
Abstract
Pontin (Pont), also known as Tip49, encodes a member of the AAA+ (ATPases Associated with Diverse Cellular Activities) superfamily and plays pivotal roles in cell proliferation and growth, yet its function in cell death has remained poorly understood. Here we performed a genetic screen for dominant modifiers of Eiger-induced JNK-dependent cell death in Drosophila, and identified Pont as a negative regulator of JNK-mediated cell death. In addition, loss of function of Pont is sufficient to induce cell death and activate the transcription of JNK target gene puc. Furthermore, the epistasis analysis indicates that Pont acts downstream of Hep. Finally, we found that Pont is also required for JNK-mediated thorax development and acts as a negative regulator of JNK phosphorylation. Together, our data suggest that pont encodes a negative component of Egr/JNK signaling pathway in Drosophila through negatively regulating JNK phosphorylation, which provides a novel role of ATPase in Egr-JNK signaling.
Similar content being viewed by others

Introduction
Pontin (Pont), also known as Tip49, Tip49a, NMP238, TAP54α, Ruvbl1, Rvb1, pontin521,2,3,4,5, belongs to the superfamily of AAA+ ATPases (ATPases Associated with Diverse Cellular Activities), which is the extension of the known AAA family6,7. Pont family proteins are evolutionally conserved from yeast to humans8, and have been reported to play vital roles in regulating gene transcription9,10, cell proliferation11,12 and growth13,14,15. However, the role of Pont in regulating cell death in development has remained elusive.
The c-Jun N-terminal kinase (JNK) pathway is evolutionarily conserved from fruit flies to humans, and plays diverse biological functions including stress response, cell death, proliferation, tumor metastasis, longevity and sleep control16,17,18,19,20,21,22,23,24,25. JNK pathway is also involved in neurodegenerative diseases such as Parkinson’s disease26,27 and Alzheimer’s disease28,29,30. In Drosophila, the tumor necrosis factor ortholog Eiger (Egr) binds to its receptors Wengen31 or Grindelwald32 to activate the conserved dTAK1 (JNKKK)–Hep (JNKK)–Bsk (JNK) kinase cascade33,34, which triggers cell death through downstream transcription factors like AP1 and FoxO35,36,37. We have previously performed a genetic screen for dominant modifiers of Egr-induced cell death, and have identified additional factors that regulate JNK-mediated cell death17,38,39,40,41,42,43.
In this report, we characterized the ATPase Pont as a negative regulator of Egr-JNK signaling in Drosophila. We found that loss of function of pont enhances Egr-induced JNK-mediated cell death, while gain of function of pont suppresses it. Furthermore, we showed that loss of function of pont activated JNK target gene puc transcription and Pontin acted downstream of Hep in the Egr-JNK pathway. Third, we found that Pontin was required for the growth of the scutellum in the developing thorax. Finally, we demonstrated that loss of function of pont was sufficient to elevate the phosphorylation of JNK in vivo. Collectively, our genetic work clarifies a role of ATPase Pontin in regulating Egr-JNK signaling during the development of the Drosophila.
Results and discussion
Loss of function of pont promotes Egr-induced cell death in Drosophila
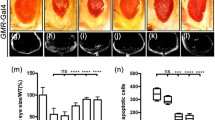
Ectopic expression of Egr in the developing eye driven by GMR-Gal4 triggers JNK-mediated cell death38,39 and produces dosage-dependent eye phenotypes—a rough eye from weak Egr expression (GMR>EgrW, Fig. 1b–m) and a small eye from strong Egr expression (GMR>EgrS, Fig. 5b–i). We have performed a genetic screen for dominant modifiers of the GMR>Egr phenotypes, and have identified additional factors that modulate Egr-induced JNK-mediated cell death40,42.

