
Abstract
The juxtaglomerular renin-producing cells (RPC) of the kidney are referred to as the major source of circulating renin. Renin is the limiting factor in renin-angiotensin system (RAS), which represents a proteolytic cascade in blood plasma that plays a central role in the regulation of blood pressure. Further cells disseminated in the entire organism express renin at a low level as part of tissue RASs, which are thought to locally modulate the effects of systemic RAS. In recent years, it became increasingly clear that the renal RPC are involved in developmental, physiological, and pathophysiological processes outside RAS. Based on recent experimental evidence, a novel concept emerges postulating that next to their traditional role, the RPC have non-canonical RAS-independent progenitor and renoprotective functions. Moreover, the RPC are part of a widespread renin lineage population, which may act as a global stem cell pool coordinating homeostatic, stress, and regenerative responses throughout the organism. This review focuses on the RAS-unrelated functions of RPC – a dynamic research area that increasingly attracts attention.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Background: The Classical View on Renal Renin-Producing Cells (RPC) Within Renin-Angiotensin System (RAS)
Circulating renin-angiotensin system (RAS) is a major endocrine factor that governs arterial blood pressure acting both in a short-term scale on vascular tone and in a long-term scale on salt, water, and volume homeostasis. Within RAS, the proteases renin and angiotensin-converting enzyme (ACE) sequentially cleave the plasma protein angiotensinogen to angiotensin (Ang) I and then to Ang II. Ang II is the main effector hormone of the system. Acting primarily through Ang II type 1 (AT1) receptors, it induces strong vasoconstriction and maintains sodium and water balance by regulating the production of aldosterone and antidiuretic hormone, respectively. The multifaceted activity of Ang II results in complex cross talk between its different modes of action. Particularly in the kidney, the functional integrity between Ang II effects on afferent/efferent arteriole tone and tubular reabsorption is decisive for the maintenance of glomerular (as well as renal) hemodynamics and water/electrolyte excretion. In fact, RAS is more complicated because it also includes AT2 receptor, prorenin, and its receptor, as well as a protective arm with ACE2, Ang 1–7, and Mas receptor. These further members are responsible for the fine-tuning of RAS under physiological conditions. They acquire a substantial role in patients with cardiovascular diseases in particular when the AT1-mediated Ang II effects of RAS are pharmacologically inhibited. The detailed description of RAS is beyond the scope of this review. Recent reviews summarizing the state of the art on the intricate RAS aspects in health and disease are available elsewhere (Ames et al. 2019; Arendse et al. 2019; Fournier et al. 2012; Santos et al. 2018; Sparks et al. 2014).
The renin-producing cells (RPC) of the adult mammalian kidney are located in the wall of the afferent arterioles next to the glomerular vascular poles. Therefore, the RPC are also dubbed juxtaglomerular (JG) cells. The JG cells are very few – they amount to only 4–8 cells per afferent arteriole. Renin is the rate-limiting factor of circulating systemic RAS since it primarily determines the rate of generated Ang II and thus the overall biological activity of the system. The reason for the key regulatory status of renin in RAS is the tight control of renin synthesis and secretion by systemic and local factors including perfusion pressure, renal sympathetic nerve activity, macula densa mechanism, and autocrine cues (nitric oxide, prostanoids, and adenosine). The function of renal RPC in steering RAS activity under physiological and pathophysiological conditions has been characterized very well and elucidated in excellent reviews (Castrop et al. 2010; Damkjaer et al. 2013; Friis et al. 2013; Schweda and Kurtz 2011; Sparks et al. 2014).
2 RPC Beyond RAS
For about 10 years, we have being interested whether the renal RPC might be more than a mere source of renin. Initially, there were two lines of evidence justifying this hypothesis. First, the number of RPC in the afferent arterioles of the adult kidney is not constant but changes in response to homeostatic fluctuations in salt intake, intravascular volume, arterial tone, or adrenergic input. Therein, the RPC do not seem to proliferate. Instead, the renin gene is switched on upstream in the afferent arterioles in some vascular smooth muscle cells (VSMC) thus changing their phenotype from contractile to secretory (Cantin et al. 1977; Dominick et al. 1990). This process is reversible and known as recruitment or metaplastic transformation (Gomez et al. 1988, 1990). The latter term, however, is confusing because it implies pathology when in fact it represents a physiological process. The phenotype plasticity of the RPC in adult life is particularly interesting with regard to the localization of these cells within the JG apparatus (JGA), which includes macula densa and the vascular glomerular pole with the adherent parts of the afferent and efferent arterioles. JGA essentially coordinates renal perfusion, glomerular filtration rate (GFR), and tubular reabsorption into integrative mechanistic responses to provide optimal kidney function. While modulation of RAS activity is part of these responses, it was worth arguing that the phenotypic flexibility of the RPC is of further functional and structural importance for the kidney. Second, the prevalence of RPC in the renal vasculature during kidney development (nephrogenesis) is higher than in adult life (Gomez et al. 1986; Sauter et al. 2008). In addition, knockout of renin in mice leads to kidney malformations and perinatal death (Takahashi et al. 2005; Yanai et al. 2000). Importantly, an identical phenotype has been reported in human fetuses with inactivating mutations in the renin gene (Michaud et al. 2011; Zivna et al. 2009). These observations provided a crucial hint that the RPC should be important for the normal development of the kidney. Based on these findings, a role of RPC as a progenitor cell pool during nephrogenesis was originally proposed. However, experimental evidence in rats and frogs suggested a fundamental role of RAS activity in terms of Ang II effects on kidney morphogenesis and vascular development (Tufro-McReddie et al. 1995). In line with these initial findings, studies in mice and humans demonstrated that the very same detrimental renal phenotype is observed when RAS is inactivated by genetic mutations not only in renin gene but also in its further key component genes, namely, angiotensinogen, ACE, and AT1 (Gribouval et al. 2005, 2012; Gubler and Antignac 2010; Hibino et al. 2015; Hilgers et al. 1997; Kim et al. 1995; Krege et al. 1995; Oliverio et al. 1998; Takahashi et al. 2005; Yanai et al. 2000). Moreover, antihypertensive treatment with RAS inhibitors (ACE inhibitors, AT1 blockers, etc.) is counter-indicated during pregnancy because they cause fetal malformations (Broughton Pipkin et al. 1982; Daikha-Dahmane et al. 2006; Friberg et al. 1994; Grove et al. 1995; Knott et al. 1989; Landing et al. 1994; McCausland et al. 1997; Polifka 2012). RAS is believed to be important in embryonic life for not only maintaining blood pressure and organ perfusion but also because Ang II exerts growth factor-like effects in the developing kidney (Chen et al. 2004; Iosipiv and Schroeder 2003; Madsen et al. 2010; McCausland et al. 1997; Niimura et al. 1995; Tufro-McReddie et al. 1995). Altogether, these findings indicated that active RAS is necessary for normal ontogenesis and nephrogenesis, in particular, thus questioning the role of RPC themselves as progenitors.
Starting from these initial observations, experimental evidence accumulated to favor a concept where RPC do have RAS independent functions (Fig. 1). These non-canonical RAS functions will be discussed in more detail below. Notwithstanding, modern transgenic animal models provided decisive evidence that independently from RAS, the RPC do serve as a progenitor niche in embryonic and adult kidney.

Novel concept on the dual role of the renin-producing cells (RPC) in kidney highlighting their new RAS-independent functions. RAS renin-angiotensin system, ACE angiotensin-converting enzyme
2.1 Progenitor Cell Pool
2.1.1 Nephrogenesis
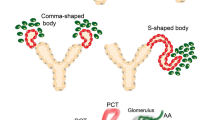
The original observations that the RPC population is spread throughout the growing renal arterial tree during nephrogenesis before shrinking to the glomerular proximity of the adult kidney came out more than 30 years ago (Gomez et al. 1986; Robillard and Nakamura 1988). This distribution has been demonstrated in multiple studies with mice, rats, pigs, and humans (Celio et al. 1985; Egerer et al. 1984; Gomez et al. 1991; Graham et al. 1992; Molteni et al. 1974). The high interspecies similarity of renal RPC ontogenesis legitimated the widespread use of transgenic animals, mostly mice, as experimental models with high translational capacity and potential clinical relevance. RPC appear in the fetal kidney around embryonic day 16 in mice (gestational week 6 in humans) shortly after the onset of renal vascular development (Celio et al. 1985; Hickmann et al. 2017; Sauter et al. 2008). The renin production is first detected in arcuate arteries and moves next to interlobular arteries and then to the afferent arterioles, i.e., proximal to distal in the growing vascular tree (Fig. 2a). The classic RPC position close to the glomeruli within the wall of terminal afferent arterioles is patterned at the end of nephrogenesis around postpartal day 10 in rodents and gestational week 35 in humans (Graham et al. 1992; Sauter et al. 2008; Sequeira Lopez and Gomez 2011). The RPC in the kidney vessels originate from a subset of cells in the stroma of the metanephric mesenchyme that expresses transcription factor FoxD1 (Pierrou et al. 1994; Lin et al. 2014). In general, the FoxD1 stromal progenitors give rise to intramural and interstitial cells (Hatini et al. 1996; Humphreys et al. 2010; Sequeira Lopez and Gomez 2011). Next to the RPC, renal VSMC, pericytes, and fibroblast-like cells are also of FoxD1 lineage. Therein, cell fate tracing experiments revealed interesting relations (Fig. 2b).

Renin-producing cells (RPC) in kidney development. (a) Localization of RPC (green) in the renal vascular tree at different developmental stages of the maturing kidney. (b) RPC as a progenitor niche in nephrogenesis. AA afferent arteriole, IA interlobular artery, ArcA arcuate artery, G glomerulus, JG juxtaglomerular, VSMCs vascular smooth muscle cells
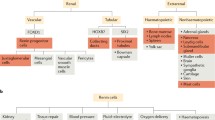
The RPC appear to be exclusively of FoxD1 descent (Gomez and Sequeira-Lopez 2018; Sequeira-Lopez et al. 2015a, b). Within the FoxD1 lineage, the RPC serve as a more differentiated precursor pool giving rise to the JG cells as well as to subpopulations of VSMC and pericytes (Sequeira Lopez et al. 2004). Pericytes are mural cells that stabilize the microvascular tree (Birbrair et al. 2015). In the kidney, mesangial cells of the glomerular capillary tuft and peritubular interstitial cells are regarded as specialized pericyte populations (Stefanska et al. 2013). Thus, in the adult mammalian kidney, a considerable part of the vasculature originates from fetal RPC progenitors. Consistent with the function of RPC as a progenitor pool during kidney development, interference with their intracellular regulatory networks such as Gs-alpha/cAMP, Notch signaling, or micro-RNA leads to adverse renal phenotype (Castellanos Rivera et al. 2011, 2015; Chen et al. 2010a, 2007; Gomez et al. 2009; Medrano et al. 2012; Neubauer et al. 2009; Sequeira-Lopez et al. 2010). As a rule, intervening in these cellular signaling pathways derails the renin production in RPC. Therefore, it remains difficult to completely separate RAS-specific and RAS-independent aspects of RPC in nephrogenesis. Importantly, two lines of recent experimental data shed light on this issue. First, renin cell ablation and renin deficiency (renin gene “knockout”) during ontogenesis result in different kidney phenotypes (Oka et al. 2017; Pentz et al. 2004; Sequeira Lopez and Gomez 2011; Takahashi et al. 2005). The most striking difference has been observed in the renal arterioles, which in “classical” renin knockout mice are with narrowed lumen due to hyperplasia of mural cells. Interestingly, these cells represent RPC descendants depleted of renin (Oka et al. 2017). On the other hand, the renal arterioles in mice with embryonic ablation of RPC have a thin vascular wall and perivascular fibrosis (Pentz et al. 2004). Second, activating molecular hypoxic signaling by conditional deletion of von Hippel-Lindau protein in RPC converts them into renin-negative fibroblast-like erythropoietin (EPO)-producing cells without leading to any further major kidney phenotype (Kurt et al. 2013, 2015). EPO is the master hormone regulating erythropoiesis. In adult mammals, it is produced almost exclusively by interstitial cells in the renal cortex (Bachmann et al. 1993; Kurtz 2017, 2019; Maxwell et al. 1997). The surprising finding that RPC could transform into EPO-expressing cells implicates that during nephrogenesis, at least part of the EPO-producing peritubular pericytes may derive from RPC. This exciting possibility awaits definitive experimental confirmation. Importantly, activating hypoxic molecular pathways in the entire FoxD1 lineage cell pool resulted in renal malformations, indicating that intact oxygen signaling in early embryonic life (before the emergence of renal RPC) is essential for the normal development of the stromal compartment of the adult kidney (Gerl et al. 2017).
