N-terminus of this protein and are suggested to increase conformational destabilisation and oligomerisation which in turn favours protein aggregation and deposition [15–18]. However, studies have also revealed that these mutations not only increase the risk of alpha-synuclein misfolding and aggregating but also increase the overall abundance of wild-type alpha-synuclein. Elevated levels of wild-type alpha-synuclein were found to be sufficient to cause protein aggregation and development of early-onset PD (age of onset 35 years) in patients with a duplication of the alpha-synuclein locus [19, 20].
Studies into the effects of alpha-synuclein on cells in culture have revealed a multitude of adverse effects. It has been shown that alpha-synuclein can pass through membranes and associate with mitochondria within 8 min of exposure highlighting potentially devastating effects to cellular bioenergetics and supporting the propagation hypothesis [3, 21].
2.1 Cellular Effects of Alpha-Synuclein and Its Role in Neurodegeneration
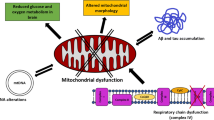
In vivo and in vitro studies have shown that overexpression of alpha-synuclein alters plasma membrane ion permeability, and exogenous oligomeric alpha-synuclein can easily pass across plasma membranes. This altered plasma membrane ion permeability has been shown to increase calcium influx from the extracellular space with detrimental consequences [3, 21, 22]. Intracellular calcium levels are tightly controlled by the endoplasmic reticulum (ER; main intracellular calcium store) and mitochondria (responsible for “fine-tuning” calcium transients) (for details see Chap. 6). Chronic calcium overload or altered handling of calcium fluxes may ultimately damage mitochondria, causing their dysfunction and ultimately the cellular bioenergetic status (e.g., reducing ATP production). This is particularly important for dopaminergic neurons which have a particularly high energy demand (for details see Chap. 6).
In addition to being the major cellular calcium store, the ER plays an important role in protein synthesis, folding, post-translational modification, and transport. Disturbances in ER function (ER stress) can have detrimental effects for the neuron and its survival. In PD, the increased levels of (misfolded) alpha-synuclein are reported to induce ER stress which in turn triggers downstream activation of the unfolded protein response [23]. This response can trigger attenuation of protein translation, altered expression of ER chaperones, and ER-associated degradation which counteracts the accumulation of misfolded proteins within the ER to protect against stress and potentially cell death [24]. ER-associated degradation allows misfolded proteins to be translocated into the cytosol where they are degraded by cytosolic proteasomes as part of the ubiquitin-proteasome system (UPS). In PD, a downregulation of the UPS and autophagy-lysosome pathways (ALP) has been proposed (see Chaps. 11 and 12), which impacts on the cellular clearance of misfolded alpha-synuclein. For example, expression of mutant alpha-synuclein (A30P and A53T) in rat PC12 and neuroblastoma cells was shown to downregulate the UPS [25–27]. The exact mechanism by which alpha-synuclein oligomers interact with and downregulate the proteasome is yet to be fully unravelled, but an interaction of alpha-synuclein with either the 20S β subunit or 19S subunits has been proposed [25, 28]. Alpha-synuclein has also been reported to downregulate ALP via an inhibition of autophagosome formation or a reduction of chaperone-mediated autophagy [29, 30]. Additional support for the importance of the relationship between intracellular protein clearance and alpha-synuclein in PD pathogenesis comes from studies involving the lysosomal enzyme, glucocerebrosidase. Mutations in this gene can cause Gaucher’s disease, an autosomal recessive disease, characterised by the accumulation of glucosylceramide (lipid) into fibrils. Gaucher’s disease patients may suffer from liver and spleen swelling, osteoporosis, and neurological defects including myoclonus, cognitive impairments, and convulsions (depending on the disease type). Furthermore, Gaucher’s disease has been reported to increase the risk of developing PD. It should also be noted that there is a well-documented decrease in the activity of the UPS as well as ALP with age leading to overall slower protein clearance. This reduction in protein clearance and therefore alpha-synuclein may explain partly the late onset of symptoms in sporadic Parkinson’s disease (see Chap. 12 for details).