Light images of adult eyes are shown (a–l). GMR-Gal4 was used as a control (a), or to drive the expression of Egr (b-h, j, k) or pont-IR #1 (d, g, h, i), pont-IR #2 (e, j, k, l), GFP-IR #1 (c). GMR>Egr-induced rough eye phenotype (b) was enhanced by expression of pont-IR (d, e) or by pont heterozygous mutant (f) and not by expression of a random GFP-IR (c). pont-IR-enhancing Egr-induced eye phenotype was suppressed by expression of Pont (d, e, g, j, n) or Puc (d, e, h, k, n). Expression of pont-IR alone produced no evident eye phenotype (i, l, n). m Statistical analysis of the eye size in (a–f). Error bars means ± SEM, ***P ≤ 0.001, **P ≤ 0.01, n.s. not significant. n Statistical analysis of the eye size of the indicated genotypes. Error bars means ± SEM, ***P ≤ 0.001. Genotypes: GMR-Gal4/+ (a); UAS-EgrW/+; GMR-Gal4/+ (b); UAS-EgrW/ UAS-GFP-IR; GMR-Gal4/+ (c); UAS-EgrW/+; GMR-Gal4/UAS-pont-IR#1 (d); UAS-EgrW/ UAS-pont-IR #2; GMR-Gal4/+ (e); UAS-EgrW/+; GMR-Gal4/ pont5.1 (f); UAS-EgrW/UAS-Pont; GMR-Gal4/UAS-pont-IR #1 (g); UAS-EgrW/+; GMR-Gal4/UAS-pont-IR #1 UAS-Puc (h); GMR-Gal4/UAS-pont-IR #1 (i); UAS-EgrW/ UAS-pont-IR #2; GMR-Gal4/UAS-Pont (j); UAS-EgrW/ UAS-pont-IR #2; GMR-Gal4/UAS-Puc (k); UAS-pont-IR #2/+; GMR-Gal4/+(l)
Pont was identified as a suppressor from the screen as knockdown of pont dramatically enhanced GMR>EgrW-induced eye phenotype, producing rather small eyes (Fig. 1d–m), reminiscent of strong Egr expression in the eye (Fig. 5b)17. pont encodes a member of the AAA+ family of helicases (ATPases Associated with Diverse Activities)6,7. To further examine the role of pont in Egr-induced cell death, we used a second pont-IR (Fig. S1), as expected, which also exacerbated Egr-induced cell death in the eye (Fig. 1e–m), while expression of pont-IR alone failed to produce any obvious phenotype in the eye (Fig.1i, l, n). Consistently, when removing one copy of pont11, GMR>Egr-induced rough eye phenotype was also significantly enhanced (Fig. 1f–m), while deleting one copy of endogenous pont alone had no obvious phenotype in the eye (data not shown). To exclude the possible competition for Gal4 protein by the UAS lines, GFP-IR was adopted. Expression of GFP-IR failed to duplicate pont-IR effect in Egr-induced eye phenotype (Fig. 1c–m). Furthermore, loss of pont enhancing Egr-induced cell death phenotype was restored by the overexpression of Pont (Fig. S2) in the eye (Fig. 1d, e, g, j, n). Collectively, the data indicate loss of function of pont function promotes Egr-induced cell death in the developing eye and pont acts as a negative regulator of JNK signaling pathway.
Loss of function of pont triggers JNK-mediated cell death
We further characterized the role of Pont in regulating cell death and Egr-JNK signaling activation. RNA interference (RNAi)-mediated knockdown of Pont by en-Gal4 in the developing wing disc provoked strong cell death in the wing discs (Fig. 2b–g) compared with the control (Fig. 2a–g). The same results were obtained by expressing of pont-IR in the wing pouch driven by sd-Gal4 (Fig. S3a, b, c and g). To examine if loss of function of pont-induced cell death was due to the activation of caspase signaling, we checked the immunostaining of cleaved caspase-3 in the developing wing disc. Though expression of pont-IR triggered strong cell death in the wing disc (Fig. S3a, b, c and g), it failed to activate caspase signaling (Fig S4a, b and c). Taken together, the data indicate pont is required for the regulation of cell death and loss of function of pont triggers caspase-independent cell death. To investigate the physiological role of pont in JNK activation, we examined the expression pattern of puckered (puc) in pont loss-of-function background. puc encodes a JNK phosphatase whose expression is positively regulated by the Egr-JNK pathway17,44,45. Here, we used the puc-LacZ expression of the pucE69 enhancer-trap allele as a readout of the JNK activity in vivo46. The en>pont-IR strongly activated puc transcription in the posterior compartment of the wing compared with en-Gal4 (Fig. 2d, e). As a positive control, expression of Hemipterous (Hep), a JNK kinase33,34,47, also triggered puc activation in the wing disc (Fig. S5a and b). The same results were also obtained in sd>pont-IR, which activated puc transcription in the whole wing pouch (Fig. S3d, e and f). Collectively, these data suggest that endogenous pont is required for regulating the transcription of puc. To further clarify the role of Pont in regulating JNK signaling, we checked if loss of JNK signaling could abolish pont-IR induced cell death and puc activation. As expected, pont-IR induced cell death and puc activation were significantly suppressed by expression of a JNK phosphatase Puc17,44,45 (Fig. 2c, f, g). Consistently, pont-IR enhancing GMR>Egr-induced small eye phenotype was also restored by expression of Puc (Fig. 1d, e, h, k, n). Collectively, the data indicate loss of function of pont induced JNK activation and initiated JNK-mediated cell death.

