
Abstract
The World Health Organization (WHO) classification of acute myeloid leukemia (AML) attempts to integrate the biologic aspects of AML derived from cytogenetic and molecular testing with a more conventional morphology-based system. A major area of emphasis in the 2016 revision of the WHO classification is on molecular genetics in cytogenetic normal AMLs (CN-AML). In this context, insight into the underlying causative driver mutations with concurrent genetic modifiers in acute myeloid leukemia, not otherwise specified (AML, NOS ), acute myeloid leukemia with myelodysplasia-related changes (AML-MRC), and therapy-related myeloid neoplasms (t-MNs), a large but genetically heterogeneous AML subtype accounting for over 50% of all AMLs, paves the way for a more precise classification for diagnostic and prognostic purposes. This chapter summarizes the diagnostic criteria; morphologic and immunophenotypic features; cytogenetic alterations in AML, NOS , AML-MRC, and t-MNs; molecular abnormalities identified in over 95% of CN-AMLs; molecular lesions enriched in secondary AMLs, including AML-MRC and t-MNs; as well as their clinical relevance, and the development of novel and more specific therapies targeting these disease-causing mutations.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Acute myeloid leukemia, not otherwise specified
- Myelodysplasia-related changes
- Therapy-related myeloid neoplasm
- Karyotype
- Fluorescence in situ hybridization
- Cytogenetics
- Somatic mutations
- Next-generation sequencing
- Targeted therapy
Introduction
Acute Myeloid Leukemia with Myelodysplasia-Related Changes
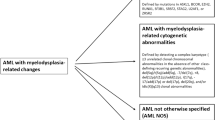
In 2016, the updated World Health Organization (WHO) classification of hematopoietic neoplasms revised this entity minimally, and it remains a subcategory of acute myeloid leukemias (AMLs) entitled “acute myeloid leukemia with myelodysplasia-related changes (AML-MRC).” AML-MRC (Table 3.1) is defined as AML (≥20% blasts in peripheral blood or bone marrow) with a history of myelodysplastic syndrome (MDS) or myelodysplastic/myeloproliferative neoplasm (MDS/MPN), or specific MDS-related cytogenetic abnormalities (Table 3.2), or morphologic features of multilineage myelodysplasia (≥50% dysplastic cells in at least two cell lineages), in the absence of NPM1 or biallelic CEBPA mutations , because the morphologic dysplasia in AML cases with NPM1 or biallelic CEBPA mutations appears not to impact patient outcome [1,2,3,4]. AMLs with NPM1 or biallelic CEBPA mutations have been upgraded from provisional entities in the 2008 WHO classification to full entities in the 2016 WHO classification and are now included amongst AMLs with recurrent genetic abnormalities.
Therapy-Related Myeloid Neoplasm
Therapy-related myeloid neoplasms (t-MNs), including AML, MDS, and MDS/MPN, occur after cytotoxic insults by chemotherapy or radiation therapy (Table 3.3) for a neoplastic or nonneoplastic disorder [5,6,7]. Although t-MNs can be further subclassified to t-MDS or t-AML based on blast counts, all therapy-related neoplasms are best considered as one clinical syndrome [4]. Associated cytogenetic and molecular abnormalities, independent of blast count, are better predictors of disease prognosis and therapy response, and these abnormalities should be incorporated into the final diagnosis [4].
Acute Myeloid Leukemia, Not Otherwise Specified (AML, NOS )
All AML cases that do not fulfill the diagnostic criteria for AML with recurrent genetic abnormalities including NPM1 and biallelic CEBPA mutations, AML-MRC, therapy-related myeloid neoplasm, or myeloid neoplasm with germline predisposition are classified as AML, not otherwise specified (NOS). The basis for subclassification within this category is primarily the morphologic features of blasts, although these morphologic subtypes lack clinical or biologic significance [8, 9]. In the updated 2016 WHO classification, the blast percentage of all AMLs including acute erythroid leukemia is based on total marrow nucleated cells, not nonerythroid cells. The attempt to achieve consistency in calculating blast percentage across all myeloid neoplasms can, at least in part, avoid overdiagnosis of AML primarily due to the low reproducibility of nonerythroid blasts counts. Further, cases previously subcategorized as acute erythroid leukemia, erythroid/myeloid type, share common clinical manifestations, morphologic, and cytogenetic features with cases of MDS [10,11,12,13]. Therefore, this subcategory of acute erythroid leukemia (defined as ≥50% BM erythroid precursors and ≥20% myeloblasts among nonerythroid cells) is removed from acute erythroid leukemia . The majority of such cases are now classified as MDS, especially MDS with excess blasts [4].
Epidemiology
AML-MRC is more common in older patients, with a median age of 68 years, and shows a slight male predominance [14]. The diagnosis of AML-MRC accounts for approximately 25–35% of adult AMLs [8, 15,16,17]. AML-MRC more commonly occurs de novo [14] and may also arise from preexisting myelodysplasia (MDS or MDS/MPN). AML-MRC is generally associated with a worse prognosis and a lower rate of achieving complete remission in response to current chemotherapies compared to other types of AML. The prognosis for AML-MRC is also dependent on other individual factors, such as the patient’s age, blast count [18], cytogenetic abnormalities [19, 20], specific molecular mutations [19, 20], and different therapies [5, 20, 21]. Patients with lower blast counts (20–29%) in the absence of MDS-related cytogenetic abnormalities, specifically in children, have relatively better clinical outcomes [21,22,23]. Recent studies also suggest that allogeneic hematopoietic stem cell transplantation may overcome the poor prognosis for AML-MRC, especially in patients who are 50 years old or younger [18].
Therapy-related myeloid neoplasms account for approximately 10% of AML and 20% of MDS, and are associated with a uniformly poor prognosis with a median survival of 6 months [5,6,7]. Overall, the median time to develop t-MDS/AML following cytotoxic therapy is approximately 3–5 years, with a marked risk reduction after the first decade [6, 7]. There are two types of t-MNs based on their causative therapeutic exposures (Table 3.4): a topoisomerase II inhibitor-related type and an alkylating agent/radiation-related type. Therapy-related myeloid neoplasms associated with chemotherapeutic administration of topoisomerase II inhibitors develop 1–3 years after exposure, and often show no morphologic dysplasia. Alkylating agent/radiation–related t-MNs occur 5–7 years after exposure to cytotoxic agents [6, 7]. Approximately two-thirds of these patients develop a myelodysplastic phase lasting months to years before overt AML. The remainder often presents with AML with myelodysplastic features [5, 6] in addition to myelodysplasia-related cytogenetic abnormalities involving chromosomes 5 (−5/del[5q]) and 7 (−7/del[7q]).
AML, NOS , encompasses a large heterogeneous group, representing 25–30% of adult AML. It occurs at a younger age than AML-MRC, and has an intermediate prognosis [14]. Generally, there are no common clinical features or consistent chromosomal abnormalities identified with this category, and this diagnosis is made by exclusion of other subcategories of AML.
Clinical Features
AML-MRC displays substantial heterogeneity in presentation and clinical course. Adults with AML-MRC often present with severe pancytopenia with a rapid increase in blasts. In contrast, children with a blast percentage between 20% and 29% and preexisting MDS (especially RAEB) experience relatively slow progression and stable blast counts for weeks to months [23]. Therapy-related myeloid neoplasms occurring with a short latency often show abnormalities of either KMT2A at 11q23 or RUNX1 at 21q22, and are often associated with use of topoisomerase II inhibitors (Table 3.4). Longer latency t-MNs often have a prolonged MDS phase, multilineage dysplasia, and MDS-associated karyotypic abnormalities, and these features are more likely to be related to alkylating agents or ionizing radiation (Table 3.4). Patients with AML, NOS usually present with evidence of bone marrow failure and pancytopenias. Beyond medullary symptoms, extramedullary manifestations including soft tissue masses, cutaneous and gingival infiltration, and central nervous system (CNS) involvement are more common in acute monoblastic and monocytic leukemia. Patients with acute basophilic leukemia often have cutaneous involvement, organomegaly, lytic bone lesions associated with hyperhistaminemia, and bone marrow failure.
Morphology and Immunophenotype
Most cases of AML-MRC and t-MNs have morphologic evidence of multilineage dysplasia best evaluated with peripheral blood smears and bone marrow aspirates. Significant dysplastic features are defined as involving at least 50% of the cells of each particular lineage. Blood smears may be particularly useful in identifying myeloid dysplasia, while erythroid and megakaryocytic dysplasia are best evaluated with bone marrow aspirates and biopsies. Many of these dysplastic morphologic features in blood smears and bone marrow are nonspecific for AML-MRC/t-MNs and may be seen in a variety of other neoplastic and nonneoplastic hematologic disorders. Myeloid dysplasia is evidenced, for instance, by hypogranulation and abnormal nuclear segmentation. Dyserythropoiesis is the most common change seen in the bone marrow and includes bizarre erythroid precursors and ring sideroblasts. Dyserythropoiesis is also manifested by karyorrhexis, profound nuclear budding and irregularity, multinucleation, marked megaloblastoid maturation, and cytoplasmic vacuolization. Dysmegakaryopoiesis includes hypolobulated micromegakaryocytes, nonlobulated nuclei or odd numbers of nuclei, and widely spaced nuclei.
t-MN cases following alkylating agents/ionizing radiation often have increased blasts with associated multilineage dysplasia. Approximately 20–30% of t-MN cases, associated with topoisomerase II inhibitors and balanced recurrent chromosomal translocations involving 11q23 (KMT2A/MLL) or 21q22 (RUNX1), show morphologic features similar to those identified in de novo acute monoblastic leukemia or myelomonocytic leukemia (Fig. 3.1).

