
Abstract
Histamine is a biogenic amine playing a central role in allergy and peripheral inflammatory reactions and acts as a neurotransmitter and neuromodulator in the brain. In the adult, histamine is produced mainly by mast cells and hypothalamic neurons, which project their axons throughout the brain. Thus, histamine exerts a range of functions, including wakefulness control, learning and memory, neurogenesis, and regulation of glial activity. Histamine is also known to modulate innate immune responses induced by brain-resident microglia cells and peripheral circulating monocytes, and monocyte-derived cells (macrophages and dendritic cells). In physiological conditions, histamine per se causes mainly a pro-inflammatory phenotype while counteracting lipopolysaccharide-induced inflammation both in microglia, monocytes, and monocyte-derived cells. In turn, the activation of the innate immune system can profoundly affect neuronal survival and function, which plays a critical role in the onset and development of brain disorders. Therefore, the dual role of histamine/antihistamines in microglia and monocytes/macrophages is relevant for identifying novel putative therapeutic strategies for brain diseases. This review focuses on the effects of histamine in innate immune responses and the impact on neuronal survival, function, and differentiation/maturation, both in physiological and acute (ischemic stroke) and chronic neurodegenerative conditions (Parkinson’s disease).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Overview of the Functions of Histamine in the Brain
Histamine is an endogenous biogenic amine commonly known as an inflammatory mediator of allergic reactions. Studies suggest that these conditions may affect brain function and contribute to neurodegenerative processes (Klein et al. 2016; Sarlus et al. 2012). Histamine is formed by decarboxylation of the essential amino acid L-histidine in a reaction catalyzed by L-histidine decarboxylase (HDC). Several stimuli such as injury, day/night cycle, inflammation, among others, regulate histamine production. Besides the endogenous production, diet (e.g., fermented food, chocolate, wine) provides an exogenous histamine (or L-histidine) source. Histamine degradation occurs by methylation, catalyzed by histamine N-methyltransferase (HNMT), or oxidation, catalyzed by diamine oxidase (DAO), which depends on the species and tissues. In the brain, most histamine is N-methylated by HNMT, and the product N-methylhistamine is further oxidized by monoamine oxidase-B (MAO-B), which is expressed in histaminergic neurons and astrocytes (Brown et al. 2001). This metabolic pathway is particularly relevant in the context of brain diseases where MAO-B inhibitors are used for therapy, such as Parkinson’s disease (PD; discussed in Sect. 3.1). Histamine mediates its actions by G protein-coupled receptors, the histamine receptors H1-4 (H1-4R) (Brown et al. 2001). H1R and H2R are low-affinity receptors expressed in the central nervous system and periphery and mediate excitatory actions. H1R recruits Gq/11, which leads to the activation of phospholipase C, the formation of inositol triphosphate (IP3), and diacylglycerol (DAG), which induces calcium release from internal stores and activation of protein kinase C (PKC); while H2R recruits Gs proteins that activate the adenylyl cyclase and protein kinase A (PKA) (Brown et al. 2001). The activation of H1R is mainly associated with allergic reactions. The most well-described physiological function of H2R is in controlling the release of gastric acid, but recent data showed that it is also involved in cell differentiation and immune reactions. H3R and H4R are high-affinity receptors with predominant inhibitory effects. H3R are abundant in the central and peripheral nervous systems and recruit Gαi/o proteins inhibiting adenylyl cyclase and PKA activation. H3R acts as a presynaptic receptor, inhibiting the release of histamine or other neurotransmitters (e.g., glutamate, noradrenaline, serotonin, dopamine, GABA, acetylcholine). Recently it was shown that H3R also forms heterodimers with dopamine receptors D1 and D2 and adenosine A2A receptors, therefore modulating dopaminergic and purinergic neurotransmission, respectively (Márquez-Gómez et al. 2018; Moreno et al. 2011; Moreno-Delgado et al. 2020). The best-known physiological actions modulated by H3R include food intake, nociception, cognition, and sleep-wake control (Brown et al. 2001; Nieto-Alamilla et al. 2016; Ito et al. 2018). H4R is mainly expressed by peripheral immune cells and recruits Gαi/o proteins that inhibit cAMP production via adenylyl cyclase inhibition and activate the mitogen-activated protein kinase (MAPK) signaling. H4R can also activate the Gβγ subunits that activate phospholipase C and increase intracellular calcium concentration. H4R is mainly involved in inflammatory reactions. Several inflammatory and injury stimuli regulate the expression of histaminergic receptors in a temporal and spatial (cells, tissues)-specific manner, which impacts the functional effects induced by histamine.
In the brain, histamine is produced mainly by mast cells and hypothalamic neurons in the tuberomammillary nucleus (TMN). Mast cells are mainly located in the area postrema, the choroid plexus, hypothalamus, hippocampus, the parenchymal border of the blood–brain barrier, thalamus, and in the meninges (Silverman et al. 2000; Mattila et al. 2011). These immune cells react quickly to several stimuli, releasing histamine and other inflammatory and vasoactive mediators from intracellular secretory granules (Chikahisa et al. 2013). On the other side, histaminergic neurons project ramifications and release histamine throughout the entire brain, allowing histamine to be involved in a broad range of physiological functions, such as sleep-wake control, emotions, learning and memory, neuronal survival, and neurogenesis (Panula and Nuutinen 2013; Bernardino et al. 2012; Saraiva et al. 2019; Rocha et al. 2016; Ferreira et al. 2012). In the healthy brain, histamine is found at nanomolar levels (Soya et al. 2008; Croyal et al. 2011; Bourgogne et al. 2012). However, several brain pathological conditions may be associated with changes in circulating histamine levels (blood and cerebrospinal fluid) and histaminergic neuronal innervations, suggesting that histamine plays a role in regulating neuronal survival, function, and behavior. Alterations in histamine levels and metabolism depend on the specific injury/pathology. For example, an increase of histaminergic innervations was found in substantia nigra of PD patients (see Sect. 3.1), and elevated cerebrospinal fluid histamine levels were found in multiple sclerosis patients (Vizuete et al. 2000; Kallweit et al. 2013; Anichtchik et al. 2000). In contrast, no or residual changes of histamine or histamine metabolite levels were found in Alzheimer’s disease patients (Gabelle et al. 2017; Motawaj et al. 2010).
The histaminergic system is involved in the proliferation and commitment of neuronal precursor cells in the embryonic and adult brain. Embryonic and adult neural stem and progenitor cells express histamine receptors (H1R, H2R, H3R) (Agasse et al. 2008; Escobedo-Avila et al. 2014), being H1R responsible for the increase of intracellular calcium levels in immature cells (Escobedo-Avila et al. 2014; Molina-Hernández et al. 2013; Grade et al. 2010) and neuronal differentiation (Bernardino et al. 2012; Molina-Hernández et al. 2013; Molina-Hernández and Velasco 2008; Rodríguez-Martínez et al. 2012). Histamine is one of the first molecules to appear in the rodent embryonic brain, reaching its maximal value at embryonic day 14, when neurogenesis of deep layers occurs in the cerebral cortex. Indeed, histamine increased proliferation and differentiation of FOXP2 deep layer cortical neuronal cells via H1R activation in cerebrocortical neural precursor cultures and infused in the cerebral ventricles through intrauterine injection (Molina-Hernández et al. 2013; Rodríguez-Martínez et al. 2012). Histamine also affected dopaminergic lineage during development by reducing the proliferation and survival of embryonic ventral mesencephalon dopaminergic precursors via H1R activation in vitro and in vivo. Neural progenitors (E10 and E12) were exceptionally responsive to histamine actions, while differentiated dopaminergic neurons (E14 and E16) were mainly spared (Escobedo-Avila et al. 2014). The same research group showed that the systemic administration of the H1R antagonist/inverse agonist chlorpheniramine increased dopaminergic differentiation in embryos (E16) while in 21-day-old pups reduced the number of dopaminergic neurons in the substantia nigra pars compacta and dorsal striatum, reduced dopamine levels in the striatum, and induced motor impairments. This suggests that histamine inhibited embryonic dopaminergic differentiation at E14-E16, while having the opposite actions in the post-natal period, with H1R being responsible for these effects (Márquez-Valadez et al. 2019). We showed that histamine induces neuronal commitment and axonogenesis of neonatal subventricular zone (SVZ) neural stem cells through upregulation of the expression of the proneurogenic genes Mash1, Dlx2, and Ngn1 and activation of JNK MAPKinase, respectively, in vitro (Bernardino et al. 2012). Moreover, the intracerebroventricular (i.c.v.) administration of adult mice with histamine increased the number of SVZ-derived neuroblasts capable of migrating towards the olfactory bulb which differentiate into mature neurons (Eiriz et al. 2014). We have also developed histamine-releasing microparticles, which were highly efficient in inducing neuronal differentiation. SVZ cells pretreated with histamine-loaded microparticles and then grafted into the dentate gyrus of hippocampal organotypic slice cultures or the dentate gyrus and striatum in vivo showed increased neuronal differentiation compared with non-treated ones (Bernardino et al. 2012). Recent studies suggest that histamine also modulates hippocampal neurogenesis via H1R or H3R activation (Ambrée et al. 2014; Guilloux et al. 2017). H1R knockout (KO) mice showed a reduced number of proliferative cells in the hippocampal dentate gyrus (DG) together with pronounced deficits in spatial learning and memory (Ambrée et al. 2014). Additionally, the chronic treatment for 28 days with S 38093, a brain-penetrant antagonist/inverse agonist of H3R, increased hippocampal neurogenesis in young (3-month-old) and aged (16-month-old) mice. In aged mice, S 38093 increased the expression of brain-derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF) and improved the cognitive performance in a context discrimination task (Guilloux et al. 2017). These studies indicate that histamine potentiates hippocampal neurogenesis, which correlates with hippocampus-related behaviors. We found that the intrahippocampal administration of histamine induces a slight increase of neuronal differentiation while decreasing the volume and complexity of DG immature neurons. Notably, histamine counteracted the negative impact induced by lipopolysaccharide (LPS) on DG neurogenesis (Saraiva et al. 2019). Altogether, these results emphasize the multidimensional effects of histamine in the modulation of SVZ and SGZ neurogenic niches, which may be differentially responsive due to particular characteristics of each niche and/or differential expression of histamine receptors.
