
Abstract
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by intrinsic genetic complexity, present at diagnosis and/or evolving during the disease course. Due to the significant prognostic role of genetic changes in AML, characterization of molecular and phenotypic profiles is essential to designing patient- and disease-specific strategies aimed at preventing disease relapse and improving long-term outcome. In recent years, deep biological knowledge emerging in myeloid neoplasms led to the revised edition of the World Health Organization (WHO) classification in 2016. Accordingly, the rules for AML classification require collection of the patient’s history, including previous cytotoxic therapies, which define “therapy-related myeloid neoplasms,” or a prior history of MDS or MPN, defining “AML with myelodysplasia-related changes” (“AML-MRC”). The second field of investigation for classifying a case of AML is the presence of specific gene mutations or rearrangements defining the category of “AML with recurrent genetic abnormalities”. The detection of balanced or unbalanced cytogenetics aberrations considered associated with MDS and/or detection of multilineage dysplasia by morphology, defines the disease as “AML-MRC.” When the disease cannot be classified in another category, the morphologic exam of bone marrow and peripheral blood is the only parameter useful in the subcategorization of “AML, not otherwise specified" ("AML-NOS").
Recently, high-throughput next-generation techniques have indeed showed the accumulation of multiple genetic abnormalities in leukemic blasts. Not only do somatic mutations affect disease pathogenesis as single events, but also their combination plays a significant role. In this line, the European Leukemia Net (ELN) defined the first genetic-based stratification system for AML in 2010, and published a revised version in 2017. Three prognostic subgroups have been identified (favorable, intermediate, and adverse), where in addition to karyotype assessment, NPM1 mutation and evaluation of FLT3-ITD allelic burden, together with assessment of TP53, RUNX1, and ASXL1 mutations, are mandatory for proper AML stratification. In this chapter, we will also review the integrated diagnostic algorithm for AML diagnosis, nowadays an important challenge in the context of precision medicine, mandatory for the design of targeted-treatment approaches.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Introduction
Acute myeloid leukemia (AML) is the most common acute hematological malignancy in adults, with an estimated annual incidence rate of 4.2/100000 persons/year (5.2 in males and 3.5 in females) (data from SEER 2016) (Arber et al. 2016; Papaemmanuil et al. 2013). AML is a disease of the elderly, with a median age of 68 years at diagnosis. Recently, significant improvements have been made in the understanding of AML biology and genetics, and in 2016, the World Health Organization (WHO) published an update of the classification of myeloid neoplasms and acute leukemias, integrating clinical features, morphology, immunophenotype, and cytogenetics with new molecular genetic alterations to better define disease entities (Arber et al. 2016). The complete 2016 WHO classification of AML is reported in Table 2.1.
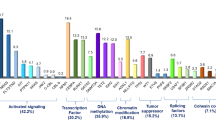
In the last few decades, efforts have been made to study the genomic landscape of AML: the result is a progressive shift from a morphologic classification, to one based on genetic/cytogenetic profiles, also taking into consideration the impact of genetic lesions on prognosis (Papaemmanuil et al. 2013). On this basis, first in 2010 and later in 2017, an international working group, on behalf of the European Leukemia Net (ELN), drew a risk-stratification model based on genetic and cytogenetic characteristics, that divided AML in three categories: favorable, intermediate, and adverse (Table 2.2) (Dohner et al. 2017). The 2017 update was required by the advancements in the definition of the mutational landscape in AML (Fig. 2.1), as well as by the development of novel antileukemic agents (Stone et al. 2017; Heuser et al. 2019; Döhner et al. 2010). Correct patient and disease stratification requires an integrated diagnostic process, including evaluation of morphology, immunophenotype, cytogenetics, and molecular changes. This is particularly important in the context of a modern personalized medicine approach, which is facilitated by the recent identification of targeted treatments. This applies also in cases of relapsed or refractory AML, where the same diagnostic algorithm must be used, due to the possibility of clonal evolution and emergence of “new” genetic alterations. Often, these alterations may be present at the time of initial diagnosis at the subclonal level, undetectable by conventional approaches (Ottone et al. 2013; Angelini et al. 2015).

Molecular classes of AML and concurrent gene mutations in adult patients. (From Dohner et al. 2017)
In this chapter, we will discuss recent guidelines for the diagnostic and prognostic stratification of AML. Diagnosis and monitoring of acute promyelocytic leukemia (APL) will be treated in a separate paragraph, due to the distinct clinical characteristics of this AML subtype, and the indications for prompt diagnosis and treatment start.
2.2 Diagnostic Procedures for AML Diagnosis
Figure 2.2 shows an algorithm for AML diagnosis.

Diagnostic tests required for AML (Adapted from Dohner et al. 2017)
2.2.1 Morphology
Morphology remains the basic diagnostic tool to assess the number and morphology of blasts in peripheral blood (PB) and bone marrow (BM). Starting from 2001, according to the WHO classification system, the diagnosis of AML requires ≥20% myeloblasts in the BM or PB, with some exceptions (Arber et al. 2016). Morphological evaluation of the BM aspirate or trephine biopsy, in cases with a dry tap (punctio sicca), represents the first indispensable tools for the routine diagnostic work-up for patients with a suspected AML. Marrow or PB smears are examined following May-Grünwald-Giemsa or Wright-Giemsa staining (Piaton et al. 2015). Myeloblasts, monoblasts, and megakaryoblasts must be included in the blast count. In AML with monocytic differentiation, monoblasts and promonocytes are counted as blast equivalents. The diagnosis of AML requires a BM blast count of 20% or more, except for AML with t(15;17), t(8;21), and inv(16), or t(16;16). In these AML subtypes, the genetic abnormality defines AML also in cases with BM blasts <20%. To identify lineage involvement, immunophenotyping is used with evaluation of myeloid differentiation markers, including myeloperoxidase (MPO). Cytochemistry with staining for nonspecific esterase (NSE), together with expression of lysozyme and monocytic markers, is required in cases with a mixed-phenotype AML (Grimwade 2001).
2.2.2 Immunophenotyping
Immunophenotyping using multiparameter flow cytometry (MFC) is a powerful tool to characterize cell surface and cytoplasmic markers, essential features for classification of AML subtypes. Common leucocyte antigen (CD45) and side scatter (SSC) gating are used to identify the blast population, (Borowitz et al. 1993) while expression of other lineage specific markers is useful for the phenotypic characterization of the blast population. The recommended panel includes the following antibodies: CD34, HLA-DR, TdT (stem cell/hematopoietic precursors), cMPO, CD13, CD33, CD117, CD15 (myeloid markers), monocytic markers (CD64, CD14, CD11b,CD11c), erythroid (CD71, CD235a), and megakaryocytic markers (CD41, CD61, CD36) (Venditti et al. 2019; Buccisano et al. 2018a; Maurillo et al. 2008). In addition, MFC is to identify monoblastic/monocytic AML (CD14+, CD64+, and CD36+), acute megakaryoblastic leukemia (CD41+ and CD61+), and pure erythroid leukemia (CD235a+ or CD36+ in the absence of CD64, MPO, or other myeloid-associated antigens) (Fig. 2.2) (Dohner et al. 2017; Del Principe et al. 2019).
2.2.3 Conventional and Molecular Cytogenetics
The WHO first added cytogenetic features to classify AML in 2001, while molecular subtypes were included in 2008 (Vardiman et al. 2009), in addition to morphologic and immunophenotypic features (Arber et al. 2016). The identification of recurrent cytogenetic abnormalities is mandatory for the diagnosis of AML, to define AML subtypes and prognostic groups, and to correctly address therapeutic strategies (Dohner et al. 2017; O’Donnell et al. 2013; Grimwade et al. 2010). Techniques used for cytogenetic analysis include karyotyping, analysis of G-banded chromosomes, and other cytogenetic banding techniques (Fig. 2.3a, b), such as fluorescent in situ hybridization (FISH) (Fig. 2.3c). In AML, chromosome abnormalities are detected in approximately 55% of patients (Grimwade 2001; Mrozek et al. 2004) and eight recurrent balanced translocations and inversions are recognized in the WHO category “AML with recurrent genetic abnormalities” (Arber et al. 2016) (Table 2.1). A minimum of 20 metaphases are required to define normal or abnormal karyotype. If the cytogenetic analysis fails, FISH is an optional approach to detect translocations, gene rearrangements, and partial or complete chromosome losses (Fig. 2.2). AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2) has been recently included in the WHO classification as a distinct type of leukemia, associated with resistance to conventional chemotherapy (Weisser et al. 2007). A new provisional entity “AML with BCR/ABL1” has been introduced to recognize AML patients with this abnormality, candidates for tyrosine kinase inhibitors. Clinical and molecular factors useful to differentiate AML with BCR/ABL1 from blast crisis of chronic myeloid leukemia (CML) are shown in Table 2.3.

Cytogenetic analysis in AML. (a) G-banding of a cytogenetically normal male karyotype (46,XY). (b) G-banded analysis in a patient with complex karyotype (45,XY,-2,der(2)t(2;?),der(7)t(7;?),der(16)t(16;?),-21,+mar). (c) Interphase FISH showing a fusion signal between chromosome 15 and 17 in a patient with APL. The PML gene on chromosome 15 is labeled red, the RARA gene on chromosome 17 is labelled green and the PML/RARA fusion gene is yellow. Cells were counterstained with DAPI II
2.2.4 Molecular Genetic Testing
In recent years, due to the availability of advanced technologies, in particular next-generation sequencing (NGS), several somatic mutations in myeloid genes have been identified in AML, some with diagnostic significance, others with prognostic or therapeutic relevance. The role of modern diagnostic in AML is to dissect these profiles, to accurately define individual entities, targetable by specific inhibitors, in the context of personalized medicine.
The genetic algorithm of newly diagnosed AML patients according to ELN criteria (Dohner et al. 2017) should include screening by RT-PCR for core-binding factor (CBF) leukemias [AML with t(8;21)(q22;q22.1), with RUNX1/RUNX1T1 rearrangement or inv(16)(p13.1q22)/t(16;16)(p13.1;q22), with CBFB/MYH11 rearrangement]. This not only allows for the identification of patients with favorable outcome, but defines the specific rearrangement type, which can be used for measurable residual disease (MRD) monitoring. Indeed, positivity of molecular MRD currently represents a powerful marker to predict early relapse (Corbacioglu et al. 2010; Willekens et al. 2016). In acute promyelocytic leukemia, rapid genetic confirmation of the t(15;17)(q22;q12) translocation (detection of PML/RARA fusion transcripts) is mandatory in cases of suspected APL, to allow for a prompt initiation of tailored therapy and supportive care (Sanz et al. 2019). Fatal hemorrhage is the most common cause of early death in patients with APL. To prevent these deaths occurring prior to the start of treatment, individuals with suspected APL should be immediately hospitalized and managed as a medical emergency. The diagnosis must be confirmed at the genetic level by experienced reference laboratories (Sanz et al. 2019). Additional analyses mandatory in all patients, and in particular for those with a normal karyotype, include screening for mutations in NPM1, CEBPA, ASLX1, TP53, and RUNX1 genes, which represent specific prognostic categories in the revised version of the ELN guidelines (Dohner et al. 2017). AML with NPM1 and CEBPA biallelic (biCEBPA) mutations have become full entities, while the new provisional entity “AML with mutated RUNX1” has been added.
NPM1 mutations occur in approximately 30% of adult AML cases, and in 50–60% of AML cases with normal karyotype (NK-AML), which makes NPM1 mutations the most frequent genetic lesions so far identified in de novo AML (Grisendi et al. 2006; Grimwade et al. 2016; Chang and Olson 1990). AML with NPM1 mutations has distinctive genetic, immunophenotypic, and clinical features. Therefore, this type of leukemia was recognized as a distinct entity (Arber et al. 2016). Mutations in the NPM1 gene predict favorable prognosis and represent a well-established marker for MRD-monitoring (Dohner et al. 2005). NPM1 is a nucleolar phosphoprotein that belongs to the nucleoplasmin/nucleophosmin family of nuclear chaperones (Schmidt-Zachmann et al. 1987; Eirín-López et al. 2006) and maps on chromosome band 5q35 in humans (Chang and Olson 1990). NPM1 mutations are mostly found in exon 12 of the NPM1 gene, leading to cytoplasmic expression of the protein (normally found in the nucleolus), due to the generation of a novel nuclear export signal (Falini et al. 2009). Currently, more than 50 different mutations located within exon 12 of the NPM1 gene have been described, and more than 95% of these involve an insertion of four nucleotides. The mutation types A, B, and D represent about 90% of NPM1 mutations (Dohner et al. 2005) and the identification of the specific NPM1 mutation by Sanger sequencing is particularly important for MRD monitoring. Figure 2.4 shows an example of PCR reaction for the detection of NPM1 mutations, followed by capillary electrophoresis (Lin et al. 2006).