Altogether, these findings demonstrated a role of RPC as a progenitor pool during ontogenesis irrespective of renin and RAS activity. At the same time, fine-tuning of the complex functions of RPC is indispensable for proper nephrogenesis.
2.1.2 Adult Kidney
The role of RPC as a progenitor niche during fetal life inspired us to address the possibility if these cells have a similar function in the adult kidney. To this end, we used reversible injury models such as intravenous infusion of anti-mesangial cell serum or renal artery perfusion with concanavalin A/anti-concanavalin A antibody to target primarily the glomeruli in the kidney (Hohenstein et al. 2008; Sradnick et al. 2016; Yo et al. 2003). We concentrated our studies on the regenerative phase after the initial acute damage. This approach was based on the general paradigm that developmental cellular mechanisms, which are active during embryogenesis, may be reenacted to regenerate damaged organs in adulthood. Since RPC serve as progenitors for glomerular cells during nephrogenesis (e.g., mesangial cells; see Sect. 2.1.1) and since glomerulopathies are a primary cause for end-stage renal disease (Jha et al. 2013), we have been focusing on glomerular injury models. We also developed a novel transgenic mouse model by combining Cre-loxP recombination with tet-on system (Schonig et al. 2002; Urlinger et al. 2000) to pulse-label and fate-trace RPC in adulthood (Kessel et al. 2019; Lachmann et al. 2017; Ruhnke et al. 2018; Starke et al. 2015; Steglich et al. 2019).
We first found that during the regenerative phase after initial damage induced by anti-mesangial cell serum, the RPC migrate away from their classical JG position into the diseased glomerulus (Starke et al. 2015). Thereby, the migrating cells differentiate in a way that they lose the renin expression and acquire alpha-8-integrin as well as platelet-derived growth factor receptor-beta proteins which are specific for mesangial cells in the glomerulus. Hence, our hypothesis received experimental confirmation because RPC transdifferentiation in the mature kidney recapitulated nephrogenesis, where mesangial cells originate from RPC (Gomez and Sequeira-Lopez 2018; Sequeira Lopez et al. 2004). However, in adult life, this is a regenerative process observed only after organ damage with cell loss and not under physiological conditions. Throughout development and adulthood, the RPC progenitors maintain cell-specific plasticity, i.e., they transdifferentiate to the very same cell types (Fig. 3). Moreover, RPC descendants did not regenerate the endothelium after depletion of glomerular endothelial cells in reversible endothelial cell injury model induced by renal artery perfusion with concanavalin A/anti-concanavalin A antibody (Ruhnke et al. 2018). Instead, RPC transdifferentiated to exclusively replace glomerular mesangial cells, which seemed to be damaged secondary to the endothelial injury.

Plasticity (transdifferentiation) of the renin-producing cells (RPC) in adult kidney recapitulates embryonic patterns. RPC serve as progenitors, which repopulate the glomerulus to replace damaged mesangial cells after injury. RPC could also reversibly switch between secretory and contractile phenotype to regulate renin production under physiological conditions through metaplastic transformation. See also Sects. 2, 2.1.1, and 2.2 and Figs. 2 and 4. AA afferent arteriole, G glomerulus, VSMC vascular smooth muscle cell, MC mesangial cell, PT proximal tubule

Recruitment and neogenesis of renin-producing cells (RPC). (a) Schematic presentation of the difference between recruitment and neogenesis. Metaplastic transformation is a phenotype switch within the developmental RPC pool (see also Fig. 2) where recruitment specifically refers to the transformation of VSMC into RPC. In neogenesis, an unrelated cell enters the renin lineage population by starting to express renin for the first time (see also Sect. 3). (b) Role of recruitment and neogenesis in the maintenance of the RPC population. The role of proliferation therein is not well understood. Dashed arrows represent secondary role. VSMC vascular smooth muscle cell
Other investigators used an alternative transgenic mouse model for inducible labeling and tracing of RPC in a model of focal segmental glomerulosclerosis (Kaverina et al. 2017b; Lichtnekert et al. 2016; Pippin et al. 2015). They found that RPC repopulate the glomerular Bowman’s capsule and differentiate not only to mesangial cells but also to podocytes and parietal epithelial (PE) cells. The reasons for the partial discrepancy compared to our results lay most probably on the different animal or injury models used. For instance, the experimental setups we used do not lead to podocyte depletion (Ruhnke et al. 2018). In any case, it is somewhat counterintuitive that RPC could replace damaged podocytes or PE cells because these are not of FoxD1 origin and thus do not belong to the RPC stromal mesenchyme descendants in the adult kidney (see Sect. 3). Therefore, it is plausible to conclude that after severe glomerular injury the progenitor potential of RPC is utilized beyond the developmentally based transdifferentiation programs of cell type-specific regeneration. Thus, the RPC seem to universally protect damaged tissue as a part of integrative stress response of the organism. Anyway, more experimental support is needed to utterly validate this hypothesis.
The communication modes between RPC and (damaged) intraglomerular cells in the mature kidney are still poorly understood. With this regard, it is unlikely that soluble messenger molecules are transported with blood flow because the RPC are upstream to the glomerulus. Cell-cell contacts are important for the physiological signaling within the JGA, and therefore they may be involved in regenerative signaling (Just et al. 2009; Kurtz 2015; Wagner et al. 2007). Moreover, the RPC are featured by a specific gap junction protein expression signature where connexin 40 is central for their function and morphology (Kurtz et al. 2009; Wagner and Kurtz 2013). Hence, future studies on the role of cell-cell contacts in transmitting signals from damaged glomeruli to the precursor RPC niche in the afferent arterioles are an attractive perspective. However, the advancements in this strategic area of interest are hampered by a deficit of appropriate experimental models. For instance, efforts to identify mesangial-specific gene(s) fail until now, thus making the generation of transgenic animals with selectively targeted mesangial cells impossible.
Immune cells could also be considered as paramount players mediating cues from injured and regenerating glomeruli to RPC. According to the modern concept of necroinflammation, necrotic organ damage inevitably goes together with an inflammatory component (Linkermann 2019; Sarhan et al. 2018). Conversely, inflammation and release of damage-associated molecular patterns (DAMPs) do not only augment tissue damage but also initiate regenerative reactions (Sarhan et al. 2018). These processes are mediated by resident cells of innate immunity like macrophages, dendritic cells, and natural killer cells that orchestrate local and systemic mechanisms with adaptive relevance in host-pathogen interactions, metabolic responses to tissue injury and degeneration, or allogeneic transplantation. Importantly, we and others have demonstrated that the RPC are equipped with the molecular machinery (including tumor necrosis factor receptors, functional NF-kappa B and STAT signaling, etc.) necessary for acquisition and transduction of innate immunity signals like inflammatory cytokines (Baumann et al. 2000; Desch et al. 2012; Liu et al. 2006; Petrovic et al. 1997; Todorov et al. 2002, 2005, 2004). Therefore, we favor the immune-mediated processes as the most feasible mechanism(s) responsible for signal delivery from damaged glomeruli to RPC and vice versa. There is already indirect evidence in support of such an assumption. Thus, both necroinflammation and RPC progenitor function in adulthood are specific for pathophysiological conditions (Linkermann 2019; Ruhnke et al. 2018; Starke et al. 2015).
At present, little is known about how RPC set up to differentiate and migrate upon glomerular damage. First data from us demonstrated that when renin is switched off by interfering with Gs-alpha/cAMP signaling, the RPC descendants produce increased amounts of tissue remodeling factors (Steglich et al. 2019). The latter could loosen cells and potentiate their migration (see also Sect. 2.3). With this regard, proteins involved in the control of cellular mechanics like small GTPases are particularly interesting targets for future studies, since cytoskeleton rearrangement generally accompanies cell motility and differentiation. The transcription factor Wilms’ tumor suppressor 1 (WT1), which belongs to this group of proteins and is expressed in RPC, has already been reported to affect their transformation and glomerular migration after podocyte depletion (Kaverina et al. 2017a).
Collectively, although it is now established that the RPC operate as a progenitor cell niche in the adult mammalian kidney, we are still at the onset of exciting findings that should elaborate the understanding of underlying molecular mechanisms and pathophysiological relevance. Therein large-scale “omics” analyses of the global transcriptional/translational landscape of RPC would be of exceptional importance (Martinez et al. 2018; Steglich et al. 2019; Wang et al. 2018).
2.2 Neogenesis and Recruitment
The discovery of a progenitor function of RPC inspired studies on their maintenance as a stable population in adulthood. This issue is also critical with regard to the classical role of RPC in RAS – if upon glomerular injury all JG cells differentiate to renin-negative descendants, RAS would be switched off, thus disturbing the general control of circulation and overall homeostasis. Moreover, (self)renewal is an archetypal feature of stem cell niches (Seaberg and van der Kooy 2003). Therefore, it is somewhat surprising that there is no conclusive data demonstrating proliferation of RPC. Early pulse labeling studies with DNA intercalating agents in the rat anti-Thy1 model indicated that repopulation of the glomerulus during repair after mesangial injury involves migration and proliferation of precursors from the JGA (Hugo et al. 1996, 1997). Recent findings did not show prominent RPC proliferation in an equivalent mouse model (Starke et al. 2015). This is consistent with other studies (Cantin et al. 1977; R. Ariel Gomez, personal communication). In contrast, proliferating RPC have been detected after continuous loading with the DNA intercalating agent bromdesoxyuridin (BrdU) for lineage tracing of dividing cells for up to 27 days (Lichtnekert et al. 2016; Owen et al. 1994). Altogether, these findings inferred that mechanisms beyond proliferation are necessary to maintain the RPC precursor niche, particularly after injury when the JG cells transdifferentiate to replace damaged glomerular cells. Progenitor cell niches (such as the RPC) could also be filled up by differentiating pluripotent stem cells through a process termed neogenesis or de novo differentiation. For RPC (and renin lineage cells in general; see Sect. 3), neogenesis is the process where a cell that is not related to the renin lineage (i.e., never expressed renin before and does not originate from cells that express/ed renin) starts to express renin for a very first time (Hickmann et al. 2017). We were lucky to make use of a versatile transgenic mouse model for neogenesis tracing (Muzumdar et al. 2007; Xiao et al. 2013), which was modified for RPC targeting. During nephrogenesis, RPC emerge almost exclusively via neogenesis (Hickmann et al. 2017). Although proliferation should also play a role in the establishment of the RPC population in the developing kidney, de novo differentiation seems to be the superior mechanism. In adulthood, the RPC neogenesis is featured by three hallmarks: it is intrarenal, lifelong, and regulated (Hickmann et al. 2017). Thus, the kidney appears to be able to effectively preserve and renew a local progenitor cell population. Moreover, the process of RPC neogenesis is counteracted by rate-matched apoptotic cell death, which explains why the adult kidney is not overfilled with RPC (Hickmann et al. 2017). Even more important is the fact that RPC neogenesis is stress-inducible. During injury when RPC repopulate damaged glomeruli, de novo differentiation is upregulated, leading to a moderate increase in RPC number (Hickmann et al. 2017). This mechanism safeguards not only the RPC precursor pool, which is primed to replace the depleted glomerular cells, but also the RAS activity. This is an important aspect demonstrating that the RPC act as a progenitor niche without impeding their classical role within RAS. The origin of the de novo differentiated RPC in the adult kidney remains open. We experimentally excluded the option that the renal RPC are bone marrow-derived (Hickmann et al. 2017). FoxD1-positive precursors are the source for RPC during development (see Sect. 2.1.1), but FoxD1 is not expressed in the mature kidney (Humphreys et al. 2010). Preliminary observations from us suggest a pericyte origin for new RPC after nephrogenesis is finished, but more studies are needed to identify the adult precursor renal niche for RPC.