Fluorescent images of acridine orange staining of the third-instar larva wing disc (a–c) and β-galactosidase immunostaining for puc-LacZ (d–f) are shown. Knockdown of pont in the posterior compartment of the wing disc driven by en-Gal4 (a, d) triggered cell death (b) and puc activation (e), which were suppressed by expressing of Puc (c, f). The lower panels are the magnification of the boxed area in the upper panels. g Statistical analysis of acridine orange-positive cells in (a–c). Error bars means ± SEM, ***P ≤ 0.001. Scale bar for a–c, 200 μm. Scale bar for d–f, 100 μm. Genotypes en-Gal4/+ (a); en-Gal4/UAS-pont-IR #2 (b); en-Gal4 UAS-pont-IR #2/+; UAS-Puc/+ (c); en-Gal4/+; pucE69/+ (d); en-Gal4/UAS-pont-IR #2; pucE69/+ (e); en-Gal4 UAS-pont-IR #2/+; pucE69/UAS-Puc (f)
Loss of function of pont produces JNK-mediated cell death phenotype in adults
Targeted knockdown of endogenous pont function in the thorax driven by pannier-Gal4 (pnr-Gal4) induced strong cell death and generated a reduced scutellum phenotype (Fig. 3a, b, i), mimicking Egr and Hep activation (Figs. 6b and 7g)17,40. Furthermore, pnr>pont-IR induced small scutellum phenotype was fully reverted by co-expressing Pont (Fig. 3b, c, i). The data indicate Pont is required for the regulation of the thorax development. To investigate the exact relationship between Pont and JNK signaling, we checked if pnr>pont-IR-induced defect was dependent on JNK pathway. As expected, pnr>pont-IR-induced developing defect in the scutellum was significantly exacerbated in the puc heterozygous mutant background (Fig. 3b, d, i), while depleting one copy of puc showed no evident phenotype in the the thorax (Fig. 3e–i). Reducing JNK signaling by removing one copy of endogenous bsk48 moderately suppressed pnr>pont-IR-induced defects in the notum (Fig. 3b, f, i). The same suppression effect was observed in another bsk mutant background48 (Fig. 3b, g, i). Upon stress, JNK translocates into the nucleus to phosphorylate the transcription factor Fos, and thus regulates cell death, tumor invasion and dorsal closure35,37,49,50. We found that fos heterozygous mutant compromised pnr>pont-IR-induced small scutellum phenotype (Fig. 3b, h, i). Based on the above data, we conclude that Pont regulates thorax development through JNK-Fos signaling.

Light images of Drosophila thoraxes (a–h) are shown. pnr-Gal4 was used as a control (a), or to drive the expression of pont-IR (b–d, f–h) or UAS-Pont (c). pnr>pont-IR induced small scutellum phenotype (b, i), was suppressed by expression of Pont (c, i) and enhanced in puc heterozygous mutant background (d, i), while puc heterozygous mutant alone showed no effect on the scutellum (e, i). pnr>pont-IR-induced small scutellum phenotype (b, i) was suppressed by in bsk heterozygous mutant (f, g, i) and fos heterozygous mutant (h, i) background. i Statistical analysis of the scutellum size in (a–h). Error bars means ± SEM, ***P ≤ 0.001, **P ≤ 0.01. Genotypes: pnr-Gal4/+ (a); UAS-pont-IR #2/+; pnr-Gal4/+ (b); UAS-pont-IR #2/UAS-Pont; pnr-Gal4/ UAS-Pont (c); UAS-pont-IR #2/+; pnr-Gal4/pucE69 (d); pnr-Gal4/pucE69 (e); UAS-pont-IR #2/+; pnr-Gal4/bsk1/+(f); UAS-pont-IR #2/+; pnr-Gal4/bsk2/+(g); UAS-pont-IR #2/+; pnr-Gal4/fos1/+(h)
Loss of function of pont triggers JNK phosphorylation
The above data demonstrate a role of Pont in negatively regulating Egr-JNK signaling. To elucidate how Pont regulates Egr-JNK in development, we checked the immunostaining of phosphorylation of JNK (pJNK), which represents the direct activation of the JNK signaling40. Compared with the en-Gal4 control (Fig. 4a–a”’), RNAi-mediated downregulation of pont in the posterior compartment of the wing disc resulted in strong cell death (Fig. 2b, g) and JNK phosphorylation (Fig. 4b–b”’), which was suppressed by expression of JNK phosphatase Puc (Figs. 2c–g and 4c–c”’). Collectively, the data indicate Pont is a negative regulator of JNK phosphorylation during development.