Genescan electropherograms of PCR reactions for NPM1 mutations. (a) AML with NPM1 wild-type gene. (b) AML mutated for NPM1. Normal amplicon sizes of NPM1 wild-type allele correspond to 236 bp, while an additional PCR fragment amplification with an insertion of 4 bp corresponds to the NPM1 mutated allele. PCR fragments are shown in blue (FAM) and GENESCAN-400HD (ROX) size markers in red
Other gene mutations are important clinico-pathological features of AML. The FLT3 gene is located on chromosome 13 at band q12 (Rosnet et al. 1991) and encodes for a receptor normally expressed on the surface of hematopoietic progenitor cells, and expression is lost upon cell maturation. FLT3 is mutated in about 30% of adult AML (Stirewalt et al. 2001). Mutations in this gene result in constitutive activation of signaling through downstream pathways, leading to uncontrolled cell proliferation and survival. Two types of FLT3 alterations have been reported: FLT3-ITD represents the most common mutation and corresponds to an internal tandem duplication (FLT3-ITD) in the cytoplasmic juxtamembrane (JM) region (exons 14 and 15) of the gene. The other FLT3 mutation is located in the tyrosine kinase domain (FLT3-TKD), is located in the activation loop of FLT3, and includes the D835 point mutations or deletions of I836 (Gary Gilliland and Griffin 2002) (exon 20). Size of duplicated nucleotides in FLT3-ITD mutations vary from three to more than 400 base pairs, and are in-frame mutations caused by the duplication of various fragments from the JM domain of the FLT3 receptor. The FLT3-ITD receptor can homodimerize with mutant receptors or heterodimerize with wild-type receptors, independent of ligand stimulation, leading to distinct signaling responses to the ligand depending on the ratio of the wild-type to the mutant receptors (Gary Gilliland and Griffin 2002). Since the mutation is in-frame, the protein kinase domain remains functional (Kiyoi et al. 2002; Stirewalt and Radich 2003). Identification of the FLT3-ITD and TKD mutations requires a semi-quantitative assessment, using PCR followed by fragment length analysis are amplified by PCR (Thiede et al. 2002). Figure 2.5 shows representative electropherograms of FLT3-ITD, FLT3-TKD, and FLT3 wild-type cases. Testing for FLT3-ITD and -TKD mutations is recommended by the ELN due to the unfavorable prognosis of these patients, who have increased risk of relapse and shorter overall survival (OS), as compared with patients without these mutations. Outcome in FLT3-ITD-positive patients is particularly unfavorable in cases with high allelic burden, who benefit from intensive consolidation treatments (Stone et al. 2017; Stirewalt and Radich 2003; Gale et al. 2008). For this reason, in addition to the presence of FLT3-ITD, which defines an adverse AML subtype in the 2010 edition of the ELN classification (Döhner et al. 2010), the revised ELN guidelines proposed that the FLT3-ITD allelic ratio (AR) is used for AML stratification, in particular in NPM1-mutated patients (Table 2.2). In these patients, a low FLT3-ITD AR (below 0.5) defines favorable risk AMLs, while a high FLT3-ITD AR (≥0.5) is associated with increasingly unfavorable prognosis, defining intermediate-risk AML if it is associated with NPM1 mutations and high-risk AML if NPM1 is wild-type. In addition to FLT3-ITD mutations, ELN also recommends that FLT3-TKD mutations at codons D835 and I836 should be assessed, although the prognostic impact of these mutations is less clear. Identification of FLT3 mutations is not only of prognostic relevance, but these mutations may be targeted with the FLT3 tyrosine kinase inhibitor, as midostaurin and quizartinib (Stone et al. 2017; Perl 2019), which have significantly improved the outcome of these patients (Sutamtewagul and Vigil 2018).

Genescan electropherograms of PCR reactions for FLT3 mutations. (a) AML without the FLT3-ITD mutation. Normal amplicon sizes of FLT3 wild-type allele correspond to 330 bp. (b) AML mutated for FLT3-ITD. An additional PCR fragment amplification of a mutated allele corresponding to 367 bp. The FLT3-ITD allelic ratio (AR) in this case is 0.83. (c) AML with two FLT3-ITD mutations. Additional PCR fragments amplification of two mutated alleles corresponding to 347 and 514 bp. The FLT3-ITD AR is 0.36 in this case. (d) AML without a FLT3-TKD mutation. Normal amplicon sizes of FLT3 wild-type allele correspond to 80 bp. (e) AML mutated for FLT3-TKD. An additional PCR fragment amplification of a mutated allele corresponding to 128 bp
CEBPA is a transcription factor upregulated during granulocytic differentiation (Koschmieder et al. 2009). Mutations in the CEBPA gene are reported in ∼10–15% of NK-AML patients (Fasan et al. 2014) and may occur on the entire coding region. However, several studies showed an in-frame-shift mutation cluster in the N-terminal domain and in-frame insertions/deletions in the C-terminal region of the gene (Fasan et al. 2014). The mutated CEBPA protein inhibits the function of the full-length protein by a dominant negative mechanism and disrupt its DNA-binding ability. CEPBA-mutation may occur as single (single-mutated CEPBAsm) or as double (double-mutated CEPBA, CEBPAdm) events, in the N-terminal and C-terminal domains of the gene. When the mutations are biallelic, wild-type CEBPA is not expressed. Several reports showed a significantly improved outcome of patients with CEPBAdm as compared with CEPBAsm, and only biallelic CEBPA mutations define a distinct genetic entity (Fasan et al. 2014). Mutational analysis of CEBPA requires PCR sequencing of the entire CEBPA coding region, using four overlapping primer pairs. Technical details have been reported elsewhere (Frohling et al. 2004).
The RUNX1 gene encodes for a myeloid transcription factor involved in the regulation differentiation of myeloid, megakaryocytic, and lymphocytic lineages (Ichikawa et al. 2004). RUNX1 is mutated in 10% of de novo AML and is associated with unfavorable overall survival and rapid disease progression (Gaidzik et al. 2011). Missense and nonsense, or frameshift mutations in the RUNX1 gene have been reported in AML; they are distributed throughout the entire gene and their identification requires a targeted next-generation sequencing (NGS) approach (Kohlmann et al. 2013).
Further gene mutations in combination with chromosome abnormalities are used for risk stratification and therapeutic decisions, and among these, ASXL1 and TP53 mutations have been included as adverse prognostic factors in the 2017 ELN recommendations. ASXL1 is an epigenetic regulator, whose mutations represent early events in leukemogenesis. They have been described in 10% of AML patients (Devillier et al. 2015) and localize in exon 12, resulting in a truncated protein, with loss of the PHD domain (Pratcorona et al. 2012). These alterations are associated with marrow dysgranulopoiesis and have been frequently identified in intermediate-risk AML, where they predict inferior survival (Devillier et al. 2015). ASLX1 mutations may be investigated by PCR amplification and Sanger sequencing or, more frequently, by NGS (Pratcorona et al. 2012). TP53 is one of the most frequently mutated genes in human cancers, with a central role in aging, senescence, and DNA repair. In AML, TP53 alterations are rare events, but are frequently associated with increased genomic instability, as observed in elderly and therapy-related AML/MDS. TP53 mutations are mostly associated with complex karyotype and predict poor outcome (Devillier et al. 2015). The majority of TP53 mutations are localized in exons 5–8, and NGS analysis is commonly used to investigated the molecular status of the TP53 gene (Leroy et al. 2013).
Following the discovery of the genomic landscape of AML (Papaemmanuil et al. 2016), other gene alterations have been shown to have prognostic relevance in AML, in particular epigenetic regulators such as IDH1 and IDH2. IDH mutations are mostly described in the intermediate-risk karyotype, are often associated with NPM1 mutation, (Abbas et al. 2010) and are mutually exclusive with TET2 alterations (Gaidzik et al. 2012). Some AML patients with IDHs mutations, mainly IDH2R172, respond poorly to standard chemotherapy and have a higher relapse rate (Largeaud et al. 2019). IDH1 and IDH2 analysis may be performed by Sanger sequencing and Fig. 2.6 shows some electropherograms. Recently, the IDH inhibitors enasidenib and ivosidenib have shown activity in R/R AML with IDH2 and IDH1 mutations, respectively. Therefore, characterization of IDHs’ molecular status represents an important step toward the use of individualized treatments.

Sequence chromatograms for IDH1 and IDH2 mutations. (a) DNA sequence traces showing IDH1 wild-type, IDH1R132C and IDH1R132H -mutated AML. The arrows indicate the nucleotide position (c.394 and c.395) of each missense mutations. (b) DNA sequence traces showing IDH2 wild-type, IDH2R140Q and IDH2R172K mutated AML patients. The arrows indicate the nucleotide position (c.419 and c.515) of each missense mutations
In addition to the identification of novel driver mutations, NGS has highlighted the existence of multiple disease clones within a single AML case. Indeed, the genetic architecture of AML is extremely dynamic, and disease evolution/progression is mainly driven by the phenomenon of clonal evolution, characterized by the expansion/emergence of specific clones during the disease course (Ding et al. 2012; Genovese et al. 2014; Jaiswal et al. 2014). Interestingly, clonal evolution studies also indicate that mutations in genes involved in the regulation of DNA methylation and of chromatin state (i.e., DNMT3A, TET2, and ASXL1) may be present in pre-leukemic stem cells and may persist after therapy, leading to clonal expansion during remission, and eventually disease relapse. Large population-based cohorts have recently identified these pre-leukemic mutations in approximately 10% of elderly and healthy subjects; this phenomenon, termed “clonal hematopoiesis of indeterminate potential” (CHIP), has been associated with increased risks of hematologic neoplasms (Jongen-Lavrencic et al. 2018).
2.3 Measurable Residual Disease (MRD) in AML and Available Technologies
MRD analysis represents a dynamic evaluation of disease course and has an independent prognostic value, important for risk stratification and treatment design, in combination with other well-established clinical, cytogenetic, and molecular data evaluated at AML diagnosis. Several techniques may be needed, but the results should be integrated in a final laboratory report that covers the different methodologies and maximizes clinically useful information, with the final goal of better addressing personalized treatment approaches.
In this chapter, we will focus on recent methodological advances in MRD assessment in AML, and their inclusion in the decision-making process for personalized treatment (Fig. 2.7) (Schuurhuis et al. 2018).

Methods for detection of minimal residual disease (MRD) in AML. (Adapted from 2018 ELN MRD Working Party documents (Schuurhuis et al. 2018))
2.3.1 RT-qPCR
In AML, molecular MRD evaluation includes the quantification of PML-RARA (Cicconi and Lo-Coco 2016; Sanz et al. 2009), RUNX1-RUNX1T1 (Jourdan et al. 2013), CBFB-MYH11 (Corbacioglu et al. 2010), and mutated-NPM1 (Ivey et al. 2016; Schnittger et al. 2005; Gorello et al. 2006). RT-qPCR methods for the above fusion genes have been standardized by the Europe Against Cancer (EAC) consortium (Gabert et al. 2003). Currently, clinical importance of MRD assessment has been best established in APL, where achievement of molecular remission in BM after consolidation therapy is regarded as a treatment objective (Sanz et al. 2009) and a useful predictor of disease relapse (Grimwade et al. 2009). As of CBF fusion transcripts (RUNX1-RUNX1T1 and CBFB-MYH11), several studies have reported the prognostic value of MRD assessment and quantification after induction therapy (Corbacioglu et al. 2010; Jourdan et al. 2013; Yin et al. 2012). NPM1 mutations are a reliable marker of the disease course and represent an ideal leukemia-specific target for MRD monitoring (Ivey et al. 2016; Krönke et al. 2013; Ossenkoppele and Schuurhuis 2016). In particular, it has been shown that the positivity of NPM1 transcripts after the second chemotherapy cycle has clinical relevance and is associated with a significantly higher relapse risk, independent of other known prognostic factors, when compared to persistent NPM1mut negativity, which is indeed associated with prolonged leukemia-free survival (Ivey et al. 2016).
Based on these findings, the ELN Working Party consensus document on MRD in AML (Dohner et al. 2017) indicates that molecular assessment for NPM1 mutations, RUNX1-RUNX1T1, CBFB-MYH11, and PML-RARA fusion transcripts, should be performed at diagnosis, at least after two cycles of induction/consolidation therapy, and every 3 months, for 24 months after the end of treatment.