Increased de novo differentiation of RPC has also been observed in a physiological model of decreased blood pressure (Hickmann et al. 2017). Arterial hypotension is the main stimulus for renin production, and the renin production is scaled up and down by changing the number of RPC, rather than the renin expression rate in the single cell (Castrop et al. 2010; Hackenthal et al. 1990; Rasch et al. 1998). The adjustment of RPC number to the functional status of the organism is a versatile process termed recruitment or metaplastic transformation (see Sect. 2). Recruitment is one of the early described physiological manifestations of RPC plasticity in the adult mammalian kidney (Cantin et al. 1977). It represents a reversible phenotypic switch between VSMC and RPC in the wall of the afferent arterioles upstream to their JG parts. Lineage tracing revealed that the switching VSMC in the afferent arterioles originate from RPC during nephrogenesis meaning that the recruitment is a recapitulation of a developmental process (Gomez et al. 2014; Kurt and Kurtz 2015; Martini and Danser 2017; Sequeira Lopez et al. 2004) in adulthood (see Sect. 2.1.1). It should be emphasized that recruitment is not to be mixed up with neogenesis although both are hallmarks of RPC plasticity. Recruitment (metaplastic transformation) is a reversible phenotype change of a cell within the developmental RPC pool (referred also in Figs. 2 and 3), while neogenesis (de novo differentiation) is the differentiation of a cell to join the renin lineage (Fig. 4a; see the definition of neogenesis above in this section). In addition, neogenesis is involved primarily in RPC renewal and progenitor function after injury, whereas recruitment is responsible for the physiological control of renin production and RAS activity (Fig. 4b; see also Fig. 3).
Thus, the RPC are not only featured by plasticity but also represent a dynamic population predestined to be involved in complex physiological and pathophysiological processes.
Antihypertensive drugs and particularly RAS inhibitors boost the renin production and the RPC count (due to negative feedback within RAS linking blood pressure to renin expression) demonstrating that recruitment and neogenesis could be targeted by therapeutic strategies (Azizi and Menard 2004; Hickmann et al. 2017; Mooser et al. 1990; Nussberger et al. 2007; Pugh et al. 2019). Although an increased number of RPC correlates with the renoprotective effect of the RAS inhibitors, it is still controversial whether this increase might be beneficial (NB! only when RAS activity is inhibited in terms of AT1-dependent Ang II effects) or not (Moniwa et al. 2013).
2.3 Protection of Renal Microvascular Endothelium
Lately, our studies revealed that RPC are quite surprisingly involved in the maintenance of renal capillary endothelium in adulthood. This novel function of RPC has been discovered when characterizing an inducible transgenic model with compromised Gs-alpha/cAMP signaling pathway (Lachmann et al. 2017). Gs-alpha is the stimulatory subunit of membrane receptor coupled trimeric G-proteins (Spiegel et al. 1992; Weinstein et al. 2001). It catalyzes the generation of cAMP upon ligand binding to the associated receptor. The RPC are equipped with Gs-alpha coupled IP/EP2/EP4 and beta-adrenergic receptors mediating effects of prostanoids and catecholamines, respectively (Boivin et al. 2001; Churchill et al. 1983; Facemire et al. 2011; Friis et al. 2005; Jensen et al. 1996; Karger et al. 2018; Kim et al. 2007; Schweda et al. 2004). Blood pressure fluctuations seem to modulate renal prostanoid production and sympathetic nerve activity to regulate renin production by adjusting the RPC number (Chen et al. 2010b; Kim et al. 2012). Therefore, Gs-alpha/cAMP signaling is central for renin synthesis and secretion, thus essentially determining the RPC phenotype (see also Sect. 2.1.1). It was predictable that the induction of Gs-alpha knockout in RPC of adult mice would result in renin deficiency and arterial hypotension (Kessel et al. 2019; Lachmann et al. 2017). However, these changes were transient because the drop in blood pressure served as a stimulus to recruit RPC in upstream parts of the afferent arterioles (see Sect. 2.2) that were not affected by the pulse induction of the Gs-alpha knockout. Although normal renin production and blood pressure persisted after these initial fluctuations, the JG-specific Gs-alpha-deficient mice developed chronic kidney disease. The adverse renal phenotype most unexpectedly included microvascular endothelial damage featured by morphological and systemic signs of thrombotic microangiopathy (Benz and Amann 2010; Lachmann et al. 2017). Remarkably, kidney injury developed on the background of normalized renin production, indicating that RPC exhibit protective effects on renal microvascular endothelium-independent on their role in the control of RAS activity (Lachmann et al. 2017). The mode of action of RPC on endothelial cells is multifaceted, and the implicated interactions are just beginning to be unraveled. One mechanism involves the production of angiogenic factors by RPC, which has been displayed in several studies (Brunskill et al. 2011; Haltia et al. 1996; Lachmann et al. 2017; Sequeira Lopez and Gomez 2011). Within the RPC-derived proangiogenic cues, vascular endothelial growth factor (VEGF) appears to be particularly relevant for two reasons. First, both pharmacological inhibition and genetically induced intrarenal deficiency of VEGF result in thrombotic microangiopathy (Eremina et al. 2008; Sison et al. 2010). Second, VEGF is a cAMP-regulated gene and its expression in RPC is induced by cAMP (Bradbury et al. 2005; Han et al. 2013; Lachmann et al. 2017). Another factor contributing to the proangiogenic capacity of RPC appears to be their secretory phenotype per se. This conclusion is also based on findings in adult RPC-specific Gs-alpha-deficient mice, where the RPC have been found to persist after Gs-alpha knockout as renin-negative cells (Steglich et al. 2019). In the persisting RPC progeny, a pool of 16 genes involved in tissue remodeling with vascular damage and fibrosis including collagens and TGF-beta were upregulated. Simultaneously, genes conferring the identity of RPC such as renin itself, connexin 40, and aldo-keto reductase family 1 member B7 (Akr1b7) was downregulated. Ablation of RPC resulted in renin deficiency but failed to produce any sign of endothelial injury (Steglich et al. 2019). This observation additionally confirmed that Gs-alpha-negative RPC descendants are necessary for the development of endothelial dysfunction. In total, the inactivation of Gs-alpha/cAMP signaling seems to switch the RPC phenotype from “protective/secretory” to “damaging/profibrotic” (Fig. 5).

Current view on the non-canonical effects of renin-producing cells (RPC) on renal microvascular endothelium
The discovery of a protective role of RPC on renal microvasculature elicits some very exciting questions to be addressed in the future. As discussed above in this section, RPC recruitment is a cAMP-regulated process. Therefore, it would be interesting to know whether the recruitable VSMC in the afferent arterioles may exert profibrotic remodeling effects when not switched to the renin-producing phenotype. It is now established that renal VSMC are of heterogeneous embryonic origin (see Sect. 2.1.1) implying that RPC-derived and non-RPC-derived VSMC may have different functional characteristics. Vice versa, a decrease of RPC number when renin production is suppressed, for instance, in some forms of arterial hypertension, could be an independent risk factor for renal damage. Extrapolating these considerations would suggest that the renoprotective effects of pharmacological RAS inhibitors rely on cAMP-dependent RPC recruitment on top of the Ang II antagonism. Therefore, discoveries of further mechanisms involved in the complex interactions between RPC and kidney microvasculature with potential pathophysiological and therapeutic significance are anticipated.
3 Renin Lineage Cells
Studies in adult mice, rats, and humans unequivocally demonstrated that many organs and specialized tissues express renin mRNA albeit to a much lesser level than the JG cells. The non-JG renin-expressing cells are part of local or tissue-specific RASs (Paul et al. 2006). However, the JG cells appear to be the sole source for plasma renin because circulating renin disappears after bilateral nephrectomy (Fordis et al. 1983; Hannon et al. 1969; Katz et al. 1997). The generation of renin from its inactive precursor prorenin upon binding to the prorenin receptor does not essentially contribute to the plasma renin activity (Nguyen et al. 2014, 2002; Rosendahl et al. 2014).
Systemic and local RASs functionally integrate into concerted Ang II-mediated effects in a way that the local RASs modulate the action of systemic RAS (Paul et al. 2006). Accordingly, fate-tracing experiments demonstrated that a marked number of cells in kidneys, spleen, bone marrow, and endocrine pancreas of the adult organism could be attributed to the renin lineage (Belyea et al. 2014; Desch et al. 2011; Glenn et al. 2014; Hickmann et al. 2017; Sequeira Lopez et al. 2004). These findings prompted the definition of a renin lineage cell (RLC) population which comprises all cells collectively featured by the expression of renin gene and/or the presence of a renin-expressing cell in their ancestry (Hickmann et al. 2017). The existence of local RAS infers that RLC build a huge population spread throughout the whole body. Such a conclusion appears to diverge from the paradigm that the JG cells are the sole RPC niche in adulthood. Indeed, there is still some controversy on the systemic availability of renin from source(s) other than the JG cells. The current view is a kind of reasonable compromise postulating that in certain diseases such as diabetes mellitus, high-grade obesity, or some subsets of arterial hypertension, renin from non-JG origin would be of pathophysiological relevance. The major issue provoking conflicting interpretations is that with very few exceptions such as pituitary, adrenal glands, or testes (Lee et al. 2005; Naruse et al. 1985), renin protein is detectable exclusively in JG cells of the kidney. The JG cells are the only cell type in the adult organism equipped with molecular machinery to store renin in secretory vesicles, which greatly facilitates their identification as renin-positive cells (Hackenthal et al. 1990; Lacasse et al. 1985; Mendez 2014; Taugner et al. 1984, 1985). In all other renin-expressing cell types, the translated (pro)renin protein is constitutively sequestered to the extracellular compartment. Thus, the lack of secretory granules together with the much lower renin mRNA expression provides a reasonable explanation for why renin is hardly observed in non-JG cells.
What about RAS independent functions for RLC? As already discussed in Sect. 2.1.1, the progenitor RPC in the kidney, which should be regarded as a subpopulation within the RLC pool, give rise not only to JG cells but also to further mural cells in the mature kidney. However, lineage-tracing experiments reproducibly demonstrated that cells in the Bowman’s capsule (PE cells), proximal tubules, and collecting ducts of the adult kidney are also mapped to the renin lineage (Hickmann et al. 2017; Sequeira-Lopez et al. 2004, 2015b). Altogether, the renal RLC represent around 10% of the renal cell mass in adult mice, and it is feasible to extrapolate this data to humans. Similar to RPC, the RLC are also maintained by neogenesis balanced by apoptosis, and thus they represent a dynamic population persisting throughout adult life (Hickmann et al. 2017). Therefore, RLC appear to be fundamental for the morphological architecture of the adult mammalian kidney. On the other hand, the renal RLC are not a homogeneous population with regard to their embryonic origin. RPC and their descendants originate from FoxD1-expressing cells in the stromal compartment of the metanephric mesenchyme (see Fig. 2b and Sect. 2.1), PE cells and proximal tubules from the core layer of the metanephric mesenchyme (known as cap mesenchyme), and collecting ducts from the ureteric bud (Humphreys et al. 2010; Kobayashi et al. 2008; Kress et al. 1990; Nelson et al. 1998; Srinivas et al. 1999). The most probable scenario for the development of FoxD1-unrelated renal RLC is that the renin gene is switched on in their transcriptional programs at some time point of embryonic differentiation. The functional importance of this event remains elusive. Tubular renin is dispensable during nephrogenesis (Sequeira-Lopez et al. 2015b). The role of collecting duct as a source of renin in diabetes mellitus and hypertensive disease is disputed (Gonzalez et al. 2011; Liu et al. 2011; Prieto-Carrasquero et al. 2004, 2005, 2009; Ramkumar et al. 2014; Song et al. 2016; Tang et al. 2019).