Bone marrow aspirates in a patient with acute myelomonocytic leukemia. Bone marrow aspirates in a patient who is diagnosed with t-MN with KMT2A translocation
No specific immunophenotypic feature typifies AML-MRC and t-MNs due to the heterogeneity of underlying causes and cytogenetic abnormalities [14]. Nonetheless, there is a general immunophenotypic overlap with AML-MRC and t-MNs, and blasts are CD34+ and CD117+ and express the pan-myeloid markers such as CD13 and CD33 [14, 24]. Aberrant expression of CD7, CD10, and CD56 in blasts is not uncommon. In secondary AMLs with monocytic differentiation, blasts often express CD4 and CD14 and lack CD34. Cases of AML-MRC and t-MNs with abnormalities of chromosomes 5 and 7 may show aberrant expression of TDT and CD7.
Most morphologic subtypes of AML, NOS , are defined by previous FAB criteria, and a 20% marrow blast cell count is sufficient for a diagnosis of acute leukemia [24]. This category includes many cases of AML with a normal karyotype , and mutation analysis, rather than morphologic or immunophenotypic features, and is probably the most predictive prognostic marker in this group.
Cytogenetic Abnormalities
Cytogenetic studies including conventional karyotyping remain a mainstream technology and are required in the evaluation of acute myeloid leukemia and other hematologic and nonhematologic disorders. The value of cytogenetic studies in AML can be divided into four main aspects: diagnosis, prognosis, prediction of therapeutic response, and monitoring minimal residual disease or early recurrence. Conventional karyotyping , although considered somewhat outdated compared to more specific and sensitive methods such as fluorescence in situ hybridization (FISH) , provides a comprehensive view of the genome that cannot be obtained with FISH or other more specific molecular studies, particularly in AMLs with a complex karyotype. Consequently, both conventional karyotyping and FISH remain the routine studies performed for AML diagnoses in clinical laboratories.
Cytogenetic Abnormalities in AML-MRC
The presence of MDS-related cytogenetic abnormalities is sufficient to make a diagnosis of AML with MRC (Table 3.1). A complex karyotype, seven unbalanced chromosomal abnormalities, and nine balanced chromosomal abnormalities are considered diagnostic of AML-MRC (Table 3.2).
Complex Karyotype
A complex karyotype (Fig. 3.2) is defined as three or more unrelated chromosomal abnormalities often leading to loss of genetic material at 5q, 14q33, 7q32q35, and 17p13, thus translating into decreased expression of genes at these chromosomal loci. It is invariably associated with a poor prognosis [25] (Fig. 3.3). Recent studies demonstrated that autosomal monosomy, defined as the presence of two or more distinct autosomal chromosome monosomies or a single autosomal monosomy with one or more other chromosomal abnormalities, is a better predictor for poor prognosis than a complex karyotype in AML [26] (Fig. 3.4). A monosomal karyotype (Fig. 3.5), occurring in 5–10% of AMLs is associated with a dismal prognosis, with a 5-year overall survival of less than 5%. Monosomies 5 and 7, included by MDS-related cytogenetic abnormality and among the most frequent autosomal monosomies in AML, are associated with poor clinical outcomes, which appear to be independent for each specific monosomic chromosome [26].

Bone marrow karyotype from a patient with AML-MRC. Bone marrow from a patient shows a complex aberrant karyotype. A diagnosis of AML-MRC was made based on the cytogenetic findings in addition to morphologic features. *Indicates translocation; * with arrow head indicates absence of the chromosome; arrowhead indicates additional aberrant chromosomal materials

Overall survival of patients with de novo AMLs. Patients with a complex karyotype (red) show a dismal clinical outcome indicated by overall survival in comparison to those with AML with balanced abnormalities or with a normal karyotype (Graph modified from [25])

Overall survival of patients with AMLs and noncore binding factor chromosomal abnormalities. Patients with a monosomal karyotype (MK+, −7, gray and red, 4 year OS 27% vs MK-, yellow and blue, 4-year OS 0–6%, p < 0.001) show a reduced overall survival regardless complex karyotypes (CK). Therefore, a single autosomal monosomy is a better predictor for very poor prognosis than a complex karyotype (Graph modified from [26])

Bone marrow karyotype from a patient with AML-MRC. Bone marrow karyotype from a patient shows a monosomy 7 (* and arrowhead) in addition to deletion 5q (arrow)
Unbalanced Chromosomal Abnormalities
Unbalanced chromosomal abnormalities include, in order of decreasing frequency: deletion of 5q, loss of 7q, loss of 17p, loss of 13q, loss of 11q, and loss of 12p. They result in the loss of genomic integrity (Fig. 3.6). These lesions are the most common genetic aberrance in AML with MRC associated with poor clinical outcomes. In the updated WHO classification, del(9q) is no longer considered a defining MDS-related cytogenetic abnormality for AML-MRC [4]. Del(9q) is among the most frequent abnormalities associated with AML carrying NPM1 or biallelic CEBPA mutations, and the presence of del(9q) is not an independent prognostic factor [2,3,4, 27]. Further, common recurring chromosomal abnormalities in MDS, such as trisomy 8, del(20q), and loss of chromosome Y, are not considered as defining abnormalities in AML-MRC, since they are also associated with AML carrying NPM1 or biallelic CEBPA mutations and they lack prognostic significance in those settings [2,3,4, 27].

Bone marrow karyotype and fluorescence in situ hybridization from a patient with AML-MRC. (a, b) Bone marrow karyotype (a) from a patient shows a deletion 5q (*) and this karyotypic finding is confirmed by FISH employing specific probes (red and green) targeting 5q (b , a a cell with normal karyotype; b a cell with del 5q)
Balanced Chromosomal Abnormalities
Nine balanced chromosomal abnormalities diagnostic of AML-MRC include, in order of decreasing frequency: t(11;16), t(3;21), t(1;3), t(2;11), t(5;12), t(5;7), t(5;17), t(5;10), and t(3;5). Among these balanced gene rearrangements, t(11;16), t(3;21), and t(2;11) are also commonly seen in t-MNs (Fig. 3.7). A previous history of cytotoxic therapy including chemotherapy or radiation warrants a diagnosis of therapy-related myeloid neoplasm instead of AML-MRC. 5q33 is involved in four of the nine gene rearrangements and rearrangement of this locus leads to activation of the platelet-derived growth factor receptor-β (PDGFRB), a member of class III tyrosine kinase receptors (RTKIII) [28]. Two other rearrangements involve the EVI1 locus at 3q26 and the GATA2 and RPN1 loci at 3q21 [26, 29,30,31]. Moreover, two other rearrangements involving a myeloid lymphoid leukemia (MLL or KMT2A) locus at 11q23 (Fig. 3.8), rather than AML with t(9;11), are also diagnostic of AML-MRC [4]. There are over 80 different chromosomal translocations involving KMT2A, and over 50 translocation partner genes have been characterized in adult and pediatric acute leukemias [32,33,34,35,36]. Translocations involving MLLT3 (AF9), resulting predominantly in AML, are the most common. Other KMT2A translocations resulting in AML include MLLT1 (ENL), MLLT10 (AF10), MLLT4 (AF6), and ELL as partner genes. Up to one-third of KMT2A translocations in AMLs are detectable by conventional karyotyping, and FISH or other molecular studies may be necessary to identify these variant translocations [32, 33]. The remaining t(3;5)(q25;q35) rearrangement is relatively unique in AML-MRC, and the overall incidence is less than 0.5% of AMLs [19, 20, 37]. This gene rearrangement leads to the fusion transcript NPM1-MLF1 [38] and tends to occur in young adults. These patients may respond well to stem cell transplants [8, 39].

Bone marrow karyotype from a patient with AML-MRC. Bone marrow karyotype from a patient shows t(2;11)(p21;q23), indicated by arrows, in addition to del(5q)*

Bone marrow fluorescence in situ hybridization from a patient with therapy related myeloid neoplasm (t-MNs). FISH study shows 11q23 rearrangement involving KMT2A gene with different translocations employing break-apart rearrangement probes. 11q23 rearrangement is revealed by segregation of red and green fluorescence labeled probes
Cytogenetic Abnormalities in t-MNs
Therapy-induced AML secondary to alkylating agents is a prototypic AML, in which successive genetic hits occur and result in numerous genetic aberrations. t-MNs associated with alkylating cytotoxic agents/radiation and relatively long latency have abnormal cytogenetics most similar to those identified in AML-MRC [40]. As discussed earlier, these cytogenetic abnormalities are often associated with loss of chromosomes, particularly -5q or -7q, often in the setting of a complex karyotype [6, 7, 40].
In contrast, t-MNs associated with topoisomerase II inhibitors and short latency usually have balanced chromosomal abnormalities involving the KMT2A locus at 11q23 and the RUNX1 locus at 21q22. Translocations involving the KMT2A locus at chromosome 11q23 are found in approximately 6% of t-MN cases.
Some patients with therapy-related AML have karyotypic changes identical to those of de novo AML, including types similar to those of the core binding factor leukemias, inv(16) or APL [41, 42]. In contrast to the dismal prognosis of most therapy-related AMLs, some studies suggest that cases with t(15;17) or inv(16) may have good prognoses similar to their de novo counterparts [41]. A summary of cytogenetic abnormalities in patients with t-MNs is listed in Table 3.5 [43].
