
Abstract
Solid-phase extraction (SPE) procedure followed by derivatization and gas chromatography electron capture detection was evaluated for the determination of trace amounts of chlorophenols (CPs) in waters samples. Different parameters affecting extraction efficiency such as, volume of elution solvent, volume and pH of water sample, quantity of sorbent phase were studied and optimized. SPE was carried out on polystyrene–divinylbenzene (Bond Elut ENV) and high recoveries were obtained using 1000 mg of this cartridge for the treatment of 500 mL of acidified water sample. The described method was then tested on spiked tap, mineral, ground and surface water samples. The overall procedure provided limits of detection lower than 20 ng L−1, recoveries of 70 %–106 % and an enrichment factor of 500 for the examined CPs in 500 mL water samples. Among the studied compounds, pentachlorophenol was detected in tap water at a concentration level of 0.06 µg L−1.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Chlorophenols are synthetic organic compounds, obtained on large, industrial and commercial scales by chlorinating phenol or hydrolysing chlorobenzenes (Czaplicka 2004). 19 congeners are possible, ranging from monochlorophenols (MCP) to the fully chlorinated pentachlorophenol (PCP). These compounds have been found in many real samples such as water (wastewater, ocean waters, coastal seawater and natural freshwater), wood and cork, food (fruits, honey, wines and clam tissues) and solid materials (soils, sediments, sludge and ash) (De Moraisa et al. 2012).
The sources of CPs to the environment are related to their widespread use as pesticides, leather or wood impregnation agents and in various industries (De Moraisa et al. 2012). They are present in the aquatic environment, during their production and use, or due to the decomposition of author chemicals products such as chlorinated phenoxy acid herbicides (Davì and Gnudi 1999; Quintana et al. 2007). CPs may also be formed as by reaction with humic substances during disinfection of drinking water by chlorination (Michalowicz and Duda 2007; Quintana et al. 2007).
Since 1984, the use of CPs has been restricted (ATSDR 2013) and five of the chlorinated phenols (2-chlorophenol, 2,4-dichlorophenol, 4-chloro-3-methylphenol, 2,4,6-trichlorophenol, and pentachlorophenol) have been considered as priority pollutants by the U.S. Environmental Protection Agency (1995). The European Community Directive 80/778/EEC states that the maximum admissible concentration of phenolic compounds in drinking water should be 0.5 μg L−1 for the total content and 0.1 μg L−1 for the individual content (Drinking Water Directive 80/778/EEC 1980). In fact, these substances represent serious healths hazards due to their toxicity and carcinogenicity (Goodman 2001; ATSDR 2013) which depends on the degree of chlorination and the position of chlorine atoms relative to the hydroxyl group (Czaplicka 2004; ATSDR 2013). Moreover, they cause earthy-musty off-flavour problems in water for µg L−1 concentration level (Malleret et al. 2003).
Many methods based on chromatographic techniques have been used for the trace-level analysis of chlorophenols in aqueous samples, such as: gas chromatography (GC) in coupling with electron capture detection (ECD), mass spectrometry (MS) and flame ionisation detection (FID) (Jin and Yang 2006; Portillo et al. 2006; Fattahi et al. 2007), and high-performance liquid chromatography (HPLC) associated with ultraviolet detection (UV), diode-array detection (DAD), electrochemical (EC) or mass spectrometry detection (Sarrión et al. 2002; Vidal et al. 2004; Jin and Yang 2006; Saraji and Marzban 2010; Al-Janabi et al. 2011). GC is often preferred, for the analysis of phenol compounds, offering unrivalled high resolution and easy coupling with sensitive and selective detectors (Rodríguez et al. 1996; Bagheri and Saraji 2001).