2.3.2 Next-Generation Sequencing (NGS)
NGS is an important approach to the molecular dissection of AML at the time of initial diagnosis, mainly in cytogenetically normal AML (Ley et al. 2008). Indeed, different clones, characterized by specific mutations or their combinations, may show variable sensitivity to therapy and distinct relapse tendency. The NGS-based MRD assessment can also identify potentially important changes occurring at the subclonal level during the disease course (Press et al. 2019; Thol et al. 2012; Ravandi 2018). Targeted NGS sequencing provides for profiling of genes of interest and is clinically relevant to dissect the impact of combined gene alterations as potential targets for MRD monitoring (Papaemmanuil et al. 2013, 2016). Indeed, MRD positivity at the time of complete remission (CR) represents an independent prognostic factor for survival (Schlenk 2016). This has been demonstrated by Jongen-Lavrencic and colleagues (Jongen-Lavrencic et al. 2018), who analyzed by targeted-NGS 482 AML patients, at diagnosis and in CR after induction therapy. Mutations persisted in about 50% of patients at the time of CR, and the presence of most mutations was associated with an increased risk of relapse. However, some of the persisting mutations such as DNMT3A, ASXL1, and TET2 (Jongen-Lavrencic et al. 2018), collectively termed DTA, known to be associated with CHIP (Genovese et al. 2014; Zink et al. 2017), did not have a prognostic role. Novel molecular alterations are currently evaluated as targets for MRD assessment. Kohlmann and colleagues quantified RUNX1 gene mutations in a large cohort of AML patients, using an amplicon-based NGS. RUNX1-mutated transcript levels correlated to clinical outcome (Kohlmann et al. 2013). RUNX1-MRD longitudinal assessment could be particularly useful in monitoring disease progression from a myelodysplastic syndrome to secondary AML (Kohlmann et al. 2013; Dicker et al. 2010).
2.3.3 Digital Droplet PCR (ddPCR)
Digital droplet PCR (ddPCR) is a molecular assay with great potential for MRD monitoring due to its high sensitivity and specificity. It is a high-throughput technology that, unlike conventional RT-qPCR, produces an absolute quantification, by amplifying the target genes without a reference standard curve (Coltoff et al. 2018; Ravandi et al. 2018). Indeed, although RT-qPCR assays are nowadays carefully standardized for accurate molecular quantifications (Gabert et al. 2003), since PCR amplification bias can influence reaction efficiency, leading to imprecise genetic quantification. NPM1-mutated monitoring is sometimes difficult due to the presence of several frameshift insertions and lack of information on the mutated sequence at diagnosis. A recent study showed that ddPCR can be used to monitor MRD using multiple NPM1 mutation-specific primers (Mencia-Trinchant et al. 2017). The multiplex assay has an overall excellent concordance with single mutation-specific ddPCR assays, as well as with conventional RT-qPCR. In addition, although the prognostic value of conventional RT-qPCR in APL is well established (Brunetti et al. 2017), ddPCR may also be used to monitor patients at high risk of relapse. In particular, a ddPCR approach may detect mutations associated with arsenic trioxide (ATO) resistance such as the PMLA216V mutation (Alfonso et al. 2019). The identification of the PMLA216V mutation by ddPCR in APL cases at the time of molecular relapse may in the future help anticipate treatment decisions in ATO-resistant patients.
2.3.4 Multiparametric Flow-Cytometry (MFC)
Multiparameter flow cytometry (MFC) represents a great opportunity for MRD monitoring since it is applicable to virtually all patients (>90% of AML) (Buccisano et al. 2010). MFC can significantly contribute to risk assessment of patients with AML during and after treatment, and allows clinicians to consider alternative strategies. The harmonization of the analytical strategies has been recommended by the ELN group (Schuurhuis et al. 2018) and may overcome the concerns about the immunophenotypical shifts that make MRD by MFC a moving target in AML (Zeijlemaker et al. 2014). The application of panels including at least eight colors and the acquisition of a proper number of events minimize the possibility of missing minor populations present at diagnosis that may eventually generate relapse (Schuurhuis et al. 2018). The panel of the ELN MRD working party suggests that to achieve a reliable estimation with a threshold set at 0.1%, the amount of residual leukemic cells by MFC should be determined on a denominator of at least 0.5–1 × 106 cells, excluding debris and CD45 negative cells (Schuurhuis et al. 2018; Buccisano et al. 2018b).
2.4 Classification of Acute Myeloid Leukemia
2.4.1 Background and History
In 1976, the French-American-British (FAB) Cooperative Group set up the first classification of AML that divided AML in seven categories, according to the morphologic and cytochemical features of blasts, coherently with their grade of maturation/differentiation. (Bennett et al. 1976)
With the improvement of diagnostic techniques, the description of the cytogenetic and genetic profiles of the disease was progressively included into the criteria for classifying AML. In 2001, the third edition of the WHO divided AML in four categories using for the first time a combination of clinical, morphologic, immunophenotypic, cytogenetic, and genetic features (Vardiman et al. 2002). The four categories were “AML with recurrent genetic abnormalities,” “AML with multilineage dysplasia,” “AML/MDS therapy-related (t-AML and t-MDS),” and “AML not otherwise categorized (NOC).” In the category of “AML with recurrent genetic abnormalities,” four entities were included, three of them (AML with t(8;21)(q22;q22), with inv(16)(p13q22) or t(16;16)(p13;q22), and APL with t(15;17)(q22;q12)) characterized by a strict correlation between genetic and morphologic features, while abnormalities of 11q23 did not identify a particular morphologic subtype. The diagnosis of “AML with multilineage dysplasia” was based on a documented history of myelodysplastic syndrome (MDS) or a myelodysplastic/myeloproliferative disease (MDS/MPD), present for at least 6 months prior to the onset of AML, or on the presence of at least 50% of dysplastic cells in two or more myeloid lineages. The category of “therapy-related AML/MDS” also included MDS due to the aggressive clinical behavior of MDS in this setting. It was divided in two sub-groups according to the type of previous therapy received to treat the primary tumor or the autoimmune disease, including alkylating agents or radiation therapy, versus topoisomerase II inhibitors. The first type is usually preceded by MDS or may onset as AML with dysplastic features, and presents frequent abnormalities of chromosomes 5 or 7 and poor outcome. Therapy-related MDS/AML following treatment with topoisomerase II inhibitors is often associated with balanced translocations involving chromosome bands 11q23 or 21q22, or other translocations such as inv(16)(p13q22) or t(15;17)(q22;q12). Later editions of the WHO classification erased these subgroups, but we think that it is important to underline that the two subgroups are indeed characterized by distinct biologic features, despite the fact that modern oncologic treatments include combinations of different drugs and new agents. The remaining 2001 WHO category consisted of “AML not otherwise categorized (NOC)” and was divided into different subgroups, mostly following the FAB morphologic classification criteria.
A profound change introduced in 2001 was the reduction in the blast threshold necessary for AML diagnosis from 30 to 20% in the peripheral blood or bone marrow, as a result of a number of studies showing similar clinical behavior of 20–30%-blast MDS and AML. In addition, the recurrent cytogenetic abnormalities t(8;21)(q22;q22), inv(16)(p13q22) or t(16;16)(p13;q22), and t(15;17)(q22;q12) were defined as diagnostic of AML, regardless of the blast percentage.
The fourth edition of WHO Classification of Myeloid Neoplasms and Acute Leukemia published in 2008 added three new categories and brought important changes into the four preexisting ones (Vardiman et al. 2009). The threshold of 20% of blasts and the diagnostic role of one of the abovementioned balanced translocations regardless of the blast percentage were confirmed. In the category of “AML with recurrent genetic abnormalities,” the group of AML with 11q23 abnormalities was better defined as AML with t(9;11)(p22;q23) (MLLT3-MLL rearrangement), while other rearrangements involving the MLL gene identified different biological entities. In APL with t(15;17)(q22;q12) (PML-RARA), variant RARA translocations with partner genes other than PML were recognized as different diseases, particularly for the resistance to all-trans retinoic acid (ATRA). Moreover, three new recurrent abnormalities, including AML with t(6;9)(p23;q34) (DEK-NUP214), AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2) (RPN1-EVI1), and AML (megakaryoblastic) with t(1;22)(p13;q13) (RBM15-MKL1), were recognized as full entities despite their low frequency. Two new provisional entities were added to this category, consistent with the multiple evidences of the prognostic significance of mutations in the NPM1 gene, especially in combination with FLT3-ITD, and CEBPA mutations. The second 2008 category was renamed as “AML with myelodysplasia-related changes (AML-MRC)”. AMLs were included in this group in case of (1) a previous history of MDS or MDS/MPN, and evolution to AML, (2) the presence of myelodysplasia-related cytogenetic abnormalities, or (3) the presence of 50% or more dysplastic cells in at least two myeloid lineages. Concerning the category of “therapy-related myeloid neoplasms,” as previously mentioned, the division into subgroups according to the type of previous therapy was no longer recommended. In parallel, improvements in the diagnostic tools for AML diagnosis reduced the number of cases classifiable as “not otherwise specified (NOS).” Furthermore, three additional categories were included: “myeloid sarcoma,” a tumor mass composed of myeloid blasts, occurring at an anatomical site different form bone marrow and that modifies the normal tissue architecture, “myeloid proliferations related to Down syndrome,” and “blastic plasmacytoid dendritic cell neoplasm.” Myeloid proliferations related to Down syndrome are characterized by specific clinical, morphologic, immunophenotypic, and molecular profiles, including mutation of the GATA1 gene. The inclusion of the “blastic plasmacytoid dendritic cell neoplasm” was due to the recognition of its derivation from precursors of a specialized subset of dendritic cells, the plasmacytoid dendritic cells. For this reason, they were re-classified as AML, as opposed to the third edition of WHO classification, in which they were classified as “blastic NK-cell lymphoma/leukemias.”
2.4.2 The 2016 Revision of the WHO Classification of AML
The 2016 revision of WHO classification of myeloid neoplasms and acute leukemia was an update necessary to incorporate the advancements in the molecular characterization of AML, occurred from 2010 on (Arber et al. 2016). As shown in Table 2.1, the 2016 revision introduced major changes including (Arber et al. 2016) the acknowledgement of AML with mutated NPM1 and AML with biallelic mutations of CEBPA as full entities; and (Papaemmanuil et al. 2013) the introduction of two provisional entities: AML with BCR-ABL1, which must be distinguished from a blastic transformation of CML, and may benefit from tyrosine-kinase inhibitors (TKI) treatment, and AML with mutated RUNX1, associated with poor prognosis. Criteria for defining “AML-MRC” were confirmed, but two points deserve our attention. First, AML with mutated NPM1 or biallelic mutation of CEBPA, associated with multilineage dysplasia, must be classified according to the mutation, since the presence of dysplasia does not affect prognosis in these cases (Falini et al. 2010); second, the cytogenetic abnormality del(9q) has been removed from the AML-MRC category because of its frequent association with mutations of NPM1 and CEBPA. However, in the presence of other MDS-related abnormalities, del(9q) is still included in the “AML-MRC” group (see Table 2.4).
Some changes have also been introduced in the “AML, NOS” category. The erythroleukemia, erythroid/myeloid subtype (previously defined by the presence of ≥50% erythroid precursors counted as proportion of bone marrow nucleated cells, and of ≥20% myeloblasts in non-erythroid cells) has been removed because of similar clinical and genetic features with cases of MDS or AML-MRC. In contrast, pure erythroid leukemia has been maintained as a subtype of “AML, NOS,” defined by the presence of >80% (with ≥30% proerythroblasts) immature erythroid precursors, and myeloblasts <20% of bone marrow nucleated cells (Grossmann et al. 2013).
Minor nomenclature changes concern the definition of the category of “AML with recurrent genetic abnormalities”: (1) APL with t(15;17)(q22;q12) was renamed APL with PML-RARA to emphasize the unique features of this gene fusion; (2) MLL was renamed KMT2A; and (3) inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2), which does not appear to produce a fusion gene, but implies the repositioning of the GATA2 enhancer, driving to deregulation of GATA2 and MECOM genes. The categories of “therapy-related myeloid neoplasms,” “myeloid sarcoma,” “myeloid proliferations related to Down syndrome,” and “blastic plasmacytoid dendritic cell neoplasm” did not change in 2016.
The background and the recent criteria for classification of acute leukemia (AL) of ambiguous lineage will be dealt with in a distinct paragraph.
2.4.3 Rules for AML Classification According to WHO 2016
Sometimes, different entities may overlap in the same patient: the heart of the matter is to prioritize a criterion (clinical, morphologic, immunophenotypic, cytogenetic, or genetic) in order to assign the disease to the right category (Arber 2019).
The first criterion to be taken into consideration to correctly classify AML is patient history. A prior chemotherapy or radiotherapy supersedes every other feature, leading to classification of the disease as a “therapy-related myeloid neoplasm.” In fact, regardless of the genetic/cytogenetic profile, these patients appear to generally have a worse prognosis than those with a corresponding de novo AML (Rowley and Olney 2002), with the exception of CBF-AML (Kayser et al. 2011), and t-APL, whose clinical course is similar to that of de novo APL (Kayser et al. 2017). The same applies to a prior history of MDS or MPN, defining “AML-MRC,” except for AML with inv(3)(q21.3q26.2)/t(3;3)(q21.3;q26.2) or t(6;9)(p23;q34.1), which are classified as AML with recurrent genetic abnormalities.
