
Abstract
The mineralocorticoid receptor (MR) was the last member of the nuclear receptor superfamily to evolve. It is responsible for the maintenance of the water and salt homeostasis. Like most ligand-activated transcription factors of this superfamily, it is activated by ligand binding. The MR exists as a large heterocomplex assembled with the heat-shock protein of 90-kDa chaperone, Hsp90, and other associated chaperones and cochaperones. The composition of this heterocomplex is affected by the nature of the bound steroid. MR biological responses are also affected by the redox status of the cell or due to protein phosphorylation. In this chapter, the conformational requirements of the steroid to become an optimal MR ligand, the role of the Hsp90-based heterocomplex, and the influence of MR modifications by oxidation and phosphorylation is discussed. These properties are analyzed in the light of the relevance of this nuclear receptor as a key pharmacological target for disorders mostly related to the hydroelectrolytic homeostasis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Mineralocorticoid receptor
- Aldosterone
- Steroid conformation
- Heat-shock protein of 90-kDa
- Immunophilins
- Dynein
- Phosphorylation
- Glutathion
1.1 An Overview of the MR Physiology
The conserved steroid receptor subfamily is comprised within the nuclear receptor superfamily. They are counted among the first members of the nuclear receptor superfamily to be cloned and structurally characterized [1]. The last two steroid receptors that emerged during evolution are the close-related partners GR [2] and MR [3]. The high homology between these two receptors led to confirm the close kinship that was hypothesized previously due to the cross-talk of their biochemical and pharmacological properties. This is particularly remarkable since the GR is a receptor that can replace MR in some functions. Simply to begin with, the main glucocorticoid ligand, cortisol, shows the same affinity for MR as the natural mineralocorticoid ligand, aldosterone [4]. In view of the higher plasma levels of circulating cortisol (2–3 orders of magnitude higher than those of aldosterone), there is a problem for the specificity of the biological response since a priori, the MR should be permanently activated by cortisol. In other words, the typical response to aldosterone (sodium- and water-retention and potassium and proton elimination) can be triggered by cortisol.
In most epithelial tissues and exceptionally in a few non-epithelial tissues such as vessel walls and nucleus tractus solitarius of the medulla oblongata, the MR is protected from activation by cortisol due to the action of the microsomal enzyme 11βHSD2 (11β-hydroxysteroid dehydrogenase type-2), which is co-expressed in the same cells where MR is expressed, and converts cortisol into the receptor-inactive oxidized metabolite cortisone [5]. When the enzyme expression is deficient or blocked by drugs or natural products such as liquorice, this protective mechanism fails, and cortisol is available to bind and activate MR. Consequently, a pseudohyper-aldosteronism syndrome is developed, i.e., a paradoxical syndrome of hyperaldosteronism showing hypertension and high levels of sodium retention but also normal or low plasma levels of aldosterone (see [6] for a comprehensive review).
In most non-epithelial cells, remarkably in the brain, the MR is not protected by that enzymatic activity since there is no 11βHSD2 co-expression in these cells. Inversely, in the nervous system, there is a considerable expression of MR coexisting with high levels of GR in the same cell types. Remarkably, the intranuclear distribution of MR and GR in the same hippocampal neurons shows a distinctive individual distribution, i.e., specific speckles that exclusively contain MR or GR, but not colocalization of both of them [7]. This clearly indicates that there are specific nuclear sites capable to specifically recognize each receptor. Does it mean that the biological response is redundant for both receptors? It does not seem to be the case since, for example, salt-intake is still aldosterone-regulated, and it is not a cortisol-dependent phenomenon [8, 9]. MR activation has also been related to behavioral responses, including memory-related events and affection feelings [10]. Interestingly, the MR is also expressed in the granulosa cells of the ovary, one of its proven specific roles being the regulation of progesterone synthesis [11], a steroid with MR antagonistic action. The reasons for the exclusion of alternative ligands and the specificity for aldosterone action in these tissues are still subject of intensive investigation and speculations.
The most important and relevant pathology associated with the biology of MR is the hypertension syndrome that results from high plasma levels of aldosterone and consequently hypernatremia and water retention. Therefore, patients with primary hyperaldosteronism have higher cardiovascular risk profiles [12] and greater evidence of cardiovascular damage [13]. Furthermore, the MR has also been associated with other pathological situations such as inflammation processes, organ fibrosis, oxidative stress, adipocyte metabolism, and aging [14]. It has been documented that MR activation induces the proliferation of smooth muscle cells in pulmonary arteries, which is a contributing factor for the development of pulmonary arterial hypertension [15]. Retina is also a target of mineralocorticoid action, such that the use of MR antagonists has shown beneficial effects in retinal diseases [16].
1.2 Evolutionary Profile of the MR
It is accepted that the first life forms originated in the sea. Because of this origin, it is thought that the circulating fluids of today living beings resemble the composition of the sea water of some millions of years ago, when life began. However, the composition of the primitive Archean Ocean has been gradually changing since that time. This was the consequence of the permanent precipitation of salts on the seabed and the washing-down of compounds from the land that were deposited in the sea by the erosive action of rivers. Today animals are unquestionably consequence of a slow but constant evolutionary adaptation to that new environment during this long period of time. Despite the biological divergence, the composition of their blood is remarkably alike in ionic composition. This fact suggests that the life conditions should be highly restricted, and it is likely that they remained relatively constant during this evolutionary process. Thus, animal life has been regulated by mechanisms whose main purpose was the maintenance of an inner optimal environment or the continued life of its constitutive cells. The Dobzhansky’s aphorism “nothing in biology makes sense except in the light of evolution” [17] is quite appropriate for the case of the MR.
It is reasonable to postulate that when the first life forms abandoned the salty waters of the sea, they must solve an additional difficulty for keeping the osmotic pressure of their blood above that of the surrounding fresh water. Furthermore, when animals moved on to the land from the waters, far-reaching adjustments of their regulatory mechanisms became a mandatory condition simply because the limits of tolerance were even narrower as a consequence of the influence of previously inexistent variables, for example, evaporation and perspiration. This is the point where both the ligand aldosterone and the receptor MR emerged simultaneously during the evolutionary process, i.e., when amphibians jumped from the waters to land. Interestingly, most fish lack both aldosterone and the enzyme responsible for its synthesis [18]. Actually, the main corticosteroid produced by the fish interrenal tissue is cortisol, which is the steroid that manages not only the metabolism but also the regulation of salt and water balance in these animals.
The biosynthetic pathway of aldosterone provides some insights into the evolution the MR ligands. Aldosterone is at the end of the pathway, i.e., it is the youngest ligand. As a matter of fact, the late developing of the MR along with CYP11B2 (aldosterone synthase) offers a clear example of a co-evolutionary process to preserve the intracellular milieu from environmental changes. Also, the six related steroid receptors expressed in vertebrates—GR, MR, estrogen receptors (ER) α and β, progesterone receptor (PR), and androgen receptor (AR)—evolved thanks to a series of gene duplications from a common ancestral receptor gene [19]. It is regarded that the first steroid receptor was ER, followed by PR. More recently, AR and corticosteroid receptors appeared. There is a common ancestor of the two youngest members of the steroid receptor subfamily, GR and MR—the CR [20]. Like the GR and MR, CR is promiscuous in the sense that it is activated by both mineralocorticoids and glucocorticoids. Descendants of this ancestral receptor are still found in jawless fish, lampreys, and hagfish (along with the expression of ER and PR), which evolved about 530 million years ago and are located at the base of the vertebrate line. These species do not produce neither cortisol nor aldosterone, but 11-deoxycortisol and 11-deoxycorticosterone, which represent their respective biosynthetic precursors and are present at physiologically relevant levels [21]. Recently, it was suggested [22] that 11-deoxycortisol is the main steroid that controls the hydromineral balance in sea lamprey, an organism that represents the most basal osmoregulating vertebrate. GR and MR are derived from that CR in cartilaginous fishes about 450 million years ago, and the consensus is that 11-deoxycorticosterone, corticosterone, and cortisol were all of the ligands for MR before the CYP11B2 enzyme required to make aldosterone from DOC evolved, following the divergence of those two receptors [23, 24].
