
Abstract
A procedure is developed for the simultaneous highly sensitive determination of eight chlorophenols in sea water based on a combination of isotope dilution, solid-phase extraction on an octadecyl adsorbent, acetylation, and determination by gas chromatography–tandem mass spectrometry with electron ionization. The attained limits of detection for the analytes are in the range 0.06−0.26 ng/L, which is 2−3 orders of magnitude lower than the standards established for the maximum permissible concentration. The duration of analysis is 15 min. The application of a deuterated internal standard ensures the high accuracy and reproducibility of the results obtained. The developed procedure was successfully used for the analysis of real water samples from the Barents Sea; the found total chlorophenol concentration in them was 15–24 ng/L. The main pollutant of the studied sea water samples is 2,4-dichlorophenol, which comprised about 30% of the total concentration of chlorophenols.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Chlorophenols belong to priority environmental pollutants of human origin because of their high toxicity, trend to accumulate in fat tissue and various organs and, as a result, in food chains. The sources of chlorophenols are enterprises of organic synthesis, pulp and paper industry (chlorine bleaching of cellulose), water purification and water disposal stations (disinfection of tap water and sewages by chlorine and chlorine-containing reagents), and the production and application of antiseptic compositions and plant protection agents [1, 2]. The real danger of the ingress of chlorophenols to the environment is substantially determined by their ability to dimerize under certain conditions with the formation of superecotoxicants, polychlorinated dioxins and dibenzofurans [3–5], possessing high carcinogenic activity. The toxicity of chlorophenols considerably increases with an increase in the number of chlorine atoms in the molecule. The World Health Organization standardized four compounds of this class (2-chlorophenol, 2,4-dichlorophenol, 2,4,6-trichlorophenol, and pentachlorophenol) of the 19 existing ones. The maximum permissible concentration of the first three compounds was established at the level 0.1 μg/L for drinking waters [6–8]. For pentachlorophenol, in view of its high carcinogenic activity, the value of the maximum permissible concentration was reduced to 0.03 μg/L [9].
Because of the growing interest to the study of environmental conditions of ecosystems of Arctic seas, extremely sensitive to the ingress of xenobiotics, problems of the determination of organochlorine compounds in sea water at lower concentration levels have gained special importance. A specific feature of sea water as a matrix with a high concentration of salts and various organic impurities and also the necessity of work with ultratrace amounts of ecotoxicants impose very strict requirements on the methods for the determination of chlorophenols and stimulate the development of new approaches to preconcentration, derivatization, and instrumental aspects of analysis. In the present-day analytical practice, such problems are solved by methods of liquid and gas chromatography (Table 1). An advantage of HPLC is a possibility of the direct determination of chlorophenols; however, gas-chromatographic methods have not lost their importance because of a possibility of the separation of a great number of compounds in the analysis of complex matrixes and the weaker effect of matrix components on the result of analysis. Two types of detectors, ensuring the high sensitivity of the method, i.e., mass spectrometry [10, 11, 14, 15] and electron-capture [12, 13, 16], are used in combination with the gas-chromatographic separation of chlorophenols.
The preliminary extraction and preconcentration of analytes are performed by various methods of extraction, including liquid−liquid [14], solid-phase (SPE) [10, 11, 13] and solid-phase microextraction [12, 17]. To increase the volatility of chlorophenols, suppress undesirable reactions, and improve separation, analysts use their acetylation by acetic anhydride [10, 15–17].
It should be noted that the application of electron-capture detection ensures the high sensitivity of analysis mainly for compounds with a great number of halogen atoms in the structure, whereas for mono- and dichlorophenols, mass spectrometry demonstrates a considerable gain in limits of detection (LOD). Nevertheless, even using mass spectrometry detection, the values of the limits of detection for chlorophenols at the level of several nanograms per liter were attained only in some works [11, 15]. The further increase of the sensitivity of analysis is possible through an increase in the rate of sample preconcentration and an increase in the selectivity of detection. In our opinion, this problem can be successfully solved through the application of the method of gas chromatography–tandem mass spectrometry (GC−MS/MS) combined with the isotope dilution of samples.
The aim of this work was the development of a procedure for the simultaneous determination of chlorophenols in aqueous solutions, including preconcentration by SPE, preparation of volatile esters by acetylation, and the determination of derivatives by GC−MS/MS, and also the application of the developed procedure to the analysis of sea water.
