
Abstract
An increasing wealth of data indicates a close relationship between the presynaptic protein alpha-synuclein and Parkinson’s disease (PD) pathogenesis. Alpha-synuclein protein levels are considered as a major determinant of its neurotoxic potential, whereas secreted extracellular alpha-synuclein has emerged as an additional important factor in this regard. However, the manner of alpha-synuclein degradation in neurons remains contentious. Both the ubiquitin–proteasome system (UPS) and the autophagy–lysosome pathway (ALP)—mainly macroautophagy and chaperone-mediated autophagy—have been suggested to contribute to alpha-synuclein turnover. Additionally, other proteases such as calpains, neurosin, and metalloproteinases have been also proposed to have a role in intracellular and extracellular alpha-synuclein processing. Both UPS and ALP activity decline with aging and such decline may play a pivotal role in many neurodegenerative conditions. Alterations in these major proteolytic pathways may result in alpha-synuclein accumulation due to impaired clearance. Conversely, increased alpha-synuclein protein burden promotes the generation of aberrant species that may impair further UPS or ALP function, generating thus a bidirectional positive feedback loop leading to neuronal death. In the current review, we summarize the recent findings related to alpha-synuclein degradation, as well as to alpha-synuclein-mediated aberrant effects on protein degradation systems. Identifying the factors that regulate alpha-synuclein association to cellular proteolytic pathways may represent potential targets for therapeutic interventions in PD and related synucleinopathies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As highlighted in other manuscripts in this volume, the aggregation of alpha-synuclein has emerged as the most important player in the neurodegenerative process that occurs in the context of Parkinson’s disease (PD) and other neurodegenerative diseases, collectively termed synucleinopathies. One of the critical factors controlling the aggregation process of alpha-synuclein is the protein levels, which are regulated by a balanced equilibrium between the synthesis, degradation, and secretion of the protein. Multiple lines of evidence implicate altered alpha-synuclein handling accompanied by decreased degradation of the protein as the major factor leading to PD pathogenesis. Failure of the two major intracellular proteolytic systems, the ubiquitin–proteasome system (UPS) or/and the autophagy–lysosome pathway (ALP), is widely considered to contribute to the accumulation of aggregated alpha-synuclein species that consequently influence fundamental cellular pathways. The UPS is considered to degrade mostly short-lived, soluble proteins [1], while the ALP is responsible for the bulk degradation of longer-lived macromolecules, cytosolic components, and dysfunctional organelles [2]. These proteolytic systems are functionally connected, such that impairment of one influences the other. Both the UPS and the ALP have been suggested to be responsible for alpha-synuclein degradation in various cell culture systems, while other proteolytic systems, such as calpains [3, 4], kallikrein-6 (neurosin) [5, 6], or metalloproteinases [7, 8], have also been proposed to play a role in alpha-synuclein processing. Of particular recent interest is the possibility that such proteolytic systems may operate on the degradation of extracellular alpha-synuclein, which is increasingly assuming pathogenic significance [7–9].
During the last decade, research in the field was focused towards two interrelated themes: deciphering the manner of alpha-synuclein degradation and identifying the targets of the aberrant effects of alpha-synuclein on protein degradation pathways. Deregulation of the major proteolytic systems may result either in accumulation of total alpha-synuclein, or of specific species that are targeted for degradation through the pathway in question; on the other hand, increased levels of alpha-synuclein may promote the generation of aberrant species that may further impair UPS or ALP function, inhibiting their own degradation as well as the degradation of other substrates and generating thus a vicious cycle of neurotoxicity. The mechanisms governing the turnover of alpha-synuclein are a critical aspect of disease mechanisms and represent potential therapeutic targets for the treatment of PD and related synucleinopathies.
Proteolysis Through the Ubiquitin–Proteasome System
Protein degradation through the UPS, a major system for degradation of short-lived proteins, consists of two distinct and consecutive steps. The first step involves the covalent attachment of the highly conserved 76-residue protein ubiquitin to surface-exposed lysine residues of the target protein [10]. Ubiquitylation is accomplished via a three-step cascade mechanism, where each step is catalyzed by ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligase (E3) enzymes. Following the initial conjugation of ubiquitin, polyubiquitylation of the substrate occurs through the sequential transfer of ubiquitin molecules—minimum of four ubiquitin moieties—generating a polyubiquitin chain [10–12]. In the second step, this polyubiquitin chain is recognized by the proteolytic core engine of the UPS, the 26S proteasome complex which in turn degrades the substrate protein into defined oligopeptides with release of free and reusable ubiquitin. The removal of the ubiquitin molecule is mediated by ubiquitin recycling enzymes [10, 13]. The 26S proteasome comprises of two multimeric protein complexes, the 20S barrel-shaped core particle, where actual proteolysis occurs, and two 19S regulatory particles that regulate the function of the 20S [14–16]. The substrate specificity and selectivity of the proteasome is achieved by two distinct groups of proteins, the E3 ligases that catalyze substrate recognition prior to their ubiquitination and the proteasome ancillary proteins [10]. Proteasome regulation is tightly controlled by the ATPase subunits of the 19S complex [17]. Alternatively, degradation of target proteins may take place without the addition of polyubiquitin chains, with the participation of regulatory non-ATPase complexes, such as the PA28 activator, resulting in ubiquitin-independent proteolysis [17].
Proteolysis Through the Autophagy–Lysosome Pathway
If the proteasome is the wandering assassin of cell proteins, the lysosome is surely protein purgatory. The major differences between the UPS and the ALP proteolytic machineries rely on the fact that the ALP is responsible for the vesicle-mediated degradation of long-lived proteins, and that, through the process of macroautophagy, the ALP can also degrade cellular organelles. Within the acidic confines of the lysosomal lumen, a plethora of proteases, lipases, and nucleases deconstruct most cellular constituents down to their composite parts. In lysosomes, degradation of cytoplasmic components is achieved through three subtypes of autophagic pathways: microautophagy, macroautophagy, and chaperone-mediated autophagy (CMA) [2, 18]. In microautophagy, direct delivery and digestion of the cellular constituents (including organelles, lipids, or proteins) is accomplished through invaginations at the level of the lysosomal membrane which eventually round up into vesicles [19].
Macroautophagy is a tightly regulated process, in which double-membrane structures (cup- or rod-shaped) called phagophores are formed and engulf organelles and/or other intracellular constituents, generating the autophagic vacuoles (AVs). Sequentially, the AVs fuse with the lysosome creating the autophagolysosome, wherein eventually the vesicular components are being degraded. In neurons, AVs can be found at long distances from the lysosomes (which are mainly located perinuclearly), for example along neuronal processes [20]; they then move to a perinuclear location to fuse with the lysosomes. Thus, efficient functioning of this pathway requires proper cellular transport machinery. There is also convergence with the endocytic pathway [21]. In general, macroautophagy consists of several sequential steps: (1) formation of AVs, which includes initiation, nucleation, cargo recognition, expansion, and completion; (2) maturation, transport, and fusion of AVs with lysosomes; and (3) degradation of the constituents in the acidic environment of the autophagolysosomes by lysosomal proteases [22]. A plethora of autophagy-related genes (ATG) involved in the above processes has been well characterized in yeast and more recently in mammals [23, 24]. Macroautophagy occurs constitutively in cells (basal macroautophagy), but is markedly induced under certain circumstances (induced macroautophagy). Amino acid deprivation and insulin/growth factor signals are thought to converge on the serine/threonine kinase mammalian target of rapamycin (mTOR), which is a master regulator of nutrient signaling [25]. However, it is important to note that macroautophagy can be induced by mTOR-dependent and mTOR-independent pathways [26, 27].
Recently, macroautophagy has been shown to mediate selective degradation of various targets such as aggregated proteins and damaged organelles. In this process, p62 (sequestosome 1, SQSTM1) and perhaps HDAC6 proteins play a vital role and are considered to be the missing link between the UPS and the ALP systems. p62 serves to selectively link ubiquitinated proteins to the autophagic machinery and enable their degradation in the lysosome [28]. HDAC6 has the capacity to bind both polyubiquitinated misfolded proteins and dynein motors, targeting thus polyubiquitinated aggregates and damaged mitochondria to aggresomes for degradation [29]. Moreover, several other proteins (“autophagy receptors”) linking the autophagic machinery to its substrates have been identified [30–33] reinforcing the idea that macroautophagy can be a rather selective process.