The second important parameter is the presence of a balanced translocation or gene mutation, characterizing the nine full entities belonging to the category of “AML with recurrent genetic abnormalities.”
In the absence of a history of cytotoxic therapy, or of a recurrent cytogenetic abnormality, detection of balanced or unbalanced aberrations considered associated with MDS defines the disease as “AML-MRC,” which is the third criterion (Table 2.4).
At this point, the role of morphology becomes significant, both for its capability of forewarning of the presence of particular genetic/cytogenetic abnormalities, and the detection of multilineage dysplasia, which, even in the absence of prior MDS or an MDS-related cytogenetic abnormality, leads to the diagnosis of “AML-MRC” (Rozman et al. 2014). Last, when the disease cannot be classified in another category, the morphologic exam of bone marrow and peripheral blood is the only parameter useful in the subcategorization of “AML, NOS” (Walter et al. 2013).
2.4.4 Acute Myeloid Leukemia with Recurrent Genetic Abnormalities
2.4.4.1 AML with t(8;21)(q22;q22.1);RUNX1-RUNX1T1
AML with t(8;21)(q22;q22.1) accounts for 4–8% of cases. This balanced translocation is commonly found in younger patients and in cases with granulocytic maturation, and is associated with a good outcome when treated with intensive consolidation therapy (Al-Harbi et al. 2020).
Usually, the percentage of bone marrow blasts is ≥20%; rarely it could be inferior, but the presence of this translocation is diagnostic for AML, independent of blast percentage. The typical morphologic features are those of the M2 subtype of FAB classification, with large size blasts, and abundant basophilic cytoplasm with the presence of numerous azurophilic granules and perinuclear clearing (hofs). In some cases, blasts show very large granules (pseudo-Chediak-Higashi granules) and Auer rods (Fig. 2.7). Dysplasia is a common finding, but usually it does not affect erythroblasts or megakaryocytes. The percentage of eosinophils, basophils, and mast cells could be increased. The immunophenotype follows the granulocytic differentiation: blasts usually express CD15 and/or CD65, together with immaturity markers such as CD34, MPO, HLA-DR, and CD13. Maturation asynchrony may be observed in the same blast population. Expression of lymphoid-aberrant antigens such as CD19, PAX5, and CD79a is frequent and expression of CD56 has been reported, correlating with worse prognosis (Baer et al. 1997). A cytogenetic analysis may demonstrate co-existing abnormalities, including loss of chromosome X (Chen et al. 2020) or Y (Zhou et al. 2020), del(9q), and trisomy 8.
The t(8;21)(q22;q22.1) generates a chimeric fusion gene, involving the RUNX1 gene on chromosome 21 and the RUNX1T1 gene on chromosome 8. RUNX1, the alpha subunit of the core-binding factor, is a key transcriptional factor crucial for hematopoietic differentiation and myeloid development, while RUNX1T1 is a transcriptional corepressor. In this way, RUNX1-RUNX1T1 works as a repressor for all RUNX1-regulated hematopoietic genes to disrupt normal hematopoietic differentiation and promote a preleukemic state (Goyama and Mulloy 2011). The t(8;21)(q22;q22.1);RUNX1-RUNX1T1 seems to be an early event, and secondary genetic events are needed to develop leukemia. Many other genes are involved in the process of leukemogenesis: 96% of t(8;21) AML cases carry additional cytogenetic or genetic abnormalities (Duployez et al. 2016). The most frequent association is with c-KIT mutations: reported in up to 46% of patients with t(8;21) AML, and associated with unfavorable outcome (Cairoli et al. 2006). FLT3 mutations have been reported in up to 16% of t(8;21) patients, although evidence for their impact on prognosis appears controversial: while FLT3-ITD mutations with a high allelic burden have been associated with poor prognosis, FLT3-TKD mutations seem associated with improved outcome (Christen et al. 2019). Other possible additional mutations concern NRAS/KRAS, CBL, JAK2, and PTPN11 genes, and also epigenetic regulators such as TET2, ASXL1, and ASXL2 (Al-Harbi et al. 2020).
2.4.4.2 AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22);CBFB-MYH11
The inv(16)(p13.1q22) or t(16;16)(p13,1;q22) are found in 5–8% of younger patients with AML, with decreasing prevalence in elderly adults. This AML subtype is characterized by granulocytic and monocytic differentiation, good response to intensive chemotherapy, and low incidence of relapse. Also in these cases, detection of ˂20% bone marrow blasts is infrequent, but similar to t(8;21), the presence of CBFB-MYH11 defines AML per se, independent of blast proportion.
The bone marrow morphologic examination shows typical features of the M4Eo subtype of the FAB classification. Blasts are characterized by myelomonocytic features, in addition to a relevant infiltration of eosinophils at all stages of maturation, without maturation arrest. The immature eosinophilic granules are larger and thicker than normal, and have a typical intense purple-violet color (Swerdlow et al. 2017).
The immunophenotypic evaluation often shows the presence of multiple blast populations, one characterized by immaturity markers such as CD34 and CD117, and others with features belonging to the granulocytic (CD13, CD33, CD15, CD65, and MPO) and/or the monocytic differentiation (CD14, CD4, CD11b, CD11c, CD64, CD36, and lysozyme). Maturation asynchrony may be observed in the same blast population. One antigen aberration frequently detected in this type of AML is the co-expression of CD2 with myeloid markers.
Additional cytogenetic abnormalities have been documented in approximately 40% of cases, including trisomy of chromosomes 22 and 8 (each occurring in 10–15% of cases), and less frequently del(7q) and trisomy of chromosome 21 (Marcucci et al. 2005). Co-existing trisomy 22 seems to be associated with improved outcome, while trisomy 8 has been associated with a worse prognosis.
The translocation or, most frequently, the pericentric inversion of chromosome 16 generates the chimeric fusion gene CBFB-MYH11. The gene MYH11 encodes for the myosin heavy chain, while CBFB encodes for the beta subunit of core-binding factor. The fusion gene encodes for a protein called CBFβ–SMMHC, acting as a dominant negative regulator of transcription, increasing the viability of pre-leukemic myeloid cells, and enhancing their resistance to genotoxic stress (Kuo et al. 2006). As in AML with t(8;21)(q22;q22.1), secondary gene mutations are present in >90% of cases. Mutations of c-KIT are the most frequent, being observed in 30–40% of cases of this AML subtype; other mutations include NRAS (in 45% of cases), KRAS (in 13%), and FLT3 (in 14%), the last one associated with decreased prognosis (Paschka et al. 2013).
2.4.4.3 Acute Promyelocytic Leukemia (APL) with PML/RARA
APL is a distinct subtype of AML, representing 5–8% of AML cases. The median age of APL onset is 35–40 years, but it can occur at any age. The genetic hallmark of APL is the balanced reciprocal t(15;17) translocation, which results in the fusion between the promyelocytic leukemia (PML) and the retinoic acid receptor α (RARA) genes. The disease presentation is frequently associated with a life-threatening coagulopathy that can cause fatal hemorrhages and thrombosis. APL is stratified according to the risk of relapse, based on initial white blood (WBC) and platelet counts at diagnosis. Low/intermediate-risk categories include patients with WBC count ≤10 × 109/L and platelet count <40 × 109/L or > 40 × 109/L in low and intermediate risk, respectively; in the high-risk group, patients present with WBC >10 × 109/L (Sanz et al. 2000).
A rapid diagnosis of APL and the institution of adequate anti-leukemic and supportive care are of relevant importance in preventing early death, which is currently considered the most important issue in the final cure of this disease (Cicconi and Lo-Coco 2016). Morphologically, it is identified as AML-M3 by the French-American-British (FAB) classification (Bennett et al. 1976) and is characterized by a differentiation block resulting in accumulation in the BM of immature, hypergranular promyelocytes with abundant cytoplasm, irregular nuclei with fine azurophilic granules, and Auer rods, often piled up (Faggott cells) in 90% of cases. Morphologically, there are three possible presentations: the classical hypergranular variant, the microgranular variant (hypogranular), and the hyperbasophilic variant. Classical APL promyelocytes are hypergranular, with the possible observation of giant granules that tend to invade all the cytoplasm; the nucleus is bilobed, but sometimes not easily visible due to the high prevalence of granules. Auer rods are frequent (Fig. 2.8). The microgranular variant of APL also presents a bilobed nucleus, while cytoplasm is hypogranular, with a nude perinuclear zone representing the Golgi zone. However, although not frequent, some hypergranular promyelocytes containing Auer rods may be present. The third type of APL, the hyperbasophilic variant, presents with a poor and basophilic cytoplasm, characterized by the presence of blebs (Bain and Bene 2019). In the majority of cases, the diagnosis of APL is suggested by the characteristic morphology of leukemic blasts (Cicconi and Lo-Coco 2016; Sanz et al. 2009). Immunophenotypic evaluation often shows a typical image called “flame-like” in the SSC/CD45 plot: this reflects the morphologic/immunophenotypic features of abnormal promyelocytes that are characterized by hypergranular cytoplasm, and express intermediate levels of CD45. Usually CD34 and HLA-DR antigens are absent or low, while CD13, CD33, CD117, and MPO are strongly expressed (Rahman et al. 2018). Approximately 10% of APL cases express CD56, which has been associated with a decreased outcome. Cytogenetics detects the t(15;17)(q22;q12) translocation in most of cases, leading to PML-RARA fusion gene, between the RARA and PML gene. In some cases, a submicroscopic insertion of RARA into PML has been described: the result is a PML-RARA transcript detectable by molecular studies, but not by cytogenetics. These cases are considered to have cryptic or masked t(15;17)(q22;q12), and are included in the category of APL with PML-RARA (Swerdlow et al. 2017), different from other variant translocations described below. Coexisting cytogenetical abnormalities are frequent and present in almost 40% of cases, with trisomy 8 as the most frequent.

AML with t(8;21)(q22;q22.1);RUNX1-RUNX1T1. Typical large size myeloblasts with abundant basophilic cytoplasm with the presence of numerous azurophilic granules and single Auer rods
Some rare cytogenetic variant involving the RARA gene has been observed. The variant fusion partners may include PLZF at 11q23.2, NPM1 at 5q35.1, NUMA1 at 11q13.4, and STATSB at 17q21.2. Cases with these variant translocations are not true APL and should be classified as “AML with a variant RARA translocation,” since they have different treatment indications and worse prognosis compared to APL.
Confirmation of genetic diagnosis with a rapid PML/RARA genetic test is crucial for patient management. Current methods for genetic confirmation of APL diagnosis include RT-PCR, RT-qPCR, RT-QLAMP, and FISH approaches (Sanz et al. 2019). However, a rapid diagnosis of APL could be confirmed by analyzing the immunocytochemical pattern of the PML protein, using the anti-PML PG-M3 monoclonal antibody (Falini et al. 1997). This assay analyses the nuclear distribution of the PML protein, differentiating the typical “microspeckled pattern” associated with PML/RARA-positivity from the “nuclear body pattern,” characteristic of other leukemias and normal hematopoietic cells (Fig. 2.9). This assay is cheap and useful for rapid diagnosis, available within 2 h (Dimov et al. 2010). However, as reported by ELN guidelines for APL diagnosis (Sanz et al. 2019), RT-PCR represents the “gold standard” for genetic confirmation of APL, as it allows for the identification of the specific PML/RARA isoform (Van Dongen et al. 1999). This information is important for subsequent molecular monitoring of minimal residual disease. Depending on PML breakpoint, usually located in intron 6, exon 6, or intron 3, different PML/RARA transcript isoforms may be generated, that is, long (bcr1), variant (bcr2), and short (bcr3), respectively (Pandolfi et al. 1992). The long and short isoforms are detectable in 95% of APL cases, whereas only 5% harbor the variant form. In contrast, RARA breakpoints are always located within intron 2 (Borrow et al. 1990). The FISH methodology is highly specific and sensitive, and less expensive and time-consuming than karyotyping on G-banded metaphases; thus, it is preferred at diagnosis (Sanz et al. 2009). Once the correct PML/RARA fusion transcript has been identified, RT-qPCR allows for a sensitive assessment of the response to therapy through MRD monitoring during follow-up and early identification of molecular relapse (Gabert et al. 2003; Grimwade et al. 2009). In this setting, APL represents a model for MRD-driven therapy, since molecular relapse is an indication for salvage treatment. Currently, the use of all-trans retinoic acid (ATRA), combined with arsenic trioxide (ATO) or with chemotherapy, induces long-term remissions in at least 85%–90% of patients. However, some patients relapse after ATRA-ATO-based treatments and the mechanisms associated with resistance to these agents are still poorly understood. The A216V mutation in the PML gene has been shown to prevent ATO binding, inhibiting degradation of the oncoprotein, thus hindering oligomerization into nuclear bodies (Zhu et al. 2014). The PMLA216V mutation may be efficiently identified by ddPCR, and PMLA216V is associated with ATO resistance. Additional genetic aberrations such as FLT3 mutations are frequently found in APL: FLT3-ITD occurs in 40% of patients, while FLT3-TKD has been observed in 8% of cases (Breccia et al. 2013). In both cases, a correlation with hyperleukocytosis has been described, and the presence of FLT3-ITD mutations results in the context of ATRA/chemotherapy is associated to reduced response rates and shorter overall survival (Breccia et al. 2013; Picharski et al. 2019). In contrast, the ATRA-ATO combination abrogates the adverse prognostic role of FLT3-ITD mutations in standard-risk APL (Cicconi et al. 2016).