Fluorescent images of the immunostaining for JNK phosphorylation (pJNK) (a–a”’, b–b”’, c–c”’) are shown. en-Gal4 was used as a control (a–a”’), or to drive the expression of pont-IR (b–b”’, c–c”’) or UAS-Puc (c–c”’). Compared with the en-Gal4 alone (a–a”’), en>pont-IR induced strong JNK phosphorylation (b’, b”, b”’, indicated by white arrow in b’ and red arrow in b”), which was abolished by expression of Puc (c–c”’). Scale bar, 200 μm. Genotypes: en-Gal4 UAS-GFP/+ (a–a”’); en-Gal4 UAS-GFP /UAS-pont-IR #2 (b–b”’); en-Gal4 UAS-GFP /UAS-pont-IR #2; UAS-Puc/+ (c–c”’)
Expression of Pont inhibits Egr-induced cell death
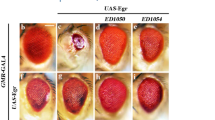
The above data argue a role of Pont in regulating Egr-JNK-induced cell death and JNK activation. Next we checked the overexpression of Pont to see if gain of Pont could suppress Egr-induced cell death phenotype. Targeted expression of a strong form of Egr in the eye disc driven by GMR promoter induced strong cell death (Fig. 5f–j) and produced a small eye phenotype with reduced eye tissues (Fig. 5a, b, i). Consistent with our hypothesis, ectopic expression of Pont (Fig. S2) showed a strong suppression effect on the Egr-induced small eye phenotype (Fig. 5c–i) and cell death in the eye disc (Fig. 5g–j), while no effect was observed in control groups (Fig. 5d, h, i, j), indicating Pont is a negative regulator of Egr in the developing eye.

Light images of adult eyes (a–d) and fluorescent images of acridine orange staining (e–h) are shown. GMR-Gal4 was used as a control (a, e) or to drive the expression of Egr (b–d, f–h) or UAS-Pont (c, g), UAS-LacZ (d, h). GMR>Egr-induced small eye phenotype (b) and cell death (f) were suppressed by expression of Pont (c, g) not by expression of LacZ (d, h). i Statistical analysis of the eye size in (a–d). Error bars means ± SEM, ***P ≤ 0.001, n.s. not significant. j Statistical analysis of acridine orange-positive cells in (e–h). Error bars means ± SEM, ***P ≤ 0.001, n.s. not significant. Scale bar, 100 μm. Genotypes: GMR-Gal4/+ (a, e); UAS-EgrS/+; GMR-Gal4/+ (b, f); UAS-EgrS/UAS-Pont; GMR-Gal4/ UAS-Pont (c, g); UAS-EgrS/+; GMR-Gal4/ UAS-LacZ (d, h)
Next we wanted to know if the genetic interaction between Egr and Pont is tissue specific. Egr-induced cell death phenotypes in the thorax and wing were examined. Expression of Egr in the wing disc initiated by patched-Gal4 (ptc-Gal4)40,51, which is expressed along the anterior/posterior (A/P) boundary and shows a strong expression pattern in the wing tips which develop into the notum of the Drosophila, induced strong cell death and almost abolished the scutellum (Fig. 6a, b, j). Consistently, ectopic expression of Pont significantly restored ptc>Egr-induced small scutellum phenotype to that of the control group (Fig. 6a, b, c, j, lower panels). In the adult wing, ptc>Egr produced a loss of anterior cross vein (ACV) and a notching phenotype in the wing margin (Fig. 6d, e, k), which was immensely suppressed by gain of Pont function (Fig. 6f–k). Collectively, the data suggest that Pont is a negative regulator of Egr-induced cell death phenotype in Drosophila. To understand how Pont regulates Egr signaling, we checked Egr target gene puc expression. Expression of Egr along the A/P boundary induced strong cell death (Fig. 6b, e, j, k, data not shown) and puc activation (Fig. 6g, h). Consistent with the role of Pont in suppressing Egr-induced cell death phenotype in the wing and thorax, ectopic Egr-induced puc activation was largely blocked by expression of Pont (Fig. 6h, i). Taken together, our observation suggests that Pont is required for the endogenous Egr-JNK signaling pathway.