CMA is a high selective mechanism in which cytosolic proteins bearing the specific pentapeptide KFERQ motif or a biochemically related one are recognized by a complex of chaperones and co-chaperones in the cytosol. Sequentially, they are translocated one by one to the lysosomal membrane, where they bind to another complex and through this binding they are finally threaded into the lysosomes and degraded [34–38]. The complex involved in substrate recognition comprises of the heat shock cognate protein of 70 kDa (Hsc70) and Hsc70 co-chaperones. The main element in the lysosomal membrane complex is the lysosome-associated membrane protein type 2a (LAMP-2a), an alternatively spliced form of the Lamp2 gene, and the only one known to participate in CMA [39]. The substrate protein is degraded after unfolding and translocation into the lysosomal lumen, with the help of lysosomal Hsc70. CMA activity is directly dependent on the levels of LAMP-2a at the lysosomal membrane and on lys-hsc70 levels in the lysosomal lumen [40–42]. Basal levels of CMA activity are detectable—although variable—in most cells and tissues, but CMA is often up-regulated under conditions of stress or nutrient deprivation [43]. Macroautophagy and CMA are interconnected; experimental blockage of one up-regulates the other, revealing a close cross-talk between the two major mammalian autophagic pathways [44–47]. Importantly, CMA activity declines with age in almost all tissues analyzed so far, mainly as a consequence of decreased LAMP-2a levels at the lysosomal membrane [48]. In various cell systems, overexpression of LAMP-2a is able to ameliorate CMA function, indicating that increased LAMP-2a is sufficient for this effect [49, 50] and stressing the importance of LAMP-2a as a rate-limiting step in the pathway.
What Is the Main Pathway Responsible for Alpha-synuclein Degradation?
Notwithstanding the escalating number of publications, the exact mechanism responsible for the degradation of alpha-synuclein remains controversial and varies depending on the system studied. In in vitro isolated purified systems, both proteasomes and lysosomes were shown to be capable of degrading recombinant alpha-synuclein [51, 52]. However, the mechanism that decides whether alpha-synuclein will be degraded by the UPS or the autophagy pathway remains puzzling (Fig. 1).

Proteolytic pathways implicated in alpha-synuclein processing. The proteasome and the lysosome are the two major pathways for the degradation of intracellular proteins. In the ubiquitin–proteasome system (UPS), substrate proteins are tagged with ubiquitin molecules and transferred to the 26S proteasome complex where they are degraded into defined oligopeptides with release of free and reusable ubiquitin. Both ubiquitin-dependent (A) and ubiquitin-independent (B) mechanisms have been proposed to degrade alpha-synuclein (wild type and mutant). In mammalian cells, three different lysosomal pathways have been described: microautophagy, macroautophagy, and chaperone-mediated autophagy (CMA). In microautophagy, substrates are directly delivered into the lumen through invaginations of the lysosomal membrane. In macroautophagy, intracellular constituents (proteins, organelles) are sequestered by a double membrane (phagophore) that finally generates the autophagic vacuole, which then fuses with the lysosome. In CMA, selective substrate proteins (bearing a KFERQ-like motif) are threaded into the lysosomes one by one after binding to the lysosomal receptor LAMP-2a. Both CMA (C) and macroautophagy (D) have been proposed to degrade alpha-synuclein, with CMA being responsible for the monomeric wild-type alpha-synuclein and macroautophagy being capable of degrading various forms (wild type, mutant, oligomeric). Besides the proteasome and the lysosome, other proteases such as calpains (E) and neurosin (F) have been implicated in the cleavage of normal or aggregated forms of intracellular alpha-synuclein. Such cleavage may promote the generation of truncated alpha-synuclein species with pathogenic significance. Moreover, secreted neurosin (G) and metalloproteinases (H) have been found to cleave at selective sites extracellular alpha-synuclein, generating fragments with increased tendency to aggregate
The primary indication for a potential role of the UPS in the degradation of alpha-synuclein, and in PD in general, arose from the finding that two UPS-related proteins, Parkin and UCH-L1, were encoded by genes with genetic links to PD [53–56] and from molecular, cellular, and animal data that link these genes to UPS-dependent processing of alpha-synuclein [57, 58]. Initial studies suggested that alpha-synuclein, in some cases in a polyubiquitinated form, accumulated in neuronal cells upon proteasomal inhibition, suggesting that the UPS is responsible for alpha-synuclein degradation (Fig. 1a) [59–62]. Subsequent studies in purified systems and in neuronal cells showed that alpha-synuclein does not need to be ubiquitinated to be degraded by the proteasome (Fig. 1b) [63, 64]. Machiya et al. recently showed that phosphorylated alpha-synuclein at Ser129 can be rapidly turned over via the proteasomal pathway in a ubiquitin-independent manner in addition to undergoing dephosphorylation [65], implicating the UPS in the biogenesis of Ser129-phosphorylated alpha-synuclein-rich Lewy bodies (LBs). In this study, only phosphorylated, but not total unmodified alpha-synuclein, was found to be stabilized by proteasomal inhibition.
Other studies, including our own, have failed to detect accumulation of endogenous or overexpressed alpha-synuclein with proteasomal inhibition in various cellular systems [66–69]. One factor that might explain the observed controversy is the assembly state and pools of alpha-synuclein utilized, since large oligomeric forms can be cleared only by the ALP but not by the UPS [70]. In this framework, we have found that only very specific species of alpha-synuclein, comprising of small, soluble oligomers, are degraded through the UPS. Interestingly, the same species appear to cause proteasomal dysfunction in their attempt to be degraded [69] (see below). A candidate molecular switch involved in the decision between the UPS and autophagy pathways was initially suggested to be CHIP (co-chaperone carboxyl terminus of heat-shock protein 70-interacting protein), which was found to co-localize with alpha-synuclein and Hsp70 in LBs from human subjects [71]. Towards the same direction, it was demonstrated that in the presence of proteolytic inhibitors, monoubiquitination of alpha-synuclein by the E3 ubiquitin-ligase SIAH led to a marked increase in alpha-synuclein aggregation and formation of toxic inclusions [72, 73], while recently, it was reported that the deubiquitinase USP9X interacts and deubiquitinates alpha-synuclein both in vitro and in vivo [74]. In search for molecules that determine the fate of alpha-synuclein, Rott et al. surmised that USP9X regulates alpha-synuclein partitioning between the proteasomal and autophagy systems favoring the latter; in the absence of proteolytic impairment, monoubiquitination promotes alpha-synuclein degradation through the proteasome, while deubiquitination targets the protein almost entirely to ALP [74]. USP9X cytosolic levels were found to be decreased in dementia with Lewy bodies (DLB) and PD nigral tissues, indicating lower deubiquitinase activity and subsequent accumulation and aggregation of monoubiquitinated alpha-synuclein in LBs [74]. Moreover, Tofaris et al. identified Nedd4 as the E3 ligase involved in alpha-synuclein degradation via the endosomal pathway and speculated that this pathway is specific for the degradation of alpha-synuclein membrane-associated pools, whereas autophagy degrades the oligomeric forms [75].
Two in vivo reports support the involvement of the UPS in alpha-synuclein degradation. Bedford et al. demonstrated that conditional ablation of the Psmc1 subunit of the 26S proteasome in nigral or forebrain (e.g., cortex, hippocampus, and striatum) neurons caused intraneuronal LB-like inclusions immunoreactive for ubiquitin and alpha-synuclein and extensive neurodegeneration [76], suggesting that the UPS is an important route for alpha-synuclein proteolysis [76]. However, an alternative explanation could be that, as in cortical neuron cell culture studies, alpha-synuclein gets “trapped” after the fact in formed inclusions [77]. More recently, an elegant study suggested that the UPS and the ALP possess distinct roles for the degradation of alpha-synuclein in the living mouse brain [78]. Using selective pharmacological inhibitors against the UPS and the ALP and multiphoton imaging in different alpha-synuclein transgenic mouse models, the authors proposed that the pathway recruited to degrade alpha-synuclein depends on the protein burden inside the cell. According to their findings, the UPS degrades alpha-synuclein under conditions of endogenous and increased protein burden, whereas ALP takes over in pathologic conditions when alpha-synuclein levels are elevated [78]. The authors also presented evidence that a functional coupling between the UPS and the ALP exists, but only in the scenario of elevated alpha-synuclein protein burden, and surmised that the intracellular alpha-synuclein levels modulate the potential cross-talk between the two degradation pathways.