EXPERIMENTAL
Test samples. The samples of surface sea water were taken in the Barents Sea along the coast of the Novaya Zemlya archipelago during sea studies on the research vessel Professor Molchanov in June, 2015 within the joint project of Rosgidromet and the Northern (Arctic) Federal University “Arctic Floating University”. The samples were collected to 1-L glass vessels, preserved with a sodium thiosulphate additive [18] to the concentration 80 mg/L and, stored at 4°C before extraction.
Reagents and materials. The analytes were eight commercially available compounds: 2-chlorophenol, 3-chlorophenol, 4-chlorophenol, 2,4-dichlorophenol, 2,6-dichlorophenol, 2,4,5-trichlorophenol, 2,4,6-trichlorophenol, and pentachlorophenol. The solutions were prepared from preparations of purity > 99% purchased from Merck (Germany). 2-Chlorophenol-3,4,5,6-d4, 98 at. % D (Aldrich, Germany) was used as an isotopically labeled internal standard.
Samples were prepared using methanol, dichloromethane, acetone of HPLC grade (Merck, Germany), grade 0 hexane (Kriokhrom, Russia), formic acid “ACS Reagent, puriss. p. a.” (Sigma-Aldrich, United States), sodium hydroxide of high-purity grade (Nevareaktiv, Russia), and high-purity water obtained using a Milli-Q system (Millipore, France).
Model solutions of chlorophenol in sea water were prepared using a sample of Barents Sea water free from chlorophenols (concentrations were lower than the limits of detection). To obtain it, a water sample containing the minimum amount of analytes was additionally purified by solid-phase extraction and boiling.
Preparation of solutions. Stock solutions of chlorophenol and of the internal standard in acetone with the concentration 10 mg/mL were prepared by dissolving precisely weighed portions and stored at –18°C for no more than one month. By mixing stock solutions of analytes and consecutively diluting them with acetone, we prepared working solutions of a mixture of chlorophenols with the concentration of each component 0.01 mg/mL. The solution obtained was stored at 4°C for no more than one week.
Model and calibration solutions of analytes were prepared immediately before the experiment by diluting the working solution with a 0.1 M solution of NaOH to the required concentration.
Solid-phase extraction. Samples of sea water were extracted with a vacuum system on Strata C18 cartridges (Phenomenex, United States) of the volume 6 mL, which contained 500 mg of a reversed-phase adsorbent. The adsorbent was prepared according to the procedure [19]: washed with 10 mL of dichloromethane and dried, after which 10 mL of methanol and 10 mL of a 0.5% aqueous solution of HCOOH were consecutively passed through it. A 25-μL portion of the internal standard solution was added to 100 mL of filtered sea water; the mixture was acidified to pH 2 by adding 1 mL of formic acid and then passed through a cartridge at a rate of about 5 mL/min. Then the cartridge was washed with 10 mL of a 1% aqueous solution of HCOOH, dried with a nitrogen flow within 30 min, and the adsorbed analytes were eluted with 10 mL of dichloromethane. The extract was evaporated at 60°C in a nitrogen flow to the volume 1 mL and back extracted with 0.5 mL of a 0.1 M NaOH solution. The extract obtained was acetylated and analyzed by chromatography−mass spectrometry.
Preparation of derivatives. Acetylation was performed similar to the procedure [20] adapted for use of microvolumes of reagents. A 250-μL portion of hexane and 25 μL of each of acetic anhydride and methanol (for the better layering of the solution) were added to 500 μL of an alkaline extract of sea water or model solutions in a microcentrifuge test tube. In the case of model solutions, a solution of the internal standard was added before the introduction of the reagents. The mixture was stirred on a shaker at room temperature within 20 min. After that, 20 μL of HCOOH (for the precipitation of organic acids from the sample matrix) was added and the mixture was stirred and centrifuged for 5 min. The upper hexane layer was collected with a pipetter, transferred to conical glass inserts for chromatographic vials of the volume 200 μL and evaporated in a nitrogen flow at 60°C to a volume of about 25 μL, and then analyzed by chromatography−mass spectrometry.
