
Abstract
The Hippo pathway is an established pathway that regulates apoptosis. The earliest characterisations of the mammalian MST1/2 kinases indicated that they were potent inducers of apoptosis in response to a wide range of stimuli. Elucidation of pathway components via genetic screens in Drosophila revealed that signalling through the Hippo pathway is required for the induction of apoptosis during development. Central to control of developmental apoptosis in Drosophila is the regulation of the transcriptional co-activator Yki, whose interaction with transcription factors including Sd, Mad and Tsh/Hth drives the transcription of potent apoptotic inhibitors including Diap-1 and the microRNA Bantam. In mammals it is clear that the core MST1/2-LATS1/2 kinase cassette has various downstream components which lead to apoptosis including the transcription of pro-apoptotic target genes via multiple transcription factors, caspase activation and histone modification. The LATS1/2 kinases and Yap function in a complex network with p53 and its associated regulatory proteins from the ASPP family which, through association with Yap, can have opposing effects on apoptosis. While it is clear that Yap is an important inhibitor of apoptosis in mammals and is subject to similar regulation to that of Yki, Yap also promotes the transcription of pro-apoptotic target genes via association with p73. Evidence suggests that the tumour suppressor RASSF1A is an important determinant in mediating Yap pro-apoptotic activities through regulation of Yap transcription factor interactions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Apoptosis, or programmed cell death, is an important process that removes superfluous or damaged cells during development and maintains organism homeostasis (Fuchs and Steller 2011). The Hippo pathway is an evolutionary conserved pathway that regulates tissue size during development by responding to upstream signals generated from cell-cell contacts and spatial development of the organ. This allows the organ to achieve correct cell number and facilitates the removal of excess cells, which are present during developmental stages. Studies from the model organism Drosophila melanogaster have demonstrated that the Hippo pathway invokes apoptosis in the developing organism through inhibition of the transcriptional co-activator Yki, which mediates expression of pro-proliferative and anti-apoptotic genes. Close co-ordination and integration of cell death and cell growth shapes the course of tissue development. In Drosophila activation of the Hippo pathway is required to inactivate Yki, and failure to do so results in an inability to induce apoptosis in superfluous cells resulting in gross tissue overgrowth (Halder and Johnson 2011; Pan 2010; Staley and Irvine 2012; Zhao et al. 2011).
In the mammalian system, the Hippo pathway and components of the core kinase unit (MST1/2-LATS1/2) are recognised as important regulators of apoptosis. As is the case in Drosophila, the mammalian pathway regulates the Yki homologues YAP and TAZ, which also promote the expression of pro-proliferative and anti-apoptotic genes (Huang et al. 2005). However, growing evidence now suggests that in mammals YAP has a dual role and can both induce and inhibit apoptosis. The pro-apoptotic activity of YAP is due to its ability to activate p73 (Strano et al. 2005), a member of the p53 family, and promote expression of pro-apoptotic members of the bcl-2 family (Matallanas et al. 2007). The expression of these proteins commits a cell to apoptosis as they promote mitochondrial permeabilisation, resulting in the release of cytochrome C and the formation of the apoptosome, a scaffolding platform required for caspase activation (Fuchs and Steller 2011). As outlined below, many apoptotic stimuli activate the mammalian Hippo homologues MST1 and MST2, which in turn drive many classical features of apoptosis such as caspase activation, chromatin condensation and DNA fragmentation. Furthermore, it is also becoming apparent that the pro-apoptotic activities of this pathway are opposed by the activity of AKT, a kinase which is frequently linked with the inhibition of apoptosis. It is the aim of this chapter to outline the stimuli, mechanisms and pathways that have been shown to require Hippo pathway members for the induction of apoptosis, with the aim to demonstrate how they may be involved in development, and also how they induce apoptosis in response to extrinsic stimuli.
2 The Hippo Pathway and Regulation of Developmental Apoptosis in Drosophila
The upstream signalling elements of the Hippo pathway that transmit information regarding cell-cell contact and cell polarity regulate the activity of the Hippo kinase, which in turn regulates the expression of genes that govern apoptosis. A recurring observation made in the examination of flies with deletions of Hippo pathway components is a failure to induce sufficient apoptosis in the developing tissue, which in part facilitates the tissue overgrowth phenotypes observed. These can range in subtle increase in cell number to massive overgrowth of organs such as the eye or wing depending on the nature of the deletion (Halder and Johnson 2011; Pan 2010; Staley and Irvine 2012). A common mechanism that has emerged from these studies is a failure to inhibit the expression of anti-apoptotic genes whose transcription is promoted by the transcriptional co-activator Yki. Yki itself has no DNA-binding activity but, as outlined below, enhances the activity of several transcription factors which are responsible for driving the transcription of both anti-apoptotic genes and a microRNA that in turn regulates the expression of pro-apoptotic genes.
3 The Core Hippo Signalling Module and the Yki-Dependent Inhibition of Apoptosis
All upstream components of the pathway (Fig. 7.1a) converge on the core Hippo signalling module which consists of the Ser/Thr kinases Warts and Hippo, and the scaffold proteins Sav and Mats that enhance their activity. The deletion of either Wts (Justice et al. 1995; Xu et al. 1995), Hpo (Harvey et al. 2003; Pantalacci et al. 2003; Udan et al. 2003; Wu et al. 2003), Sav (Harvey et al. 2003; Tapon et al. 2002) or Mats (Lai et al. 2005) result in the most severe tissue overgrowth phenotypes described, with accompanying decreases in apoptosis as determined by classical markers of apoptosis including decreased TUNEL staining, decreased activation of the Drosophila Caspase-3 like protein DrICE, decreased expression of pro-apoptotic proteins and increased expression of the Drosophila inhibitor of apoptosis (IAP) Diap-1. Furthermore, deletion of Hpo Mats, Wts or Sav reduces apoptosis promoted by the overexpression of Hid, Grm or Rpr, which are members of the Reaper Bcl-2 family and are potent pro-apoptotic genes within the intrinsic cell death pathway (Fuchs and Steller 2011; Harvey et al. 2003; Lai et al. 2005; Pantalacci et al. 2003; Tapon et al. 2002). In contrast, the overexpression of Hpo, Sav or Wts, or activation of the pathway by constitutively targeting Mob to the membrane, results in the induction of apoptosis (Ho et al. 2010; Udan et al. 2003; Verghese et al. 2012). The ability of the core Hippo unit to induce apoptosis in Drosophila is dependent on the inhibition of the transcriptional co-activator Yki, which when overexpressed results in tissue overgrowth and defective apoptosis, and can even block apoptosis due to overexpression of the Dronc initiator caspase and its co-factor Dark (Huang et al. 2005; Verghese et al. 2012).

Hippo pathway-mediated regulation of apoptosis in Drosophila. (a) Multiple upstream signalling inputs including the atypical cadherin FAT, the Expanded-Merlin-Kibra complex, Crumbs and the Lgl, Scrib Dlg complex positively regulate the activity of the core Hippo kinase cassette. dRASSF negatively regulates Hpo activity. Wts phosphorylates Yki and inhibits its activity by promoting relocalisation from the nucleus to the cytoplasm blocking the transcription of anti-apoptotic genes such as Diap-1. (b) Yki promotes the transcription of Diap-1 and the microRNA Bantam which protect against apoptosis. Diap-1 inhibits the Drosophila caspases Dronc and DrICE and Bantam represses the translation of Hid. Hid is a member of the Bcl2 family (which includes Rpr and Grm) of anti-apoptotic proteins and inhibits Diap-1 binding to Dronc and DrICE
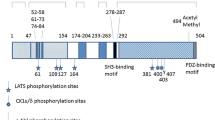
Inhibition of Yki transcriptional activity is mediated by Wts-dependent phosphorylation. Wts phosphorylates Yki on multiple sites which share the consensus motif (HX(R/H/K)XX(S/T)) including Ser111, Ser250 and Ser168 (the equivalent of Ser127 in YAP) (Dong et al. 2007; Huang et al. 2005; Oh and Irvine 2008, 2009; Ren et al. 2010; Zhao et al. 2007). Phosphorylation of Yki on Ser168 creates a binding site for 14-3-3 proteins which sequester Yki in the cytoplasm (Dong et al. 2007; Oh and Irvine 2008; Ren et al. 2010) where it is prevented from interacting with its transcription factor (TF)-binding partners including Scalloped (Sd), (Goulev et al. 2008; Wu et al. 2008; Zhang et al. 2008; Zhao et al. 2008), Homothorax (Hth), Teashirt (Tsh), (Peng et al. 2009) and Mad (Oh and Irvine 2011). The transcriptional output of Yki/Sd, Yki/Tsh/Hth or Yki/Mad complexes is an important determinant in promoting the inhibition of apoptosis in different tissues. However, it has recently been suggested that Yki may have additional roles in inhibiting apoptosis which are independent of the Hippo pathway. Yki has been shown to enhance the anti-apoptotic activity of the non-receptor tyrosine kinase ACK. Although the molecular targets are not defined, this does not require Yki-mediated transcriptional regulation or nuclear localisation (Schoenherr et al. 2012).
4 Yki/TF Complexes and the Regulation of Apoptosis
The Yki/Sd complex has been shown to bind to enhancer elements in the Diap-1 promoter resulting in elevated Diap-1 expression (Goulev et al. 2008; Wu et al. 2008; Zhang et al. 2008). Indeed, Sd was shown to bind to a minimal 26 bp Hippo response element that drove Hippo-dependent up-regulation of Diap-1 (Wu et al. 2008). Diap-1 is a member of the IAP family and is an important modulator of apoptosis in developing Drosophila tissue (Kornbluth and White 2005; Steller 2008). In the Drosophila embryo, loss of Diap-1 induces ubiquitous apoptosis through inappropriate caspase activation (Goyal et al. 2000; Wang et al. 1999). Diap-1 is an E3 ligase that ubiquitinates and inhibits the Drosophila initiator caspase Dronc (caspase 9 homologue) via proteasomal degradation (Wilson et al. 2002), and also the effector caspase drICE (Caspase3 homologue) (Ditzel et al. 2008) via a mechanism involving non-degradative polyubiquitination (Fig. 7.1b). Thus, the enhanced Yki-dependent transcription of Diap-1 in Hippo pathway mutants is a potent anti-apoptotic mechanism that facilitates tissue overgrowth. It is interesting to note that the minimal Hippo response element characterised in the Diap-1 promoter contains non-Sd-binding sites that are absolutely required Diap-1 expression, indicating that additional uncharacterised Yki/TF complexes may regulate Diap-1 expression (Wu et al. 2008).