Taking into account that local RAS are ubiquitous, extrarenal RLC are still extremely underrepresented in current research. Such status-quo is quite amazing because the few available studies on non-kidney RLC yielded very exciting results. Brain RLC seem to protect from arterial hypertension. The mechanistic explanation is that in brain RLC a specific renin isoform is transcribed from an alternative start codon producing a truncated protein, which remains intracellularly and controls neurotransmitter release (Shinohara et al. 2016). Unfortunately, there are no studies focusing on a putative role of brain RLC after injury in the central nervous system.
RLC are detectable in the bone marrow, peripheral blood, and spleen (Belyea et al. 2014), where they are also maintained by neogenesis (Hickmann et al. 2017). The majority of the immune system RLC are B-lymphocytes. Defects in the developmental transcriptional program induced by RLC-specific knockout of the Notch pathway transcriptional effector RBP-J (recombination signal binding protein for immunoglobulin kappa J region) or p53 and retinoblastoma protein (Rb) result in B-cell leukemia or pancreatic neuroendocrine carcinoma, respectively (Belyea et al. 2014; Glenn et al. 2014). These findings imply that the embryonic differentiation of extrarenal RLC is under tight control to ensure proper organogenesis. At present, further roles for RLC in adulthood are not known.
4 Future Perspectives
This review tells a story where after entering the textbooks in the context of RAS, nowadays the renal RPC revive as a hot topic in research for their novel RAS-independent functions. Progenitor and protective features are the main aspects delineating the new non-canonical profile of RPC (see also Fig. 1). While already expanding the paradigm, the novel insights on RPC summarized above provide also an inspiration for future work. The questions to be addressed could be grouped into three prospective experimental pipelines:
-
1.
Deciphering the molecular “motors” driving the complex protective activity of renal RPC in health and in response to tissue injury
-
2.
Characterization of the RPC niche as a dynamic population with regard to origin, refilling and cell death
-
3.
Unravelling the role of RLC beyond RAS
The last group includes several independent intriguing aspects. One is the role of renin in RLC – is it simply an eponymous marker or it has some non-conventional function(s)? Since renin is a protease, we hypothesize that it might be involved in the remodeling of neighboring tissue by cleavage of extracellular matrix proteins, which is a fundamental process in organ growth and regeneration. Support for RAS-unrelated protease activity of renin provided the recent observation that it activates the complement cascade by triggering C3 proteolysis (Bekassy et al. 2018). Another aspect is the idea that RLC represent stress-inducible stem cells (SISC). SISC have been recently defined as a subpopulation of progenitor cells that are particularly prone to stress-related signals (Bornstein et al. 2019). SISC are featured by coordinated signaling pathways and transcriptional signature, which enable them to be an efficient integrative part of general adaptive stress responses in the entire organism. Accordingly, in distress situations, SISC function would be dysregulated leading to maladaptive reactions and disease. It has been postulated that SISC are ubiquitous and should be present in virtually any organ and specialized tissue. Nestin and Notch1 serve as general markers of SISC populations. RLC fulfill all these criteria thus perfectly fitting into the definition for SISC (Hanner et al. 2008) (see also this section above). Stress signals influencing RLC and particularly renal RPC include acute drop in arterial blood pressure (e.g., due to hemorrhage), chronic salt depletion, extreme dehydration, hyperactivation of the sympathetic nervous system by environmental natural and social factors, etc. Therefore, it is feasible to assume that RLC build at least part of the SISC niche and that the renin lineage might be one further universal SISC marker. Being an attractive unifying concept, the presence of interrelated organ- and tissue-specific RLC (including RPC) with coordinated stress-regulated functions during development and adulthood awaits empirical support. Since transgenic animal models applicable in experiments addressing the issues formulated above already exist, a further expansion of our knowledge on renal RPC and RLC is pending.
Abbreviations
- AA:
-
Afferent arteriole
- ACE:
-
Angiotensin-converting enzyme
- Akr1b7:
-
Aldo-keto reductase family 1 member B7
- Ang:
-
Angiotensin
- ArcA:
-
Arcuate artery
- AT1:
-
Angiotensin II type 1 receptors
- AT2:
-
Angiotensin II type 2 receptors
- BrdU:
-
Bromdesoxyuridin
- cAMP:
-
3′,5′-Cyclic adenosine monophosphate
- Cre:
-
Cre-recombinase
- DAMPs:
-
Damage-associated molecular patterns
- DNA:
-
Deoxyribonucleic acid
- EP:
-
Prostaglandin EP receptor
- EPO:
-
Erythropoietin
- FoxD1:
-
Forkhead box D1
- G:
-
Glomerulus
- GFR:
-
Glomerular filtration rate
- Gs:
-
G-protein stimulatory subunit
- GTPases:
-
Guanosine-5′-triphosphate hydrolases
- IA:
-
Interlobular artery
- IP:
-
Prostacyclin I2 receptor
- JG:
-
Juxtaglomerular
- JGA:
-
JG apparatus
- loxP:
-
Locus of X-over P1
- MC:
-
Mesangial cell
- NB:
-
Nota bene
- NF-kappa B:
-
Nuclear factor “kappa-light-chain-enhancer” of activated B-cells
- PE:
-
Parietal epithelial
- PT:
-
Proximal tubule
- RAS:
-
Renin-angiotensin system
- Rb:
-
Retinoblastoma protein
- RBP-J:
-
Recombination signal binding protein for immunoglobulin kappa J region
- RLC:
-
Renin lineage cells
- RNA:
-
Ribonucleic acid
- RPC:
-
Renin-producing cells
- SISC:
-
Stress-inducible stem cells
- STAT:
-
Signal transducers and activators of transcription
- tet:
-
Tetracycline
- TGF:
-
Transforming growth factor
- Thy1:
-
Thymocyte antigen 1
- VEGF:
-
Vascular endothelial growth factor
- VSMC:
-
Vascular smooth muscle cells
- WT1:
-
Wilms’ tumor suppressor 1
References
Ames MK, Atkins CE, Pitt B (2019) The renin-angiotensin-aldosterone system and its suppression. J Vet Intern Med 33(2):363–382. https://doi.org/10.1111/jvim.15454
Arendse LB, Danser AHJ, Poglitsch M, Touyz RM, Burnett JC Jr, Llorens-Cortes C, Ehlers MR, Sturrock ED (2019) Novel therapeutic approaches targeting the renin-angiotensin system and associated peptides in hypertension and heart failure. Pharmacol Rev 71(4):539–570. https://doi.org/10.1124/pr.118.017129
Azizi M, Menard J (2004) Combined blockade of the renin-angiotensin system with angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists. Circulation 109(21):2492–2499. https://doi.org/10.1161/01.CIR.0000131449.94713.AD
Bachmann S, Le Hir M, Eckardt KU (1993) Co-localization of erythropoietin mRNA and ecto-5′-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin. J Histochem Cytochem 41(3):335–341. https://doi.org/10.1177/41.3.8429197
Baumann H, Wang Y, Richards CD, Jones CA, Black TA, Gross KW (2000) Endotoxin-induced renal inflammatory response. Oncostatin M as a major mediator of suppressed renin expression. J Biol Chem 275(29):22014–22019. https://doi.org/10.1074/jbc.M002830200
Bekassy ZD, Kristoffersson AC, Rebetz J, Tati R, Olin AI, Karpman D (2018) Aliskiren inhibits renin-mediated complement activation. Kidney Int 94(4):689–700. https://doi.org/10.1016/j.kint.2018.04.004
Belyea BC, Xu F, Pentz ES, Medrano S, Li M, Hu Y, Turner S, Legallo R, Jones CA, Tario JD, Liang P, Gross KW, Sequeira-Lopez ML, Gomez RA (2014) Identification of renin progenitors in the mouse bone marrow that give rise to B-cell leukaemia. Nat Commun 5:3273. https://doi.org/10.1038/ncomms4273
Benz K, Amann K (2010) Thrombotic microangiopathy: new insights. Curr Opin Nephrol Hypertens 19(3):242–247. https://doi.org/10.1097/MNH.0b013e3283378f25
Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O (2015) Pericytes at the intersection between tissue regeneration and pathology. Clin Sci 128(2):81–93. https://doi.org/10.1042/CS20140278
Boivin V, Jahns R, Gambaryan S, Ness W, Boege F, Lohse MJ (2001) Immunofluorescent imaging of beta 1- and beta 2-adrenergic receptors in rat kidney. Kidney Int 59(2):515–531. https://doi.org/10.1046/j.1523-1755.2001.059002515.x
Bornstein SR, Steenblock C, Chrousos GP, Schally AV, Beuschlein F, Kline G, Krone NP, Licinio J, Wong ML, Ullmann E, Ruiz-Babot G, Boehm BO, Behrens A, Brennand A, Santambrogio A, Berger I, Werdermann M, Sancho R, Linkermann A, Lenders JW, Eisenhofer G, Andoniadou CL (2019) Stress-inducible-stem cells: a new view on endocrine, metabolic and mental disease? Mol Psychiatry 24(1):2–9. https://doi.org/10.1038/s41380-018-0244-9
Bradbury D, Clarke D, Seedhouse C, Corbett L, Stocks J, Knox A (2005) Vascular endothelial growth factor induction by prostaglandin E2 in human airway smooth muscle cells is mediated by E prostanoid EP2/EP4 receptors and SP-1 transcription factor binding sites. J Biol Chem 280(34):29993–30000. https://doi.org/10.1074/jbc.M414530200
Broughton Pipkin F, Symonds EM, Turner SR (1982) The effect of captopril (SQ14,225) upon mother and fetus in the chronically cannulated ewe and in the pregnant rabbit. J Physiol 323:415–422. https://doi.org/10.1113/jphysiol.1982.sp014081
Brunskill EW, Sequeira-Lopez ML, Pentz ES, Lin E, Yu J, Aronow BJ, Potter SS, Gomez RA (2011) Genes that confer the identity of the renin cell. J Am Soc Nephrol 22(12):2213–2225. https://doi.org/10.1681/ASN.2011040401
Cantin M, Araujo-Nascimento MD, Benchimol S, Desormeaux Y (1977) Metaplasia of smooth muscle cells into juxtaglomerular cells in the juxtaglomerular apparatus, arteries, and arterioles of the ischemic (endocrine) kidney. An ultrastructural-cytochemical and autoradiographic study. Am J Pathol 87(3):581–602