The ER and mitochondria are also known to interact to form the mitochondria-associated endoplasmic reticulum membrane (MAM) which regulates essential calcium and lipid exchange between these organelles [31, 32]. This interaction supports the tricarboxylic acid cycle, electron transport chain, and ultimately ATP production. A recent study by Guardia-Laguarta et al. [33] provided evidence of wild-type alpha-synuclein co-localisation with the MAM, which together with the calcium dysregulation caused by alpha-synuclein is likely to have an effect on energy generation and mitochondrial health. In healthy cells, the ER and mitochondria are distributed throughout axons and dendrites. Considering the multitude of cellular functions supported by MAM interactions, in particular maintenance of the ETC and ATP production, it is not surprising that disruption of MAM will affect cells that have a high energy demand such as dopaminergic neurons. Indeed, reduced MAM interactions have been reported by Guardia-Laguarta et al. in cells that have been exposed to alpha-synuclein [33]. This loss of MAM interactions leads not only to a reduction in MAM function but also in mitochondrial fragmentation, suggesting an important role not only in mitochondrial functioning but also in morphology. These findings warrant further studies into the interaction of alpha-synuclein and its effect on these organelles.
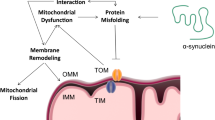
Alpha-synuclein has also been shown to co-localise with the inner mitochondrial membrane which harbours the ETC complexes (Fig. 10.1) [34]. In fact, it has long been known that alpha-synuclein can interact with complex I of the ETC which in turn induces elevated reactive oxygen species (ROS) production leading to oxidative stress [35, 36]. ROS can induce the formation of the permeability transition pore (PTP) which increases the permeability of the inner mitochondrial membrane allowing ions and solutes to pass more freely. This in turn leads to vast bioenergetic disturbances, ATP depletion and eventually cell death [37]. Furthermore, increased ROS can induce mitochondrial DNA mutations leading to mitochondrial damage and may ultimately lead to cell death [38] (see Fig. 10.1).

Neurotoxic effects of alpha-synuclein, tau, β-amyloid, and huntingtin proteins. Many of the proteins which aggregate in neurodegenerative diseases are known to interact with various intracellular components and different complexes within the mitochondria. (a) Both β-amyloid (βA) and alpha-synuclein interact with the endoplasmic reticulum (ER) resulting in ER stress and altered mitochondria-associated membrane interaction. (b) Alpha-synuclein (in PD) and beta-amyloid (in AD) are known to interact with complex I of the electron transport chain. (c) Mitochondrial health can also be compromised by β-amyloid and tau interacting with complex IV. Similarly huntingtin (Htt (in HD)) has been shown to interact with mitochondria through complex IV. (d) Dysregulation of calcium homeostasis can be induced by protein inclusions. (e) Htt can also interact with the nucleus itself. These proteins have all been shown to interact with mitochondria and induce superoxide production which can ultimately lead to PTP opening and neuronal cell death
3 Huntingtin Protein
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by an abnormal expansion of a CAG repeat located in exon 1 of the huntingtin gene. Wild-type huntingtin protein (Htt) localises to the nucleus as well as the cytoplasm, whereas mutant Htt has been found to localise to the mitochondria [39, 40] (see Fig. 10.1). Studies have shown that wild-type Htt plays a vital role in apoptosis, vesicle trafficking, and secretion pathways [41, 42]. It has been found that the N-terminal cleavage product of mutant Htt is vital to disease progression as the cleavage product has been shown to induce cellular changes, whereas inhibition of caspase-6-dependent proteolysis of full-length mutant Htt protected mutant Htt mice against neurodegeneration [43].
3.1 Cellular Effects of the Huntingtin Protein and Its Role in Neurodegeneration
The htt knockout mouse model is not viable (death occurs at embryonic day 8), providing evidence of the importance of this gene product [44]. Mutations in the htt locus are reported to either result in a gain or loss of function affecting a multitude of signalling pathways [45]. Vulnerability and degeneration of excitatory cortical and striatal connections is the first obvious sign of early grade HD.