According to our own work, and that of others, a significant portion of the degradation of alpha-synuclein in neuronal systems occurs through the lysosomal pathways of CMA and macroautophagy; consequently, we and others have suggested that dysfunction of these degradation pathways may be a contributing factor to PD pathogenesis [44, 45, 79]. Paxinou et al. were the first to show that in cultured cells alpha-synuclein accumulated with lysosomal, but not proteasomal inhibition [80]. The Rubinsztein lab found that alpha-synuclein was indeed located within lysosomal compartments of cultured neuronal cells and that the degradation of PD-linked mutant, but not of wild-type alpha-synuclein was attenuated by application of macroautophagy inhibitors [62]. Importantly, from a therapeutic point of view, the mTOR inhibitor and macroautophagy enhancer rapamycin promoted the degradation of both wild-type and mutant alpha-synuclein [75]. This approach was also taken by the Masliah lab, who showed that pharmacological and molecular enhancement of macroautophagy led to significant reduction in wild-type alpha-synuclein levels in cell culture systems, and extended such studies to the in vivo level, where reduction of alpha-synuclein-related neuropathological effects in a transgenic mouse model was observed with viral transduction of the macroautophagy-inducing protein beclin-1 [81]. The group of Lee found that general lysosomal inhibition via bafilomycin led to a specific build-up of soluble oligomeric, but not of bulk monomeric or fully fibrillar forms of overexpressed wild-type alpha-synuclein in neuronal cell lines [70]. This enrichment, however, did not occur with the selective macroautophagy inhibitor 3-MA. In our own work, 3-MA application led to a significant increase of both endogenous and overexpressed wild-type alpha-synuclein in PC12 cells and of endogenous alpha-synuclein in cultured cortical and ventral midbrain dopaminergic neurons [44]. Alvarez-Erviti et al. in agreement with the Rubinsztein group found that 3-MA inhibited only the degradation of mutant A53T, but not wild-type, alpha-synuclein, whereas general lysosomal inhibition affected both [79]. The discrepancies among these studies are clear, but also the general impression that macroautophagy is important for the degradation of at least some species of alpha-synuclein; perhaps more importantly, forced induction of macroautophagy may have therapeutic potential against synucleinopathies (but see caveats below).
Some of the above studies were indicative of an alternative lysosomal pathway being the primary workhorse in alpha-synuclein turnover, as in certain instances general lysosomal inhibitors had more profound effects than selective macroautophagy inhibition. An explanation for this particular conundrum had actually been in the works since the late 1980s, near single-handedly through the work of Fred Dice’s lab, which, through a series of elegant experiments, proved the existence of CMA [82]. In pivotal experiments, Ana Maria Cuervo and colleagues showed, using mainly cell-free systems, that wild-type alpha-synuclein, which contained the appropriate motif, was indeed a CMA substrate, whereas the A30P and A53T alpha-synuclein PD-linked mutants bound strongly to LAMP-2a but were not internalized and degraded and thus acted as inhibitors of CMA degradation of other substrates [52]. This seminal discovery has spurred interest for the association between CMA and PD pathology.
In subsequent studies, by using siRNAs against LAMP-2a and expressing forms of alpha-synuclein lacking the CMA recognition motif, we confirmed that CMA is an important pathway for normal alpha-synuclein turnover in neuronal cell lines and primary cortical and ventral midbrain neurons [44]. Moreover, detergent-insoluble or high molecular weight oligomeric alpha-synuclein conformations also increased following CMA inhibition, suggesting that CMA dysfunction could eventually lead to alpha-synuclein-related pathology [44]. The data implicating CMA in wild-type alpha-synuclein degradation in neuronal cellular systems have recently been confirmed using similar approaches [79]. Moreover, Mak et al. demonstrated that mice exposed to the mitochondrial toxin paraquat or alpha-synuclein transgenic mice up-regulate lysosomal LAMP-2a and degrade alpha-synuclein more efficiently, providing indirect evidence that CMA is a main mechanism for alpha-synuclein degradation in vivo, at least under conditions in which alpha-synuclein levels are induced above a certain threshold [83].
As for the proteolysis of alpha-synuclein itself, evidence suggests that the primary lysosomal protease involved is cathepsin D, as its expression correlates with the levels and neurotoxic potential of alpha-synuclein [84–86]. More particularly, Qiao et al. reported accumulation of high molecular weight but not monomeric alpha-synuclein species in the cortex of cathepsin D-deficient mice, in the absence of an increase of alpha-synuclein mRNA expression [85]. Conversely, up-regulation of cathepsin D, but not cathepsin B or L reduced human overexpressed alpha-synuclein aggregation and subsequent neurotoxicity, both in dopaminergic cell lines and in Caenorhabditis elegans [85]. In addition, Cullen et al. demonstrated a reduction—rather than an increase—in soluble alpha-synuclein levels and an increase in insoluble high molecular weight alpha-synuclein oligomers/aggregates in cathepsin D−/− mouse brains, compared to their control littermates [86]. Furthermore, the authors reported alpha-synuclein accumulation in human postmortem brains from three infants diagnosed with cathepsin D deficiency altered alpha-synuclein immunostaining in mutant cathepsin D sheep brains and enhanced alpha-synuclein toxicity in Drosophila flies expressing human alpha-synuclein in a cathepsin D-null background [86]. These in vivo studies which point out increased cathepsin D “synucleinase” activity and show formation of alpha-synuclein aggregates without significant accumulation of the monomer in a cathepsin D-null background suggest that in an in vivo scenario the lysosomes are responsible mainly for the degradation of aggregated or aggregated-prone species. This could be consistent with the findings mentioned above suggesting a lysosomal-dependent degradation specifically of oligomeric aggregation-prone species or of excess levels of alpha-synuclein [78]. Alternatively, this might indicate that in certain circumstances—perhaps favored in in vivo settings—even small increases in alpha-synuclein levels lead to its deposition within inclusions.
In view of the recent findings supporting the notion that alpha-synuclein exists physiologically as a tetramer with decreased aggregation propensity [87], while the PD-linked mutants (A30P, E46K, and A53T) markedly decrease the stability of the tetramer and shift alpha-synuclein equilibrium towards the “toxic” monomer [88], the clarification of the mechanisms controlling the clearance of this putative physiologic form will also be of great interest.
Degradation of Alpha-synuclein by Other Proteases
Besides the UPS and the ALP systems, several lines of evidence implicate other proteases, including calpain-1 [3, 4], neurosin (kallikrein-6) [5, 6], and matrix metalloproteinases [7, 8, 89], in the cleavage of normal or aggregated forms of alpha-synuclein in vitro (Fig. 1e–h). Such cleavage might lead to generation of alpha-synuclein truncated species with pathogenic significance, in view of the fact that they can promote the ability of the full length protein to aggregate and that similar truncated species are found in LBs of sporadic PD and DLB brains [90, 91].
Alpha-synuclein is predominantly a presynaptic protein, suggesting that it may be a substrate for soluble or membrane-associated proteases such as the calcium-activated neutral protease calpain I (Fig. 1e). Mishizen-Eberz et al. initially demonstrated in a cell-free system that calpain I cleaved wild-type alpha-synuclein primarily within the NAC region and at amino acid 57, while the cleavage of fibrillized alpha-synuclein occurred mainly at amino acid 120, close to the C-terminal, generating fragments similar to the ones that confer susceptibility to dopamine toxicity and oxidative stress [4]. The major cleavage between amino acids 57 and 58 did not occur in A53T alpha-synuclein, indicating that this cleavage might be protective and that lack of this calpain-induced proteolysis might increase the stability of A53T protein and enhance its accumulation [4]. Furthermore, Dufty et al. showed that calpain I proteolytic processing of alpha-synuclein leads to the formation of aggregated high molecular weight species and adoption of a β-sheet structure and that similar calpain-induced C-terminal fragments were present in mouse models of cerebral ischemia and PD [3]. Moreover, calpain-cleaved N- or C-terminal fragments of alpha-synuclein were identified using two site-directed calpain cleavage antibodies within nigral LBs and Lewy neurites and were co-localized with activated calpain in neurons of PD and DLB brains [3]. These findings implicate calpain I in the disease-linked aggregation of alpha-synuclein, through generation of the C-terminal truncated pathogenic species and could thus represent a target for therapy. Recently, it was shown that passive immunization with antibodies against the C-terminal of alpha-synuclein was able to reduce memory/learning deficits and promote clearance of cortical and hippocampal alpha-synuclein aggregates in human alpha-synuclein transgenic mice, by reducing the accumulation of calpain I-cleaved and oligomerized alpha-synuclein aggregates [92].