Light images of Drosophila adult thoraxes (a–c), Drosophila wings (d–f) and X-gal staining of third-instar larva wing disc (g–i) are shown. ptc-Gal4 was used as a control (a, d, g), or to drive the expression of Egr (b, c, e, f, h, i) or UAS-Pont (c, f, i), or UAS-LacZ (b, e). The lower panels are magnification of the boxed area in the upper panels. Compared with the ptc-Gal4 control (a, d, g), ptc>Egr-induced small scutellum (b), loss of anterior cross vein (ACV) phenotype (e) and puc activation (h) were suppressed by expression of Pont (c, f, i). j Statistical analysis of the scutellum size in (a–c). Error bars means ± SEM, ***P ≤ 0.001. k Statistical analysis of the presence of ACV in (d–f). Error bars means ± SEM, ***P ≤ 0.001. Genotypes: ptc-Gal4/+; pucE69/+ (a, d, g); ptc-Gal4 UAS-EgrW/+; pucE69/UAS-LacZ (b, e); ptc-Gal4 UAS-EgrW/+; pucE69/+ (h); ptc-Gal4 UAS-EgrW /UAS-Pont; pucE69/ UAS-Pont (c, f, i)
Pont acts downstream of Hep in the Egr-JNK pathway
To genetically locate the epistasis of Pont in the Egr-JNK pathway, we examined the genetic interaction between Hep (JNKK) and Pont in the developing eye, thorax and wing. Expression of a constitutive active form of Hep in the developing eye (GMR>HepCA) triggers JNK-mediated cell death and gives rise to small eyes with a reduced photoreceptor cells phenotype (Fig. 7a, b, e)40. Expression of Pont compromised GMR>HepCA-induced phenotype (Fig. 7c–e), while expression of randomly inserted LacZ transgene showed no effect (Fig. 7d, e). Similar to ectopic Egr (ptc>Egr) producing a small scutellum and loss of ACV phenotype under the control of ptc-promoter, the expression of wild type of Hep (ptc>Hep) also generates such phenotype (Fig. 7f, g, j, k, l, o). Consistent with the observation in the eye, expression of Pont but not LacZ shows a strong suppression effect of ptc>Hep-induced small scutellum and loss of ACV phenotype (Fig. 7h–j, m–o). Combining the above data, we conclude that Pont modulates Egr-JNK pathway at downstream of Hep through the phosphorylation of Bsk.

Light images of Drosophila adult eyes (a–d), thoraxes (f–i) and Drosophila wings (k–n) are shown. GMR-Gal4 was used as a control (a) or to drive the expression of Hep (b–d) or UAS-Pont (c) or UAS-LacZ (d). GMR>Hep-induced cell death phenotype (b) was suppressed by expression of Pont (c) and not by expression of LacZ (d). ptc-Gal4 was used as a control (f, k) or to drive the expression of Hep (g–i, l–n) or UAS-Pont (h, m) or UAS-LacZ (i, n). ptc>Hep-induced small scutellum phenotype (g) and loss of ACV phenotype (l) were suppressed by expression of Pont (h, m) and not by expression of LacZ (i, n). The lower panels are the magnification of boxed area in the upper panels. e Statistical analysis of the eye size in (a–d). Error bars means ± SEM, ***P ≤ 0.001, **P ≤ 0.01, n.s. not significant. j Statistical analysis of the scutellum size in (f–i). Error bars means ± SEM, ***P ≤ 0.001, n.s. not significant. o Statistical analysis of the presence of ACV in (k–n). Error bars means ± SEM, ***P ≤ 0.001, n.s. not significant. Genotypes: GMR-Gal4/+ (a); GMR-Gal4 UAS-HepCA/+ (b); UAS-Pont/+; GMR-Gal4 UAS-HepCA/UAS-Pont (c); GMR-Gal4 UAS-HepCA/UAS-LacZ (d); ptc-Gal4/+; pucE69/+ (f, k); ptc-Gal4/UAS-HepWT; pucE69/+ (g, l); ptc-Gal4 UAS-HepWT/UAS-Pont; UAS-Pont/ pucE69 (h, m); ptc-Gal4 UAS-HepWT/+; UAS-LacZ/ pucE69 (i, n)
Conclusion
In the present work, we have identified ATPase Pontin as a crucial modulator of the conserved Egr-JNK signaling during the development of Drosophila. Our genetic evidence revealed that Pontin is a negative regulator of Egr-JNK cascade. We showed that loss of function of Pontin promoted while gain of function of Pontin suppressed Egr-induced cell death. Consistent with our observation, dominant-negative mutant of Pontin potentiates the apoptotic activity of c-Myc and E2F152 and in HCC cells the knockdown of Pont also led to spontaneous apoptosis53. We further showed that Pontin acted downstream of Hep in the Egr-JNK pathway to induce JNK-mediated puc activation and scutellum development. Finally, we demonstrated that Pontin was a negative regulator of JNK phosphorylation, for loss of function of Pontin was sufficient to induce JNK phosphorylation in vivo. The work proposes a novel role of ATPase in the modulating of JNK pathway in vivo and further study should clarify if other ATPases also interact with the JNK cascade.