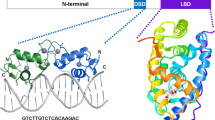
MR is the largest protein among all human steroid receptors and the last to evolve. Like the other members of the nuclear receptor superfamily, it shows three major functional domains (Fig.1.1a): The N-terminal domain (∼603 amino acids), which has the most variable comparative sequence compared to the other steroid receptors (≤15%). It is classically known as the transactivation domain (TD). The central DNA-binding domain (DBD) shows the highest homology with other members of the subfamily, especially with GR (∼94% identity across the 66 amino acid domain). It contains two Zn-finger protrusions responsible for the recognition of the DNA promoter sequence of the target genes. The C-terminal domain (∼253 amino acids) comprises the ligand-binding domain (LBD) where the steroid binds. Between the LBD and the DBD, there is a “hinge” domain (HD), a region of ∼62 amino acids that it is thought to play a role in receptor dimerization.

(a) Structural domains of human MR. Black dots show potential phosphorylation sites based on the consensus sequence. (Modified from Ref. [92]). These sites correspond to serines in position 8, 129, 183, 250, 255, 259, 262, 274, 283, 299, 311, 361, 424, 543, 703, and 843. (b) Mature heterocomplex of MR with the Hsp90-based chaperone machinery. The black crescent of the immunophilin (IMM) represents its TPR domain, and the bay in the Hsp90 dimer represents the TPR-acceptor site
There are two possible evolutionary reasons that may lead to that particular intramolecular organization of these receptors. Perhaps different domains showing different origins such that those related to the regulation of metabolism became fused to a DNA-binding motif to generate a novel transcription factor. Alternatively, a multi-domain precursor that at first may mediate a simple signal transduction pathway could have acquired increasingly complex functions during the evolution. Based on the analysis of protein sequences and the evolutionary trees, the second model is more likely and the one accepted by general consensus. However, regardless of how the organization of nuclear receptors had taken place during evolution, these proteins only stand for a part of the tale; the other significant part corresponds to the steroid. Therefore, the information encrypted in the hormonal response is dictated neither by the steroid nor the receptor exclusively, but it is complementary written in both modules of a multifaceted operational unit. In turn, this functional unit is subject of other kinds of non-hormonal- and non-receptor-dependent regulations such as receptor modifications by post-transcriptional modifications stimulated by ligand binding, association with other proteins that may conduct to trans-repression mechanisms, or the competitive action of metabolizing enzymes that sequester active ligands from the medium making them unavailable for the receptor.
1.3 The Hsp90-Based Heterocomplex
Steroid receptors exist as oligomeric structures with the Hsp90-based chaperone heterocomplex (Fig.1.1b). The assembly of the oligomeric structure has been well characterized for GR [25], PR [26], and MR [27] and appears to be quite representative for the assembly of most Hsp90-client proteins associated to the same oligomeric complex. The chaperone Hsp90 always function as a dimer, such that the stoichiometry of the mature receptor•(Hsp90)2 complex shows one molecule of Hsp70, one molecule of p23, and a TPR (tetratricopeptide repeat)-domain cochaperone bound to the TPR accept or site of the Hsp90 dimer [28,29,30,31]. The final heterocomplex depicted in Fig. 1.1b must pass through a maturation cycle in the cell cytosol [32].
Due to the high hydrophobicity the steroid binding cleft of the LBD of MR, this domain is collapsed and consequently unable to bind aldosterone, unless the Hsp90-based heterocomplex is bound to the receptor. When this happens, the steroid binding cleft of the LBD becomes thermodynamically more stable and steroid binding does occur. It is accepted that the minimal composition for the assembly of MR that permits aldosterone binding is the cytosolic complex named “foldosome,” which already exists folded in the cytoplasm. It includes (Hsp90)2•Hop•Hsp70/Hsp40•p23. Nonetheless, a step-by-step mechanism primed by binding of Hsp70 to the receptor followed by Hop and Hsp90 binding is also viable [25, 33]. Note that Hsp70 is associated with Hsp40, which is required in sub-stoichiometric quantities to enhance the intrinsic ATPase activity of Hsp70. The foldosome is transferred to the receptor in an ATP-, K+-, and Mg2+-dependent manner, and the resultant complex is now able to bind steroid.
Although the MR is biologically inactive in the sense that it does not bind hormone, it should be pointed out that the Hsp90-based chaperone system binds to a structure that shows a stable tertiary structure rather than to a denatured protein. The TPR-domain protein Hop (formerly called p60) is important because it brings together Hsp90 and Hsp70, two chaperones that are essential for the complex, but they are incapable to associate by themselves spontaneously. It occurs that Hsp90 dimers are in a dynamic equilibrium between an open (ADP-bound) and closed (ATP-bound) conformation [34]. Hop first stabilizes the open (V-shaped) conformation of the dimer and consequently prevents the intrinsic ATPase activity of Hsp90. Then, the small acidic cochaperone of p23 is recruited to the Hsp90 dimer. This step is critical for two reasons: first because p23 stabilizes the MR•Hsp90 association and, second, because p23 binding favors the release of Hop from the TPR-acceptor site of Hsp90 since the dimer closes its open conformation. This weakens Hop binding [35, 36]. In other words, even though Hop is required for priming the folding of the heterocomplex, it is not present in the final, mature form of the oligomer. Nevertheless, some Hop can always be recovered co-immunoprecipitated with MR, but it merely represents the intermediate complexes.
When Hop is released, the TPR-acceptor site of Hsp90 dimers is empty and can be occupied by other TPR-domain co-chaperone such as a TPR-domain immunophilin. Because there is only one acceptor site per dimer [29, 37], these TPR proteins compete one another for binding to Hsp90 in a mutually exclusive manner [38,39,40]. The most frequent members of the immunophilin family that can interact with Hsp90 in steroid receptor complexes are FKBP51, FKBP52, CyP40, and PP5 [41,42,43]. They are also found associated with cytoskeleton shaping the phenotype of the cell [44,45,46]. In the cases of MR and GR, the presence of CyP40 in the final mature heterocomplex is unusual in biological samples. CyP40 is more frequently found associated with PR and ER [47].
Binding of the Hsp90-based chaperone complex to the MR is mediated by the C-terminal of the hinge region [48]. The dissociation of Hsp90 or its functional disruption by drugs leads to the polyubiquitylation and consequent proteasomal degradation of MR via the ubiquitin-protein ligase CHIP (C-terminus of Hsp70-Interacting Protein) [49]. This E3 ligase is also shared with the GR [50].