In spite of recent progress in analytical instrumentation, sample pretreatment steps prior to the chromatographic analysis are usually required to clean up or to enrich the target species. Solid phase extraction has been increasingly applied for the preconcentration and clean-up of aqueous environmental samples, due to its advantages over liquid–liquid extraction. Silica-based C18, graphitized carbon and polymeric sorbents are the most used phases for trapping phenolic compounds, especially for water analysis. It has generally been concluded that styrene–divinylbenzene polymer-based phases provide best overall performance for this application (Lacorte et al. 1999; Rodríguez et al. 2000). More recently, solid-phase microextraction (SPME) coupled to GC has been used for determination of CPs in water (Llompart et al. 2002; Portillo et al. 2006). This process is significantly simpler than conventional techniques, thereby reducing analyte loss during extraction. SPME sensitivity and precision are generally as good as or better than standard methods. Nevertheless, the SPME fiber is usually damaged when analyzing complex matrices.
The main purpose of the present study was to evaluate an analytical procedure, based on SPE for pre-concentration setups and GC–ECD for the detection and quantification of di, tri, tetra, and pentachlorophenol in water samples. For this purpose, styrene–divinylbenzene (SDVB) cartridges have been evaluated under different experimental conditions: (1) volume of elution solvent, (2) pH and volume of the treated sample, (3) amounts of sorbent phase. The performance of this method was finally checked with tap, mineral, ground and surface water collected from the region of Bizerte (North of Tunisia).
Materials and Methods
The chlorophenol standards 3,5-dichlorophenol (3,5-DCP), 2,3-dichlorophenol (2,3-DCP), 2,3,5-trichlorophenol (2,3,5-TCP), 2,4,5-trichlorophenol (2,4,5-TCP), 2,3,5,6-tetrachlorophenol (2,3,5,6-TeCP) and pentachlorophenol (PCP) used in this study were supplied by Supelco (USA) and had a purity of at least 97 %. Stock solutions were prepared for each of the standards in methanol (MeOH) at a concentration level of 2 mg mL−1. Working solutions used in further studies were prepared by diluting different amounts of each stock standard solution with MeOH. Standard and work solutions were stored in a refrigerated environment at 4°C and kept in darkness. The reagents used, acetic anhydride, anhydrous sodium sulfate, sodium hydroxide and potassium carbonate were of analytical reagent grade and obtained from Prolabo (France). Acetone, n-Hexane, methanol, and sulfuric acid were pesticide quality and were purchased from Fluka (Buchs, Switzerland).
SPE cartridges for sample concentration used in this study were Bond Elut Env (styrene-divinylbenzene, 200 and 1000 mg) and were purchased from Varian (USA). Prior to the preconcentration step cartridges were conditioned with MeOH (1 mL for 100 mg cartridges) and activated with distilled water at pH 2. The pH of sample was adjusted to 2 with sulfuric acid. A known volume of distilled, tap, mineral, ground or river water was spiked with CP standards and was subsequently passed through a preconditioned SPE cartridge at a flow-rate of 10–12 mL min−1. In the case of 200 mg cartridges, retained analytes were desorbed with 3 mL of MeOH at the flow-rate of 0.2 mL min−1.
Acetylation was chosen as the derivatization method since it is one of the most efficient, simplest and fastest reactions for chlorophenolic species (Rodríguez et al. 1996). This reaction makes the compounds less polar by replacing the hydroxyl group with an acetate group, avoiding the problems associated with chromatography. The process was carried out at basic pH using acetic anhydride as the derivatization reagent as described by Rodriguez et al. (1996). In brief, one milliliter of a methanol solution containing CPs was mixed with 2 mL of 5 % K2CO3 and 200 µL of acetic anhydride and the mixture was shaken for 1 min. After that, 1 mL of n-hexane was added and mixed for another minute. The organic phase containing acetylated compounds was separated, and the aqueous phase was reextracted with 1 mL of n-hexane. The two n-hexane portions were collected, mixed and dried over anhydrous sodium sulfate. The final extract was concentrated to 1 mL under a gentle stream of nitrogen before injection into the GC–ECD system.