One of the most well-known functions induced by histamine is its involvement in wakefulness regulation. The neuronal production of histamine shows diurnal fluctuations in healthy individuals, with increased levels of histamine found during the day. Recently, it was demonstrated that mice displaying chronic histamine depletion, induced by adeno-associated virus expressing Cre recombinase microinjected into the TMN of HDC flox adult mice, exhibited a decrease in wakefulness and increased in non-rapid eye movement sleep throughout the day. Moreover, these mice showed induced depression-like behavior, decreased locomotor activity, and impaired aversive memory (Yamada et al. 2020). Neuronal histamine fluctuations are also altered in patients with neurodegenerative diseases, which in turn impact circadian rhythms. Healthy adult subjects showed high HDC mRNA levels during the daytime, suggesting a role for neuronal histamine in regulating day-night rhythms. In contrast, the HDC mRNA day-night fluctuation was markedly distinct in the TMN of patients with neurodegenerative diseases such as PD, Alzheimer’s disease, and Huntington’s disease (Shan et al. 2012a).
Aging, the leading risk factor for most neurodegenerative diseases, is also accompanied by alterations in histamine levels, signaling, and metabolism. Aged mice (24-month-old) showed lower expression of H1R mRNA in the cortex, hypothalamus, hippocampus, and medulla relative to adult (3-month-old) animals. Age-related changes in H2R mRNA levels were restricted to the pons and cerebellum, and decreased H3R mRNA was found only in the medulla. Histamine levels were increased while HNMT activity significantly decreased in the hypothalamus, midbrain, and cortex of 12 versus 3-month-old rats (Terao et al. 2004; Mazurkiewicz-Kwilecki and Prell 1984). Further studies should address whether these changes of the histaminergic system during aging contribute to the etiology and/or progression of neurodegenerative diseases.
The histaminergic system is also gender-dependent (Acevedo et al. 2006). Histamine levels and the cortical levels of H1R and H2R are higher in female animals than in males (Lebel et al. 1980; Ghi 1999). In particular, higher levels of H1R were found in the median eminence and neurohypophysis of aged rodent females (Cacabelos et al. 2016). The mast cells’ degranulation and histamine release are also gender-dependent, with mast cells from rat females being more susceptible than males to the effects of sex steroids (Muñoz-Cruz et al. 2015). In humans, the levels of the metabolites of histamine, tele-methylhistamine (t-MH), and tele-methylimidazoleacetic acid (t-MIAA) were higher in cerebrospinal fluid from older subjects, being higher in females than in males (Prell et al. 1990). A general experimental procedure to investigate the impact of gender in function and behavior is by removing the reproductive organs. It was found that ovariectomized female mice were more sensitive to the arousal-reducing effects of the histamine H1R antagonist pyrilamine than castrated males (Easton et al. 2004). Moreover, HDC KO female mice did not show impairment in object recognition as reported in HDC KO males while showed impairments in spatial learning and memory compared with the males that showed increased water-maze acquisition and memory retention (Acevedo et al. 2006; Dere 2003). Moreover, female rats are more sensitive than males to histidine-induced anorexia (Kasaoka et al. 2005). These reports raise the importance of considering age and gender aspects in studying the impact of the histaminergic system on brain function and behavior.
2 The Functions of Histamine in Innate Immune Cells
Histamine plays a crucial role in the modulation of the activity of innate immune cells. The innate immune system is the first line of defense against pathogens. Contrary to the adaptive immune system, this response is non-specific and immediately prevents the spread of pathogens. The following sections focus on the effects of histamine on brain-resident microglia and circulating peripheral monocytes and macrophages, the most well-described innate immune cells.
2.1 Microglia
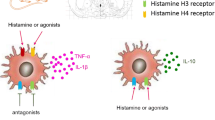
Microglial cells, the resident innate immune cells in the brain, can patrol and protect brain parenchyma against injuries or infections. Lesion or degeneration activates microglial cells becoming amoeboid, phagocytic, capable of migrating to the injury site and releasing inflammatory factors (Prinz et al. 2019). In vitro, microglia express HDC, HNMT and can release histamine (Iida et al. 2015; Katoh et al. 2001). Moreover, microglial cells express all four types of histamine receptors (H1-4R) in vitro and in vivo (Zhang et al. 2020), whose expression is regulated by several inflammatory conditions (Shan et al. 2019). At the functional level, histamine increases microglia mobility through signaling pathways involving α5β1 integrin, p-38, and Akt (Ferreira et al. 2012) and promotes phagocytosis by the activation of H1R (Rocha et al. 2016). Moreover, it promotes the release of pro-inflammatory mediators, namely tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), IL-1β, and IL-10, and the production of reactive oxygen species (ROS) (Zhang et al. 2020; Dong et al. 2014). These pro-inflammatory actions are mediated mainly by H1R or H4R activation (Rocha et al. 2016; Ferreira et al. 2012; Zhou et al. 2019). Other evidence supported the role of microglial H3R for brain homeostasis by showing that JNJ10181457, an H3R inverse agonist, suppressed ATP-induced microglial migration in hippocampal slices, inhibited microglial engulfment of dead neurons induced by N-methyl-d-aspartate in hippocampal slices and prefrontal cortex, and reduced the LPS-induced upregulation of microglial pro-inflammatory cytokines and improved depression-like behavior in vivo (Iida et al. 2017). In contrast, under an inflammatory context mimicked by LPS, histamine acts as an anti-inflammatory and neuroprotective agent (Saraiva et al. 2019; Barata-Antunes et al. 2017). Some evidence suggest that this effect may be due to the interaction of H4R with tumor necrosis factor receptor-associated factor 6 (TRAF6), which decreased TRAF6-mediated ubiquitination of K63, inhibited NF-kB activation ultimately resulting in an inhibition of the release of inflammatory cytokines in LPS-induced microglial cells (Shan et al. 2019). Other evidence suggests that histamine and imetit (H3R agonist) inhibited microglial chemotaxis, phagocytosis, and LPS-induced cytokine production, probably reducing forskolin-induced cAMP accumulation and ATP-induced intracellular calcium transients in vitro (Iida et al. 2015). Recently, a study showed that histamine 2/3 receptor agonists inhibited exploratory laparotomy-induced and LPS-induced cognitive decline, microglia activation, and the release of inflammatory factors (TNF-α, IL-1β, IL-10) and signaling (NF-κB) by activating the PI3K/AKT/FoxO1 pathway (Chen et al. 2020). We also demonstrated that histamine could revert LPS-induced hippocampal neuroinflammation by decreasing the expression of markers for activated glial cells (Iba-1, HMGB1), and markers correlated with neuronal functionality and synaptic strength (CREB, PSD-95), indicating a reversion of LPS-induced cognitive decline in the adult hippocampus (Saraiva et al. 2019). Thus, histamine seems to have a dual role in microglial functions depending on the microenvironment, the activation state of cells, and which histamine receptor is activated. Table 1 summarizes the studies mentioned above supporting the role of histamine in microglial cells.
2.2 Monocytes/Macrophages
In contrast to microglia, monocytes are short-lived cells generated throughout life from bone marrow resident hematopoietic stem cells. Circulating monocytes patrol the bloodstream and, upon recruitment to tissues, give rise to tissue-resident macrophages or dendritic cells. Human monocytes and monocyte-derived macrophages express H1R and H2R at the mRNA and protein levels, while the protein expression of H4R is controversial due to limitations in the specificity of available antibodies (Werner et al. 2014a; Werner et al. 2014b; Triggiani et al. 2007). The expression of histamine receptors may depend on the inflammatory milieu (Capelo et al. 2016). Moreover, differentiation of monocytes into macrophages or dendritic cells is associated with changes in histamine receptor expression, specifically an increase of H1R and downregulation of H2R (Mommert et al. 2018; Triggiani et al. 2007; Wang et al. 2000). This change in histamine receptors’ expression through differentiation may explain the differential effects of histamine in these immune cell populations.
Histamine induces monocytic expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) and its receptor CCR2 and the endothelial expression of adhesion molecules (Kimura et al. 2004), which facilitate transmigration. Histamine is involved in the reactions of human monocytes to allergens. Monocytes from allergic rhinitis patients stimulated with allergen extracts of house dust mite release IFN-γ via H4R activation and IL-6 via H1R activity. This study suggests that a combination of H1R and H4R antagonists should be more effective in blocking the inflammatory allergic response (Peng et al. 2019). In bone marrow-derived macrophages of BALB/c mice and on RAW 264.7 cells, the activation of H4R induces chemotaxis and phagocytosis (Czerner et al. 2014).