1.4 MR Trafficking
In the absence of ligand, the MR is primarily cytoplasmic, and rapidly translocates into the nucleus upon steroid binding [51,52,53]. For decades, it has been heuristically accepted that Hsp90 anchors MR to cytoplasmic structures, such that its release from the complex was thought to be a requirement to permit the nuclear localization of the receptor. However, it has been proved that the Hsp90•FKBP52 complex is necessary for the active retrotransport of cytoplasmic receptors on cytoskeletal tracks, the motor protein dynein powering this transport (see Fig. 1.2). This model was first demonstrated for the GR [54, 55] and then for the MR [38, 40, 56]. A similar model was also reported for the transcription factor NF-κB, but in this complex Hsp90 in not an interactor and the binding of the immunophilin occurs directly to the p65/RelA [57].

Transportosome model. In the absence of steroid, MR forms cytoplasmic complexes with Hsp90, Hsp70, p23, and FKBP51. Upon hormone (H) binding, MR undergoes a conformational change, and FKBP51 is exchanged by FKBP52, an immunofilin that recruits dynein in its PPIase domain. MR is actively transported to the nucleus, passes intact through the nuclear pore complex (NPC), is “transformed” in the nucleoplasm, and dimerizes and binds to the promoter sequences of target genes. The Hsp90-based heterocomplex can be recycled. The black crescent represents the TPR domain of the immunophilins
In unstimulated cells, the immunophilin FKBP51 is primarily bound to the MR•Hsp90 complex. Upon steroid binding, FKBP51 is exchanged by FKBP52, an immunophilin that shares 75% similarity with FKBP51 and is capable to interact with the dynein/dynactin motor protein machinery via its PPIase domain (i.e., a domain that has enzymatic activity of peptidylprolyl isomerase). Immunophilins FKBP52, CyP40, and PP5 can associate dynein via their respective PPIase domains, but not FKBP51 [58]. When the PPIase domains of FKBP51 and FKBP52 were exchanged in chimera constructs and assayed in intact cells, the properties of both immunophilins were also exchanged, i.e., FKBP51, but not FKBP52 was capable to favour the retrotransport of GR via dynein [55]. Interestingly, FKBP51 has also been reported as a mitochondrial protein [59, 60] and is also complexed with mitochondrial GR in identical oligomers as that depicted for cytosolic GR in Fig.1.1b.
Because of its biological relevance in the receptor retrotransport, we named the (Hsp90)2•FKBP52•dynein functional unit as “transportosome.” The active, transportosome-dependent movement occurs on microtubules filaments [40, 44, 58, 61]. When the MR reaches the nuclear pore complex, the entire transportosome passes intact through the nuclear pore, the chaperones and immunophilins being interacting factors with the nucleoporins and importins of the pore complex [40, 62, 63]. The permeability barrier of the pore is in part due to a sieve structure created by the reversible cross-linking between Phe and Gly (FG)-rich nucleoporin repeats, which create a three-dimensional meshwork with hydrogel-like properties [64]. According to the novel model, nuclear transport receptors overcome the size limit of the sieve and catalyze their own nuclear pore passage by a competitive disruption of adjacent inter-repeat contacts, which transiently opens adjoining meshes. The chaperone complex would enhance the capability of the MR to overcome the resistance of the meshwork simply by accomplishing its standard role of chaperones. Therefore, the MR•Hsp90 complex dissociates in the nucleus rather than in the cytoplasm as it has always been thought. The receptor dimerizes in the nucleoplasm [65,66,67] and becomes activated to acquire its main biological role, i.e., to be a transcription factor. In contrast to the classic model of action posited for steroid receptors years ago, all these mechanistic steps are not heuristic and have been experimentally supported for each individual step.
An additional relevance of the presence of FKBP52 associated with the MR is the capability of this immunophilin to anchor the receptor to nuclear matrix structures [63]. Actually, the overexpression of FKBP51 expels MR from the nuclear compartment, perhaps due to competition with FKBP52 for the nuclear anchoring sites [40]. Similar observations and conclusions were also achieved for the role of FKBP52 in the mechanism of action of NF-κB [57, 68].
1.5 Agonist Structure-Activity Relationship
Binding of the cognate ligand to its specific receptor is the primordial first step to trigger cellular events that lead to the final biological effect in the body, i.e., binding of aldosterone to MR have profound effects in the electrolyte balance of the body, plasma osmolarity, blood pressure, heart rate, adiponectins activity, slow wave sleep, salt appetite, interoception, emotionality, etc. Therefore, the proper recognition of both components of the ligand-receptor functional unit is essential. Decades ago, Duax et al. [69,70,71] summarized the minimal conformational requirements on ring A of steroid hormones for optimal binding to different receptors. According to that study, the optimal conformation for the MR would be a 1α-envelope to a 1α,2β-half-chair containing the 3-keto-4-ene function. Better affinity ratios for the MR were also measured when those substituents that show the tendency to bend the A-ring toward the α face of the steroid molecule were eliminated, for example, for steroids lacking the C11-hydroxy function or the C19-methyl group [72]. An equivalent result is also observed upon introduction of ketalic bridges that flatten the overall structure, for example, in the cases of aldosterone itself and related 18-oxygenated analogues [73].
Although several compounds have been synthesized for all the other members of the steroid receptor family (and many of them have even replaced the natural ligands in many clinical treatments), only one synthetic MR agonist showing no cross-reaction with the other members of the steroid receptor subfamily is currently available to study the agonist mineralocorticoid function—11,19-oxidoprogesterone [74, 75] (see its structure in Fig. 1.3). In vivo assays in rats demonstrated that this steroid shows identical activity to the naturally occurring mineralocorticoid 11-deoxycorticosterone at low doses and becomes undistinguishable from aldosterone at as low dose as 10μg/100 g [75]. On the other hand, the bent conformers 6,19-oxidoprogesterone and its 21-hydroxylated derivative are devoid a mineralocorticoid effect and show no binding to the MR [75, 76]. Similar observations can be made for other pairs of steroids that share similar or identical functional groups but show different conformational structures (Fig. 1.3 depicts some practical examples). Based on these facts, it has been postulated that the essential requirement of a ligand to become a mineralocorticoid ligand is to possess an overall planarity of the steroidal frame, and its ability to preserve it in vivo is critical to confer a steroid mineralocorticoid activity [75, 77, 78]. The last statement refers to putative chemical modifications the steroid may suffer because of the metabolism or associations with other molecules or proteins that may affect its conformational structure according to its molecular flexibility. Thus, A/BCD angle for progesterone is not greatly different from that of aldosterone (−24.0° vs −21.4°, respectively), but progesterone is a highly flexible steroid, whereas the presence of a hemiketalic ring that involves the C18-aldhehyde in aldosterone (from which the name of the steroid derives) favors that aldosterone can preserve its overall conformational flatness due to the rigidity of the molecule. A similar property is conferred by the presence of the C11-O- C19 bridge in the synthetic agonist 11,19-oxidoprogesterone.