The analysis of CPs was performed on an Agilent Model 6890 Gas Chromatograph equipped with a 63Ni electron capture detector (GC–ECD). Separations were carried out using a PTE-5 capillary column (30 m × 0.32 mm i.d. and 0.32 µm film thickness). Helium was used as carrier gas at a constant flow of 1.5 mL min−1 and N2 as make up gas at a 60 mL min−1 flow rate. Injections (1 µL volume) were performed in the splitless mode. Oven temperature was programmed as follows: 60°C initial (1 min) to 115°C at 15°C min−1 and to 175°C at 3°C min−1 and to 250°C at 30°C min−1 and finally 10 min at 250°C. The temperature of the injector and detector were fixed to 250 and 300°C respectively. A Chemstation data system was used to obtain the chromatogram and perform data calculations. The data presented in this paper were obtained using the PTE-5 column and an SPB™-1701 capillary column (30 m × 0.32 mm i.d., 0.25 µm film thickness) with different polarity was used as second column to confirm the identification of CP compounds in water samples.
Results and Discussions
Figure 1 shows the chromatographic separation on a PTE-5 capillary column of acetylated chlorophenols at 50 µg L−1 when using the conditions already detailed above. chromatographic resolution was higher than 1 for 3,5-DCP, 2,3-DCP, 2,3,5,6-TeCP and PCP but not exceed 1 for 2,3,5-TCP and 2,4,5-TCP (peaks of these two later compounds overlap). Nevertheless, this chromatographic separation is the best one after several changes in the chromatographic conditions such as temperature program and carrier gas flow. In addition, better chromatographic response was performed by varying the initial temperature column (50, 60 and 70°C) and testing different duration of closure of the split less injection valve (0.3, 0.4, 0.5, 0.7 and 1 min). Best results were observed for 50 or 60°C and 1 min respectively. The repeatability evaluated by the relative standard deviations (RSD) of seven consecutive injections originating from a standard solution at 50 µg L−1 concentration level, lay between 3.4 % for 2,3,5,6-TeCP and 5.5 % for 2,4,5-TCP. Calibration curves with excellent linearity were obtained for each CPs in the concentration range of 1 up to 50 µg L−1 (Fig. 2). The regression coefficients obtained were superior to 0.998. Detection limits (LOD), based on a signal-to-noise ratio (S/N) of 3, were 6.6 µg L−1 (3,5-DCP), 6.2 µg L−1 (2,3-DCP), 4 µg L−1 (2,3,5-TCP), 2.6 µg L−1 (2,4,5-TCP), 2.9 µg L−1 (2,3,5,6-TeCP) and 0.5 µg L−1 (PCP).

GC-ECD Chromatogram obtained for the standard solution of CPs at 50 µg L−1. Peaks identification: 1 3,5-DCP; 2 2,3-DCP; 3 2,3,5-TCP; 4 2,4,5-TCP; 5 2,3,5,6-TeCP and 6 PCP

Calibration curves for standard solutions of (2,3-DCP; 3,5-DCP; 2,3,5-TCP; 2,4,5-TCP; 2,3,5,6-TeCP and PCP) in the concentration range 1–50 µg L−1
There are a number of factors that can influence the recovery of CPs during SPE. The first step is to select an appropriate eluent and essentially the volume of this eluent to desorb these compounds from cartridge. Elution of phenols from ENV cartridge is usually performed with methanol, ethanol, acidified ethanol, acetonitrile, methanol–acetonitrile and ethyl acetate. In this study we have selected methanol as elution solvent due to its compatibility with the subsequent acetylation reaction and capability to elute organic compounds from reversed-phase sorbents. To find the required volume of methanol to elute all CPs from the cartridge, a volume of 100 mL of distilled water (pH 2) containing 0.5 µg L−1 of the analytes was analyzed for two quantity of SDVB phase (200 and 1000 mg). Elution volumes up to 12 mL were examined. Table 1 shows the obtained recoveries of selected CPs. In the case of 200 mg of cartridge, we note that with 1 and 2 mL of elution solvent, recoveries were less than 85 %. As soon as the elution volume reaches or exceeds 3 mL, total extraction of the trapped pollutants was achieved. However, this volume becomes not sufficient for 1000 mg of cartridge. As indicate in Table 1, total extraction of the analyzed compounds was achieved using 8 mL of MeOH with the exception of PCP higher volume of elution solvent was needed. These relatively low volumes of MeOH able to elute quantitatively all compounds from the cartridge exclude others solvent from any further examination.