In human monocytes and macrophages, histamine suppressed LPS-induced pro-inflammatory cytokine secretion (TNF-α, IFN-α, IL-18), whereas it enhanced anti-inflammatory IL-10 (Østerud and Olsen 2014; Frei et al. 2013). Several studies suggest that the activation of H2R is responsible for these effects (Takahashi et al. 2004; Azuma et al. 2001). Inline, histamine decreased the expression of CD14 (cell surface receptor that binds to the LPS-LBP complex) in human monocytes via H2R activation, which may explain the inhibitory effects induced by histamine on LPS-induced TNF-α production (Takahashi et al. 2003). Other studies showed that histamine and the H4R agonist ST-1006 decreased the IFN-γ and LPS-induced CCL4 expression in differentiated M1 macrophages. These data suggest that histamine may counteract inflammatory reactions via H4R activation (Mommert et al. 2018). This dual role of histamine is also observed when an additional stimulus is given in combination with LPS. Accordingly, histamine inhibited LPS or the combination of LPS and TNF-α-induced LPS-induced tissue factor (TF) activity in human monocytes. In contrast, when monocytes were incubated with LPS and PMA, histamine induced a significant rise in TF activity. These data suggest that histamine induces an anti-inflammatory effect on LPS and LPS/TNF-α stimulated monocytes while having a pro-inflammatory effect in the presence of LPS and PMA (Østerud and Olsen 2014). Histamine may also modulate the response of monocytes to other inflammatory stimuli besides LPS. High mobility group box 1 (HMGB1) is a conserved nuclear protein that induces adhesion molecules and inflammatory factors (e.g., IFN-γ, TNF-α) on monocytes. It was shown that histamine inhibited pro-inflammatory effects induced by HMGB1 in human peripheral blood mononuclear cells (PBMCs) via PKA activation (Takahashi et al. 2013). Histamine also inhibited the pro-inflammatory responses induced by advanced glycation end products (AGEs) in human monocytes via H2R activation and the cAMP/PKA pathway (Zhang et al. 2010). Moreover, histamine prevented human monocytic apoptosis induced by serum deprivation, CD95/Fas ligation, or dexamethasone via H2R. Monocytes cultured with anti-IL-10 mAb and histamine did not exhibit an inhibitory effect on apoptosis, suggesting a role for IL-10 in this effect (Soga et al. 2007). Like microglia, histamine may induce pro- or anti-inflammatory reactions in monocytes and tissue-derived cells, depending on the microenvironment. Table 1 summarizes the studies mentioned above supporting the role of histamine and its receptors in monocytes and macrophages.
3 The Role of Histamine in Neurodegenerative Diseases
Accumulating evidence support the relevance of the histaminergic system for several brain diseases, including PD, stroke, Alzheimer’s disease, neuropsychiatric disorders, epilepsy, multiple sclerosis, and amyotrophic lateral sclerosis. Histamine and histamine receptor levels change in a disease-specific pattern, explaining the differential effects of histamine in each context. This review focuses on the impact of histamine in PD and ischemic stroke, as prototypical chronic and acute brain diseases, respectively. There is a substantial amount of data about the role of histamine in these diseases and the putative crosstalk between innate immune cells and neuronal survival and function, which sustain the focus on these two diseases in the following sections.
3.1 Parkinson’s Disease
Parkinson’s disease is characterized by the progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta leading to striatal dopamine depletion and the accumulation of α-synuclein aggregates known as Lewy bodies (Spillantini et al. 1997; Damier 1999). The key symptoms that clinically define PD are rigidity, tremor, bradykinesia, and postural instability, among other non-motor manifestations (e.g., olfactory impairment, gastrointestinal dysfunction) preceding motor symptoms.
The innate immune system plays a crucial role in PD pathology. Increased numbers of microglial cells within the substantia nigra, and increased levels of pro-inflammatory mediators (TNF-α, IL-1β, IL-6) were found in the blood, cerebrospinal fluid, and brains of PD patients and animal models of the disease (McGeer et al. 1988; Harms et al. 2013; Watson et al. 2012; Nagatsu et al. 2000). Microglia activation precedes dopaminergic degeneration (Krashia et al. 2019; Sanchez-Guajardo et al. 2010; Gerhard et al. 2006), which may contribute to dopaminergic degeneration in later stages of the disease (Harms et al. 2021). Interestingly, the substantia nigra contains a higher density of microglia than other brain regions (Kim et al. 2000), rendering DA neurons more sensitive to an immune challenge. Inline, the intracerebral or systemic administration of LPS induced microgliosis and reduced tyrosine hydroxylase-positive neurons in the substantia nigra (Kim et al. 2000; Qin et al. 2007). Therefore, these and other studies raised the notion that microglial activation leads to dopaminergic neuronal degeneration and disease progression. On the contrary, recent studies showed that Cx3cr1-deficiency mice, which display deficient microglia function, had exacerbated dopaminergic degeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and α-synuclein-A53T, suggesting that microglia plays a protective role in PD (Castro-Sánchez et al. 2018; Parillaud et al. 2017). These discrepancies may depend on the experimental models and the stage of development of the disease. Alterations in the peripheral innate immune system, particularly in monocytes, were also shown in PD. Classical monocytes expressing CCR2 and CCL2 are enriched in the blood, and CSF isolated from PD patients (Funk et al. 2013; Wijeyekoon et al. 2020). Monocytes from PD patients have impaired pro-inflammatory cytokine production, impaired phagocytic function, and had high expression of genes related to PD, such as Snca or Lrrk2 (Raj et al. 2014; Grozdanov et al. 2014; Hasegawa et al. 2000; Gardai et al. 2013). On the other hand, other reports showed that monocytes from PD patients are hyperactive to LPS stimulation and showed increased phagocytic capacity (Grozdanov et al. 2014; Grozdanov et al. 2019; Wijeyekoon et al. 2018). These contradictory data are most likely due to differences in the isolation and culture procedures, cohorts, and methodologies used. Notably, the genetic deletion of CCR2 was neuroprotective, suggesting a deleterious role for infiltrating monocytes in PD (Harms et al. 2018). Taken together, these studies illustrate the critical influence of microglia and peripheral myeloid cell actions on PD pathogenesis.
The histaminergic system is affected in PD. Several reports have shown increased histaminergic fibers and local histamine levels in the substantia nigra of PD postmortem human brain and animal models (Panula and Nuutinen 2013; Anichtchik et al. 2000; Rinne et al. 2002; Nowak et al. 2009). Moreover, a Thr105Ile polymorphism of HNMT was shown to be associated with PD, suggesting that lower HNMT activity plays a role in the pathogenesis of PD (Palada et al. 2012). On the contrary, Shan and colleagues showed no alterations in HDC mRNA levels among different clinical or Braak-PD stages, despite the accumulation of Lewy bodies and Lewy neurites in the TMN of PD patients (Shan et al. 2012b). Interestingly, the same authors showed that the mRNA expression of H3R decreased in the SN in PD, while H4R expression increased in the caudate nucleus and putamen. Moreover, increased mRNA levels of HNMT were found in the SN and the putamen in PD patients (Shan et al. 2012c). Altogether these data suggest that changes in the histaminergic system, particularly increased histamine levels, altered histamine metabolism, and expression of histamine receptors, may contribute to PD pathology.
Some studies have been focusing on the role of endogenous histamine in PD pathology. By using HDC KO mice, it was found that histamine deficiency increased amphetamine-induced rotation induced by the neurotoxin 6-hydroxydopamine (6-OHDA) injection in the medial forebrain bundle (MFB) but did not affect levodopa-induced dyskinesia (LID), increased striatal expression of D1 and D2 receptors and H3R mRNA, and increased dopamine release. Therefore, there is an interplay between histaminergic and dopaminergic neurotransmission within the nigrostriatal pathway, impacting motor behavior (Koski et al. 2020). On the other side, the administration of histamine in the substantia nigra or systemic administration of histidine (a precursor of histamine) leads to dopaminergic neuronal death and aggravated motor behavior in vivo (Rocha et al. 2016; Vizuete et al. 2000; Liu et al. 2007; Liu et al. 2008). These neurotoxic effects may depend on the activation of microglia phagocytosis and oxidative signaling pathways (Rocha et al. 2016). Moreover, the i.c.v. administration of the specific H4R antagonist JNJ7777120 inhibited microglial activation and TNF-α release, reduced apomorphine-induced rotational behavior, prevented dopaminergic neuron degeneration, and reduced Lewy body-like neuropathology in a rotenone-induced PD rat model (Zhou et al. 2019; Fang et al. 2021). This suggests that histamine may have a detrimental effect on DA survival via H4R activation. In addition, H2R antagonists have also been reported to improve the motor symptoms of PD patients and to exert neuroprotective effects, suggesting that depending on the model, histamine may also induce DA degeneration by H2R activation. Indeed, ranitidine (an H2R antagonist), protected against rotenone-induced apoptosis, inhibiting phosphorylation of JNK and P38, promoting the phosphorylation of extracellular signal-regulated protein kinase (ERK), and suppressed CASP3 enzyme activity in an human dopaminergic cell line (Park et al. 2009). These studies suggest that histamine may play a major role in inducing DA degeneration via H4R or H2R activation.
Interestingly, dopaminergic neurons are susceptible to LPS-induced inflammatory response, being LPS widely used to mimic PD in rodent models (Qin et al. 2007; Zhao et al. 2018). Noteworthy, we showed that histamine prevented LPS-induced microglial activation and dopaminergic neuronal death (Rocha et al. 2014). Thus, histamine per se induces the activation of microglial responses and dopaminergic degeneration while, when given in combination with other inflammatory stimuli (LPS), triggers an anti-inflammatory and neuroprotective response.
Therapeutic options for PD include levodopa (dopamine precursor), dopamine agonists, and MAO-B inhibitors. The therapeutic choice depends on the stage of the disease (de Bie et al. 2020). MAO-B is involved in the degradation of an extensive range of biogenic and dietary amines, including dopamine and the histaminergic metabolite N-methylhistamine (see Sect. 1). Therefore, MAO-B inhibitors increase dopamine and N-methylhistamine levels (Riederer and Laux 2011). Increased levels of dopamine improve motor control in patients. The inhibition of histamine metabolism may impact histaminergic neurotransmission and dopaminergic function/survival; however, no studies have been described so far focusing on this putative relation.