Somatic Mutations and Prognosis
The molecular dissection of many nonrandom recurrent gene rearrangements originally detected by conventional karyotyping and FISH has paved the way for the development of more specific and sensitive molecular tools in the diagnosis of AML. Compared to cytogenetic studies, these new approaches, particularly polymerase chain reaction (PCR) and sequencing-based methods, are more timely, sensitive, accurate, and quantitative, and overcome some of the limitations of karyotyping, such as the requirement of fresh material and viable dividing cells. Increasingly, next-generation sequencing (NGS) technologies , also known as high-throughput sequencing, have superseded traditional sequencing methods such as Sanger sequencing and pyrosequencing in clinical laboratories. NGS-based assays have numerous technical advantages over traditional methods, with the capacity to fully and rapidly sequence many genes in a single reaction that requires less DNA input. NGS can detect a large variety of genetic lesions in hematologic malignancies, including point mutations, small and intermediate-size deletions and insertions, copy number variants, and even translocations. The use of NGS-based assays in clinical and research laboratories has led to the discovery of a vast number of new mutations in myeloid neoplasms.
Common Mutations in Cytogenetically Normal Acute Myeloid Leukemia (CN-AML)
A large number of somatic, point, and insertion/deletion mutations have been identified, in addition to those cytogenetically detectable lesions described previously, and these mutations require distinct molecular genetic analyses beyond cytogenetic studies for sensitive and accurate detection. The genes likely to have clinical and prognostic significance in CN-AMLs include FLT3 abnormalities ; KMT2A partial tandem duplications (KMT2A PTD); NPM1, CEBPA, RUNX1, ASXL1/2, IDH1/2, KIT, TET2, RAS, CBL, ND4, and DNMT3A mutations ; and overexpression of a large number of different genes (Table 3.6). Most, but not all, of these mutations are enriched in CN-AMLs and, to a lesser degree, in AML-MRC and t-MNs (Table 3.6). Indeed, almost all AMLs have mutations in one of the eight categories of genes that are highly relevant for myeloid tumorigenesis, including (1) myeloid transcription-factor fusions, (2) the gene encoding nucleophosmin (NPM1), (3) multifunctional tumor-suppressor genes, (4) DNA methylation-related genes, (5) signal transduction genes, (6) chromatin-modifying genes, (7) spliceosome-complex genes, and (8) cohesin-complex genes. Among these genes, some mutations are either cooperative or mutually exclusive, and these specific patterns suggest biologic relationships among many of the genes and categories [44,45,46]. For instance, mutations of FLT3, DNMT3A, and NPM1 are often present simultaneously, while other mutations of NPM1, RUNX1, CEBPA, and TP53 are almost always mutually exclusive both at diagnosis and at the time of disease transformation (Fig. 3.9) [47,48,49,50]. Further, the AML morphologic phenotype might be driven by a more complex pattern of concurrent mutations rather than individual genes.

Molecular heterogeneity and complexity of AML. (a) Circos plots demonstrate the molecular heterogeneity of de novo AMLs and the specific patterns of cooperation and mutual exclusivity. Colored lines indicate concurrent genetic abnormalities. (b) The outer circular segments indicate the particular molecular maturations and the length of each segment indicates their relative frequencies in AML. Outer segments indicate a particular subcohort being positive for the given marker. Ribbon widths indicate relative frequencies of co-occurrences (Graph modified from [50, 143])
A major new area of emphasis in the revised 2016 WHO classification is on molecular genetics in cytogenetically normal AML. The three genes with the mutations most commonly detected and clinically relevant in CN-AMLs are FLT3, NPM1, and CEBPA (Table 3.6). Unlike FLT3 mutations, which can also be identified in AML with recurrent genetic abnormalities and many other AML subtypes, AML with mutations in either NPM1 or biallelic CEBPA are defined as distinct biologic subtypes of AML, and they are considered full AML entities in the most updated WHO classification: AML with mutated NPM1, and AML with biallelic mutations of CEBPA. Mutations in NPM1 and biallelic CEBPA, diagnostic for entities are discussed elsewhere.
Fms-Like Tyrosine Kinase 3 (FLT3)
FLT3 mutations are identified in many AML subtypes (including cases with karyotypic abnormalities), thus FLT3 mutations alone do not define a distinct biologic subtype [46]. Interestingly, mutations in FLT3 and in genes encoding other kinases including tyrosine kinases, serine–threonine kinases, protein tyrosine phosphatases, and RAS family proteins are mutually exclusive in AMLs [46]. FLT3 is a class III receptor tyrosine kinase and Ig receptor superfamily member that is expressed by hematopoietic progenitor cells and downregulated during differentiation. Independent of ligand binding, phosphorylation of regions in the juxtamembranous (JM) domain of FLT3, resulting in a persistent “on” signal, leads to uncontrolled proliferation and inhibition of differentiation and apoptosis via STAT5 and mitogen-activated protein kinase (MAPK) signaling [51,52,53] in transformed leukemic cells. There are two fundamental types of FLT3 abnormalities: internal tandem duplication (ITD) of the JM domain, and a missense mutation at Asp835 (TKD) [54, 55]. FLT3-ITD is relatively more common [54], and occurs in 12–28% of AML, NOS , approximately 8% of AML-MRC, and about 7% of t-MNs (Table 3.6). Point mutations of FLT3-TKD are reported in about 7% of all AML, NOS [54]. Overall, FLT3 mutations, among the most commonly identified somatic abnormalities in AML, are seen in approximately 35% of all AMLs, and are enriched in up to 50% of AMLs with normal cytogenetics [54, 55]. Overactivation of the FLT3 signaling pathway appears to be the single most important prognostic factor for overall survival in AML patients younger than 60 years [53]. It is correlated with a poor prognosis, and this correlation appears to be independent of the karyotypically aberrant AML groups especially in AMLs with mutant NPM1 and DNMT3A [53, 54, 56]. In addition, FLT3-TKD mutations with high allele fraction (>10%), similar to KIT mutations, are associated with a higher cumulative incidence of relapse in core binding factor AML patients [57].
KIT (CD117)
KIT is a member of a type III tyrosine kinase receptor (RTK) family. Mutations in RTKs and their associated pathways in AML result in constitutive activation of downstream signaling cascades. KIT activation is triggered by binding of its ligands, stem cell factors, and receptor dimerization, which in turn facilitates receptor autophosphorylation. Intracellular signal transduction is mediated through multiple signaling pathways, including RAS–RAF–MAPK, JAK-STAT, and PI3K-AKT pathways. Mutations in KIT result in either KIT overexpression or constitutively active tyrosine kinase receptors. This gain of function confers a proliferative and survival advantage to hematopoietic progenitors thus promoting malignant transformation in myeloid cells. Similar to KRAS and NRAS, the critical components of its downstream signaling pathways, KIT mutations can be seen in AMLs with normal cytogenetics (Table 3.6) but are particularly common in core binding factor AMLs, occurring in 7–46% of AMLs with t(8;21) or inv(16) [58]. These mutations tend to be associated with a poor prognosis in these cytogenetically favorable AMLs. In AML, KIT mutations are found in the extracellular domain (exon 8), kinase domain (exon 17, especially KIT D816V mutations), and rarely in transmembrane (exon 10) and juxtamembrane domains (exon 11) [59].
RAS
The RAS (rat sarcoma) gene family encodes approximately 50 structurally homologous proteins which contain a consensus guanosine triphosphate-binding motif, hence the name, small G-protein. RAS proteins, located on the cell surface, function as conduits for RTK signaling through downstream cascades to nuclear transcription factors regulating cell growth and cell-cycling proteins. Mutations in both NRAS (neuroblastoma RAS) and KRAS (Kirsten RAS) have been reported in CN-AML and AML with MRC (Table 3.6). Single-point mutations in RAS lead to constitutive activation by locking RAS into its active conformation or reducing its sensitivity to the GTPase-activating proteins (GAPs), thus resulting in overall gain-of-function effects [60, 61], and almost exclusively occur in “hot spot” codons 12, 13, and 61 [61]. RAS mutations are detected by PCR followed by pyrosequencing and NGS in most clinical laboratories.
Histone-Lysine N-Methyltransferase 2A (KMT2A or MLL)
Partial tandem duplication (PTD) of the KMT2A gene, also known as acute lymphoblastic leukemia 1 (ALL-1) or myeloid/lymphoid or mixed-lineage leukemia (MLL), is another example of a biologically relevant genetic abnormality in AML with normal karyotype. KMT2A is a histone methyltransferase mediating chromatin modification and, as a positive regulator of gene expression for known targets such as HOX genes, plays an essential role in regulating gene expression during embryonic development and hematopoiesis. KMT2A PTD, containing a varied number of exons 5–12 duplicated and inserted before exon 12 giving rise to an in-frame repetition and an elongated protein [62], is associated with an unfavorable outcome [63] in AMLs. Although ~90% of cases with trisomy 11 are associated with KMT2A PTD, it is also present in ~10% of AMLs with normal cytogenetics [62, 64]. This mutation is readily detected by RT-PCR amplifying exons 2–12 in most clinical laboratories [62].