Most stable conformers for some pairs of steroids. Under physiologic conditions, all ligands on the left column exhibit better Na+-retaining activity and higher relative affinity for MR than the bent partners depicted on the right column
An interesting property of the dose-response curves for Na+-retention is that most agonists exhibit a parabolic shape [74, 75], that is, a maximal antinatriuretic action at certain doses and then, a clear reversion at higher doses, a feature that is less evident for the most active ligands versus the less active steroids. Such a biphasic function of the dose-response curves makes unsuitable the concept of a conventional ED50 to quantify properly the entire function of the biological effect. Nevertheless, a good correlation could be observed if the overall function is considered as a second-order polynomial function defined by the equation y = ax2 + bx + c. Thus, the second-order coefficient “a” is a direct measure of the concavity of the polynomials and quantifies the biopharmacological parameters of the dose-response curve. Therefore, the most potent mineralocorticoid action corresponds to the lowest “a” value. Figure 1.4 shows the excellent correlation between this coefficient and the relative affinity of the steroid for the MR (Fig.1.4a) and the overall flatness of the conformers estimated as the angle between the C3-carbonyl and the middle plane of the D ring (Fig.1.4b).

(a) Mineralocorticoid response measured as the co-efficient “a” of the parabolic dose-response curves (low “a” means high response, see text). RBA: Relative binding affinity relative to [3H]-aldo-sterone. (●) 21-hydroxyste-roids (○) 21-deoxysteroids. (b) Steroid flatness improves the biological effect (‘a)
1.6 MR Antagonism
Aldosterone antagonists that are capable to impair the activation of the MR have a cardinal importance in the treatment of cardiovascular diseases [79]. Consequently, a considerable effort has been made by several laboratories and companies for the development and safe clinical use of synthetic anti-mineralocorticoid steroids, particularly during RALES (Randomized Aldactone Evaluation Study), EPHESUS (Eplerenone Post-AMI Heart Failure Efficacy and Survival Study), and EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization And Survival Study in Heart Failure) pioneer past trails, and most recently the ARTS (Arterial Revascularization Therapies Study) series [80, 81].
During the 1960s, spironolactone (Aldactone®) became the first synthetic anti-mineralocorticoid approved for massive human use. This synthetic steroid is indicated in cases of primary hyperaldosteronism, congestive heart failure, edematous conditions, hepatic cirrhosis, and nephrotic syndrome [82], among the most common pathologies. With time, it was also indicated for cases of severe heart failure and hypokalemia when standard alternative treatments were not well tolerated or are ineffective. Eplerenone (Inspra®) was approved in the year 2002. Even though it shows lower pharmacologic potency than spironolactone as an MR antagonist, it has other advantages such as longer half-life and does not generate active metabolites [83].
From the physiologic perspective, it should be pointed out that at the renal level, progesterone behaves as an MR antagonist in most vertebrates [84]. Progesterone shows equivalent affinity to aldosterone for the MR, a property conserved among mammalian species, suggesting the potential existence of unexplored roles for this ligand bound to MR. During pregnancy, progesterone raises plasma levels up to one order of magnitude higher than those of aldosterone, perhaps it is a self-protective mechanism since aldosterone also increases its concentration. Interestingly, it has been reported that a single nucleotide mutation (S810L) in the gene encoding the human MR and creates an MR that responds to progesterone resulting in early-onset hypertension, which is very much exacerbated during pregnancy [85].
The crystal structure of MR associated with aldosterone and antagonists [86] unrevealed key structural characteristics of the MR for the further development of synthetic antagonists. In combination with mutational studies, it was evidenced that Asn770 is essential for MR activation and also that the interactions of Thr945 in helix 10 are critical for the activation of the receptor. The crystal structure of MR with the natural antagonist progesterone evidenced that the orientation of the Thr945 side chain is somehow vague because of competition between the ligand and intramolecular hydrogen bond acceptors. This decrease of both number of hydrogen bonds and the strength of the effective hydrogen bonds, plus the lack interaction with the Asn770 residue is the most likely molecular reason by which progesterone produces a weak activation (if any) of the MR despite the fact that its affinity for the receptor is high.
As a consequence of these studies, various possible antagonistic mechanisms have been postulated for the MR [86], i.e., competitive antagonism where there are no conformational changes induced in the LBD (such as in the case of spironolactone binding); impairment of MR dimerization; ligands whose binding favors MR degradation; and the case of selective ligands (antagonists or agonists) of trans-repression, this being a similar situation to that already reported for GR antagonists [87]. As it was stated above, the distinction between an MR antagonist and an MR agonist could be subtle since the substitution of a single amino acid (S810L) can make the difference and transforms not only progesterone, but also the antagonist spironolactone and the endogenous cortisone in strong MR agonists [85, 88].
1.7 MR Regulation by Phosphorylation and Redox Potential
Like most members of the nuclear receptor superfamily, the MR is a phosphoprotein. The first evidence was obtained when hMR was expressed in Spodoptera frugiperda cells grown in the presence of 32P [89]. Then, it was demonstrated that rat kidney MR is a phosphoprotein in a physiologic milieu [90, 91]. In that early work, a treatment of native MR with alkaline phosphatase resulted in the loss of aldosterone-binding capacity and dramatic changes of MR hydrodynamic properties in sucrose density gradients, causing a strong shift from the 8.8 S (untransformed, Hsp90-bound) isoform to the 5.1 S, transformed isoform. During the late 1990s, it was postulated that Ser/Thr-phosphatases of the PP1/PP2A family are involved in the mechanism of activation of MR and that this fact enhances its capacity to interact with the promoter sequences of target genes in the DNA [90, 91]. It was also hypothesized that the phosphorylated forms of MR are not beneficial for aldosterone-binding capacity [51, 90]. These early findings were confirmed years later by the Lifton lab [92, 93] in a study where it was demonstrated that phosphorylation at S843 in the MR LBD prevents aldosterone binding. In line with this, an MR phosphomimetic mutant (S843E) revealed showed that steroid binding capacity is severely impairs, increasing the dissociation constant by more than 100-fold [93]. This phosphorylated form of the MR was found in intercalated cells of the distal nephron. Importantly, angiotensin II signalling decreases phospho-MR levels, a phenomenon dependent on the activity of a PP1 protein-phosphatase, just as it had been predicted in the early studies [90, 91]. This increases the aldosterone-dependent biological response of the cells. More recently it was demonstrated that the effect of angiotensin II is due to inhibition of the Ser/Thr-protein-kinase ULK1 (Unc-51 Like-autophagy-activating Kinase 1), which results in decreased MR phosphorylation via mTOR [94].
Phosphorylation of MR is also related to the regulation of the receptor by the redox potential of the cell, glutathione (GSH) being the most prevalent and abundant (mM range) intracellular reducer thiol. GSH is not required in the diet, but synthesized by the sequential actions of two enzymes: Q-glutamyl-cysteine synthetase and GST [94]. GSH is exported continuously and degraded extracellularly. Therefore, in vivo GSH deficiency can achieved with the inhibitor of the enzyme that generate GSH and BSO (L-buthionine-(S,R)-sulfoximine). When adrenalectomized rats were treated with BSO [95], the low redox potential generated exerted drastic and uncompensated inhibition of the MR-dependent response with loss of the mineralocorticoid response (i.e. Na+-retention, kaliuresis, low aldosterone-binding capacity) accompanied by a higher level of receptor phosphorylation. The loss of steroid binding capacity was assigned to the oxidation of essential cysteine groups of the MR but also due to an inefficient synthesis of MR due to failures at the elongation/termination step during the receptor translation, mimicking the observations made with rats along the ageing process. There are several other variables that may affect the MR-dependent response by influencing the redox milieu. For example, the use of drugs that are designed for unrelated applications, but they may affect the redox potential of the cell. It is known that melatonin affects the GR nuclear translocation due to unknown reasons [95], and its influence on the close-related partner MR has not been studied to date.