Materials and methods
Fly stocks
All stocks were raised on standard Drosophila media and crosses were performed at 25 °C unless otherwise indicated. UAS-pont-IR #1 and UAS-pont-IR #2 were obtained from NIG stock center. bsk1/+, bsk2/+, fos1/+, UAS-LacZ, UAS-GFP-IR and en-Gal4 were obtained from the Bloomington Drosophila Stock Center. UAS-Pont, pont5.1/TM3, Ser11, pucE69, UAS-Egrw, UAS-EgrR 17, UAS-HepWT 40, UAS-HepCA 42, UAS-Puc40, GMR-Gal4, ptc-Gal4, sd-Gal4, pnr-Gal4 and ap-Gal451 have been previously described. The third-instar larvae were heated-shocked at 37 °C for 1 h and allowed to recover for 2 h at 25 °C.
Light image
Flies of indicated genotypes were collected and immediately frozen in −80 °C. Flies were placed on the 1% agarose plate before images taking. Wings were dissected and mounted on the slide in the alcohol/glycerol (1:1) medium and flies were mounted on the 1% agarose plate in the alcohol/glycerol medium for the image taking of thoraxes. Light images of wings were collected with Olympus microscope BX51, and light images of thoraxes were collected with OLYMPUS stereo microscope SZX16.
AO staining
Wing discs were dissected from the third-instar larvae in 1% phosphate-buffered saline (PBS) buffer and stained for acridine orange as previously described40. Each genotype was dissected with 20 discs for statistics.
X-gal staining
Wing discs were dissected from the third-instar larvae in 1% PBS buffer and stained for β-galactosidase (β-gal) activity as previously described54.
Immunohistochemistry
The third-instar larvae of indicated genotypes were collected and dissected in 1% PBS buffer. The antibody staining of imaginal discs was conducted as previously described18. The following antibodies were used: mouse anti-β-gal (1:400, Developmental Studies Hybridoma Bank), rabbit anti-phospho-JNK (1:200, Calbiochem) and rabbit anti-cleaved caspase-3 (1:400, Cell Signaling and Technology); secondary antibodies were anti-rabbit-Alexa (1:1000, Cell Signaling and Technology) and anti-mouse-Cy3 (1:1000, Jackson ImmunoResearch).
Change history
10 July 2019
Due to a technical error, content intended for publication in Volume 4 (2018) published in Volume 5 (2019). The content has been moved into the correct volume, and the citation information was updated accordingly.
References
Huber, O. et al. Pontin and reptin, two related ATPases with multiple roles in cancer. Cancer Res. 68, 6873–6876 (2008).
Huen, J. et al. Rvb1-Rvb2: essential ATP-dependent helicases for critical complexes. Biochem. Cell Biol. 88, 29–40 (2010).
Kanemaki, M. et al. Molecular cloning of a rat 49-kDa TBP-interacting protein (TIP49) that is highly homologous to the bacterial RuvB. Biochem. Biophys. Res. Commun. 235, 64–68 (1997).
Shen, X., Mizuguchi, G., Hamiche, A. & Wu, C. A chromatin remodelling complex involved in transcription and DNA processing. Nature 406, 541–544 (2000).
Bauer, A. et al. Pontin52 and reptin52 function as antagonistic regulators of beta-catenin signalling activity. EMBO J. 19, 6121–6130 (2000).
Iyer, L. M., Leipe, D. D., Koonin, E. V. & Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 146, 11–31 (2004).
Neuwald, A. F., Aravind, L., Spouge, J. L. & Koonin, E. V. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43 (1999).
Baek, S. H. When ATPases pontin and reptin met telomerase. Dev. Cell 14, 459–461 (2008).