The pH of the sample is a significant factor, which may affect the CPs extraction recovery in water samples. To increase the extraction efficiency, it is necessary to acidify the sample prior the SPE preconcentration (Puig and Barceló 1995; Bagheri and Saraji 2001). When the pH is low, the acid–base equilibrium for the acidic phenols shifts significantly toward the neutral forms which have greater affinities to the sorbent. The pH effect on the CPs extraction from water samples was studied within the range of 2–6 using sulfuric acid (H2SO4) and sodium hydroxide (NaOH). The recoveries of the CPs were obtained by treating 100 mL of distilled water spiked with a mixture of studied CPs at 0.5 μg L−1 and analyzed according the complete procedure. As indicated in Table 2, for pH 2 and 3, extraction recoveries exceed 95 % for all studied compounds. When pH of sample increase to 4 a loss of about 20 % was observed for PCP. For pH greater than 4 a significant decrease in the extraction efficiency was observed for both PCP and 2,3,5,6-TeCP compounds. However a slightly decrease in the extraction recoveries was observed on the pH in the range 2–6 for the others compounds. This can be explained by the higher acidity of PCP and 2,3,5,6-TeCP compared to other CPs. Thus, for the rest of this work pH of the sample is maintained equal to 2.
In order to determine the volume of the sample that can be concentrated in 200 mg of sorbent with the acceptable levels of recoveries, different volumes (100, 200, 500, and 1000 mL) of distilled water at pH 2 were spiked with a solution containing phenolic compounds at the 0.5; 0.25; 0.1; and 0.05 µg L−1 level (containing the same quantity of analyte). The trapped compounds on the SPE cartridge were, then, eluted with 3 mL of MeOH; followed by derivatization and extraction with a total of 2 mL of n-hexane and gentle evaporation to 1 mL, an aliquot of 1 µL was injected into the GC system. The recoveries of phenolic compounds and the repeatability for the different volumes are given in Table 3. Good recoveries were obtained for all studied compounds using 100 or 200 mL sample volumes. Recuperation is slightly lower for 500 mL of water. However, when the volume of the sample reaches 1 L the losses of analyte exceed 28 % with the exception of 2,3,5,6-TeCP and PCP. For these two later compounds the recovery is higher than 95 %. Therefore, the breakthrough volume using 200 mg of cartridge is about 500 mL for CPs having a chlorination degree below than three.
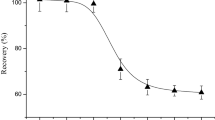
In order to improve the enrichment factor achieved in the SPE step, a higher amount of Bond Elut Env sorbent was considered in further experiments. Distilled water samples of 500 and 1000 mL spiked with 0.05 µg of both studied compounds, were treated using 1000 mg of cartridge (elution of chlorophenols were achieved with 12 mL of MeOH). As illustrate in Fig. 3, an increasing in the amount of sorbent improve the extraction efficiency for both treated volume. Recoveries become higher than 95 % when treating 500 mL of water sample (excepted 3,5-DCP for this later extraction recoveries are less than 90 %) and higher than 70 % when treating 1000 mL of water sample. As this SPE procedure will be applied to the analyses of environmental water samples which are more charged in organic compounds than distilled water, a 1000 mg of sorbent phase and a volume of 500 mL were, therefore, selected as optimized parameters.

Variation in the recovery (%) of selected CPs extracted by the Bond Elut Env (200 and 1000 mg) from a 500 mL and b 1000 mL of distilled water spiked at 0.1 and 0.05 μg L−1 respectively. Asterisk Mean value of three determinations
The efficiency of the presented method was evaluated by determining the CPs concentration in tap, mineral, ground and river water samples. River water was collected from Joumine River (Bizerte governorate), while tap and ground water samples were collected from Bizerte city (Tunisia). The samples were analyzed by GC–ECD, after the performance of the SPE procedure. 500 mL of each sample was acidified to pH 2 with H2SO4, spiked with the CPs standard solutions at 0.1 µg L−1 levels and treated with 1000 mg of Bond Elut Env cartridges to assess the matrix effects. A decrease in recovery efficiencies was observed for all studied compounds from different water samples compared with distilled water. In fact the organic substances such as humic and fulvic substances present in natural water particularly river water are coextracted with CPs through SDVB phase as a result a decrease in recovery efficiencies. Nevertheless, extraction recoveries were in the range of 70 %–106 % for all treated water samples and this shows that humic and fulvic acids have little effect on the present method. Moreover, chromatograms from the river water and the spiked river water samples at the concentration level of 0.1 µg L−1 do not show any interfering peaks with studied compounds (Fig. 4) as also observed for mineral, tap and ground waters. Among the studied compounds, trace levels of PCP were found in tap water at a concentration ± standard deviation (SD) of 0.06 ± 0.0025 µg L−1 (Fig. 5).