AML with PML/RARA: classic variant. Promyelocytes are characterized by a hypergranular cytoplasm, with the presence of giant granules that tend to invade all the cytoplasm, and multiple Auer rods
2.4.4.4 AML with t(9;11)(p21.3;q23.3);MLLT3-KMT2A
This recurrent genetic abnormality accounts for 9–12% of pediatric and 2% of adult AML cases. Morphologic and immunophenotypic features often follow monoblastic/monocytic differentiation, with overexpression of CD33, CD65, CD4, and HLA-DR, whereas the expression of CD13, CD34, and CD14 is usually low.
The (9;11)(p21.3;q23.3) (MLLT3-KMT2A) translocation involves the KMT2A gene, which encodes for a histone methyltransferase that regulates gene transcription via chromatin remodeling, and the MLLT3 gene, which encodes for AF9, a protein involved in cell growth and maintenance. Secondary additional cytogenetic abnormalities are common, and the most frequent is trisomy of chromosome 8, without clear prognostic significance (Mrozek et al. 1997).
2.4.4.5 AML with t(6;9)(p23;q34.1);DEK-NUP214
AML with t(6;9)(p23;q34.1) (DEK-NUP214) is a rare disease, more frequent in children and younger adults, accounting for 0.7–1.8% of AML cases. It is characterized by poor outcome. Morphologically, this entity may present as an AML with maturation, or sometimes as acute myelomonocytic leukemia. Both peripheral blood and bone marrow are often (44–62% of cases) characterized by an increase in the basophil proportion (≥2%), and signs of multilineage dysplasia can be observed.
The immunophenotypic profile is characterized by high expression of MPO, CD9, CD13, CD33, CD38, CD123, and HLA-DR. The basophil population can be detected and separated for its positivity for CD123, CD33, and CD38, and negativity for HLA-DR (Swerdlow et al. 2017).
The t(6;9) translocation involves the DEK gene at 6p22, and the NUP214 gene (formerly known as CAN), located at 9q34, creating the DEK-NUP214 fusion gene, which acts as an aberrant transcription factor and alters nuclear transport by binding soluble transportins. Moreover, DEK-NUP214 has been reported to enhance protein synthesis in myeloid cells. In most of cases, there are no other cytogenetic abnormalities, but a minor percentage of patients present a complex karyotype. FLT3-ITD has been observed in 42–69% of pediatric and 73–90% of adult AML patients (Kayser et al. 2020).
2.4.4.6 AML with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2);GATA2, MECOM
AML with inv(3)(q21,3q26.2) or t(3;3)(q21.3;q26.2) accounts for 1–2% of all AML and is more common in the adult population. It may often present with normal or even increased platelet counts, and it must be considered a poor prognosis disease.
The morphologic features of bone marrow blasts reflect those of AML without maturation, acute myelomonocytic leukemia or acute megakaryoblastic leukemia. A frequent finding is multilineage dysplasia of non-blast bone marrow cells, especially in megakaryocytes, which are often small non-lobated or bilobated. Megakaryocytic differentiation, when present, may be confirmed by the expression of CD41, CD42, and/or CD61 on blasts. In other cases, markers of immaturity like CD34, CD117, CD13, and CD33 are expressed by the blast population, together with CD7, CD11c, CD11b, and CD123 (Bain and Bene 2019).
The inv(3)(q21.3q26.2) and t(3;3)(q21.3;q26.2) involve the MECOM oncogene at 3q26.2, and a distal GATA2 enhancer, located at 3q21.3. These abnormalities result in the activation of MECOM expression and in GATA2 haploinsufficiency at the same time.
Frequently, these cytogenetic abnormalities are associated with other adverse-risk anomalies, as monosomy of chromosome 7, del(5q), or complex karyotype. The association with BCR-ABL1 positive CML has been described, and it must be considered a marker of accelerated phase or blastic transformation of the disease. Secondary gene mutations are found in almost all cases of AML with inv(3) or t(3;3), with high frequency of NRAS mutations (45.0%), followed by SF3B1 (15.0%), GATA2 (15.0%), FLT3-ITD (10.0%), c-KIT/D816 (5.0%), and CEBPA (5.0%) (Gong et al. 2019).
2.4.4.7 AML (Megakaryoblastic) with t(1;22)(p13.3;q13.3);RBM15-MKL1
AML with t(1;22)(p13,3;q13.1) accounts for <1% of all cases of AML and is typical of infants and young children, with the highest incidence in the first 6 months of life. It is characterized by megakaryoblastic differentiation and hepatosplenomegaly at onset, and it must be considered an aggressive disease.
Morphological examination of bone marrow aspirate usually shows megakaryoblasts with a basophilic agranular cytoplasm and numerous blebs; signs of dysplasia of the other cell lines are infrequent. Fibrosis is a common finding, so that a bone marrow biopsy results helpful or even mandatory for diagnosis.
Immunophenotyping may confirm the megakaryoblastic differentiation through expression of CD41, CD42, and/or CD61. The myeloid-associated markers CD33 and CD13 may also be positive, while CD45, CD34, and HLA-DR are often negative.
In most cases, t(1;22)(p13.3;q13.1) is the sole karyotypic abnormality. Rarely, trisomy of chromosome 21, 19, or 8, may be present, without clear prognostic significance (Inaba et al. 2015).
2.4.4.8 AML with Mutated NPM1
Mutations of the NPM1 gene occur in 2–8% of childhood, and 27%–35% of adult AML, as well as in 45–64% of adult cases with normal karyotype (Swerdlow et al. 2017). Initially described as a favorable-risk entity, in the last few years, AML with mutated NPM1 showed heterogeneous outcome, primarily depending on the presence of co-mutations, and on the MRD status post-consolidation treatment (Ivey et al. 2016).
Most cases of AML with mutated NPM1 present morphologic features of acute myelomonocytic leukemia or acute monocytic leukemia, but characteristics of AML with or without maturation have also been described. The bone marrow is often hypercellular with signs of multilineage dysplasia that, as mentioned above, does not affect prognosis. The immunophenotypic profiling identifies two subgroups: one expressing antigens of monocytic differentiation (CD36, CD64, CD11b, and CD14), and the other with a pattern of myeloblastic differentiation (CD33, CD117, and MPO). CD34 is usually negative and, in a minor percentage, HLA-DR may also be absent (Bain and Bene 2019). Presence of CD34+/CD25+/CD123+/CD99+ blasts is predictive for the presence of FLT3-ITD mutations (Angelini et al. 2015).
AML with mutated NPM1 is usually de novo and has a normal karyotype. However, 5–15% of cases show additional chromosomal abnormalities, including gain of chromosome 8 and del(9q), and adverse-risk karyotypes, which impact prognosis (Angenendt et al. 2019).
Secondary mutations are common in AML with mutated NPM1 and most frequently involve the FLT3 gene (ITD or TKD mutations) and, in 70% of cases, genes regulating DNA methylation, such as DNMT3A (50% of cases), TET2, IDH1, and IDH2 (each occurring in 15% of cases) (Mason et al. 2019). The combination of NPM1 and FLT3-ITD mutations, quantified in terms of ITD allelic ratios >0.5 or <0.5, identifies patients with significantly different outcome, and has been included in the 2017 ELN genetic/cytogenetic risk stratification (Dohner et al. 2017).
2.4.4.9 AML with Biallelic Mutations of CEBPA
Mutations in the CEBPA gene occur in 5–10% cases of AML, mostly in children and younger adults. Biallelic mutations are typically associated with de novo AML, normal karyotype, and favorable outcome.
AML with biallelic mutation of CEBPA does not have typical morphologic features. Similar to AML with mutated NPM1, a possible finding is multilineage dysplasia, without adverse prognostic significance. Cytological features are not specific, but the immunophenotypic profile may be suggestive of this AML subtype. Recently, Mannelli et al. identified a pattern of antigens predictive of CEBPA biallelic mutation, with overexpression of CD34, CD117, HLA-DR, and MPO in blasts, and asynchronous CD15 and CD65 expression. CD64 has also been found overexpressed, not only by blasts but also by granulocytes, and patterns of erythroblast dysplasia with CD117 and CD105 expression associated with low levels of CD36 and CD71 have been described (Mannelli et al. 2017). This immunophenotypic profile suggests further investigation of CEBPA mutations.
Most cases of AML with biallelic mutation of CEBPA have a normal karyotype, but in some patients, other cytogenetic abnormalities may be found, usually del(9q), which has no prognostic impact. Co-mutations of GATA2 and FLT3-ITD occur in 39% and 5–9% cases of AML with biallelic CEBPA mutations, respectively (Swerdlow et al. 2017).
2.4.4.10 AML with t(9; 22)(q34.1;q11.2);BCR-ABL1
AML with BCR-ABL1 is a provisional entity, firstly introduced in 2016 WHO Classification revision but not yet recognized as a full entity. This new group includes de novo AML cases with BCR-ABL1 rearrangements without evidence of a previous CML. The incidence of BCR-ABL1 de novo AML ranges from 0.5 to 3% (Konoplev et al. 2013).
There are no specific morphologic features of myeloblasts, while the presence of peripheral blood basophilia is usually lower than those observed in cases of blastic transformation of CML. Immunophenotypic features include positivity for myeloid antigens of immaturity and lineage aberrations, like CD7, CD19, or TdT. In these cases, it is recommended to exclude the diagnosis of MPAL with BCR-ABL1 (Bain and Bene 2019).
The cytogenetic/genetic profile shows the presence of the translocation t(9;22)(q34.1;q11.2) and/or the BCR-ABL1 fusion gene, in both p210 and p190 types. Other secondary abnormalities include gain or loss of chromosomes or the presence of a complex karyotype. Moreover, cases of AML with BCR-ABL1 and NPM1 or FLT3-ITD mutations have been described. Being a provisional entity, the eventual presence of another recurrent abnormality supersedes in the classification the detection of BCR-ABL1. Treatment strategies in these cases of AML should include the use of tyrosine kinase inhibitors (TKI) (Swerdlow et al. 2017; Neuendorff et al. 2016).
2.4.4.11 AML with Mutated RUNX1
This is the second de novo provisional entity introduced with the 2016 revision of the WHO Classification of AML and is associated with poor prognosis.
RUNX1 gene mutations occur in 6–18% of AML cases. They are also found in about 28% of AML secondary to MDS, and they are often associated with prior radiotherapy or chemotherapy. These latter must be classified, as “AML-MRC” and “therapy-related myeloid neoplasms,” respectively (Yokota et al. 2020). The cytological features often follow those of AML with minimal differentiation, but not exclusively. Immunophenotypic evaluation usually shows expression of markers of immaturity, as CD34, CD13, and HLA-DR, while markers of differentiation, such as CD33 and CD15, are less common (Bain and Bene 2019). The cytogenetic profile is often characterized by alterations of karyotype, including trisomy 8 and trisomy 13 in most cases; additional mutations have been described in 41–95% of AML with RUNX1 mutations, mostly involving FLT3, NRAS, MLL, ASXL1, IDH1/IDH2, TET2, BCOR, DNMT3A, SRSF2, SF3B1, and WT1 genes (Yokota et al. 2020).
2.4.5 AML with Myelodysplasia-Related Changes (AML-MRC)
“AML-MRC” is a WHO category that includes cases with a documented history of MDS or MDS/MPN, or with MDS-related cytogenetic abnormalities, and/or cases with multilineage dysplasia. It accounts for 24–35% of AML with a higher incidence in elderly patients, and is considered a category with poor outcome for its frequent resistance to therapy.
Multilineage dysplasia is defined by the observation of over 50% of dysplastic non-blast cells in two or more hematopoietic cell lineages in bone marrow and/or peripheral blood smears. Features of dysgranulopoiesis include the presence of hyposegmented nuclei and hypogranular cytoplasm, while features of dysmegakaryopoiesis include the presence of normal/large megakaryocytes with non-lobated or multiple nuclei, or micromegakaryocites. Cytological features defining dyserythropoiesis are fragmentation/irregularity of nuclei, megaloblastosis, karyorrhexis, and the presence of ring sideroblasts (Fig. 2.10) (Swerdlow et al. 2017). Hypogranularity of neutrophils, studied with MFC-SSC, is one of the immunophenotypic features considered suggestive of the diagnosis of MDS and “AML-MRC.” Other immunophenotypic characteristics suggesting dysplasia are aberrant differentiation patterns with expression of antigens belonging to different maturative stages, reduction of hematogones, and aberrant expression of lineage-infidelity markers (LIM), such as CD7 and CD56 (Porwit et al. 2014). However, immunophenotype characteristics are not formally included in the diagnostic criteria of AML-MRC.