The initial suggestion for a potential role of the serine protease neurosin (kallikrein-6) in alpha-synuclein degradation (Fig. 1f) arose through the finding that neurosin immunoreactivity was present in LBs [93]. Neurosin is preferentially expressed in the brain, especially in neurons and oligodendrocytes [94]. Iwata et al. showed that alpha-synuclein-degrading activity from HEK-293 cells was dose-dependently inhibited by a kallikrein inhibitor, suggesting that alpha-synuclein fragments were generated by a protease of the kallikrein family, such as neurosin [5]. Neurosin was found to be mostly present in the mitochondria and translocated to the cytoplasm upon conditions of cellular stress co-localizing with alpha-synuclein, not only in the healthy mouse brain but also in synucleinopathy models, where it accumulated in the core of LB-like structures [5]. A subsequent in vitro study in a cell-free system estimated that the major cleavage site of neurosin was localized between Lys80 and Thr81, within the NAC region, indicating that this cleavage may preclude alpha-synuclein polymerization [6]. Moreover, phosphorylated (at Ser129) alpha-synuclein was found to be more resistant to degradation by neurosin than nonphosphorylated forms, and the A30P mutant was more resistant to degradation than the wild-type or the A53T and E46K alpha-synuclein mutants [6], supporting the idea that posttranslational modifications or PD-associated mutations might alter its degradation by neurosin. This study indicated that neurosin plays a role in the metabolism of alpha-synuclein, but this is not yet verified in a cell culture system; in fact, the main site of action of neurosin may be extracellular (Fig. 1g). Kallikreins are synthesized as pre-/pro-peptides and are secreted as inactive pro-forms that are subsequently processed extracellularly to their active form via removal of the pro-peptide. Neurosin was also found present as an inactive pro-enzyme in the human cerebrospinal fluid and plasma, suggesting that it can act in the extracellular space [95, 96]. Tatebe et al. analyzed the localization of neurosin and its ability to degrade intracellular and extracellular alpha-synuclein, in HeLa or HEK293T cells transiently transfected with human neurosin and/or alpha-synuclein. The authors found that transiently expressed neurosin was secreted from HeLa or HEK293T cells, activated in the extracellular space, and degraded extracellular alpha-synuclein [9]. Intracellularly, neurosin was localized predominantly in the ER but rarely within the mitochondria or the lysosomes but, in contrast with previous findings [5], was not able to cleave alpha-synuclein within cells [9].
In addition to neurosin, matrix metalloproteinases (MMPs), which also act extracellularly, have been shown to cleave recombinant alpha-synuclein in a selective manner (Fig. 1h), suggesting that this specific cleavage might play a role in PD pathogenesis [7, 8]. MMPs are zinc-dependent endopeptidases that degrade extracellular matrix and nonmatrix proteins. They are up-regulated upon oxidative and nitrative stress that accompanies various neurodegenerative conditions, and this up-regulation is associated with blood–brain barrier breakdown, demyelization, inflammation, and neuronal death [97]. Sung et al. reported that, in SK-N-BE human dopaminergic (DA) neuroblastoma cells, transiently transfected alpha-synuclein was cleaved extracellularly most efficiently by MMP-3 at its C-terminal end with at least four cleavage sites within the NAC region; additionally, MMP-14, MMP-2, MMP-1, and MMP-9 could also digest alpha-synuclein, but to a lesser extent [7]. Compared with the intact form, the aggregation propensity of alpha-synuclein was remarkably facilitated in the presence of the MMP-3-produced proteolytic fragments, and these aggregates were more toxic to the recipient cells [7]. Importantly, MMP-3 levels were also found to be elevated significantly in the rat substantia nigra upon intracerebral injection of 6-hydroxydopamine [7], suggesting that increased levels of MMP-3 could be associated with the pathogenesis of PD through generation of specific aggregation-prone alpha-synuclein fragments resulting from limited proteolysis. Moreover, Levin et al. characterized four additional cleavage sites of MMP-3 related to the NAC domain of alpha-synuclein and reported that limited digestion by MMP-1 and MMP-3, but not by MMP-9, increased the tendency of alpha-synuclein to aggregate [8].
Further evidence for in vitro and in vivo cleavage of alpha-synuclein by MMP-3 was provided by a recent study that demonstrated co-localization of the two proteins in the majority of the LBs in nigral DA neurons from postmortem brains of PD patients, while MMP-3 expression was barely detectable in control subjects [89]. Incubation of alpha-synuclein with the catalytic domain of MMP-3 resulted in generation of several peptides, which were different between the wild-type and the PD-linked A53T mutant form; the A53T alpha-synuclein produced significantly higher quantities of these peptides. Overexpression of these C-terminal truncated peptides via AAVs in the rat substantia nigra resulted in DA loss without LB-like aggregate formation [89], supporting further the hypothesis that MMP-3 cleavage might modulate alpha-synuclein aggregation, LB formation, and neurotoxicity. Previously, it was reported that intracellularly active MMP-3 participated in apoptosis of DA cells in the presence of toxic stimuli [98], while active MMP-3 digested DJ-1, abolishing its antioxidant function and leading to dopaminergic neuronal degeneration [99]. The above observations indicate that MMP-3 activity is not limited in the extracellular space, but that intracellular MMP-3 is active as an endopeptidase and can interact with available substrates inside the cells, making an interaction of active MMP-3 with alpha-synuclein possible, leading to a potential cleavage of the latter.
To add further complexity in the connection between alpha-synuclein and degradation systems, it was reported that alpha-synuclein not only can be processed by metalloproteinases, but conversely, can induce MMPs up-regulation evoking an inflammatory response. More particularly, alpha-synuclein was found to up-regulate MMP-9 and down-regulate tissue plasminogen activator activity in rat primary astrocytes and microglia in an ERK1/2-dependent fashion [100]. Moreover, alpha-synuclein applied extracellularly in rat primary microglia cultures induced the mRNA expression of MMP-1, MMP-3, MMP-8, and MMP-9 and the protein expression of MMP-3 and MMP-9 [101]. These MMPs in turn stimulated microglia by activating protease-activated receptor-1 in an autocrine or paracrine manner, amplifying alpha-synuclein-induced inflammatory responses [101]. These observations add further importance to the potential role of extracellular alpha-synuclein in the DA neurodegeneration and PD pathogenesis.
Impact of Alpha-synuclein in Proteolytic Systems: a Reciprocal Relationship
A common denominator of synucleinopathies and other neurodegenerative conditions is the presence of ubiquitinated inclusions, indicative of failure not only of the UPS, but also of the ALP protein degradation systems, as selective genetic inhibition of macroautophagy in mouse brain leads to accumulation of polyubiquitinated proteins and ubiquitinated aggregates [102–104]. Furthermore, accumulating evidence suggests a more generalized lysosomal dysfunction in various synucleinopathy models, characterized by the presence of increased numbers of autophagic vacuoles, indicative of excessive induction or failure of macroautophagy completion and CMA impairment [105]. In addition, another important contributing factor is aging, which represents the best-established risk factor for PD. Alpha-synuclein protein levels are significantly elevated in aged human substantia nigra [106], while both UPS and ALP activities decline with age [107], implicating a potential dysfunction of these proteolytic systems in the disease process. Whether malfunction of the intracellular degradation pathways is the primary cause of synucleinopathies or occurs secondarily, in part through impairment of these pathways by aberrant alpha-synuclein species, remains to be deciphered.