Another standard therapeutic option is levodopa (or L-DOPA) which shows high efficacy in the early stage of the disorder. Over time levodopa loses effectiveness and causes dyskinesias and severe psychiatric complications. Several studies have studied the role of the histaminergic system in levodopa-induced dyskinesia (LID). The mechanisms underlying LID in PD may involve H2R. H2R are highly expressed in the input (striatum) and output (globus pallidus, SN) regions of the basal ganglia, particularly in the GABAergic striatopallidal and striatonigral pathways. Several H2R antagonists (e.g., famotidine, ranitidine) could inhibit LID in PD models in vivo (Ahmed et al. 2019; Lim et al. 2015; Yang et al. 2013). This effect may be due to normalized levels of GRK3, reduced ERK activation, and FosB accumulation in the lesioned striatum, and reduced Arc and proenkephalin levels in dyskinetic animals (Ahmed et al. 2019). The ability of famotidine to counteract LID was also observed in MPTP-lesioned macaques but not in clinical trials with PD patients (Mestre et al. 2014; Johnston et al. 2010). Another study focused on the role of H3R agonists immepip or imetit in LID, in rats lesioned with 6-OHDA in the SN or MPTP-lesioned marmoset models for PD. The chronic administration of the H3R agonist immepip alongside L-DOPA decreased LID compared with L-DOPA alone (Gomez-Ramirez et al. 2006; Avila-Luna et al. 2019). Immepip also decreased GABA and glutamate content in the striatum (Avila-Luna et al. 2019).
Deep brain stimulation is also used in some PD patients, in more advanced stages of the disorder, with successful suppression of motor symptoms; however, it does not stop the disease progression. The subthalamic nucleus is an effective therapeutic target for deep brain stimulation, and histamine levels are elevated in the basal ganglia in PD patients. Zhuang et al. demonstrated that histamine levels rise in the subthalamic nucleus to compensate for abnormal firing patterns. Injection of histamine into the subthalamic nucleus restored normal firing patterns and ameliorated Parkinsonian motor deficits via H2R activation. Moreover, deep brain stimulation regularized neuronal firing through endogenous histamine release under Parkinsonian conditions. These data suggest a role of histamine in the basal ganglia circuitry that regularizes subthalamic nucleus neuronal firing patterns and ameliorates motor dysfunction (Zhuang et al. 2018). Table 2 summarizes the main effects induced by histamine in PD models.
3.2 Stroke
Stroke is characterized by the sudden onset of focal neurological deficits of variable nature and severity caused by cerebrovascular dysfunction. Nearly 85% of strokes are ischemic, meaning that thrombosis or an embolism causes a sudden interruption of blood flow, which may lead to paralysis, impaired speech, and loss of vision, or other neurological signs and symptoms. The remaining percentage of cases is triggered by hemorrhage (Moskowitz et al. 2010).
Innate immune responses mediated by microglia and monocytes play a crucial role in the pathology of ischemic stroke (Schilling et al. 2003; Zrzavy et al. 2018). Both microglia and monocytes trigger pro- and anti-inflammatory roles, which depend on the type and severity of the injury, brain area affected, the window of time post-stroke, and methodologies. Microglial cells became activated within minutes after the onset of ischemic injury, proliferate, migrate to the site of injury, release inflammatory mediators (TNF-α, IL-1β, IL-6, among others) and phagocyte cellular debris (Clausen et al. 2008; Lambertsen et al. 2012). The pharmacological or genetic depletion of microglia induced either protection or toxicity in ischemic injury. Depletion caused by the treatment with colony-stimulating factor 1 (CSF1)/c-kit inhibitor, minocycline, or by using the CX3CR1 KO mice exacerbated inflammation, infiltration of peripheral immune cells, and augmented ischemic brain injury, suggesting endogenous defense mechanism induced by microglia in ischemic stroke (Jin et al. 2017; Szalay et al. 2016; Faustino et al. 2011; Tsuji et al. 2020). On the contrary, Li and colleagues showed that the selective elimination of microglia in the early phases of ischemic injury induced anti-inflammatory and decreased pro-inflammatory factors, decreased ischemic infarct volume, while improved motor performance (Li et al. 2021). These data corroborates that microglia has a detrimental phenotype at the early stages of ischemic injury while at later stages is involved in the repair program post-ischemia. Microglia activation precedes infiltration of peripheral monocytes into the ischemic brain. The leaky BBB allows monocytes to enter into the ischemic injury within the first hours post-ischemia and peaks at 3–7 days (Chu et al. 2014). Monocytes also release several inflammatory mediators and interact with parenchymal cells. The functional role of monocytes has been investigated using CCR2 pharmacological inhibitors or CCR2-deficient mice. Studies using these strategies reveal that CCR2 deficiency reduced angiogenesis and worse behavioral performance suggesting a pro-regenerative action of infiltrating monocytes (Pedragosa et al. 2020; Perego et al. 2016). This effect may be time-dependent, as other studies showed that using CCR2 KO mice and the CCR2 pharmacological inhibitors or neutralizing antibodies resulted in smaller infarct size and lower mortality at 3 days post-stroke while from 5 to 28 days after stroke, treated or KO mice had higher mortality and showed no functional recovery (Fang et al. 2018; Wattananit et al. 2016). These data suggest that monocytes/macrophages mainly polarized to a pro-inflammatory phenotype at the early stage but gradually switched to anti-inflammatory at later stages post-stroke.
Ischemic stroke is caused by a blockage of the cerebral blood supply resulting in death or dysfunction of brain cells, activation of microglia and astrocytes, and mobilization of monocytes (Garcia-Bonilla et al. 2018). Evidence showed that cerebral mast cells releasing histamine also accumulate in the ischemic core and penumbra after injury (Biran et al. 2008). The levels of histamine receptors are also altered, as shown in an ischemia-reperfusion injury model in vivo. In particular, H1R mRNA expression was increased in the caudate-putamen, a decrease in H2R binding densities in the caudate-putamen was observed, H3R mRNA expression was raised in the caudate-putamen of the postischemic brain but was decreased in the globus pallidus and the thalamus; in association with this, H3R binding densities were increased in the cortex, caudate-putamen, globus pallidus, and hippocampus. These data suggest that histamine receptor expression and ligand binding are altered in brain ischemia in distinct areas and may participate in neuroprotection and/or ischemia-associated neuronal damage (Lozada et al. 2005).
The endogenous histamine was found to be essential for hypoxic preconditioning stroke tolerance in mice (Fan et al. 2011). By using HDC KO mice, it was shown that histamine is a critical mediator in hypoxic preconditioning, likely due to enhancing hypoxia-induced VEGF expression (Fan et al. 2011). Moreover, enhancement of histaminergic activity suppresses inflammatory cell recruitment after ischemic events through H2R, which may be a mechanism underlying the protective effect of L-histidine (Hiraga et al. 2007; Motoki et al. 2005; Adachi et al. 2005).
Several contradictory data were reported about the role of histamine receptors in stroke. Regarding the involvement of H3R, it was shown that thioperamide, an H3R antagonist, promotes neurogenesis (SVZ and SGZ) and protects against neuronal death and cognitive impairments in brain ischemic stroke models in vitro and in vivo (Wang et al. 2020). These effects may be due to increased phosphorylation of cAMP-response element-binding (CREB) and upregulation of the expression and release of BDNF (Wang et al. 2020). Another study showed that H3R blockade protects against ischemic/reperfusion injury by histamine-independent mechanisms that involve autophagy mechanisms (Yan et al. 2014). Nevertheless, both studies agree that H3R inhibition is a therapeutic target for cerebral ischemia.
On the other hand, clemastine, an H1R antagonist, reduced cerebral hematoma volume, decreased cerebral edema, lowered rates of neuronal apoptosis, improved behavioral scores in an acute intracerebral hemorrhage murine model. These effects were accompanied by reduced microglia activation and reduced pro-inflammatory effectors, and increased anti-inflammatory effectors post-lesion (Zhi et al. 2021). Similar results were observed in a hypoxic-ischemic brain injury mimicked by a bilateral common carotid artery occlusion (BCCAO) rat model where clemastine can improve hypomyelination by suppressing the activated microglia and promoting the maturation of oligodendrocyte progenitor cells by restraining the upregulation of IL-1β and NLRP3 in the corpus callosum (Xie et al. 2020). These data suggest that the activation of H1R may trigger detrimental effects in ischemic stroke.
The involvement of H4R has also been suggested to play a crucial role in the modulation of ischemia mechanisms. Chronic intraperitoneal administration with the H4R antagonist, JNJ7777120, protected from the neurological deficit in a rat model of focal ischemia induced by transient MCAo. At short-term (2 days post-lesion), JNJ7777120 reduced granulocyte infiltration in the ischemic area, while at long-term (7 days post-lesion), it was able to reduce the ischemic cortical and striatal lesion, the number of activated microglia and astrocytes in the ischemic cortex, and striatum and decreased the plasma levels of IL-1β and TNF-α, while increased the levels of IL-10. This may suggest that H4R is also a valuable pharmacological target after focal brain ischemia (Dettori et al. 2018).
Administration of H2R antagonists has been reported to produce contradictory results. The administration of H2R antagonists has also shown to be protective in ischemic-induced neuronal lesions in vitro (Malagelada et al. 2004), while in vivo, the administration of ranitidine, an H2R antagonist, antagonized the protective effects mediated by the i.c.v. administration of histamine in transient occlusion of the right middle cerebral artery in rats (Hamami et al. 2004). The last outcome may be due to a suppression of the ischemic release of excitatory neurotransmitters (dopamine, glutamate) (Hamami et al. 2004; Adachi et al. 2004).
All these reports support the role of the histaminergic system in the modulation of ischemic injury. Better comprehension and in-depth analysis of the experimental paradigms and lesion models applied is essential to provide more substantial proofs to proceed into clinical trials. Table 3 summarizes the main effects induced by histamine and its receptors in ischemic stroke.