Isocitrate Dehydrogenase 1 and 2 (IDH1/2)
IDH1 and IDH2 are key metabolic enzymes involved in the biosynthesis of central metabolites in the TCA cycle, the major pathway for cellular NADPH generation, and the pentose phosphate pathway. Loss of IDH1/2 function in malignancies impairs oxidative detoxification mechanisms, leading to DNA damage and genomic instability, and thus promoting tumorigenesis, especially in hypoxic setting [65, 66]. IDH converts αKG to (D)-2-hydroxyglutarate (2-HG) using NADPH as cofactors, and 2-HG in turns inhibits histone demethylation by suppressing αKG-dependent dioxygenases. Mutations in IDH are uniformly associated with elevated 2-HG in a vast number of cancers. Inhibition of histone demethylation (hypermethylated histone) and altered DNA methylation patterns can block the differentiation of nontransformed cells and facilitate myeloid neoplastic transformation [67]. The prognostic value of IDH mutations in AML remains unknown since results vary across studies and a recent retrospective analysis found no impact of IDH mutation status on overall survival in patients with AMLs [68].
Additional Sex Combs Like 1 (ASXL1)
The ASXL1 gene encodes a human homologue of the additional sex combs (Asx) gene of Drosophila, located on chromosome 20q11. ASXL1 protein function in human remains largely unknown, although recent studies suggest that ASXL1 may be involved in the regulation of histone methylation and that ASXL1 mutations are an early event contributing to leukemogenesis [69, 70]. ASXL1 mutations are found in 5–12% of CN-AML and are enriched in secondary AMLs (Table 3.6), especially in older patients [71, 72]. ASXL1 mutations, including nonsense or frameshift mutations, are almost exclusively in the “hot spot,” exon 12, leading to disruption of the carboxy-terminal plant homeodomain (PHD) finger domain. Indeed, the mutation c.1934dupG (G646WfsX12) represents half of all ASXL1 mutations and can be easily detected by sequencing. ASXL1 mutations are also closely associated with other cytogenetic and molecular lesions, such as isolated trisomy 8 and RUNX1, and appear to be an independent adverse prognostic factor [73, 74].
DNA Methyltransferases 3A (DNMT3A)
DNA methyltransferases (DNMTs) transfer a methyl group from the universal methyl donor, S-adenosyl-methionine (SAM), to carbon-5 (C5) of cytosine and are predominately responsible for the maintenance of genomic DNA methylation patterns. DNMT3A plays an important role in the maintenance of methylation patterns during primordial germ cell and early embryonic development [75]. Although DNMT3A was originally considered an oncogene in human tumorigenesis, recent studies identified several inactivating DNMT3A mutations and loss of DNMT3A activity in myeloid neoplasms, suggesting that DNMT3A may also behave as a tumor suppressor [76, 77]. Mutations include missense, nonsense, frameshift, and in-frame alterations throughout the gene across all functional domains, although mutations at arginine 882 (R882) in the catalytic domain resulting in impaired enzyme activity are the most common. However, most of the specific mutations identified in AMLs, presumably preleukemic lesions, have not been functionally characterized. DNMT3A mutations rarely occur alone in AMLs, and concurrent mutations in FLT3 /ITD, NPM1, and IDH are common. These DNMT3A mutations show a negative correlation with AMLs with t(8;21), t(15;17), or inv(16). In addition, DNMT3A R882 mutations are associated with advanced age, high leukocyte and blast counts, and morphologic features of monocytic differentiation (FAB M4/M5) [78]. DNMT3A mutations also predict an adverse risk and poor outcome in AML patients [79] and can be detected by sequencing. Further, DNMT3A mutations at R882 can also be potential markers for minimal residual disease studies .
Tet Methylcytosine Dioxygenase 2 (TET2)
As a member of the TET family, TET2 proteins convert 5-methylcytosine to 5-hydroxymethylcytosine (5-hmC) in DNA, with ferrous iron and α-KG as cofactors. By regulating DNA methylation and gene expression of downstream targets, TET2 inhibits cell proliferation, self-renewal, and differentiation. TET2 mutations were first identified in myeloid malignancies via single-nucleotide polymorphism array and comparative genomic-hybridization arrays and confirmed by NGS . Deletion of TET2 on chromosome 4q24 is common in AMLs [80, 81]. TET2 mutations, including frameshift, nonsense, and deletion mutations, are detected in 8–30% of patients with CN-AML, and lead to loss of function, resulting in uncontrolled myeloid proliferation and abnormal differentiation [80, 81] (Table 3.6). The presence of single or double allelic TET2 mutations is positively correlated with mutations of NPM1, DNMT3A , ASXL1 , and RUNX1, but virtually mutually exclusive with IDH mutations. TET2 mutations are also closely associated with older age, higher leukocyte count, normal karyotype, intermediate-risk cytogenetics, and isolated trisomy 8. Although studies regarding the prognostic impact of TET2 mutations in different AML subgroups showed inconsistent results, a meta-analysis of over 2500 patients with de novo, secondary, or therapy-related AML revealed that TET2 mutations appear to be an adverse prognostic factor and independently predict the risk of relapse [82, 83].
Gene Mutations Enriched in AML-MRC and t-MNs
As previously mentioned, secondary AMLs, either AML secondary to a preexisting MDS or to cytogenetic toxic therapy, contain successive genetic hits and are characterized by numerous genetic aberrations. A subset of molecular mutations including SRSF2 , SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, STAG2, and TP53 are relatively specific for this group of secondary AMLs (Table 3.7) compared to those genes described above [19, 20, 40, 84,85,86,87,88,89,90,91,92]. These secondary AMLs have mutations in one of five categories of genes that are relevant for tumorigenesis and possibly for clinical prognosis, including (1) spliceosomes regulating mRNA splicing; (2) chromatin modifying genes; (3) transcription factors; (4) cohesin complex; and (5) TP53, which is a multifunctional tumor suppressor and particularly associated with t-MNs. Almost all of these mutations are detectable by sequencing. Among these genes, mutations in ASXL1 are most common and their clinical significance has been discussed.
Mutations in Splicing Factors
Mutations in spliceosome factor genes are most common in myeloid neoplasms including MDS, AMLs, and chronic lymphocytic leukemia. They are also present in several solid tumors, although at lower frequencies. Dysregulation in alternative mRNA splicing, potentially impacting more than 20,000 human genes, results in malignant transformation and tumorigenesis in various cell types. Prototypic examples of genes with alternative splicing impaired by spliceosome mutations include BCL2 and BCL-X, CD44, and GSK3B [93]. These mutations promote cell proliferation and inhibit apoptosis, thus promoting tumorigenesis. Recent studies demonstrated that somatic mutations in not only downstream targets but also the splicing machinery itself are common mutations driving myeloid neoplasms including AMLs. SRSF2, SF3B1, and U2AF1 frequently acquire heterozygous missense mutations in specific codons leading to gain of function. In contrast, ZRSR2 mutations largely include loss-of-function variants. Commonly identified in MDS and AML with MRC, mutations in one of four genes, which are mutually exclusive accounting for 5–10% of all AMLs [56]. The clinical or prognostic significance of mutations in each individual gene remains unknown. However, lesions in these genes as a group, particularly in combination with accumulated somatic mutations in chromatin regulators, are frequently associated with a low overall survival rate in AMLs. Further, AMLs with mutations in regulators of chromatin, splicing, and transcription show a distinct clinical presentation including dysplastic features in older age and dismal clinical outcome. Therefore, these common and clinically significant mutations should be considered for incorporation into diagnostic and prognostic classification guidelines [56] (Fig. 3.10).

Overall survival of patients with cytogenetic normal AMLs and following five driver mutations. Patients with AML and NPM1 (blue) and biallelic CEBPA mutations (green) show optimal overall survival. A subgroup of AMLs with TP53-aneuploidy has a distinct dismal long term outcome (purple). As compared with other subgroups, patients in the subgroup with chromatin-spliceosome mutations show a poor overall survival (red) and the long term outlook in the IDH2R172 subgroup is similar to that in patients with NPM1-mutated AML (Graph modified from [56])
Serine/Arginine-Rich Splicing Factor 2 Gene (SRSF2)
SRSF2 is an important component in the spliceosome, regulating many steps in RNA-related processes, including spliceosome assembly of the U1 snRNP, U2 snRNP binding at the branch point, and mRNA stabilization [94]. SRSF2 contains an RNA recognition motif and an RS domain rich in arginine and serine residues. Somatic mutations of SRSF2 are predominantly missense mutations at a specific hotspot, codon Pro95. Proline at this codon is replaced by histidine, leucine, or arginine through missense mutations, or disrupted by in-frame insertions and deletions. Commonly identified in MDS (15%) and in chronic myelomonocytic leukemia (CMML, approximately 50%) [95], SRSF2 mutations also preferentially occur in acute myeloid leukemia with preceding MDS or myelodysplastic morphology [90]. All patients with an initial mutation retain the same mutation after AML transformation, and approximately 15% of patients who are initially mutation-negative acquire this type mutation after AML progression. There appears to be no significant difference in the time to leukemic transformation between mutation-positive and -negative patients at the initial diagnosis of MDS [90].
Splicing Factor 3B1 (SF3B1)
This gene encodes subunit 1 of the splicing factor 3b protein complex, which is an essential member of the U2 small ribonucleoprotein complex responsible for branch site recognition near the 3′ end of premessenger RNA. SF3B1 mutations are frequently identified in a specific MDS subtype with refractory anemia and ring sideroblasts (RARS), and in other subtypes of MDS or MDS/MPN overlap syndromes, such as MDS/MPN with ring sideroblasts and thrombocytosis, a new full entity in the updated WHO classification. Ring sideroblasts are a morphological feature especially associated with SF3B1 mutations. Missense substitutions are concentrated at codon 700, in the middle of four contiguous HEAT domains, and result in a gain of function or dominant negative activity, in which the mutant protein suppresses its wild-type counterpart. SF3B1 mutations are associated with a lower rate of transformation to AML and appear to predict an indolent disease in MDS, while the same mutations alone have no detectable prognostic impact in secondary AMLs [91, 96]. In contrast, mutations in splicing factors in combination with SRSF2 , ZRSR2, and U2AF1 mutations appear to predict unfavorable prognosis in patients with de novo acute myeloid leukemia [97].