In addition to the regulation of Diap-1, several Yki/TF complexes including Yki/Sd (Zhang et al. 2008), Yki/Hth (Peng et al. 2009) and Yki/Mad (Oh and Irvine 2011) promote the transcription of Bantam, a microRNA which regulates cell proliferation and apoptosis (Brennecke et al. 2003). When overexpressed Bantam rescues cells from apoptosis induced by overexpression of Hpo, and can promote growth in cells expressing decreased levels of Yki. In contrast, deletion of Bantam blocks Yki-driven overgrowth (Nolo et al. 2006; Thompson and Cohen 2006). Although a limited number of Bantam microRNA targets have been identified, the pro-apoptotic gene Hid, whose expression is repressed by Bantam (Fig. 7.1b), appears to have the clearest impact on apoptosis (Brennecke et al. 2003). Notably, Yki mutant clones which have severely reduced Yki expression have elevated expression of Hid (Nolo et al. 2006).
Hid is a member of the Reaper family of Bcl-2 like pro-apoptotic proteins, which also includes Reaper (Rpr), Sickle (Skl) and Grim (Grm). Hid binding via its IBM domain antagonises Diap-1 function by competing with Drosophila caspases for binding to the BIR domain of Diap-1, which would otherwise associate with and inhibit caspase activity (Fig. 7.1b). In addition, Hid also promotes the auto-ubiquitination and proteolytic degradation of Diap-1 (Yoo et al. 2002) allowing the removal of this potent caspase inhibitor. Thus, Hid promotes elevated caspase activity and induction of apoptosis (Fuchs and Steller 2011; Kornbluth and White 2005; Steller 2008).
Hpo overexpression has also been reported to specifically induce Hid, resulting in Hid-dependent cell death. Interestingly, Hpo overexpression does not up-regulate the expression of Reaper family members Grm or Rpr, suggesting that the Hpo pathway regulates the expression of a specific subset of proteins that control apoptosis in Drosophila (Udan et al. 2003). Given the role of Yki in regulating Hid expression through Bantam, it is likely that the elevated Hid expression reported in Hpo overexpressing clones is the result of sustained Yki inhibition. However, it is also possible that Hid expression may be controlled by other Hpo-regulated mechanisms (Fig. 7.1b). To date no other pro-apoptotic targets which are negatively regulated by Bantam have been identified.
The activity of Hpo itself has also been suggested as an important regulator of apoptosis in Drosophila. Hpo has been reported to phosphorylate Diap-1, resulting in decreased Diap-1 protein stability (Pantalacci et al. 2003). However, overexpression of Hpo within imaginal discs does not appear to have an effect on Diap-1 levels (Verghese et al. 2012). overexpression of Hpo increases the transcription and activity of Dronc and furthermore, Hpo synergises with Dronc overexpression leading to a more dramatic apoptotic response. In contrast, overexpression of Yki, or deletion of Wts, blocks Dronc transcription suggesting that signalling through the Hippo pathway balances the expression and activity of pro- and anti-apoptotic signalling to determine cell fate (Verghese et al. 2012).
Overexpression or deletion of the upstream signalling inputs to the Hippo pathway that provide information on cell-cell contact and cell polarity also influence the induction of apoptosis through the negative regulation of Yki. The Expanded-Merlin-Kirba complex and the recently characterised interacting proteins PEZ and Echinoid (Baumgartner et al. 2010; Bennett and Harvey 2006; Cho et al. 2006; Genevet et al. 2010; Hamaratoglu et al. 2006; Pellock et al. 2007; Poernbacher et al. 2012; Silva et al. 2006; Tyler and Baker 2007; Yu et al. 2010; Yue et al. 2012), Fat and its ligand Dachous (Bennett and Harvey 2006; Cho et al. 2006; Rogulja et al. 2008; Silva et al. 2006; Tyler and Baker 2007; Willecke et al. 2006, 2008), the Lgl, Scrib Dlg complex (Grzeschik et al. 2010), and Crumbs (Chen et al. 2010; Grzeschik et al. 2010; Ling et al. 2010; Robinson et al. 2010) have all been shown to regulate Yki-dependent Diap-1 transcription/expression. However, despite up-regulation Diap-1 in single mutant tissue most large-scale developmental apoptosis proceeds and the observed decrease in apoptosis is often more subtle. This is in contrast to the more dramatic effect of the core Hippo cassette which is universally required to induce apoptosis in the tissues examined to date. The discrepancy may be that the level of Diap-1 induced is insufficient to protect against apoptosis or as recently reviewed (Halder and Johnson 2011; Pan 2010; Staley and Irvine 2012), the role of each of these upstream complexes in regulating developmentally controlled apoptosis is complicated by the observations that these upstream elements function with partial redundancy in different tissues and are often required at different developmental stages (Milton et al. 2010). Thus fine tuning and the strength of signal from upstream elements to induce apoptosis in an Hpo-dependent manner is also tissue specific and developmentally regulated.
The literature suggests that the role of Yki transcription factor complexes in the regulation of developmentally driven apoptosis is tissue specific (Staley and Irvine 2012). Indeed, the Yki-binding partners Sd (Goulev et al. 2008) and Hth/Tsh (Peng et al. 2009) have distinct predominant roles in the wing disc and anterior eye disc respectively. In contrast, it has been suggested that Mad serves as a general TF for Yki in the maintenance of growth control as it is ubiquitously expressed (Oh and Irvine 2011). This regulated selection of transcription factor use presumably allows the expression of a specific subset of genes in a tissue-specific manner and reflects the input from additional signal pathways in order for faithful organ development through co-ordinated spatial growth and apoptosis. It will thus be of interest to determine if there are further anti-apoptotic targets regulated by Yki in a tissue-specific manner or indeed if other Yki transcription factor complexes contribute to the anti-apoptotic transcription profile of Yki.
5 Regulation of Apoptosis in Mammalian Cells by the MST-LATS Kinase Cassette
5.1 Regulation of Apoptosis via the MST1 and MST2 Kinases
The mammalian orthologues of the Hippo kinase are MST1 and MST2, themselves named as the mammalian homologues of the yeast Sterile Twenty kinase (Creasy and Chernoff 1995a, b; Taylor et al. 1996). Prior to the recognition of Hpo in developmental models, MST1 and MST2 have long been recognised as pro-apoptotic kinases that induce apoptosis (Fig. 7.2) in response to a range of stimuli including sodium arsenite, etoposide, cyclohexamide, staurospaurine, H2O2, FAS, TNFα cytotrienin A, and bisphonates (Graves et al. 1998; Kakeya et al. 1998; Lee et al. 2001; Reszka et al. 1999; Taylor et al. 1996). Furthermore, MST1/2 are required for apoptosis induced by constitutively active KRasV12 and function in a tumour suppressive apoptotic pathway (Khokhlatchev et al. 2002; Matallanas et al. 2011). In vivo experiments with mice harbouring genetic deletions of both MST1 and MST2 demonstrated that cells within the liver of mice were more resistant to TNFα and FAS-induced apoptosis (Song et al. 2010; Zhou et al. 2009). Furthermore, MST1 has also been reported to mediate cardiomyocyte apoptosis in the heart in response to tissue damage induced by various insults including myocardial infarction, ischemic reperfusion injury and chronic pressure overload (Del Re et al. 2010; Odashima et al. 2007; Yamamoto et al. 2003), further highlighting the pro-apoptotic role of MST1/2 in response to a variety of stimuli.

Multiple pathways of MST1/2-mediated apoptosis. MST1/2 regulate multiple apoptotic pathways in response to wide range of pro-apoptotic stimuli (see text for details). MST1/2 activity is regulated by the RASSF family of scaffold proteins and by caspase activation. Additionally, phosphorylation by other kinases regulates MST1/2 activity. MST1 phosphorylates histones H2B and H2AX, which promotes chromatin condensation and DNA fragmentation. Phospho H2B also binds and sequesters RCC1 and RAN-GTP onto chromatin-blocking nuclear transport. Transcription of pro-apoptotic genes is also promoted by MST in response to apoptotic stimuli. Activation of MST1/2 also activates a number of downstream kinases which have been associated with the induction of apoptosis although the targets of some of these kinases remain to be identified
In response to apoptotic stimuli MST1/2 are cleaved by caspases which removes a C-terminal auto-inhibitory domain and the resulting kinase domain displays greatly enhanced activity and altered substrate specificity (Figs. 7.2 and 7.3) (Anand et al. 2008; Graves et al. 1998, 2001; Lee et al. 1998, 2001). MST1 is cleaved by caspase 3 and 7 at Asp326 and Asp349, whereas MST2 is cleaved at Asp322 (Song and Lee 2008). Although the Asp residues corresponding to 326/322 in MST1/2 are evolutionary conserved between MST1/2 homologues in mammals, Drosophila and nematodes (Lee et al. 2001), residues C-terminal to the Asp influence MST cleavage. In mammals this Asp residue is flanked by an Ser (Ser327) which has been identified as a potential MST1/2 autophosphorylation site (Glantschnig et al. 2002; Graves et al. 2001) and mutation to Asp inhibits caspase 3-mediated cleavage of MST1 (Glantschnig et al. 2002; Graves et al. 2001). In contrast to MST1/2, Hippo contains a flanking Asp residue which is thought to prevent Hippo from being cleaved by DrICE (Wu et al. 2003). Thus this may be an evolutionary difference that allows a distinction to be made between apoptotic and non-apoptotic functions of MST1/2 in the cell. Overexpression of MST1 has also been demonstrated to induce caspase 3 and 8 activity in a JNK-dependent manner, suggesting that MST can also directly activate caspase activity suggesting a positive feedback loop (Fig. 7.2) (Ura et al. 2001b). Caspase-mediated cleavage of MST1/2 also removes two C-terminal nuclear export sequences allowing the truncated kinase domain to traffic to the nucleus resulting in enhanced nucleosomal DNA fragmentation, nuclear condensation and membrane blebbing, all of which are hallmarks of apoptosis (Lee et al. 2001; Lin et al. 2002; Ura et al. 2001a).