As is was detailed in the first section, when 11βHSD2 activity is deficient or blocked, its protective mechanism on the MR against cortisol activation fails, such that cortisol activates principal cell MR. In tubular intercalated cells, MR but not 11βHSD2 is expressed. However, the MR is protected from cortisol activation by phosphorylation at S843. When MR becomes dephosphorylated in response to angiotensin II, MR can be stimulated by both steroids, aldosterone or cortisol, but more likely by the latter given its 2–3 orders of magnitude higher of plasma levels compared to aldosterone [11]. In addition to converting cortisol to cortisone, this enzyme also produces NADH from NAD+. Interestingly, it has been postulated that what appears to hold cortisol-MR complexes inactive is the high levels of NADH generated [96].
In the renal target cells, the enzyme 11βHSD2 can debulk intracellular cortisol by 90%, i.e., to levels ∼tenfold those of aldosterone. This implies that when 11βHSD2 is functional, most epithelial MR pool can be still occupied by cortisol, but it is not active. When intracellular redox state changes due to inhibition of 11βHSD2 (no NADH is produced), the increased production of ROS and oxidized glutathione (GSSG) could transform cortisol from an MR antagonist to an MR agonist. Thus, it was reported [96, 97] that when rabbit cardiomyocytes are patch clamped and treated with 10 nM aldosterone, ion-influx is increased tenfold (as expected), whereas 100 nM cortisol shows no effect. When both steroids are added together, the aldosterone action is 90% antagonized. If 5 mM GSSG is instilled intracellularly (i.e., to mimic the redox state under tissue damage conditions) cortisol becomes an MR agonist and similar effect as that measured with aldosterone alone is observed. Cardiomyocytes do not express 11βHSD2, so MR is “unprotected” and overwhelmingly occupied by cortisol, previously shown not to mimic the effects of aldosterone via MR in neonatal rat cardiomyocytes [98]. In line with these observations, in other study it was also demonstrated that corticosterone action via MR in rat ventricular cardiomyocytes requires an oxidized milieu [99]. Therefore, oxidative stress experienced after a postischemic reperfusion would favor glucocorticoid activation of the MR and the potentiation of the GR response, such that both receptors could contribute to remodelling the functional properties of ventricular cardiomyocytes. This makes them prone to spontaneous contractions and consequently, increasing the deadly risk of ventricular arrhythmias.
Importantly, oxidative stress can be transmitted through a glutathione-S-transferase (GST) “switch” connecting to kinase cascades influencing cell signalling. High ROS can cause a disassociation of the GST•JNK complex, thus activating JNK pathways [100]. PKC can also be activated, a protein-kinase that has been related to phosphorylation of MR [101, 102]. Aldosterone-dependent activation of MR induces the expression of the immunophilin FKBP51 [103], a phenomenon also reported for angiotensin II stimulation of smooth muscle cells via MR where PKC activation is also involved [102]. ROS effects are not entirely surprising since several studies have already shown that many kinases affect their activity upon the onset of oxidative stress such as JNK, p70-S6 kinase, Akt/PKB, PDK1, and SGK, among many other examples [104, 105]. Interestingly, the last two kinases are linked to the MR-dependent response. SGK affects the activation of the epithelial sodium channel and is in turn regulated PDK1 [106].
1.8 Conclusions
The MR and its two most potent physiologic ligands aldosterone and 11-deoxycorticosterone have evolved together under the evolutionary pressure of maintaining the internal homeostatic balance of water and electrolytes in an environment where especially the offer of water is frequently limited. On the other hand, glucocorticoids took on the task of ensuring energy homeostasis. Both receptors, MR and GR, may often work in concert or in counterpoint to meet the constant presence of new and varied environmental challenges. Therefore, the MR/GR ratio of selective activation is critical for normal function of the body. This is particularly relevant for the brain, where the highest concentrations of MR per gram of tissue are expressed. Within the most conventional diseases, hypertension is perhaps the best known when there is unequal or inappropriate MR/GR occupation and activation, as well as it is the case of metabolic syndrome and depressive disorders.
In pathologic situations, it is regarded that MR can be often be occupied by glucocorticoids rather than aldosterone. Moreover, the required concentration of local ligand necessary to activate the MR could be quite different from their plasma concentrations, especially in the nervous system and other systems where both aldosterone and glucocorticoids could be produced locally in an reduced or increased manner under pathologic situations.
It should be kept in mind that the transactivation activity of the MR is crucially dependent on the nature of the bound ligand. Agonist or antagonist ligands are capable to induce a unique conformational change that drives interactions of the MR with several coregulators and tissue-specific transcriptional factors. Therefore, each ligand•MR complex surely shows distinct and often opposite tissue-specific target genes and therefore distinct downstream biological effects depending on how the steroid has been accommodated in the ligand binding pocket.
The uncovered mechanism of action of the MR to date show a complex picture of multifunctional systems that require additional studies to unravel several poorly understood events such as the protection of the MR against nonspecific activation, by its several binding ligands, or the influence of the cellular context for its activation. Its cognate endogenous ligand aldosterone is a key therapeutic target in hypertension [81, 107] and chronic heart failure [108, 109]. Accumulating data also indicate that MR antagonists can be protective against the chronic kidney disease [109, 110]. After several years of intensive research, the development of new therapeutic approaches and the development of novel cardiovascular drugs is a fact based on studies that began explaining the basics of the molecular mechanism of action of the youngest member of the nuclear receptor superfamily. Nonetheless, more detailed characterization of the molecular mechanisms regulating MR function in the kidney, heart, brain, and other tissues may reveal new targets that might be exploited for therapeutic purposes.
References
Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240(4854):889–95.
Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318(6047):635–41.
Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237(4812):268–75.
Funder JW. Glucocorticoid and mineralocorticoid receptors: biology and clinical relevance. Annu Rev Med. 1997;48:231–40.
Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242(4878):583–5.
Sabbadin C, Andrisani A, Ambrosini G, Bordin L, Dona G, Manso J, et al. Aldosterone in gynecology and its involvement on the risk of hypertension in pregnancy. Front Endocrinol. 2019;10:575.
van Steensel B, van Binnendijk EP, Hornsby CD, van der Voort HT, Krozowski ZS, de Kloet ER, et al. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J Cell Sci. 1996;109(Pt 4):787–92.
Magarinos AM, Coirini H, De Nicola AF, McEwen BS. Mineralocorticoid regulation of salt intake is preserved in hippocampectomized rats. Neuroendocrinology. 1986;44(4):494–7.
de Kloet ER, Joels M. Brain mineralocorticoid receptor function in control of salt balance and stress-adaptation. Physiol Behav. 2017;178:13–20.
Joels M, de Kloet ER. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: The brain mineralocorticoid receptor: a saga in three episodes. J Endocrinol. 2017;234(1):T49–66.
Fru KN, VandeVoort CA, Chaffin CL. Mineralocorticoid synthesis during the periovulatory interval in macaques. Biol Reprod. 2006;75(4):568–74.