4 Conclusions/Perspectives
Histamine plays a key role in the modulation of neuronal activities and behavioral functions. It has been shown that it also modulates innate immune cells, both in the brain (microglia) and in the periphery. In turn, the activation of peripheral immune cells can profoundly affect microglia activity, which plays a critical role in the onset and development of brain disorders. Therefore, unraveling the multiple actions of histamine might lead to the development of anti-inflammatory and regenerative therapies for both acute brain pathologies and neurodegenerative diseases. Besides the accumulating evidence supporting the role of the histaminergic system in these mechanisms, there is controversy about the most promising experimental strategy, histamine receptor, and dose to use in the context of brain diseases. The lack of consensus about the effects of histamine modulators can be due to the diversity of models and experimental paradigms used. Studies using animal models of disease should use complementary models, mimicking different aspects of the pathology. Choosing experimental models more representative of the human condition is also an upset. More comprehensive analysis, using in vivo and experimental models using human-derived cells (e.g., induced pluripotent stem cells—iPSC), is needed to advance in the knowledge and translational potential of histamine and its receptors. For the experiments using human monocytes, it is also crucial to pay attention to the cohort selection, methods for selecting and cultivating immune cells, which may introduce many variables that impact the effects induced by histamine and histamine receptor modulators.
Brain diseases are accompanied by alterations in histamine levels and the expression of its receptors. Disclosing whether these alterations are a consequence or a cause of neurodegeneration is crucial. Chronic exposure paradigms or histamine depletion (on a temporal and cell-specific basis) before neuronal lesions could help to address this issue. Moreover, additional studies are necessary to correlate the effects of histamine in innate immune cells and the functional consequence for neurons. The use of conditional KO mice targeting the histaminergic system (e.g., HDC, HR) in specific cell populations is relevant to address this question.
The specificity of HR agonists/antagonists is another aspect that hampers robust conclusions. Many HR agonists/antagonists were reported in the literature, but only a few have been studied in humans. Thus, the development of more specific pharmacological modulators is needed. Recent studies showed that H3R forms heteromers with other receptors (D1R, A2A). Thus, it is of utmost relevance to develop novel dual receptor modulators that interact with different neurotransmitter systems, thus exerting more robust effects. The benefits of interacting with dual systems should be further explored in the context of brain diseases. Besides this need to improve a better outcome, there is much evidence supporting more research in this field, which may significantly impact novel therapeutic strategies for brain diseases.
References
Acevedo SF, Pfankuch T, Ohtsu H, Raber J (2006) Anxiety and cognition in female histidine decarboxylase knockout (Hdc−/−) mice. Behav Brain Res 168:92–99
Adachi N, Liu K, Arai T (2004) Alleviation of ischemic neuronal damage by postischemic loading with histidine in the rat striatum. Brain Res 998:136–138
Adachi N, Liu K, Arai T (2005) Prevention of brain infarction by postischemic administration of histidine in rats. Brain Res 1039:220–223
Agasse F, Bernardino L, Silva B, Ferreira R, Grade S, Malva JO (2008) Response to histamine allows the functional identification of neuronal progenitors, neurons, astrocytes, and immature cells in subventricular zone cell cultures. Rejuvenation Res 11:187–200
Ahmed MR, Jayakumar M, Ahmed MS, Zamaleeva AI, Tao J, Li EH, Job JK, Pittenger C, Ohtsu H, Rajadas J (2019) Pharmacological antagonism of histamine H2R ameliorated L-DOPA–induced dyskinesia via normalization of GRK3 and by suppressing FosB and ERK in PD. Neurobiol Aging 81:177–189
Ambrée O, Buschert J, Zhang W, Arolt V, Dere E, Zlomuzica A (2014) Impaired spatial learning and reduced adult hippocampal neurogenesis in histamine H1-receptor knockout mice. Eur Neuropsychopharmacol 24:1394–1404
Anichtchik OV, Rinne JO, Kalimo H, Panula P (2000) An altered histaminergic innervation of the substantia Nigra in Parkinson’s disease. Exp Neurol 163:20–30
Avila-Luna A, Ríos C, Gálvez-Rosas A, Montes S, Arias-Montaño J-A, Bueno-Nava A (2019) Chronic administration of the histamine H3 receptor agonist immepip decreases l-Dopa-induced dyskinesias in 6-hydroxydopamine-lesioned rats. Psychopharmacology (Berl) 236:1937–1948
Azuma Y, Shinohara M, Wang P-L, Hidaka A, Ohura K (2001) Histamine inhibits chemotaxis, phagocytosis, superoxide anion production, and the production of TNFα and IL-12 by macrophages via H2-receptors. Int Immunopharmacol 1:1867–1875
Barata-Antunes S, Cristóvão AC, Pires J, Rocha SM, Bernardino L (2017) Dual role of histamine on microglia-induced neurodegeneration. Biochim Biophys Acta Mol Basis Dis 1863:764–769
Bernardino L, Eiriz MF, Santos T, Xapelli S, Grade S, Rosa AI, Cortes L, Ferreira R, Bragança J, Agasse F, Ferreira L, Malva JO (2012) Histamine stimulates neurogenesis in the rodent subventricular zone. Stem Cells 30:773–784
Biran V, Cochois V, Karroubi A, Arrang JM, Charriaut-Marlangue C, Héron A (2008) Stroke induces histamine accumulation and mast cell degranulation in the neonatal rat brain. Brain Pathol 18:1–9
Bourgogne E, Mathy F-X, Boucaut D, Boekens H, Laprevote O (2012) Simultaneous quantitation of histamine and its major metabolite 1-methylhistamine in brain dialysates by using precolumn derivatization prior to HILIC-MS/MS analysis. Anal Bioanal Chem 402:449–459
Brown RE, Stevens DR, Haas HL (2001) The physiology of brain histamine. Prog Neurobiol 63:637–672
Cacabelos R, Torrellas C, Fernández-Novoa L, Aliev G (2016) Neuroimmune crosstalk in CNS disorders: the histamine connection. Curr Pharm Des 22:819–848
Capelo R, Lehmann C, Ahmad K, Snodgrass R, Diehl O, Ringleb J, Flamand N, Weigert A, Stark H, Steinhilber D, Kahnt AS (2016) Cellular analysis of the histamine H4 receptor in human myeloid cells. Biochem Pharmacol 103:74–84
Castro-Sánchez S, García-Yagüe ÁJ, López-Royo T, Casarejos M, Lanciego JL, Lastres-Becker I (2018) Cx3cr1-deficiency exacerbates alpha-synuclein-A53T induced neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease. Glia 66:1752–1762
Chen Y-N, Sha H-H, Wang Y-W, Zhou Q, Bhuiyan P, Li N-N, Qian Y-N, Dong H-Q (2020) Histamine 2/3 receptor agonists alleviate perioperative neurocognitive disorders by inhibiting microglia activation through the PI3K/AKT/FoxO1 pathway in aged rats. J Neuroinflammation 17:217
Chikahisa S, Kodama T, Soya A, Sagawa Y, Ishimaru Y, Séi H, Nishino S (2013) Histamine from brain resident MAST cells promotes wakefulness and modulates behavioral states. PLoS One 8:e78434
Chu HX, Kim HA, Lee S, Moore JP, Chan CT, Vinh A, Gelderblom M, Arumugam TV, Broughton BR, Drummond GR, Sobey CG (2014) Immune cell infiltration in malignant middle cerebral artery infarction: comparison with transient cerebral ischemia. J Cereb Blood Flow Metab 34:450–459
Clausen BH, Lambertsen KL, Babcock AA, Holm TH, Dagnaes-Hansen F, Finsen B (2008) Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation 5:46
Croyal M, Dauvilliers Y, Labeeuw O, Capet M, Schwartz J-C, Robert P (2011) Histamine and tele-methylhistamine quantification in cerebrospinal fluid from narcoleptic subjects by liquid chromatography tandem mass spectrometry with precolumn derivatization. Anal Biochem 409:28–36
Czerner CP, Klos A, Seifert R, Neumann D (2014) Histamine induces chemotaxis and phagocytosis in murine bone marrow-derived macrophages and RAW 264.7 macrophage-like cells via histamine H4-receptor. Inflamm Res 63:239–247
Damier P (1999) The substantia nigra of the human brain: II Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122:1437–1448
de Bie RMA, Clarke CE, Espay AJ, Fox SH, Lang AE (2020) Initiation of pharmacological therapy in Parkinson’s disease: when, why, and how. Lancet Neurol 19:452–461
Dere E (2003) Histidine-decarboxylase knockout mice show deficient nonreinforced episodic object memory, improved negatively reinforced water-maze performance, and increased neo- and ventro-striatal dopamine turnover. Learn Mem 10:510–519
Dettori I, Gaviano L, Melani A, Lucarini L, Durante M, Masini E, Pedata F (2018) A selective histamine H4 receptor antagonist, JNJ7777120, is protective in a rat model of transient cerebral ischemia. Front Pharmacol 9:1231