Mutant Htt co-localises to mitochondria and studies have reported a negative effect on electron transport chain activity and mitochondrial function. Expression of an N-terminal fragment of mutant Htt (82 CAG repeats) in mice resulted in a decreased neuronal mitochondrial membrane potential, suggesting altered mitochondrial health. A lower mitochondrial membrane potential may indicate that one or more of the electron transport chain complexes are not working correctly. In fact, it was found that both the expression and activity of complex II and complex IV were reduced [46–48] (see Fig. 10.1). The lower mitochondrial membrane potential reported in HD results in lowered mitochondrial calcium buffering and efflux. This leads to a prolonged calcium exposure which increases cellular stress and therefore susceptibility to excitotoxic (excitatory-mediated cell death) insults. Whether the mitochondrial calcium dysregulation is a primary or secondary effect caused by the malfunctioning electron transport chain is yet to be unravelled [49].
Interestingly, the total peroxisome proliferator-activated receptor gamma coactivator-1 α RNA level (PGC-1α) is decreased in postmortem brain tissue from HD patients. PGC-1α is a transcriptional coactivator that regulates mitochondrial biogenesis and respiration, and mutant Htt (111 CAG repeats) was found to interact with the PGC-1α promoter in striatal cells. Furthermore, overexpression of PHC-1α rescued not only the mutant Htt-associated mitochondrial phenotypes but also protected mutant Htt primary striatal cultures from cell death making PGC-1α a key player in early HD pathogenesis [50].
4 Beta-Amyloid
The amyloid precursor protein (APP) is cleaved by β- and γ-secretase generating a range of β-amyloid (βA) peptides between 39 and 43 amino acid residues long. It is the hydrophobic nature of βA1-40 and βA1-42 which promotes self-aggregation and neurotoxicity revealing the importance of these forms in neurodegenerative disease. A series of conformational changes may occur causing βA aggregation via dimers, oligomers, protofibrils, and fibrils leading ultimately to the formation and deposition of amyloid into characteristic plaques. This deposition of βA is thought to play a central role in the development of Alzheimer’s disease (AD). Accumulation of βA in extracellular neuronal plaques is the defining feature for a diagnosis of Alzheimer’s disease in postmortem brain tissue [51].
4.1 Cellular Effects of β-Amyloid and Its Role in Neurodegeneration
Recently, it has been shown that APP/βA, in addition to its known interactions with the plasma membrane and ER, may also be targeted to mitochondria [52] again suggesting an effect on mitochondrial health (see Fig. 10.1).
Dysregulation of calcium homeostasis has been demonstrated in AD, with βA causing increased cytoplasmic calcium levels, mitochondrial calcium overload, and dysfunction. Mitochondrial dysfunction has been shown to be one of the earliest pathological signs, appearing before neurofibrillary tangles can be detected [53]. Studies have described mitochondrial deficiency in cultured cells overexpressing APP [54] or the spliced form APP751 [55]. A reduction in complex IV activity has been demonstrated in mitochondria from the hippocampus and platelets of patients with AD, as well as in AD animal models and AD cybrid cells [56]. In cortical neurons, βA causes mitochondrial dysfunction, reducing ATP levels [57], mostly through inhibition of complex I, causing both mitochondrial depolarisation and a loss of mitochondrial mass [58], while in isolated mitochondria, βA induced respiratory inhibition mostly through inhibition of complex IV [59, 60]. It has also been proposed that βA may increase mitochondrial ROS production [61] which in turn causes further mitochondrial impairment [62].
Application of exogenous βA to mixed cultures of neurons and astrocytes induced two types of mitochondrial depolarisation in astrocytes in the first hours of incubation – a slow and progressive loss and a transient loss of mitochondrial membrane potential [63]. Both types of βA-induced mitochondrial membrane potential change induced ROS production/oxidative stress through interaction with NADPH oxidase and can be blocked by inhibitors of this enzyme [63–65].