Diop, S. B. et al. Reptin and Pontin function antagonistically with PcG and TrxG complexes to mediate Hox gene control. EMBO Rep. 9, 260–266 (2008).
Kim, J. H. et al. Transcriptional regulation of a metastasis suppressor gene by Tip60 and beta-catenin complexes. Nature 434, 921–926 (2005).
Bellosta, P. et al. Myc interacts genetically with Tip48/Reptin and Tip49/Pontin to control growth and proliferation during Drosophila development. Proc. Natl. Acad. Sci. USA 102, 11799–11804 (2005).
Etard, C., Gradl, D., Kunz, M., Eilers, M. & Wedlich, D. Pontin and Reptin regulate cell proliferation in early Xenopus embryos in collaboration with c-Myc and Miz-1. Mech. Dev. 122, 545–556 (2005).
Jonsson, Z. O. et al. Rvb1p and Rvb2p are essential components of a chromatin remodeling complex that regulates transcription of over 5% of yeast genes. J. Biol. Chem. 276, 16279–16288 (2001).
King, T. H., Decatur, W. A., Bertrand, E., Maxwell, E. S. & Fournier, M. J. A well-connected and conserved nucleoplasmic helicase is required for production of box C/D and H/ACA snoRNAs and localization of snoRNP proteins. Mol. Cell. Biol. 21, 7731–7746 (2001).
Lim, C. R. et al. The Saccharomyces cerevisiae RuvB-like protein, Tih2p, is required for cell cycle progression and RNA polymerase II-directed transcription. J. Biol. Chem. 275, 22409–22417 (2000).
Davis, R. J. Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 (2000).
Xue, L. et al. Tumor suppressor CYLD regulates JNK-induced cell death in Drosophila. Dev. Cell 13, 446–454 (2007).
Igaki, T., Pagliarini, R. A. & Xu, T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 16, 1139–1146 (2006).
Enomoto, M. & Igaki, T. Src controls tumorigenesis via JNK-dependent regulation of the Hippo pathway in Drosophila. EMBO Rep. 14, 65–72 (2013).
Igaki, T. Correcting developmental errors by apoptosis: lessons from Drosophila JNK signaling. Apoptosis 14, 1021–1028 (2009).
Takahama, K. et al. Pan-neuronal knockdown of the c-Jun N-terminal Kinase (JNK) results in a reduction in sleep and longevity in Drosophila. Biochem. Biophys. Res. Commun. 417, 807–811 (2012).
Wang, M. C., Bohmann, D. & Jasper, H. JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev. Cell 5, 811–816 (2003).
Twumasi-Boateng, K. et al. An age-dependent reversal in the protective capacities of JNK signaling shortens Caenorhabditis elegans lifespan. Aging Cell 11, 659–667 (2012).
Biteau, B., Karpac, J., Hwangbo, D. & Jasper, H. Regulation of Drosophila lifespan by JNK signaling. Exp. Gerontol. 46, 349–354 (2011).
Oh, S. W. et al. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc. Natl. Acad. Sci. USA 102, 4494–4499 (2005).
Wilhelm, M., Xu, Z., Kukekov, N. V., Gire, S. & Greene, L. A. Proapoptotic Nix activates the JNK pathway by interacting with POSH and mediates death in a Parkinson disease model. J. Biol. Chem. 282, 1288–1295 (2007).
Pan, J. et al. Expression of FasL and its interaction with Fas are mediated by c-Jun N-terminal kinase (JNK) pathway in 6-OHDA-induced rat model of Parkinson disease. Neurosci. Lett. 428, 82–87 (2007).
Zhu, X. et al. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the ‘two hit’ hypothesis. Mech. Ageing Dev. 123, 39–46 (2001).
D’Ambrosio, C. et al. Hyperphosphorylation of JNK-interacting protein 1, a protein associated with Alzheimer disease. Mol. Cell. Proteomics 5, 97–113 (2006).
Lagalwar, S., Guillozet-Bongaarts, A. L., Berry, R. W. & Binder, L. I. Formation of phospho-SAPK/JNK granules in the hippocampus is an early event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 65, 455–464 (2006).
Kanda, H., Igaki, T., Kanuka, H., Yagi, T. & Miura, M. Wengen, a member of the Drosophila tumor necrosis factor receptor superfamily, is required for Eiger signaling. J. Biol. Chem. 277, 28372–28375 (2002).
Andersen, D. S. et al. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature 522, 482–486 (2015).
Igaki, T. et al. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 21, 3009–3018 (2002).