Negative regulation of MST activity and apoptosis. The activity of MST1/2 and AKT acts antagonistically. MST1/2 inhibit AKT and MST activity is negatively regulated by AKT. AKT phosphorylates MST1 on Thr120 and Thr 387 and MST2 on Thr117 and Thr384. Phosphorylation on these sites promotes association with Raf1 which sequesters MST1/2 into an inhibitory complex, blocks MST1/2 dimerisation which is required for autophosphorylation and caspase-mediated cleavage and activation of MST1/2. The inhibition of MST1 by AKT is opposed by the PHLPP phosphatase. RASSF1A disrupts the RAF1/MST1/2 inhibitory complex, prevents PP2A-mediated dephosphorylation of MST1/2 and promotes MST1/2 dimerisation. Phosphorylation of MST1/2 has been shown to inhibit a number of pro-apoptotic pathways
It is interesting to note that in quiescent tissue, caspase-mediated cleavage of MST has a function independent of apoptosis (Zhou et al. 2009). In the murine liver MST1/2 are present almost exclusively as the truncated N-terminal kinase domain. Previous studies demonstrated that a truncated species of MST2 was detected in quiescent cells (Wang and Fecteau 2000), and that caspase 3-mediated cleavage of MST2 was required for muscle differentiation (Fernando et al. 2002). Caspases are now being recognised as having many non-apoptotic roles (Fuchs and Steller 2011) and thus it will be of interest to determine what MST-regulated substrates are contributing to maintenance of quiescence.
In addition to the caspase-mediated activation of MST1/2, the best characterised positive upstream regulators of the MST kinases are the RASSF family of scaffold proteins, in particular RASSF1A and NORE1 (Fig. 7.2). RASSF1A and NORE1 have both been shown to enhance MST1/2 activity and apoptosis following oxidative stress, treatment with TNF, FAS or oncogenic RAS and RASSF1A is required to activate the downstream targets of MST1/2, NDR and LATS1/2 (Khokhlatchev et al. 2002; Matallanas et al. 2007; Oh et al. 2006; Park et al. 2010; Vichalkovski et al. 2008; Yuan et al. 2009). Furthermore Nore1 −/− MEFs are resistant to TNF-induced activation of MST1 and apoptosis, and display reduced activation of JNK and p38 MAP kinase. Interestingly NORE1 only enhances MST activity in response to a limited number of stimuli (Park et al. 2010). Both RASSF1A and NORE1 promote MST1/2 dimerisation and auto-activation which is thought to be required before caspase-mediated cleavage can occur (Matallanas et al. 2007; O’Neill et al. 2004; Praskova et al. 2004; Romano et al. 2010).
6 Downstream Effectors of the MST1/2 Kinases
Overexpression of MST1 has been shown to result in apoptosis with accompanying activation of the stress-activated MAP kinases, JNK and p38 MAP kinase (Fig. 7.2) (Glantschnig et al. 2002; Graves et al. 1998; Qiao et al. 2010; Ura et al. 2007). Indeed MST1 −/−, MST2 −/− null MEFs display reduced JNK activation following TNFα stimulation (Song et al. 2010), suggesting that JNK is natural downstream target of MST1/2 in response to death receptor signalling. Although dependent on the upstream JNK activators MKK4 and MKK7 (Ura et al. 2007), the mechanism as to how MST1 activates JNK or p38 has not been defined. As outlined below, JNK signalling is important in MST1-induced apoptosis mediated by the FOXO transcription factors. Furthermore, JNK activation also contributes to MST1 activation. JNK phosphorylates MST1 on Ser82 and mutation of this site, or treatment with JNK inhibitors, restricts MST1 activation, caspase-mediated cleavage and nuclear translocation (Bi et al. 2010).
An additional target of MST1 is the histone H2B, which is phosphorylated on Ser14 in response to a number of stimuli (Fig. 7.2) (Cheung et al. 2003; Teraishi et al. 2006; Wong et al. 2009; Yun et al. 2011). Phosphorylation acts as a trigger for DNA fragmentation during apoptosis which is a biochemical hallmark of late apoptosis. Indeed phospho-Ser14-H2B is abundant in degraded chromatin (Ajiro et al. 2010). Phosphorylation of Ser14 occurs with similar kinetics to that of MST1 cleavage during apoptosis (Cheung et al. 2003) and interestingly, H2B is a better substrate for cleaved MST1 compared to full length (Anand et al. 2008), suggesting that cleavage of MST1 may aid in committing a cell to apoptosis. Additionally, cleaved MST1-mediated phosphorylation of H2B also sequesters RanGTP and its associated GTP-activating protein, RCC1, on the chromatin, collapsing the RanGTP-RanGDP gradient (Fig. 7.2), which is required for the import of nuclear proteins (Wong et al. 2009). Phosphorylation of H2B is evolutionary conserved, and yeast homologue ste20 also phosphorylates H2B at the equivalent residue (Ser10) (Ahn et al. 2005). In addition, MST1 also phosphorylates the H2A variant H2AX on Ser139 (Fig. 7.2) in response to apoptotic inducing stimuli (Teraishi et al. 2006; Wen et al. 2010). This occurs with similar kinetics to caspase 3 cleavage of MST1 and is required for chromatin condensation and DNA fragmentation. These studies would suggest that MST1 kinase activity can commit the cell to apoptosis through modulation of histone and chromatin structure which leads to DNA fragmentation.
7 MST1/2-Mediated Transcriptional Regulation of Pro-apoptotic Target Genes
MST1 induces apoptosis in neuronal cells through the regulation of FOXO transcription factors (Fig. 7.2). This is also a conserved pathway as the Caenorhabditis elegans MST orthologue cst1 also regulates the activity of the FOXO orthologue daf-16 (Lehtinen et al. 2006). FOXO transcription factors are inhibited via AKT-mediated phosphorylation, which promotes 14-3-3 proteins-mediated sequestration in the cytoplasm, preventing up-regulation of pro-apoptotic genes or genes involved in cell cycle arrest (Greer and Brunet 2008). MST1 phosphorylates FOXO3 on Ser207 and FOXO1 on Ser212, within the forkhead domain, which blocks 14-3-3 binding and promotes the nuclear translocation and accumulation of FOXO1/3, leading to transcription of the pro-apoptotic proteins BIM and NOXA (Lehtinen et al. 2006; Valis et al. 2011; Yuan et al. 2009). The pro-apoptotic activities of FOXO transcription factors are also positively regulated by JNK in response to oxidative stress (Greer and Brunet 2008) and as JNK phosphorylation of MST1 on Ser82 enhances FOXO3 Ser207 phosphorylation (Bi et al. 2010) these two kinases act co-operatively to induce FOXO-mediated transcription of pro-apoptotic genes (Fig. 7.2). Additionally, inhibition of MST1 polyubiquitination and proteasomal degradation by c-ABL phosphorylation of Tyr433 enhanced FOXO3-mediated BIM expression (Xiao et al. 2011). This site is not conserved on MST2 and may represent a specific route of MST1 activation. Interestingly, MST1 regulation of FOXO in mammals is not always pro-apoptotic. In naive T-cells MST1 is required to protect T-cells from elevated levels of reactive oxygen species (ROS) through FOXO-mediated transcription of ROS detoxifying enzymes, catalase and SOD2 (Choi et al. 2009). Thus MST1-mediated regulation of FOXO transcriptional output may be cell or stress dependent and it remains to be determined what other signalling inputs determine cell fate through FOXO transcriptional output.
MST1 also affects the activity of p53 and induces apoptosis in a p53-dependant manner. In response to DNA-damaging agents MST1 phosphorylates SIRT1, an NAD+-dependent deacetylase, which antagonises p53 activity (Fig. 7.2). Overexpression of MST1 inhibits SIRT1-dependent p53 acetylation, as well as that of FOXO3, resulting in increased p53 transcriptional activity and cell death (Yuan et al. 2011). P53 also indirectly activates MST1 and drives MST1-induced apoptosis. In response to oxidative stress Peroxiredoxin (PRX1) oligomerises in a p53-dependent manner and is a key intermediate in the induction of MST1 activity (Morinaka et al. 2011). In light of MST1-mediated activation of p53, this may represent a feedback mechanism for amplification of MST1 activity (Fig. 7.2). Additionally, MST1 may activate p53 via other scaffold proteins such as death-associated protein 4 (DAP4) which has been reported to mediate p53-dependent MST1-induced apoptosis (Lin et al. 2002). DAP4 induces MST1 nuclear translocation and was shown to bind p53 directly. One possibility may be that DAP4 helps to scaffold MST1 to other p53-interacting proteins such as SIRT1, although the molecular details of how DAP4 induces MST1-mediated apoptosis are unclear.
A complex of MST2 and WW45 has also been shown to mediate RUNX3-induced cell death (Min et al. 2012). MST2 phosphorylates RUNX3 on Ser17, Thr16, Ser71, Ser77 and Ser81 within the Runt domain. MST2 phosphorylation of RUNX3 promotes translocation to the nucleus by preventing association of the E3 ligase, SMURF1, which otherwise targets RUNX3 for degradation. The targets of MST2/RUNX3-mediated cell death remain to be identified (Fig. 7.2), although it is interesting to note that RUNX3 cooperates with FOXO3 to induce apoptosis and as such the MST kinases may coordinate the output of multiple transcription factors in order to commit the cell to apoptosis. (Yamamura et al. 2006). The MST2/RUNX3 interaction is conserved in Drosophila (Min et al. 2012) and it will be interesting to determine if this is involved in Hippo pathway-mediated developmental apoptosis.
7.1 Negative Regulation of MST1/2-Mediated Apoptosis
As MST1/2 are pro-apoptotic kinases, their activity must be regulated in order to prevent inappropriate cell death. The mechanisms reported to regulate MST1/2 activity include dephosphorylation, sequestration into inhibitory complexes, phosphorylation via AKT, binding to heat shock proteins and down-regulation via microRNA (Fig. 7.3).
Inhibitory complexes with RAF-1 or A-RAF prevent MST2 dimerisation and auto-activation as well as activation of the downstream effectors p38 and JNK (Matallanas et al. 2007; O’Neill et al. 2004; Rauch et al. 2010; Romano et al. 2010). Indeed knockdown of MST2 rescues Raf-1 null cells from apoptotic stimuli such as FAS and in normal cells it is the RASSF1A-dependent disruption of RAF1 complexes which promotes MST2 dimerisation and auto-activation (Fig. 7.3) (Matallanas et al. 2007; O’Neill et al. 2004; Romano et al. 2010). Early studies demonstrated that binding of MST1/2 to RAF1 recruited the PP2A phosphatase to this complex which dephosphorylated and inactivated MST2 (O’Neill et al. 2004). Recently it has been shown that RASSF1A protects MST1/2 from PP2A-mediated inhibition through dephosphorylation of Thr183/180 (Guo et al. 2011) and is likely to occur through dissociation of the RAF1-MST complex (Fig. 7.3). In an analogous fashion, Hpo is also negatively regulated by dephosphorylation (Ribeiro et al. 2010). The association of MST2 with RAF1 is also disrupted by c-ABL phosphorylation of MST2 on Tyr81 which in turn promotes MST2 dimerisation and auto-activation (Liu et al. 2012).