Further, it has been shown that βA-induced oxidative stress leads to activation of the DNA repair enzyme poly(ADP-ribose) polymerase (PARP). PARP consumes nicotinamide adenine dinucleotide which in turn decreases substrate availability for mitochondrial complex I of the ETC, resulting in a bioenergetic collapse and cell death [66, 67]. Further, provision of mitochondrial substrates (pyruvate and methyl succinate) reversed βA-induced mitochondrial depolarisation and prevented cell death [63, 65].
Studies have linked βA directly with cyclophilin D which is a key regulator of the PTP. Cyclophilin D is essential for PTP formation since inhibition was shown to inhibit PTP opening. PTP opening depletes mitochondrial calcium, elevates the tosolic calcium, and increases ROS levels, ultimately leading to depletion of cellular ATP and cell death [68]. βA was found to reduce the threshold for mPTP opening and molecular inhibition of cyclophilin D was able to rescue this phenotype. In deed, inhibition of cyclophilin D was able to improve mitochondrial function and learning/memory in an ageing Alzheimer’s disease mouse model [69].
5 Tau Protein
Tau is a microtubule-associated protein (MAP), encoded by the MAPT gene, and known to interact with α- and β-tubulin to facilitate microtubule assembly. With advancing age tau becomes enriched and prone to aggregation in axons and dendrites [70], while in the brain of AD patients, tau is hyperphosphorylated. This process of hyperphosphorylation is known to promote self-assembly, leading to the formation of oligomers and fibrils which eventually leads to neurofibrillary tangle (NFT) aggregation. Hyperphosphorylation of tau results also in microtubule destabilisation and compromised axonal transport which can, together with βA, lead to the pathologies observed in AD.
5.1 Cellular Effects of Tau and Its Role in Neurodegeneration
Gomez-Ramos et al. (2006) have shown that application of exogenous tau to neuronal cells leads to an increase in intracellular calcium levels via a muscarinic receptor-mediated mechanism which ultimately results in cell death [71]. In addition, limited studies have provided evidence of a tau-mitochondria interaction but it is thought that tau may act on mitochondria synergistically with βA as well as independently. For example, while βA is known to affect mitochondrial complex IV, tau has been linked to complex I inhibition suggestive of an influence on mitochondrial dynamics and health [72] (see Fig. 10.1).
Quintanilla et al. [73] proposed that the N-terminal fragment of tau may cause mitochondrial dysfunction, while truncated tau (Asp-421) induces mitochondrial fragmentation and elevated mitochondrial superoxide production in immortalised cortical neurons. Furthermore, a truncated tau N-terminal fragment (NH2-26–44) was found to act on the adenine nucleotide translocator (ANT) which is responsible for the translocation of mitochondrial ATP to the cytosol in exchange of ADP. In addition, Atlante et al. [74] found that the interaction of tau and ANT leads to an impairment of oxidative phosphorylation and therefore a decrease in ATP production. It should be noted that these studies established that it is the truncated N-terminal tau fragment which exerts these deleterious effects on mitochondrial health rather than the full-length tau protein. Overall, these studies suggest that through a disruption of mitochondrial function and therefore neuronal health, tau is a major contributor to the pathological changes and neurodegeneration central to the development of AD.
6 Conclusion
Recent studies have provided strong evidence that protein intermediates such as alpha-synuclein oligomers are more toxic to the cell than protein aggregates. Toxic protein intermediates can affect mitochondria and the energetic status of the cell. Disturbances in the energetic status are detrimental to neurons which have a high energy demand. This highlights the importance of continued research to develop therapeutic agents that target these early changes and cellular disruptions that occur before neuronal death and other pathological changes. To achieve early disease intervention, studies must provide robust biological marker analysis as well as develop therapeutic agents that are able to cross the blood-brain barrier to prevent the damage to and changes within the structures of the proteins described above. Most of the modern therapeutic strategies aim to prevent peptide aggregation or indeed protect cells with antibodies against oligomeric forms. However, there is an increasing amount of research being undertaken to prevent/modulate the interactions between these proteins and cellular organelles to delay or prevent subsequent disruption of mitochondrial dysfunction and hence neuronal loss. Despite this, very few therapeutic approaches have undergone clinical trials and even fewer have shown evidence of efficacy making further research into disease onset and progression essential.