Milliez P, Girerd X, Plouin PF, Blacher J, Safar ME, Mourad JJ. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol. 2005;45(8):1243–8.
Stowasser M, Sharman J, Leano R, Gordon RD, Ward G, Cowley D, et al. Evidence for abnormal left ventricular structure and function in normotensive individuals with familial hyperaldosteronism type I. J Clin Endocrinol Metab. 2005;90(9):5070–6.
Jaisser F, Farman N. Emerging roles of the mineralocorticoid receptor in pathology: toward new paradigms in clinical pharmacology. Pharmacol Rev. 2016;68(1):49–75.
Preston IR, Sagliani KD, Warburton RR, Hill NS, Fanburg BL, Jaffe IZ. Mineralocorticoid receptor antagonism attenuates experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2013;304(10):L678–88.
Daruich A, Matet A, Dirani A, Bousquet E, Zhao M, Farman N, et al. Central serous chorioretinopathy: recent findings and new physiopathology hypothesis. Prog Retin Eye Res. 2015;48:82–118.
Dobzhansky T. Nothing in biology makes sense except in the light of evolution. Am Biol Teach. 1973;35(3):125–9.
Rossier BC, Baker ME, Studer RA. Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiol Rev. 2015;95(1):297–340.
Thornton JW, DeSalle R. A new method to localize and test the significance of incongruence: detecting domain shuffling in the nuclear receptor superfamily. Syst Biol. 2000;49(2):183–201.
Thornton JW. Evolution of vertebrate steroid receptors from an ancestral estrogen receptor by ligand exploitation and serial genome expansions. Proc Natl Acad Sci U S A. 2001;98(10):5671–6.
Rai S, Szeitz A, Roberts BW, Christie Q, Didier W, Eom J, et al. A putative corticosteroid hormone in Pacific lamprey, Entosphenus tridentatus. Gen Comp Endocrinol. 2015;212:178–84.
Shaughnessy CA, Barany A, McCormick SD. 11-Deoxycortisol controls hydromineral balance in the most basal osmoregulating vertebrate, sea lamprey (Petromyzon marinus). Sci Rep. 2020;10(1):12148.
Gomez-Sanchez E, Gomez-Sanchez CE. The multifaceted mineralocorticoid receptor. Compr Physiol. 2014;4(3):965–94.
Kassahn KS, Ragan MA, Funder JW. Mineralocorticoid receptors: evolutionary and pathophysiological considerations. Endocrinology. 2011;152(5):1883–90.
Pratt WB, Galigniana MD, Morishima Y, Murphy PJ. Role of molecular chaperones in steroid receptor action. Essays Biochem. 2004;40:41–58.
Kosano H, Stensgard B, Charlesworth MC, McMahon N, Toft D. The assembly of progesterone receptor-hsp90 complexes using purified proteins. J Biol Chem. 1998;273(49):32973–9.
Galigniana MD, Echeverria PC, Erlejman AG, Piwien-Pilipuk G. Role of molecular chaperones and TPR-domain proteins in the cytoplasmic transport of steroid receptors and their passage through the nuclear pore. Nucleus. 2010;1(4):299–308.
Pratt WB, Silverstein AM, Galigniana MD. A model for the cytoplasmic trafficking of signalling proteins involving the hsp90-binding immunophilins and p50cdc37. Cell Signal. 1999;11(12):839–51.
Murphy PJ, Kanelakis KC, Galigniana MD, Morishima Y, Pratt WB. Stoichiometry, abundance, and functional significance of the hsp90/hsp70-based multiprotein chaperone machinery in reticulocyte lysate. J Biol Chem. 2001;276(32):30092–8.
Molinari AM, Machado-Rada MY, Mazaira GI, Erlejman AG, Galigniana MD. Molecular basis of mineralocorticoid receptor action in the nervous system. CNS Neurol Disord Drug Targets. 2013;12(8):1163–74.
Mazaira G, Lagadari M, Erlejman AG, Galigniana MD. Molecular chaperones shape steroid receptor action and pharmacologic strategies. J J Cell Mol Biol. 2015;1(4):1–7.
Pratt WB, Galigniana MD, Harrell JM, DeFranco DB. Role of hsp90 and the hsp90-binding immunophilins in signalling protein movement. Cell Signal. 2004;16(8):857–72.
Murphy PJ, Morishima Y, Chen H, Galigniana MD, Mansfield JF, Simons SS Jr, et al. Visualization and mechanism of assembly of a glucocorticoid receptor.Hsp70 complex that is primed for subsequent Hsp90-dependent opening of the steroid binding cleft. J Biol Chem. 2003;278(37):34764–73.
Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017;18(6):345–60.
Li J, Richter K, Buchner J. Mixed Hsp90-cochaperone complexes are important for the progression of the reaction cycle. Nat Struct Mol Biol. 2011;18(1):61–6.
Kanelakis KC, Murphy PJ, Galigniana MD, Morishima Y, Takayama S, Reed JC, et al. hsp70 interacting protein Hip does not affect glucocorticoid receptor folding by the hsp90-based chaperone machinery except to oppose the effect of BAG-1. Biochemistry. 2000;39(46):14314–21.
Silverstein AM, Galigniana MD, Kanelakis KC, Radanyi C, Renoir JM, Pratt WB. Different regions of the immunophilin FKBP52 determine its association with the glucocorticoid receptor, hsp90, and cytoplasmic dynein. J Biol Chem. 1999;274(52):36980–6.
Gallo LI, Ghini AA, Piwien Pilipuk G, Galigniana MD. Differential recruitment of tetratricorpeptide repeat domain immunophilins to the mineralocorticoid receptor influences both heat-shock protein 90-dependent retrotransport and hormone-dependent transcriptional activity. Biochemistry. 2007;46(49):14044–57.
Sivils JC, Storer CL, Galigniana MD, Cox MB. Regulation of steroid hormone receptor function by the 52-kDa FK506-binding protein (FKBP52). Curr Opin Pharmacol. 2011;11(4):314–9.
Galigniana MD, Erlejman AG, Monte M, Gomez-Sanchez C, Piwien-Pilipuk G. The hsp90-FKBP52 complex links the mineralocorticoid receptor to motor proteins and persists bound to the receptor in early nuclear events. Mol Cell Biol. 2010;30(5):1285–98.
Zgajnar NR, De Leo SA, Lotufo CM, Erlejman AG, Piwien-Pilipuk G, Galigniana MD. Biological actions of the Hsp90-binding Immunophilins FKBP51 and FKBP52. Biomol Ther. 2019;9(2):52.
Erlejman AG, Lagadari M, Harris DC, Cox MB, Galigniana MD. Molecular chaperone activity and biological regulatory actions of the TPR-domain immunophilins FKBP51 and FKBP52. Curr Protein Pept Sci. 2014;15(3):205–15.
Storer CL, Dickey CA, Galigniana MD, Rein T, Cox MB. FKBP51 and FKBP52 in signaling and disease. Trends Endocrinol Metab. 2011;22(12):481–90.
Quinta HR, Galigniana NM, Erlejman AG, Lagadari M, Piwien-Pilipuk G, Galigniana MD. Management of cytoskeleton architecture by molecular chaperones and immunophilins. Cell Signal. 2011;23(12):1907–20.