Alpha-synuclein Impairs UPS Function
Numerous studies link alpha-synuclein to UPS dysfunction, even though it remains dubious whether the proteasome is responsible for the degradation of the protein [59, 67]. In vitro work, either with purified proteins or in cell culture systems, notwithstanding some important differences, has demonstrated that expression of mutant or wild-type alpha-synuclein, in particular in soluble oligomeric and aggregated conformations, inhibits the activity of the 20S or 26S proteasome [108–112]. A proposed mechanism mediating these effects may be a direct binding of alpha-synuclein to the S6′ or the Rpt5 subunit of the 19S proteasome or to the β5 subunit of the 20S proteasome [111, 113, 114]. Tanaka et al. showed that inducible expression of mutant A30P alpha-synuclein in PC12 cells led to a reduction of proteasomal function, as measured by incubation of total cell lysates with synthetic substrates for the individual proteolytic activities of the 20S proteasome [110]. We have shown that stable expression of A53T, but not wild-type alpha-synuclein in PC12 cells led to a 25–35 % inhibition of chymotrypsin-like proteasomal activity, accompanied by accumulation of poorly soluble polyubiquitinated species in punctuate aggregates, further confirming a defect of the UPS at the level of the proteasome [108]. Moreover, Petrucelli et al. reported accumulation of the GFPu reporter of UPS activity in neuroblastoma cell lines overexpressing mutant A30P or A53T, but not wild-type alpha-synuclein [109], whereas Snyder et al. found that even wild-type alpha-synuclein inhibited the enzymatic proteolytic activity of the proteasome when stably overexpressed in neuroblastoma cells [111]. Additionally, Chen et al. found that expression of wild-type or A30P alpha-synuclein in yeast markedly inhibited proteasome-dependent degradation through changes in composition of the proteasome, even though there was only a small effect on the chymotrypsin-like activity in cell lysates [115]. In contrast to these studies, Martin-Clemente et al. failed to detect UPS dysfunction in PC12 cells or even in transgenic mice overexpressing wild-type or mutant alpha-synuclein [116].
In search for a mechanism through which alpha-synuclein affects proteasome function, we have addressed, in a cellular context, the identity of alpha-synuclein species that are implicated in UPS dysfunction [69]. Our results indicated that specific oligomeric alpha-synuclein species of intermediate size are targeted to and impair the 26S proteasome, possibly through a functional interaction [117]. In particular, we have found that stable overexpression of A53T mutant alpha-synuclein in PC12 cells significantly reduced 26S enzymatic proteasomal activity, without altering the levels of proteasome subunits or the assembly of the 26S complex [117]. Moreover, a small amount of alpha-synuclein, migrating between 150 and 450 kDa, and thus distinct from monomeric or recombinant alpha-synuclein, co-eluted specifically in the 26S proteasome-containing fractions. These species of alpha-synuclein were removed upon in vitro incubation with the amyloid-disrupting agent Congo Red or upon cellular application of an inducer of heat shock-related chaperone activity, further confirming their oligomeric, β-sheet-pleated structure. Removal of these species was associated with restoration of proteasomal activity, indicating that these particular species were responsible for proteasomal dysfunction [69]. Importantly, similar species were detected in cortex homogenates of homozygous human A53T alpha-synuclein transgenic mice and were also associated with 26S proteasomal dysfunction [118].
Up to date, the nature of the interaction between alpha-synuclein and the 26S proteasome remains to be deciphered (see Fig. 2). One possibility could be a direct binding of alpha-synuclein oligomers to the active sites of the 20S β-subunits, although this would require translocation of the oligomers into the catalytic cylinder through a narrow open-gated channel (Fig. 2a) [119]. Lindersson et al. have proposed a related model in which this interaction occurs on the outer part of the 20S complex and secondarily affects 20S proteolytic function [113]. Alternatively, alpha-synuclein oligomers may interfere with processes controlled by the 19S complex; this could occur either because of aberrant binding of alpha-synuclein oligomers to specific 19S subunits that leads to alteration of 19S function, or because of steric hindrance induced by bulky alpha-synuclein oligomers, inhibiting further interactions of other substrates with the proteasome (Fig. 2b, c). Such a mechanism has also been proposed in the case of oligomeric prion protein [120] and would be consistent with our own data, as the same oligomeric species that were responsible for proteasomal dysfunction were selectively degraded by the proteasome [81].

Proposed mechanisms through which alpha-synuclein might impair proteasomal function. A Unfolding and translocation of alpha-synuclein into the interior of the 20S cylinder (i) results in alpha-synuclein binding to one or more of the catalytic β-subunits. Such changes could interfere with the active site(s) thus decreasing the overall proteolytic activity of the proteasome. Alternatively, this interaction might occur on the outer part of the 20S complex (ii) and secondarily affect 20S proteolytic function. B Oligomeric alpha-synuclein can directly bind to a subunit of the 19S complex inhibiting substrate recognition or gate opening. C Alpha-synuclein oligomers can transiently interact with the 19S particle possibly by preventing 19S subunits from obtaining the appropriate conformation, therefore “clogging” the 19S cap. In all the above possibilities, the resultant proteasome dysfunction may lead to cytoplasmic accumulation of proteasomal substrates, among which the central role possesses toxic alpha-synuclein species (oligomeric, phosphorylated). Alpha-synuclein oligomers may further impair proteasomal function, generating thus a positive feedback loop. Ubiquitinated, nondegraded proteins are present in Lewy bodies and their accumulation may have deleterious consequences on neuronal viability
Alpha-synuclein Impairs ALP Function
Dysfunctions in macroautophagy and CMA have been linked with intracellular alpha-synuclein accumulation in several studies using both cellular and animal models (Fig. 3) [105, 121]. We have previously shown that stable expression of the A53T mutant alpha-synuclein in PC12 cells induced a marked accumulation of autophagic vacuoles, accompanied by a dramatic decrease of lysosomal acidification in these cells [108]. Actually, we have observed that the ability of lysosomes to degrade long-lived proteins was severely impaired in the A53T-expressing cells [52], which could have led to the dramatic macroautophagy phenotype that was observed [108]. The increase in AVs in cells and transgenic mice overexpressing mutant, or, in some cases, wild-type alpha-synuclein (Fig. 3c) has now been reported in numerous studies [81, 122, 123]. In subsequent work in neuronally differentiated human neuroblastoma cells inducibly expressing alpha-synuclein and in rat cortical neurons adenovirally transduced with alpha-synuclein, we observed consistent CMA dysfunction, AV accumulation, and neuronal toxicity [124]. The observed macroautophagic alterations and toxicity were secondary to CMA impairment, as forms of alpha-synuclein that were not targeted to CMA did not induce AV accumulation and induced death to a lesser extent [124]. There thus appear to be two main mechanisms through which mutant alpha-synuclein can induce AV accumulation (Fig. 3c): through generalized lysosomal dysfunction, in which case macroautophagy would be nonproductive (i.e., not lead to enhanced proteolysis), and specifically through CMA impairment, in which case macroautophagy is productive (i.e., it does lead to enhanced protelolysis). Whether the observed AV accumulation may also have other causes, such as alterations of converging signaling pathways or calcium homeostasis, remains to be deciphered.