AKT is a negative regulator of apoptosis (Duronio 2008) and inhibits MST1/2 activity. Growth factor receptor signalling which activates AKT has been shown to inhibit MST1/2 (Fig. 7.3) (Creasy and Chernoff 1995a; Jang et al. 2007; Kim et al. 2010; Matallanas et al. 2011; Romano et al. 2010). AKT phosphorylates MST1/2 on two sites within the canonical AKT substrate recognition motif RXRXXS/T; MST1 is phosphorylated on Thr120 (Yuan et al. 2010b) and Thr387 (Jang et al. 2007) and MST2 is phosphorylated on Thr117 and Thr384 (Kim et al. 2010; Romano et al. 2010). AKT-mediated phosphorylation of MST1 inhibits MST1 kinase activity, FOXO3 Ser207 phosphorylation and caspase-mediated MST1/2 cleavage. Furthermore, autocrine signalling via EGFR and activation of AKT inhibit MST2-mediated LATS1 activation and induction of p53-dependent apoptosis (Matallanas et al. 2011). In addition, phosphorylation of MST2 by AKT also increases with association of MST with the negative regulator, RAF-1 (Romano et al. 2010). Interestingly, Thr387 is also dephosphorylated by the tumour suppressor pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP) which also antagonises AKT activity (Fig. 7.3) (O’Neill et al. 2012). MST1, but not MST2, interacts with PHLPP and dephosphorylation of MST1 phospho-Thr387 by PHLPP increases MST1, p38 and JNK activity with accompanying increases in apoptosis (Qiao et al. 2010). MST1/2 have also been reported to be direct inhibitors of AKT1 (Fig. 7.3) (Cinar et al. 2007). The antagonistic relations relation between MST and AKT is also conserved in Drosophila, and Hpo inhibits Yki-mediated expression of AKT (Ye et al. 2012). Recently MST2 was shown to be a target of the tumour-promoting micoRNA 133b which promotes elevated AKT1 and ERK activity (Qin et al. 2012). Thus a complex network exists whereby signalling via MST1/2 and AKT can balance the output of each other in order to determine cell fate. MST1 activity is also regulated by the heat shock protein Hsp70 (Ren et al. 2008). Hsp70-mediated inhibition of MST1 activity is dependent on the proteasomal degradation of MST1 and requires the E3 ligase CHIP for targeting MST1 to the proteasome.
8 LATS1/2-Mediated Induction of Apoptosis
Overexpression of LATS1/2 has been shown to induce apoptosis with reported increases in p53 and Bax expression as well as caspase activation and down-regulation of the anti-apoptotic proteins BCL2 and BCLxl (Ke et al. 2004; Xia et al. 2002; Yang et al. 2001). Furthermore, overexpression of LATS2 increased the processing and activation of caspase 9 (Ke et al. 2004). As important regulators of mitosis and the spindle checkpoint (Visser and Yang 2010), LATS1/2 can induce apoptosis in response to failure to satisfy the spindle checkpoint or faithful segregation of the genome. LATS1 has been shown to induce apoptosis in response to spindle damage mediated by the microtubule poison nocodazole (Iida et al. 2004) and LATS2 promotes apoptosis of cells with polyploidy genomes (Aylon et al. 2009, 2010).
The kinase activity of LATS1 is required for the activation of proteins that drive processing and initiation of apoptotic pathways. The C-terminus of LATS1 binds to the PDZ domain of Omi/HtrA2 when it is released from the mitochondria, leading to the processing and activation of this protease in a kinase-dependant manner (Kuninaka et al. 2005). LATS2 does not interact with Omi/HtrA2 (Kuninaka et al. 2007). Omi/HtrA2 is an inhibitor of mammalian IAPS including XIAP and cIAP and binds to IAPs via the IBM domain. Omi/HtrA2 induces degradation of the mammalian IAPs and hence leads to caspase activation (Vande Walle et al. 2008). Depletion of LATS1 leads to elevated IAP expression (Kuninaka et al. 2005). Thus in an analogous manner to the Hippo pathway in Drosophila, inhibition of IAP activity is an important downstream target in mammalian Hippo-regulated apoptosis. Interestingly, LATS1 is also an Omi/HtrA2 substrate and Omi/HtrA2 was shown to mediate LATS1 control of the G1/S checkpoint (Kuninaka et al. 2007). It would therefore be interesting to determine if this is also negative feedback mechanism that regulates LATS1-induced apoptosis.
9 YAP and TAZ Apoptotic Target Genes and Their Regulation
The LATS1/2-mediated inhibition of YAP transcriptional activity is conserved in mammals. In addition, LATS1/2 also negatively regulates the activity of the YAP-related protein TAZ, which can also promote the transcription of many genes regulated by YAP via a common interaction with the transcription factor TEAD (Zhang et al. 2009; Zhao et al. 2008). LATS1/2 inhibits YAP/TAZ activity in an analogous manner to Wts-mediated inhibition of Yki. LATS1/2 phosphorylates YAP on Ser127 (also Ser61, 104, 164 and 381) and TAZ on Ser89 (also Ser66, 117 and 311) which are within the HX(R/H/K)XX(S/T) LATS consensus motif. Phosphorylation of YAP-Ser127 or TAZ-Ser89 promotes 14-3-3 binding and relocation to the cytoplasm (Dong et al. 2007; Lei et al. 2008; Zhao et al. 2007).
Like Yki, YAP up-regulates the expression of anti-apoptotic genes including the mammalian IAPs BIRC5, BIRC2 and the Bcl2 family member MCL1 (Dong et al. 2007; Liu-Chittenden et al. 2012; Zhao et al. 2008), and down-regulates the expression of the pro-apoptotic proteins BIM and BAX (Vigneron et al. 2010; Zhao et al. 2008). Microarray analysis suggests TAZ can also up-regulate the expression of BIRC5 (Zhang et al. 2009). Furthermore, YAP and TAZ can also protect the cell against anoikis, and LATS-mediated inactivation of YAP helps drive this cell death pathway (Vigneron et al. 2010; Zhao et al. 2012). In a transgenic mouse model, inducible overexpression of YAP was shown to protect the liver from FAS receptor agonist-induced apoptosis, and YAP-dependent BIRC5 expression enhanced colony growth in soft agar (Dong et al. 2007; Liu-Chittenden et al. 2012). In contrast, genetic deletion of Yap1 in the liver results in increased hepatocyte apoptosis (Zhang et al. 2010). BIRC2 and BIRC5 expression and YAP1 nuclear localisation are enhanced in the heart when WW45 is conditionally deleted (Heallen et al. 2011). However, despite the increase in the expression of these two genes, there is no difference in apoptosis compared to the control mice, suggesting that the ability of YAP to protect against apoptosis may be tissue/context dependant. Expression of constitutively active YAP-S127A in the skin protects keratinocytes from apoptosis (Zhang et al. 2011a), and loss of WW45 in epithelial tissue results in decreased apoptosis within the developing mouse as a result of elevated YAP activity (Lee et al. 2008). In addition, genetic deletion of NF2 in the liver which lies upstream of YAP promotes elevated YAP nuclear localisation and the enhanced transcription of BIRC5 (Liu-Chittenden et al. 2012; Zhang et al. 2010).
Although many YAP-interacting transcription factors have been identified (Pan 2010; Zhao et al. 2011), the YAP/TEAD complex is the best characterised (Zhao et al. 2008). In both human and mouse cells, overexpression of TAZ and YAP induces BIRC5 expression (Zhang et al. 2009; Zhao et al. 2008). YAP/TEAD is found on the promoters of BIRC family members such as BIRC7 and likely mediates the inhibition of BIM expression (Vigneron et al. 2010; Zhao et al. 2008). Indeed the role of TEAD transcription factors in mediating BIRC5 transcription was demonstrated in a recent study where expression of a dominant negative version of TEAD blocked BIRC5 transcription and enhanced apoptosis in response to treatment with a FAS agonist antibody (Liu-Chittenden et al. 2012).
In contrast to Wts, LATS1/2 has an additional mode of YAP inhibition. LATS1/2 phosphorylation of Ser381 primes YAP for an additional phosphorylation by CK1 δ/ε which in turn recruits the E3 ubiquitin ligase SCFβ-TRCP, leading to ubiquitin-dependent proteasomal degradation (Zhao et al. 2010). A similar mechanism exists for TAZ which requires LATS2-mediated phosphorylation on Ser311 (Liu et al. 2010).
10 A Complex Relationship Between LATS, Yap and the p53 Family of Tumour Suppressors
Unlike Yki, YAP activity is not strictly anti-apoptotic and pro-proliferative. Indeed growing evidence suggests that YAP contributes to the induction of apoptosis and that this phenotype is mediated by the p53 family of tumour suppressors namely p63 and p73. Furthermore, proteins which regulate p53 have also been reported to regulate YAP activity. As discussed below, LATS2 is also a p53 target gene and contributes to enhanced p53 activity, suggesting that in mammalian cells a complex network exists between the Hippo pathway the pro-apoptotic activities of p53 and its related family members.
10.1 LATS1/2 and p53
The LATS1/2 kinases have been identified as positive regulators of p53, a tumour suppressor with potent pro-apoptotic activity (Vousden and Prives 2009). In response to oncogenically activated K-RAS or H-RAS, both LATS1 and LATS2 have been shown to induce p53 stabilisation and transcription of pro-apoptotic target genes, including p21, BAX, PIG3 and inhibition of BIRC3 expression (Aylon et al. 2006, 2009, 2010; Matallanas et al. 2011). Both LATS1 (Matallanas et al. 2011) and LATS2 (Aylon et al. 2006) promote p53 stabilisation by binding and inhibiting the E3 ligase MDM2, which targets p53 for ubiquitination and proteasomal degradation. Oncogenic K-RAS-driven activation of LATS1 and p53 is mediated by RASSF1A and MST2. In colorectal cancer, apoptosis induced by K-RAS-driven MST2 and LATS1 activation creates a selective pressure to bypass this apoptotic pathway through inactivation of MST2 expression (Matallanas et al. 2011).
LATS2-mediated activation of p53 induces apoptosis in response to a limited range of stimuli that induce genomic instability and the formation of polyploidy genomes (Aylon et al. 2006, 2009, 2010). The selective LATS2-dependent apoptotic response of polyploidy cells requires p53 and as discussed below is promoted by ASSP1, a known p53 regulator (Fig. 7.4). LATS2-mediated activation of p53 removes polyploidy cells through cell cycle checkpoint activation and the induction of pro-apoptotic p53 target genes including BAX, and down-regulation of the apoptotic inhibitor BIRC3 (Aylon et al. 2009, 2010). Interestingly LATS2 is itself a p53 target gene (Aylon et al. 2006; Kostic and Shaw 2000), creating a positive feedback loop between these two tumour suppressors.