Castellanos Rivera RM, Monteagudo MC, Pentz ES, Glenn ST, Gross KW, Carretero O, Sequeira-Lopez ML, Gomez RA (2011) Transcriptional regulator RBP-J regulates the number and plasticity of renin cells. Physiol Genomics 43(17):1021–1028. https://doi.org/10.1152/physiolgenomics.00061.2011
Castellanos-Rivera RM, Pentz ES, Lin E, Gross KW, Medrano S, Yu J, Sequeira-Lopez ML, Gomez RA (2015) Recombination signal binding protein for Ig-kappaJ region regulates juxtaglomerular cell phenotype by activating the myo-endocrine program and suppressing ectopic gene expression. J Am Soc Nephrol 26(1):67–80. https://doi.org/10.1681/ASN.2013101045
Castrop H, Hocherl K, Kurtz A, Schweda F, Todorov V, Wagner C (2010) Physiology of kidney renin. Physiol Rev 90(2):607–673. https://doi.org/10.1152/physrev.00011.2009
Celio MR, Groscurth P, Inagami T (1985) Ontogeny of renin immunoreactive cells in the human kidney. Anat Embryol 173(2):149–155. https://doi.org/10.1007/bf00316297
Chen Y, Lasaitiene D, Gabrielsson BG, Carlsson LM, Billig H, Carlsson B, Marcussen N, Sun XF, Friberg P (2004) Neonatal losartan treatment suppresses renal expression of molecules involved in cell-cell and cell-matrix interactions. J Am Soc Nephrol 15(5):1232–1243. https://doi.org/10.1097/01.asn.0000123690.75029.3f
Chen L, Kim SM, Oppermann M, Faulhaber-Walter R, Huang Y, Mizel D, Chen M, Lopez ML, Weinstein LS, Gomez RA, Briggs JP, Schnermann J (2007) Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gsalpha in juxtaglomerular cells. Am J Physiol Renal Physiol 292(1):F27–F37. https://doi.org/10.1152/ajprenal.00193.2006
Chen L, Faulhaber-Walter R, Wen Y, Huang Y, Mizel D, Chen M, Sequeira Lopez ML, Weinstein LS, Gomez RA, Briggs JP, Schnermann J (2010a) Renal failure in mice with Gsalpha deletion in juxtaglomerular cells. Am J Nephrol 32(1):83–94. https://doi.org/10.1159/000314635
Chen L, Kim SM, Eisner C, Oppermann M, Huang Y, Mizel D, Li L, Chen M, Sequeira Lopez ML, Weinstein LS, Gomez RA, Schnermann J, Briggs JP (2010b) Stimulation of renin secretion by angiotensin II blockade is Gsalpha-dependent. J Am Soc Nephrol 21(6):986–992. https://doi.org/10.1681/ASN.2009030307
Churchill PC, Churchill MC, McDonald FD (1983) Evidence that beta 1-adrenoceptor activation mediates isoproterenol-stimulated renin secretion in the rat. Endocrinology 113(2):687–692. https://doi.org/10.1210/endo-113-2-687
Daikha-Dahmane F, Levy-Beff E, Jugie M, Lenclen R (2006) Foetal kidney maldevelopment in maternal use of angiotensin II type I receptor antagonists. Pediatr Nephrol 21(5):729–732. https://doi.org/10.1007/s00467-006-0070-1
Damkjaer M, Isaksson GL, Stubbe J, Jensen BL, Assersen K, Bie P (2013) Renal renin secretion as regulator of body fluid homeostasis. Pflugers Arch 465(1):153–165. https://doi.org/10.1007/s00424-012-1171-2
Desch M, Harlander S, Neubauer B, Gerl M, Germain S, Castrop H, Todorov VT (2011) cAMP target sequences enhCRE and CNRE sense low-salt intake to increase human renin gene expression in vivo. Pflugers Arch 461(5):567–577. https://doi.org/10.1007/s00424-011-0956-z
Desch M, Hackmayer G, Todorov VT (2012) Identification of ATF2 as a transcriptional regulator of renin gene. Biol Chem 393(1–2):93–100. https://doi.org/10.1515/BC-2011-157
Dominick MA, Bobrowski WF, Metz AL, Gough AW, MacDonald JR (1990) Ultrastructural juxtaglomerular cell changes in normotensive rats treated with quinapril, an inhibitor of angiotensin-converting enzyme. Toxicol Pathol 18(3):396–406. https://doi.org/10.1177/019262339001800306
Egerer G, Taugner R, Tiedemann K (1984) Renin immunohistochemistry in the mesonephros and metanephros of the pig embryo. Histochemistry 81(4):385–390. https://doi.org/10.1007/bf00514334
Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE (2008) VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358(11):1129–1136. https://doi.org/10.1056/NEJMoa0707330
Facemire CS, Nguyen M, Jania L, Beierwaltes WH, Kim HS, Koller BH, Coffman TM (2011) A major role for the EP4 receptor in regulation of renin. Am J Physiol Renal Physiol 301(5):F1035–F1041. https://doi.org/10.1152/ajprenal.00054.2011
Fordis CM, Megorden JS, Ropchak TG, Keiser HR (1983) Absence of renin-like activity in rat aorta and microvessels. Hypertension 5(5):635–641. https://doi.org/10.1161/01.hyp.5.5.635
Fournier D, Luft FC, Bader M, Ganten D, Andrade-Navarro MA (2012) Emergence and evolution of the renin-angiotensin-aldosterone system. J Mol Med 90(5):495–508. https://doi.org/10.1007/s00109-012-0894-z
Friberg P, Sundelin B, Bohman SO, Bobik A, Nilsson H, Wickman A, Gustafsson H, Petersen J, Adams MA (1994) Renin-angiotensin system in neonatal rats: induction of a renal abnormality in response to ACE inhibition or angiotensin II antagonism. Kidney Int 45(2):485–492. https://doi.org/10.1038/ki.1994.63
Friis UG, Stubbe J, Uhrenholt TR, Svenningsen P, Nusing RM, Skott O, Jensen BL (2005) Prostaglandin E2 EP2 and EP4 receptor activation mediates cAMP-dependent hyperpolarization and exocytosis of renin in juxtaglomerular cells. Am J Physiol Renal Physiol 289(5):F989–F997. https://doi.org/10.1152/ajprenal.00201.2005
Friis UG, Madsen K, Stubbe J, Hansen PB, Svenningsen P, Bie P, Skott O, Jensen BL (2013) Regulation of renin secretion by renal juxtaglomerular cells. Pflugers Arch 465(1):25–37. https://doi.org/10.1007/s00424-012-1126-7
Gerl K, Steppan D, Fuchs M, Wagner C, Willam C, Kurtz A, Kurt B (2017) Activation of hypoxia signaling in stromal progenitors impairs kidney development. Am J Pathol 187(7):1496–1511. https://doi.org/10.1016/j.ajpath.2017.03.014
Glenn ST, Jones CA, Sexton S, LeVea CM, Caraker SM, Hajduczok G, Gross KW (2014) Conditional deletion of p53 and Rb in the renin-expressing compartment of the pancreas leads to a highly penetrant metastatic pancreatic neuroendocrine carcinoma. Oncogene 33(50):5706–5715. https://doi.org/10.1038/onc.2013.514
Gomez RA, Sequeira-Lopez MLS (2018) Renin cells in homeostasis, regeneration and immune defence mechanisms. Nat Rev Nephrol 14(4):231–245. https://doi.org/10.1038/nrneph.2017.186
Gomez RA, Chevalier RL, Sturgill BC, Johns DW, Peach MJ, Carey RM (1986) Maturation of the intrarenal renin distribution in Wistar-Kyoto rats. J Hypertens 4(Suppl. 5):S31–S33
Gomez RA, Lynch KR, Chevalier RL, Everett AD, Johns DW, Wilfong N, Peach MJ, Carey RM (1988) Renin and angiotensinogen gene expression and intrarenal renin distribution during ACE inhibition. Am J Phys 254(6 Pt 2):F900–F906. https://doi.org/10.1152/ajprenal.1988.254.6.F900
Gomez RA, Chevalier RL, Everett AD, Elwood JP, Peach MJ, Lynch KR, Carey RM (1990) Recruitment of renin gene-expressing cells in adult rat kidneys. Am J Phys 259(4 Pt 2):F660–F665. https://doi.org/10.1152/ajprenal.1990.259.4.F660
Gomez RA, Pupilli C, Everett AD (1991) Molecular and cellular aspects of renin during kidney ontogeny. Pediatr Nephrol 5(1):80–87. https://doi.org/10.1007/bf00852854
Gomez RA, Pentz ES, Jin X, Cordaillat M, Sequeira Lopez ML (2009) CBP and p300 are essential for renin cell identity and morphological integrity of the kidney. Am J Physiol Heart Circ Physiol 296(5):H1255–H1262. https://doi.org/10.1152/ajpheart.01266.2008
Gomez RA, Belyea B, Medrano S, Pentz ES, Sequeira-Lopez ML (2014) Fate and plasticity of renin precursors in development and disease. Pediatr Nephrol 29(4):721–726. https://doi.org/10.1007/s00467-013-2688-0
Gonzalez AA, Liu L, Lara LS, Seth DM, Navar LG, Prieto MC (2011) Angiotensin II stimulates renin in inner medullary collecting duct cells via protein kinase C and independent of epithelial sodium channel and mineralocorticoid receptor activity. Hypertension 57(3):594–599. https://doi.org/10.1161/HYPERTENSIONAHA.110.165902
Graham PC, Kingdom JC, Raweily EA, Gibson AA, Lindop GB (1992) Distribution of renin-containing cells in the developing human kidney: an immunocytochemical study. Br J Obstet Gynaecol 99(9):765–769. https://doi.org/10.1111/j.1471-0528.1992.tb13881.x
Gribouval O, Gonzales M, Neuhaus T, Aziza J, Bieth E, Laurent N, Bouton JM, Feuillet F, Makni S, Ben Amar H, Laube G, Delezoide AL, Bouvier R, Dijoud F, Ollagnon-Roman E, Roume J, Joubert M, Antignac C, Gubler MC (2005) Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet 37(9):964–968. https://doi.org/10.1038/ng1623
Gribouval O, Moriniere V, Pawtowski A, Arrondel C, Sallinen SL, Saloranta C, Clericuzio C, Viot G, Tantau J, Blesson S, Cloarec S, Machet MC, Chitayat D, Thauvin C, Laurent N, Sampson JR, Bernstein JA, Clemenson A, Prieur F, Daniel L, Levy-Mozziconacci A, Lachlan K, Alessandri JL, Cartault F, Riviere JP, Picard N, Baumann C, Delezoide AL, Belar Ortega M, Chassaing N, Labrune P, Yu S, Firth H, Wellesley D, Bitzan M, Alfares A, Braverman N, Krogh L, Tolmie J, Gaspar H, Doray B, Majore S, Bonneau D, Triau S, Loirat C, David A, Bartholdi D, Peleg A, Brackman D, Stone R, DeBerardinis R, Corvol P, Michaud A, Antignac C, Gubler MC (2012) Spectrum of mutations in the renin-angiotensin system genes in autosomal recessive renal tubular dysgenesis. Hum Mutat 33(2):316–326. https://doi.org/10.1002/humu.21661
Grove KL, Mayo RJ, Forsyth CS, Frank AA, Speth RC (1995) Fosinopril treatment of pregnant rats: developmental toxicity, fetal angiotensin-converting enzyme inhibition, and fetal angiotensin II receptor regulation. Toxicol Lett 80(1–3):85–95. https://doi.org/10.1016/0378-4274(95)03346-m
Gubler MC, Antignac C (2010) Renin-angiotensin system in kidney development: renal tubular dysgenesis. Kidney Int 77(5):400–406. https://doi.org/10.1038/ki.2009.423
Hackenthal E, Paul M, Ganten D, Taugner R (1990) Morphology, physiology, and molecular biology of renin secretion. Physiol Rev 70(4):1067–1116. https://doi.org/10.1152/physrev.1990.70.4.1067
Haltia A, Solin ML, Jalanko H, Holmberg C, Miettinen A, Holthofer H (1996) Mechanisms of proteinuria: vascular permeability factor in congenital nephrotic syndrome of the Finnish type. Pediatr Res 40(5):652–657. https://doi.org/10.1203/00006450-199611000-00002