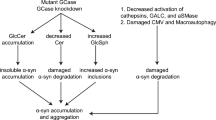
Aberrant effects of alpha-synuclein on lysosomal function. A Mutated and dopamine-modified wild-type alpha-synuclein bind stronger to the LAMP-2a receptor with resistance to translocation, inhibiting their own uptake and that of other CMA-substrates. B The cytosolic aggregation of one such substrate, the survival factor MEF2D, can be detrimental to neuronal health. C Aberrant productive macroautophagic activation, due to disrupted CMA, might result in accumulation of autophagic vacuoles, jeopardizing neuronal survival. D Macroautophagy up-regulation may promote neuronal toxicity through mitochondrial dysfunction, due to enhanced mitophagy. E Moreover, alpha-synuclein-mediated inhibition of the GTPase Rab1a may impair the early formation of the phagophore, causing lysosomal hydrolases to escape to the cytosol and promoting neuronal death. F Additionally, oligomeric alpha-synuclein may disrupt the trafficking of glucocerebrocidase (GCase) to the lysosomal lumen, causing intralysosomal build-up of its substrate, glucosylceramide (GlcCer) that in turn stabilizes additional alpha-synuclein oligomers, generating thus a vicious cycle of cytotoxicity
A previous study from the Cuervo group reported that only wild-type alpha-synuclein monomers and dimers, but not oligomers, are degraded via CMA, while oxidation and nitration of the protein inhibit slightly its degradation by CMA [125]. On the contrary, phosphorylation and dopamine modification of the protein almost completely prevented the CMA-dependent degradation (Fig. 3a); of these, only dopamine modification was able to significantly inhibit the degradation of other CMA substrates [125]. Consistent with the above findings, we have shown that, in dopaminergic neuroblastoma cells, even wild-type alpha-synuclein mediated CMA dysfunction, followed by neuronal death, in a dopamine-dependent fashion [124]. This is especially important, as it implicates CMA dysfunction as a pathogenic PD mechanism also in the sporadic disease, which is much more common than the familial forms. As free dopamine is abundant in neurons of the substantia nigra, this mechanism could serve to explain the selective vulnerability of this region. Moreover, a postmortem study of brain tissue from human PD patients recently revealed significant reductions of LAMP-2a and Hsc70 in nigral dopaminergic neurons, suggestive of overall reduced CMA in the parkinsonian brain [79]. In rats, an age-dependent decrease of LAMP-2a levels has also been identified—in good agreement with the proposed role of age as the primary risk factor in sporadic PD [48, 126]. In total, such studies suggest the notion that CMA dysfunction, either primary or secondary to aberrant alpha-synuclein, may be important in PD pathogenesis.
As mentioned above, in our studies, alpha-synuclein-induced CMA dysfunction was followed by a compensatory up-regulation of macroautophagy, supportive of a potential cross-talk between the two major autophagic pathways [124], as previously reported [45]. In our experiments, up-regulation of macroautophagy was detrimental, as pharmacological or molecular inhibition of macroautophagy was protective [124]. A recent report has confirmed this finding and has demonstrated that mutant alpha-synuclein exerted toxicity in primary neurons through induction of mitochondrial autophagy, providing a potential link between alpha-synuclein accumulation and mitochondrial dysfunction (Fig. 3d) [127]. Apart from excess mitochondrial consumption, aberrant productive macroautophagy may lead to cell death through surplus consumption of other vital cellular constituents, release of lysosomal hydrolases into the cytosol, or enhanced production of damaging molecules within AVs or autophagosomes. These data for a potential deleterious role of macroautophagy induction are in contradiction to several reports in the field suggesting macroautophagy augmentation as the treatment strategy against synucleinopathies or other neurodegenerative conditions [81, 122, 123]. Our view is that in the case of synucleinopathies, macroautophagy induction strategies should be undertaken with caution, as excessive aberrant macroautophagy may form part of the disease process. Ideally, means to promote “healthy” macroautophagy and reduce aberrant macroautophagy could be developed.
Another interesting possibility was raised by the Rubinsztein group, who showed that wild-type alpha-synuclein could inhibit an early point in autophagosome formation, through an interaction with Rab1a (Fig. 3e) [128]. These apparent conflicting results could be attributed to timing (i.e., alpha-synuclein may inhibit somewhat macroautophagy early on, but later, when cells are already compromised, and CMA is impaired, aberrant macroautophagy occurs) or to differences in the particular models used.
Apart from indirect aberrant induction of macroautophagy, CMA impairment induced by alpha-synuclein may also lead to neuronal dysfunction and death through direct disruption of CMA-dependent proteolysis (Fig. 3a, b). Consistent with this idea, wild-type and mutant A53T alpha-synuclein interfered with the CMA-dependent degradation of the neuronal survival factor MEF2D (Fig. 3b), leading to its enhanced cytosolic localization and reduction of its protective nuclear form; interestingly, a similar picture emerged in PD brains and in A53T transgenic mice [129].
Other lines of evidence have also implicated the Gaucher disease (GD)-associated lysosomal enzyme glucocerebrosidase (GCase) in the pathogenesis of PD and related synucleinopathies. Following the discovery that mutations in GBA1—the GCase encoding gene—constitute a significant genetic risk factor for PD [130], several publications have investigated the potential mechanisms surrounding this relationship. Pharmacological inhibition of GCase was reported to cause increased levels of intracellular alpha-synuclein in neuroblastoma cells and mouse nigra [131]. This suggested that loss of GCase function led to lysosomal dysfunction and to secondary build-up of alpha synuclein (Fig. 3f). This concept was further elaborated in a seminal study where, in various cellular models, including human iPS neurons derived from GD fibroblasts, down-regulation of GCase activity compromised lysosomal degradation and evoked alpha-synuclein accumulation/aggregation followed by neuronal toxicity [132]. In the same study, it was shown that GCase dysfunction led to accumulation of glucosylceramide, which enhanced further oligomeric alpha-synuclein intermediates and reinforced the pathogenic process [132]. Importantly, alpha-synuclein overexpression inhibited the intracellular trafficking of wild-type GCase, leading to its decreased lysosomal activity, and thus into a feed-forward pathogenic amplification loop of alpha-synuclein accumulation and GCase dysfunction [132]. Furthermore, restoration of wild-type GCase expression via adeno-associated viruses in a mouse GD model ameliorated both alpha-synuclein/ubiquitin accumulation in hippocampal neurons, as well as related histopathological and behavioral aberrations [133]. In further support of an association between alpha-synuclein and GCase in a lysosomal context, both wild-type and the GD-related N370S mutant GCase were found to interact physically and selectively with alpha-synuclein in vitro, in human tissue and in neuronal cultures under lysosomal solution conditions, with the mutant GCase displaying reduced affinity for alpha-synuclein compared to the wild-type enzyme [134].
Arguing against the notion that loss of GCase activity provides the pathogenic link, Cullen et al. found that overexpression of various mutant GBA forms promoted marginally, but significantly, human alpha-synuclein accumulation in cellular and in vivo conditions, in a dose- and time-dependent manner, without altering GCase activity [135]. The results of this study are therefore more consistent with a gain of aberrant function conferred by mutant GCase.
In conclusion, though the causative agent remains unknown, evidence exists for a relationship between lysosomal failure and alpha-synuclein pathology. As one seems to reinforce the other, it seems that an equilibrium exists in the disease-free brain that needs to be reinstated in the parkinsonian brain to counteract the downward spiral.
Conclusions
A major factor in idiopathic PD and related synucleinopathies pathogenesis is the accumulation of alpha-synuclein, either because of enhanced transcription, reduced degradation, or specific posttranslational alterations that favor select toxic species. The pathways of alpha-synuclein degradation remain controversial. We conjecture that the decision in alpha-synuclein clearance is likely dependent upon the overall protein burden, the assembly state of the protein, the homeostatic environment of the particular cell type, and the stage of the pathogenic process. Alpha-synuclein and intracellular proteolytic systems interact with each other in a self-feeding cascade leading to neurodegeneration. Future therapeutic strategies targeting specific degradation pathways can be applied to counteract alpha-synuclein pathology. Such strategies have the advantage of “killing two birds with one stone,” in a way that they would theoretically not only enhance excess alpha-synuclein clearance, but also ameliorate its aberrant effects on protein degradation pathways, which have been observed in various synucleinopathy models.