Salatino M, Beguelin W, Peters MG, Carnevale R, Proietti CJ, Galigniana MD, et al. Progestin-induced caveolin-1 expression mediates breast cancer cell proliferation. Oncogene. 2006;25(59):7723–39.
Quintá HR, Maschi D, Gomez-Sanchez C, Piwien-Pilipuk G, Galigniana MD. Subcellular rearrangement of hsp90-binding immunophilins accompanies neuronal differentiation and neurite outgrowth. J Neurochem. 2010;115(3):716–34.
Ratajczak T, Ward BK, Cluning C, Allan RK. Cyclophilin 40: an Hsp90-cochaperone associated with apo-steroid receptors. Int J Biochem Cell Biol. 2009;41(8-9):1652–5.
Huyet J, Pinon GM, Fay MR, Rafestin-Oblin ME, Fagart J. Structural determinants of ligand binding to the mineralocorticoid receptor. Mol Cell Endocrinol. 2012;350(2):187–95.
Faresse N, Ruffieux-Daidie D, Salamin M, Gomez-Sanchez CE, Staub O. Mineralocorticoid receptor degradation is promoted by Hsp90 inhibition and the ubiquitin-protein ligase CHIP. Am J Physiol Renal Physiol. 2010;299(6):F1462–72.
McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8(4):303–8.
Galigniana MD. Functional regulation of corticosteroid receptors by phosphorylation and redox potential. Curr Topics Steroid Res. 2000;3:1–22.
Galigniana MD, Piwien Pilipuk G, Kanelakis KC, Burton G, Lantos CP. Molecular mechanism of activation and nuclear translocation of the mineralocorticoid receptor upon binding of pregnanesteroids. Mol Cell Endocrinol. 2004;217(1-2):167–79.
Pearce D, Naray-Fejes-Toth A, Fejes-Toth G. Determinants of subnuclear organization of mineralocorticoid receptor characterized through analysis of wild type and mutant receptors. J Biol Chem. 2002;277(2):1451–6.
Galigniana MD, Radanyi C, Renoir JM, Housley PR, Pratt WB. Evidence that the peptidylprolyl isomerase domain of the hsp90-binding immunophilin FKBP52 is involved in both dynein interaction and glucocorticoid receptor movement to the nucleus. J Biol Chem. 2001;276(18):14884–9.
Wochnik GM, Ruegg J, Abel GA, Schmidt U, Holsboer F, Rein T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem. 2005;280(6):4609–16.
Piwien Pilipuk G, Vinson GP, Sanchez CG, Galigniana MD. Evidence for NL1-independent nuclear translocation of the mineralocorticoid receptor. Biochemistry. 2007;46(5):1389–97.
Erlejman AG, De Leo SA, Mazaira GI, Molinari AM, Camisay MF, Fontana V, et al. NF-kappaB transcriptional activity is modulated by FK506-binding proteins FKBP51 and FKBP52: a role for peptidyl-prolyl isomerase activity. J Biol Chem. 2014;289(38):26263–76.
Galigniana MD, Harrell JM, Murphy PJ, Chinkers M, Radanyi C, Renoir JM, et al. Binding of hsp90-associated immunophilins to cytoplasmic dynein: direct binding and in vivo evidence that the peptidylprolyl isomerase domain is a dynein interaction domain. Biochemistry. 2002;41(46):13602–10.
Gallo LI, Lagadari M, Piwien-Pilipuk G, Galigniana MD. The 90-kDa heat-shock protein (Hsp90)-binding immunophilin FKBP51 is a mitochondrial protein that translocates to the nucleus to protect cells against oxidative stress. J Biol Chem. 2011;286(34):30152–60.
Toneatto J, Guber S, Charo NL, Susperreguy S, Schwartz J, Galigniana MD, et al. Dynamic mitochondrial-nuclear redistribution of the immunophilin FKBP51 is regulated by the PKA signaling pathway to control gene expression during adipocyte differentiation. J Cell Sci. 2013;126(Pt 23):5357–68.
Harrell JM, Murphy PJ, Morishima Y, Chen H, Mansfield JF, Galigniana MD, et al. Evidence for glucocorticoid receptor transport on microtubules by dynein. J Biol Chem. 2004;279(52):54647–54.
Echeverria PC, Mazaira G, Erlejman A, Gomez-Sanchez C, Piwien Pilipuk G, Galigniana MD. Nuclear import of the glucocorticoid receptor-hsp90 complex through the nuclear pore complex is mediated by its interaction with Nup62 and importin beta. Mol Cell Biol. 2009;29(17):4788–97.
Mazaira GI, Echeverria PC, Galigniana MD. Nucleocytoplasmic shuttling of the glucocorticoid receptor is influenced by tetratricopeptide repeat-containing proteins. J Cell Sci. 2020;133(12):jcs238873.
Frey S, Richter RP, Gorlich D. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science. 2006;314(5800):815–7.
Grossmann C, Ruhs S, Langenbruch L, Mildenberger S, Stratz N, Schumann K, et al. Nuclear shuttling precedes dimerization in mineralocorticoid receptor signaling. Chem Biol. 2012;19(6):742–51.
Presman DM, Ogara MF, Stortz M, Alvarez LD, Pooley JR, Schiltz RL, et al. Live cell imaging unveils multiple domain requirements for in vivo dimerization of the glucocorticoid receptor. PLoS Biol. 2014;12(3):e1001813.
Galigniana MD. Steroid receptor coupling becomes nuclear. Chem Biol. 2012;19(6):662–3.
Lagadari M, De Leo SA, Camisay MF, Galigniana MD, Erlejman AG. Regulation of NF-kappaB signalling cascade by immunophilins. Curr Mol Pharmacol. 2016;9(2):99–108.
Duax WL, Cody V, Griffin JF, Rohrer DC, Weeks CM. Molecular conformation and protein binding affinity of progestins. J Toxicol Environ Health. 1978;4(2-3):205–27.
Duax WL, Griffin JF, Strong PD, Funder JW, Ulick S. Molecular structure of 18-deoxyaldosterone and its relationship to receptor binding and activity. J Am Chem Soc. 1982;104(25):7291–3.
Duax WL, Hauptman H. The crystal structure and molecular conformation of aldosterone. J Am Chem Soc. 1972;94(15):5467–71.
Yamakawa M, Ezumi K, Shiro M, Nakai H, Kamata S, Matsui T, et al. Relationships of the molecular structure of aldosterone derivatives with their binding affinity for mineralocorticoid receptor. Mol Pharmacol. 1986;30(6):585–9.
Fattah DI, Whitehouse BJ, Vinson GP. Proceedings: synthesis of 18-oxygenated steroids in the rat adrenal cortex. J Endocrinol. 1975;65(3):50P–1P.
Burton G, Galigniana M, De Lavallaz S, Brachet-Cota AL, Sproviero EM, Ghini AA, et al. Sodium-retaining activity of some natural and synthetic 21-deoxysteroids. Mol Pharmacol. 1995;47(3):535–43.
Piwien-Pilipuk G, Kanelakis KC, Galigniana MD. Correlation between pregnanesteroid conformation, receptor affinity, and anti-natriuretic effect. Eur J Pharmacol. 2002;454(2-3):131–43.