Patterns of PML nuclear staining. (a) Typical “microspeckled pattern” of two PML/RARA-positive APL samples. (b) “Nuclear bodies pattern” of two PML/RARA-negative samples
Multilineage dysplasia is a sufficient criterion for defining AML-MRC, unless mutations of NPM1 or CEBPA are detected. These cases are then classified as “AML with recurrent genetic abnormalities.” Conversely, detection of an MDS-related karyotype (see Table 2.4) is sufficient to define “AML-MRC,” even in the presence of these mutations. However, NPM1 and CEBPA mutations are very uncommon in this category of AML, while other mutations, such as ASXL1 and TP53, are often observed. Mutations of TP53 occur in up to 70% of cases with complex-karyotype AML, explaining why TP53-mutated cases are included in the AML-MRC category. TP53 mutations typically lead to chemo-resistance and are one of the most important unfavorable prognostic factors in AML (Vardiman and Reichard 2015).
2.4.6 Therapy-Related Myeloid Neoplasms (t-MN)
This category includes both MDS and AML developing after radiation therapy, chemotherapy, or immunomodulating treatment for a previous tumor or autoimmune disease. The definition does not include any criterion of time-to-exposure. It accounts for 10–20% of all AML cases, median age at diagnosis is 64 years, and it generally has a poor outcome, with the exception of CBF-AML and APL (McNerney et al. 2017).
Morphologic, immunophenotypic, and cytogenetic features are often similar to those observed in “AML-MRC,” especially in cases following radiation therapy and/or alkylating agents. These characteristics include multilineage dysplasia, expression of LIM, and aberrations of differentiation antigens, and cytogenetic alterations, mostly affecting chromosomes 5 and 7, or complex karyotype. Other cases, usually preceded by therapy with topoisomerase II inhibitors, are characterized by various morphologic features, including monoblastic or myelomonocytic presentation, with heterogeneous immunophenotypes. Balanced translocations have also been reported in t-MN, mostly involving 11q23 or 21q22.1 rearrangements, but also cases with inv(16) or t(16;16) and t(15;17) have been described. This latter defines APL with PML/RARA, although the correct classification is t-AML with PML/RARA (Swerdlow et al. 2017). As for the genetic profile, mutations of the TP53 gene are very common and have been detected in 80% of cases with del(5q); instead, alterations affecting the RAS pathway are frequently associated with −7/del(7q) cases (Side et al. 2004). TP53 mutations are strongly associated with chemo-resistance and a very poor outcome; other genes frequently mutated are TET2, PTPN11, IDH1/2, NRAS, and FLT3.
2.4.7 AML, Not Otherwise Specified (NOS)
To define the diagnosis of “AML NOS,” it is necessary to rule out other WHO categories according to medical history, and morphology, immunophenotype, and genetics: “AML NOS” includes cases that do not fulfill the criteria for any of the other categories. Morphology and immunophenotyping are crucial for the diagnosis and subclassification, since these features are different for each entity belonging to this category, and indicate the major lineages involved and their degree of maturation/differentiation.
AML with minimal differentiation coincides with FAB classification M0: most commonly, blasts are medium size with agranular cytoplasm and round or indented nuclei, with dispersed chromatin and presence of nucleoli. Cytochemical staining demonstrates the negativity for MPO and Sudan Black B; immunophenotypic features include the expression of markers of immaturity as CD34 and HLA-DR, while antigens of monocytic maturation are absent. Immunophenotypic evaluation is helpful in identifying those cases that are morphologically indistinguishable from acute lymphoblastic leukemias or acute leukemias of ambiguous lineage. About 16–22% FLT3 mutations have been described.
AML without maturation coincides with FAB classification M1 and requires <10% maturing cells of the granulocytic lineage counted as proportion of all the nucleated bone marrow cells. Blasts may have azurophilic granules or may be agranular, looking like lymphoblasts, but MPO and Sudan Black B are positive in about 3% of blasts. Immunophenotypic features include expression of myeloblastic differentiation markers (CD33, CD13, and CD117) and markers of immaturity (CD34 and HLA-DR), while antigens of granulocytic and monocytic maturation are absent; it is possible to find lineage aberration antigens, as CD7, CD2, CD19, or CD56.
AML with maturation coincides with FAB classification M2: for diagnosis, ≥10% maturing cells of the granulocytic lineage and <20% cells with monocytic differentiation counted as proportion of bone marrow cells are required. Morphologic features of blasts are the same described for AML with t(8;21)(q22;q22.1), and this balanced translocation must be excluded. Immunophenotypic characteristics include the expression of myeloid-associated markers with granulocytic differentiation antigens (CD13, CD33, CD65, CD11b, and CD15 positivity); some cases have shown aberrant expression of CD7.
Acute myelomonocytic leukemia coincides with FAB classification M4, and ≥20% cells with granulocytic differentiation and ≥20% with monocytic differentiation are necessary for diagnosis. Morphologic examination shows the same features described for AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22), and this recurrent abnormality has to be excluded by genetic/cytogenetic evaluation for a correct classification. Cytochemical staining with MPO and NSE may be helpful in the diagnosis since these reactions are positive in most of the cases. Immunophenotyping typically shows more than one blast population: one expressing granulocytic differentiation antigens and another expressing monocytic differentiation markers, while in some cases it is possible to identify a third group of blasts expressing immaturity antigens; positivity for CD7 may be revealed.
Acute monoblastic/monocytic leukemia coincides with FAB M5 classification, with >80% of blasts belonging to monocytic lineage, including monoblasts, promonocytes, and monocytes. Monoblasts are typically large, with abundant basophilic cytoplasm and round nuclei with lacy chromatin, and one or more large prominent nucleoli; pseudopods may be observed. Promonocytes have a less basophilic, more granulated cytoplasm, and irregular and delicately convoluted nuclear configuration, sometimes hypersegmented. NSE reaction is positive in 80–90% of cases. Immunophenotyping usually shows expression of myeloid antigens and monocytic differentiation markers, while aberrant presence of CD7 and/or CD56 may be observed. The t(8;16)(p11.2;p13.3) translocation has been associated with acute monocytic leukemia (but also with acute myelomonocytic leukemia), and in most cases, the clinical presentation includes hemophagocytosis by leukemic cells and coagulopathy. Acute monoblastic/monocytic leukemia, in general, may present with bleeding disorders and extramedullary infiltration, especially in the central nervous system (CNS), cutis, and gingiva (Swerdlow et al. 2017).
Pure erythroid leukemia coincides with FAB classification M6 and is characterized by the presence of >80% (with ≥30% proerythroblasts) immature erythroid precursors, and myeloblasts <20% of bone marrow nucleated cells. Pathological erythroblasts have basophilic agranular cytoplasm, round nuclei with fine chromatin, and frequently cytoplasmatic elongated vacuoles that are often positive for periodic acid-Schiff (PAS) reaction. Immunophenotypic features include the expression of CD235a (glycophorin A), CD36, and strong CD71, while CD34 and HLA-DR are usually negative. The prognosis of this entity is particularly poor.
Acute megakaryoblastic leukemia coincides with FAB classification M7 and, for diagnosis, >50% of bone marrow blasts must belong to the megakaryocyte lineage. Morphologic aspects include the presence of megakaryoblasts with blebs and moderately basophilic, agranular cytoplasm; also micromegakaryocytes may be observed, but they must not be included in the leukemic cell count. Since aspiration often results in a dry tap, bone marrow biopsy may be necessary for diagnosis. Immunophenotyping typically shows expression of CD41, CD42b, and/or CD61, and in some cases, aberrant expression of CD7 has been described. For diagnosis, the t(1;22) balanced translocation must be excluded.
Acute basophilic leukemia is a very rare AML in which the primary differentiation of blasts is toward basophils. This entity can be easily recognized by cytological features: the blast cytoplasm results basophilic since it contains a variable number of coarse basophilic granules that are positive for metachromatic staining with toluidine blue. The immunophenotypic profile shows expression of CD123, CD203c, and CD11b in addition to other myeloid antigens, while CD117 is not expressed.
Acute panmyelosis with myelofibrosis (APMF) is a very rare form of de novo AML, associated by definition with the presence of medullary fibrosis: for this reason, bone marrow biopsy with immunohistochemistry is required for diagnosis. The term panmyelosis indicates the presence of a hypercellular bone marrow with an increase in multiple cell lines (erythroid precursors, granulocytic precursors, and megakaryocytes): the multilineage nature of the proliferation may be confirmed by immunohistochemistry, using a panel of antibodies including MPO, lysozyme, megakaryocytic, and erythroid markers (Bain and Bene 2019).
2.4.8 Myeloid Sarcoma
Myeloid sarcoma is a rare AML manifestation. It is defined as a tumor mass composed of myeloid blasts, occurring at an anatomical site different from bone marrow and that modifies the normal tissue architecture, which distinguishes myeloid sarcoma from other types of AML with infiltration by myeloid blasts. Myeloid sarcoma may present without an underlying AML or other myeloid neoplasms in about 25% of cases; more commonly, it may precede or coincide with AML onset or with acute blastic transformation of MDS, MDS/MPN, or MPN. It may also represent the first manifestation of relapse in a patient with previously diagnosed AML, as well as one of the possible complications of allogenic hematopoietic stem cell transplantation (allo-HSCT) (Almond et al. 2017).
About 90% of myeloid sarcoma cases involve a unique site, commonly skin, lymph nodes, gastrointestinal tract, bone, soft tissue, peritoneum, and testes. The diagnosis is based on histological and immunohistochemical evaluation: the absence of a significant blast population assessed by morphologic and immunohistochemical studies, brings to the diagnosis of extramedullary hematopoiesis (myeloid metaplasia), and excludes a myeloid sarcoma. Morphology usually presents blasts with myeloblastic, myelomonocytic, or monoblastic/monocytic features. Frequently, the blastic population mimics a metastatic carcinoma by forming cohesive nests, and/or a linear stretch, surrounded by fibrotic septa. Immunohistochemistry is helpful in distinguishing myeloid sarcoma from solid tumors or lymphomas: CD68-KP1 is the most commonly expressed marker, followed by MPO, CD117, CD99, CD68/PG-M1, lysozyme, CD34, TdT, CD56, CD61, CD30, glycophorin A, and CD4 (Magdy et al. 2019). Cytogenetic alterations have been reported in more than 50% of cases, balanced or unbalanced, including 11q23 rearrangements, t(8;21), monosomy of chromosomes 7 or 16, trisomy of chromosomes 8, 11, or 4, inv(16), and the deletion of (16q), (5q), or (20q). About 16% of cases of myeloid sarcoma stains for NPM1 at the nuclear and cytoplasmic level, reflecting the presence of NPM1 gene mutations; these mutations seem more frequent when studied by NGS, reaching more than 50% of cases (Swerdlow et al. 2017).
2.4.9 Myeloid Proliferations Related to Down Syndrome
In general, individuals affected by Down syndrome have an increased risk of leukemia at all ages. However, in these patients, the probability of developing an AML is high during childhood, and 1–2% of children affected by Down syndrome develop AML before the age of 5 years. Most cases (70%) of Down syndrome associated myeloid leukemia (ML-DS) correspond to a specific subtype of acute megakaryoblastic leukemia, characterized by distinct clinical, morphological, immunophenotypic, and genetic features, including transcription factor GATA1 mutations (absent in the other forms of acute megakaryoblastic leukemia) (Swerdlow et al. 2017).
The other disorder included in this category is transient abnormal myelopoiesis (TAM); it is a pre-leukemic condition that onsets in 10–15% of neonates affected by Down syndrome, spontaneously resolving in most cases within some months. Further 10–20% of patients will develop an ML-DS within the first 5 years of life. Few patients go through life-threatening or fatal complications. GATA1 mutations acquired during fetal life lead to the development of TAM in Down syndrome newborns; in a second phase, GATA1 mutated cells tend to acquire additional transforming mutations in other oncogenes, resulting in ML-DS onset (Labuhn et al. 2019). Both entities are characterized by morphologic and immunophenotypic features belonging to megakaryoblastic differentiation leukemia. In patients affected by ML-DS, additional cytogenetic abnormalities have been described, such as trisomy 8, trisomy 11, del(6q), del(7p), del(16q), and dup(1p) (Bhatnagar et al. 2016).