References
Goldberg AL (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426:895–899
Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721
Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, Oxford JT, Feany MB, Masliah E, Rohn TT (2007) Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol 170:1725–1738
Mishizen-Eberz AJ, Guttmann RP, Giasson BI, Day GA 3rd, Hodara R, Ischiropoulos H, Lee VM, Trojanowski JQ, Lynch DR (2003) Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro. J Neurochem 86:836–847
Iwata A, Maruyama M, Akagi T, Hashikawa T, Kanazawa I, Tsuji S, Nukina N (2003) Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum Mol Genet 12:2625–2635
Kasai T, Tokuda T, Yamaguchi N, Watanabe Y, Kametani F, Nakagawa M, Mizuno T (2008) Cleavage of normal and pathological forms of alpha-synuclein by neurosin in vitro. Neurosci Lett 436:52–56
Sung JY, Park SM, Lee CH, Um JW, Lee HJ, Kim J, Oh YJ, Lee ST, Paik SR, Chung KC (2005) Proteolytic cleavage of extracellular secreted {alpha}-synuclein via matrix metalloproteinases. J Biol Chem 280:25216–25224
Levin J, Giese A, Boetzel K, Israel L, Hogen T, Nubling G, Kretzschmar H, Lorenzl S (2009) Increased alpha-synuclein aggregation following limited cleavage by certain matrix metalloproteinases. Exp Neurol 215:201–208
Tatebe H, Watanabe Y, Kasai T, Mizuno T, Nakagawa M, Tanaka M, Tokuda T (2010) Extracellular neurosin degrades alpha-synuclein in cultured cells. Neurosci Res 67:341–346
Glickman MH, Ciechanover A (2002) The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82:373–428
Layfield R, Tooth D, Landon M, Dawson S, Mayer J, Alban A (2001) Purification of poly-ubiquitinated proteins by S5a-affinity chromatography. Proteomics 1:773–777
Gallastegui N, Groll M (2010) The 26S proteasome: assembly and function of a destructive machine. Trends Biochem Sci 35:634–642
Ardley HC, Robinson PA (2005) E3 ubiquitin ligases. Essays Biochem 41:15–30
Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278:43628–43635
Crews CM (2003) Feeding the machine: mechanisms of proteasome-catalyzed degradation of ubiquitinated proteins. Curr Opin Chem Biol 7:534–539
Benaroudj N, Zwickl P, Seemuller E, Baumeister W, Goldberg AL (2003) ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol Cell 11:69–78
Demartino GN, Gillette TG (2007) Proteasomes: machines for all reasons. Cell 129:659–662
Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463–477
Muller O, Sattler T, Flotenmeyer M, Schwarz H, Plattner H, Mayer A (2000) Autophagic tubes: vacuolar invaginations involved in lateral membrane sorting and inverse vesicle budding. J Cell Biol 151:519–528
Hollenbeck PJ (1993) Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol 121:305–315
Mehrpour M, Esclatine A, Beau I, Codogno P (2010) Overview of macroautophagy regulation in mammalian cells. Cell Res 20:748–762
Xilouri M, Stefanis L (2010) Autophagy in the central nervous system: implications for neurodegenerative disorders. CNS Neurol Disord Drug Targets 9:701–719
Ohsumi Y (2001) Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2:211–216
Klionsky DJ (2005) The molecular machinery of autophagy: unanswered questions. J Cell Sci 118:7–18
Jung CH, Ro SH, Cao J, Otto NM, Kim DH (2010) mTOR regulation of autophagy. FEBS Lett 584:1287–1295
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90:1383–1435
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93
Ichimura Y, Kominami E, Tanaka K, Komatsu M (2008) Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 4:1063–1066
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115:727–738
Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F (2009) The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 10:1215–1221
Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dotsch V, Ney PA, Dikic I (2009) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11:45–51
Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ (2009) Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 17:98–109
Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 33:505–516
Chiang HL, Terlecky SR, Plant CP, Dice JF (1989) A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246:382–385
Cuervo AM, Dice JF, Knecht E (1997) A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem 272:5606–5615
Cuervo AM, Dice JF (1996) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273:501–503
Agarraberes FA, Dice JF (2001) A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci 114:2491–2499
Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM (2008) The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 28:5747–5763
Hatem CL, Gough NR, Fambrough DM (1995) Multiple mRNAs encode the avian lysosomal membrane protein LAMP-2, resulting in alternative transmembrane and cytoplasmic domains. J Cell Sci 108(Pt 5):2093–2100
Cuervo AM, Dice JF (2000) Regulation of lamp2a levels in the lysosomal membrane. Traffic 1:570–583
Cuervo AM, Knecht E, Terlecky SR, Dice JF (1995) Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol 269:C1200–C1208
Agarraberes FA, Terlecky SR, Dice JF (1997) An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol 137:825–834
Martinez-Vicente M, Cuervo AM (2007) Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol 6:352–361
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L (2008) Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 283:23542–23556
Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM (2006) Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 103:5805–5810
Wang Y., Singh R., Massey A.C., Kane S.S., Kaushik S., Grant T., Xiang Y., Cuervo A.M., and Czaja M.J. (2007) Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem
Kaushik S, Massey AC, Mizushima N, Cuervo AM (2008) Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 19:2179–2192
Cuervo AM, Dice JF (2000) Age-related decline in chaperone-mediated autophagy. J Biol Chem 275:31505–31513
Cuervo AM, Dice JF (2000) Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci 113(Pt 24):4441–4450
Zhang C, Cuervo AM (2008) Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 14:959–965
Liu CW, Corboy MJ, DeMartino GN, Thomas PJ (2003) Endoproteolytic activity of the proteasome. Science 299:408–411
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295
Leroy E, Anastasopoulos D, Konitsiotis S, Lavedan C, Polymeropoulos MH (1998) Deletions in the Parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson's disease. Hum Genet 103:424–427
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH (1998) The ubiquitin pathway in Parkinson's disease. Nature 395:451–452
Maraganore DM, Lesnick TG, Elbaz A, Chartier-Harlin MC, Gasser T, Kruger R, Hattori N, Mellick GD, Quattrone A, Satoh J, Toda T, Wang J, Ioannidis JP, de Andrade M, Rocca WA (2004) UCHL1 is a Parkinson's disease susceptibility gene. Ann Neurol 55:512–521
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ (2001) Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science 293:263–269
Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT Jr (2002) The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell 111:209–218
Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM (1999) Degradation of alpha-synuclein by proteasome. J Biol Chem 274:33855–33858
Imai Y, Soda M, Takahashi R (2000) Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem 275:35661–35664
McLean PJ, Kawamata H, Hyman BT (2001) Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 104:901–912
Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC (2003) Alpha-synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278:25009–25013
Tofaris GK, Layfield R, Spillantini MG (2001) Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 509:22–26
Nakajima T, Takauchi S, Ohara K, Kokai M, Nishii R, Maeda S, Takanaga A, Tanaka T, Takeda M, Seki M, Morita Y (2005) Alpha-synuclein-positive structures induced in leupeptin-infused rats. Brain Res 1040:73–80
Machiya Y, Hara S, Arawaka S, Fukushima S, Sato H, Sakamoto M, Koyama S, Kato T (2010) Phosphorylated alpha-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J Biol Chem 285:40732–40744
Ancolio K, Alves da Costa C, Ueda K, Checler F (2000) Alpha-synuclein and the Parkinson's disease-related mutant Ala53Thr-alpha-synuclein do not undergo proteasomal degradation in HEK293 and neuronal cells. Neurosci Lett 285:79–82
Rideout HJ, Larsen KE, Sulzer D, Stefanis L (2001) Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem 78:899–908
Rideout HJ, Stefanis L (2002) Proteasomal inhibition-induced inclusion formation and death in cortical neurons require transcription and ubiquitination. Mol Cell Neurosci 21:223–238
Emmanouilidou E, Stefanis L, Vekrellis K (2010) Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging 31:953–968
Lee HJ, Khoshaghideh F, Patel S, Lee SJ (2004) Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 24:1888–1896
Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ (2005) The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem 280:23727–23734
Liani E, Eyal A, Avraham E, Shemer R, Szargel R, Berg D, Bornemann A, Riess O, Ross CA, Rott R, Engelender S (2004) Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson's disease. Proc Natl Acad Sci U S A 101:5500–5505
Lee JT, Wheeler TC, Li L, Chin LS (2008) Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum Mol Genet 17:906–917
Rott R, Szargel R, Haskin J, Bandopadhyay R, Lees AJ, Shani V, Engelender S (2011) alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc Natl Acad Sci U S A 108:18666–18671
Tofaris GK, Kim HT, Hourez R, Jung JW, Kim KP, Goldberg AL (2011) Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal–lysosomal pathway. Proc Natl Acad Sci U S A 108:17004–17009
Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, Gray T, Topham I, Fone K, Rezvani N, Mee M, Soane T, Layfield R, Sheppard PW, Ebendal T, Usoskin D, Lowe J, Mayer RJ (2008) Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci 28:8189–8198
Rideout HJ, Dietrich P, Wang Q, Dauer WT, Stefanis L (2004) Alpha-synuclein is required for the fibrillar nature of ubiquitinated inclusions induced by proteasomal inhibition in primary neurons. J Biol Chem 279:46915–46920
Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ, Unni VK (2011) Distinct roles in vivo for the ubiquitin–proteasome system and the autophagy–lysosomal pathway in the degradation of alpha-synuclein. J Neurosci 31:14508–14520
Alvarez-Erviti L., Rodriguez-Oroz M.C., Cooper J.M., Caballero C., Ferrer I., Obeso J.A., and Schapira A.H. (2010) Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol
Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, Ischiropoulos H (2001) Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci 21:8053–8061
Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E (2009) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci 29:13578–13588
Dice JF, Chiang HL, Spencer EP, Backer JM (1986) Regulation of catabolism of microinjected ribonuclease A. Identification of residues 7–11 as the essential pentapeptide. J Biol Chem 261:6853–6859
Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA (2010) Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem 285:13621–13629
Sevlever D, Jiang P, Yen SH (2008) Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47:9678–9687
Qiao L., Hamamichi S., Caldwell K.A., Caldwell G.A., Yacoubian T.A., Wilson S., Xie Z.L., Speake L.D., Parks R., Crabtree D., Liang Q., Crimmins S., Schneider L., Uchiyama Y., Iwatsubo T., Zhou Y., Peng L., Lu Y., Standaert D.G., Walls K.C., Shacka J.J., Roth K.A., and Zhang J. (2008) Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain 1: p. 17
Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, Boston H, Saftig P, Woulfe J, Feany MB, Myllykangas L, Schlossmacher MG, Tyynela J (2009) Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain 2:5
Bartels T, Choi JG, Selkoe DJ (2011) alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477:107–110
Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ (2011) A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A 108:17797–17802
Choi DH, Kim YJ, Kim YG, Joh TH, Beal MF, Kim YS (2011) Role of matrix metalloproteinase 3-mediated alpha-synuclein cleavage in dopaminergic cell death. J Biol Chem 286:14168–14177
Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T (1998) Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol 152:879–884
Liu CW, Giasson BI, Lewis KA, Lee VM, Demartino GN, Thomas PJ (2005) A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J Biol Chem 280:22670–22678
Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, Mueller-Steiner S, Seubert P, Barbour R, McConlogue L, Buttini M, Games D, Schenk D (2011) Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 6:e19338
Ogawa K, Yamada T, Tsujioka Y, Taguchi J, Takahashi M, Tsuboi Y, Fujino Y, Nakajima M, Yamamoto T, Akatsu H, Mitsui S, Yamaguchi N (2000) Localization of a novel type trypsin-like serine protease, neurosin, in brain tissues of Alzheimer's disease and Parkinson's disease. Psychiatry Clin Neurosci 54:419–426
Petraki CD, Karavana VN, Skoufogiannis PT, Little SP, Howarth DJ, Yousef GM, Diamandis EP (2001) The spectrum of human kallikrein 6 (zyme/protease M/neurosin) expression in human tissues as assessed by immunohistochemistry. J Histochem Cytochem 49:1431–1441
Okui A, Kominami K, Uemura H, Mitsui S, Yamaguchi N (2001) Characterization of a brain-related serine protease, neurosin (human kaillikrein 6), in human cerebrospinal fluid. Neuroreport 12:1345–1350
Diamandis EP, Yousef GM, Soosaipillai AR, Grass L, Porter A, Little S, Sotiropoulou G (2000) Immunofluorometric assay of human kallikrein 6 (zyme/protease M/neurosin) and preliminary clinical applications. Clin Biochem 33:369–375
Yong VW (2005) Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci 6:931–944
Choi DH, Kim EM, Son HJ, Joh TH, Kim YS, Kim D, Flint Beal M, Hwang O (2008) A novel intracellular role of matrix metalloproteinase-3 during apoptosis of dopaminergic cells. J Neurochem 106:405–415
Choi DH, Hwang O, Lee KH, Lee J, Beal MF, Kim YS (2010) DJ-1 cleavage by matrix metalloproteinase 3 mediates oxidative stress-induced dopaminergic cell death. Antioxid Redox Signal 14:2137–2150
Joo SH, Kwon KJ, Kim JW, Hasan MR, Lee HJ, Han SH, Shin CY (2009) Regulation of matrix metalloproteinase-9 and tissue plasminogen activator activity by alpha-synuclein in rat primary glial cells. Neurosci Lett 469:352–356
Lee EJ, Woo MS, Moon PG, Baek MC, Choi IY, Kim WK, Junn E, Kim HS (2010) Alpha-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J Immunol 185:615–623
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131:1149–1163
Xilouri M, Stefanis L (2011) Autophagic pathways in Parkinson disease and related disorders. Expert Rev Mol Med 13:e8
Chu Y, Kordower JH (2007) Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: is this the target for Parkinson's disease? Neurobiol Dis 25:134–149
Ward WF (2002) Protein degradation in the aging organism. Prog Mol Subcell Biol 29:35–42
Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA (2001) Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 21:9549–9560
Petrucelli L, O'Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR (2002) Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36:1007–1019
Tanaka Y., Engelender S., Igarashi S., Rao R.K., Wanner T., Tanzi R.E., Sawa A., V L.D., Dawson T.M., and Ross C.A. (2001) Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet 10:919–926
Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B (2003) Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem 278:11753–11759
Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA (2005) Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet 14:3801–3811
Lindersson E, Beedholm R, Hojrup P, Moos T, Gai W, Hendil KB, Jensen PH (2004) Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem 279:12924–12934
Ghee M, Fournier A, Mallet J (2000) Rat alpha-synuclein interacts with Tat binding protein 1, a component of the 26S proteasomal complex. J Neurochem 75:2221–2224
Chen Q, Thorpe J, Keller JN (2005) Alpha-synuclein alters proteasome function, protein synthesis, and stationary phase viability. J Biol Chem 280:30009–30017
Martin-Clemente B, Alvarez-Castelao B, Mayo I, Sierra AB, Diaz V, Milan M, Farinas I, Gomez-Isla T, Ferrer I, Castano JG (2004) Alpha-synuclein expression levels do not significantly affect proteasome function and expression in mice and stably transfected PC12 cell lines. J Biol Chem 279:52984–52990
Emmanouilidou E., Stefanis L., and Vekrellis K. (2010) Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging
Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM (2002) Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34:521–533
Pickart CM, Cohen RE (2004) Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol 5:177–187
Kristiansen M, Deriziotis P, Dimcheff DE, Jackson GS, Ovaa H, Naumann H, Clarke AR, van Leeuwen FW, Menendez-Benito V, Dantuma NP, Portis JL, Collinge J, Tabrizi SJ (2007) Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell 26:175–188
Cuervo AM, Wong ES, Martinez-Vicente M (2010) Protein degradation, aggregation, and misfolding. Mov Disord 25(Suppl 1):S49–S54
Yu WH, Dorado B, Figueroa HY, Wang L, Planel E, Cookson MR, Clark LN, Duff KE (2009) Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol 175:736–747
Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E (2010) Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 5:e9313
Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L (2009) Aberrant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One 4:e5515
Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM (2008) Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118:777–788
Collier TJ, Kanaan NM, Kordower JH (2011) Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non-human primates. Nat Rev Neurosci 12:359–366
Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A (2011) Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 286:10814–10824
Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC (2010) alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol 190:1023–1037
Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, Mao Z (2009) Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 323:124–127
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 361:1651–1661
Manning-Bog AB, Schule B, Langston JW (2009) Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology 30:1127–1132
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146(1):37–52
Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, Passini MA, Grabowski GA, Schlossmacher MG, Sidman RL, Cheng SH, Shihabuddin LS (2011) CNS expression of glucocerebrosidase corrects {alpha}-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A 108:12101–12106
Yap TL, Gruschus JM, Velayati A, Westbroek W, Goldin E, Moaven N, Sidransky E, Lee JC (2011) {alpha}-Synuclein interacts with glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J Biol Chem 286:28080–28088
Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, Kolodziej P, Kahn I, Saftig P, Woulfe J, Rochet JC, Glicksman MA, Cheng SH, Grabowski GA, Shihabuddin LS, Schlossmacher MG (2011) Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol 69:940–953
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xilouri, M., Brekk, O.R. & Stefanis, L. Alpha-synuclein and Protein Degradation Systems: a Reciprocal Relationship. Mol Neurobiol 47, 537–551 (2013). https://doi.org/10.1007/s12035-012-8341-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-012-8341-2