References
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–8.
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83(13):4913–7.
Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell. 2012;149(5):1048–59.
Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14(1):38–48.
El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003;17(13):1945–7.
Kordower JH, Dodiya HB, Kordower AM, Terpstra B, Paumier K, Madhavan L, et al. Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol Dis. 2011;43(3):552–7.
Recasens A, Dehay B. Alpha-synuclein spreading in Parkinson’s disease. Front Neuroanat. 2014;8:159.
Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19(14):2040–1.
Labbadia J, Morimoto RI. Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci. 2013;38(8):378–85.
Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8(8):2804–15.
Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, et al. alpha-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004;23(22):4506–16.
Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci. 2014;34(28):9364–76.
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–12.
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 1998;95(11):6469–73.
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18(2):106–8.
Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury Jr PT. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97(2):571–6.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–7.
Kruger R, Kuhn W, Leenders KL, Sprengelmeyer R, Muller T, Woitalla D, et al. Familial parkinsonism with synuclein pathology: clinical and PET studies of A30P mutation carriers. Neurology. 2001;56(10):1355–62.
Kojovic M, Sheerin UM, Rubio-Agusti I, Saha A, Bras J, Gibbons V, et al. Young-onset parkinsonism due to homozygous duplication of alpha-synuclein in a consanguineous family. Mov Disord. 2012;27(14):1827–9.
Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet. 2004;364(9440):1169–71.
Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, et al. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27(34):9220–32.
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106(31):13010–5.
Bellucci A, Navarria L, Zaltieri M, Falarti E, Bodei S, Sigala S, et al. Induction of the unfolded protein response by alpha-synuclein in experimental models of Parkinson’s disease. J Neurochem. 2011;116(4):588–605.
Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11(4):372–80.
Xilouri M, Brekk OR, Stefanis L. alpha-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol. 2013;47(2):537–51.
Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21(24):9549–60.
Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, et al. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36(6):1007–19.
Lindersson E, Beedholm R, Hojrup P, Moos T, Gai W, Hendil KB, et al. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem. 2004;279(13):12924–34.
Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010;190(6):1023–37.
Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010;67(12):1464–72.
Flis VV, Daum G. Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb Perspect Biol. 2013;5(6):a013235.
Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta. 2013;1833(1):213–24.
Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, et al. alpha-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci. 2014;34(1):249–59.
Robotta M, Gerding HR, Vogel A, Hauser K, Schildknecht S, Karreman C, et al. Alpha-synuclein binds to the inner membrane of mitochondria in an alpha-helical conformation. Chembiochem. 2014;15(17):2499–502.
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283(14):9089–100.
Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci. 2008;65(7–8):1272–84.
Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol. 2013;4:95.
Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev. 2012;2012:428010.
Rockabrand E, Slepko N, Pantalone A, Nukala VN, Kazantsev A, Marsh JL, et al. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet. 2007;16(1):61–77.
Hoogeveen AT, Willemsen R, Meyer N, de Rooij KE, Roos RA, van Ommen GJ, et al. Characterization and localization of the Huntington disease gene product. Hum Mol Genet. 1993;2(12):2069–73.
Brandstaetter H, Kruppa AJ, Buss F. Huntingtin is required for ER-to-Golgi transport and for secretory vesicle fusion at the plasma membrane. Dis Model Mech. 2014;7(12):1335–40.
Rigamonti D, Bauer JH, De-Fraja C, Conti L, Sipione S, Sciorati C, et al. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci. 2000;20(10):3705–13.
Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125(6):1179–91.
Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81(5):811–23.