Han DY, Cho JS, Moon YM, Lee HR, Lee HM, Lee BD, Baek BJ (2013) Effect of prostaglandin e2 on vascular endothelial growth factor production in nasal polyp fibroblasts. Allergy Asthma Immunol Res 5(4):224–231. https://doi.org/10.4168/aair.2013.5.4.224
Hanner F, von Maltzahn J, Maxeiner S, Toma I, Sipos A, Kruger O, Willecke K, Peti-Peterdi J (2008) Connexin45 is expressed in the juxtaglomerular apparatus and is involved in the regulation of renin secretion and blood pressure. Am J Physiol Regul Integr Comp Physiol 295(2):R371–R380. https://doi.org/10.1152/ajpregu.00468.2007
Hannon RC, Deruyck RP, Joossens JV, Ameryak AK (1969) Disappearance rate of endogenous renin from the plasma after bilateral nephrectomy in humans. J Clin Endocrinol Metab 29(11):1420–1424. https://doi.org/10.1210/jcem-29-11-1420
Hatini V, Huh SO, Herzlinger D, Soares VC, Lai E (1996) Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev 10(12):1467–1478. https://doi.org/10.1101/gad.10.12.1467
Hibino S, Sasaki H, Abe Y, Hojo A, Uematsu M, Sekine T, Itabashi K (2015) Renal function in angiotensinogen gene-mutated renal tubular dysgenesis with glomerular cysts. Pediatr Nephrol 30(2):357–360. https://doi.org/10.1007/s00467-014-3007-0
Hickmann L, Steglich A, Gerlach M, Al-Mekhlafi M, Sradnick J, Lachmann P, Sequeira-Lopez MLS, Gomez RA, Hohenstein B, Hugo C, Todorov VT (2017) Persistent and inducible neogenesis repopulates progenitor renin lineage cells in the kidney. Kidney Int 92(6):1419–1432. https://doi.org/10.1016/j.kint.2017.04.014
Hilgers KF, Reddi V, Krege JH, Smithies O, Gomez RA (1997) Aberrant renal vascular morphology and renin expression in mutant mice lacking angiotensin-converting enzyme. Hypertension 29(1 Pt 2):216–221. https://doi.org/10.1161/01.hyp.29.1.216
Hohenstein B, Braun A, Amann KU, Johnson RJ, Hugo CP (2008) A murine model of site-specific renal microvascular endothelial injury and thrombotic microangiopathy. Nephrol Dial Transplant 23(4):1144–1156. https://doi.org/10.1093/ndt/gfm774
Hugo C, Hugo C, Pichler R, Gordon K, Schmidt R, Amieva M, Couser WG, Furthmayr H, Johnson RJ (1996) The cytoskeletal linking proteins, moesin and radixin, are upregulated by platelet-derived growth factor, but not basic fibroblast growth factor in experimental mesangial proliferative glomerulonephritis. J Clin Invest 97(11):2499–2508. https://doi.org/10.1172/JCI118697
Hugo C, Shankland SJ, Bowen-Pope DF, Couser WG, Johnson RJ (1997) Extraglomerular origin of the mesangial cell after injury. A new role of the juxtaglomerular apparatus. J Clin Invest 100(4):786–794. https://doi.org/10.1172/JCI119592
Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176(1):85–97. https://doi.org/10.2353/ajpath.2010.090517
Iosipiv IV, Schroeder M (2003) A role for angiotensin II AT1 receptors in ureteric bud cell branching. Am J Physiol Renal Physiol 285(2):F199–F207. https://doi.org/10.1152/ajprenal.00401.2002
Jensen BL, Schmid C, Kurtz A (1996) Prostaglandins stimulate renin secretion and renin mRNA in mouse renal juxtaglomerular cells. Am J Phys 271(3 Pt 2):F659–F669. https://doi.org/10.1152/ajprenal.1996.271.3.F659
Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang AY, Yang CW (2013) Chronic kidney disease: global dimension and perspectives. Lancet 382(9888):260–272. https://doi.org/10.1016/S0140-6736(13)60687-X
Just A, Kurtz L, de Wit C, Wagner C, Kurtz A, Arendshorst WJ (2009) Connexin 40 mediates the tubuloglomerular feedback contribution to renal blood flow autoregulation. J Am Soc Nephrol 20(7):1577–1585. https://doi.org/10.1681/ASN.2008090943
Karger C, Machura K, Schneider A, Hugo C, Todorov VT, Kurtz A (2018) COX-2-derived PGE2 triggers hyperplastic renin expression and hyperreninemia in aldosterone synthase-deficient mice. Pflugers Arch 470(7):1127–1137. https://doi.org/10.1007/s00424-018-2118-z
Katz SA, Opsahl JA, Lunzer MM, Forbis LM, Hirsch AT (1997) Effect of bilateral nephrectomy on active renin, angiotensinogen, and renin glycoforms in plasma and myocardium. Hypertension 30(2 Pt 1):259–266. https://doi.org/10.1161/01.hyp.30.2.259
Kaverina NV, Eng DG, Largent AD, Daehn I, Chang A, Gross KW, Pippin JW, Hohenstein P, Shankland SJ (2017a) WT1 is necessary for the proliferation and migration of cells of renin lineage following kidney podocyte depletion. Stem Cell Rep 9(4):1152–1166. https://doi.org/10.1016/j.stemcr.2017.08.020
Kaverina NV, Kadoya H, Eng DG, Rusiniak ME, Sequeira-Lopez ML, Gomez RA, Pippin JW, Gross KW, Peti-Peterdi J, Shankland SJ (2017b) Tracking the stochastic fate of cells of the renin lineage after podocyte depletion using multicolor reporters and intravital imaging. PLoS One 12(3):e0173891. https://doi.org/10.1371/journal.pone.0173891
Kessel F, Steglich A, Tschongov T, Gembardt F, Ruhnke L, Stumpf J, Behrendt R, Cohrs C, Kopaliani I, Todorov V, Gerlach M, Hugo C (2019) New automatic quantification method of immunofluorescence and histochemistry in whole histological sections. Cell Signal 62:109335. https://doi.org/10.1016/j.cellsig.2019.05.020
Kim HS, Krege JH, Kluckman KD, Hagaman JR, Hodgin JB, Best CF, Jennette JC, Coffman TM, Maeda N, Smithies O (1995) Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci U S A 92(7):2735–2739. https://doi.org/10.1073/pnas.92.7.2735
Kim SM, Chen L, Faulhaber-Walter R, Oppermann M, Huang Y, Mizel D, Briggs JP, Schnermann J (2007) Regulation of renin secretion and expression in mice deficient in beta1- and beta2-adrenergic receptors. Hypertension 50(1):103–109. https://doi.org/10.1161/HYPERTENSIONAHA.107.087577
Kim SM, Briggs JP, Schnermann J (2012) Convergence of major physiological stimuli for renin release on the Gs-alpha/cyclic adenosine monophosphate signaling pathway. Clin Exp Nephrol 16(1):17–24. https://doi.org/10.1007/s10157-011-0494-1
Knott PD, Thorpe SS, Lamont CA (1989) Congenital renal dysgenesis possibly due to captopril. Lancet 1(8635):451. https://doi.org/10.1016/s0140-6736(89)90058-5
Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP (2008) Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3(2):169–181. https://doi.org/10.1016/j.stem.2008.05.020
Krege JH, John SW, Langenbach LL, Hodgin JB, Hagaman JR, Bachman ES, Jennette JC, O’Brien DA, Smithies O (1995) Male-female differences in fertility and blood pressure in ACE-deficient mice. Nature 375(6527):146–148. https://doi.org/10.1038/375146a0
Kress C, Vogels R, De Graaff W, Bonnerot C, Meijlink F, Nicolas JF, Deschamps J (1990) Hox-2.3 upstream sequences mediate lacZ expression in intermediate mesoderm derivatives of transgenic mice. Development 109(4):775–786
Kurt B, Kurtz A (2015) Plasticity of renal endocrine function. Am J Physiol Regul Integr Comp Physiol 308(6):R455–R466. https://doi.org/10.1152/ajpregu.00568.2013
Kurt B, Paliege A, Willam C, Schwarzensteiner I, Schucht K, Neymeyer H, Sequeira-Lopez ML, Bachmann S, Gomez RA, Eckardt KU, Kurtz A (2013) Deletion of von Hippel-Lindau protein converts renin-producing cells into erythropoietin-producing cells. J Am Soc Nephrol 24(3):433–444. https://doi.org/10.1681/ASN.2012080791
Kurt B, Gerl K, Karger C, Schwarzensteiner I, Kurtz A (2015) Chronic hypoxia-inducible transcription factor-2 activation stably transforms juxtaglomerular renin cells into fibroblast-like cells in vivo. J Am Soc Nephrol 26(3):587–596. https://doi.org/10.1681/ASN.2013111152
Kurtz A (2015) Connexins, renin cell displacement and hypertension. Curr Opin Pharmacol 21:1–6. https://doi.org/10.1016/j.coph.2014.11.009
Kurtz A (2017) Endocrine functions of the renal interstitium. Pflugers Arch 469(7–8):869–876. https://doi.org/10.1007/s00424-017-2008-9
Kurtz A (2019) Nobel prize 2019 pays tribute to translational physiology on oxygen sensing. Pflugers Arch 471(11–12):1341–1342. https://doi.org/10.1007/s00424-019-02328-6
Kurtz L, Janssen-Bienhold U, Kurtz A, Wagner C (2009) Connexin expression in renin-producing cells. J Am Soc Nephrol 20(3):506–512. https://doi.org/10.1681/ASN.2008030252
Lacasse J, Ballak M, Mercure C, Gutkowska J, Chapeau C, Foote S, Menard J, Corvol P, Cantin M, Genest J (1985) Immunocytochemical localization of renin in juxtaglomerular cells. J Histochem Cytochem 33(4):323–332. https://doi.org/10.1177/33.4.3884706
Lachmann P, Hickmann L, Steglich A, Al-Mekhlafi M, Gerlach M, Jetschin N, Jahn S, Hamann B, Wnuk M, Madsen K, Djonov V, Chen M, Weinstein LS, Hohenstein B, Hugo CPM, Todorov VT (2017) Interference with gsalpha-coupled receptor signaling in renin-producing cells leads to renal endothelial damage. J Am Soc Nephrol 28(12):3479–3489. https://doi.org/10.1681/ASN.2017020173
Landing BH, Ang SM, Herta N, Larson EF, Turner M (1994) Labeled lectin studies of renal tubular dysgenesis and renal tubular atrophy of postnatal renal ischemia and end-stage kidney disease. Pediatr Pathol 14(1):87–99. https://doi.org/10.3109/15513819409022029
Lee G, Makhanova N, Caron K, Lopez ML, Gomez RA, Smithies O, Kim HS (2005) Homeostatic responses in the adrenal cortex to the absence of aldosterone in mice. Endocrinology 146(6):2650–2656. https://doi.org/10.1210/en.2004-1102
Lichtnekert J, Kaverina NV, Eng DG, Gross KW, Kutz JN, Pippin JW, Shankland SJ (2016) Renin-angiotensin-aldosterone system inhibition increases podocyte derivation from cells of renin lineage. J Am Soc Nephrol 27(12):3611–3627. https://doi.org/10.1681/ASN.2015080877
Lin EE, Sequeira-Lopez ML, Gomez RA (2014) RBP-J in FOXD1+ renal stromal progenitors is crucial for the proper development and assembly of the kidney vasculature and glomerular mesangial cells. Am J Physiol Renal Physiol 306(2):F249–F258. https://doi.org/10.1152/ajprenal.00313.2013
Linkermann A (2019) Death and fire-the concept of necroinflammation. Cell Death Differ 26(1):1–3. https://doi.org/10.1038/s41418-018-0218-0