U2-Complex Auxiliary Factor 1 (U2AF1)
U2AF1 encodes a subunit of the U2 spliceosome required for the binding of U2 snRNP to the pre-mRNA branch site. The encoded protein contains four major functional domains: two zinc-finger regions, an arginine-serine domain, and a U2AF homology domain. U2AF1 binds directly to several other splicing factors important for myeloid neoplastic transformation, including U2AF2, SRSF2, and SF1. There are two hotspots at codons S34 and Q157 where several missense substitutions occur [95]. Small in-frame insertions and deletions around codon 157 have also been identified but at a much lower frequency. These mutations lead to oncogenic gain-of-function phenotypes and directly interfere with RNA binding, resulting in splicing changes [98]. As with other spliceosome components, U2AF1 mutations are associated with AML-MRC and morphologic trilineage dysplasia. Further, U2AF1 mutations alone are an independent prognostic factor and associated with poor clinical outcomes in patients with AMLs [99].
Zinc Finger, RNA-Binding Motif and Serine/Arginine Rich 2 Gene (ZRSR2)
ZRSR2, frequently mutated in myeloid malignancies, is located on the X chromosome and encodes a serine/arginine-rich splicing factor regulating the recognition of the 3′ splice acceptor site as a component of the U2 auxiliary factor heterodimer. In contrast to other splicing factors described above, ZRSR2 mutations include out-of-frame insertions and deletions, splice-site mutations, and nonsense mutations, resulting in prematurely truncated proteins. Missense mutations across all exons also occur. These mutations result in loss of function, and occur in about 5% of patients with MDS, T-cell acute lymphoblastic leukemia, and plasmacytoid dendritic cell neoplasms [100,101,102]. Recent studies demonstrated no direct impact of ZRSR2 mutations on clinical outcomes in patients with MDS, and the clinical relevance of ZRSR2 mutations alone in patients with AMLs remains unknown .
Enhancer of Zeste Homolog 2 (EZH2)
The polycomb repressive complex 2 (PRC2) maintains transcriptional silencing through posttranslational histone modification and regulation of homeotic gene expression [103], and is essential for hematopoiesis and lymphopoiesis in human. Comprising the catalytic subunit of PRC2 is EZH2 or EZH1, which serves as a H3K27 methyltransferase in the complex [103]. Overexpression of EZH2 is frequently identified in solid tumors such as prostate, breast, and endometrial cancers. Gain-of-function mutations in the catalytic domain are also commonly detected in patients with follicular lymphoma and diffuse large B-cell lymphoma [104]. In contrast, loss-of-function mutations across all 20 exons of EZH2 are commonly detected in myeloid malignancies, especially MDS and acute myeloid leukemia. Inactivation of EZH2 leading to loss of H3K27 trimethylation contributes to myeloid neoplasms including MDS, MPN, and MDS/MPN overlap syndrome, but appears to attenuate its predisposition to leukemic transformation secondary to MDS [105,106,107] in animal models. Although frequently found in other myeloid neoplasms associated with a poor prognosis and clinical outcome, the prognostic relevance of EZH2 mutations remains largely unknown in AMLs due to the low detection rate [78, 108].
BCL6 Corepressor (BCOR)
The BCOR gene encodes a POZ/zinc finger transcriptional repressor, also known as an interacting corepressor of BCL6, which is required for germinal center formation and may influence apoptosis in lymphoid tissue. BCOR is a key component in the polycomb repressive complex 1 (PRC1) variant, which inhibits transcriptional activity and regulates early embryonic development, mesenchymal stem cell function, and hematopoiesis [89]. Recent studies demonstrated that BCOR mutations occur in 3.8% of unselected CN-AML patients and are enriched in a substantial fraction (17.1%) of CN-AML patients showing the same genotype as the AML index subjected to whole-exome sequencing. Disruptive somatic BCOR mutations include out-of-frame mutations, small insertions and deletions, nonsense mutations, and splice-site mutations along the BCOR coding exons and exon/intron junctions. BCOR mutations are associated with decreased BCOR mRNA levels, absence of full-length BCOR proteins, and low expression of a truncated BCOR protein. Further mutations in BCOR are closely associated with mutations in other histone modifiers such as DNMT3A , suggesting cooperativity among these genetic alterations, while BCOR mutations are virtually mutually exclusive with NPM1 mutations. BCOR mutations are associated with an unfavorable outcome in patients with CN-AML [89].
Cohesin Complex
The cohesin complex forms a ring structure regulating appropriate chromosomal segregation during mitosis and cell division, double-stranded DNA repair, and transcription. Genes encoding the cohesin complex in somatic vertebrate cells are SMC1A, SMC3, RAD21 (SCC1), STAG2 (SA-2), and STAG1 (SA-1) [109]. Mutations in STAGs or other components in this complex result in chromosomal instability and contribute to myeloid malignant transformation [88]. Indeed, disruptive mutations of the core cohesin subunit STAG2 occurs in a variety of human tumors, including glioblastoma, Ewing sarcoma, melanoma, cervical carcinoma, and hematologic cancers, as STAG2 mutations lead to aneuploidy in a variety of human cancer cells [110]. Mutations throughout all coding exons of STAG2 are common in antecedent MDS, secondary AML with predominantly normal cytogenetics, and de novo AMLs. Concurrent mutations in NPM1, FLT3, DNMT3A, and PTPN11 correlate with STAG2 mutation status. Cohesin complex or STAG2 mutations alone are not independent predictive factors for overall survival or remission rates in AMLs [88, 111].
Tumor Protein 53 (TP53)
The tumor protein 53 (TP53) is a transcription factor and prototypical tumor suppressor that arrests the cell cycle, promotes apoptosis, and coordinates DNA damage repair in response to various cellular stresses and cytotoxic insults. TP53 mutations result in resistance to protein degradation through MDM2, and in abnormal nuclear accumulation detectable by immunohistochemistry. TP53 mutations are found in up to 40% of therapy-related AML [40] and in over 50% of AML with complex cytogenetics. Somatic TP53 mutations are also detected in 10–15% of de novo AML and MDS [91]. A large variety of TP53 mutations have been identified to date, including the point mutations and loss of the TP53 locus at chromosome 17p commonly seen in AMLs. These TP53 mutations can be identified by cytogenetic studies and sequencing. It has been suggested that TP53 mutations are not directly induced by cytotoxic chemotherapy. Rather, they are likely to reflect rare age-related mutations that are resistant to chemotherapy and which expand preferentially following treatment. TP53 mutations are directly correlated with a poor prognosis in patients with MDS and AML, with resistance to chemotherapy and reductions in overall and disease-free survival, even in patients undergoing stem cell transplantation [91]. Moreover, TP53 mutations impart similarly poor prognoses in other malignancies as well. Recent studies suggest that a mutation in TP53 with aneuploidy may represent an acquired causative driver mutation involving discrete evolution pathways in the tumorigenesis of acute myeloid leukemia. AML patients with TP53 mutations and aneuploidy had dismal clinical outcomes, and beyond the current WHO classification , AML with TP53-aneuploidy may be defined as a distinct subtype [56, 87, 99].
Standard and Targeted Therapy
As the molecular landscape of AML becomes more detailed, and we understand how various cytogenetic abnormalities and somatic mutations affect the development of AML, the role of targeted therapies will become more critical. While standard therapies for AML include induction with cytarabine and an anthracycline drug such as daunorubicin followed by consolidation therapy with cytarabine and/or stem cell transplant, here we discuss mainly targeted therapies, again which are gaining momentum in use.
In the case of DNMT3A , which is commonly mutated in AML, nonspecific inhibitors of DNMT, are already commonly used, such as azacitidine and decitabine, and, in preliminary studies, provide better responses in older patients harboring mutations in epigenetic modifiers including DNMT3A [112, 113]. DNMT inhibitors may be a superior treatment option for older patients ineligible for intensive chemotherapy. Additional large-scale studies are warranted.