Vicent GP, Monteserin MC, Veleiro AS, Burton G, Lantos CP, Galigniana MD. 21-Hydroxy-6,19-oxidoprogesterone: a novel synthetic steroid with specific antiglucocorticoid properties in the rat. Mol Pharmacol. 1997;52(4):749–53.
Galigniana MD, Piwien PG. Activation of the ligand-mineralocorticoid receptor functional unit by ancient, classical, and novel ligands. Structure-activity relationship. Vitam Horm. 2004;69:31–68.
Gomez-Sanchez CE, Gomez-Sanchez EP, Galigniana MD. Aldosterone receptors and their renal effects: molecular biology and gene regulation. In: Singh AK, Williams GH, editors. Textbook of nephro-endocrinology. London, UK: Academic Press; 2009. p. 329–48.
Pitt B, Pedro Ferreira J, Zannad F. Mineralocorticoid receptor antagonists in patients with heart failure: current experience and future perspectives. Eur Heart J Cardiovasc Pharmacother. 2017;3(1):48–57.
Funder JW. Mineralocorticoid receptor antagonists: emerging roles in cardiovascular medicine. Integr Blood Pressure Control. 2013;6:129–38.
Kolkhof P, Barfacker L. 30 YEARS OF THE MINERALOCORTICOID RECEPTOR: Mineralocorticoid receptor antagonists: 60 years of research and development. J Endocrinol. 2017;234(1):T125–T40.
Nordqvist A, Granberg KL. Mineralocorticoid receptor antagonists. Vitam Horm. 2019;109:151–88.
Parthasarathy HK, Menard J, White WB, Young WF Jr, Williams GH, Williams B, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens. 2011;29(5):980–90.
Baker ME, Katsu Y. Progesterone: an enigmatic ligand for the mineralocorticoid receptor. Biochem Pharmacol. 2020;177:113976.
Geller DS, Farhi A, Pinkerton N, Fradley M, Moritz M, Spitzer A, et al. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science. 2000;289(5476):119–23.
Bledsoe RK, Madauss KP, Holt JA, Apolito CJ, Lambert MH, Pearce KH, et al. A ligand-mediated hydrogen bond network required for the activation of the mineralocorticoid receptor. J Biol Chem. 2005;280(35):31283–93.
De Bosscher K, Haegeman G, Elewaut D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr Opin Pharmacol. 2010;10(4):497–504.
Rafestin-Oblin ME, Souque A, Bocchi B, Pinon G, Fagart J, Vandewalle A. The severe form of hypertension caused by the activating S810L mutation in the mineralocorticoid receptor is cortisone related. Endocrinology. 2003;144(2):528–33.
Alnemri ES, Maksymowych AB, Robertson NM, Litwack G. Characterization and purification of a functional rat glucocorticoid receptor overexpressed in a baculovirus system. J Biol Chem. 1991;266(6):3925–36.
Galigniana MD. Native rat kidney mineralocorticoid receptor is a phosphoprotein whose transformation to a DNA-binding form is induced by phosphatases. Biochem J. 1998;333(Pt 3):555–63.
Piwien-Pilipuk G, Galigniana MD. Tautomycin inhibits phosphatase-dependent transformation of the rat kidney mineralocorticoid receptor. Mol Cell Endocrinol. 1998;144(1-2):119–30.
Shibata S. Context-dependent mechanisms modulating aldosterone signaling in the kidney. Clin Exp Nephrol. 2016;20(5):663–70.
Shibata S, Rinehart J, Zhang J, Moeckel G, Castaneda-Bueno M, Stiegler AL, et al. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab. 2013;18(5):660–71.
Shibata S, Ishizawa K, Wang Q, Xu N, Fujita T, Uchida S, et al. ULK1 phosphorylates and regulates mineralocorticoid receptor. Cell Rep. 2018;24(3):569–76.
Presman DM, Hoijman E, Ceballos NR, Galigniana MD, Pecci A. Melatonin inhibits glucocorticoid receptor nuclear translocation in mouse thymocytes. Endocrinology. 2006;147(11):5452–9.
Funder JW. Mineralocorticoid receptors: distribution and activation. Heart Fail Rev. 2005;10(1):15–22.
Mihailidou AS, Funder JW. Nongenomic effects of mineralocorticoid receptor activation in the cardiovascular system. Steroids. 2005;70(5-7):347–51.
Sato A, Funder JW. High glucose stimulates aldosterone-induced hypertrophy via type I mineralocorticoid receptors in neonatal rat cardiomyocytes. Endocrinology. 1996;137(10):4145–53.
Rossier MF, Lenglet S, Vetterli L, Python M, Maturana A. Corticosteroids and redox potential modulate spontaneous contractions in isolated rat ventricular cardiomyocytes. Hypertension. 2008;52(4):721–8.
Townsend DM, Findlay VL, Tew KD. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005;401:287–307.
Le Moellic C, Ouvrard-Pascaud A, Capurro C, Cluzeaud F, Fay M, Jaisser F, et al. Early nongenomic events in aldosterone action in renal collecting duct cells: PKCalpha activation, mineralocorticoid receptor phosphorylation, and cross-talk with the genomic response. J Am Soc Nephrol. 2004;15(5):1145–60.
Lu Q, Davel AP, McGraw AP, Rao SP, Newfell BG, Jaffe IZ. PKCdelta mediates mineralocorticoid receptor activation by angiotensin II to modulate smooth muscle cell function. Endocrinology. 2019;160(9):2101–14.
Newfell BG, Iyer LK, Mohammad NN, McGraw AP, Ehsan A, Rosano G, et al. Aldosterone regulates vascular gene transcription via oxidative stress-dependent and -independent pathways. Arterioscler Thromb Vasc Biol. 2011;31(8):1871–80.
Ong GS, Young MJ. Mineralocorticoid regulation of cell function: the role of rapid signalling and gene transcription pathways. J Mol Endocrinol. 2017;58(1):R33–57.
Dever TE. Translation initiation: adept at adapting. Trends Biochem Sci. 1999;24(10):398–403.
Pearce D. The role of SGK1 in hormone-regulated sodium transport. Trends Endocrinol Metab. 2001;12(8):341–7.
Williams B, MacDonald TM, Morant S, Webb DJ, Sever P, McInnes G, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet. 2015;386(10008):2059–68.
Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348(14):1309–21.
Filippatos G, Anker SD, Bohm M, Gheorghiade M, Kober L, Krum H, et al. A randomized controlled study of finerenone vs. eplerenone in patients with worsening chronic heart failure and diabetes mellitus and/or chronic kidney disease. Eur Heart J. 2016;37(27):2105–14.
Bakris GL, Agarwal R, Chan JC, Cooper ME, Gansevoort RT, Haller H, et al. Effect of Finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial. JAMA. 2015;314(9):884–94.
Acknowledgments
The author is indebted for the financial support received from Universidad de Buenos Aires (UBACYT 20020170100558BA) and Agencia Nacional de Promoción Científica y Tecnológica (PICT-2016-0545 and PICT-2018-0546).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Galigniana, M.D. (2021). Molecular Pharmacology of the Youngest Member of the Nuclear Receptor Family: The Mineralocorticoid Receptor. In: Badr, M.Z. (eds) Nuclear Receptors. Springer, Cham. https://doi.org/10.1007/978-3-030-78315-0_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-78315-0_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-78314-3
Online ISBN: 978-3-030-78315-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)