Liu X, Shi Q, Sigmund CD (2006) Interleukin-1beta attenuates renin gene expression via a mitogen-activated protein kinase kinase-extracellular signal-regulated kinase and signal transducer and activator of transcription 3-dependent mechanism in As4.1 cells. Endocrinology 147(12):6011–6018. https://doi.org/10.1210/en.2006-0129
Liu L, Gonzalez AA, McCormack M, Seth DM, Kobori H, Navar LG, Prieto MC (2011) Increased renin excretion is associated with augmented urinary angiotensin II levels in chronic angiotensin II-infused hypertensive rats. Am J Physiol Renal Physiol 301(6):F1195–F1201. https://doi.org/10.1152/ajprenal.00339.2011
Madsen K, Marcussen N, Pedersen M, Kjaersgaard G, Facemire C, Coffman TM, Jensen BL (2010) Angiotensin II promotes development of the renal microcirculation through AT1 receptors. J Am Soc Nephrol 21(3):448–459. https://doi.org/10.1681/ASN.2009010045
Martinez MF, Medrano S, Brown EA, Tufan T, Shang S, Bertoncello N, Guessoum O, Adli M, Belyea BC, Sequeira-Lopez MLS, Gomez RA (2018) Super-enhancers maintain renin-expressing cell identity and memory to preserve multi-system homeostasis. J Clin Invest 128(11):4787–4803. https://doi.org/10.1172/JCI121361
Martini AG, Danser AHJ (2017) Juxtaglomerular cell phenotypic plasticity. High Blood Press Cardiovasc Prev 24(3):231–242. https://doi.org/10.1007/s40292-017-0212-5
Maxwell PH, Ferguson DJ, Nicholls LG, Iredale JP, Pugh CW, Johnson MH, Ratcliffe PJ (1997) Sites of erythropoietin production. Kidney Int 51(2):393–401. https://doi.org/10.1038/ki.1997.52
McCausland JE, Bertram JF, Ryan GB, Alcorn D (1997) Glomerular number and size following chronic angiotensin II blockade in the postnatal rat. Exp Nephrol 5(3):201–209
Medrano S, Monteagudo MC, Sequeira-Lopez ML, Pentz ES, Gomez RA (2012) Two microRNAs, miR-330 and miR-125b-5p, mark the juxtaglomerular cell and balance its smooth muscle phenotype. Am J Physiol Renal Physiol 302(1):F29–F37. https://doi.org/10.1152/ajprenal.00460.2011
Mendez M (2014) Renin release: role of SNAREs. Am J Physiol Regul Integr Comp Physiol 307(5):R484–R486. https://doi.org/10.1152/ajpregu.00175.2014
Michaud A, Bur D, Gribouval O, Muller L, Iturrioz X, Clemessy M, Gasc JM, Gubler MC, Corvol P (2011) Loss-of-function point mutations associated with renal tubular dysgenesis provide insights about renin function and cellular trafficking. Hum Mol Genet 20(2):301–311. https://doi.org/10.1093/hmg/ddq465
Molteni A, Rahill WJ, Koo JH (1974) Evidence for a vasopressor substance (renin) in human fetal kidneys. Lab Investig 30(2):115–118
Moniwa N, Varagic J, Ahmad S, VonCannon JL, Simington SW, Wang H, Groban L, Brosnihan KB, Nagata S, Kato J, Kitamura K, Gomez RA, Lopez ML, Ferrario CM (2013) Hemodynamic and hormonal changes to dual renin-angiotensin system inhibition in experimental hypertension. Hypertension 61(2):417–424. https://doi.org/10.1161/HYPERTENSIONAHA.112.201889
Mooser V, Nussberger J, Juillerat L, Burnier M, Waeber B, Bidiville J, Pauly N, Brunner HR (1990) Reactive hyperreninemia is a major determinant of plasma angiotensin II during ACE inhibition. J Cardiovasc Pharmacol 15(2):276–282. https://doi.org/10.1097/00005344-199002000-00015
Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L (2007) A global double-fluorescent Cre reporter mouse. Genesis 45(9):593–605. https://doi.org/10.1002/dvg.20335
Naruse K, Murakoshi M, Osamura RY, Naruse M, Toma H, Watanabe K, Demura H, Inagami T, Shizume K (1985) Immunohistological evidence for renin in human endocrine tissues. J Clin Endocrinol Metab 61(1):172–177. https://doi.org/10.1210/jcem-61-1-172
Nelson RD, Stricklett P, Gustafson C, Stevens A, Ausiello D, Brown D, Kohan DE (1998) Expression of an AQP2 Cre recombinase transgene in kidney and male reproductive system of transgenic mice. Am J Phys 275(1):C216–C226. https://doi.org/10.1152/ajpcell.1998.275.1.C216
Neubauer B, Machura K, Chen M, Weinstein LS, Oppermann M, Sequeira-Lopez ML, Gomez RA, Schnermann J, Castrop H, Kurtz A, Wagner C (2009) Development of vascular renin expression in the kidney critically depends on the cyclic AMP pathway. Am J Physiol Renal Physiol 296(5):F1006–F1012. https://doi.org/10.1152/ajprenal.90448.2008
Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD (2002) Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109(11):1417–1427. https://doi.org/10.1172/JCI14276
Nguyen G, Blanchard A, Curis E, Bergerot D, Chambon Y, Hirose T, Caumont-Prim A, Tabard SB, Baron S, Frank M, Totsune K, Azizi M (2014) Plasma soluble (pro)renin receptor is independent of plasma renin, prorenin, and aldosterone concentrations but is affected by ethnicity. Hypertension 63(2):297–302. https://doi.org/10.1161/HYPERTENSIONAHA.113.02217
Niimura F, Labosky PA, Kakuchi J, Okubo S, Yoshida H, Oikawa T, Ichiki T, Naftilan AJ, Fogo A, Inagami T et al (1995) Gene targeting in mice reveals a requirement for angiotensin in the development and maintenance of kidney morphology and growth factor regulation. J Clin Invest 96(6):2947–2954. https://doi.org/10.1172/JCI118366
Nussberger J, Gradman AH, Schmieder RE, Lins RL, Chiang Y, Prescott MF (2007) Plasma renin and the antihypertensive effect of the orally active renin inhibitor aliskiren in clinical hypertension. Int J Clin Pract 61(9):1461–1468. https://doi.org/10.1111/j.1742-1241.2007.01473.x
Oka M, Medrano S, Sequeira-Lomicronpez MLS, Gomez RA (2017) Chronic stimulation of renin cells leads to vascular pathology. Hypertension 70(1):119–128. https://doi.org/10.1161/HYPERTENSIONAHA.117.09283
Oliverio MI, Kim HS, Ito M, Le T, Audoly L, Best CF, Hiller S, Kluckman K, Maeda N, Smithies O, Coffman TM (1998) Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci U S A 95(26):15496–15501. https://doi.org/10.1073/pnas.95.26.15496
Owen RA, Molon-Noblot S, Hubert MF, Kindt MV, Keenan KP, Eydelloth RS (1994) The morphology of juxtaglomerular cell hyperplasia and hypertrophy in normotensive rats and monkeys given an angiotensin II receptor antagonist. Toxicol Pathol 22(6):606–619. https://doi.org/10.1177/019262339402200605
Paul M, Poyan Mehr A, Kreutz R (2006) Physiology of local renin-angiotensin systems. Physiol Rev 86(3):747–803. https://doi.org/10.1152/physrev.00036.2005
Pentz ES, Moyano MA, Thornhill BA, Sequeira Lopez ML, Gomez RA (2004) Ablation of renin-expressing juxtaglomerular cells results in a distinct kidney phenotype. Am J Physiol Regul Integr Comp Physiol 286(3):R474–R483. https://doi.org/10.1152/ajpregu.00426.2003
Petrovic N, Kane CM, Sigmund CD, Gross KW (1997) Downregulation of renin gene expression by interleukin-1. Hypertension 30(2 Pt 1):230–235. https://doi.org/10.1161/01.hyp.30.2.230
Pierrou S, Hellqvist M, Samuelsson L, Enerback S, Carlsson P (1994) Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J 13(20):5002–5012
Pippin JW, Kaverina NV, Eng DG, Krofft RD, Glenn ST, Duffield JS, Gross KW, Shankland SJ (2015) Cells of renin lineage are adult pluripotent progenitors in experimental glomerular disease. Am J Physiol Renal Physiol 309(4):F341–F358. https://doi.org/10.1152/ajprenal.00438.2014
Polifka JE (2012) Is there an embryopathy associated with first-trimester exposure to angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists? A critical review of the evidence. Birth Defects Res A Clin Mol Teratol 94(8):576–598. https://doi.org/10.1002/bdra.23027
Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, Ozawa Y, Hering-Smith KS, Hamm LL, Navar LG (2004) Enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Hypertension 44(2):223–229. https://doi.org/10.1161/01.HYP.0000135678.20725.54
Prieto-Carrasquero MC, Kobori H, Ozawa Y, Gutierrez A, Seth D, Navar LG (2005) AT1 receptor-mediated enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Am J Physiol Renal Physiol 289(3):F632–F637. https://doi.org/10.1152/ajprenal.00462.2004
Prieto-Carrasquero MC, Botros FT, Kobori H, Navar LG (2009) Collecting duct renin: a major player in angiotensin II-dependent hypertension. J Am Soc Hypertens 3(2):96–104. https://doi.org/10.1016/j.jash.2008.11.003
Pugh D, Gallacher PJ, Dhaun N (2019) Management of hypertension in chronic kidney disease. Drugs 79(4):365–379. https://doi.org/10.1007/s40265-019-1064-1
Ramkumar N, Stuart D, Rees S, Hoek AV, Sigmund CD, Kohan DE (2014) Collecting duct-specific knockout of renin attenuates angiotensin II-induced hypertension. Am J Physiol Renal Physiol 307(8):F931–F938. https://doi.org/10.1152/ajprenal.00367.2014
Rasch R, Jensen BL, Nyengaard JR, Skott O (1998) Quantitative changes in rat renin secretory granules after acute and chronic stimulation of the renin system. Cell Tissue Res 292(3):563–571. https://doi.org/10.1007/s004410051085
Robillard JE, Nakamura KT (1988) Neurohormonal regulation of renal function during development. Am J Phys 254(6 Pt 2):F771–F779. https://doi.org/10.1152/ajprenal.1988.254.6.F771
Rosendahl A, Niemann G, Lange S, Ahadzadeh E, Krebs C, Contrepas A, van Goor H, Wiech T, Bader M, Schwake M, Peters J, Stahl R, Nguyen G, Wenzel UO (2014) Increased expression of (pro)renin receptor does not cause hypertension or cardiac and renal fibrosis in mice. Lab Investig 94(8):863–872. https://doi.org/10.1038/labinvest.2014.83
Ruhnke L, Sradnick J, Al-Mekhlafi M, Gerlach M, Gembardt F, Hohenstein B, Todorov VT, Hugo C (2018) Progenitor Renin Lineage Cells are not involved in the regeneration of glomerular endothelial cells during experimental renal thrombotic microangiopathy. PLoS One 13(5):e0196752. https://doi.org/10.1371/journal.pone.0196752
Santos RAS, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, Campagnole-Santos MJ (2018) The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1-7). Physiol Rev 98(1):505–553. https://doi.org/10.1152/physrev.00023.2016
Sarhan M, Land WG, Tonnus W, Hugo CP, Linkermann A (2018) Origin and consequences of necroinflammation. Physiol Rev 98(2):727–780. https://doi.org/10.1152/physrev.00041.2016
Sauter A, Machura K, Neubauer B, Kurtz A, Wagner C (2008) Development of renin expression in the mouse kidney. Kidney Int 73(1):43–51. https://doi.org/10.1038/sj.ki.5002571