2.4.10 Blastic Plasmacytoid Dendritic Cell Neoplasm
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a rare type of AML, particularly aggressive, derived from precursors of plasmacytoid dendritic cells; the median age of incidence is 60–70 years old, but it may present at any age, with a prevalence in males. The clinical presentation includes the involvement of cutis, with single or disseminated nodular/popular lesions, and bone marrow in almost all cases; other sites that may be infiltrated are lymph nodes, soft tissue, and SNC (Pagano et al. 2013).
Morphologic features of blasts are very heterogeneous and both myeloid-like and lymphoid-like characteristics are possible findings. In most of cases, blasts are medium sized with basophilic agranular cytoplasm, characterized by the presence of gray zones, with a “granite” or “cloudy sky” coloration; nucleus may be rounded or irregular, peripheral, and containing small nucleoli. A circumferential nuclear rimming by vacuoles (pearl neck appearance), and the presence of pseudopod cytoplasmic extensions may be evident. Immunophenotyping is mandatory to confirm the diagnosis of BPDCN. Blasts usually express CD4 and CD56, but their negativity (infrequent) does not rule out the diagnosis if other PDC-associated antigens (such as CD123, IL3 alpha-chain receptor), CD303, TCL1A, CD2AP, and SPIB) are expressed. Expression of isolated myeloid markers (CD33, CD117, or CD13) and aberrant expression of isolated lymphoid antigens (CD7, CD2, CD22, or CD79a) have been described; in contrast, MPO, CD14, CD64, cCD3, and CD19 are typically negative. As mentioned above, almost all cases of BPDCN present with cutaneous manifestations: histopathological evaluation of cutaneous lesions is an important complementary tool, using PDC-associated markers, such as TCL1, CD2AP, SPIB, TCF4, and MX1 (Garnache-Ottou et al. 2019). More than 50% patients have an altered cytogenetic profile, and in most of cases, it is characterized by abnormalities of chromosomes 5, 6, 9, 11, 12, 13, 15, or complex karyotype. TET2 is the most commonly mutated gene in BPDCN; other mutations affect NPM1, ASXL1, NRAS, ATM, KRAS, IDH2, KIT, ARC, RB1, VHL, BRAE, MLH1, TP53, and RET genes (Swerdlow et al. 2017).
2.5 Classification of Acute Leukemias (AL) of Ambiguous Lineage
AL of ambiguous lineage is a heterogenous group of diseases, including two possible scenarios: (Arber et al. 2016) the absence of lineage-specific (myeloid, B-lymphoid, and T-lymphoid) antigens on blasts, or (Papaemmanuil et al. 2013) the expression of markers of more than one lineage on leukemic cells, resulting in the impossibility to assign the AL to a specific lineage-related category.
The 2008 edition of the WHO classification placed AL of ambiguous lineage in a chapter distinct from AML and ALL, and introduced new criteria to defining the largest subset of cases expressing antigens related to more than one lineage. Cases without lineage-specific markers are named “acute undifferentiated leukemia” (AUL), while the term “mixed-phenotype acute leukemia” (MPAL) has been introduced to collectively include entities previously defined “bi-phenotypic AL” (BAL) and “acute bilineal leukemia” (ABL) (Vardiman et al. 2009).
In the 2016 revision of the WHO classification, the category of AL of ambiguous lineage includes seven subgroups, according to the presence of specific-lineage antigens and genetic abnormalities: AL undifferentiated (AUL); MPAL with t(9;22)(q34.1;q11.2) (BCR-ABL1); MPAL with t(v;11q23.3) (KMT2A rearranged); MPAL, B/myeloid, NOS; MPAL, T/myeloid, NOS; MPAL, NOS, rare types; and AL of ambiguous lineage, NOS (Arber et al. 2016).
This category accounts for only <4% of all AL cases and it is considered an aggressive group of leukemias, with worse prognosis than AML or acute lymphoid leukemia (ALL). MFC is the method of choice to diagnose AUL and MPAL, and a recommended minimum panel of antibodies to is: (1) anti-CD3; (2) anti-CD19 and three other B-specific markers (CD22, CD79a, CD10); (3) anti-MPO and two to three markers associated with the monocytic lineage (CD14, CD11c, CD64, CD36, or anti-lysozyme) (Matutes et al. 2011).
The immunophenotypic criteria for lineage assignment are:
-
1.
myeloid lineage: MPO (by flow cytometry, immunohistochemistry, or cytochemistry) OR monocytic differentiation (>2 of the following: NSE, CD11c, CD14, CD64, lysozyme);
-
2.
T-cell lineage: cytoplasmic CD3 (by flow cytometry with antibodies to CDS epsilon chain; immunohistochemistry using polyclonal anti-CD3 antibody may detect the CD3 zeta chain, which is not T-cell-specific) OR surface CD3 (rare in mixed-phenotype acute leukemias);
-
3.
B-cell lineage (multiple antigens required): strong CD19 expression, with >1 of the following strongly expressed: CD79a, cytoplasmic CD22, CD10 OR weak CD19 with >2 of the following strongly expressed: CD79a, cytoplasmic CD22, CD10.
Immunophenotypic criteria for lineage assignment are used to identify the subgroup of MPAL, B/myeloid, NOS and MPAL, T/myeloid, NOS. Conversely, AUL blasts often express HLA-DR, CD34, and/or CD38, and may be positive for TdT, but by definition, they lack the T-cell and myeloid markers cCD3 and MPO, and also lack B-cell markers such as cCD22, cCD79a, or CD19. Moreover, they do not express the specific antigens of other lineages, such as megakaryocytes or plasmacytoid dendritic cells (Swerdlow et al. 2017).
Genetic and cytogenetic analyses assume an important role in identifying two separated entities: MPAL with t(9;22)(q34.1;q11.2) (BCR-ABL1) and MPAL with t(v;11q23.3) (KMT2A rearranged), with the first one benefiting from TKI-based treatments. In the presence of a recurrent genetic abnormality different from t(9;22)(q34.1;q11.2) and t(v;11q23.3), the AML must be classified following the balanced translocation or mutation (Khan et al. 2018) (Fig. 2.11).

AML with myelodysplasia-related changes (AML-MRC). One myeloblast surrounded by three granulocytes with evident signs of digranulopyesis (hyposegmented nuclei and hypogranular cytoplasm)
References
Abbas S et al (2010) Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116:2122–2126
Alfonso V et al (2019) Early and sensitive detection of PML-A216V mutation by droplet digital PCR in ATO-resistant acute promyelocytic leukemia. Leukemia 33(6):1527–1530. https://doi.org/10.1038/s41375-018-0298-3
Al-Harbi S et al (2020) An update on the molecular pathogenesis and potential therapeutic targeting of AML with t(8;21)(q22;q22.1);RUNX1-RUNX1T1. Blood Adv 4:229–238
Almond LM et al (2017) Myeloid sarcoma: presentation, diagnosis, and treatment. Clin Lymphoma Myeloma Leuk 17:263–267
Angelini DF et al (2015) A leukemia-associated CD34/CD123/CD25/CD99+immunophenotype identifies FLT3-mutated clones in acute myeloid leukemia. Clin Cancer Res 21:3977–3985
Angenendt L et al (2019) Chromosomal abnormalities and prognosis in NPM1-mutated acute myeloid leukemia: a pooled analysis of individual patient data from nine international cohorts. J Clin Oncol 37:2632–2642
Arber DA (2019) The 2016 WHO classification of acute myeloid leukemia: what the practicing clinician needs to know. Semin Hematol 56:90–95
Arber DA et al (2016) The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia. Blood 127:2391–2405
Baer MR et al (1997) Expression of the neural cell adhesion molecule CD56 is associated with short remission duration and survival in acute myeloid leukemia with t(8;21)(q22;q22). Blood 90:1643–1648
Bain BJ, Bene MC (2019) Morphological and immunophenotypic clues to the WHO categories of acute myeloid leukaemia. Acta Haematol 141:232–244
Bennett J et al (1976) Proposals for the classification of the acute Leukaemias. Br J Haematol 33:451–458
Bhatnagar N et al (2016) Transient abnormal myelopoiesis and AML in Down syndrome: an update. Curr Hematol Malig Rep 11:333–341
Borowitz MJ et al (1993) Immunophenotyping of acute leukemia by flow cytometric analysis. Use of CD45 and right-angle light scatter to gate on leukemic blasts in three-color analysis. Am J Clin Pathol 100:534–540
Borrow J et al (1990) Molecular analysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science 249:1577–1580
Breccia M et al (2013) FLT3-ITD confers poor prognosis in patients with acute promyelocytic leukemia treated with AIDA protocols: long-term follow-up analysis. Haematologica 98:e161–e163
Brunetti C et al (2017) Droplet digital PCR is a reliable tool for monitoring minimal residual disease in acute promyelocytic leukemia. J Mol Diagn 19:437–444
Buccisano F et al (2010) Cytogenetic and molecular diagnostic characterization combined to postconsolidation minimal residual disease assessment by flow cytometry improves risk stratification in adult acute myeloid leukemia. Blood 116(13):2295–2303
Buccisano F et al (2018a) Role of minimal (measurable) residual disease assessment in older patients with acute myeloid leukemia. Cancers (Basel) 10
Buccisano F et al (2018b) Minimal residual disease as a biomarker for outcome prediction and therapy optimization in acute myeloid leukemia. Expert Rev Hematol 11:307–313
Cairoli R et al (2006) Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood 107:3463–3468
Chang JH, Olson MO (1990) Structure of the gene for rat nucleolar protein B23. J Biol Chem 265:18227–18233
Chen G et al (2020) Loss of X chromosome predicts favorable prognosis in female patients with t(8;21) acute myeloid leukemia. Leuk Lymphoma 61(5):1168–1177. https://doi.org/10.1080/10428194.2019.1709836
Christen F et al (2019) Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): an international study on 331 patients. Blood 133:1140–1151
Cicconi L, Lo-Coco F (2016) Current management of newly diagnosed acute promyelocytic leukemia. Ann Oncol 27:1474–1481
Cicconi L et al (2016) PML-RAR? Kinetics and impact of FLT3-ITD mutations in newly diagnosed acute promyelocytic leukaemia treated with ATRA and ATO or ATRA and chemotherapy. Leukemia 30:1987–1992
Coltoff A et al (2018) Role of minimal residual disease in the management of acute myeloid leukemia-a case-based discussion. Ann Hematol 97:1155–1167
Corbacioglu A et al (2010) Prognostic impact of minimal residual disease in CBFB-MYH11-positive acute myeloid leukemia. J Clin Oncol 28:3724–3729
Cuneo A et al (1996) Philadelphia chromosome-positive acute myeloid leukemia: cytoimmunologic and cytogenetic features. Haematologica 81:423–427
Del Principe MI et al (2019) Applications and efficiency of flow cytometry for leukemia diagnostics. Expert Rev Mol Diagn 19:1089–1097
Devillier R et al (2015) Role of ASXL1 and TP53 mutations in the molecular classification and prognosis of acute myeloid leukemias with myelodysplasia-related changes. Oncotarget 6:8388–8396
Dicker F et al (2010) Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia 24:1528–1532
Dimov ND et al (2010) Rapid and reliable confirmation of acute promyelocytic leukemia by immunofluorescence staining with an antipromyelocytic leukemia antibody: The M. D. Anderson cancer center experience of 349 patients. Cancer 116:369–376
Ding L et al (2012) Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481:506–510
Dohner K et al (2005) Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 106:3740–3746
Döhner H et al (2010) Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115:453–474
Dohner H et al (2017) Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129:424–447
Duployez N et al (2016) Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 127:2451–2459
Eirín-López JM et al (2006) Long-term evolution and functional diversification in the members of the nucleophosmin/nucleoplasmin family of nuclear chaperones. Genetics 173:1835–1850
Falini B et al (1997) Immunocytochemical diagnosis of acute promyelocytic leukemia (M3) with the monoclonal antibody PG-M3 (anti-PML). Blood 90:4046–4053
Falini B et al (2009) Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia 23:1731–1743
Falini B et al (2010) Multilineage dysplasia has no impact on biologic, clinicopathologic, and prognostic features of AML with mutated nucleophosmin (NPM1). Blood 115:3776–3786
Fasan A et al (2014) The role of different genetic subtypes of CEBPA mutated AML. Leukemia 28:794–803
Frohling S et al (2004) CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol 22:624–633
Gabert J et al (2003) Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—a Europe against cancer program. Leukemia 17:2318–2357
Gaidzik VI et al (2011) RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol 29:1364–1372
Gaidzik VI et al (2012) TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. J Clin Oncol 30:1350–1357
Gale RE et al (2008) The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 111:2776–2784
Garnache-Ottou F et al (2019) How should we diagnose and treat blastic plasmacytoid dendritic cell neoplasm patients? Blood Adv 3:4238–4251
Gary Gilliland D, Griffin JD (2002) The roles of FLT3 in hematopoiesis and leukemia. Blood 100:1532–1542
Genovese G et al (2014) Clonal hematopoiesis and blood-Cancer risk inferred from blood DNA sequence. N Engl J Med 371:2477–2487
Gong X et al (2019) Unusual findings of acute myeloid leukemia with inv(3)(q21q26.2) or t(3;3)(q21;q26.2): a multicenter study. Int J Lab Hematol 41:380–386