Determination by chromatography−mass spectrometry was performed in the mode of selected reaction monitoring (SRM) using an Agilent 7890/7000B GC-MS/MS system (Agilent, United States), which consisted of an Agilent 7890A gas chromatograph equipped with an Agilent 7693A autosampler and an Agilent 7000B tandem mass spectrometry detector with triple quadrupole. Separation was performed on an HP-5ms capillary column (Agilent, United States), 30 m × 0.25 mm, thickness of stationary phase layer 0.25 μm. We used the following parameters of work of the chromatograph−mass spectrometer: carrier gas helium (brand 6.0), pressure control of gas flow rate (flow rate through the column 1 mL/min), electron ionization (70 eV), temperature of the interface and ion source 230C°, voltage on the detector 1.2 kV (automatic adjustment). Nitrogen was used as a collision gas and helium as a buffer gas in the collision cell. As was found in preliminary experiments, the change in the flow rate of buffer gas in the range 0.5−2 mL/min had no significant effect on the sensitivity of analyte determination; in this regard, this parameter was set at the level 1 mL/min, recommended by the manufacturer. The variation of the flow rate of collision gas within the same limits showed that, for all of the chosen analytes, the maximum signal intensity in the SRM mode was also reached at the value 1 mL/min, which was used in the further experiments. We used a time program of the registration of ion transitions; for all of the studied compounds, the time of detection of each selected reaction was 30 ms. The control of the chromatograph−mass spectrometer and data collection and processing were performed using the MassHunter software (Agilent, United States).
RESULTS AND DISCUSSION
Mass spectrometry detection. The electron ionization of chlorophenol derivatives was accompanied by easy deacetylation, which resulted in the prevalence of signals of molecular ions of corresponding chlorophenols in the mass spectra. In this regard, they were chosen as precursor ions for mass spectrometry detection in the SRM mode. Because of the isotope distribution of chlorine, the maximum intensity for pentachlorophenol is typical for the isotope peak of the ion \({{[{{{\text{C}}}_{6}}{{{\text{H}}}^{{35}}}{\text{Cl}}_{4}^{{37}}{\text{ClO]}}}^{{\centerdot + }}},\) which we chose as the precursor instead of the monoisotopic ion. The study of tandem mass spectra on varying collision energy in the range from 10 to 50 eV with a step of no more than 3 eV allowed us to choose an analytical (with the most intense signal) and confirming ion transitions for each analyte and to determine the optimum values of collision energy for them (Table 2). As an additional criterion of the reliability of analyte identification in the analysis of real samples, we determined the intensity ratio of the analytical and confirming ion transitions, the admissible deviation for which from the established value was accepted at a level of 20%.
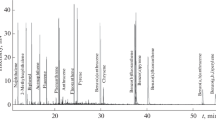
Chromatographic separation. The critical pair of analytes for the optimization of chromatographic separation was comprised by 3-chlorophenol and 4-chlorophenol, which were characterized by very close retention times on a nonpolar stationary phase. At the initial temperature of the thermostat 100°C, chosen based on the necessity of the elimination of the effect of solvent on the separation of low-boiling components, the almost complete separation of the acetyl derivatives of two chlorophenols was attained at the rate of temperature increase no more than 10 K/min. To shorten the duration of the analysis, after the elution of peaks of 3-and 4-chlorophenol, temperature gradient was increased to 40 K/min. The use of an optimized temperature program with a two-step gradient (storage at 100°C for 2 min, raise to 150°C at a rate of 10 of K/min, raise to 250°C at a rate of 40 of K/min, storage at 250°C for 3 min) allowed us to almost completely separate the target compounds (for 3-chlorophenol and 4-chlorophenol Rs = 1.51) and to provide an acceptable duration of the separation, 12.5 min. (Fig. 1). Taking into account the necessity of cleaning the chromatographic column after the determination at 300°C within 2.5 min, the total duration of an analytical cycle was 15 min.

A chromatogram of a standard solution containing 5 mg/L of each of (1) 2-chlorophenol, (2) 3-chlorophenol, (3) 4-chlorophenol, (4) 2,6-dichlorophenol, (5) 2,4-dichlorophenol, (6) 2,4,6-trichlorophenol, (7) 2,4,5-trichlorophenol, and (8) pentachlorophenol in the selected reaction monitoring mode.
It was found that the best separation of the critical pair of analytes at the maintaining of the maximum sensitivity was reached at flow splitting of 1 : 10 and the volume of the injected sample of 2 μL. Taking into account the low concentration of analytes in the real samples, and also a necessity of the study of a matrix with the high concentration of salts and impurities of organic substances, an important role in obtaining reliable results and the achievement of high sensitivity is played by the proper choice of a liner. The highest signal-to-noise ratio at the maintaining of the ability to capture nonvolatile sample components is provided by liner with a Jennings cup, deactivated and without glass wool.