The activity of LATS1/2 is differentially regulated by members id the ASSP family. The ASPP family of proteins which regulate p53 can differentially regulate LATS1/2 activity. Oncogenic H-Ras or nocodazole-mediated damage of the mitotic spindle promotes activation of LATS2 which phosphorylates ASPP1 and drives the nuclear translocation of LATS2 and ASPP1. The LATS2/ASPP1 complex binds p53 and promotes the selective activation of pro-apoptotic target genes and is required to prevent cells acquiring polyploid genomes. This interaction is blocked by YAP. In contrast, cells with high levels of cytoplasmic ASPP1 enhance YAP/TAZ activity by inhibiting the association of LATS1 with YAP, which prevents LATS1-dependent phosphorylation of YAP-Ser127 and nuclear accumulation of YAP. This promotes YAP-mediated repression of BIM, preventing the induction of apoptosis. YAP also negatively regulates the expression of LATS2 and indirectly inhibits p53. ASPP2 has also been shown to activate TAZ by promoting the dephosphorylation of TAZ and also preventing the binding of TAZ to the SCF E3 ubiquitin ligase which targets TAZ for proteasomal degradation
The activation of p53 by LATS2 in response to oncogenic H-RAS requires the translocation of LATS2 from the cytoplasm to the nucleus in an ATR-CHK1-dependant manner (Aylon et al. 2009). In response to oncogenic H-RAS, ASPP1 undergoes a LATS2-induced phosphorylation event. Interestingly, LATS1 also interacts with ASPP1 (Vigneron et al. 2010). ASSP1 is a member of the ankyrin-repeat-, SH3-domain- and proline-rich-region-containing family of proteins which specifically regulates p53 apoptotic activity (Sullivan and Lu 2007). Phosphorylation of ASPP1 by LATS2 promotes the cytoplasmic to nuclear translocation of ASPP1, and the ASPP1-LATS2 complex enhances p53 recruitment onto the promoters of pro-apoptotic genes including BAX, CD95, PUMA and GADD45a while reducing p53 recruitment onto non-apoptotic p53 targets such as p21 (Aylon et al. 2010) (Fig. 7.4).
Interestingly ASSP1-LATS2-induced apoptosis is inhibited by YAP. YAP binds to both ASSP1 (Aylon et al. 2010; Vigneron et al. 2010) and ASSP2 (Espanel and Sudol 2001), and YAP antagonises LATS2-ASPP1-induced apoptosis in response to constitutively active H-RAS. YAP inhibits LATS2-ASPP1 translocation to the nucleus through competition with LATS2 for binding to ASPP1 (Aylon et al. 2010) (Fig. 7.4). Furthermore, ASSP1 and YAP also exhibit anti-apoptotic activity in a mechanism which is dependent on the cellular localisation of ASSP1 (Vigneron et al. 2010). In cell lines where ASPP1 is predominately cytoplasmic, ASPP1 contributes to the transcriptional output of YAP. ASPP1 competes with LATS1 for association with YAP, preventing LATS1-mediated phosphorylation of YAP, resulting in increased YAP stability and accumulation in the nucleus. Perhaps the observed increased YAP stability and decreased YAP-S127 phosphorylation is due to ASPP1-dependent phosphatase recruitment. An analogous mechanism has been described for ASPP2-PP1-mediated dephosphorylation of Ser89 in TAZ, and PP1A-mediated dephosphorylation of YAP has been demonstrated to positively regulate YAP activity (Liu et al. 2011; Schlegelmilch et al. 2011) (Fig. 7.4).
The anti-apoptotic phenotype mediated by ASPP1 and YAP is dependent on repression of Bim and knockdown of either ASPP1 or YAP enhances apoptosis in response to hydroxyurea treatment. Overexpression of ASPP1 and YAP antagonises p53 activity, inhibiting the expression of p53 pro-apoptotic target genes including, BAX, PUMA and PHLDA3 (Vigneron and Vousden 2012). LATS2 expression is repressed by cytoplasmic ASPP1, as ASPP1 enhances YAP recruitment to the LATS2 promoter (Fig. 7.4). The enhanced YAP stability and activity conferred by cytoplasmic ASPP1 (Vigneron et al. 2010), coupled with the inhibition of LATS2 expression by YAP (Vigneron and Vousden 2012), indirectly promotes p53 inhibition by preventing LATS2 recruitment to p53 target genes, which when overexpressed with ASPP1 was shown to enhance the expression of pro-apoptotic p53 targets (Aylon et al. 2010) (Fig. 7.4). Unanswered questions remain as to what additional proteins or post-translational modifications can control the switch between ASPP anti- and pro-apoptotic activities, and how in an untransformed cell a balance is achieved between ASPP-YAP-mediated growth and ASPP-LATS mediated cell death.
10.2 YAP, p73 and ΔNp63α
To date the best characterised pro-apoptotic effector of YAP is the p53 family member p73, which transcriptionally regulates many p53 pro-apoptotic target genes (Pietsch et al. 2008). The YAP-p73 complex has been shown to induce apoptosis in response to ionising radiation, chemically induced DNA damage, treatment with FAS ligand or TNF and accumulation of the amyloid β peptide (Basu et al. 2003; Hamilton et al. 2009; Matallanas et al. 2007; Park et al. 2010; Strano et al. 2005; Yee et al. 2012; Zhang et al. 2011b). YAP binds to p63α and the p73α and β isoforms through a conserved WW domain and PPXY motif interaction and selectively enhances the recruitment of the YAP/p73 complex onto the promoters of pro-apoptotic genes in response to a range of DNA-damaging agents (Strano et al. 2001, 2005). Interestingly, neuronal cells expressing truncated variants of YAP lacking the C-terminal transactivation domain block p73-mediated cell death (Hoshino et al. 2006). Co-expression of YAP and p73 enhances the p73-dependent expression of pro-apoptotic genes and endogenous YAP-p73 are recruited to the promoters of Bax, p53AIP1, Killer/DR5, PDCD5 and PIG3 following treatment with DNA-damaging agents (Basu et al. 2003; Levy et al. 2008; Matallanas et al. 2007; Strano et al. 2001, 2005). Knockdown of YAP inhibits the expression of p73 target genes and subsequent cell death (Basu et al. 2003; Levy et al. 2008; Strano et al. 2005). There is a mutual requirement for YAP and p73 in the induction of apoptosis (Levy et al. 2008; Strano et al. 2005) as p73 is required for YAP translocation to the nucleus and YAP is necessary for p300-mediated acetylation of p73 which in turn promotes the induction of p73 pro-apoptotic target genes (Fig. 7.5a) (Costanzo et al. 2002; Strano et al. 2005). Additionally, post-translational modification of YAP also promotes the enrichment of the YAP-p73 transcriptional complex on the promoters of p73 pro-apoptotic genes. In response to DNA-damaging agents c-ABL phosphorylates YAP on Tyr375 (Levy et al. 2008). Phosphorylation of Tyr375 enhances YAP stability and promotes an increased association with p73. In addition, phosphor-Tyr375 is an important determinant in driving the selective activation of pro-apoptotic p73 target genes (Levy et al. 2008).

The pro-apoptotic functions of YAP are promoted by RASSF1A. (a) RASSF1A is required for the formation of the YAP-p73 complex following DNA damage. Phosphorylation of RASSF1A via ATM promotes the MST2-LATS1-dependent formation of YAP-p73 and the expression of pro-apoptotic target genes. In addition, YAP-p73 is also subject to additional regulatory inputs through Tyr phosphorylation by c-Abl which promotes the selective activation of YAP-73 while inhibiting non-apoptotic YAP transcription factor complexes. MST1 also phosphorylates the H2A variant H2AX which is commonly phosphorylated by ATM following DNA damage. Thus MST1 and ATM may co-operate to amplify phosphorylation of H2AX which is required for the DNA damage response pathway. (b) The scaffold protein RASSF1A is an important determinant in regulating the transcription factor repertoire of YAP. Expression of YAP reduces the association of YAP and TEAD and if other binding partners of YAP such as RUNX2 are lost, RASSF1A further enhances the association of YAP with p73 and the inhibition of cell growth
YAP is also required for the enhanced stability and accumulation of p73, and mutants of p73 that cannot bind YAP fail to be stabilised in response to DNA-damaging agents (Levy et al. 2007; Strano et al. 2005). YAP stabilises p73 by competing with the E3 ligase ITCH, which targets p73 for ubiquitination and proteasomal degradation. ITCH binds to p73 through the PPXY motif and YAP stabilises p73 by competing with ITCH for binding to this site (Levy et al. 2007). Furthermore, additional proteins have also been shown to be important in the promotion of YAP-p73-induced apoptosis. Promyelocytic leukaemia protein (PML) influences YAP-p73-induced apoptosis. PML is required for YAP-p73-mediated apoptosis and importantly as it is itself a YAP-p73 target, amplifies the induction of YAP-p73 target genes (Lapi et al. 2008; Strano et al. 2005). PML has several activities in this regard. It is required for YAP translocation to nuclear bodies (nuclear substructures with transcriptionally active regions of chromatin), and enhances p73 activity where it is detected in complex with YAP-p73 on the promoters of Bax and p53AIP1 (Lapi et al. 2008; Strano et al. 2005) (Fig. 7.5). Moreover, PML enhances YAP stability, by promoting sumoylation of YAP on Lys97 and Lys242 and inhibiting YAP polyubiquitination and proteasomal-mediated degradation of YAP.
Although signalling through the core Hippo signalling cassette inhibits YAP in response to cell polarity, evidence suggests that under certain conditions, components of the core MST-LATS kinase cassette can induce apoptosis through the formation and stabilisation of the Yap-p73 transcriptional complex (Fig. 7.5a) (Donninger et al. 2011; Hamilton et al. 2009; Kawahara et al. 2008; Matallanas et al. 2007; Park et al. 2010; Yee et al. 2012). In leukemic cells, loss of LATS2 expression prevents p73 stabilisation and the induction of target genes including p21 and BAX, resulting in resistance to chemotherapeutic drugs (Kawahara et al. 2008).