Gorello P et al (2006) Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia 20:1103–1108
Goyama S, Mulloy JC (2011) Molecular pathogenesis of core binding factor leukemia: current knowledge and future prospects. Int J Hematol 94:126–133
Grimwade D (2001) The clinical significance of cytogenetic abnormalities in acute myeloid leukaemia. Best Pract Res Clin Haematol 14:497–529
Grimwade D et al (2009) Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J Clin Oncol 27:3650–3658
Grimwade D et al (2010) Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 116:354–365
Grimwade D et al (2016) Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 127:29–41
Grisendi S et al (2006) Nucleophosmin and cancer. Nat Rev Cancer 6:493–505
Grossmann V et al (2013) Acute erythroid leukemia (AEL) can be separated into distinct prognostic subsets based on cytogenetic and molecular genetic characteristics. Leukemia 27:1940–1943
Heuser M et al (2019) How precision medicine is changing acute myeloid leukemia therapy. Am Soc Clin Oncol Educ book 39:411–420
Ichikawa M et al (2004) AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med 10:299–304
Inaba H et al (2015) Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: a retrospective international study. Blood 126:1575–1584
Ivey A et al (2016) Assessment of minimal residual disease in standard-risk AML. N Engl J Med 374:422–433
Jaiswal S et al (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371:2488–2498
Jongen-Lavrencic M et al (2018) Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N Engl J Med 378:1189–1199
Jourdan E et al (2013) Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood 121:2213–2223
Kang Z-J et al (2016) The Philadelphia chromosome in leukemogenesis. Chin J Cancer 35:48
Kayser S et al (2011) The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood 117:2137–2145
Kayser S et al (2017) Characteristics and outcome of patients with therapy-related acute promyelocytic leukemia front-line treated with or without arsenic trioxide. Leukemia 31:2347–2354
Kayser S et al (2020) Allogeneic hematopoietic cell transplantation improves outcome of adults with t(6;9) acute myeloid leukemia: results from an international collaborative study. Haematologica 105:161–169
Khan M et al (2018) An update on classification, genetics, and clinical approach to mixed phenotype acute leukemia (MPAL). Ann Hematol 97:945–953
Kiyoi H et al (2002) Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene 21:2555–2563
Kohlmann A et al (2013) Monitoring of residual disease by next-generation deep-sequencing of RUNX1 mutations can identify acute myeloid leukemia patients with resistant disease. Leukemia 28:129
Konoplev S et al (2013) Molecular characterization of de novo Philadelphia chromosome-positive acute myeloid leukemia. Leuk Lymphoma 54:138–144
Koschmieder S et al (2009) Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J Clin Oncol 27:619–628
Krönke J et al (2013) Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood 122:100–108
Kuo Y-H et al (2006) Cbf beta-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell 9:57–68
Labuhn M et al (2019) Mechanisms of progression of myeloid preleukemia to transformed myeloid leukemia in children with Down syndrome. Cancer Cell 36:123–138.e10
Largeaud L et al (2019) Outcome of AML patients with IDH2 mutations in real world before the era of IDH2 inhibitors. Leuk Res 81:82–87
Leroy B et al (2013) The TP53 website: an integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res 41:D962–D969
Ley TJ et al (2008) DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456:66–72
Lin L-I et al (2006) A novel fluorescence-based multiplex PCR assay for rapid simultaneous detection of CEBPA mutations and NPM mutations in patients with acute myeloid leukemias. Leukemia 20:1899–1903
Magdy M et al (2019) Myeloid sarcoma. Oncol Res Treat 42:224–229
Mannelli F et al (2017) CEBPA-double-mutated acute myeloid leukemia displays a unique phenotypic profile: a reliable screening method and insight into biological features. Haematologica 102:529–540
Marcucci G et al (2005) Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): a cancer and leukemia group B study. J Clin Oncol 23:5705–5717
Mason EF, Hasserjian RP, Aggarwal N, Seegmiller AC, Pozdnyakova O (2019) Blast phenotype and comutations in acute myeloid leukemia with mutated NPM1 influence disease biology and outcome. Blood Adv 3:3322–3332
Matutes E et al (2011) Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood 117:3163–3171
Maurillo L et al (2008) Toward optimization of postremission therapy for residual disease-positive patients with acute myeloid leukemia. J Clin Oncol 26:4944–4951
McNerney ME et al (2017) Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer 17:513–527
Mencia-Trinchant N et al (2017) Minimal residual disease monitoring of acute myeloid leukemia by massively multiplex digital PCR in patients with NPM1 mutations. J Mol Diagn 19:537–548
Mrozek K et al (1997) Adult patients with de novo acute myeloid leukemia and t(9; 11)(p22; q23) have a superior outcome to patients with other translocations involving band 11q23: a cancer and leukemia group B study. Blood 90:4532–4538
Mrozek K et al (2004) Cytogenetics in acute leukemia. Blood Rev 18:115–136
Nacheva EP et al (2013) Does BCR/ABL1 positive acute myeloid leukaemia exist? Br J Haematol 161:541–550
Neuendorff NR et al (2016) BCR-ABL-positive acute myeloid leukemia: a new entity? Analysis of clinical and molecular features. Ann Hematol 95:1211–1221
O’Donnell MR et al (2013) Acute myeloid leukemia, version 2.2013. J Natl Compr Canc Netw 11:1047–1055
Ossenkoppele G, Schuurhuis GJ (2016) MRD in AML: does it already guide therapy decision-making? Hematology Am Soc Hematol Educ Program 2016:356–365
Ottone T et al (2013) Identification of emerging FLT3 ITD-positive clones during clinical remission and kinetics of disease relapse in acute myeloid leukaemia with mutated nucleophosmin. Br J Haematol 161:533–540
Pagano L et al (2013) Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation: an Italian multicenter study. Haematologica 98:239–246
Pandolfi PP et al (1992) Genomic variability and alternative splicing generate multiple PML/RAR alpha transcripts that encode aberrant PML proteins and PML/RAR alpha isoforms in acute promyelocytic leukaemia. EMBO J 11:1397–1407
Papaemmanuil E et al (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368:2209–2221
Papaemmanuil E et al (2016) Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 374:2209–2221
Paschka P et al (2013) Secondary genetic lesions in acute myeloid leukemia with inv(16) or t(16;16): a study of the German-Austrian AML study group (AMLSG). Blood 121:170–177
Perl AE (2019) Availability of FLT3 inhibitors: how do we use them? Blood 134:741–745
Piaton E, et al (2015) [Technical recommendations and best practice guidelines for May-Grunwald-Giemsa staining: literature review and insights from the quality assurance]. Ann Pathol 35:294–305
Picharski GL et al (2019) The impact of Flt3 gene mutations in acute promyelocytic leukemia: a meta-analysis. Cancers (Basel) 11:1311
Porwit A et al (2014) Revisiting guidelines for integration of flow cytometry results in the WHO classification of myelodysplastic syndromes-proposal from the International/European LeukemiaNet Working Group for flow cytometry in MDS. Leukemia 28:1793–1798
Pratcorona M et al (2012) Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica 97:388–392
Press RD et al (2019) Next-generation sequencing-defined minimal residual disease before stem cell transplantation predicts acute myeloid leukemia relapse. Am J Hematol 94:902–912
Rahman K et al (2018) The triple-negative (CD34-/HLA-DR-/CD11b-) profile rapidly and specifically identifies an acute promyelocytic leukemia. Int J Lab Hematol 40:144–151
Ravandi F (2018) Is it time to routinely incorporate MRD into practice? Best Pract Res Clin Haematol 31:396–400
Ravandi F et al (2018) Evaluating measurable residual disease in acute myeloid leukemia. Blood Adv 2:1356–1366
Rosnet O et al (1991) Isolation and chromosomal localization of a novel FMS-like tyrosine kinase gene. Genomics 9:380–385
Rowley JD, Olney HJ (2002) International workshop on the relationship of prior therapy to balanced chromosome aberrations in therapy-related myelodysplastic syndromes and acute leukemia: overview report. Genes Chromosomes Cancer 33:331–345
Rozman M et al (2014) Multilineage dysplasia is associated with a poorer prognosis in patients with de novo acute myeloid leukemia with intermediate-risk cytogenetics and wild-type NPM1. Ann Hematol 93:1695–1703
Sanz MA et al (2000) Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: a joint study of the PETHEMA and GIMEMA cooperative groups. Blood 96:1247–1253
Sanz MA et al (2009) Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 113:1875–1891
Sanz MA et al (2019) Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood 133:1630–1643
Schlenk RF (2016) Is there justification for 4 cycles of consolidation therapy in AML? Best Pract Res Clin Haematol 29:341–344
Schmidt-Zachmann MS et al (1987) A constitutive nucleolar protein identified as a member of the nucleoplasmin family. EMBO J 6:1881–1890
Schnittger S et al (2005) Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 106:3733–3739
Schuurhuis GJ et al (2018) Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD working party. Blood 131:1275–1291
Side LE et al (2004) RAS, FLT3, and TP53 mutations in therapy-related myeloid malignancies with abnormalities of chromosomes 5 and 7. Genes Chromosomes Cancer 39:217–223
Soupir CP et al (2007) Philadelphia chromosome-positive acute myeloid leukemia: a rare aggressive leukemia with clinicopathologic features distinct from chronic myeloid leukemia in myeloid blast crisis. Am J Clin Pathol 127:642–650
Stirewalt DL, Radich JP (2003) The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer 3:650–665
Stirewalt DL et al (2001) FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood 97:3589–3595
Stone RM et al (2017) Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 377:454–464
Sutamtewagul G, Vigil CE (2018) Clinical use of FLT3 inhibitors in acute myeloid leukemia. Onco Targets Ther 11:7041–7052
Swerdlow SH et al (2017) WHO classification of tumors of hematopoietic and lymphoid tissues. IARC
Thiede C et al (2002) Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99:4326–4335
Thol F et al (2012) Next-generation sequencing for minimal residual disease monitoring in acute myeloid leukemia patients with FLT3-ITD or NPM1 mutations. Genes Chromosomes Cancer 51:689–695
Van Dongen JJ et al (1999) Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 concerted action: investigation of minimal residual disease in acute leukemia. Leukemia 13:1901–1928
Vardiman J, Reichard K (2015) Acute myeloid leukemia with myelodysplasia-related changes. Am J Clin Pathol 144:29–43
Vardiman JW et al (2002) The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100:2292–2302
Vardiman JW et al (2009) The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114:937–951
Venditti A et al (2019) GIMEMA AML1310 trial of risk-adapted, MRD-directed therapy for young adults with newly diagnosed acute myeloid leukemia. Blood 134:935–945
Walter RB et al (2013) Significance of FAB subclassification of ‘acute myeloid leukemia, NOS’ in the 2008 WHO classification: analysis of 5848 newly diagnosed patients. Blood 121:2424–2431
Weisser M et al (2007) Advanced age and high initial WBC influence the outcome of inv(3) (q21q26)/t(3;3) (q21;q26) positive AML. Leuk Lymphoma 48:2145–2151
Willekens C et al (2016) Prospective long-term minimal residual disease monitoring using RQ-PCR in RUNX1-RUNX1T1-positive acute myeloid leukemia: results of the French CBF-2006 trial. Haematologica 101:328–335
Yin JAL et al (2012) Minimal residual disease monitoring by quantitative RT-PCR in core binding factor AML allows risk stratification and predicts relapse: results of the United Kingdom MRC AML-15 trial. Blood 120:2826–2835
Yokota A et al (2020) The clinical, molecular, and mechanistic basis of RUNX1 mutations identified in hematological malignancies. Mol Cells 43(2):145–152
Zeijlemaker W et al (2014) Tumor heterogeneity makes AML a ‘moving target’ for detection of residual disease. Cytometry B Clin Cytom 86:3–14
Zhou W et al (2020) Loss of the Y chromosome predicts a high relapse risk in younger adult male patients with t(8;21) acute myeloid leukemia on high-dose cytarabine consolidation therapy: a retrospective multicenter study. Leuk Lymphoma 61(4):820–830. https://doi.org/10.1080/10428194.2019.1683734
Zhu H-H et al (2014) Resistance to arsenic therapy in acute promyelocytic leukemia. N Engl J Med 370:1864–1866
Zink F et al (2017) Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130:742–752
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Voso, M.T., De Bellis, E., Ottone, T. (2021). Diagnosis and Classification of AML: WHO 2016. In: Röllig, C., Ossenkoppele, G.J. (eds) Acute Myeloid Leukemia . Hematologic Malignancies. Springer, Cham. https://doi.org/10.1007/978-3-030-72676-8_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-72676-8_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-72675-1
Online ISBN: 978-3-030-72676-8
eBook Packages: MedicineMedicine (R0)