Schonig K, Schwenk F, Rajewsky K, Bujard H (2002) Stringent doxycycline dependent control of CRE recombinase in vivo. Nucleic Acids Res 30(23):e134. https://doi.org/10.1093/nar/gnf134
Schweda F, Kurtz A (2011) Regulation of renin release by local and systemic factors. Rev Physiol Biochem Pharmacol 161:1–44. https://doi.org/10.1007/112_2008_1
Schweda F, Klar J, Narumiya S, Nusing RM, Kurtz A (2004) Stimulation of renin release by prostaglandin E2 is mediated by EP2 and EP4 receptors in mouse kidneys. Am J Physiol Renal Physiol 287(3):F427–F433. https://doi.org/10.1152/ajprenal.00072.2004
Seaberg RM, van der Kooy D (2003) Stem and progenitor cells: the premature desertion of rigorous definitions. Trends Neurosci 26(3):125–131. https://doi.org/10.1016/S0166-2236(03)00031-6
Sequeira Lopez ML, Gomez RA (2011) Development of the renal arterioles. J Am Soc Nephrol 22(12):2156–2165. https://doi.org/10.1681/ASN.2011080818
Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA (2004) Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell 6(5):719–728. https://doi.org/10.1016/s1534-5807(04)00134-0
Sequeira-Lopez ML, Weatherford ET, Borges GR, Monteagudo MC, Pentz ES, Harfe BD, Carretero O, Sigmund CD, Gomez RA (2010) The microRNA-processing enzyme dicer maintains juxtaglomerular cells. J Am Soc Nephrol 21(3):460–467. https://doi.org/10.1681/ASN.2009090964
Sequeira-Lopez ML, Lin EE, Li M, Hu Y, Sigmund CD, Gomez RA (2015a) The earliest metanephric arteriolar progenitors and their role in kidney vascular development. Am J Physiol Regul Integr Comp Physiol 308(2):R138–R149. https://doi.org/10.1152/ajpregu.00428.2014
Sequeira-Lopez ML, Nagalakshmi VK, Li M, Sigmund CD, Gomez RA (2015b) Vascular versus tubular renin: role in kidney development. Am J Physiol Regul Integr Comp Physiol 309(6):R650–R657. https://doi.org/10.1152/ajpregu.00313.2015
Shinohara K, Liu X, Morgan DA, Davis DR, Sequeira-Lopez ML, Cassell MD, Grobe JL, Rahmouni K, Sigmund CD (2016) Selective deletion of the brain-specific isoform of renin causes neurogenic hypertension. Hypertension 68(6):1385–1392. https://doi.org/10.1161/HYPERTENSIONAHA.116.08242
Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, Quaggin SE (2010) Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol 21(10):1691–1701. https://doi.org/10.1681/ASN.2010030295
Song K, Stuart D, Abraham N, Wang F, Wang S, Yang T, Sigmund CD, Kohan DE, Ramkumar N (2016) Collecting duct renin does not mediate DOCA-salt hypertension or renal injury. PLoS One 11(7):e0159872. https://doi.org/10.1371/journal.pone.0159872
Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM (2014) Classical renin-angiotensin system in kidney physiology. Compr Physiol 4(3):1201–1228. https://doi.org/10.1002/cphy.c130040
Spiegel AM, Shenker A, Weinstein LS (1992) Receptor-effector coupling by G proteins: implications for normal and abnormal signal transduction. Endocr Rev 13(3):536–565. https://doi.org/10.1210/edrv-13-3-536
Sradnick J, Rong S, Luedemann A, Parmentier SP, Bartaun C, Todorov VT, Gueler F, Hugo CP, Hohenstein B (2016) Extrarenal progenitor cells do not contribute to renal endothelial repair. J Am Soc Nephrol 27(6):1714–1726. https://doi.org/10.1681/ASN.2015030321
Srinivas S, Goldberg MR, Watanabe T, D’Agati V, al-Awqati Q, Costantini F (1999) Expression of green fluorescent protein in the ureteric bud of transgenic mice: a new tool for the analysis of ureteric bud morphogenesis. Dev Genet 24(3–4):241–251. https://doi.org/10.1002/(SICI)1520-6408(1999)24:3/4<241::AID-DVG7>3.0.CO;2-R
Starke C, Betz H, Hickmann L, Lachmann P, Neubauer B, Kopp JB, Sequeira-Lopez ML, Gomez RA, Hohenstein B, Todorov VT, Hugo CP (2015) Renin lineage cells repopulate the glomerular mesangium after injury. J Am Soc Nephrol 26(1):48–54. https://doi.org/10.1681/ASN.2014030265
Stefanska A, Peault B, Mullins JJ (2013) Renal pericytes: multifunctional cells of the kidneys. Pflugers Arch 465(6):767–773. https://doi.org/10.1007/s00424-013-1263-7
Steglich A, Kessel F, Hickmann L, Gerlach M, Lachmann P, Gembardt F, Lesche M, Dahl A, Federlein A, Schweda F, Hugo CPM, Todorov VT (2019) Renin cells with defective Gsalpha/cAMP signaling contribute to renal endothelial damage. Pflugers Arch 471(9):1205–1217. https://doi.org/10.1007/s00424-019-02298-9
Takahashi N, Lopez ML, Cowhig JE Jr, Taylor MA, Hatada T, Riggs E, Lee G, Gomez RA, Kim HS, Smithies O (2005) Ren1c homozygous null mice are hypotensive and polyuric, but heterozygotes are indistinguishable from wild-type. J Am Soc Nephrol 16(1):125–132. https://doi.org/10.1681/ASN.2004060490
Tang J, Wysocki J, Ye M, Valles PG, Rein J, Shirazi M, Bader M, Gomez RA, Sequeira-Lopez MS, Afkarian M, Batlle D (2019) Urinary renin in patients and mice with diabetic kidney disease. Hypertension 74(1):83–94. https://doi.org/10.1161/HYPERTENSIONAHA.119.12873
Taugner R, Buhrle CP, Nobiling R (1984) Ultrastructural changes associated with renin secretion from the juxtaglomerular apparatus of mice. Cell Tissue Res 237(3):459–472. https://doi.org/10.1007/bf00228430
Taugner R, Whalley A, Angermuller S, Buhrle CP, Hackenthal E (1985) Are the renin-containing granules of juxtaglomerular epithelioid cells modified lysosomes? Cell Tissue Res 239(3):575–587. https://doi.org/10.1007/bf00219236
Todorov V, Muller M, Schweda F, Kurtz A (2002) Tumor necrosis factor-alpha inhibits renin gene expression. Am J Physiol Regul Integr Comp Physiol 283(5):R1046–R1051. https://doi.org/10.1152/ajpregu.00142.2002
Todorov VT, Volkl S, Muller M, Bohla A, Klar J, Kunz-Schughart LA, Hehlgans T, Kurtz A (2004) Tumor necrosis factor-alpha activates NFkappaB to inhibit renin transcription by targeting cAMP-responsive element. J Biol Chem 279(2):1458–1467. https://doi.org/10.1074/jbc.M308697200
Todorov VT, Volkl S, Friedrich J, Kunz-Schughart LA, Hehlgans T, Vermeulen L, Haegeman G, Schmitz ML, Kurtz A (2005) Role of CREB1 and NF{kappa}B-p65 in the down-regulation of renin gene expression by tumor necrosis factor {alpha}. J Biol Chem 280(26):24356–24362. https://doi.org/10.1074/jbc.M502968200
Tufro-McReddie A, Romano LM, Harris JM, Ferder L, Gomez RA (1995) Angiotensin II regulates nephrogenesis and renal vascular development. Am J Phys 269(1 Pt 2):F110–F115. https://doi.org/10.1152/ajprenal.1995.269.1.F110
Urlinger S, Baron U, Thellmann M, Hasan MT, Bujard H, Hillen W (2000) Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc Natl Acad Sci U S A 97(14):7963–7968. https://doi.org/10.1073/pnas.130192197
Wagner C, Kurtz A (2013) Distribution and functional relevance of connexins in renin-producing cells. Pflugers Arch 465(1):71–77. https://doi.org/10.1007/s00424-012-1134-7
Wagner C, de Wit C, Kurtz L, Grunberger C, Kurtz A, Schweda F (2007) Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ Res 100(4):556–563. https://doi.org/10.1161/01.RES.0000258856.19922.45
Wang Y, Eng DG, Pippin JW, Gharib SA, McClelland A, Gross KW, Shankland SJ (2018) Sex differences in transcriptomic profiles in aged kidney cells of renin lineage. Aging 10(4):606–621. https://doi.org/10.18632/aging.101416
Weinstein LS, Yu S, Warner DR, Liu J (2001) Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev 22(5):675–705. https://doi.org/10.1210/edrv.22.5.0439
Xiao X, Chen Z, Shiota C, Prasadan K, Guo P, El-Gohary Y, Paredes J, Welsh C, Wiersch J, Gittes GK (2013) No evidence for beta cell neogenesis in murine adult pancreas. J Clin Invest 123(5):2207–2217. https://doi.org/10.1172/JCI66323
Yanai K, Saito T, Kakinuma Y, Kon Y, Hirota K, Taniguchi-Yanai K, Nishijo N, Shigematsu Y, Horiguchi H, Kasuya Y, Sugiyama F, Yagami K, Murakami K, Fukamizu A (2000) Renin-dependent cardiovascular functions and renin-independent blood-brain barrier functions revealed by renin-deficient mice. J Biol Chem 275(1):5–8. https://doi.org/10.1074/jbc.275.1.5
Yo Y, Braun MC, Barisoni L, Mobaraki H, Lu H, Shrivastav S, Owens J, Kopp JB (2003) Anti-mouse mesangial cell serum induces acute glomerulonephropathy in mice. Nephron Exp Nephrol 93(3):e92–e106. https://doi.org/10.1159/000069551
Zivna M, Hulkova H, Matignon M, Hodanova K, Vylet’al P, Kalbacova M, Baresova V, Sikora J, Blazkova H, Zivny J, Ivanek R, Stranecky V, Sovova J, Claes K, Lerut E, Fryns JP, Hart PS, Hart TC, Adams JN, Pawtowski A, Clemessy M, Gasc JM, Gubler MC, Antignac C, Elleder M, Kapp K, Grimbert P, Bleyer AJ, Kmoch S (2009) Dominant renin gene mutations associated with early-onset hyperuricemia, anemia, and chronic kidney failure. Am J Hum Genet 85(2):204–213. https://doi.org/10.1016/j.ajhg.2009.07.010
Acknowledgments
We are greatly indebted to all present and past members of the Experimental Nephrology for their devoted scientific work.
We would also like to thank Armin Kurtz from the University of Regensburg and Maria Luisa Sequeira Lopez and R. Ariel Gomez from the University of Virginia for the inspiring discussions.
The authors’ work is continuously supported by Deutsche Forschungsgemeinschaft (DFG) Projects SFB 699/B1, TO 679/1-1, TO 679/2-1, TO 679/3-1 (to V.T.T.), HU 600/6-1, HU 600/8-1, HU 600/11-1, HU 600/12-1, HU 600/13-1 (to C.H.). A.L. is supported by the DFG Heisenberg-Professorship Programme (Project number 324141047). A. S. received grant from the Graduate Academy of the Technical University, Dresden.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Steglich, A., Hickmann, L., Linkermann, A., Bornstein, S., Hugo, C., Todorov, V.T. (2020). Beyond the Paradigm: Novel Functions of Renin-Producing Cells. In: Pedersen, S.H.F. (eds) Reviews of Physiology, Biochemistry and Pharmacology . Reviews of Physiology, Biochemistry and Pharmacology, vol 177. Springer, Cham. https://doi.org/10.1007/112_2020_27
Download citation
DOI: https://doi.org/10.1007/112_2020_27
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-61494-2
Online ISBN: 978-3-030-61495-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)