Dong H, Zhang W, Zeng X, Hu G, Zhang H, He S, Zhang S (2014) Histamine induces upregulated expression of histamine receptors and increases release of inflammatory mediators from microglia. Mol Neurobiol 49:1487–1500
Easton A, Norton J, Goodwillie A, Pfaff DW (2004) Sex differences in mouse behavior following pyrilamine treatment: role of histamine 1 receptors in arousal. Pharmacol Biochem Behav 79:563–572
Eiriz MF, Valero J, Malva JO, Bernardino L (2014) New insights into the role of histamine in subventricular zone-olfactory bulb neurogenesis. Front Neurosci 8:142
Escobedo-Avila I, Vargas-Romero F, Molina-Hernández A, López-González R, Cortés D, De Carlos JA, Velasco I (2014) Histamine impairs midbrain dopaminergic development in vivo by activating histamine type 1 receptors. Mol Brain 7:58
Fan Y-Y, Hu W-W, Dai H-B, Zhang J-X, Zhang L-Y, He P, Shen Y, Ohtsu H, Wei E-Q, Chen Z (2011) Activation of the central histaminergic system is involved in hypoxia-induced stroke tolerance in adult mice. J Cereb Blood Flow Metab 31:305–314
Fang W, Zhai X, Han D, Xiong X, Wang T, Zeng X, He S, Liu R, Miyata M, Xu B, Zhao H (2018) CCR2-dependent monocytes/macrophages exacerbate acute brain injury but promote functional recovery after ischemic stroke in mice. Theranostics 8:3530–3543
Fang Q, Xicoy H, Shen J, Luchetti S, Dai D, Zhou P, Qi X-R, Martens GJM, Huitinga I, Swaab DF, Liu C, Shan L (2021) Histamine-4 receptor antagonist ameliorates Parkinson-like pathology in the striatum. Brain Behav Immun 92:127–138
Faustino JV, Wang X, Johnson CE, Klibanov A, Derugin N, Wendland MF, Vexler ZS (2011) Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci 31:12992–13001
Ferreira R, Santos T, Gonçalves J, Baltazar G, Ferreira L, Agasse F, Bernardino L (2012) Histamine modulates microglia function. J Neuroinflammation 9:90
Frei R, Ferstl R, Konieczna P, Ziegler M, Simon T, Rugeles TM, Mailand S, Watanabe T, Lauener R, Akdis CA, O’Mahony L (2013) Histamine receptor 2 modifies dendritic cell responses to microbial ligands. J Allergy Clin Immunol 132:194–204
Funk N, Wieghofer P, Grimm S, Schaefer R, Bühring H-J, Gasser T, Biskup S (2013) Characterization of peripheral hematopoietic stem cells and monocytes in Parkinson’s disease. Mov Disord 28:392–395
Gabelle A, Jaussent I, Hirtz C, Vialaret J, Navucet S, Grasselli C, Robert P, Lehmann S, Dauvilliers Y (2017) Cerebrospinal fluid levels of orexin-a and histamine, and sleep profile within the Alzheimer process. Neurobiol Aging 53:59–66
Garcia-Bonilla L, Brea D, Benakis C, Lane DA, Murphy M, Moore J, Racchumi G, Jiang X, Iadecola C, Anrather J (2018) Endogenous protection from ischemic brain injury by preconditioned monocytes. J Neurosci 38:6722–6736
Gardai SJ, Mao W, Schüle B, Babcock M, Schoebel S, Lorenzana C, Alexander J, Kim S, Glick H, Hilton K, Fitzgerald JK, Buttini M, Chiou S-S, McConlogue L, Anderson JP, Schenk DB, Bard F, Langston JW, Yednock T, Johnston JA (2013) Elevated alpha-Synuclein impairs innate immune cell function and provides a potential peripheral biomarker for Parkinson’s disease. PLoS One 8:e71634
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21:404–412
Ghi P (1999) Sex differences in memory performance in the object recognition test. Possible role of histamine receptors. Pharmacol Biochem Behav 64:761–766
Gomez-Ramirez J, Johnston TH, Visanji NP, Fox SH, Brotchie JM (2006) Histamine H3 receptor agonists reduce L-dopa-induced chorea, but not dystonia, in the MPTP-lesioned nonhuman primate model of Parkinson’s disease. Mov Disord 21:839–846
Grade S, Agasse F, Bernardino L, Silva CG, Cortes L, Malva JO (2010) Functional identification of neural stem cell-derived oligodendrocytes by means of calcium transients elicited by thrombin. Rejuvenation Res 13:27
Grozdanov V, Bliederhaeuser C, Ruf WP, Roth V, Fundel-Clemens K, Zondler L, Brenner D, Martin-Villalba A, Hengerer B, Kassubek J, Ludolph AC, Weishaupt JH, Danzer KM (2014) Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropathol 128:651–663
Grozdanov V, Bousset L, Hoffmeister M, Bliederhaeuser C, Meier C, Madiona K, Pieri L, Kiechle M, McLean PJ, Kassubek J, Behrends C, Ludolph AC, Weishaupt JH, Melki R, Danzer KM (2019) Increased immune activation by pathologic α-Synuclein in Parkinson’s disease. Ann Neurol 86:593–606
Guilloux J-P, Samuels BA, Mendez-David I, Hu A, Levinstein M, Faye C, Mekiri M, Mocaer E, Gardier AM, Hen R, Sors A, David DJ (2017) S 38093, a histamine H3 antagonist/inverse agonist, promotes hippocampal neurogenesis and improves context discrimination task in aged mice. Sci Rep 7:42946
Hamami G, Adachi N, Liu K, Arai T (2004) Alleviation of ischemic neuronal damage by histamine H2 receptor stimulation in the rat striatum. Eur J Pharmacol 484:167–173
Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, Cron RQ, Shacka JJ, Raman C, Standaert DG (2013) MHCII is required for synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci 33:9592–9600
Harms AS, Thome AD, Yan Z, Schonhoff AM, Williams GP, Li X, Liu Y, Qin H, Benveniste EN, Standaert DG (2018) Peripheral monocyte entry is required for alpha-Synuclein induced inflammation and neurodegeneration in a model of Parkinson disease. Exp Neurol 300:179–187
Harms AS, Ferreira SA, Romero-Ramos M (2021) Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol 141:527–545
Hasegawa Y, Inagaki T, Sawada M, Impaired SA (2000) Impaired cytokine production by peripheral blood mononuclear cells and monocytes macrophages in Parkinson s disease. Acta Neurol Scand 101:159–164
Hiraga N, Adachi N, Liu K, Nagaro T, Arai T (2007) Suppression of inflammatory cell recruitment by histamine receptor stimulation in ischemic rat brains. Eur J Pharmacol 557:236–244
Iida T, Yoshikawa T, Matsuzawa T, Naganuma F, Nakamura T, Miura Y, Mohsen AS, Harada R, Iwata R, Yanai K (2015) Histamine H 3 receptor in primary mouse microglia inhibits chemotaxis, phagocytosis, and cytokine secretion. Glia 63:1213–1225
Iida T, Yoshikawa T, Kárpáti A, Matsuzawa T, Kitano H, Mogi A, Harada R, Naganuma F, Nakamura T, Yanai K (2017) JNJ10181457, a histamine H3 receptor inverse agonist, regulates in vivo microglial functions and improves depression-like behaviours in mice. Biochem Biophys Res Commun 488:534–540
Ito T, Kimura Y, Seki C, Ichise M, Yokokawa K, Kawamura K, Takahashi H, Higuchi M, Zhang M-R, Suhara T, Yamada M (2018) Histamine H3 receptor density is negatively correlated with neural activity related to working memory in humans. EJNMMI Res 8:48
Jin W-N, Shi SX-Y, Li Z, Li M, Wood K, Gonzales RJ, Liu Q (2017) Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab 37:2224–2236
Johnston TH, van der Meij A, Brotchie JM, Fox SH (2010) Effect of histamine H 2 receptor antagonism on levodopa-induced dyskinesia in the MPTP-macaque model of Parkinson’s disease. Mov Disord 25:1379–1390
Kallweit U, Aritake K, Bassetti CL, Blumenthal S, Hayaishi O, Linnebank M, Baumann CR, Urade Y (2013) Elevated CSF histamine levels in multiple sclerosis patients. Fluids Barriers CNS 10:19
Kasaoka S, Kawahara Y, Inoue S, Tsuji M, Kato H, Tsuchiya T, Okuda H, Nakajima S (2005) Gender effects in dietary histidine-induced anorexia. Nutrition 21:855–858
Katoh Y, Niimi M, Yamamoto Y, Kawamura T, Morimoto-Ishizuka T, Sawada M, Takemori H, Yamatodani A (2001) Histamine production by cultured microglial cells of the mouse. Neurosci Lett 305:181–184
Kim W-G, Mohney RP, Wilson B, Jeohn G-H, Liu B, Hong J-S (2000) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci 20:6309–6316
Kimura S, Wang K-Y, Tanimoto A, Murata Y, Nakashima Y, Sasaguri Y (2004) Acute inflammatory reactions caused by histamine via monocytes/macrophages chronically participate in the initiation and progression of atherosclerosis. Pathol Int 54:465–474
Klein B, Mrowetz H, Thalhamer J, Scheiblhofer S, Weiss R, Aigner L (2016) Allergy enhances neurogenesis and modulates microglial activation in the Hippocampus. Front Cell Neurosci 10:169
Koski SK, Leino S, Panula P, Rannanpää S, Salminen O (2020) Genetic lack of histamine upregulates dopamine neurotransmission and alters rotational behavior but not levodopa-induced dyskinesia in a mouse model of Parkinson’s disease. Neurosci Lett 729:134932
Krashia P, Cordella A, Nobili A, La Barbera L, Federici M, Leuti A, Campanelli F, Natale G, Marino G, Calabrese V, Vedele F, Ghiglieri V, Picconi B, Di Lazzaro G, Schirinzi T, Sancesario G, Casadei N, Riess O, Bernardini S, Pisani A, Calabresi P, Viscomi MT, Serhan CN, Chiurchiù V, D’Amelio M, Mercuri NB (2019) Blunting neuroinflammation with resolvin D1 prevents early pathology in a rat model of Parkinson’s disease. Nat Commun 10:3945
Lambertsen KL, Biber K, Finsen B (2012) Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab 32:1677–1698