GC-ECD chromatograms obtained after SPE of a 500 mL river water and b 500 mL river water spiked with selected CPs at 0.1 μg L−1: 1 3,5-DCP; 2 2,3-DCP; 3 2,3,5-TCP; 4 2,4,5-TCP; 5 2,3,5,6-TeCP and 6 PCP

GC-ECD chromatograms of a unspiked and b spiked (0.1 μg L-1) tap water samples (500 mL) with selected CPs: 1 3,5-DCP; 2 2,3-DCP; 3 2,3,5-TCP; 4 2,4,5-TCP; 5 2,3,5,6-TeCP and 6 PCP
This work describes simple and robust analytical protocols to determine chlorophenols from water samples. An off-line SPE with Bond Elut ENV cartridges was optimized for the preconcentration step and gas chromatography with electron capture detector (GC–ECD) was performed for identification and quantification of studied compounds. Chromatographic results show that GC–ECD using capillary column in moderately polar phase allowed the separation of six acetylated CPs with acceptable chromatographic resolution. The regression coefficients of the calibration curves were superior to 0.998 across a concentration range of 1–50 µg L−1, representing good linearity of the method. The optimized extraction step uses minimal amounts of methanol 3 and 12 mL for the quantitative elution of CPs retained in 200 and 1000 mg of cartridges respectively. The pH of the sample was optimised in the range of 2–6 and best extraction recoveries were observed for pH less than 4. The estimated breakthrough volume using 200 mg of cartridge is about 500 mL for the compounds having less than three chlorine atoms. The extraction efficiencies was improved using 1000 mg of cartridge and up to 1000 mL of sample can be treated with acceptable recoveries for all studied compounds. The final SPE method yielded recoveries of 70 %–106 % for all treated water samples with RSD less than 12 %. Preconcentrating 500 mL of sample, method detection limits were lower than 20 ng L−1. The method has been successfully applied to tap, mineral, ground and river water samples. From the analyzed compounds pentachlorophenol was detected at nanogram level in tap water samples.
References
Al-Janabi KWS, Alazawi FN, Mohammed MI, Kadhum AAH, Mohamad AB (2011) Chlorophenols in Tigris River and drinking water of Baghdad, Iraq. Bull Environ Contam Toxicol 87:106–112. doi:10.1007/s00128-011-0315-y
ATSDR (2013) Agency for toxic substances and disease registry, addendum for chlorophenols. Supplement to the 1999 toxicological profile for chlorophenols, Atlanta, GA 30333. http://www.atsdr.cdc.gov/toxprofiles/chlorophenols_addendum.pdf. Accessed in 08/12/2014
Bagheri H, Saraji M (2001) New polymeric sorbent for the solid-phase extraction of chlorophenols from water samples followed by gas chromatography–electron-capture detection. J Chromatogr A 910:87–93
Czaplicka M (2004) Sources and transformations of chlorophenols in the natural environment. Sci Total Environ 322:21–39
Davì ML, Gnudi F (1999) Phenolic compounds in surface water. Wat Res 33:3213–3219
De Moraisa P, Stoichev T, Basto MC, Vasconcelos MT (2012) Extraction and preconcentration techniques for chromatographic determination of chlorophenols in environmental and food samples. Talanta 89:1–11
Drinking Water Directive 80/778/EEC (1980) Commission of the European Communities. Brussels
EPA Method 8041 (1995) Phenols by gas chromatography: capillary column technique. US Environmental Protection Agency, Washington, pp 1–28
Fattahi N, Samadi S, Assadi Y, Hosseini MRM (2007) Solid-phase extraction combined with dispersive liquid–liquid microextraction-ultra preconcentration of chlorophenols in aqueous samples. J Chromatogr A 1169:63–69