Quantitative analysis. To attain the maximum sensitivity of analysis, one should ensure the high recoveries of analytes in SPE from the matrix of sea water. A comparison of the chromatographic peak areas of analytes for model solutions with the concentration 2000 ng/L (without preconcentration) and for sea water with the concentration of chlorophenols 10 ng/L after 200-fold SPE-preconcentration followed by acetylation showed that the used procedure ensures the recoveries of analytes in the range 70–100%.
Based on the analysis of a series of model solutions with different concentrations of analytes and of the deuterated standard, it was found that the dependences of the ratio of chromatographic peak areas of chlorophenol and the internal standard (Si/Sst) on the corresponding ratio of their concentrations (ci/cst) were linear in the wide range of concentrations, covering not less than 3 orders of magnitude, and was described by the equation y = ax with the coefficients of correlation (R2) higher than 0.99 for the majority of analytes (Table 3). It should be noted that, for 2-chlorophenol, coefficient a was not equal to unity, as it should be expected based on the identity of the chemical properties of this analyte and its deuterated analog used as an internal standard. This fact can be explained by the difference in the efficiency of electron ionization and collision-activated dissociation of these compounds because of the difference in energies of C–H and C–D bonds [21].
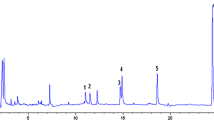
The values of the limits of detection, LOD and lower limits of quantitation, LOQ were determined by extrapolating linear dependences to the achievement of the signal-to-noise ratio to 3 and 10, respectively. The obtained values were confirmed and refined by analyzing a model solution of chlorophenol in sea water with the concentrations of analytes close to the LOQ value (Fig. 2). The attained values of the limits of detection were almost 10 times lower in comparison to those in HPLC with preliminary solid-phase extraction [11] and 2–3 orders of magnitude lower than in the majority of the known gas-chromatographic procedures (Table 1).

A chromatogram of a model solution of sea water with 1 ng/L additives of (1) 2-chlorophenol, (3) 4-chlorophenol, (5) 2,4-dichlorophenol and 0.5 ng/L additives of (2) 3-chlorophenol, (4) 2,6-dichlorophenol, (6) 2,4,6-trichlorophenol, (7) 2,4,5-trichlorophenol, and (8) pentachlorophenol.
The results of testing the developed procedure by the spike-recovery test (Table 4) indicate that the level of accuracy of the results of analysis is high because of the use of the internal standard. The values of this parameter for the studied analytes lie in the range 85–107%, even for the very low concentration about 10 ng/L. The maximum random error of the results of analysis was observed for pentachlorophenol and was as high as 16% (n = 9, P = 0.95), which may be considered acceptable in the determination of trace concentrations, significantly lower than the established standards.
Analysis of sea water. The developed procedure was successfully tested in the analysis of real samples of sea water, which were selected during the sea studies near the Novaya Zemlya archipelago from the continental coast to its northernmost tip (Cape Zhelaniya). The results of analysis (Table 5) indicate that the attained sensitivity level is sufficient for the determination of the majority of chlorophenols in sea waters with the low level of man-made pollution. The found concentrations of analytes lie in the range up to 10 ng/L and cannot be reliably determined using the majority of the earlier described procedures. The total concentrations of chlorophenols in all of the studied water samples differ slightly and lie in the range 15–24 ng/L, which is, probably, because of the fare distance of sampling regions from the sources of pollution and a good stirring of sea waters. The main pollutant in the studied group of compounds is 2,4-dichlorophenol; it comprises about 30% of the total amount of chlorophenols. The concentrations of 2,4,5-trichlorophenol and pentachlorophenol in all samples do not exceed the LOQ values; nevertheless, the first compound was found in the majority of samples and the second, in the water sample selected at the northernmost tip of the Novaya Zemlya archipelago.
REFERENCES
Lur’e, Yu.Yu., Analiticheskaya khimiya promyshlennykh stochnykh vod (Analytical Chemistry of Industrial Wastewater), Moscow: Khimiya, 1984.
Grushko, Ya.M., Vrednye organicheskie soedineniya v promyshlennykh stochnykh vodakh: Spravochnik (Harmful Organic Compounds in Industrial Wastewater: Handbook), Leningrad: Khimiya, 1982.