Many targeted therapies focus on mutations in kinase proteins (FLT3 , KIT). For instance, FLT3 is considered a potentially important target for leukemia therapy [54]. Although no FLT3-specific inhibitor has been developed to date, many tyrosine kinase inhibitors (TKIs) appear to inhibit FLT3, and several potential FLT3 inhibitors are being evaluated in clinical trials [54, 114,115,116,117]. Midostaurin (PKC412, Novartis Pharmaceuticals, East Hanover, NJ, USA), a benzoylstaurosporine, is a first-generation FLT3 inhibitor, and inhibits the kinase activities of both FLT3/ITDs and FLT3/KDMs, as well as wild-type FLT3 [118]. Phase 2 studies of PKC412, alone or in combination with conventional chemotherapeutic agents in patients with AML or MDS, have shown that patients with FLT3 mutations but not previously exposed to other FLT3 inhibitors realize the greatest benefit, although the duration of remission was short in some patients [119,120,121]. United States Food and Drug Administration (FDA) granted Breakthrough Therapy designation to PKC412 (midostaurin) recently primarily based upon the positive results from the Phase III RATIFY (CALGB 10603) clinical trial. Patients who received PKC412 (midostaurin) and standard induction and consolidation chemotherapy experienced a significant improvement in overall survival (OS) (hazard ratio = 0.77, P = 0.0074) compared with those who received standard induction and consolidation chemotherapy alone. The median OS for patients in the PKC412 (midostaurin) treatment group was 74.7 months (95% confidence interval [CI]: 31.7, not attained), versus 25.6 months (95% CI: 18.6, 42.9) for patients in the placebo group. Unfortunately, PKC412 alone has insufficient activity in AML patients, and off-target effects such as pulmonary edema are concerning. CEP-701 (Cephalon, Frazer, PA, USA), another relatively-specific first-generation inhibitor targeting FLT3 kinases, showed a transient decrease in circulating blasts [122] in patients with relapsed or refractory acute myeloid leukemia without achieving a complete remission [122, 123]. In recent years, a new generation of more potent and specific FLT3 inhibitors has been developed, with AC220 [124] and G-749 [125] as leading examples. These newer agents have shown significant promise in early phases of clinical investigation including animal models and are currently in more advanced clinical trials [123,124,125].
TKIs have been used in combination with conventional chemotherapy in KIT-positive relapsed/refractory AML and are in clinical trials [126]. Imatinib combined with traditional chemotherapy reagents including mitoxantrone, etoposide, and cytarabine in patients with KIT-positive relapsed/refractory AML achieved a complete response in approximately 60% of patients [127, 128]. A multicenter phase 2 trial for older patients with KIT-positive AML, who are not optimal candidates for intensive induction therapy, showed that imatinib combined with low-dose cytarabine achieved an 11% hematologic response [129]. However, in most of these studies, the KIT mutation status was evaluated by flow cytometry rather than molecular studies. In contrast, dasatinib alone as a maintenance therapy in patients with high-risk AMLs who achieved complete remission 1 (CR1) showed a poor 2-year disease-free survival rate of 25% [130].
Other targeted therapies may be exquisitely protein specific such as IDH inhibitors [65, 66], which are currently being assessed in clinical trials. Other therapies may be somewhat indirect and target a step in the pathway involving the gene of interest. For instance, RAS is another gene mutated highly in AML and since RAS activity is dependent on posttranslational farnesylation, specific inhibitors targeting farnesyltransferase have been examined in clinical trials involving patients with AML [131]. Overall, responses to farnesyltransferase inhibitors in AMLs have not been encouraging. In a trial of 348 elderly patients with AML (aged ≥70 years) who received tipifarnib, less than 20% achieved a complete remission [132,133,134]. RAS signaling also activates downstream targets, most notably mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)–AKT through a variety of pathways to promote cell proliferation. Combination therapy with inhibitors targeting the downstream RAS effectors, MEK and AKT, is currently being investigated in clinical trials in AML patients with RAS mutations [135, 136].
In the case of MLL/KMT2A, it is known that DOT1L, a histone methyltransferase, is required for the development and maintenance of KMT2A-rearranged leukemias [137]. Preclinical studies have shown potential clinical utility of DOT1L inhibition in AMLs [137,138,139], and small-molecule and competitive inhibitors of DOT1L have been developed (EPZ-5676; Epizyme Inc., Cambridge, MA, USA) which preliminarily show some promising responses, including complete morphologic and cytogenetic remissions, and resolution of leukemia cutis and treatment-related increases in neutrophils and/or monocytes [140].
Finally, modulation of the spliceosome complex may provide a new therapeutic approach in patients with MDS or AML containing spliceosome gene mutations; but studies on specific inhibitors targeting splicing factors in AMLs remain in the preclinical stage. E7107, a structurally distinct splicing inhibitor, is the only compound that specifically inhibits SF3B1, and has been tested in clinical trials. Preliminary data suggest that SF3B1 inhibition has therapeutic potential for the treatment of solid tumors with SF3B1 mutations. Additional clinical studies with new inhibitors targeting splicing factors for patients with myeloid neoplasms are needed [141, 142].
Conclusions
As our understanding of the molecular landscape of AML continues to grow, so too will our subclassification of this disease. Additionally, while such detailed understanding drives our diagnostic categorization of this disease, perhaps more importantly it also informs us of the mechanisms driving AML. Importantly, with an understanding of the mechanisms of this disease, continual development of specific targeted therapies is possible which will continue to positively change the course of this disease.
References
Schlenk RF, Taskesen E, van Norden Y, Krauter J, Ganser A, Bullinger L, et al. The value of allogeneic and autologous hematopoietic stem cell transplantation in prognostically favorable acute myeloid leukemia with double mutant CEBPA. Blood. 2013;122(9):1576–82.
Haferlach C, Mecucci C, Schnittger S, Kohlmann A, Mancini M, Cuneo A, et al. AML with mutated NPM1 carrying a normal or aberrant karyotype show overlapping biologic, pathologic, immunophenotypic, and prognostic features. Blood. 2009;114(14):3024–32.
Falini B, Macijewski K, Weiss T, Bacher U, Schnittger S, Kern W, et al. Multilineage dysplasia has no impact on biologic, clinicopathologic, and prognostic features of AML with mutated nucleophosmin (NPM1). Blood. 2010;115(18):3776–86.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Pedersen-Bjergaard J, Andersen MK, Christiansen DH. Therapy-related acute myeloid leukemia and myelodysplasia after high-dose chemotherapy and autologous stem cell transplantation. Blood. 2000;95(11):3273–9.
Sill H, Olipitz W, Zebisch A, Schulz E, Wölfler A. Therapy-related myeloid neoplasms: pathobiology and clinical characteristics. Br J Pharmacol. 2011;162(4):792–805.
Bhatia S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin Oncol. 2013;40(6):666–75.
Arber DA, Stein AS, Carter NH, Ikle D, Forman SJ, Slovak ML. Prognostic impact of acute myeloid leukemia classification. Importance of detection of recurring cytogenetic abnormalities and multilineage dysplasia on survival. Am J Clin Pathol. 2003;119(5):672–80.
Tallman MS, Kim HT, Paietta E, Bennett JM, Dewald G, Cassileth PA, et al. Acute monocytic leukemia (French-American-British classification M5) does not have a worse prognosis than other subtypes of acute myeloid leukemia: a report from the eastern cooperative oncology group. J Clin Oncol. 2004;22(7):1276–86.
Hasserjian RP, Zuo Z, Garcia C, Tang G, Kasyan A, Luthra R, et al. Acute erythroid leukemia: a reassessment using criteria refined in the 2008 WHO classification. Blood. 2010;115(10):1985–92.
Liu W, Hasserjian RP, Hu Y, Zhang L, Miranda RN, Medeiros LJ, Wang SA, et al. Pure erythroid leukemia: a reassessment of the entity using the 2008 World Health Organization classification. Mod Pathol. 2011;24(3):375–83.
Porwit A, Vardiman JW. Acute myeloid leukemia with expanded erythropoiesis. Haematologica. 2011;96(9):1241–3.
Grossmann V, Bacher U, Haferlach C, Schnittger S, Pötzinger F, Weissmann S, et al. Acute erythroid leukemia (AEL) can be separated into distinct prognostic subsets based on cytogenetic and molecular genetic characteristics. Leukemia. 2013;27(9):1940–3.
Weinberg OK, Seetharam M, Ren L, Seo K, Ma L, Merker JD, et al. Clinical characterization of acute myeloid leukemia with myelodysplasia-related changes as defined by the 2008 WHO classification system. Blood. 2009;113(9):1906–8.
Haferlach T, Schoch C, Löffler H, Gassmann W, Kern W, Schnittger S, et al. Morphologic dysplasia in de novo acute myeloid leukemia (AML) is related to unfavorable cytogenetics but has no independent prognostic relevance under the conditions of intensive induction therapy: results of a multiparameter analysis from the German AML cooperative group studies. J Clin Oncol. 2003;21(2):256–65.
Miyazaki Y, Kuriyama K, Miyawaki S, Ohtake S, Sakamaki H, Matsuo T, et al. Cytogenetic heterogeneity of acute myeloid leukaemia (AML) with trilineage dysplasia: Japan adult Leukaemia study group-AML 92 study. Br J Haematol. 2003;120(1):56–62.
Yanada M, Suzuki M, Kawashima K, Kiyoi H, Kinoshita T, Emi N, et al. Long-term outcomes for unselected patients with acute myeloid leukemia categorized according to the World Health Organization classification: a single-center experience. Eur J Haematol. 2005;74(5):418–23.
Ikegawa S, Doki N, Kurosawa S, Yamaguchi T, Sakaguchi M, Harada K, et al. Allogeneic hematopoietic stem cell transplant overcomes poor prognosis of acute myeloid leukemia with myelodysplasia-related changes. Leuk Lymphoma. 2016;57(1):76–80.
Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013;122(25):4021–34.
Malcovati L, Ambaglio I, Elena C. The genomic landscape of myeloid neoplasms with myelodysplasia and its clinical implications. Curr Opin Oncol. 2015;27(6):551–9.
Boogaerts MA. Stem cell transplantation and intensified cytotoxic treatment for myelodysplasia. Curr Opin Hematol. 1998;5(6):465–71.
Emanuel PD. Myelodysplasia and myeloproliferative disorders in childhood: an update. Br J Haematol. 1999;105(4):852–63.