The tumour suppressor RASSF1A is a key determinant in driving the formation of the YAP-p73 complex via signalling through the MST1/2-LATS1/2 kinase cassette. RASSF1A promotes MST1/2 activation by releasing MST from the inhibitory RAF-1 complex and preventing its dephosphorylation (Guo et al. 2011; Matallanas et al. 2007; O’Neill et al. 2004; Romano et al. 2010). In response to DNA-damaging agents, RASSF1A promotes the YAP-p73-dependent expression of PUMA and BAX. (Hamilton et al. 2009; Matallanas et al. 2007; Yee et al. 2012). Indeed phosphorylation of RASSF1A Ser131 by ATM, a key kinase activated following DNA damage, is required for YAP-p73-induced gene expression in response to DNA damage (Hamilton et al. 2009). RASSF1A requires activation of MST2 and LATS1 for the expression of p73 target genes and siRNA-mediated knockdown of MST2; LATS1- or YAP blocks the expression of BAX and PUMA, inhibiting apoptosis. Interestingly RASSF1A also activates p73 in a manner that is partially independent of MST2 but dependent on SAV for p73 activity (Fig. 7.5a). However, RASSF1A requires MST2 for YAP nuclear localisation and stabilisation of p73 (Donninger et al. 2011). The molecular determinants of quite how RASSF1A promotes the formation of the YAP-p73 complex remain to be determined although new evidence suggests that RASSF1A is an important determinant in defining the transcription factor repertoire of YAP (Fig. 7.5b) (van der Weyden et al. 2012). RASSF1A promotes YAP-p73 at the expense of other non-apoptotic YAP transcription factor complexes including YAP-TEAD and YAP-RUNX2. In this context it is likely that RASSF1A is promoting the interaction of YAP with additional regulatory proteins or altering the post-translational modifications of YAP which ultimately determine transcription factor preference (Fig. 7.5b). Indeed one such modification has been identified. The c-ABL phosphorylation of YAP-Tyr357 enhances the affinity of YAP for p73 while decreasing its interaction with RUNX1 (Levy et al. 2008). Thus post-translational modification is a clear regulator of YAP pro-apoptotic activity.
Recent reports suggest that the expression levels of the p63 isoform ΔNp63α may antagonise YAP-p73-mediated apoptosis as ΔNp63α has been shown to inhibit p73-mediated apoptosis (Rocco et al. 2006). YAP interacts with p63 and ΔNp63α and is required for ΔNp63α-mediated resistance to apoptosis in response to cisplatin (Strano et al. 2001; Yuan et al. 2010a). In response to cisplatin, c-ABL phosphorylation of both YAP (Tyr357) and ΔNp63α (Tyr 55, Tyr137 and Tyr308) promotes the formation of the YAP/ΔNp63α complex which inhibits the induction of apoptosis (Yuan et al. 2010a). Furthermore, in response to UV irradiation, JNK-mediated phosphorylation of YAP (Ser 138, Ser317,Thr362) promotes the stabilisation of ΔNp63α, by competing with ITCH for binding to ΔNp63α in an analogous manner to YAP-mediated stabilisation of p73 (Levy et al. 2008; Tomlinson et al. 2010). Indeed, it has been suggested that the elevated expression of ΔNp63α may protect against YAP-p73-driven apoptosis (Tomlinson et al. 2010).
It is of interest to note that p63 contains the same residues that are phosphorylated by JNK and c-ABL. c-ABL-dependent phosphorylation of p63 induces the expression of NOXA and PUMA which induce apoptosis (Gonfloni et al. 2009). It will therefore be of interest to determine the role of YAP in p63-mediated apoptosis.
11 Summary
It is now clear that the Hippo pathway has an important, evolutionary conserved role in the regulation of the apoptotic response. Many parallels can be drawn between the pathway in Drosophila and mammals. The anti-apoptotic transcriptional programme of Yki and YAP shares common targets, with transcription of members of the IAP family promoting the protection of cells from apoptosis. The current understanding of Hpo and Wts in driving apoptosis in Drosophila suggests that phosphorylation-dependent inactivation of Yki and inhibition of Diap-1 and Bantam transcription are key to the induction of developmental apoptosis. Multiple regulators of Bantam, both ubiquitous and tissue specific, have been characterised and it is likely that other yet unidentified Yki transcriptional complexes are responsible for the regulation of Diap-1 transcription.
In the mammalian system the MST and LATS kinases function in multiple pro-apoptotic pathways and can regulate the activity of multiple transcription factors that are classically associated with apoptosis, resulting in the induced expression of pro-apoptotic members of the bcl-2 family. In addition, MST1/2 also promote histone-mediated chromatin condensation via H2B and H2AX phosphorylation, allowing controlled destruction of the nucleus through fragmentation of genomic DNA. Some open questions remain regarding the upstream activators of both MST and LATS and although we know that caspases and RASSF family members promote MST1/2 there are likely to be other routes to activate this kinase and importantly we do not know if apoptotic pathways regulated by LATS1/2 are dependent on MST. In the mammalian system, YAP can both inhibit and promote apoptosis and the MST-LATS kinase cassette is required for both these activities. Although we are now beginning to understand some of the players such as RASSF1A and the expression of ΔNp63α that differentiate YAP pro- or anti-apoptotic activity, the molecular details of what determines the switch in the choice of YAP in transcription factor binding remain unknown. It is now clear that in cancer, inhibition of apoptosis is a hallmark that drives tumour growth. Indeed inactivation of MST (Matallanas et al. 2011) or LATS (Aylon et al. 2009) has been proposed as a mechanism of bypassing apoptosis. However, the most common mechanism in inhibiting Hippo pathway-mediated apoptosis is through the methylation of the RASSF family proteins which are a frequent occurrence in cancer (Richter et al. 2009). Not only does loss of RASSF1A reduce the activation of MST, it also impacts on the pro-apoptotic activities of YAP. Moreover, failure to initiate apoptosis alone may lead to enhanced proliferation through promotion of YAP proliferative complexes; however recent evidence suggests that sustained proliferation of human tumours frequently requires concomitant loss of YAP differentiation cofactors (Van der weyden et al. 2012).
References
Ahn SH, Cheung WL, Hsu JY, Diaz RL, Smith MM, Allis CD. Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell. 2005;120:25–36.
Ajiro K, Scoltock AB, Smith LK, Ashasima M, Cidlowski JA. Reciprocal epigenetic modification of histone H2B occurs in chromatin during apoptosis in vitro and in vivo. Cell Death Differ. 2010;17:984–93.
Anand R, Kim AY, Brent M, Marmorstein R. Biochemical analysis of MST1 kinase: elucidation of a C-terminal regulatory region. Biochemistry. 2008;47:6719–26.
Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006;20:2687–700.
Aylon Y, Yabuta N, Besserglick H, Buganim Y, Rotter V, Nojima H, et al. Silencing of the Lats2 tumor suppressor overrides a p53-dependent oncogenic stress checkpoint and enables mutant H-Ras-driven cell transformation. Oncogene. 2009;28:4469–79.
Aylon Y, Ofir-Rosenfeld Y, Yabuta N, Lapi E, Nojima H, Lu X, et al. The Lats2 tumor suppressor augments p53-mediated apoptosis by promoting the nuclear proapoptotic function of ASPP1. Genes Dev. 2010;24:2420–9.
Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell. 2003;11:11–23.
Baumgartner R, Poernbacher I, Buser N, Hafen E, Stocker H. The WW domain protein Kibra acts upstream of Hippo in Drosophila. Dev Cell. 2010;18:309–16.
Bennett FC, Harvey KF. Fat cadherin modulates organ size in Drosophila via the Salvador/Warts/Hippo signaling pathway. Curr Biol. 2006;16:2101–10.
Bi W, Xiao L, Jia Y, Wu J, Xie Q, Ren J, et al. c-Jun N-terminal kinase enhances MST1-mediated pro-apoptotic signaling through phosphorylation at serine 82. J Biol Chem. 2010;285:6259–64.
Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36.
Chen CL, Gajewski KM, Hamaratoglu F, Bossuyt W, Sansores-Garcia L, Tao C, et al. The apical-basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc Natl Acad Sci U S A. 2010;107:15810–5.
Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003;113:507–17.
Cho E, Feng Y, Rauskolb C, Maitra S, Fehon R, Irvine KD. Delineation of a Fat tumor suppressor pathway. Nat Genet. 2006;38:1142–50.
Choi J, Oh S, Lee D, Oh HJ, Park JY, Lee SB, et al. Mst1-FoxO signaling protects naive T lymphocytes from cellular oxidative stress in mice. PLoS One. 2009;4:e8011.
Cinar B, Fang PK, Lutchman M, Di Vizio D, Adam RM, Pavlova N, et al. The pro-apoptotic kinase Mst1 and its caspase cleavage products are direct inhibitors of Akt1. EMBO J. 2007;26:4523–34.
Costanzo A, Merlo P, Pediconi N, Fulco M, Sartorelli V, Cole PA, et al. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol Cell. 2002;9:175–86.
Creasy CL, Chernoff J. Cloning and characterization of a human protein kinase with homology to Ste20. J Biol Chem. 1995a;270:21695–700.
Creasy CL, Chernoff J. Cloning and characterization of a member of the MST subfamily of Ste20-like kinases. Gene. 1995b;167:303–6.
Del Re DP, Matsuda T, Zhai P, Gao S, Clark GJ, Van Der Weyden L, et al. Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J Clin Invest. 2010;120:3555–67.
Ditzel M, Broemer M, Tenev T, Bolduc C, Lee TV, Rigbolt KT, et al. Inactivation of effector caspases through nondegradative polyubiquitylation. Mol Cell. 2008;32:540–53.
Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–33.
Donninger H, Allen N, Henson A, Pogue J, Williams A, Gordon L, et al. Salvador protein is a tumor suppressor effector of RASSF1A with hippo pathway-independent functions. J Biol Chem. 2011;286:18483–91.
Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415:333–44.
Espanel X, Sudol M. Yes-associated protein and p53-binding protein-2 interact through their WW and SH3 domains. J Biol Chem. 2001;276:14514–23.
Fernando P, Kelly JF, Balazsi K, Slack RS, Megeney LA. Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci U S A. 2002;99:11025–30.
Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–58.
Genevet A, Wehr MC, Brain R, Thompson BJ, Tapon N. Kibra is a regulator of the Salvador/Warts/Hippo signaling network. Dev Cell. 2010;18:300–8.
Glantschnig H, Rodan GA, Reszka AA. Mapping of MST1 kinase sites of phosphorylation. Activation and autophosphorylation. J Biol Chem. 2002;277:42987–96.
Gonfloni S, Di Tella L, Caldarola S, Cannata SM, Klinger FG, Di Bartolomeo C, et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med. 2009;15:1179–85.
Goulev Y, Fauny JD, Gonzalez-Marti B, Flagiello D, Silber J, Zider A. SCALLOPED interacts with YORKIE, the nuclear effector of the hippo tumor-suppressor pathway in Drosophila. Curr Biol. 2008;18:435–41.
Goyal L, McCall K, Agapite J, Hartwieg E, Steller H. Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J. 2000;19:589–97.