Maistrenko, V.N. and Klyuev, N.A., Ekologo-analiticheskii monitoring stoikikh organicheskikh zagryaznitelei (Ecological and Analytical Monitoring of Persistent Organic Pollutants), Moscow: Binom. Laboratoriya znanii, 2004.
Vorob’eva, T.V., Terletskaya, A.V., and Kushchevskaya, N.F., J. Water Chem. Technol., 2007, vol. 29, no. 4, p. 203.
Muslimova, I.M., Khizbullin, F.F., and Chernova, L.N., Khim. Tekhnol. Vody, 2000, vol. 22, no. 2, p. 198.
Guidelines for Drinking Water Quality, vol. 2: Health Criteria and Other Supporting Information, 1996.
On the approval of water quality standards for water objects of fishery importance, including standards for maximum permissible concentrations of harmful substances in the waters of water bodies of fishery importance, Order of the Russian Federal Agency for Fisheries no. 20, Moscow, 2010.
GN (Hygienic Standard) 2.1.5.689-98: Maximum Permissible Concentration (MPC) of Chemicals in Water in Water Bodies of Domestic and Drinking and Cultural Water Use, Moscow, 1998.
Pentachlorophenol in Drinking-Water: Background Document for Preparation of WHO Guidelines for Drinking-Water Quality, Geneva: WHO, 2003.
Kawaguchi, M., Ishii, Y., Sakuia, N., Okanouchi, N., Ito, R., Saito, K., and Nakazawa, H., Anal. Chim. Acta, 2005, vol. 533, p. 57.
Micong, J., Xiaohong, C., and Bingxian, P., J. Liq. Chromatogr. Relat. Technol., 2006, vol. 29, p. 1369.
Wei, M.-C. and Jen, J.-F., Chromatographia, 2002, vol. 55, nos. 11–12, p. 701.
Crespin, M.A., Ballesteros, E., Gallego, M., and Valcárcel, M., Chromatographia, 1996, vol. 43, nos. 11–12, p. 633.
Popp, P., J. Chromatogr. A, 2005, vol. 1072, no. 1, p. 37.
Welvaert, K. and Vercammen, J., J. Chromatogr. A, 2005, vol. 1071, nos. 1–2, p. 41.
Fattahi, N., Assadi, Y., Hosseini, M.R., and Jahromi, E.Z., J. Chromatogr. A, 2007, vol. 1157, nos. 1–2, p. 23.
Regueiroa, J., Becerrila, E., Garcia-Jaresa, C., and Llomparta, M., J. Chromatogr. A, 2009, vol. 1216, no. 23, p. 4693.
PND (Environmental Regulatory Document) F 14.1:2:4.225-2006: Quantitative Chemical Analysis of Waters. Method for Measuring the Mass Concentrations of Phenol and pPhenol Derivatives in Drinking, Natural, and Waste Waters by Gas Chromatography, Moscow, 2006.
Method 528. Determination of Phenols in Drinking Water by Solid Phase Extraction and Capillary Column Gas Chromatography/Mass Spectrometry (GC/MS), Cincnnnati, OH: U.S. Environ. Protection Agency, 2000.
PND (Environmental Regulatory Document) F 16.1:2.2:2.3:3.60-09: Procedure for Performing Measurements of the Mass Fractions of Phenol and Phenol Derivatives in Soils, Sediments, Wastewater Sediments, and Production and Consumption Wastes by Chromatography–Mass Spectrometry, Moscow, 2009.
Alzweiri, M., Khanfar, M., and Al-Hiari, Y., Chromatographia, 2015, vol. 78, p. 251.
ACKNOWLEDGMENTS
This work was performed using instrumentation of the “Arktika” Core Facility Center of the Northern (Arctic) Federal University (unique identifier RFMEFI59417X0013) with the financial support of the Ministry of Education and Science of the Russian Federation (state task project no. 4.2518.2017/4.6).The authors express their gratitude to the team of the joint expedition of NAFU and Rosgidromet “Arctic Floating University—2015” for their assistance in sampling sea water.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by E. Rykova
Rights and permissions
About this article

Cite this article
Pokryshkin, S.A., Kosyakov, D.S., Kozhevnikov, A.Y. et al. Highly Sensitive Determination of Chlorophenols in Sea Water by Gas Chromatography−Tandem Mass Spectrometry. J Anal Chem 73, 991–998 (2018). https://doi.org/10.1134/S1061934818100088
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934818100088