Damiano M, Galvan L, Deglon N, Brouillet E. Mitochondria in Huntington’s disease. Biochim Biophys Acta. 2010;1802(1):52–61.
Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17(4):1652–63.
Benchoua A, Trioulier Y, Diguet E, Malgorn C, Gaillard MC, Dufour N, et al. Dopamine determines the vulnerability of striatal neurons to the N-terminal fragment of mutant huntingtin through the regulation of mitochondrial complex II. Hum Mol Genet. 2008;17(10):1446–56.
Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. 2005;47(1):29–41.
Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5(8):731–6.
Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127(1):59–69.
Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, et al. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283(12):1571–7.
Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161(1):41–54.
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21(9):3017–23.
Askanas V, McFerrin J, Baque S, Alvarez RB, Sarkozi E, Engel WK. Transfer of beta-amyloid precursor protein gene using adenovirus vector causes mitochondrial abnormalities in cultured normal human muscle. Proc Natl Acad Sci U S A. 1996;93(3):1314–9.
Grant SM, Shankar SL, Chalmers-Redman RM, Tatton WG, Szyf M, Cuello AC. Mitochondrial abnormalities in neuroectodermal cells stably expressing human amyloid precursor protein (hAPP751). Neuroreport. 1999;10(1):41–6.
Du H, Yan SS. Mitochondrial medicine for neurodegenerative diseases. Int J Biochem Cell Biol. 2010;42(5):560–72.
Casley CS, Land JM, Sharpe MA, Clark JB, Duchen MR, Canevari L. Beta-amyloid fragment 25–35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol Dis. 2002;10(3):258–67.
Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80(1):91–100.
Canevari L, Clark JB, Bates TE. beta-Amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999;457(1):131–4.
Parks JK, Smith TS, Trimmer PA, Bennett Jr JP, Parker Jr WD. Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J Neurochem. 2001;76(4):1050–6.
Sheehan JP, Swerdlow RH, Miller SW, Davis RE, Parks JK, Parker WD, et al. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s disease. J Neurosci. 1997;17(12):4612–22.
Arias C, Montiel T, Quiroz-Baez R, Massieu L. beta-Amyloid neurotoxicity is exacerbated during glycolysis inhibition and mitochondrial impairment in the rat hippocampus in vivo and in isolated nerve terminals: implications for Alzheimer’s disease. Exp Neurol. 2002;176(1):163–74.
Abramov AY, Canevari L, Duchen MR. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci. 2004;24(2):565–75.
Abramov AY, Fraley C, Diao CT, Winkfein R, Colicos MA, Duchen MR, et al. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc Natl Acad Sci U S A. 2007;104(46):18091–6.
Abramov AY, Duchen MR. The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2309–14.
Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, et al. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278(20):18426–33.
Abeti R, Abramov AY, Duchen MR. Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain. 2011;134(Pt 6):1658–72.
Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6(3):337–50.
Du H, Guo L, Zhang W, Rydzewska M, Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2011;32(3):398–406.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van E J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142(3):387–97.
Gomez-Ramos A, Diaz-Hernandez M, Cuadros R, Hernandez F, Avila J. Extracellular tau is toxic to neuronal cells. FEBS Lett. 2006;580(20):4842–50.
Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci U S A. 2009;106(47):20057–62.
Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem. 2009;284(28):18754–66.
Atlante A, Amadoro G, Bobba A, de Bari L, Corsetti V, Pappalardo G, et al. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim Biophys Acta. 2008;1777(10):1289–300.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing
About this chapter
Cite this chapter
Ludtmann, M.H.R., Abramov, A.Y. (2016). Protein Misfolding and Aggregation: Implications for Mitochondrial Dysfunction and Neurodegeneration. In: Reeve, A., Simcox, E., Duchen, M., Turnbull, D. (eds) Mitochondrial Dysfunction in Neurodegenerative Disorders. Springer, Cham. https://doi.org/10.1007/978-3-319-28637-2_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-28637-2_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-28635-8
Online ISBN: 978-3-319-28637-2
eBook Packages: MedicineMedicine (R0)