Lebel B, Scheinmann P, Canu P, Burtin C (1980) Histamine levels in mouse tissues of different strains: influence of sex. Agents Actions 10:149–150
Li T, Zhao J, Xie W, Yuan W, Guo J, Pang S, Gan W-B, Gómez-Nicola D, Zhang S (2021) Specific depletion of resident microglia in the early stage of stroke reduces cerebral ischemic damage. J Neuroinflammation 18:81
Lim SAO, Xia R, Ding Y, Won L, Ray WJ, Hitchcock SA, McGehee DS, Kang UJ (2015) Enhanced histamine H2 excitation of striatal cholinergic interneurons in l-DOPA-induced dyskinesia. Neurobiol Dis 76:67–76
Liu C-Q, Chen Z, Liu F-X, Hu D-N, Luo J-H (2007) Involvement of brain endogenous histamine in the degeneration of dopaminergic neurons in 6-hydroxydopamine-lesioned rats. Neuropharmacology 53:832–841
Liu C-Q, Hu D-N, Liu F-X, Chen Z, Luo J-H (2008) Apomorphine-induced turning behavior in 6-hydroxydopamine lesioned rats is increased by histidine and decreased by histidine decarboxylase, histamine H1 and H2 receptor antagonists, and an H3 receptor agonist. Pharmacol Biochem Behav 90:325–330
Lozada A, Munyao N, Sallmen T, Lintunen M, Leurs R, Lindsberg PJ, Panula P (2005) Postischemic regulation of central histamine receptors. Neuroscience 136:371–379
Malagelada C, Xifró X, Badiola N, Sabrià J, Rodríguez-Álvarez J (2004) Histamine H 2 -receptor antagonist ranitidine protects against neural death induced by oxygen-glucose deprivation. Stroke 35:2396–2401
Márquez-Gómez R, Robins MT, Gutiérrez-Rodelo C, Arias J-M, Olivares-Reyes J-A, van Rijn RM, Arias-Montaño J-A (2018) Functional histamine H 3 and adenosine A 2A receptor heteromers in recombinant cells and rat striatum. Pharmacol Res 129:515–525
Márquez-Valadez B, Aquino-Miranda G, Quintero-Romero M-O, Papacostas-Quintanilla H, Bueno-Nava A, López-Rubalcava C, Díaz NF, Arias-Montaño J-A, Molina-Hernández A (2019) The systemic administration of the histamine H1 receptor antagonist/inverse agonist Chlorpheniramine to pregnant rats impairs the development of Nigro-striatal dopaminergic neurons. Front Neurosci 13:360
Mattila OS, Strbian D, Saksi J, Pikkarainen TO, Rantanen V, Tatlisumak T, Lindsberg PJ (2011) Cerebral mast cells mediate blood-brain barrier disruption in acute experimental ischemic stroke through perivascular gelatinase activation. Stroke 42:3600–3605
Mazurkiewicz-Kwilecki IM, Prell GD (1984) Age-related changes in brain histamine. Agents Actions 14:554–557
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1285
Mestre TA, Shah BB, Connolly BS, de Aquino C, Al Dhakeel A, Walsh R, Ghate T, Lui JP, Fox SH (2014) Famotidine, a histamine H 2 receptor antagonist, does not reduce levodopa-induced dyskinesia in Parkinson’s disease: a proof-of-concept study. Mov Disord Clin Pract 1:219–224
Molina-Hernández A, Rodríguez-Martínez G, Escobedo-Ávila I, Velasco I (2013) Histamine up-regulates fibroblast growth factor receptor 1 and increases FOXP2 neurons in cultured neural precursors by histamine type 1 receptor activation: conceivable role of histamine in neurogenesis during cortical development in vivo. Neural Dev 8:4
Molina-Hernández A, Velasco I (2008) Histamine induces neural stem cell proliferation and neuronal differentiation by activation of distinct histamine receptors. J Neurochem 106:706–717
Mommert S, Ratz L, Stark H, Gutzmer R, Werfel T (2018) The histamine H4 receptor modulates the differentiation process of human monocyte-derived M1 macrophages and the release of CCL4/MIP-1β from fully differentiated M1 macrophages. Inflamm Res 67:503–513
Moreno E, Hoffmann H, Gonzalez-Sepúlveda M, Navarro G, Casadó V, Cortés A, Mallol J, Vignes M, McCormick PJ, Canela EI, Lluís C, Moratalla R, Ferré S, Ortiz J, Franco R (2011) Dopamine D1-histamine H3 receptor heteromers provide a selective link to MAPK signaling in GABAergic neurons of the direct striatal pathway. J Biol Chem 286:5846–5854
Moreno-Delgado D, Puigdellívol M, Moreno E, Rodríguez-Ruiz M, Botta J, Gasperini P, Chiarlone A, Howell LA, Scarselli M, Casadó V, Cortés A, Ferré S, Guzmán M, Lluís C, Alberch J, Canela EI, Ginés S, McCormick PJ (2020) Modulation of dopamine D1 receptors via histamine H3 receptors is a novel therapeutic target for Huntington’s disease. eLife
Moskowitz MA, Lo EH, Iadecola C (2010) The science of stroke: mechanisms in search of treatments. Neuron 67:181–198
Motawaj M, Peoc’h K, Callebert J, Arrang J-M (2010) CSF levels of the histamine metabolite tele-methylhistamine are only slightly decreased in Alzheimer’s disease. J Alzheimers Dis 22:861–871
Motoki A, Adachi N, Semba K, Liu K, Arai T (2005) Reduction in brain infarction by augmentation of central histaminergic activity in rats. Brain Res 1066:172–178
Muñoz-Cruz S, Mendoza-Rodríguez Y, Nava-Castro KE, Yepez-Mulia L, Morales-Montor J (2015) Gender-related effects of sex steroids on histamine release and Fc ε RI expression in rat peritoneal mast cells. J Immunol Res 2015:1–10
Nagatsu T, Mogi M, Ichinose H, Togari A (2000) Changes in cytokines and neurotrophins in Parkinson’s disease. In: Advances in research on neurodegeneration. Springer, Vienna, pp 277–290
Nieto-Alamilla G, Márquez-Gómez R, García-Gálvez A-M, Morales-Figueroa G-E, Arias-Montaño J-A (2016) The histamine H 3 receptor: structure, pharmacology, and function. Mol Pharmacol 90:649–673
Nowak P, Noras Ł, Jochem J, Szkilnik R, Brus H, Körőssy E, Drab J, Kostrzewa RM, Brus R (2009) Histaminergic activity in a rodent model of Parkinson’s disease. Neurotox Res 15:246–251
Østerud B, Olsen JO (2014) Pro- and anti-inflammatory effects of histamine on tissue factor and TNFα expression in monocytes of human blood. Thromb Res 133:477–480
Palada V, Terzić J, Mazzulli J, Bwala G, Hagenah J, Peterlin B, Hung AY, Klein C, Krainc D (2012) Histamine N-methyltransferase Thr105Ile polymorphism is associated with Parkinson’s disease. Neurobiol Aging 33:836
Panula P, Nuutinen S (2013) The histaminergic network in the brain: basic organization and role in disease. Nat Rev Neurosci 14:472–487
Parillaud VR, Lornet G, Monnet Y, Privat A-L, Haddad AT, Brochard V, Bekaert A, de Chanville CB, Hirsch EC, Combadière C, Hunot S, Lobsiger CS (2017) Analysis of monocyte infiltration in MPTP mice reveals that microglial CX3CR1 protects against neurotoxic over-induction of monocyte-attracting CCL2 by astrocytes. J Neuroinflammation 14:60
Park HJ, Kim HJ, Park H-K, Chung J-H (2009) Protective effect of histamine H2 receptor antagonist ranitidine against rotenone-induced apoptosis. Neurotoxicology 30:1114–1119
Pedragosa J, Miró-Mur F, Otxoa-de-Amezaga A, Justicia C, Ruíz-Jaén F, Ponsaerts P, Pasparakis M, Planas AM (2020) CCR2 deficiency in monocytes impairs angiogenesis and functional recovery after ischemic stroke in mice. J Cereb Blood Flow Metab 40:S98–S116
Peng H, Wang J, Ye XY, Cheng J, Huang CZ, Li LY, Li TY, Li CW (2019) Histamine H4 receptor regulates IL-6 and INF-γ secretion in native monocytes from healthy subjects and patients with allergic rhinitis. Clin Transl Allergy 9:49
Perego C, Fumagalli S, Zanier ER, Carlino E, Panini N, Erba E, De Simoni M-G (2016) Macrophages are essential for maintaining a M2 protective response early after ischemic brain injury. Neurobiol Dis 96:284–293
Prell GD, Khandelwal JK, Burns RS, LeWitt PA, Green JP (1990) Influence of age and gender on the levels of histamine metabolites and pros-methylimidazoleacetic acid in human cerebrospinal fluid. Arch Gerontol Geriatr 11:85–95
Prinz M, Jung S, Priller J (2019) Microglia biology: one century of evolving concepts. Cell 179:292–311
Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong J-S, Knapp DJ, Crews FT (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55:453–462
Raj T, Rothamel K, Mostafavi S, Ye C, Lee MN, Replogle JM, Feng T, Lee M, Asinovski N, Frohlich I, Imboywa S, Von Korff A, Okada Y, Patsopoulos NA, Davis S, McCabe C, Paik H, Srivastava GP, Raychaudhuri S, Hafler DA, Koller D, Regev A, Hacohen N, Mathis D, Benoist C, Stranger BE, De Jager PL (2014) Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344:519–523
Riederer P, Laux G (2011) MAO-inhibitors in Parkinson’s disease. Exp Neurobiol 20:1–17
Rinne JO, Anichtchik OV, Eriksson KS, Kaslin J, Tuomisto L, Kalimo H, Röyttä M, Panula P (2002) Increased brain histamine levels in Parkinson’s disease but not in multiple system atrophy. J Neurochem 81:954–960
Rocha SM, Pires J, Esteves M, Graasa B, Bernardino L (2014) Histamine: a new immunomodulatory player in the neuron-glia crosstalk. Front Cell Neurosci
Rocha SM, Saraiva T, Cristóvão AC, Ferreira R, Santos T, Esteves M, Saraiva C, Je G, Cortes L, Valero J, Alves G, Klibanov A, Kim Y-S, Bernardino L (2016) Histamine induces microglia activation and dopaminergic neuronal toxicity via H1 receptor activation. J Neuroinflammation 13:137