Goodman G (2001) Pentachlorophenol. In: Krieger RI (ed) Handbook of pesticide toxicology, vol 2, 2nd edn., AgentsAcademic Press, San Diego, pp 1481–1509
Jin MC, Yang YW (2006) Simultaneous determination of nine trace mono- and di-chlorophenols in water by ion chromatography atmospheric pressure chemical ionization mass spectrometry. Anal Chim Acta 566:193–199
Lacorte S, Fraisse D, Barceló D (1999) Efficient solid-phase extraction procedures for trace enrichment of priority phenols from industrial effluents with high total organic carbon content. J Chromatogr A 857:97–106
Llompart M, Lourido M, Landín P, García-Jares C, Cela R (2002) Optimization of a derivatization–solid-phase microextraction method for the analysis of thirty phenolic pollutants in water samples. J Chromatogr A 963:137–148
Malleret L, Dugay J, Bruchet A, Hennion MC (2003) Simultaneous determination of “earthy-musty” odorous haloanisoles and their corresponding halophenols in water samples using solid-phase microextraction coupled to gas chromatography with electron-capture detection. J Chromatogr A 999:135–144
Michalowicz J, Duda W (2007) Phenols—sources and toxicity. Polish J Environ Stud 16:347–362
Portillo M, Prohibas N, Salvadó V, Simonet BM (2006) Vial position in the determination of chlorophenols in water by solid phase microextraction. J Chromatogr A 1103:29–34
Puig D, Barceló D (1995) Off-line and on-line solid-phase extraction followed by liquid chromatography for the determination of priority phenols in natural waters. Chromatographia 40:435–444
Quintana JB, Rodil R, Muniategui-Lorenzo S, López-Mahía P, Prada-Rodríguez D (2007) Multiresidue analysis of acidic and polar organic contaminants in water samples by stir-bar sorptive extraction–liquid desorption–gas chromatography–mass spectrometry. J Chromatogr A 1174:27–39
Rodríguez I, Turnes MI, Mejuto MC, Cela R (1996) Determination of chlorophenols at the sub-ppb level in tap water using derivatization, solid-phase extraction and gas chromatography with plasma atomic emission detection. J Chromatogr A 721:297–304
Rodríguez I, Llompart MP, Cela R (2000) Solid-phase extraction of phenols. J Chromatogr A 885:291–304
Saraji M, Marzban M (2010) Determination of 11 priority pollutant phenols in wastewater using dispersive liquid-liquid microextraction followed by high-performance liquid chromatography-diode-array detection. Anal Bioanal Chem 396:2685–2693. doi:10.1007/s00216-010-3496-z
Sarrión MN, Santos FJ, Galceran MT (2002) Determination of chlorophenols by solid-phase microextraction and liquid chromatography with electrochemical detection. J Chromatogr A 947:155–165
Vidal JLM, Vega AB, Frenich AG, Gonzalez FJE, Liebanas FJA (2004) Determination of fifteen priority phenolic compounds in environmental samples from Andalusia (Spain) by liquid chromatography–mass spectrometry. Anal Bioanal Chem 379:125–130. doi:10.1007/s00216-003-2459-z
Acknowledgments
We gratefully acknowledge the financial support of the Tunisian Ministry of Higher Education, Scientific Research and Technology.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
This manuscript has not been previously published and it is not under consideration by any other journal. All authors are aware of, and accept responsibility for, the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ben Hassine, S., Hammami, B., Touil, S. et al. Determination of Chlorophenols in Water Samples Using Solid-Phase Extraction Enrichment Procedure and Gas Chromatography Analysis. Bull Environ Contam Toxicol 95, 654–660 (2015). https://doi.org/10.1007/s00128-015-1570-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00128-015-1570-0