Davis KL, Marina N, Arber DA, Ma L, Cherry A, Dahl GV, et al. Pediatric acute myeloid leukemia as classified using 2008 WHO criteria: a single-center experience. Am J Clin Pathol. 2013;139(6):818–25.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–51.
Schoch C, Kern W, Kohlmann A, Hiddemann W, Schnittger S, Haferlach T. Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosomes Cancer. 2005;43(3):227–38.
Breems DA, Van Putten WL, De Greef GE, Van Zelderen-Bhola SL, Gerssen-Schoorl KB, Mellink CH, et al. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol. 2008;26(29):4791–7.
Pabst T, Eyholzer M, Fos J, Mueller BU. Heterogeneity within AML with CEBPA mutations; only CEBPA double mutations, but not single CEBPA mutations are associated with favourable prognosis. Br J Cancer. 2009;100(8):1343–6.
Heldin CH, Lennartsson J. Structural and functional properties of platelet-derived growth factor and stem cell factor receptors. Cold Spring Harb Perspect Biol. 2013;5(8):a009100.
Lugthart S, van Drunen E, van Norden Y, van Hoven A, Erpelinck CA, et al. High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood. 2008;111(8):4329–37.
Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BA, Erpelinck C, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157(2):369–81.
Yamazaki H, Suzuki M, Otsuki A, Shimizu R, Bresnick EH, Engel JD, Yamamoto M. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell. 2014;25(4):415–27.
Meyer C, Schneider B, Jakob S, Strehl S, Attarbaschi A, Schnittger S, et al. The MLL recombinome of acute leukemias. Leukemia. 2006;20(5):777–84.
Shih LY, Liang DC, Fu JF, Wu JH, Wang PN, Lin TL, et al. Characterization of fusion partner genes in 114 patients with de novo acute myeloid leukemia and MLL rearrangement. Leukemia. 2006;20(2):218–23.
Byrd JC, Mrózek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from cancer and leukemia group B (CALGB 8461). Blood. 2002;100(13):4325–36.
Forestier E, Heim S, Blennow E, Borgström G, Holmgren G, Heinonen K, et al. Cytogenetic abnormalities in childhood acute myeloid leukaemia: a Nordic series comprising all children enrolled in the NOPHO-93-AML trial between 1993 and 2001. Br J Haematol. 2003;121(4):566–77.
Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23(8):1490–9.
Miesner M, Haferlach C, Bacher U, Weiss T, Macijewski K, Kohlmann A, et al. Multilineage dysplasia (MLD) in acute myeloid leukemia (AML) correlates with MDS-related cytogenetic abnormalities and a prior history of MDS or MDS/MPN but has no independent prognostic relevance: a comparison of 408 cases classified as “AML not otherwise specified” (AML-NOS) or “AML with myelodysplasia-related changes” (AML-MRC). Blood. 2010;116(15):2742–51.
Lim G, Choi JR, Kim MJ, Kim SY, Lee HJ, Suh JT, et al. Detection of t(3;5) and NPM1/MLF1 rearrangement in an elderly patient with acute myeloid leukemia: clinical and laboratory study with review of the literature. Cancer Genet Cytogenet. 2010;199(2):101–9.
Arber DA, Chang KL, Lyda MH, Bedell V, Spielberger R, Slovak ML, et al. Detection of NPM/MLF1 fusion in t(3;5)-positive acute myeloid leukemia and myelodysplasia. Hum Pathol. 2003;34(8):809–13.
Shih AH, Chung SS, Dolezal EK, Zhang SJ, Abdel-Wahab OI, Park CY, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica. 2013;98(6):908–12.
Andersen MK, Larson RA, Mauritzson N, Schnittger S, Jhanwar SC, Pedersen-Bjergaard J. Balanced chromosome abnormalities inv(16) and t(15;17) in therapy-related myelodysplastic syndromes and acute leukemia: report from an international workshop. Genes Chromosomes Cancer. 2002;33(4):395–400.
Borthakur G, Lin E, Jain N, Estey EE, Cortes JE, O'Brien S, et al. Survival is poorer in patients with secondary core-binding factor acute myelogenous leukemia compared with de novo core-binding factor leukemia. Cancer. 2009;115(14):3217–21.
Qian Z, Joslin JM, Tennant TR, Reshmi SC, Young DJ, Stoddart A, et al. Cytogenetic and genetic pathways in therapy-related acute myeloid leukemia. Chem Biol Interact. 2010;184(1–2):50–7.
Takahashi S. Current findings for recurring mutations in acute myeloid leukemia. J Hematol Oncol. 2011;4:36.
Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127(1):42–52.
Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Larsson CA, Cote G, Quintas-Cardama A. The changing mutational landscape of acute myeloid leukemia and myelodysplastic syndrome. Mol Cancer Res. 2013;11(8):815–27.
Hou HA, Chou WC, Kuo YY, Liu CY, Lin LI, Tseng MH, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015;5:e331.
Miller CA, Wilson RK, Ley TJ. Genomic landscapes and clonality of de novo AML. N Engl J Med. 2013;369(15):1473.
Sanders MA, Valk PJ. The evolving molecular genetic landscape in acute myeloid leukaemia. Curr Opin Hematol. 2013;20(2):79–85.
Drexler HG, Meyer C, Quentmeier H. Effects of FLT3 ligand on proliferation and survival of myeloid leukemia cells. Leuk Lymphoma. 1999;33(1–2):83–91.
Meyer C, Drexler HG. FLT3 ligand inhibits apoptosis and promotes survival of myeloid leukemia cell lines. Leuk Lymphoma. 1999;32(5–6):577–81.
Voutsadakis IA. Flt3 in acute myelogenous leukemia: biology, prognosis, and therapeutic implications. Med Oncol. 2003;20(4):311–24.
Kiyoi H, Naoe T. Biology, clinical relevance, and molecularly targeted therapy in acute leukemia with FLT3 mutation. Int J Hematol. 2006;83(4):301–8.
Kiyoi H, Naoe T. FLT3 mutations in acute myeloid leukemia. Methods Mol Med. 2006;125:189–97.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21.
Duployez N, Marceau-Renaut A, Boissel N, Petit A, Bucci M, Geffroy S, et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 2016;127(20):2451–2459.
Yohe S. Molecular genetic markers in acute myeloid leukemia. J Clin Forensic Med. 2015;4(3):460–78.
Becker H, Pfeifer D, Afonso JD, Nimer SD, Veelken H, Schwabe M, Lübbert M. Two cell lines of t(8;21) acute myeloid leukemia with activating KIT exon 17 mutation: models for the ‘second hit’ hypothesis. Leukemia. 2008;22(9):1792–4.
Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE. 2004;2004(250):RE13.
Johnson DB, Smalley KS, Sosman JA. Molecular pathways: targeting NRAS in melanoma and acute myelogenous leukemia. Clin Cancer Res. 2014;20(16):4186–92.
Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7(11):823–33.
Kao HW, Liang DC, Kuo MC, Wu JH, Dunn P, Wang PN, et al. High frequency of additional gene mutations in acute myeloid leukemia with MLL partial tandem duplication: DNMT3A mutation is associated with poor prognosis. Oncotarget. 2015;6(32):33217–25.
Bacher U, Schnittger S, Haferlach T. Molecular genetics in acute myeloid leukemia. Curr Opin Oncol. 2010;22(6):646–55.
Dang L, Jin S, Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med. 2010;16(9):387–97.
Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27(4):599–608.
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–8.
DiNardo CD, Ravandi F, Agresta S, Konopleva M, Takahashi K, Kadia T, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732–6.
Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22(2):180–93.
Inoue D, Kitaura J, Togami K, Nishimura K, Enomoto Y, Uchida T, et al. Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. J Clin Invest. 2013;123(11):4627–40.
Chou WC, Huang HH, Hou HA, Chen CY, Tang JL, Yao M, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116(20):4086–94.
Paschka P, Schlenk RF, Gaidzik VI, Herzig JK, Aulitzky T, Bullinger L, et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: a study by the German-Austrian acute myeloid leukemia study group. Haematologica. 2015;100(3):324–30.
Pratcorona M, Abbas S, Sanders MA, Koenders JE, Kavelaars FG, Erpelinck-Verschueren CA, et al. Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica. 2012;97(3):388–92.
Schnittger S, Eder C, Jeromin S, Alpermann T, Fasan A, Grossmann V, et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. 2013;27(1):82–91.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–57.
Gao Q, Steine EJ, Barrasa MI, Hockemeyer D, Pawlak M, Fu D, et al. Deletion of the de novo DNA methyltransferase Dnmt3a promotes lung tumor progression. Proc Natl Acad Sci U S A. 2011;108(44):18061–6.
Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25(7):1153–8.
Hou HA, Tien HF. Mutations in epigenetic modifiers in acute myeloid leukemia and their clinical utility. Expert Rev Hematol. 2016;9(5):447–69.
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–33.
Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301.
Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838–42.
Liu WJ, et al. Prognostic significance of TET methylcytosine dioxygenase 2 (TET2) gene mutations in adult patients with acute myeloid leukemia: a meta-analysis. Leuk Lymphoma. 2014;55(12):2691–8.
Ahn JS, Kim HJ, Kim YK, Jung SH, Yang DH, Lee JJ, et al. Adverse prognostic effect of homozygous TET2 mutation on the relapse risk of acute myeloid leukemia in patients of normal karyotype. Haematologica. 2015;100(9):e351–3.
Bravo GM, Lee E, Merchan B, Kantarjian HM, García-Manero G. Integrating genetics and epigenetics in myelodysplastic syndromes: advances in pathogenesis and disease evolution. Br J Haematol. 2014;166(5):646–59.