Rodríguez-Martínez G, Velasco I, García-López G, Solís KH, Flores-Herrera H, Díaz NF, Molina-Hernández A (2012) Histamine is required during neural stem cell proliferation to increase neuron differentiation. Neuroscience 216:10–17
Sanchez-Guajardo V, Febbraro F, Kirik D, Romero-Ramos M (2010) Microglia acquire distinct activation profiles depending on the degree of α-Synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS One 5:e8784
Saraiva C, Barata-Antunes S, Santos T, Ferreiro E, Cristóvão AC, Serra-Almeida C, Ferreira R, Bernardino L (2019) Histamine modulates hippocampal inflammation and neurogenesis in adult mice. Sci Rep 9:8384
Sarlus H, Höglund CO, Karshikoff B, Wang X, Lekander M, Schultzberg M, Oprica M (2012) Allergy influences the inflammatory status of the brain and enhances tau-phosphorylation. J Cell Mol Med 16:2401–2412
Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R (2003) Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 183:25–33
Shan L, Hofman MA, van Wamelen DJ, Van Someren EJW, Bao A-M, Swaab DF (2012a) Diurnal fluctuation in histidine decarboxylase expression, the rate limiting enzyme for histamine production, and its disorder in neurodegenerative diseases. Sleep 35:713–715
Shan L, Liu C-Q, Balesar R, Hofman MA, Bao A-M, Swaab DF (2012b) Neuronal histamine production remains unaltered in Parkinson’s disease despite the accumulation of Lewy bodies and Lewy neurites in the tuberomamillary nucleus. Neurobiol Aging 33:1343–1344
Shan L, Bossers K, Luchetti S, Balesar R, Lethbridge N, Chazot PL, Bao A-M, Swaab DF (2012c) Alterations in the histaminergic system in the substantia nigra and striatum of Parkinson’s patients: a postmortem study. Neurobiol Aging 33:1488
Shan Y, Gao Y, Zhang L, Ma L, Shi Y, Liu X (2019) H4 receptor inhibits lipopolysaccharide-induced NF-κB activation by interacting with tumor necrosis factor receptor-associated factor 6. Neuroscience 398:113–125
Silverman A-J, Sutherland AK, Wilhelm M, Silver R (2000) Mast cells migrate from blood to brain. J Neurosci 20:401–408
Soga F, Katoh N, Kishimoto S (2007) Histamine prevents apoptosis in human monocytes. Clin Exp Allergy 37:323–330
Soya A, Song YH, Kodama T, Honda Y, Fujiki N, Nishino S (2008) CSF histamine levels in rats reflect the central histamine neurotransmission. Neurosci Lett 430:224–229
Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840
Szalay G, Martinecz B, Lénárt N, Környei Z, Orsolits B, Judák L, Császár E, Fekete R, West BL, Katona G, Rózsa B, Dénes Á (2016) Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 7:11499
Takahashi HK, Morichika T, Iwagaki H, Tamura R, Kubo S, Yoshino T, Mori S, Akagi T, Tanaka N, Nishibori M (2003) Histamine downregulates CD14 expression via H2 receptorson human monocytes. Clin Immunol 108:274–281
Takahashi HK, Iwagaki H, Mori S, Yoshino T, Tanaka N, Nishibori M (2004) Histamine inhibits lipopolysaccharide-induced interleukin (IL)-18 production in human monocytes. Clin Immunol 112:30–34
Takahashi H, Sadamori H, Teshigawara K, Niwa A, Liu K, Wake H, Mori S, Yoshino T, Nishibori M (2013) Histamine inhibits high mobility group box 1-induced adhesion molecule expression on human monocytes. Eur J Pharmacol 718:305–313
Terao A, Steininger TL, Morairty SR, Kilduff TS (2004) Age-related changes in histamine receptor mRNA levels in the mouse brain. Neurosci Lett 355:81–84
Triggiani M, Petraroli A, Loffredo S, Frattini A, Granata F, Morabito P, Staiano RI, Secondo A, Annunziato L, Marone G (2007) Differentiation of monocytes into macrophages induces the upregulation of histamine H1 receptor. J Allergy Clin Immunol 119:472–481
Tsuji S, Di Martino E, Mukai T, Tsuji S, Murakami T, Harris RA, Blomgren K, Åden U (2020) Aggravated brain injury after neonatal hypoxic ischemia in microglia-depleted mice. J Neuroinflammation 17:111
Vizuete ML, Merino M, Venero JL, Santiago M, Cano J, Machado A (2000) Histamine infusion induces a selective dopaminergic neuronal death along with an inflammatory reaction in rat substantia nigra. J Neurochem 75:540–552
Wang K-Y, Arima N, Higuchi S, Shimajiri S, Tanimoto A, Murata Y, Hamada T, Sasaguri Y (2000) Switch of histamine receptor expression from H2 to H1 during differentiation of monocytes into macrophages. FEBS Lett 473:345–348
Wang N, Ma J, Liu J, Wang J, Liu C, Wang H, Liu Y, Yan H, Jiang S (2020) Histamine H3 receptor antagonist enhances neurogenesis and improves chronic cerebral hypoperfusion-induced cognitive impairments. Front Pharmacol 10:1583
Watson MB, Richter F, Lee SK, Gabby L, Wu J, Masliah E, Effros RB, Chesselet M-F (2012) Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp Neurol 237:318–334
Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, Miskinyte G, Ge R, Ahlenius H, Lindvall O, Schwartz M, Kokaia Z (2016) Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci 36:4182–4195
Werner K, Neumann D, Buschauer A, Seifert R (2014a) No evidence for histamine H 4 receptor in human monocytes. J Pharmacol Exp Ther 351:519–526
Werner K, Neumann D, Seifert R (2014b) Analysis of the histamine H2-receptor in human monocytes. Biochem Pharmacol 92:369–379
Wijeyekoon RS, Kronenberg-Versteeg D, Scott KM, Hayat S, Jones JL, Clatworthy MR, Floto RA, Barker RA, Williams-Gray CH (2018) Monocyte function in Parkinson’s disease and the impact of autologous serum on phagocytosis. Front Neurol 9:870
Wijeyekoon RS, Kronenberg-Versteeg D, Scott KM, Hayat S, Kuan W-L, Evans JR, Breen DP, Cummins G, Jones JL, Clatworthy MR, Floto RA, Barker RA, Williams-Gray CH (2020) Peripheral innate immune and bacterial signals relate to clinical heterogeneity in Parkinson’s disease. Brain Behav Immun 87:473–488
Xie D, Ge X, Ma Y, Tang J, Wang Y, Zhu Y, Gao C, Pan S (2020) Clemastine improves hypomyelination in rats with hypoxic–ischemic brain injury by reducing microglia-derived IL-1β via P38 signaling pathway. J Neuroinflammation 17:57
Yamada Y, Yoshikawa T, Naganuma F, Kikkawa T, Osumi N, Yanai K (2020) Chronic brain histamine depletion in adult mice induced depression-like behaviours and impaired sleep-wake cycle. Neuropharmacology 175:108179
Yan H, Zhang X, Hu W, Ma J, Hou W, Zhang X, Wang X, Gao J, Shen Y, Lv J, Ohtsu H, Han F, Wang G, Chen Z (2014) Histamine H3 receptors aggravate cerebral ischaemic injury by histamine-independent mechanisms. Nat Commun 5:3334
Yang CG, Wang X, Yue X, Shi H, Shen X, Zhang Z (2013) Ranitidine reduced levodopa-induced dyskinesia in a rat model of Parkinson’s disease. Neuropsychiatr Dis Treat 10:39
Zhang J, Takahashi H, Liu K, Wake H, Liu R, Sadamori H, Matsuda H, Yagi T, Yoshino T, Mori S, Nishibori M (2010) Histamine inhibits adhesion molecule expression in human monocytes, induced by advanced glycation end products, during the mixed lymphocyte reaction. Br J Pharmacol 160:1378–1386
Zhang W, Zhang X, Zhang Y, Qu C, Zhou X, Zhang S (2020) Histamine induces microglia activation and the release of Proinflammatory mediators in rat brain via H1R or H4R. J Neuroimmune Pharmacol 15:280–291
Zhao Y-F, Qiong-Zhang Z, Lou J-F, Zu Z-Y, Wang H-B, Zeng Z-G, Kai-Yao W-C, Xiao B-G (2018) The synergy of aging and LPS exposure in a mouse model of Parkinson’s disease. Aging Dis 9:785
Zhi C, Zeng S, Chen Y, Liao D, Lai M, Wang Z, Wang Y, Xiao S (2021) Clemastine promotes recovery of neural function and suppresses neuronal apoptosis by restoring balance of pro-inflammatory mediators in an experimental model of intracerebral hemorrhage. Int J Med Sci 18:639–645
Zhou P, Homberg JR, Fang Q, Wang J, Li W, Meng X, Shen J, Luan Y, Liao P, Swaab DF, Shan L, Liu C (2019) Histamine-4 receptor antagonist JNJ7777120 inhibits pro-inflammatory microglia and prevents the progression of Parkinson-like pathology and behaviour in a rat model. Brain Behav Immun 76:61–73
Zhuang Q-X, Li G-Y, Li B, Zhang C-Z, Zhang X-Y, Xi K, Li H-Z, Wang J-J, Zhu J-N (2018) Regularizing firing patterns of rat subthalamic neurons ameliorates parkinsonian motor deficits. J Clin Invest 128:5413–5427
Zrzavy T, Machado-Santos J, Christine S, Baumgartner C, Weiner HL, Butovsky O, Lassmann H (2018) Dominant role of microglial and macrophage innate immune responses in human ischemic infarcts. Brain Pathol 28:791–805
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bernardino, L. (2021). Histamine in the Crosstalk Between Innate Immune Cells and Neurons: Relevance for Brain Homeostasis and Disease. In: Yanai, K., Passani, M.B. (eds) The Functional Roles of Histamine Receptors. Current Topics in Behavioral Neurosciences, vol 59. Springer, Cham. https://doi.org/10.1007/7854_2021_235
Download citation
DOI: https://doi.org/10.1007/7854_2021_235
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-16996-0
Online ISBN: 978-3-031-16997-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)