Moreno, E., Yan, M. & Basler, K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 12, 1263–1268 (2002).
Chaussepied, M. et al. Upregulation of Jun and Fos family members and permanent JNK activity lead to constitutive AP-1 activation in Theileria-transformed leukocytes. Mol. Biochem. Parasitol. 94, 215–226 (1998).
Luo, X., Puig, O., Hyun, J., Bohmann, D. & Jasper, H. Foxo and Fos regulate the decision between cell death and survival in response to UV irradiation. EMBO J. 26, 380–390 (2007).
Oo, T. F., Henchcliffe, C., James, D. & Burke, R. E. Expression of c-fos, c-jun, and c-jun N-terminal kinase (JNK) in a developmental model of induced apoptotic death in neurons of the substantia nigra. J. Neurochem. 72, 557–564 (1999).
Liu, J. et al. Analysis of Drosophila segmentation network identifies a JNK pathway factor overexpressed in kidney cancer. Science 323, 1218–1222 (2009).
Ma, X. J. & Xue, L. [Regulation of the JNK signaling pathway by dual leucine zipper kinase DLK.]. Yi Chuan 32, 785–790 (2010).
Ma, X. et al. dUev1a modulates TNF-JNK mediated tumor progression and cell death in Drosophila. Dev. Biol. 380, 211–221 (2013).
Ma, X. et al. Src42A modulates tumor invasion and cell death via Ben/dUev1a-mediated JNK activation in Drosophila. Cell Death Dis. 4, e864 (2013).
Ma, X. et al. NOPO modulates Egr-induced JNK-independent cell death in Drosophila. Cell Res. 22, 425–431 (2012).
Ma, X. et al. Bendless modulates JNK-mediated cell death and migration in Drosophila. Cell Death Differ. 21, 407–415 (2014).
Martin-Blanco, E. et al. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 12, 557–570 (1998).
McEwen, D. G. & Peifer, M. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development 132, 3935–3946 (2005).
Agnes, F., Suzanne, M. & Noselli, S. The Drosophila JNK pathway controls the morphogenesis of imaginal discs during metamorphosis. Development 126, 5453–5462 (1999).
Glise, B., Bourbon, H. & Noselli, S. hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell 83, 451–461 (1995).
Sluss, H. K. et al. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev. 10, 2745–2758 (1996).
Uhlirova, M. & Bohmann, D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 25, 5294–5304 (2006).
Zeitlinger, J. & Bohmann, D. Thorax closure in Drosophila: involvement of Fos and the JNK pathway. Development 126, 3947–3956 (1999).
Wang, X. et al. FoxO mediates APP-induced AICD-dependent cell death. Cell Death Dis. 5, e1233 (2014).
Dugan, K. A., Wood, M. A. & Cole, M. D. TIP49, but not TRRAP, modulates c-Myc and E2F1 dependent apoptosis. Oncogene 21, 5835–5843 (2002).
Wood, M. A., McMahon, S. B. & Cole, M. D. An ATPase/helicase complex is an essential cofactor for oncogenic transformation by c-Myc. Mol. Cell 5, 321–330 (2000).
Xue, L. & Noll, M. Dual role of the Pax gene paired in accessory gland development of Drosophila. Development 129, 339–346 (2002).
Acknowledgements
We thank Dr. Peter Gallant, the NIG and the Bloomington Drosophila Stock Center for fly stocks. This research was supported by the Fundamental Research Funds for the Central Universities (2000219125) to X.W. and also supported by the National Basic Research Program of China (973 Program) (2010CB944901, 2011CB943903), National Natural Science Foundation of China (31071294, 31171413, 31371490), the Specialized Research Fund for the Doctoral Program of Higher Education of China (20120072110023) and Shanghai Committee of Science and Technology (09DZ2260100, 14JC1406000).
Author information
Authors and Affiliations
Contributions
X.W. and L.X. conceived and designed the experiments; X.W, X.H. and C.W. performed the experiments and analyzed the data; X.W. and L.X. wrote the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by A. Rufini
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Wang, X., Huang, X., Wu, C. et al. Pontin/Tip49 negatively regulates JNK-mediated cell death in Drosophila. Cell Death Discov. 4, 74 (2018). https://doi.org/10.1038/s41420-018-0074-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-018-0074-1
- Springer Nature Limited
This article is cited by
-
MKK3 modulates JNK-dependent cell migration and invasion
Cell Death & Disease (2019)
-
Pontin/Tip49 acts as a novel regulator of JNK pathway
Cell Death & Disease (2018)