Karimi M, Nilsson C, Dimitriou M, Jansson M, Matsson H, Unneberg P, et al. High-throughput mutational screening adds clinically important information in myelodysplastic syndromes and secondary or therapy-related acute myeloid leukemia. Haematologica. 2015;100(6):e223–5.
Damm F, Chesnais V, Nagata Y, Yoshida K, Scourzic L, Okuno Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169–77.
Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–76.
Thol F, Bollin R, Gehlhaar M, Walter C, Dugas M, Suchanek KJ, et al. Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood. 2014;123(6):914–20.
Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153–63.
Cho YU, Jang S, Seo EJ, Park CJ, Chi HS, Kim DY, et al. Preferential occurrence of spliceosome mutations in acute myeloid leukemia with preceding myelodysplastic syndrome and/or myelodysplasia morphology. Leuk Lymphoma. 2015;56(8):2301–8.
Nardi V, Hasserjian RP. Genetic testing in acute myeloid leukemia and Myelodysplastic syndromes. Surg Pathol Clin. 2016;9(1):143–63.
Churpek JE, Marquez R, Neistadt B, Claussen K, Lee MK, Churpek MM, et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer. 2016;122(2):304–11.
Shkreta L, Bell B, Revil T, Venables JP, Prinos P, Elela SA, Chabot B. Cancer-associated perturbations in alternative pre-messenger RNA splicing. Cancer Treat Res. 2013;158:41–94.
Moon H, Cho S, Loh TJ, Oh HK, Jang HN, Zhou J, et al. SRSF2 promotes splicing and transcription of exon 11 included isoform in Ron proto-oncogene. Biochim Biophys Acta. 2014;1839(11):1132–40.
Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119(14):3203–10.
Peng J, Hasserjian RP, Tang G, Patel KP, Goswami M, Jabbour EJ, Garcia-Manero G, et al. Myelodysplastic syndromes following therapy with hypomethylating agents (HMAs): development of acute erythroleukemia may not influence assessment of treatment response. Leuk Lymphoma. 2016;57(4):812–9.
Hou HA, Liu CY, Kuo YY, Chou WC, Tsai CH, Lin CC, et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget. 2016;7(8):9084–101.
Shao C, Yang B, Wu T, Huang J, Tang P, Zhou Y, et al. Mechanisms for U2AF to define 3′ splice sites and regulate alternative splicing in the human genome. Nat Struct Mol Biol. 2014;21(11):997–1005.
Ohgami RS, Ma L, Merker JD, Gotlib JR, Schrijver I, Zehnder JL, Arber DA. Next-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutations. Mod Pathol. 2015;28(5):706–14.
Neumann M, Vosberg S, Schlee C, Heesch S, Schwartz S, Gökbuget N, et al. Mutational spectrum of adult T-ALL. Oncotarget. 2015;6(5):2754–66.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–9.
An HJ, Yoon DH, Kim S, Shin SJ, Huh J, Lee KH, Suh C. Blastic plasmacytoid dendritic cell neoplasm: a single-center experience. Ann Hematol. 2013;92(3):351–6.
Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–9.
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5.
Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–6.
Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tönnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8):665–7.
Sashida G, Harada H, Matsui H, Oshima M, Yui M, Harada Y, et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun. 2014;5:4177.
Wang X, Dai H, Wang Q, Wang Q, Xu Y, Wang Y, et al. EZH2 mutations are related to low blast percentage in bone marrow and −7/del(7q) in de novo acute myeloid leukemia. PLoS One. 2013;8(4):e61341.
Wassmann K. Sister chromatid segregation in meiosis II: deprotection through phosphorylation. Cell Cycle. 2013;12(9):1352–9.
Solomon DA, et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. 2011;333(6045):1039–43.
Solomon DA, Kim JS, Waldman T. Cohesin gene mutations in tumorigenesis: from discovery to clinical significance. BMB Rep. 2014;47:299–310.
Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrózek K, et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol. 2012;30(7):742–50.
Metzeler KH, Walker A, Geyer S, Garzon R, Klisovic RB, Bloomfield CD, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26(5):1106–7.
Levis M, Small D. FLT3 tyrosine kinase inhibitors. Int J Hematol. 2005;82(2):100–7.
DeAngelo DJ, Stone RM, Heaney ML, Nimer SD, Paquette RL, Klisovic RB, et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood. 2006;108(12):3674–81.
Pemmaraju N, Kantarjian H, Ravandi F, Cortes J. FLT3 inhibitors in the treatment of acute myeloid leukemia: the start of an era? Cancer. 2011;117(15):3293–304.
Takahashi K, Kantarjian H, Pemmaraju N, Andreeff M, Borthakur G, Faderl S, et al. Salvage therapy using FLT3 inhibitors may improve long-term outcome of relapsed or refractory AML in patients with FLT3-ITD. Br J Haematol. 2013;161(5):659–66.
Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1(5):433–43.
Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60.
Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339–45.
Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116(24):5089–102.
Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103(10):3669–76.
Fathi AT, Chen YB. Treatment of FLT3-ITD acute myeloid leukemia. Am J Blood Res. 2011;1(2):175–89.
Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114(14):2984–92.
Lee HK, Kim HW, Lee IY, Lee J, Lee J, Jung DS, et al. G-749, a novel FLT3 kinase inhibitor, can overcome drug resistance for the treatment of acute myeloid leukemia. Blood. 2014;123(14):2209–19.
Coombs CC, Tallman MS, Levine RL. Molecular therapy for acute myeloid leukaemia. Nat Rev Clin Oncol. 2016;13(5):305–18.
Brandwein JM, Hedley DW, Chow S, Schimmer AD, Yee KW, Schuh AC, et al. A phase I/II study of imatinib plus reinduction therapy for c-kit-positive relapsed/refractory acute myeloid leukemia: inhibition of Akt activation correlates with complete response. Leukemia. 2011;25(6):945–52.
Advani AS, Tiu R, Saunthararajah Y, Maciejewski J, Copelan EA, Sobecks R, et al. A phase 1 study of imatinib mesylate in combination with cytarabine and daunorubicin for c-kit positive relapsed acute myeloid leukemia. Leuk Res. 2010;34(12):1622–6.
Heidel F, Cortes J, Rücker FG, Aulitzky W, Letvak L, Kindler T, et al. Results of a multicenter phase II trial for older patients with c-kit-positive acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (HR-MDS) using low-dose Ara-C and Imatinib. Cancer. 2007;109(5):907–14.
Boissel N, Renneville A, Leguay T, Lefebvre PC, Recher C, Lecerf T, et al. Dasatinib in high-risk core binding factor acute myeloid leukemia in first complete remission: a French acute myeloid leukemia intergroup trial. Haematologica. 2015;100(6):780–5.
Rowinsky EK, Windle JJ, Von Hoff DD. Ras protein farnesyltransferase: a strategic target for anticancer therapeutic development. J Clin Oncol. 1999;17(11):3631–52.
Harousseau JL, Lancet JE, Reiffers J, Lowenberg B, Thomas X, Huguet F, et al. A phase 2 study of the oral farnesyltransferase inhibitor tipifarnib in patients with refractory or relapsed acute myeloid leukemia. Blood. 2007;109(12):5151–6.
Lancet JE, Gojo I, Gotlib J, Feldman EJ, Greer J, Liesveld JL, et al. A phase 2 study of the farnesyltransferase inhibitor tipifarnib in poor-risk and elderly patients with previously untreated acute myelogenous leukemia. Blood. 2007;109(4):1387–94.
Erba HP, Othus M, Walter RB, Kirschbaum MH, Tallman MS, Larson RA, et al. Four different regimens of farnesyltransferase inhibitor tipifarnib in older, untreated acute myeloid leukemia patients: north American intergroup phase II study SWOG S0432. Leuk Res. 2014;38(3):329–33.
Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci U S A. 2013;110(10):4015–20.
US National Library of Medicine. ClinicalTrials.gov [online], 2015. https://clinicaltrials.gov/ct2/show/NCT01907815?term=NCT01907815&rank=1.
Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013;121(13):2533–41.
Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122(6):1017–25.
Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20(1):53–65.
Stein EM, Tallman MS. Mixed lineage rearranged leukaemia: pathogenesis and targeting DOT1L. Curr Opin Hematol. 2015;22(2):92–6.
Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, et al. Erratum: modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. 2016;22(6):692.
Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. 2016;22(6):672–8.
Rose D, Haferlach T, Schnittger S, Perglerová K, Kern W, Haferlach C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia. 2017;31(1):11–7.
Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39(3):348–54.
Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15(3):152–65.
Damm F, Bunke T, Thol F, Markus B, Wagner K, Göhring G, et al. Prognostic implications and molecular associations of NADH dehydrogenase subunit 4 (ND4) mutations in acute myeloid leukemia. Leukemia. 2012;26(2):289–95.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Li, P., Ohgami, R.S. (2018). Acute Myeloid Leukemia with Myelodysplasia-Related Changes, Therapy-Related Myeloid Neoplasms, and Acute Myeloid Leukemia, Not Otherwise Specified. In: Chang, CC., Ohgami, R. (eds) Precision Molecular Pathology of Myeloid Neoplasms. Molecular Pathology Library, vol 12. Springer, Cham. https://doi.org/10.1007/978-3-319-62146-3_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-62146-3_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-62144-9
Online ISBN: 978-3-319-62146-3
eBook Packages: MedicineMedicine (R0)