Graves JD, Gotoh Y, Draves KE, Ambrose D, Han DK, Wright M, et al. Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J. 1998;17:2224–34.
Graves JD, Draves KE, Gotoh Y, Krebs EG, Clark EA. Both phosphorylation and caspase-mediated cleavage contribute to regulation of the Ste20-like protein kinase Mst1 during CD95/Fas-induced apoptosis. J Biol Chem. 2001;276:14909–15.
Greer EL, Brunet A. FOXO transcription factors in ageing and cancer. Acta Physiol (Oxf). 2008;192:19–28.
Grzeschik NA, Parsons LM, Allott ML, Harvey KF, Richardson HE. Lgl, aPKC, and Crumbs regulate the Salvador/Warts/Hippo pathway through two distinct mechanisms. Curr Biol. 2010;20:573–81.
Guo C, Zhang X, Pfeifer GP. The tumor suppressor RASSF1A prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J Biol Chem. 2011;286:6253–61.
Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development. 2011;138:9–22.
Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, et al. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8:27–36.
Hamilton G, Yee KS, Scrace S, O’Neill E. ATM regulates a RASSF1A-dependent DNA damage response. Curr Biol. 2009;19:2020–5.
Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114:457–67.
Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–61.
Ho LL, Wei X, Shimizu T, Lai ZC. Mob as tumor suppressor is activated at the cell membrane to control tissue growth and organ size in Drosophila. Dev Biol. 2010;337:274–83.
Hoshino M, Qi ML, Yoshimura N, Miyashita T, Tagawa K, Wada Y, et al. Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of YAP isoforms and p73. J Cell Biol. 2006;172:589–604.
Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell. 2005;122:421–34.
Iida S, Hirota T, Morisaki T, Marumoto T, Hara T, Kuninaka S, et al. Tumor suppressor WARTS ensures genomic integrity by regulating both mitotic progression and G1 tetraploidy checkpoint function. Oncogene. 2004;23:5266–74.
Jang SW, Yang SJ, Srinivasan S, Ye K. Akt phosphorylates MstI and prevents its proteolytic activation, blocking FOXO3 phosphorylation and nuclear translocation. J Biol Chem. 2007;282:30836–44.
Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995;9:534–46.
Kakeya H, Onose R, Osada H. Caspase-mediated activation of a 36-kDa myelin basic protein kinase during anticancer drug-induced apoptosis. Cancer Res. 1998;58:4888–94.
Kawahara M, Hori T, Chonabayashi K, Oka T, Sudol M, Uchiyama T. Kpm/Lats2 is linked to chemosensitivity of leukemic cells through the stabilization of p73. Blood. 2008;112:3856–66.
Ke H, Pei J, Ni Z, Xia H, Qi H, Woods T, et al. Putative tumor suppressor Lats2 induces apoptosis through downregulation of Bcl-2 and Bcl-x(L). Exp Cell Res. 2004;298:329–38.
Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, et al. Identification of a novel Ras-regulated proapoptotic pathway. Curr Biol. 2002;12:253–65.
Kim D, Shu S, Coppola MD, Kaneko S, Yuan ZQ, Cheng JQ. Regulation of proapoptotic mammalian ste20-like kinase MST2 by the IGF1-Akt pathway. PLoS One. 2010;5:e9616.
Kornbluth S, White K. Apoptosis in Drosophila: neither fish nor fowl (nor man, nor worm). J Cell Sci. 2005;118:1779–87.
Kostic C, Shaw PH. Isolation and characterization of sixteen novel p53 response genes. Oncogene. 2000;19:3978–87.
Kuninaka S, Nomura M, Hirota T, Iida S, Hara T, Honda S, et al. The tumor suppressor WARTS activates the Omi/HtrA2-dependent pathway of cell death. Oncogene. 2005;24:5287–98.
Kuninaka S, Iida SI, Hara T, Nomura M, Naoe H, Morisaki T, et al. Serine protease Omi/HtrA2 targets WARTS kinase to control cell proliferation. Oncogene. 2007;26:2395–406.
Lai ZC, Wei X, Shimizu T, Ramos E, Rohrbaugh M, Nikolaidis N, et al. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell. 2005;120:675–85.
Lapi E, Di Agostino S, Donzelli S, Gal H, Domany E, Rechavi G, et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol Cell. 2008;32:803–14.
Lee KK, Murakawa M, Nishida E, Tsubuki S, Kawashima S, Sakamaki K, et al. Proteolytic activation of MST/Krs, STE20-related protein kinase, by caspase during apoptosis. Oncogene. 1998;16:3029–37.
Lee KK, Ohyama T, Yajima N, Tsubuki S, Yonehara S. MST, a physiological caspase substrate, highly sensitizes apoptosis both upstream and downstream of caspase activation. J Biol Chem. 2001;276:19276–85.
Lee JH, Kim TS, Yang TH, Koo BK, Oh SP, Lee KP, et al. A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J. 2008;27:1231–42.
Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001.
Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28:2426–36.
Levy D, Adamovich Y, Reuven N, Shaul Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007;14:743–51.
Levy D, Adamovich Y, Reuven N, Shaul Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell. 2008;29:350–61.
Lin Y, Khokhlatchev A, Figeys D, Avruch J. Death-associated protein 4 binds MST1 and augments MST1-induced apoptosis. J Biol Chem. 2002;277:47991–8001.
Ling C, Zheng Y, Yin F, Yu J, Huang J, Hong Y, et al. The apical transmembrane protein Crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc Natl Acad Sci U S A. 2010;107:10532–7.
Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D, et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem. 2010;285:37159–69.
Liu CY, Lv X, Li T, Xu Y, Zhou X, Zhao S, et al. PP1 cooperates with ASPP2 to dephosphorylate and activate TAZ. J Biol Chem. 2011;286:5558–66.
Liu W, Wu J, Xiao L, Bai Y, Qu A, Zheng Z, et al. Regulation of neuronal cell death by c-Abl-Hippo/MST2 signaling pathway. PLoS One. 2012;7:e36562.
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–5.
Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, Piazzolla D, et al. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell. 2007;27:962–75.
Matallanas D, Romano D, Al-Mulla F, O’Neill E, Al-Ali W, Crespo P, et al. Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol Cell. 2011;44:893–906.
Milton CC, Zhang X, Albanese NO, Harvey KF. Differential requirement of Salvador-Warts-Hippo pathway members for organ size control in Drosophila melanogaster. Development. 2010;137:735–43.
Min B, Kim MK, Zhang JW, Kim J, Chung KC, Oh BC, et al. Identification of RUNX3 as a component of the MST/Hpo signaling pathway. J Cell Physiol. 2012;227:839–49.
Morinaka A, Funato Y, Uesugi K, Miki H. Oligomeric peroxiredoxin-I is an essential intermediate for p53 to activate MST1 kinase and apoptosis. Oncogene. 2011;30:4208–18.
Nolo R, Morrison CM, Tao C, Zhang X, Halder G. The bantam microRNA is a target of the hippo tumor-suppressor pathway. Curr Biol. 2006;16:1895–904.
O’Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 2004;306:2267–70.
O’Neill AK, Niederst MJ, Newton AC. Suppression of survival signalling pathways by the phosphatase PHLPP. FEBS J. 2012. http://onlinelibrary.wiley.com/doi/10.1111/j.1742-4658.2012.08537.x/full
Odashima M, Usui S, Takagi H, Hong C, Liu J, Yokota M, et al. Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circ Res. 2007;100:1344–52.
Oh H, Irvine KD. In vivo regulation of Yorkie phosphorylation and localization. Development. 2008;135:1081–8.
Oh H, Irvine KD. In vivo analysis of Yorkie phosphorylation sites. Oncogene. 2009;28:1916–27.
Oh H, Irvine KD. Cooperative regulation of growth by Yorkie and Mad through bantam. Dev Cell. 2011;20:109–22.
Oh HJ, Lee KK, Song SJ, Jin MS, Song MS, Lee JH, et al. Role of the tumor suppressor RASSF1A in Mst1-mediated apoptosis. Cancer Res. 2006;66:2562–9.
Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505.
Pantalacci S, Tapon N, Leopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol. 2003;5:921–7.
Park J, Kang SI, Lee SY, Zhang XF, Kim MS, Beers LF, et al. Tumor suppressor ras association domain family 5 (RASSF5/NORE1) mediates death receptor ligand-induced apoptosis. J Biol Chem. 2010;285:35029–38.
Pellock BJ, Buff E, White K, Hariharan IK. The Drosophila tumor suppressors Expanded and Merlin differentially regulate cell cycle exit, apoptosis, and Wingless signaling. Dev Biol. 2007;304:102–15.
Peng HW, Slattery M, Mann RS. Transcription factor choice in the Hippo signaling pathway: homothorax and yorkie regulation of the microRNA bantam in the progenitor domain of the Drosophila eye imaginal disc. Genes Dev. 2009;23:2307–19.
Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27:6507–21.
Poernbacher I, Baumgartner R, Marada SK, Edwards K, Stocker H. Drosophila pez acts in hippo signaling to restrict intestinal stem cell proliferation. Curr Biol. 2012;22:389–96.
Praskova M, Khoklatchev A, Ortiz-Vega S, Avruch J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem J. 2004;381:453–62.
Qiao M, Wang Y, Xu X, Lu J, Dong Y, Tao W, et al. Mst1 is an interacting protein that mediates PHLPPs’ induced apoptosis. Mol Cell. 2010;38:512–23.
Qin W, Dong P, Ma C, Mitchelson K, Deng T, Zhang L, et al. MicroRNA-133b is a key promoter of cervical carcinoma development through the activation of the ERK and AKT1 pathways. Oncogene. 2012;31(36):4067–75.
Rauch J, O’Neill E, Mack B, Matthias C, Munz M, Kolch W, et al. Heterogeneous nuclear ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells by regulating A-Raf transcription. Cancer Res. 2010;70:1679–88.
Ren A, Yan G, You B, Sun J. Down-regulation of mammalian sterile 20-like kinase 1 by heat shock protein 70 mediates cisplatin resistance in prostate cancer cells. Cancer Res. 2008;68:2266–74.
Ren F, Zhang L, Jiang J. Hippo signaling regulates Yorkie nuclear localization and activity through 14-3-3 dependent and independent mechanisms. Dev Biol. 2010;337:303–12.
Reszka AA, Halasy-Nagy JM, Masarachia PJ, Rodan GA. Bisphosphonates act directly on the osteoclast to induce caspase cleavage of mst1 kinase during apoptosis. A link between inhibition of the mevalonate pathway and regulation of an apoptosis-promoting kinase. J Biol Chem. 1999;274:34967–73.
Ribeiro PS, Josue F, Wepf A, Wehr MC, Rinner O, Kelly G, et al. Combined functional genomic and proteomic approaches identify a PP2A complex as a negative regulator of Hippo signaling. Mol Cell. 2010;39:521–34.
Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta. 2009;1796:114–28.
Robinson BS, Huang J, Hong Y, Moberg KH. Crumbs regulates Salvador/Warts/Hippo signaling in Drosophila via the FERM-domain protein Expanded. Curr Biol. 2010;20:582–90.
Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45–56.
Rogulja D, Rauskolb C, Irvine KD. Morphogen control of wing growth through the Fat signaling pathway. Dev Cell. 2008;15:309–21.
Romano D, Matallanas D, Weitsman G, Preisinger C, Ng T, Kolch W. Proapoptotic kinase MST2 coordinates signaling crosstalk between RASSF1A, Raf-1, and Akt. Cancer Res. 2010;70:1195–203.
Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, et al. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell. 2011;144:782–95.
Schoenherr JA, Drennan JM, Martinez JS, Chikka MR, Hall MC, Chang HC, et al. Drosophila activated Cdc42 kinase has an anti-apoptotic function. PLoS Genet. 2012;8:e1002725.
Silva E, Tsatskis Y, Gardano L, Tapon N, McNeill H. The tumor-suppressor gene fat controls tissue growth upstream of expanded in the hippo signaling pathway. Curr Biol. 2006;16:2081–9.
Song JJ, Lee YJ. Differential cleavage of Mst1 by caspase-7/-3 is responsible for TRAIL-induced activation of the MAPK superfamily. Cell Signal. 2008;20:892–906.
Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci U S A. 2010;107:1431–6.
Staley BK, Irvine KD. Hippo signaling in Drosophila: recent advances and insights. Dev Dyn. 2012;241:3–15.
Steller H. Regulation of apoptosis in Drosophila. Cell Death Differ. 2008;15:1132–8.
Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, et al. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J Biol Chem. 2001;276:15164–73.
Strano S, Monti O, Pediconi N, Baccarini A, Fontemaggi G, Lapi E, et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA damage. Mol Cell. 2005;18:447–59.
Sullivan A, Lu X. ASPP: a new family of oncogenes and tumour suppressor genes. Br J Cancer. 2007;96:196–200.
Tapon N, Harvey KF, Bell DW, Wahrer DC, Schiripo TA, Haber DA, et al. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell. 2002;110:467–78.
Taylor LK, Wang HC, Erikson RL. Newly identified stress-responsive protein kinases, Krs-1 and Krs-2. Proc Natl Acad Sci U S A. 1996;93:10099–104.
Teraishi F, Guo W, Zhang L, Dong F, Davis JJ, Sasazuki T, et al. Activation of sterile20-like kinase 1 in proteasome inhibitor bortezomib-induced apoptosis in oncogenic K-ras-transformed cells. Cancer Res. 2006;66:6072–9.
Thompson BJ, Cohen SM. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell. 2006;126:767–74.
Tomlinson V, Gudmundsdottir K, Luong P, Leung KY, Knebel A, Basu S. JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis. 2010;1:e29.
Tyler DM, Baker NE. Expanded and fat regulate growth and differentiation in the Drosophila eye through multiple signaling pathways. Dev Biol. 2007;305:187–201.
Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol. 2003;5:914–20.
Ura S, Masuyama N, Graves JD, Gotoh Y. Caspase cleavage of MST1 promotes nuclear translocation and chromatin condensation. Proc Natl Acad Sci U S A. 2001a;98:10148–53.
Ura S, Masuyama N, Graves JD, Gotoh Y. MST1-JNK promotes apoptosis via caspase-dependent and independent pathways. Genes Cells. 2001b;6:519–30.
Ura S, Nishina H, Gotoh Y, Katada T. Activation of the c-Jun N-terminal kinase pathway by MST1 is essential and sufficient for the induction of chromatin condensation during apoptosis. Mol Cell Biol. 2007;27:5514–22.
Valis K, Prochazka L, Boura E, Chladova J, Obsil T, Rohlena J, et al. Hippo/Mst1 stimulates transcription of the proapoptotic mediator NOXA in a FoxO1-dependent manner. Cancer Res. 2011;71:946–54.
van der Weyden L, Papaspyropoulos A, Poulogiannis G, Rust AG, Rashid M, Adams DJ, et al. Loss of Rassf1a synergizes with deregulated Runx2 signaling in tumorigenesis. Cancer Res. 2012;72(15):3517–27.
Vande Walle L, Lamkanfi M, Vandenabeele P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ. 2008;15:453–60.
Verghese S, Bedi S, Kango-Singh M. Hippo signalling controls Dronc activity to regulate organ size in Drosophila. Cell Death Differ. 2012;19(10):1664–76.
Vichalkovski A, Gresko E, Cornils H, Hergovich A, Schmitz D, Hemmings BA. NDR kinase is activated by RASSF1A/MST1 in response to Fas receptor stimulation and promotes apoptosis. Curr Biol. 2008;18:1889–95.
Vigneron AM, Vousden KH. An indirect role for ASPP1 in limiting p53-dependent p21 expression and cellular senescence. EMBO J. 2012;31:471–80.
Vigneron AM, Ludwig RL, Vousden KH. Cytoplasmic ASPP1 inhibits apoptosis through the control of YAP. Genes Dev. 2010;24:2430–9.
Visser S, Yang X. LATS tumor suppressor: a new governor of cellular homeostasis. Cell Cycle. 2010;9:3892–903.
Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–31.
Wang HC, Fecteau KA. Detection of a novel quiescence-dependent protein kinase. J Biol Chem. 2000;275:25850–7.
Wang SL, Hawkins CJ, Yoo SJ, Muller HA, Hay BA. The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell. 1999;98:453–63.
Wen W, Zhu F, Zhang J, Keum YS, Zykova T, Yao K, et al. MST1 promotes apoptosis through phosphorylation of histone H2AX. J Biol Chem. 2010;285:39108–16.
Willecke M, Hamaratoglu F, Kango-Singh M, Udan R, Chen CL, Tao C, et al. The fat cadherin acts through the hippo tumor-suppressor pathway to regulate tissue size. Curr Biol. 2006;16:2090–100.
Willecke M, Hamaratoglu F, Sansores-Garcia L, Tao C, Halder G. Boundaries of Dachsous cadherin activity modulate the Hippo signaling pathway to induce cell proliferation. Proc Natl Acad Sci U S A. 2008;105:14897–902.
Wilson R, Goyal L, Ditzel M, Zachariou A, Baker DA, Agapite J, et al. The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat Cell Biol. 2002;4:445–50.
Wong CH, Chan H, Ho CY, Lai SK, Chan KS, Koh CG, et al. Apoptotic histone modification inhibits nuclear transport by regulating RCC1. Nat Cell Biol. 2009;11:36–45.
Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–56.
Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell. 2008;14:388–98.
Xia H, Qi H, Li Y, Pei J, Barton J, Blackstad M, et al. LATS1 tumor suppressor regulates G2/M transition and apoptosis. Oncogene. 2002;21:1233–41.
Xiao L, Chen D, Hu P, Wu J, Liu W, Zhao Y, et al. The c-Abl-MST1 signaling pathway mediates oxidative stress-induced neuronal cell death. J Neurosci. 2011;31:9611–9.
Xu T, Wang W, Zhang S, Stewart RA, Yu W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development. 1995;121:1053–63.
Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, et al. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J Clin Invest. 2003;111:1463–74.
Yamamura Y, Lee WL, Inoue K, Ida H, Ito Y. RUNX3 cooperates with FoxO3a to induce apoptosis in gastric cancer cells. J Biol Chem. 2006;281:5267–76.
Yang X, Li DM, Chen W, Xu T. Human homologue of Drosophila lats, LATS1, negatively regulate growth by inducing G(2)/M arrest or apoptosis. Oncogene. 2001;20:6516–23.
Ye X, Deng Y, Lai ZC. Akt is negatively regulated by Hippo signaling for growth inhibition in Drosophila. Dev Biol. 2012;369(1):115–23.
Yee KS, Grochola L, Hamilton G, Grawenda A, Bond EE, Taubert H, et al. A RASSF1A polymorphism restricts p53/p73 activation and associates with poor survival and accelerated age of onset of soft tissue sarcoma. Cancer Res. 2012;72:2206–17.
Yoo SJ, Huh JR, Muro I, Yu H, Wang L, Wang SL, et al. Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat Cell Biol. 2002;4:416–24.
Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev Cell. 2010;18:288–99.
Yuan Z, Lehtinen MK, Merlo P, Villen J, Gygi S, Bonni A. Regulation of neuronal cell death by MST1-FOXO1 signaling. J Biol Chem. 2009;284:11285–92.
Yuan M, Luong P, Hudson C, Gudmundsdottir K, Basu S. c-Abl phosphorylation of DeltaNp63alpha is critical for cell viability. Cell Death Dis. 2010a;1:e16.
Yuan Z, Kim D, Shu S, Wu J, Guo J, Xiao L, et al. Phosphoinositide 3-kinase/Akt inhibits MST1-mediated pro-apoptotic signaling through phosphorylation of threonine 120. J Biol Chem. 2010b;285:3815–24.
Yuan F, Xie Q, Wu J, Bai Y, Mao B, Dong Y, et al. MST1 promotes apoptosis through regulating Sirt1-dependent p53 deacetylation. J Biol Chem. 2011;286:6940–5.
Yue T, Tian A, Jiang J. The cell adhesion molecule echinoid functions as a tumor suppressor and upstream regulator of the Hippo signaling pathway. Dev Cell. 2012;22:255–67.
Yun HJ, Yoon JH, Lee JK, Noh KT, Yoon KW, Oh SP, et al. Daxx mediates activation-induced cell death in microglia by triggering MST1 signalling. EMBO J. 2011;30:2465–76.
Zhang L, Ren F, Zhang Q, Chen Y, Wang B, Jiang J. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev Cell. 2008;14:377–87.
Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem. 2009;284:13355–62.
Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19:27–38.
Zhang H, Pasolli HA, Fuchs E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci U S A. 2011a;108:2270–5.
Zhang H, Wu S, Xing D. YAP accelerates Abeta(25–35)-induced apoptosis through upregulation of Bax expression by interaction with p73. Apoptosis. 2011b;16:808–21.
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–61.
Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–71.
Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010;24:72–85.
Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13:877–83.
Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012;26:54–68.
Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16:425–38.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Hamilton, G., O’Neill, E. (2013). Hippo Pathway and Apoptosis. In: Oren, M., Aylon, Y. (eds) The Hippo Signaling Pathway and Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6220-0_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6220-0_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6219-4
Online ISBN: 978-1-4614-6220-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)